

Summary
Short tandem repeat (STR) instability causes transcriptional silencing in several repeat expansion disorders. In fragile X syndrome (FXS), mutation-length expansion of a CGG STR represses FMR1 via local DNA methylation. Here, we find Megabase-scale H3K9me3 domains on autosomes and encompassing FMR1 on the X-chromosome in iPSCs, iPSC-derived neural progenitors, EBV-transformed-lymphoblasts, and FXS brain tissue with mutation-length CGG expansion. H3K9me3 domains connect via inter-chromosomal interactions and demarcate severe misfolding of TADs and loops. They harbor long synaptic genes replicating at the end of S-phase, replication stress-induced double strand breaks, and STRs prone to stepwise somatic instability. CRISPR engineering of the mutation-length CGG to premutation-length reverses H3K9me3 on the X-chromosome and multiple autosomes, refolds TADs, and restores gene expression. H3K9me3 domains can also arise in normal-length iPSCs created with perturbations linked to genome instability, suggesting their relevance beyond FXS. Our results reveal Mb-scale heterochromatinization and trans interactions among loci susceptible to instability.
Graphical Abstract
In brief
Megabase-scale H3K9me3 domains are connected by inter-chromosomal interactions, harboring long synaptic genes prone to instability, and are reversible by CGG short tandem repeat tract engineering in fragile X syndrome.
Introduction
Fragile X syndrome (FXS) is the most common form of inherited intellectual disability, affecting 1 in 4,000 males and 1 in 8,000 females. The disease manifests early in life and presents as a range of mild to severe defects in communication skills, cognitive ability, and physical appearance, as well as seizures, anxiety, and hypersensitivity to stimuli1. FXS is caused by expansion of a CGG short tandem repeat (STR) in the 5’ untranslated region (5’UTR) of the FMR1 gene2–4. CGG tract length correlates with disease severity and can be stratified into <40 (normal-length), 41–60 (intermediate), 61–199 (pre-mutation), and 200+ (mutation-length) repeats5–8. Individuals with a premutation-length FMR1 CGG tract are at risk of developing the late-stage neurodegenerative disease Fragile X-associated tremor/ataxia syndrome (FXTAS) marked by cerebellar ataxia, essential tremor, peripheral neuropathy, and cognitive decline9. Together, these data highlight the critical role for CGG STR tract length in a wide range of pathological clinical presentations.
Increases in STR tract length correlate with pathologically altered gene expression levels in a number of repeat expansion disorders10. In FXTAS, CGG expansion from normal- to premutation-length causes a 2–8-fold increase in FMR1 expression9. By contrast, expansion to mutation-length causes transcriptional inhibition of FMR1 and loss of the Fragile X Messenger Ribonucleoprotein (FMRP) it encodes10. Transcriptional silencing of FMR1 occurs via local DNA methylation and heterochromatinization of the mutation-length FMR1 CGG tract and its adjacent promoter2,11–15. Some genome-wide reports suggest that changes in DNA methylation are restricted to FMR1 in FXS16. Thus, classic models assert that local silencing of FMR1 drives FXS onset, and downstream genome-wide disruption of gene expression is thought to be a secondary consequence of FMRP loss17.
Multiple lines of evidence suggest that the onset and progression of FXS might involve additional silencing mechanisms beyond local promoter DNA methylation. Fmr1 knock-out mice only partially recapitulate FXS clinical presentations18, suggesting that the human CGG expansion event itself is important for the full range and severity of pathologic features. Long-range loop disruption around FMR1 has been reported in FXS patient-derived cell lines and post-mortem brain tissue with mutation-length CGG expansion19, indicating that chromatin dysregulation can also occur distal from the FMR1 promoter. Furthermore, blocking DNA methylation by global 5-aza-2’-deoxycytidine treatment or targeted DNA demethylation by dCas9-Tet1 does not fully de-repress FMR1 in every case, and patient cells with longer CGG tracts can be refractory to FMR1 de-repression20–23. Altogether, these data indicate that FXS might involve additional mechanisms working in conjunction with the classic model of local DNA methylation, FMR1 silencing, and gene expression changes downstream of FMRP loss.
Here, we use nanopore long-read sequencing, kilobase-resolution Hi-C, CUT&RUN, CRISPR STR engineering, and single-cell Oligopaint FISH imaging to demonstrate that Mb-sized H3K9me3 domains on autosomes and the X-chromosome are significantly more likely to occur in FXS patient-derived cell lines and brain tissue with mutation-length CGG expansion compared to matched normal-length controls. H3K9me3 domains replicate at the end of S phase and demarcate severe Mb-scale misfolding of TADs, subTADs, and loops. They harbor long synaptic genes, replication stress-induced double strand breaks, and STRs susceptible to stepwise somatic instability. H3K9me3 signal over a subset of domains on the X-chromosome and multiple autosomes can be reversed by engineering the mutation-length CGG STR to premutation-length; TADs are refolded, trans interactions untethered, and expression restored upon H3K9me3 reversal. Our results reveal BREACHes – Beacons of Repeat Expansion Anchored by Contacting Heterochromatin – linking Mb-scale H3K9me3 domains, severe chromatin misfolding in cis, long-range inter-chromosomal interactions, and instability of the repetitive genome.
Results
A five Megabase-sized H3K9me3 domain demarcates severe long-range chromatin misfolding on the X-chromosome in iPSC-derived NPCs with mutation-length CGG STR expansion
We analyzed a series of human induced pluripotent stem cell (iPSC) lines in which the CGG STR tract is normal-length (5–40 CGG triplets, NL iPSC Replicates, NL_18, NL_25, NL_27), premutation-length (61–199 CGG triplets, PM iPSC, PM_137), or mutation-length (200+ CGG triplets, ML FXS-patient derived iPSC Replicates, FXS_421, FXS_426, FXS_470) (Figure 1A). All iPSC lines were male, derived from fibroblasts, of European ancestry, and confirmed to be karyotypically normal with morphology and markers of robust pluripotency (Figure S1A–D, Table S1).
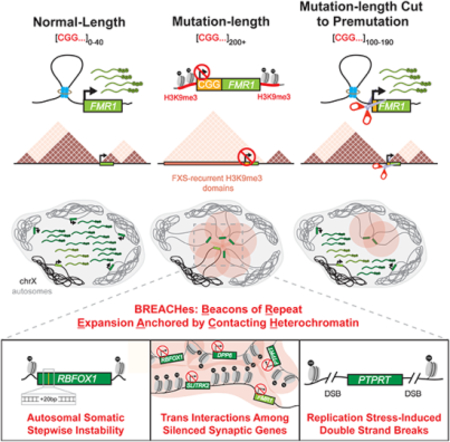
Figure 1. A Megabase-sized H3K9me3 domain spreads upstream of the FMR1 locus in iPSC-derived NPCs and post-mortem caudate nucleus brain tissue from FXS patients.
