

Summary
Eukaryotic DNA exists in chromatin, where the genomic DNA is packaged into a fundamental repeating unit known as the nucleosome. In this chromatin environment, our genomic DNA is constantly under attack by exogenous and endogenous stressors that can lead to DNA damage. Importantly, this DNA damage must be repaired to prevent the accumulation of mutations and ensure normal cellular function. To date, most in depth biochemical studies of DNA repair proteins have been performed in the context of free duplex DNA. However, chromatin can serve as a barrier that DNA repair enzymes must navigate in order find, access, and process DNA damage in the cell. To facilitate future studies of DNA repair in chromatin, we describe a protocol for generating nucleosome containing site-specific DNA damage that can be utilized for a variety of in vitro applications. This protocol describes several key steps including how to generate damaged DNA oligonucleotides, the expression and purification of recombinant histones, the refolding of histone complexes, and the reconstitution of nucleosomes containing site-specific DNA damage. These methods will enable researchers to generate nucleosomes containing site-specific DNA damage for extensive biochemical and structural studies of DNA repair in the nucleosome.
Keywords: DNA damage, base excision repair, nucleosomes, chromatin
1. Introduction
DNA is constantly exposed to endogenous and exogenous stressors that lead to the accumulation of DNA damage. Oxidative stress is a primary source of DNA damage that contributes approximately 104 lesions per cell, per day [1]. Well over 100 different forms of oxidative DNA base lesions exist including the prevalent examples of 8-oxo-7,8-dihydro-2’-deoxyguanosine (8oxoG), 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG), 8-oxox-7,8-dihydro-2’-deoxyadenosine (8oxoA), thymine glycol, and 5-hydroxy-2’-deoxycytidine (OH5C). In general, oxidative damage results in changes to the structure of the DNA base. These alterations increase the likelihood that the damaged base will form non-canonical base pairing that can result in the accumulation of genomic transversions and/or transitions [2,3]. Ultimately, these oxidative DNA lesions must be repaired to prevent the accumulation of mutations that can lead to genomic instability and several different disease states [4,5].
The base excision repair (BER) pathway is responsible for the repair of most oxidative DNA base damage, requiring efficient recognition of oxidative DNA damage [6,7]. We direct readers to other excellent reviews of BER for more details about the pathway [8–10]. Briefly, BER is initiated by one of several different DNA glycosylases, which recognize damaged DNA bases [7,11]. The DNA glycosylase then excises the damaged DNA base resulting in the formation of a baseless sugar moiety known as an apurinic/apyrimidinic site (AP-site). The resulting AP-site is then processed by AP Endonuclease 1 (APE1), resulting in a 5’-nick in the DNA backbone. The 5’-nicked substrate is then processed by two distinct enzymatic activities of Polymerase (Pol) β. The lyase activity of DNA Pol β removes the 5’-nick generating a one nucleotide gapped DNA substrate. Pol β then utilizes its DNA synthesis activity to fill the gap with a non-damaged nucleotide, generating a 3’-nick. After incorporation of the non-damaged nucleotide, the XRCC1-DNA ligase III complex seals the 3’-nick, ultimately restoring the coding potential of the DNA.
Mechanistic insight into the function of BER enzymes has largely come from in vitro studies using free duplex DNA, see reference [12] and associated references. However, cellular DNA often exists as chromatin, where genomic DNA is packaged into a fundamental repeating unit known as the nucleosome [13,14]. The nucleosome is composed of an octameric core of histone proteins: H2A, H2B, H3 and H4, that wraps ~147bp of DNA [15]. The robust interaction between histone proteins and the nucleosomal DNA can act as a significant barrier to BER proteins, which must be overcome to effectively complete repair of the damaged DNA. We direct the readers to several excellent reviews for further information on how nucleosome structure regulates BER enzymes [16–22]. Over the past twenty years, initial progress has been made to define how BER proteins repair DNA damage in the context of the nucleosome [23–66]. However, rapid progress has been hampered by the lack of comprehensive step-by-step protocols for generating nucleosomes containing site-specific DNA damage.
To overcome this challenge, we present a step-by-step protocol for the reconstitution of recombinant NCPs containing site-specific DNA damage. This protocol was adapted from the landmark method from Dyer and Luger et al. described over 15 years ago, as well as the many labs who have made significant progress on methods for generating damaged DNA substrates for the in vitro reconstitution of nucleosomes [67,65,68,64,39,69,43,70]. Initially, we describe a ligation-based method for generating a 147bp Widom 601 strong positioning DNA substrate containing site-specific DNA damage [71]. We then describe the expression and purification of human histone proteins from E. coli and the salt-dialysis method for refolding recombinant histone proteins into histone H2A/H2B dimers and histone H3/H4 tetramers. Finally, we describe a salt-dialysis method for reconstituting NCPs using the Widom 601 strong positioning DNA containing site-specific DNA damage, and how to purify these NCP substrates for downstream in vitro biochemical or structural biology experiments. Importantly, the utility of this step-by-step protocol is highlighted by recent work from our lab using this method to generate NCPs containing THF, enabling us to determine the structural basis for APE1 processing DNA damage in the NCP [72].
2. Materials
2.1. Instruments and equipment
PCR Thermal cycler
Thermo-Fisher Owl A-series horizontal gel system or equivalent
Bioreactor or Shaking incubator
QSonica Q500 Sonic dismembrator or equivalent
Stir plate
Gravity flow column with filter unit
3,000 Da MWCO Dialysis Membrane
10,000 Da MWCO Centrifugal Filter Unit
AKTA Pure protein purification system or equivalent
Superdex 200 Increase 10/300 GL or equivalent
Biocomp Gradient Master 108 or equivalent
Beckman Coulter 38.5 mL Open-Top Ultra Centrifuge tube or equivalent
SW 32 Ti Swinging-Bucket Rotor or equivalent
Optima XE-100 Ultracentrifuge or equivalent.
2.2. Reagents
Project specific oligonucleotides (design described in section 3.1.1)
Agarose
New England Biolabs T4 DNA ligase or equivalent
New England Biolabs 10X T4 DNA ligase buffer or equivalent
New England Biolabs 6x gel loading dye or equivalent
Ethidium bromide or equivalent DNA visualizing reagent
pET3a-histone H2A plasmid
pET3a-histone H2B plasmid
pET3a-histone H3 plasmid
pET3a-histone H4 plasmid
New England Biolabs BL21 (DE3) pLysS competent cells or equivalent
Novagen Rosetta™ 2(DE3) pLysS competent cells or equivalent
Luria broth (LB) - Casein Digest Peptone 10g/L, Sodium Chloride 10g/L, Yeast Extract 5g/L
LB agar plates containing 100 μg/ml ampicillin
LB agar plates containing 100 μg/ml ampicillin and 25 μg/ml chloramphenicol
Centrum multivitamin or equivalent, 1 tablet dissolved in 50 mL ddH2O
Triton X-100
Dimethyl sulfoxide (DMSO)
Cytiva Q-sepharose Fast Flow anion exchange chromatography resin or equivalent
Cytiva SP-sepharose Fast Flow cation exchange chromatography resin or equivalent
2.3. Buffers, Solutions, and Media
1x TS: 10mM Tris (pH-7.5) and 10mM NaCl
1x TBE: 90mM Tris-base, 90mM Boric Acid, and 2mM EDTA (pH-8.3)
10% denaturing Urea-PAGE gel solution: 10% 29:1 acrylamide:bis-acrylamide, 8M Urea, and 1X TBE
1x TE: 10 mM Tris (pH-7.5) and 1 mM EDTA
Gel extraction buffer: 200mM NaCl, 1mM EDTA
10x M9 Minimal media pH (7.2): 335 mM Na2HPO4•7H2O, 220 mM KH2PO4, 10 mM NaCl, 20mM NH4Cl, supplemented with 0.5% glucose, 2 mM MgSO4, 0.2 mM CaCl2, and 1.0% vitamin solution. (See Note 1).
20% Glucose solution (w/v)
1 M MgSO4
1 M CaCl2
100 mg/mL Ampicillin
25 mg/mL Chloramphenicol
1M Isopropyl β- d-1-thiogalactopyranoside (IPTG)
Histone lysis buffer: 50 mM Tris pH 7.5, 100 mM NaCl, 1 mM benzamidine, 5 mM beta mercaptoethanol, 1 mM EDTA
Guanidinium buffer: 20 mM Tris pH 7.5, 6 M Guanidinium hydrochloride, 10 mM DTT
8 M urea
8 M urea, 100 mM NaCl
8 M urea, 200 mM NaCl
8 M urea, 300 mM NaCl
8 M urea, 400 mM NaCl
8 M urea, 500 mM NaCl
8 M urea, 600 mM NaCl
8 M urea, 700 mM NaCl
High-salt reconstitution buffer: 20 mM Tris-HCL pH 7.5, 2 M NaCl, 1 mM EDTA, 0.5 mM benzamidine, 1mM DTT (added fresh).