(A) Schematic of iPSC lines used to model FMR1 CGG expansion in FXS, including normal-length (NL), premutation-length (PM), and mutation-length (FXS). (B) Representative Nanopore long-reads across the FMR1 5’UTR. Colors reflect nucleotides (orange: A, blue: T, green: C, red: G, dark green: CGG). (C) Number of CGG triplets in the FMR1 5’UTR from Nanopore long-reads. (D) FMR1 mRNA levels normalized to GAPDH via qRT-PCR. Horizontal line, mean n=2 biological replicates. (E) Proportion of 19 CpG dinucleotides methylated in the 500 bp FMR1 promoter computed from Nanopore long-reads. (F) Proportion of CGG triplets methylated within the 5’ UTR STR using STRique. Each dot, one allele. (G) Hi-C and ChIP-seq in iPSC-NPCs across a 5Mb region around FMR1. (H) Hi-C fold-change interaction frequency maps. Gained and lost contacts compared to NL_18 highlighted in red and blue, respectively. (I) SLITRK2 and SLITRK4 mRNA levels via RNA-seq. Horizontal lines, mean n=2 biological replicates. (J) H3K9me3 CUT&RUN in brain tissue from N=2 FXS patients with sex- and age-matched N=2 normal-length individuals.
To obtain precise estimates of CGG STR length, we developed a customized assay coupling Nanopore long-read sequencing with guide RNA-directed Cas9 editing around the 5’UTR of the FMR1 gene (Figure 1B–C, Table S2, STAR Methods). Consistent with previous reports, normal-length and premutation-length iPSC lines had ~18–27 and 137 CGG triplets, respectively (Figure 1B–C). All three independent FXS-patient derived iPSC lines showed a similar median of ~420–470 CGG triplets and thus represent three biological replicates of mutation-length expansion events (Figure 1B–C). Consistent with previous reports9, we observed that FMR1 mRNA levels increased in premutation-length and then decreased significantly upon mutation-length CGG expansion (Figure 1D). As previously reported, we observed DNA methylation at the FMR1 promoter and CGG tract in all three mutation-length iPSC lines (Figure 1E–F, Figure S2A–D)2,11–15. Thus, we have estimated CGG tract length and verified known molecular hallmarks of CGG expansion, including increased FMR1 mRNA levels in permutation-length iPSCs as well as local DNA methylation and FMR1 silencing in three independent iPSC lines with mutation-length CGG expansion.
To investigate higher-order chromatin folding patterns in FXS, we differentiated our iPSC lines to homogenous populations of neural progenitor cells (iPSC-NPCs) (Figure S1E–F) and generated genome-wide high-resolution Hi-C libraries (Table S3). We observed severe genome misfolding in all three mutation-length FXS iPSC-NPC lines, including the dissolution of TADs, subTADs, and loops for up to 5 Megabases (Mbs) upstream of the ~1200 bp CGG STR (Figure 1G and Figure S2E). We also observed destruction of the local TAD boundary at FMR1 (Figure 1H and Figure S2F–G) as we have previously reported in FXS EBV-transformed B-lymphoblastoid cell lines and post-mortem brain tissue using targeted 5C analysis19. Thus, chromatin misfolding is severe in FXS and encompasses additional Megabases of the X-chromosome upstream of the FMR1 CGG STR.
To gain insight into the underlying mechanisms governing genome misfolding, we used ChIP-seq to map genome-wide patterns of the repressive histone mark H3K9me3 and the architectural protein CTCF (Table S3). We observed H3K9me3 signal local to FMR1 as in previous reports12–15,24. We also unexpectedly observed H3K9me3 signal spread in a domain-like pattern for up to 5 Mb upstream of FMR1 in all three mutation-length FXS iPSC-NPC lines (Figure 1G and Figure S2E). Upon gain of H3K9me3 in FXS, we observed loss of occupancy of the majority of CTCF sites (Figure 1G–H, Figures S2E–F+S2H). Boundaries of the Mb-scale H3K9me3 domain delimit the genomic range in which chromatin is misfolded (Figure 1G–H, Figure S2E–F). These results indicate that heterochromatin encompasses FMR1, spreads up to 5 Mb upstream, and correlates with large-scale misfolding of the genome on the X-chromosome in iPSC-NPCs with mutation-length CGG expansion.
H3K9me3 silences neural adhesion genes SLITRK2 and SLITRK4 on the X-chromosome in FXS patient-derived iPSCs, iPSC-NPCs, EBV-transformed B-lymphoblasts, and brain tissue
FXS is characterized by defects in synaptic plasticity and cognitive ability25. We observed the H3K9me3 domain in FXS iPSC-NPCs spanned two additional genes, SLITRK2 and SLITRK4, linked to neuronal cell adhesion and synaptic plasticity (Figure 1G and Figure S2E). Using our Hi-C maps, we observed that FMR1 loops directly to SLITRK2 and SLITRK4 in normal-length and premutation-length iPSC-NPCs (Figure S2I–L). In FXS, the long-range gene-gene cis interactions are abolished, consistent with the spread of H3K9me3 across the locus starting at FMR1’s loop anchor (Figures S2I–L). SLITRK2 mRNA levels are decreased in all FXS iPSC-NPCs as anticipated from the reproducible spread of H3K9me3 over the locus (Figure 1I). We note that in the FXS_421 line the H3K9me3 domain spreads to encompass SLITRK4 and the gene is silenced only in this FXS line. However, SLITRK4 is not silenced and the H3K9me3 signal does not spread over the gene in the FXS_426 and FXS_470 lines (Figure 1I, Figure 1G and Figure S2E). Together, these data suggest that a Mb-scale H3K9me3 domain spreads over the X-chromosome to encompass and silence synaptic and neural cell adhesion genes in addition to FMR1 in mutation-length iPSC-NPCs from FXS patients. The lateral spread of H3K9me3 along the genome can exhibit clone-to-clone variation.
We examined the extent to which large-scale genome misfolding and the X-chromosome H3K9me3 domain would vary by cell type or in subclones from the same parent line. First, we derived a second mutation-length iPSC line, FXS_425, from the parent line FXS_421. We observed similar CGG tract length (Figure S3A), CGG tract DNA methylation (Figure S3B), genome misfolding (Figure S3C, top), H3K9me3 signal (Figure S3C, bottom), and silenced gene expression (Figure S3D) in both FXS_425 and parent-clone FXS_421. Second, we generated H3K9me3 ChIP-seq libraries in the same seven normal-length, premutation-length, and mutation-length iPSC parent lines as examined for iPSC-NPCs (Figure S3E–G). The H3K9me3 domain was nearly identical in both pluripotent iPSCs and multipotent iPSC-NPCs from the same genetic background (Figure S3E–G). Thus, the X-chromosome H3K9me3 domain signal is robust in iPSC subclones from the same FXS parent line and iPSC and iPSC-NPCs from the same genotype.
We next queried if H3K9me3 signal could be detected in brain tissue derived from post-mortem brains from N=2 male FXS patients (71 and 80 years old, respectively) and N=2 sex- and age-matched normal-length individuals (STAR Methods). Using caudate nucleus tissue previously implicated as affected in FXS neuroanatomical studies26, we performed CUT&RUN for H3K9me3. We observed spreading of H3K9me3 across the FMR1 gene, as well as SLITRK2 and SLITRK4, in FXS patient-derived caudate nucleus tissue samples (Figure 1J). Such signal was not present in matched tissue from normal-length individuals. Thus, the H3K9me3 signal encompassing FMR1, SLITRK2, and SLITRK4 in FXS patient-derived post-mortem brain tissue is unlikely to be an artifact due to iPSC reprogramming methods, clonal variation in cell lines, or tissue culture selective pressure.
Finally, we created H3K9me3 ChIP-seq and RNA-seq libraries in EBV-transformed lymphoblastoid B-cell lines (Table S3) (hereafter referred to as B-lymphoblastoid cells). In B-lymphoblastoid cells with a normal-length CGG tract, FMR1 is expressed at low levels and the neural adhesion genes SLITRK2/4 are developmentally silenced (Figure S3H). Consistent with gene expression patterns, the X-chromosome H3K9me3 domain spans silenced SLITRK2/4 in normal-length B-lymphoblastoid cells and spreads ~300 kb downstream to encompass and silence FMR1 upon mutation-length expansion (Figure S3I–J). Thus, in FXS patient-derived iPSC-NPCs, the X-chromosome H3K9me3 domain arises de novo, whereas in FXS patient-derived B-lymphoblastoid cells it spreads over the mutation-length FMR1 CGG STR. Because the neural adhesion genes SLITRK2/4 are developmentally silenced and heterochromatinized in the B-cell lineage, our working model is that H3K9me3 domains can arise during healthy development to silence genes in off-target lineages or arise in FXS via CGG STR length-dependent mechanisms.
FXS-recurrent H3K9me3 domains are acquired on autosomes and encompass silenced genes linked to synaptic plasticity, neural cell adhesion, and epithelial integrity
We unexpectedly identified ten additional genomic locations on autosomes in which large (>300 kb up to multiple Mb) H3K9me3 domains were acquired in all three of our mutation-length FXS iPSC-NPCs along with negligible HK9me3 signal in all four of our normal-length and premutation iPSC-NPCs (Figure 2A, Figure S3K). Our observation of FXS-recurrent H3K9me3 domains on autosomes is particularly unexpected given that the CGG STR expansion is on the X-chromosome. One such domain encompasses the synaptic gene DPP6 located on chromosome 7 (Figure 2B)27. Similar to the X-chromosome, we observe H3K9me3 domain deposition, TAD ablation, and loss of CTCF occupancy around DPP6 in all three FXS lines (Figure 2B). DPP6 mRNA levels decrease in all three FXS iPSC-NPCs compared to normal- and premutation-length (Figure 2C). The reproducible decrease in DPP6 in our FXS iPSC-NPCs is noteworthy because loss of DPP6 disrupts spine density and functional synapses, which is a pathological hallmark of FXS27. In aggregate for autosomal FXS-recurrent domains, we observed loss of CTCF occupancy (Figure 2D), TAD boundary disruption (Figure 2E), and a marked reduction in gene expression (Figure 2F). Our data reveal that Mb-scale H3K9me3 domains corresponding to severe genome misfolding and loss of CTCF occupancy are present on autosomes in mutation-length iPSC-NPCs.
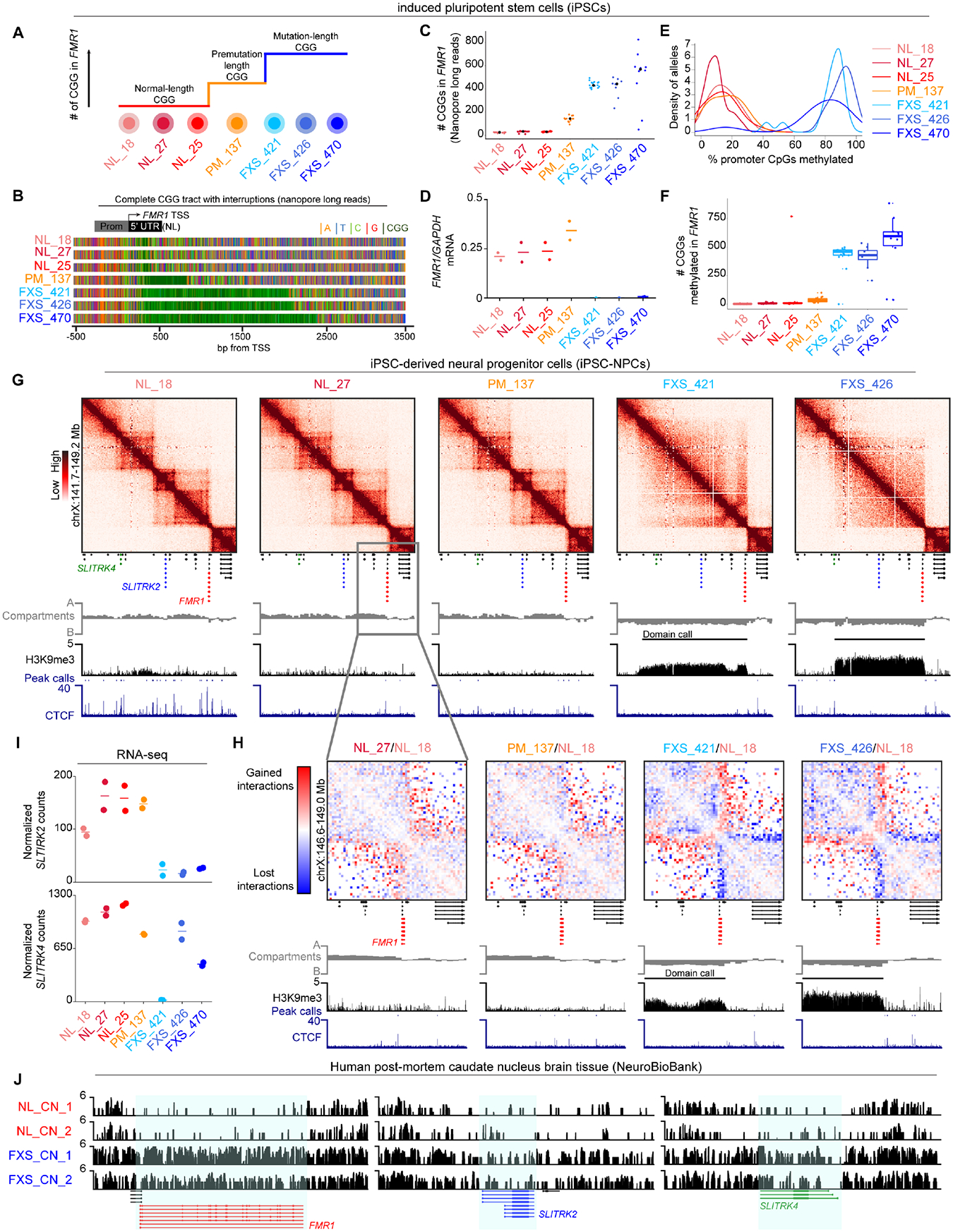
Figure 2. Heterochromatin domains and synaptic gene silencing on autosomes in FXS patient-derived iPSC-NPCs and brain tissue.
(A) Two classes of autosomal H3K9me3 domains (i) FXS-recurrent: consistently gained in all three FXS iPSC-NPCs and not in NL/PM iPSC-NPCs or (ii) Genotype-invariant: present in NL/PM/FXS iPSC-NPCs. (B) Hi-C and ChIP-seq for a 3.5 Mb region around a H3K9me3 domain encompassing DPP6. (C) DPP6 mRNA levels via RNA-seq. Horizontal lines, mean n=2 biological replicates. (D) Average H3K9me3 and CTCF ChIP-seq signal across autosomal FXS-recurrent H3K9me3 domains. (E) Boundary strength in NL_18 and FXS_426 iPSC-NPCs for one TAD boundary per autosomal FXS-recurrent H3K9me3 domain. (F) mRNA levels via RNA-seq for N=25 expressed protein-coding genes in autosomal and chrX FXS-recurrent H3K9me3 domains. Each point, mean per gene n=2 biological replicates. P-values, one-tailed MWU, where * P-value <0.05 versus NL_18. (G) Gene ontology for all N=36 protein-coding genes in autosomal and chrX FXS-recurrent H3K9me3 domains. (H) Expression of N=54 coding/noncoding genes in FXS-recurrent H3K9me3 domains across GTEX tissues. (I) Number of autosomal H3K9me3 domains arising in FXS patient-derived brain tissue compared to sex- and age-matched normal-length control tissue. (J) H3K9me3 CUT&RUN in brain tissue from N=2 FXS patients with sex- and age-matched N=2 normal-length individuals at DPP6, RBFOX1, and CSMD1.