No-salt reconstitution buffer: 20 mM Tris-HCL pH 7.5, 1 mM EDTA, 0.5 mM benzamidine, 1mM DTT (added fresh).
10% sucrose in 1X TE
40% sucrose in 1X TE
5% Native PAGE gel: 5% 59:1 acrylamide:bis-acrylamide and 0.2X TBE
3. Methods
3.1. Generation and purification of damaged DNA substrates
Several methods exist for incorporating site-specific DNA damage into the 601 strong positioning DNA sequence. These methods include oligonucleotide synthesis strategies [57,24,64,43], PCR amplification [65,37], nickase-based methods [39,40,73], and ligation-based methods [74,75,70,66]. We point the readers to an excellent and comprehensive review on the different methods for generating site-specific DNA damage for nucleosome reconstitution [76]. Here, we describe a T4-DNA ligase-based method for site-specific incorporation of the apurinic/apyrimidinic site analogue tetrahydrofuran (THF) into the Widom 601 strong positioning DNA sequence containing a 6-Carboxyfluorescein (6-FAM) label [71]. This modular methodology can be performed at large scales in a cost-effective manner, ultimately providing an optimal way of generating damaged DNA for nucleosome reconstitutions (for additional benefits of the T4 DNA ligase-based system see section 3.1.1).
3.1.1. Design of damaged DNA substrates
The T4 DNA ligase-based method described here allows for the site-specific incorporation of DNA damage within a 147 bp Widom 601 strong positioning DNA sequence. The first step is to design oligonucleotide (oligo) fragments that correspond to each strand of the 601 DNA sequence. A diagram of the oligo design can be found in Fig. 1a. For simplicity, we refer to these two DNA strands as the J-strand (non-damaged strand) and the I-strand (damaged strand, see note 2), which is modeled after the nomenclature used from the first crystal structure of a nucleosome containing the Widom 601 sequence [77]. The J-strand is split into two oligonucleotides that consist of 74 bp (J-strand oligo 1) and 73 bp (J-strand oligo 2) of DNA. The I-strand is then split into three oligonucleotides that consist of 50 bp (I-strand oligo 1), 50 bp (I-strand oligo 2), and 47 bp (I-strand oligo 3) of DNA. The length of these component oligos are kept under 100 bps to enable easy access to commonly used oligo synthesis companies, such as Integrated DNA Technologies. The sequences for each of the 5 oligos are as follows:
-
J-strand oligo 1 –
5′ATCGGATGTATATATCTGACACGTGCCTGGAGACTAGGGAGTAATCCCCTTGGCGGTTAAAACGCGGGGGACAG3′
-
J-strand oligo 2 –
5′/Phos/CGCGTACGTGCGTTTAAGCGGTGCTAGAGCTGTCTACGACCAATTGAGCGGCCTCGGCACCGGGATTCTCGAT3′
-
I-strand oligo 1 –
5′[FAM]ATCGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTC3′
-
I-strand oligo 2 –
5′/Phos/TAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCA3′
-
I-strand oligo 3 –
5′/Phos/AGGGGATTACTCCCTAGTCTCCAGGCACGTGTCAGATATAT/THF/CATCCGAT3′
Fig 1.
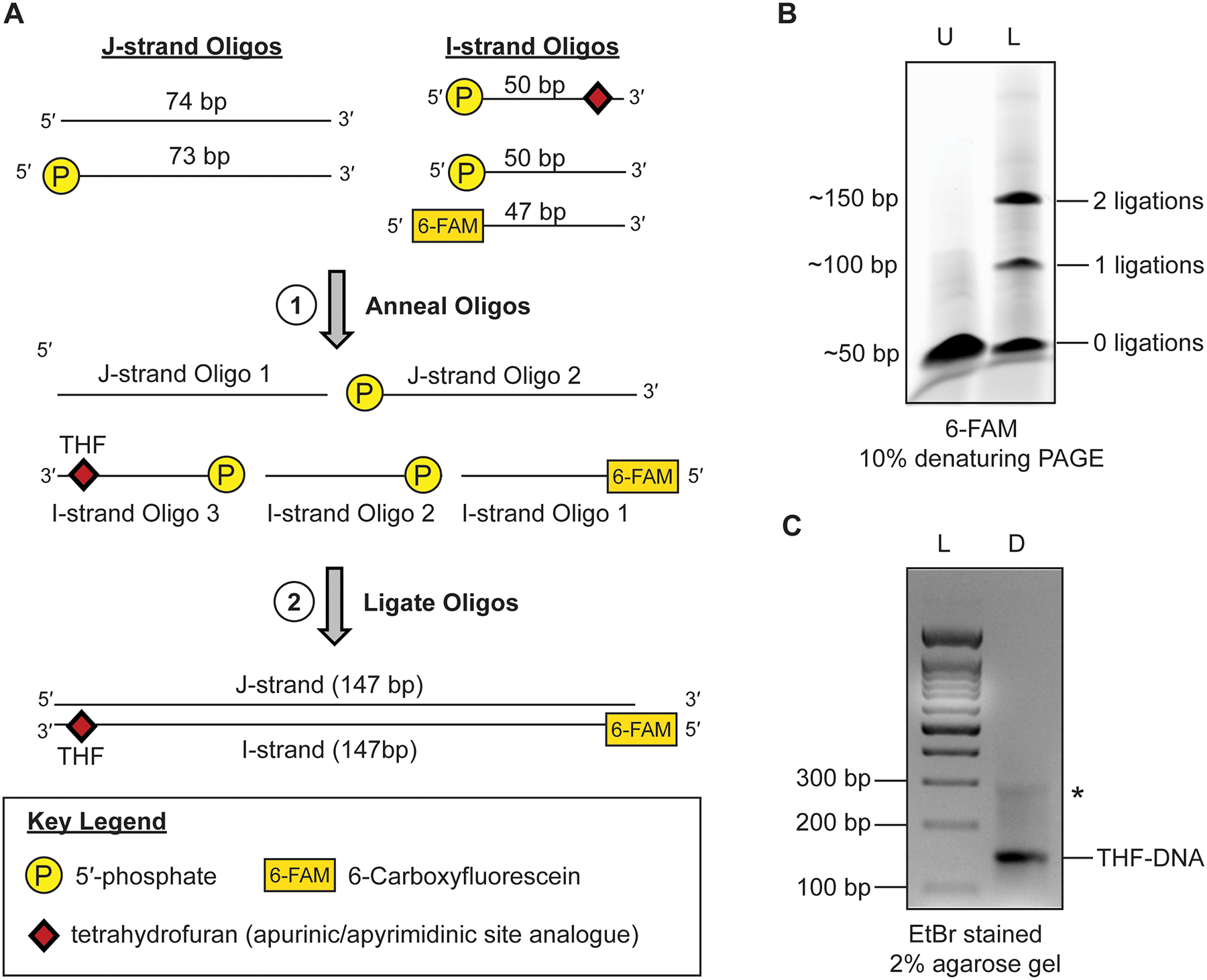
Generation and purification of damaged DNA substrates using the ligation-based method. (a) Diagram for the individual oligos generation of damaged DNA substrates. (b) A representative 10% denaturing PAGE gel for the ligation reaction of. Lane ‘U’ is the starting unligated reaction and lane ‘L’ is the ligated reaction. The DNA weas visualized using the 6-FAM label. (c) A representative 2% agarose gel of the purified and annealed damaged DNA substrate. Lane ‘L’ is a 100bp DNA ladder and lane ‘D’ is the purified and annealed damaged DNA. The DNA was visualized with ethidium bromide. The * denotes a minor contamination that is commonly seen after purification of the ligated DNA.
The oligo design is such that each oligo has at least 25 bp of complimentary DNA between every J- and I-strand DNA oligo. Importantly, J-strand oligo 2, I-strand oligo 2, and I-strand oligo 3 also all contain 5′-phosphate groups at the end of the DNA that is required for ligation (Fig. 1a). When mixed at equimolar ratios and annealed, the resulting 147bp DNA sequence contains three ligation spots with a 5′-phosphate juxtaposed to a 3′-OH. The three nicks can then be sealed by T4 DNA ligase generating the full-length 147 bp DNA sequence containing DNA damage. In the example shown in Fig. 1A, the THF is the 9th position from the end of the DNA on the I-strand oligo 3. However, this ligation-based method is built to enable movement of the THF to any location on the I-strand or J-strand of the DNA, with only minor exceptions (see note 3).
The benefits of using the T4-DNA ligase-based approach are numerous. First, the relatively small oligo size means purchasing numerous oligos containing DNA damage is cost-effective, which is not the case with full-length 147 bp DNA oligos containing DNA damage. Next, the ligation reactions and purification scheme can be performed in large-scale batches enabling the generation of large quantities of damaged DNA. Next, the modularity of the system means that moving the DNA damage to different locations in the 601 DNA sequence requires simply ordering a single additional oligo. Finally, the modularity also allows for the incorporation of various fluorescent tags, protein-attachment modifications, and additional DNA damage types. This ultimately means that nucleosome substrates containing DNA damage can be generated for a variety of BER substrates and different in vitro experimental techniques.
3.1.2. Ligation of damaged DNA substrates
Dissolve oligos with 1x TS to a final concentration of 1 nmol/μL.
Mix the oligos in a 1:1 stoichiometry at a concentration of 1 nmol/μL (see note 4).
Anneal the oligos using a thermal cycler by heating to 95 °C before cooling to 4 °C at a rate of 0.1 °C/sec.
Dilute the annealed oligos to a final volume of 190 μL with H2O and 10X T4 DNA ligase buffer (NEB).
Ligate the annealed DNA oligos by the addition of 10 μL of T4 DNA ligase (NEB) for a final reaction volume of 200 uL.
Incubate the ligation reactions at 25 °C for 24–48 hours.
Run a 10% denaturing Urea-PAGE gel (10% 29:1 acrylamide:bis acrylamide, 8M Urea, and 1x TBE) to monitor the progress of the ligation reaction. (Fig. 1b, see note 5).
3.1.3. Purification of damaged DNA substrates
Pour a 10% denaturing Urea-PAGE gel (10% 29:1 acrylamide:bis-acrylamide, 8M Urea, and 1x TBE).
Load the denaturing Urea-PAGE gel with equal volume of ligated DNA mixed with 6x gel loading dye.
Run the denaturing Urea-PAGE gel at 125 V for 1 hour.
Visualize the ligated DNA via the fluorescent tag, or by staining with ethidium bromide. An example gel of the ligation reaction before purification can be found in Fig. 1b.
Excise the ligated 147bp DNA from denaturing Urea-PAGE gel and cut the excised fragments into small pieces.
Extract the ligated 147bp DNA from the gel fragments by soaking in 20 mL of DNA extraction buffer for 24 hours. Repeat the extraction at least twice to ensure collection of all the damaged DNA.
Buffer exchange the extracted DNA with 1X TE at least 5 times using a centrifugal filter unit (10,000 Da MWCO).
Concentrate the extracted DNA in the centrifugal filter unit (10,000 Da MWCO) to a final concentration of 10 μM (see note 6)
3.1.4. Annealing and storage of damaged DNA substrates
Anneal the purified 147bp oligos using a thermal cycler by heating to 95 °C before cooling to 4 °C at a rate of 0.1 °C/sec.
Determine the concentration of resuspended 147 bp DNA spectroscopically using the theoretical extinction coefficient for the respective 147 bp DNA sequence. Typical yields are ~10–15% of starting material.
Aliquot the 147 bp damaged DNA in 5 nmol aliquots and store frozen at −20 °C indefinitely (see note 7 and 8). An example of a purified 147bp damaged DNA can be found in Fig. 1c.
3.2. Histone expression and purification
Generating recombinant nucleosomes requires milligrams quantities of histone H2A, H2B, H3 and H4 proteins. To generate these quantities, recombinant histones are expressed in E. coli and purified extensively using a protocol modified from Dyer and Luger et, al. [67]. The expression, purification, and long-term storage process for each individual histone is similar, with only minor differences highlighted below.
3.2.1. Histone Plasmid Generation
The inserts corresponding to the coding region of human histone H2A (UniProt identifier: P0C0S8), H2B (UniProt identifier: P62807), H3 C110A (UniProt identifier Q71DI3), and H4 (Uniprot identifier: P62805) genes were cloned into a tagless pet3a expression vector.
3.2.2. Histone Expression
Perform a fresh transformation using NEB BL21 (DE3) pLysS competent cells for histone H2A, H3, and H4 growths. For histone H2B, perform a fresh transformation using Novagen Rosetta™ 2 (DE3) pLysS competent cells. Perform the transformation following the manufacturers protocol for the respective cells.
- Plate the transformation on an LB agar plate with antibiotic specific for that histone.
- H2A – 100 μg/ml ampicillin
- H2B – 100 μg/ml ampicillin 25 μg/ml chloramphenicol
- H3 – 100 μg/ml ampicillin
- H4 – 100 μg/ml ampicillin
Take a streak of colonies from the transformation and inoculate 35mL of LB Broth containing antibiotics (50 ug/mL ampicillin and/or 12.5 ug/mL chloramphenicol).
Grow the inoculated culture in a shaking incubator at 37°C until turbid, which generally takes ~3–4 hours.
Inoculate 100 mL of M9 minimal media culture containing antibiotics (50 ug/mL ampicillin and/or 12.5 ug/mL chloramphenicol) with 5 mL of the turbid culture. We typically inoculate a single 100 mL M9 minimal media culture per 5 bioreactor bottles
Grow the inoculated culture in a shaking incubator at 37 °C for 12–16 hours
Inoculate each 2 L bioreactor bottle containing 1.5 L of M9 minimal media and antibiotics (50 ug/mL ampicillin and/or 12.5 ug/mL chloramphenicol) with 15 mL of overnight culture that was inoculated and grown in steps 5 & 6.
Grow the large, inoculated cultures in a bioreactor at 37°C to an OD600=0.4
- Induce expression of the histone with the following IPTG (from a 1M IPTG stock) concentrations and induction duration:
- H2A – 0.4 mM IPTG for 4 hours
- H2B – 0.4 mM IPTG for 4 hours
- H3 – 0.4 mM IPTG for 3 hours
- H4 – 0.2 mM IPTG for 3 hours
Harvest cells by centrifugation 1,500 RCF at 25 °C in a swinging bucket rotor.
Resuspend each histone pellet with histone lysis buffer (15 mL per 1.5 L culture), and store frozen at −80 °C.
3.2.3. Histone Lysis and Extraction from Inclusion Bodies
Completely thaw the resuspended histone pellet at room temperature. This generally takes approximately one hour.
Dilute the resuspended cells with histone lysis buffer to a final volume of 160 mL.
Lyse the resuspended cells via sonication (QSonica Q500) on ice for three rounds of 10 seconds on and 50 seconds off (amplitude=90%).
Repeat the sonication procedure three times.
Transfer the 160 mL of cell lysate to 4 conical tubes.
Clear the cell lysate via high-speed centrifugation using a fixed angle rotor at 24,000 xG for 20 minutes at 25 °C.
Discard the supernatant and keep the pellet.
Resuspend each pellet with 25 mL of histone lysis buffer supplemented with 1% Triton X-100.
Centrifuge the resuspended pellet in a fixed angle rotor at 24,000 xG for 20 minutes at 25 °C.
Repeat step 8 and step 9.
Discard the supernatant and resuspend each pellet in 25 mL of histone lysis buffer without Triton X-100.
Centrifuge the resuspended pellet in a fixed angle rotor at 24,000 xG for 20 minutes at 25 °C.
Discard the supernatant and consolidate each pellet into a beaker.
Add 1 mL of DMSO to the pellet and break the pellet up using a spatula.
Add a small stir bar to the beaker and stir the pellet for 30 minutes using a stir plate at 25 °C.
Add 30 mL of guanidinium buffer dropwise (in a continuous manner) to the pellet and DMSO mixture using a pasteur pipette.
Extract the histones from the pellet by stirring vigorously for 1 hour at 25 °C.
Centrifuge the solution containing extracted histones at 24,000 xG for 20 minutes at 25 °C.
Save the supernatant.
Perform a second round of histone extraction by repeating steps 14 through 19.
Combine the supernatant containing the extracted histones and dialyze against 8 M urea overnight (see note 9 and 10).