We next conducted ontology analysis on protein-coding genes in FXS-recurrent domains in iPSC-NPCs. Autosomal FXS-recurrent H3K9me3 domains, and not genotype-invariant H3K9me3 domains, are enriched for genes encoding synaptic plasticity and neural cell adhesion (Figure 2G, Figure S3L). Long genes in autosomal domains with an established role in synaptic plasticity include RBFOX1, PTPRT, CSMD1, and DPP6 (Figure S3K). Although we see both gain and loss of expression genome-wide in FXS iPSC-NPCs, the genes in the FXS-recurrent H3K9me3 domains are largely downregulated upon mutation-length expansion (Figure S3M). We also identified H3K9me3 domains present in only one FXS line (so-called FXS-variable H3K9me3 domains). Genes co-localized with FXS-variable H3K9me3 domains were also enriched for synaptic and neural cell adhesion ontology (Figure S3N). Thus, autosomal domains encompass repressed synaptic genes in FXS iPSC-NPCs, which is of particular interest given the synaptic and cognitive defects reported in FXS patients28.
Soft skin, connective tissue defects, and macroorchidism are non-neurologic clinical presentations in FXS29. We examined RNA-seq profiles for coding and non-coding genes co-localized with FXS-recurrent H3K9me3 domains across 54 tissues from the GTEX consortium. Genes localized in FXS-recurrent heterochromatin domains from iPSC-NPCs exhibit tissue-specific expression profiles indicative of testis, epithelium, and brain (Figure 2H). We also re-analyzed RNA-seq data published in human fetal brain tissue from a healthy, normal-length male and a mutation-length male FXS patient30. We found multiple synaptic genes, including FMR1, DPP6, and RBFOX1, silenced in the FXS patient-derived fetal brain tissue compared to sex-matched normal-length fetal brain tissue (Figure S4A–D, Table S4). These observations suggest that genes silenced by autosomal H3K9me3 domains could be relevant to other tissues and cell types impacted in FXS beyond NPCs.
Autosomal H3K9me3 domains occur in iPSC-NPCs, B-lymphoblastoid cells, and post-mortem brain tissue derived from FXS patients with mutation-length CGG STR expansion
To further confirm that the phenomenon of autosomal H3K9me3 domains could occur in somatic cells that have never undergone iPSC reprogramming, we created H3K9me3 ChIP-seq libraries in normal-length and mutation-length B-lymphoblastoid cells. We found that 4/10 of the H3K9me3 domains found in FXS iPSC-NPCs also arise de novo in FXS compared to NL B-lymphoblastoid cells (Figure S4E–F). We additionally found that 6/10 of the H3K9me3 domains found in FXS iPSC-NPCs – specifically the domains spanning synaptic genes - are heterochromatinized in both normal-length and mutation-length B-lymphoblastoid cells (Figure S4E–F). Importantly, we also found autosomal H3K9me3 domains that reproducibly spread (Figure S4G) or arise de novo (Figure S4H) in FXS mutation-length compared to normal-length B-lymphoblastoid cells. Such domains are specific to FXS B-lymphoblastoid cells and not present in FXS iPSC-NPCs, and they correlate with the expected decrease in autosomal gene expression (Figure S4I). Together, these results further support our working model that Mb-scale H3K9me3 domains can arise on autosomes and the X-chromosome through at least two mechanisms: (1) in neural lineages where synaptic genes are expressed, domains can arise or spread in FXS patient-derived cells with mutational-length CGG and will not be present in normal-length or (2) in off-target lineages where genes are not physiologically relevant (such as synaptic genes in B-lymphoblasts), both normal-length and mutation-length genotypes will acquire H3K9me3 domains via developmental mechanisms.
Finally, we investigated our H3K9me3 CUT&RUN data from caudate nucleus post-mortem brain t issue for the presence or absence of autosomal H3K9me3 domains. In both male FXS patients, we find domain-like H3K9me3 signal at all FXS-recurrent domain locations originally found in iPSC-NPCs, including the synaptic genes DPP6, RBFOX1, and CSMD1 (Figure 2I–J). Specifically, we find that 4/11 of the original FXS-recurrent heterochromatin domain locations reproducibly gain Mb-scale de novo domain-like H3K9me3 signal in FXS patient-derived post-mortem caudate nucleus tissue (Figure 2I–J, Figure S4J–L). There is negligible H3K9me3 signal in sex- and age-matched tissue from normal-length individuals. We also observe that 7/11 of the original FXS-recurrent heterochromatin domain locations exhibit spreading of H3K9me3 in FXS individuals (Figure S4J–L). Altogether, our data confirm that autosomal H3K9me3 domains can occur in FXS patient-derived brain tissue and are unlikely to be solely due to artifacts from tissue culture selective pressure or iPSC reprogramming.
Engineering the CGG STR from mutation-length to premutation-length reverses FXS-recurrent heterochromatin domains on the X-chromosome and a subset of the autosomes
Previous studies have reported FMR1 de-repression and local removal of H3K9me3 only at the FMR1 promoter due to excision of the CGG STR tract31,32. We sought to understand how cut-back of the mutation-length CGG STR affects the maintenance of FXS-recurrent H3K9me3 domains on the X-chromosome and on autosomes. Starting with the FXS_421 mutation-length parent iPSC line, we used a CRISPR engineering strategy to cut-back the CGG STR (STAR Methods). We screened over 900 clones to identify single-cell-derived clonal iPSC lines with FMR1 de-repression (STAR Methods). We identified 7 clones with at least 30-fold FMR1 de-repression and 7 matched single-cell-derived clones with maintained FMR1 silencing (Figure 3A–B, Figure S5). Upon evaluation of CGG STR length with our targeted Nanopore long-read assay (Figure 1), we observed that 7/7 engineered clones with de-repressed FMR1 mRNA levels also represented bona fide premutation-length (100–199 CGGs) cut-back clones (Figure 3C). It is noteworthy that no normal-length clones were recovered in our 900-clone screen using two stringent thresholds as read-outs: (i) FMR1 de-repression of at least 30-fold compared to the mutation-length parent line (FXS_421) and (ii) expression levels to within 2-fold of normal-length iPSCs. We confirmed that all 7 matched single-cell clones from the mutation-length parent line exhibited FMR1 silencing and remained at mutation-length (Figure 3C). These data demonstrate successful generation of a cohort of N=7 single-cell-derived iPSC clones exhibiting both FMR1 de-repression and bona fide premutation-length CGG STR cut-back, as well as N=7 matched single-cell-derived mutation-length iPSC clones with sustained FMR1 silencing (Figure 3A–C, Figure S5).