3.2.4. Histone Purification
Equilibrate 10 mL Q-sepharose Resin with 8M Urea.
Mix the supernatant containing the extracted histones with the equilibrated Q-sepharose resin (see note 11).
Incubate stirring for 30 minutes at 25 °C.
Add the slurry containing the extracted histones and Q-sepharose resin to a gravity-flow column.
Collect and save the Q-sepharose column flow-through containing the histone.
Wash the Q-sepharose resin with 40 mL of 8 M Urea and combine the wash with the flow-through from step 5.
Equilibrate 25 mL S-sepharose resin with 8 M Urea.
Mix the Q-sepharose flow-through and wash containing the histone with 25 mL of S-sepharose resin.
Add the slurry containing the extracted histones and S-sepharose resin to a gravity-flow column.
Wash the S-sepharose resin with 40 mL of 8 M Urea.
- Elute the histone from the S-sepharose resin by addition of 50 mL of 8 M Urea containing increasing concentrations of NaCl. Collect each NaCl elution.
- 8 M urea, 100 mM NaCl
- 8 M urea, 200 mM NaCl
- 8 M urea, 300 mM NaCl
- 8 M urea, 400 mM NaCl
- 8 M urea, 500 mM NaCl
- 8 M urea, 600 mM NaCl
- 8 M urea, 700 mM NaCl
Run a sample of each NaCl elution on a 12% SDS-PAGE gel with a protein ladder.
Pool fractions containing histone protein based on the molecular weight of each histone. A representative gel for the purification of each individual histone is shown in Fig. 2.
Pool the purified histone fractions and dialyze against 4 L of water. Exchange the water for fresh water 5 times, with at least two of those exchanges lasting overnight.
Aliquot into the purified histone fractions into 50 mL conical tubes and flash freeze using liquid nitrogen.
Lyophilize the frozen, purified histone.
Aliquot the lyophilized histone and store at −20°C.
Fig 2.
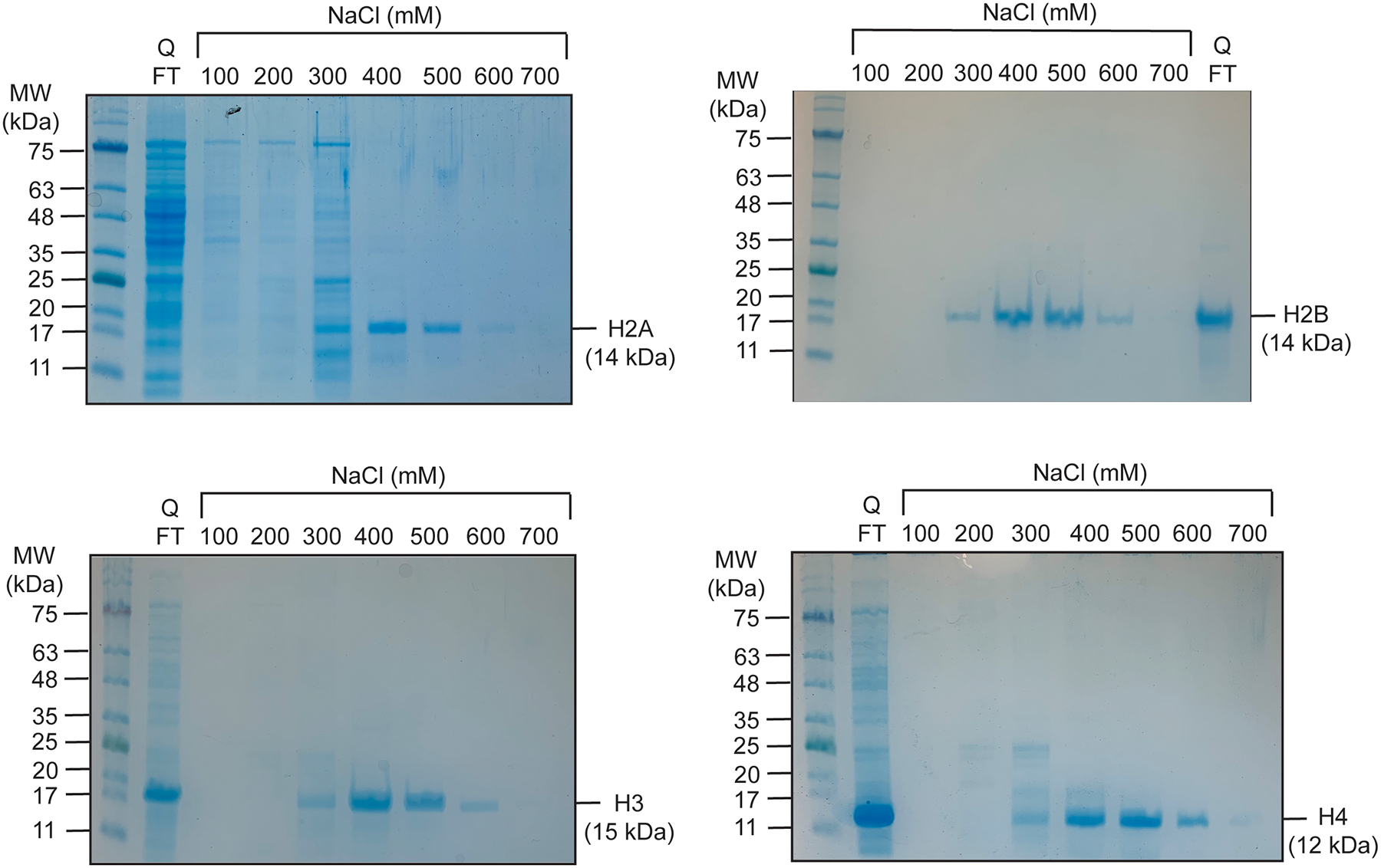
Representative 12.5% SDS-PAGE gels for the purification of histone H2A (top left), H2B (top right), H3 (bottom left), and H4 (bottom right). The proteins were visualized by coomassie blue staining.
3.3. Generation of H2A/H2B dimer and H3/H4 tetramer
After purifying each individual histone, histone octamers or histone H2A/H2B dimers and H3/H4 tetramers can be generated, purified, and stored for rapid reconstitution of NCPs. Below, we outline the salt-dialysis method for refolding H2A/H2B dimers and H3/H4 tetramers. We prefer refolding H2A/H2B dimers and H3/H4 tetramers instead of histone octamers due to the increased yield of in-tact complexes.
3.3.1. Refolding of H2A/H2B dimer
Resuspend the lyophilized H2A and H2B in guanidinium buffer to a final concentration of 2 mg/mL.
Incubate the resuspended H2A and H2B in guanidinium buffer at room temperature for 2 hours.
- Determine the concentration of resuspended H2A and H2B spectroscopically using the following theoretical molar extinction coefficients:
- H2A: 4,470 M−1cm−1
- H2B: 7,450 M−1cm−1
Mix equimolar amounts of H2A and H2B in dialysis tubing (3,000 Da MWCO).
Dialyze against 1 L of high salt refolding buffer (ice cold, 4 °C) three times, for a minimum of 8 hours each exchange.
3.3.2. Refolding of H3/H4 tetramer
Resuspend lyophilized H3 and H4 in guanidinium buffer to a final concentration of 2 mg/mL.
Incubate the resuspended H3 and H4 in guanidinium buffer at room temperature for 2 hours.
- Determine the concentration of resuspended H3 and H4 spectroscopically using the following theoretical molar extinction coefficients:
- H3: 4,470 M−1cm−1
- H4: 5,960 M−1cm−1
Mix equimolar amounts of histone H3 and H4 in dialysis tubing (3,000 Da MWCO).
Dialyze against 1 L of high salt ice cold (4 °C) refolding buffer three times, with at least two of those exchanges overnight (see note 12).
3.3.2. Purification of H2A/H2B dimer and H3/H4 tetramer
Concentrate the refolded H2A/H2B dimer and H3/H4 tetramer to ~100 μM (see note 13).
Purify H2A/H2B dimer or H3/H4 tetramer over a Superdex 200 Increase 10/300 GL gel filtration column.
Confirm the purity and stoichiometry of the H2A/H2B dimer or H3/H4 tetramer by running an SDS PAGE gel of the S200 gel filtration fractions (Fig. 3, see note 14).
Combine fractions containing purified H2A/H2B dimer or H3/H4 tetramer and concentrate using a centrifugal filter unit (10,000 Da MWCO) to at least 100 μM.
Mix the concentrated H2A/H2B dimer and H3/H4 tetramer with an equal volume of 100% glycerol and store indefinitely at −20 °C.
Fig 3.
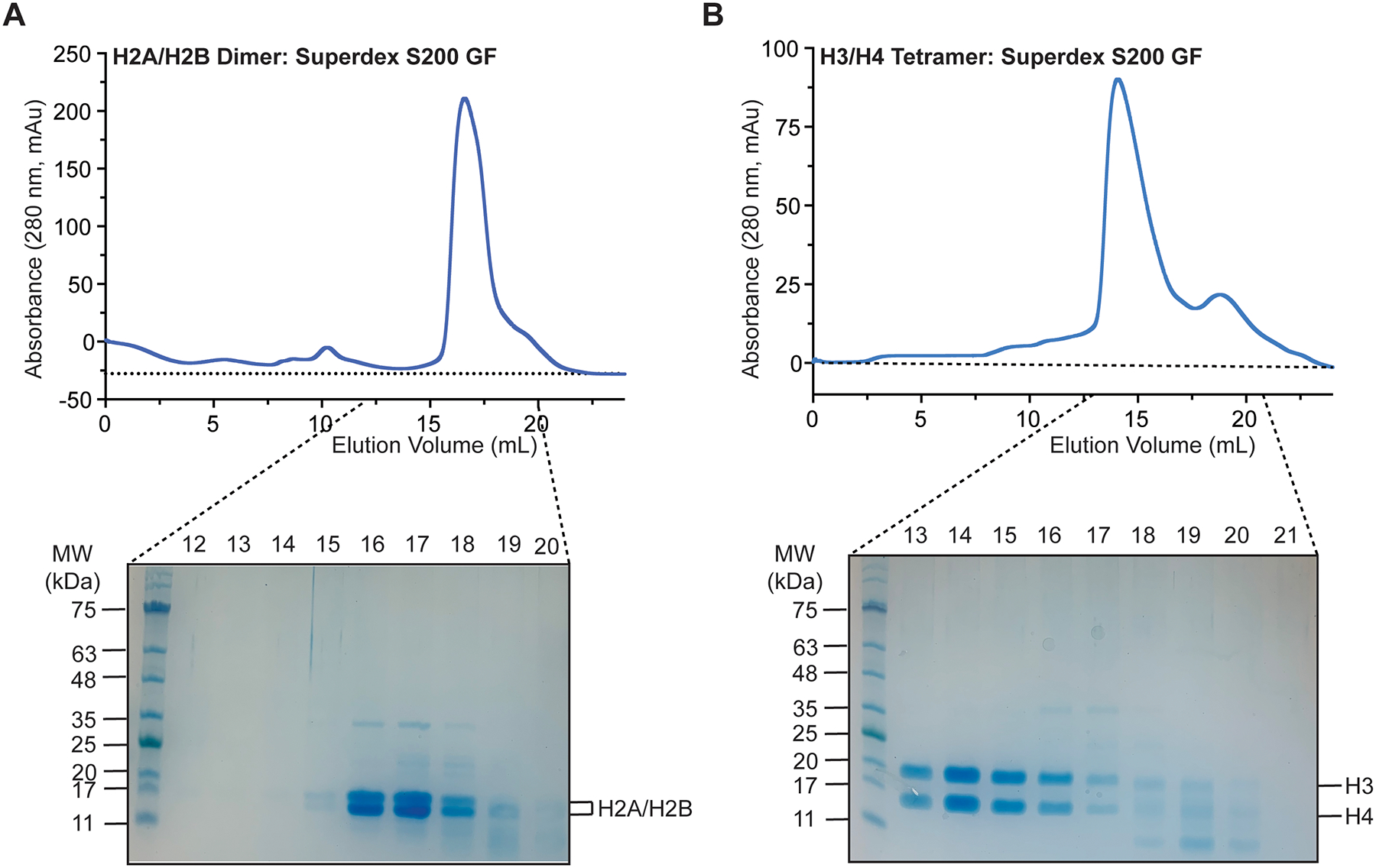
Purification of H2A/H2B dimer and H3/H4 tetramer. (a) Representative H2A/H2B dimer chromatogram from a Superdex S200 gel filtration run. The resulting fractions were resolved on a 12.5% SDS-PAGE gel and visualized by coomassie blue staining. (b) Representative H3/H4 tetramer chromatogram from a Superdex S200 gel filtration run. The resulting fractions were resolved on a 12.5% SDS-PAGE gel and visualized by coomassie blue staining.
3.4. Reconstituting nucleosomes containing DNA damage
NCP reconstitution is done via a salt dialysis method using the damaged DNA, H2A/H2B dimer, and H3/H4 tetramer previously purified. After nucleosome reconstitution, the nucleosomes containing DNA damage are purified by sucrose gradient ultracentrifugation to separate free linear DNA and other subnucleosomal species, which makes them suitable for quantitative in vitro experiments.
3.4.1. Nucleosome reconstitution
Thaw the damaged DNA, H2A/H2B dimer, and H3/H4 tetramer on ice.
Determine the concentration of the damaged DNA spectroscopically using the theoretical molar extinction coefficient for the specific DNA substrate.
Dilute the damaged DNA to a concentration of 2.5 μM using high salt buffer.
- Determine the concentration of H2A/H2B dimer and H3/H4 tetramer spectroscopically using the following theoretical molar extinction coefficients:
- H2A/H2B dimer: 11,920 M−1cm−1
- H3/H4 tetramer: 20,860 M−1cm−1
Dilute the H2A/H2B dimer to 5 μM and H3/H4 tetramer to 2.5 μM using high salt buffer.
Mix the damaged DNA, H2A/H2B dimer, and H3/H4 tetramer in a 1:2:1 molar ratio and place in dialysis tubing (10,000 Da MWCO).
Place the dialysis tubing in a beaker containing 300 mL of high salt buffer and equilibrate for 30 minutes.
- Dilute the high salt buffer with no salt buffer stepwise to the following concentrations:
- 1.5 M NaCl, 150 mL of no salt buffer, 2 hours
- 1 M NaCl, 150 mL of no salt buffer, 2 hours
- 0.75 M NaCl, 300 mL of no salt buffer, 2 hours
- 0.5 M NaCl, 300 mL of no salt buffer, 2 hours
- 0.25 M NaCl, 1200 mL of no salt buffer, overnight
- 0.125 M NaCl, 2400 mL of no salt buffer, 2 hours
3.4.2. Nucleosome Purification
Remove the reconstituted nucleosome from the dialysis tubing and place in a conical tube.
Concentrate the nucleosome down to ~0.5 mL using a centrifugal filter unit (10,000 Da MWCO).
Heat-shock the nucleosome at 55 °C for 30 minutes (see note 15). A representative gel of the nucleosome prior to purification can be seen in Fig. 4a.
Make six 10 – 40% sucrose gradients in 38.5 mL Beckman Coulter Ultra Centrifuge tubes using a Biocomp Gradient Master 108 (see note 16).
Layer the 0.5 mL of damaged nucleosome to the top of a sucrose gradient.
Ultracentrifuge the sucrose gradients at 125,000 xG for 40–42 hours in an SW 32 rotor at 4°C.
Fractionate the sucrose gradient containing nucleosome by pulling 1 mL from the top of the tube and storing in 1.5 mL eppendorf tubes.
Measure the absorbance at 260 nm for each fraction to identify those that contain nucleic acids and run the fractions containing nucleic acid on a Native PAGE gel (5% 59:1 acrylamide:bis-acrylamide and 0.2X TBE). A representative chromatogram of the nucleosome fractions after sucrose gradient ultracentrifugation can be seen in Fig. 4b.
Pool the fractions that contain purified nucleosome and buffer exchange 5 times into 1X TE using a centrifugal filter unit (10,000 Da MWCO). A representative gel of the purified nucleosome containing DNA damage can be seen in Fig. 4c.
Concentrate the nucleosome to at least 1 μM and store at 4°C (see note 17 and 18).
Fig 4.