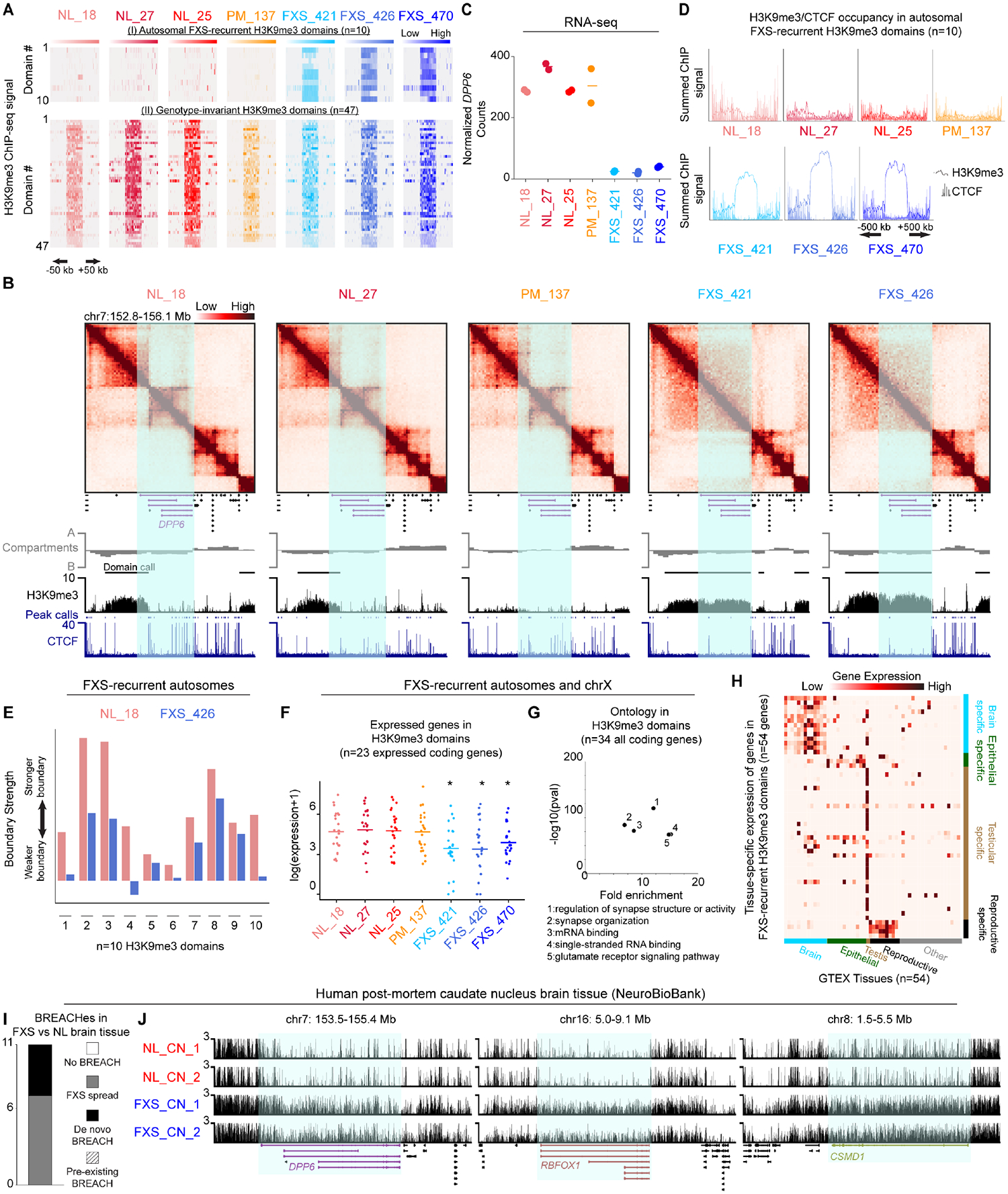
Figure 3. Engineering the mutation-length FMR1 CGG STR to premutation-length attenuates a subset of H3K9me3 domains and de-represses gene expression.
(A) Schematic of N=7 mutation-length and premutation-length single-cell-derived CGG CRISPR cut-back iPSC clones generated from the FXS_421 parent iPSC line. (B) FMR1 mRNA levels normalized to GAPDH and shown relative to FXS_421 using qRT-PCR. Error bars, standard deviation n=2 biological replicates. (C) Number of CGG triplets in the FMR1 5’UTR computed from Nanopore long-reads. (D) Average input normalized H3K9me3 signal for the chrX FXS-recurrent H3K9me3 domain. Dots represent equal sized bins (N=5) across the domain. (E) FXS-recurrent H3K9me3 domains amenable (red) and refractory (black) to reprogramming. For each domain, we measured the fraction of iPSC clones with persistent, lowered, or removed H3K9me3 signal for all mutation-length (N=7) and premutation-length (N=7) clones. (F) Hi-C and ChIP-seq for a 5 Mb region around FMR1 in FXS_421 and PMcut_scClone1 iPSCs. (G) Log2 fold change of gene expression in FXS_421 vs. PMcut_scClone1 with respect to NL_18. Each dot, one gene. P-values, one-tailed MWU.
We next investigated the H3K9me3 signal in our single-cell CGG STR tract engineered iPSC clones. We observed that the Mb-sized H3K9me3 domain on the X-chromosome is reproducibly reversed in all N=7/7 clones representing cutback to premutation-length (Figure 3D–E, Figure S5A–D). Corroborating the loss of H3K9me3, CTCF occupancy was restored, and TAD boundaries were re-instated at the broader FMR1 locus upon mutation-length to premutation cut-back (Figure 3F). The H3K9me3 reversal effect after mutation-length to premutation-length cutback was substantially higher frequency (7/7 clones) compared to random noise observed in the mutation-length cutback (1/7 clones). Our results reveal that endogenous cut-back of the mutation-length CGG STR to premutation-length can fully reverse the X-chromosome H3K9me3 domain, de-repress FMR1 gene expression, and re-fold disrupted higher-order chromatin folding patterns on the X-chromosome in FXS iPSCs.
We next sought to understand the extent to which autosomal H3K9me3 domains in FXS could be reversed upon engineering of the FMR1 CGG tract on the X-chromosome. Unexpectedly, we observed that a subset of autosomal H3K9me3 domains lost H3K9me3 signal upon engineering to the FMR1 CGG STR premutation-length (Figure 3E (left panel), Figure S5E–F). Most notably, the H3K9me3 domains on chromosome 5 (IRX2), chromosome 17 (SHISA6), and chromosome 16 (RBFOX1) were nearly completely removed across all premutation cut-back clones (Figure S5G–H). Negligible fluctuation in H3K9me3 signal was observed in the single-cell clones derived from the mutation-length parent line. Genes were in large part de-repressed within the domain segments which lost H3K9me3 signal upon CGG premutation cut-back (Figure 3G). The genes encompassed by H3K9me3 domains refractory to reprogramming include: COL22A1, CSMD1, DPP6, PTPRT, TCERG1L, TMEM132C, LINC01591, MYOM2, SHISA6, and FAM135B. Genotype-invariant H3K9me3 domains were unaffected by the CGG tract engineering (Figure S5I–J). Together, these results indicate that the mutation-length CGG tract is necessary for the maintenance of H3K9me3 signal at a subset of heterochromatin domains.
Autosomal FXS-recurrent domains spatially co-localize with FMR1 via inter-chromosomal interactions that are reversible upon removal of H3K9me3
We sought to gain insight into the extent to which genomic loci on autosomes make physical contact with FMR1. Using Hi-C, we observed unusually strong inter-chromosomal interactions connecting the FMR1 locus to autosomal H3K9me3 domains in iPSC-NPCs with mutation-length CGG expansion (Figure 4A–B). Autosomal H3K9me3 domains contact each other as well as the X-chromosome, suggesting they form multi-way subnuclear hubs with FMR1 in FXS (Figure 4C, Figure S6A–B). All seven of our iPSC lines exhibit largely normal karyotype, and do not display Mb-scale copy number variations that would artifactually cause trans interaction signal (Figure S1C–D). These data indicate that autosomal FXS-recurrent heterochromatin domains engage via trans interactions with FMR1 upon mutation-length CGG expansion.