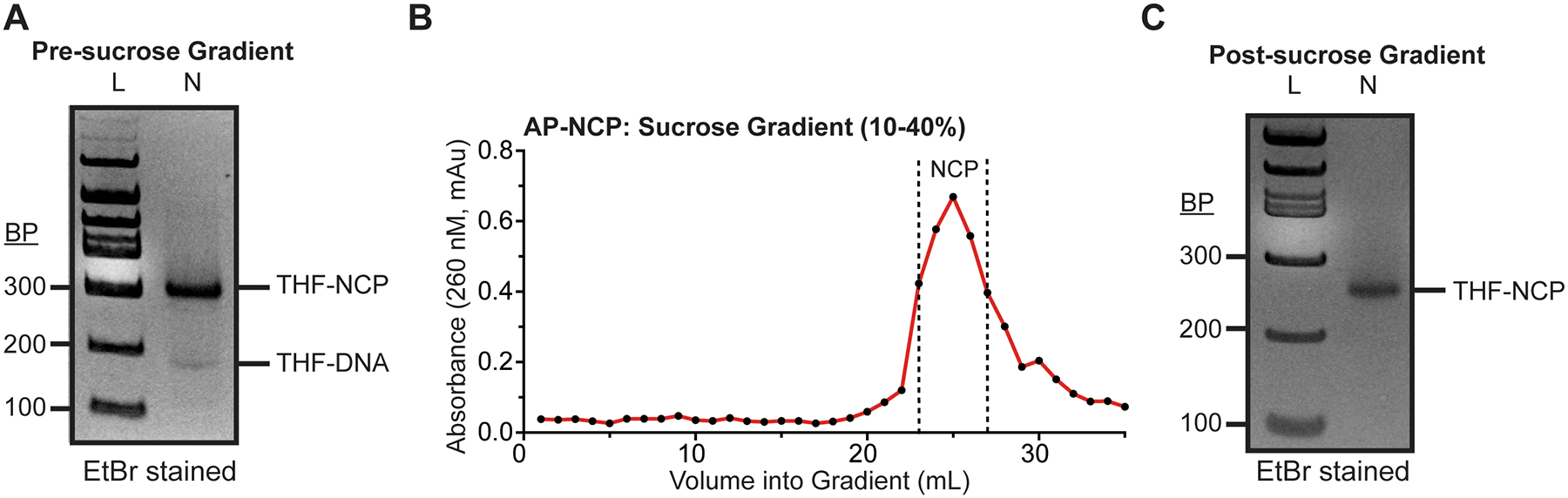
Generation and purification of nucleosomes containing site-specific DNA damage. (a) A representative 6% Native PAGE (59:1) gel of the reconstituted nucleosome before purification (b) A representative chromatogram of the nucleosome after fractionation from a 10–40% sucrose gradient. (c) A representative 6% Native PAGE (59:1) gel of the reconstituted nucleosome after purification.
4. Notes
The glucose, MgSO4, CaCl2, and vitamin solution are added to the M9 media after autoclaving as they can precipitate and/or degrade during autoclaving.
The DNA damage can be placed on either I-strand or J-strand depending on the desired location in the nucleosome. If moving the DNA damage to the J-strand, we suggest changing the J-strand to three oligos and the I-strand to two oligos
Avoid placing the THF or other DNA damage within 5 bp of any T4-DNA ligase sites. If unavoidable, the reactions will result in a significantly reduced yield of full-length ligated 147 bp DNA.
We perform each individual reaction with 10 nmols of each individual oligo, which should theoretically yield 10 nmols of final ligated product (assuming 100% yield). An average prep size is ~50–100 nmols, or 5–10 individual reactions.
The ligation reaction generally does not reach 100% completion. If desired, an additional 5uL of T4 DNA ligase can be added after the first 24 hours to ensure as much of the DNA substrate has been ligated as possible.
Concentrating the extracted DNA to 10 μM enables efficient annealing of the I- and J-strand oligos in the subsequent annealing step.
The purified damaged DNA should be stored in aliquots that match the average prep size of the downstream nucleosome reconstitution. We typically perform 5 nmol nucleosome reconstitutions.
We have stored damaged DNA substrates for up to a year without issues. After long-term storage, we suggest checking the quality of the DNA on an agarose gel before proceeding to nucleosome reconstitution.
Urea can spontaneously degrade into isocyanic acid, which reacts with lysine and arginine side chains to form carbamylation [78]. To prevent carbamylation of histones during purification, 8M urea solutions are made fresh and de-ionized with a Dowex Amberlite resin.
The extracted histones should be dialyzed against enough 8M Urea to bring the concentration of guanidinium-HCl below 250 mM. This step is critical for the subsequent purification of the histone via ion-exchange chromatography as the histones will not bind the S-sepharose resin above 300 mM NaCl.
The Q-sepharose is used to bind contaminating proteins from the histone extraction step.
It is not uncommon to see precipitant forming in the dialysis membrane after the first 24 hours spinning in the high salt buffer. This is often a result of impurities in the purification of the individual histones.
The H2A/H2B dimer and H3/H4 tetramer is concentrated to no more than 100 μM to ensure separation from contaminants during gel filtration.
It’s not uncommon to see minor contaminates in the dimer or tetramer. In our experience, these can be removed at subsequent purification steps, and do not significantly affect reconstitution of the nucleosome.
Purifying the damaged DNA via denaturing PAGE can lead to an excess of either the I- or J-strand. Excess I- or J-strand ssDNA will readily form histone octamers wrapped by single-stranded DNA [79]. The heat-shock at 55 °C will ensure the nucleosome is properly positioned on the 601 DNA, while also denaturing any ssDNA-histone octamer complexes that are difficult to purify via sucrose gradient ultracentrifugation.
Another method for purifying nucleosomes is via electrophoresis using a Bio-Rad Model 491 Prep Cell [80].
The nucleosome concentration is determined spectroscopically by diluting 2x with 4 M NaCl and measuring the absorbance at 260 nM. Theoretical extinction coefficients should be determined using the damaged DNA sequence.
The purified nucleosomes should be stored at 4°C in 1x TE at a concentration of at least 1 μM. The purified nucleosomes should be stable for at least 1 month. For longer term storage (>1 month), we suggest storing the purified nucleosomes at 4°C in 1x TE at a concentration of at least 10 μM.
Acknowledgements
This research was supported by the National Institute of General Medical Science R35-GM128562 (B.J.R., T.M.W., J.J.S., and B.D.F) and the National Institute of General Medical Science F32-GM140718 (T.M.W.). We also thank Drs. Karolin Luger, Catherine Musselman, and Michael Poirier for the histone plasmids used in this study.