Figure 4. Autosomal heterochromatin domains spatially connect with FMR1 via inter-chromosomal interactions in FXS.
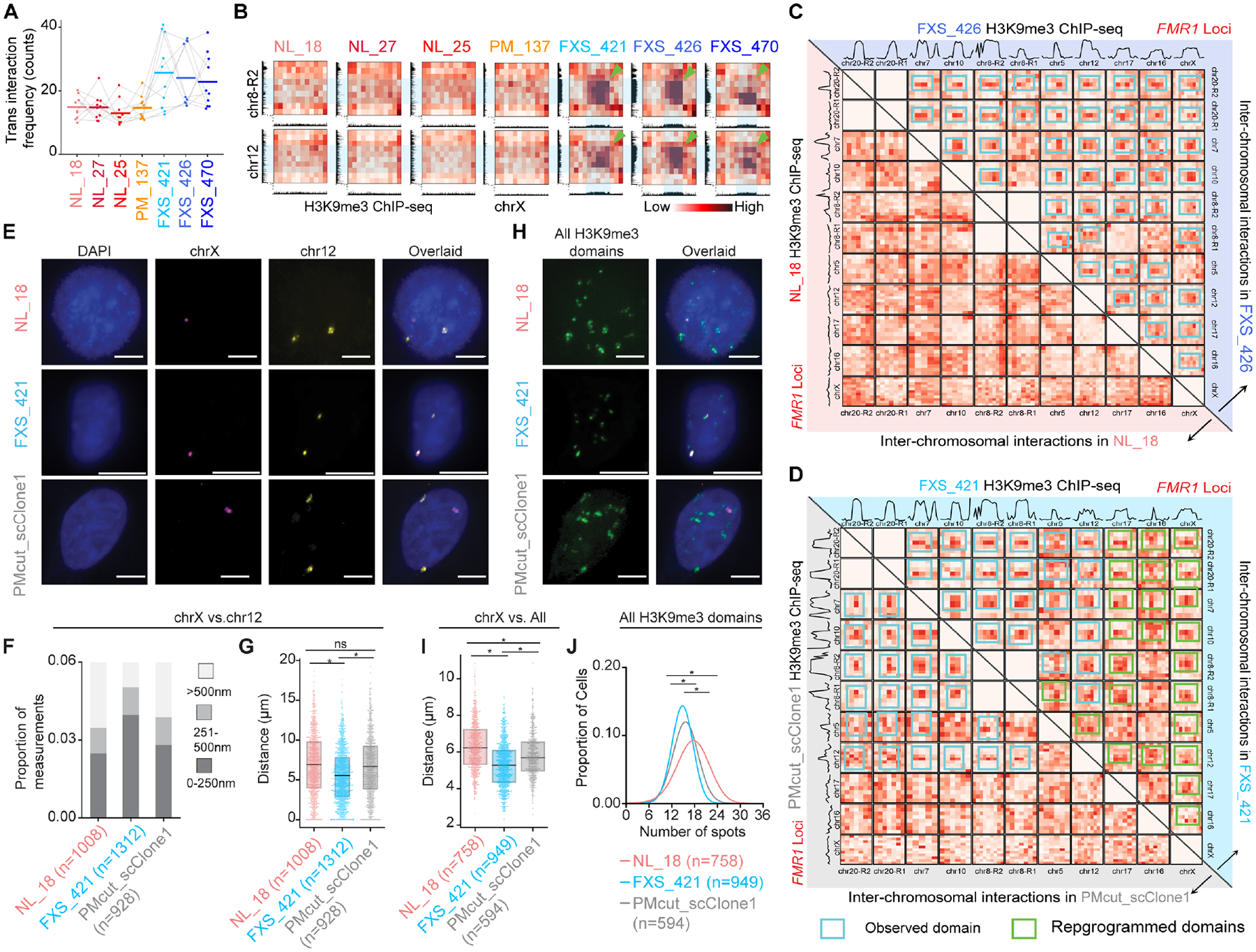
(A) Trans interactions between each of the N=10 FXS-recurrent H3K9me3 domain on autosomes and FMR1 on chrX. (B) Hi-C inter-chromosomal interaction heatmaps binned at 1 Mb resolution. Green arrows, trans interactions. (C-D) Hi-C inter-chromosomal interactions among FXS-recurrent H3K9me3 domains (C) FXS_426 (upper triangle) versus NL_18 (lower triangle) iPSC-NPCs and (D) FXS_421 (upper triangle) versus PMcut_scClone1 (lower triangle) iPSCs. H3K9me3 ChIP-seq signal plotted above Hi-C heatmaps. Blue boxes, FXS-gained trans interactions. Green boxes, attenuated trans interactions after premutation-length cutback. (E+H) DNA FISH images for the H3K9me3 domain on chrX interacting with (E) the chr12 domain or (H) all domains in NL_18, FXS_421, and PMcut_scClone1 iPSC nuclei. Scale bars, 10 μm. (F-G) Distances between chrX and chr12 H3K9me3 domains in iPSCs, including (F) proportion of measurements stratified by distance and (G) measurements directly compared with a two-tailed MWU, where * P-value <1E-6. (I) Average distance per cell between the chrX and all other FXS-recurrent H3K9me3 domains. (J) Kernel density estimation of the number of foci per nucleus. (I-J) Two-tailed MWU, where * P-value <1E-12.
We also sought to determine if the trans interactions changed upon CRISPR engineering to a premutation-length CGG tract. Although many autosomal H3K9me3 loci remained tethered in a trans interaction hub, the FMR1 locus and the subset of autosomal domains which lost their H3K9me3 signal also spatially disconnected from the other loci upon engineering of the mutation-length CGG to premutation (Figure 4D). To validate the trans interactions, we also used Oligopaint DNA FISH probes to image H3K9me3 domains in single cells (Figures 4E–J, Table S5). We observed that the H3K9me3 domains on chrX and chr12 are closer together in a higher proportion of mutation-length vs. normal-length single iPSCs (Figure 4E–G). The chrX H3K9me3 domain is closer on average to all autosomal H3K9me3 domains, and all H3K9me3 domains coalesce into fewer resolvable subnuclear hubs in mutation-length compared to normal-length iPSC nuclei (Figure 4H–J). Consistent with our Hi-C results, we observe that engineering the CGG tract to premutation-length restores the spatial distance between chrX and chr12 domains to resemble the normal-length condition (Figure 4E–G). Thus, using ensemble Hi-C and single-cell imaging methods, we demonstrate that autosomal H3K9me3 domains form CGG-length-dependent trans interactions with the FMR1 H3K9me3 domain in FXS.