References
- 1.Lindahl T, Nyberg B (1972) Rate of depurination of native deoxyribonucleic acid. Biochemistry 11 (19):3610–3618 [DOI] [PubMed] [Google Scholar]
- 2.Sekiguchi M, Tsuzuki T (2002) Oxidative nucleotide damage: consequences and prevention. Oncogene 21 (58):8895–8904 [DOI] [PubMed] [Google Scholar]
- 3.Kreutzer DA, Essigmann JM (1998) Oxidized, deaminated cytosines are a source of C→ T transitions in vivo. Proceedings of the National Academy of Sciences 95 (7):3578–3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cooke MS, Evans MD, Dizdaroglu M, Lunec J (2003) Oxidative DNA damage: mechanisms, mutation, and disease. The FASEB Journal 17 (10):1195–1214 [DOI] [PubMed] [Google Scholar]
- 5.Wallace SS, Murphy DL, Sweasy JB (2012) Base excision repair and cancer. Cancer Lett 327 (1–2):73–89. doi: 10.1016/j.canlet.2011.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krokan HE, Bjoras M (2013) Base excision repair. Cold Spring Harb Perspect Biol 5 (4):a012583. doi: 10.1101/cshperspect.a012583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim Y-J, Wilson DM III (2012) Overview of base excision repair biochemistry. Current molecular pharmacology 5 (1):3–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whitaker AM, Schaich MA, Smith MS, Flynn TS, Freudenthal BD (2017) Base excision repair of oxidative DNA damage: from mechanism to disease. Frontiers in bioscience (Landmark edition) 22:1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wallace SS (2014) Base excision repair: a critical player in many games. DNA repair 19:14–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beard WA, Horton JK, Prasad R, Wilson SH (2019) Eukaryotic base excision repair: new approaches shine light on mechanism. Annual review of biochemistry 88:137–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hegde ML, Hazra TK, Mitra S (2008) Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res 18 (1):27–47. doi: 10.1038/cr.2008.8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schermerhorn KM, Delaney S (2014) A chemical and kinetic perspective on base excision repair of DNA. Acc Chem Res 47 (4):1238–1246. doi: 10.1021/ar400275a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kornberg RD (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184 (4139):868–871 [DOI] [PubMed] [Google Scholar]
- 14.Woodcock C, Safer J, Stanchfield J (1976) Structural repeating units in chromatin: I. Evidence for their general occurrence. Experimental cell research 97 (1):101–110 [DOI] [PubMed] [Google Scholar]
- 15.Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389 (6648):251–260 [DOI] [PubMed] [Google Scholar]
- 16.Caffrey PJ, Delaney S (2020) Chromatin and other obstacles to base excision repair: potential roles in carcinogenesis. Mutagenesis 35 (1):39–50 [DOI] [PubMed] [Google Scholar]
- 17.Kennedy EE, Caffrey PJ, Delaney S (2018) Initiating base excision repair in chromatin. DNA repair 71:87–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li C, Delaney S (2019) Challenges for base excision repair enzymes: Acquiring access to damaged DNA in chromatin. The Enzymes 45:27–57 [DOI] [PubMed] [Google Scholar]
- 19.Kumar N, Raja S, Van Houten B (2020) The involvement of nucleotide excision repair proteins in the removal of oxidative DNA damage. Nucleic acids research 48 (20):11227–11243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kutuzov M, Belousova E, Ilina E, Lavrik O (2020) Impact of PARP1, PARP2 & PARP3 on the base excision repair of nucleosomal DNA. Adv Exp Med Biol 1241:47–57 [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez Y, Hinz JM, Smerdon MJ (2015) Accessing DNA damage in chromatin: Preparing the chromatin landscape for base excision repair. DNA repair 32:113–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meas R, Wyrick JJ, Smerdon MJ (2019) Nucleosomes Regulate Base Excision Repair in Chromatin. Mutat Res Rev Mutat Res 780:29–36. doi: 10.1016/j.mrrev.2017.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bilotti K, Kennedy EE, Li C, Delaney S (2017) Human OGG1 activity in nucleosomes is facilitated by transient unwrapping of DNA and is influenced by the local histone environment. DNA Repair (Amst) 59:1–8. doi: 10.1016/j.dnarep.2017.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li C, Delaney S (2019) Histone H2A Variants Enhance the Initiation of Base Excision Repair in Nucleosomes. ACS Chem Biol 14 (5):1041–1050. doi: 10.1021/acschembio.9b00229 [DOI] [PubMed] [Google Scholar]
- 25.Olmon ED, Delaney S (2017) Differential Ability of Five DNA Glycosylases to Recognize and Repair Damage on Nucleosomal DNA. ACS Chem Biol 12 (3):692–701. doi: 10.1021/acschembio.6b00921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarantino ME, Dow BJ, Drohat AC, Delaney S (2018) Nucleosomes and the three glycosylases: High, medium, and low levels of excision by the uracil DNA glycosylase superfamily. DNA Repair (Amst) 72:56–63. doi: 10.1016/j.dnarep.2018.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beard BC, Stevenson JJ, Wilson SH, Smerdon MJ (2005) Base excision repair in nucleosomes lacking histone tails. DNA Repair (Amst) 4 (2):203–209. doi: 10.1016/j.dnarep.2004.09.011 [DOI] [PubMed] [Google Scholar]
- 28.Beard BC, Wilson SH, Smerdon MJ (2003) Suppressed catalytic activity of base excision repair enzymes on rotationally positioned uracil in nucleosomes. Proc Natl Acad Sci U S A 100 (13):7465–7470. doi: 10.1073/pnas.1330328100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinz JM, Rodriguez Y, Smerdon MJ (2010) Rotational dynamics of DNA on the nucleosome surface markedly impact accessibility to a DNA repair enzyme. Proc Natl Acad Sci U S A 107 (10):4646–4651. doi: 10.1073/pnas.0914443107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meas R, Smerdon MJ (2016) Nucleosomes determine their own patch size in base excision repair. Sci Rep 6:27122. doi: 10.1038/srep27122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meas R, Smerdon MJ, Wyrick JJ (2015) The amino-terminal tails of histones H2A and H3 coordinate efficient base excision repair, DNA damage signaling and postreplication repair in Saccharomyces cerevisiae. Nucleic Acids Res 43 (10):4990–5001. doi: 10.1093/nar/gkv372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakanishi S, Prasad R, Wilson SH, Smerdon M (2007) Different structural states in oligonucleosomes are required for early versus late steps of base excision repair. Nucleic Acids Res 35 (13):4313–4321. doi: 10.1093/nar/gkm436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez Y, Duan M, Wyrick JJ, Smerdon MJ (2018) A cassette of basic amino acids in histone H2B regulates nucleosome dynamics and access to DNA damage. J Biol Chem 293 (19):7376–7386. doi: 10.1074/jbc.RA117.000358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodriguez Y, Hinz JM, Laughery MF, Wyrick JJ, Smerdon MJ (2016) Site-specific Acetylation of Histone H3 Decreases Polymerase beta Activity on Nucleosome Core Particles in Vitro. J Biol Chem 291 (21):11434–11445. doi: 10.1074/jbc.M116.725788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rodriguez Y, Smerdon MJ (2013) The structural location of DNA lesions in nucleosome core particles determines accessibility by base excision repair enzymes. J Biol Chem 288 (19):13863–13875. doi: 10.1074/jbc.M112.441444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Howard MJ, Rodriguez Y, Wilson SH (2017) DNA polymerase beta uses its lyase domain in a processive search for DNA damage. Nucleic Acids Res 45 (7):3822–3832. doi: 10.1093/nar/gkx047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rodriguez Y, Horton JK, Wilson SH (2019) Histone H3 Lysine 56 Acetylation Enhances AP Endonuclease 1-Mediated Repair of AP Sites in Nucleosome Core Particles. Biochemistry 58 (35):3646–3655. doi: 10.1021/acs.biochem.9b00433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodriguez Y, Howard MJ, Cuneo MJ, Prasad R, Wilson SH (2017) Unencumbered Pol beta lyase activity in nucleosome core particles. Nucleic Acids Res 45 (15):8901–8915. doi: 10.1093/nar/gkx593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Banerjee DR, Deckard CE 3rd, Elinski MB, Buzbee ML, Wang WW, Batteas JD, Sczepanski JT (2018) Plug-and-Play Approach for Preparing Chromatin Containing Site-Specific DNA Modifications: The Influence of Chromatin Structure on Base Excision Repair. J Am Chem Soc 140 (26):8260–8267. doi: 10.1021/jacs.8b04063 [DOI] [PubMed] [Google Scholar]
- 40.Banerjee DR, Deckard CE, Zeng Y, Sczepanski JT (2019) Acetylation of the histone H3 tail domain regulates base excision repair on higher-order chromatin structures. Scientific Reports 9 (1):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huggins CF, Chafin DR, Aoyagi S, Henricksen LA, Bambara RA, Hayes JJ (2002) Flap endonuclease 1 efficiently cleaves base excision repair and DNA replication intermediates assembled into nucleosomes. Molecular cell 10 (5):1201–1211 [DOI] [PubMed] [Google Scholar]