Autosomal FXS-recurrent H3K9me3 domains harbor long transcribed genes, replication stress-induced double strand breaks, and replicate at the end of S-phase
We sought to identify features that could provide insight into why H3K9me3 is deposited on distinct autosomal locations in iPSCs. Because heterochromatin generally protects and silences the repetitive genome33, we hypothesized that H3K9me3 marks loci susceptible to genetic instability. We first observed that autosomal H3K9me3 domains are gene poor and harbor significantly longer genes than those in size- and chromosome arm-matched random genomic intervals (Figure 5A–B). All autosomal H3K9me3 domains, as well as the domain encompassing FMR1, exhibit late replication timing at the end of S phase in normal-length iPSCs, which has previously been reported at genes susceptible to replication-associated fragile sites34 (Figure 5C). FXS-recurrent H3K9me3 domains are also strongly enriched with recurrent replication stress-mediated double strand breaks35 (Figure 5D). Such patterns are not enriched at genotype-invariant H3K9me3 domains present across all NL, PM, and FXS iPSC lines (Figure 5E–H). Several key long synaptic genes in the autosomal FXS-recurrent H3K9me3 domains, including RBFOX1, DPP6, and PTPRT, replicate at the end of S-phase and co-localize with replication stress-induced double strand breaks (Figure 5I). Finally, we also demonstrate that genes with normal-length CGG STR tracts in the first 2 kb of their promoter are significantly enriched in autosomal H3K9me3 domains (Figure 5J–K). Together, these data suggest that autosomal H3K9me3 domains in FXS iPSC-NPCs encompass genomic loci replicating at the end of S-phase and susceptible to genome instability in the form of replication stress-induced double strand breaks.
Figure 5. Autosomal H3K9me3 domains are enriched for late replicating long synaptic genes and replication stress-induced double strand breaks.
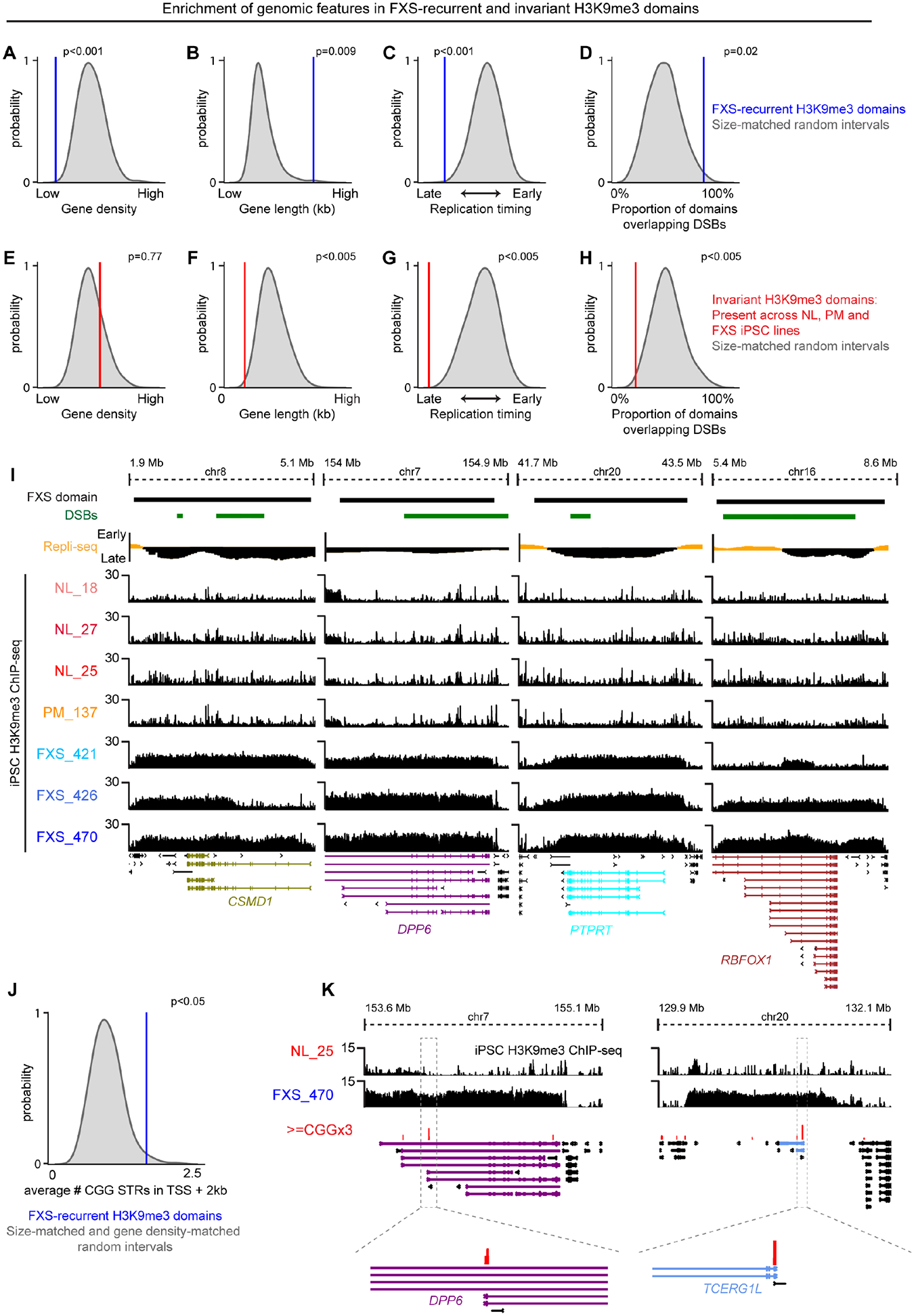
(A-H) Empirical randomization test assessing the enrichment of (A+E) gene density, (B+F) gene length, (C+G) replication timing, and (D+H) replication stress-induced double stranded breaks in (A-D) FXS-recurrent H3K9me3 domains or (E-H) genotype-invariant H3K9me3 domains compared to N=1000 draws of random genomic intervals matched by size. (I) FXS-recurrent H3K9me3 domains encompassing CSMD1 (gene length: ~2.10 Mb), DPP6 (gene length: ~1.15 Mb), PTPRT (gene length: ~1.16 Mb), and RBFOX1 (gene length: ~2.47 Mb). Replication stress-induced double strand breaks, dark green. Replication timing, yellow (early S phase) and black (late S phase). (J) Empirical randomization test assessing the enrichment of CGG tracts (>=CGGx3) in TSSs + 2kb within FXS-recurrent H3K9me3 domains compared to N=1000 draws of random genomic intervals matched by size. (K) Examples of CGG tracts in FXS-recurrent H3K9me3 domains encompassing DPP6 and TCERG1L.
Autosomal FXS-recurrent H3K9me3 domains harbor STRs prone to stepwise somatic instability in FXS iPSCs and EBV-transformed B-lymphoblasts
Stepwise instability of STR tracts on autosomes was reported recently in individuals with autism spectrum disorder (ASD) using the GangSTR and Expansion Hunter (EH) computational methods36,37. We used PCR-free whole genome sequencing coupled with GangSTR and EH to ascertain if STR instability on autosomes could be observed in our FXS iPSCs. We computed STR length for >830,000 STR tracts on autosomes genome-wide in N=3 FXS iPSC lines as well as in N=120 ancestry-, sex-, sequencing depth-, and cell type-matched non-diseased, normal-length individuals from the HipSci Consortium (Figure 6A). We formulated a statistical test (>830,000 tests, 1 per STR tract) in which we identified autosomal alleles with significantly longer STR tracts in our FXS iPSC lines compared to the expected null distribution of tract lengths in N=120 iPSCs (240 alleles) from normal-length individuals (Figure 6A, STAR Methods). We identified N=71 “FXS long STRs” on autosomes which are reproducibly called with both GangSTR and EH as significantly longer in all 3/3 FXS iPSC lines compared to the population of N=120 normal-length iPSCs (Figure 6B, Figure S6C, Table S6).
Figure 6. Autosomal FXS-recurrent H3K9me3 domains can harbor STR tracts prone to stepwise somatic instability.