- 42.Nilsen H, Lindahl T, Verreault A (2002) DNA base excision repair of uracil residues in reconstituted nucleosome core particles. The EMBO journal 21 (21):5943–5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Menoni H, Gasparutto D, Hamiche A, Cadet J, Dimitrov S, Bouvet P, Angelov D (2007) ATP-dependent chromatin remodeling is required for base excision repair in conventional but not in variant H2A.Bbd nucleosomes. Mol Cell Biol 27 (17):5949–5956. doi: 10.1128/MCB.00376-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Menoni H, Shukla MS, Gerson V, Dimitrov S, Angelov D (2012) Base excision repair of 8-oxoG in dinucleosomes. Nucleic Acids Res 40 (2):692–700. doi: 10.1093/nar/gkr761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prasad A, Wallace SS, Pederson DS (2007) Initiation of base excision repair of oxidative lesions in nucleosomes by the human, bifunctional DNA glycosylase NTH1. Mol Cell Biol 27 (24):8442–8453. doi: 10.1128/MCB.00791-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Odell ID, Barbour JE, Murphy DL, Della-Maria JA, Sweasy JB, Tomkinson AE, Wallace SS, Pederson DS (2011) Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol Cell Biol 31 (22):4623–4632. doi: 10.1128/MCB.05715-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Odell ID, Newick K, Heintz NH, Wallace SS, Pederson DS (2010) Non-specific DNA binding interferes with the efficient excision of oxidative lesions from chromatin by the human DNA glycosylase, NEIL1. DNA Repair (Amst) 9 (2):134–143. doi: 10.1016/j.dnarep.2009.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jagannathan I, Pepenella S, Hayes JJ (2011) Activity of FEN1 endonuclease on nucleosome substrates is dependent upon DNA sequence but not flap orientation. J Biol Chem 286 (20):17521–17529. doi: 10.1074/jbc.M111.229658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang C, Sengupta S, Hegde PM, Mitra J, Jiang S, Holey B, Sarker AH, Tsai MS, Hegde ML, Mitra S (2017) Regulation of oxidized base damage repair by chromatin assembly factor 1 subunit A. Nucleic Acids Res 45 (2):739–748. doi: 10.1093/nar/gkw1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fu I, Smith DJ, Broyde S (2019) Rotational and translational positions determine the structural and dynamic impact of a single ribonucleotide incorporated in the nucleosome. DNA Repair (Amst) 73:155–163. doi: 10.1016/j.dnarep.2018.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hinz JM (2014) Impact of abasic site orientation within nucleosomes on human APE1 endonuclease activity. Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis 766–767:19–24. doi: 10.1016/j.mrfmmm.2014.05.008 [DOI] [PubMed] [Google Scholar]
- 52.Hinz JM, Mao P, McNeill DR, Wilson DM 3rd (2015) Reduced Nuclease Activity of Apurinic/Apyrimidinic Endonuclease (APE1) Variants on Nucleosomes: IDENTIFICATION OF ACCESS RESIDUES. J Biol Chem 290 (34):21067–21075. doi: 10.1074/jbc.M115.665547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maher RL, Prasad A, Rizvanova O, Wallace SS, Pederson DS (2013) Contribution of DNA unwrapping from histone octamers to the repair of oxidatively damaged DNA in nucleosomes. DNA Repair (Amst) 12 (11):964–971. doi: 10.1016/j.dnarep.2013.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maher RL, Wallace SS, Pederson DS (2019) The lyase activity of bifunctional DNA glycosylases and the 3’-diesterase activity of APE1 contribute to the repair of oxidized bases in nucleosomes. Nucleic Acids Res 47 (6):2922–2931. doi: 10.1093/nar/gky1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maher RL, Marsden CG, Averill AM, Wallace SS, Sweasy JB, Pederson DS (2017) Human cells contain a factor that facilitates the DNA glycosylase-mediated excision of oxidized bases from occluded sites in nucleosomes. DNA Repair (Amst) 57:91–97. doi: 10.1016/j.dnarep.2017.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caffrey PJ, Delaney S (2021) Nucleosome Core Particles Lacking H2B or H3 Tails Are Altered Structurally and Have Differential Base Excision Repair Fingerprints. Biochemistry 60 (3):210–218 [DOI] [PubMed] [Google Scholar]
- 57.Kennedy EE, Li C, Delaney S (2019) Global repair profile of human alkyladenine DNA glycosylase on nucleosomes reveals DNA packaging effects. ACS chemical biology 14 (8):1687–1692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Caffrey PJ, Kher R, Bian K, Li D, Delaney S (2020) Comparison of the Base Excision and Direct Reversal Repair Pathways for Correcting 1, N 6-Ethenoadenine in Strongly Positioned Nucleosome Core Particles. Chemical research in toxicology 33 (7):1888–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cannan WJ, Tsang BP, Wallace SS, Pederson DS (2014) Nucleosomes suppress the formation of double-strand DNA breaks during attempted base excision repair of clustered oxidative damages. J Biol Chem 289 (29):19881–19893. doi: 10.1074/jbc.M114.571588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bennett L, Madders E, Parsons JL (2019) HECTD1 promotes base excision repair in nucleosomes through chromatin remodelling. Nucleic Acids Res. doi: 10.1093/nar/gkz1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cannan WJ, Rashid I, Tomkinson AE, Wallace SS, Pederson DS (2017) The Human Ligase IIIalpha-XRCC1 Protein Complex Performs DNA Nick Repair after Transient Unwrapping of Nucleosomal DNA. J Biol Chem 292 (13):5227–5238. doi: 10.1074/jbc.M116.736728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chafin DR, Vitolo JM, Henricksen LA, Bambara RA, Hayes JJ (2000) Human DNA ligase I efficiently seals nicks in nucleosomes. The EMBO journal 19 (20):5492–5501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kutuzov M, Belousova E, Kurgina T, Ukraintsev A, Vasil’eva I, Khodyreva S, Lavrik O (2021) The contribution of PARP1, PARP2 and poly (ADP-ribosyl) ation to base excision repair in the nucleosomal context. Scientific reports 11 (1):1–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bilotti K, Tarantino ME, Delaney S (2018) Human oxoguanine glycosylase 1 removes solution accessible 8-oxo-7, 8-dihydroguanine lesions from globally substituted nucleosomes except in the dyad region. Biochemistry 57 (9):1436–1439 [DOI] [PubMed] [Google Scholar]
- 65.Ura K, Araki M, Saeki H, Masutani C, Ito T, Iwai S, Mizukoshi T, Kaneda Y, Hanaoka F (2001) ATP‐dependent chromatin remodeling facilitates nucleotide excision repair of UV‐induced DNA lesions in synthetic dinucleosomes. The EMBO journal 20 (8):2004–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ren M, Shang M, Wang H, Xi Z, Zhou C (2021) Histones participate in base excision repair of 8-oxodGuo by transiently cross-linking with active repair intermediates in nucleosome core particles. Nucleic acids research 49 (1):257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, Luger K (2003) Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods in enzymology 375:23–44 [DOI] [PubMed] [Google Scholar]
- 68.Kosmoski JV, Smerdon MJ (1999) Synthesis and nucleosome structure of DNA containing a UV photoproduct at a specific site. Biochemistry 38 (29):9485–9494 [DOI] [PubMed] [Google Scholar]
- 69.Sczepanski JT, Wong RS, McKnight JN, Bowman GD, Greenberg MM (2010) Rapid DNA-protein cross-linking and strand scission by an abasic site in a nucleosome core particle. Proc Natl Acad Sci U S A 107 (52):22475–22480. doi: 10.1073/pnas.1012860108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Duan M-R, Smerdon MJ (2010) UV damage in DNA promotes nucleosome unwrapping. Journal of Biological Chemistry 285 (34):26295–26303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lowary P, Widom J (1998) New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. Journal of molecular biology 276 (1):19–42 [DOI] [PubMed] [Google Scholar]
- 72.Weaver TM, Hoitsma NM, Spencer JJ, Gakhar L, Schnicker NJ, Freudenthal BD (2022) Structural basis for APE1 processing DNA damage in the nucleosome. Nat Commun 13 (1):5390. doi: 10.1038/s41467-022-33057-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Deckard CE III, Sczepanski JT (2021) Reversible chromatin condensation by the DNA repair and demethylation factor thymine DNA glycosylase. Nucleic acids research 49 (5):2450–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bai J, Zhang Y, Xi Z, Greenberg MM, Zhou C (2018) Oxidation of 8-Oxo-7,8-dihydro-2’-deoxyguanosine Leads to Substantial DNA-Histone Cross-Links within Nucleosome Core Particles. Chem Res Toxicol 31 (12):1364–1372. doi: 10.1021/acs.chemrestox.8b00244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li F, Zhang Y, Bai J, Greenberg MM, Xi Z, Zhou C (2017) 5-Formylcytosine Yields DNA-Protein Cross-Links in Nucleosome Core Particles. J Am Chem Soc 139 (31):10617–10620. doi: 10.1021/jacs.7b05495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Taylor J-S (2015) Design, synthesis, and characterization of nucleosomes containing site-specific DNA damage. DNA repair 36:59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vasudevan D, Chua EY, Davey CA (2010) Crystal structures of nucleosome core particles containing the ‘601’strong positioning sequence. Journal of molecular biology 403 (1):1–10 [DOI] [PubMed] [Google Scholar]
- 78.Stark GR, Stein WH, Moore S (1960) Reactions of the cyanate present in aqueous urea with amino acids and proteins. Journal of Biological Chemistry 235 (11):3177–3181 [Google Scholar]
- 79.Adkins NL, Swygert SG, Kaur P, Niu H, Grigoryev SA, Sung P, Wang H, Peterson CL (2017) Nucleosome-like, single-stranded DNA (ssDNA)-histone octamer complexes and the implication for DNA double strand break repair. Journal of Biological Chemistry 292 (13):5271–5281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kujirai T, Arimura Y, Fujita R, Horikoshi N, Machida S, Kurumizaka H (2018) Methods for preparing nucleosomes containing histone variants. In: Histone Variants. Springer, pp 3–20 [DOI] [PubMed] [Google Scholar]