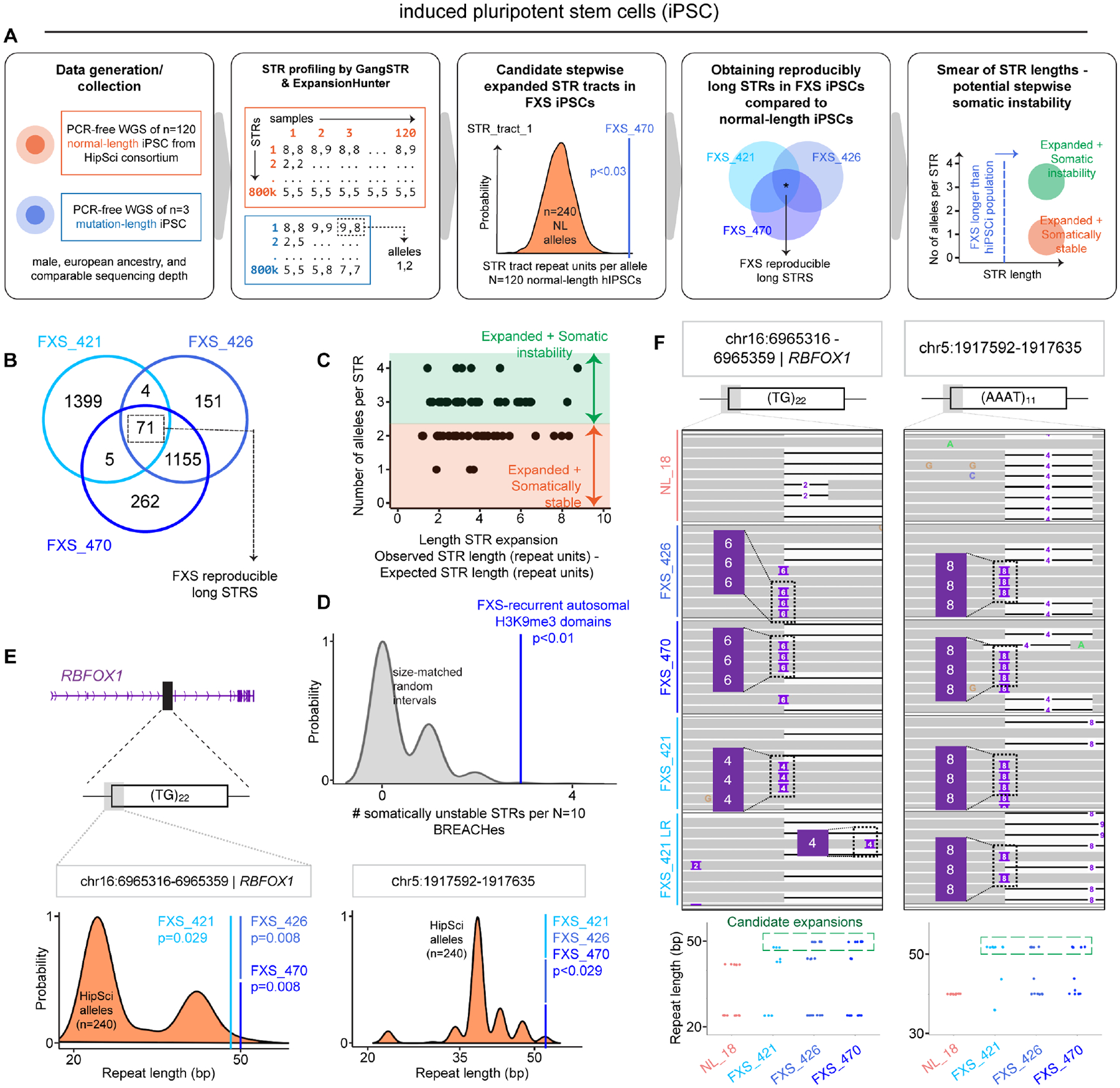
(A) Schematic depicting the pipeline for identifying candidate long STRs with potential for somatic instability using GangSTR and ExpansionHunter. (B) Venn diagram depicting “FXS long STRs” identified in FXS iPSCs as significantly longer than expected in N=120 ancestry-, sex-, sequencing depth-, and cell type-matched normal-length individuals. (C) Stratification of “FXS long STRs” into those exhibiting patterns potentially consistent with somatic instability (green: >=3 alleles per FXS iPSC line per STR) and those that do not (orange: somatically stable). (D) Empirical randomization test assessing the enrichment of FXS-reproducible stepwise somatically unstable STRs in FXS-recurrent H3K9me3 domains compared to N=1000 draws of random genomic intervals matched by size. (E) Distribution of STR tract length (bp) across N=240 alleles of ancestry-, sex-, sequencing depth-, and cell type-matched normal-length HipSci iPSC lines. Overlayed blue dashed lines indicate the maximum STR length in each of the three FXS iPSC lines. Empirical one-tailed P-value. Distributions shown for “FXS long STRs” in RBFOX1 (left) and an intergenic region on chr5 (right). (F) Representative reads for direct visualization of stepwise STR expansion events in short-reads across all 3 FXS iPSC lines as well as verified in FXS_421 with Nanopore long-reads (top). STR lengths computed directly from reads via the CIGAR string (bottom).
To test our hypothesis that “FXS long STRs” might represent candidates for potential somatic instability, we created a custom algorithm to compute the number of unique tract lengths identifiable in PCR-free sequencing reads for each individual STR (STAR Methods). We stratified our reproducible set of “FXS long STRs” into those exhibiting three or more tract lengths potentially indicative of somatic instability (‘candidate FXS somatically unstable STRs’, N=53) and those that had the expected 1–2 alleles (‘FXS long but somatically stable’, N=18) (Figure 6C, Figure S6D). We confirmed that “FXS long STRs” are significantly more associated with somatic instability in each FXS iPSC line compared to STRs which do not change in length across the normal-length HipSci population (Figure S6E). We observed that ‘candidate FXS somatically unstable STRs’ are enriched in FXS-recurrent H3K9me3 domains compared to size-matched random intervals (Figure 6D), including noteworthy examples in the long synaptic genes PTPRT38 and RBFOX139 previously linked to ASD in case-control studies (Figure 6E–F, Figure S7). Finally, we independently validated the allelic variation at these key STRs with nanopore long-read sequencing (Figure S7). Altogether, our analyses uncover candidate stepwise somatic STR instability events co-localized with Mb-scale autosomal H3K9me3 domains in FXS iPSCs, therefore we term them BREACHes - Beacons of Repeat Expansion Anchored by Contacting Heterochromatin.
BREACH-silenced genes exhibit minimal overlap with repressed genes in Fmr1 knock-out mice
We next investigated the extent to which genes silenced due to Fmr1 knock-out overlapped BREACH-silenced genes from human model systems with mutation-length CGG expansion. We re-analyzed published RNA-seq data examining the down- and up-regulation of genes in mouse embryonic neurons due to Fmr1 (and FMRP) knock-out17. We demonstrate that the genes repressed by BREACHes in FXS iPSC-NPCs with mutation-length CGG expansion are generally not repressed in embryonic neurons from Fmr1 knock-out mice (Figure 7A–C). Our data suggest that BREACH-silenced genes in cell lines with mutation-length CGG expansion might be independent of genes silenced due to the loss of FMRP and its downstream signaling pathways.
Figure 7. Specific normal-length iPSC lines made with p53 perturbation exhibit an intermediate level of H3K9me3 signal at BREACHes.
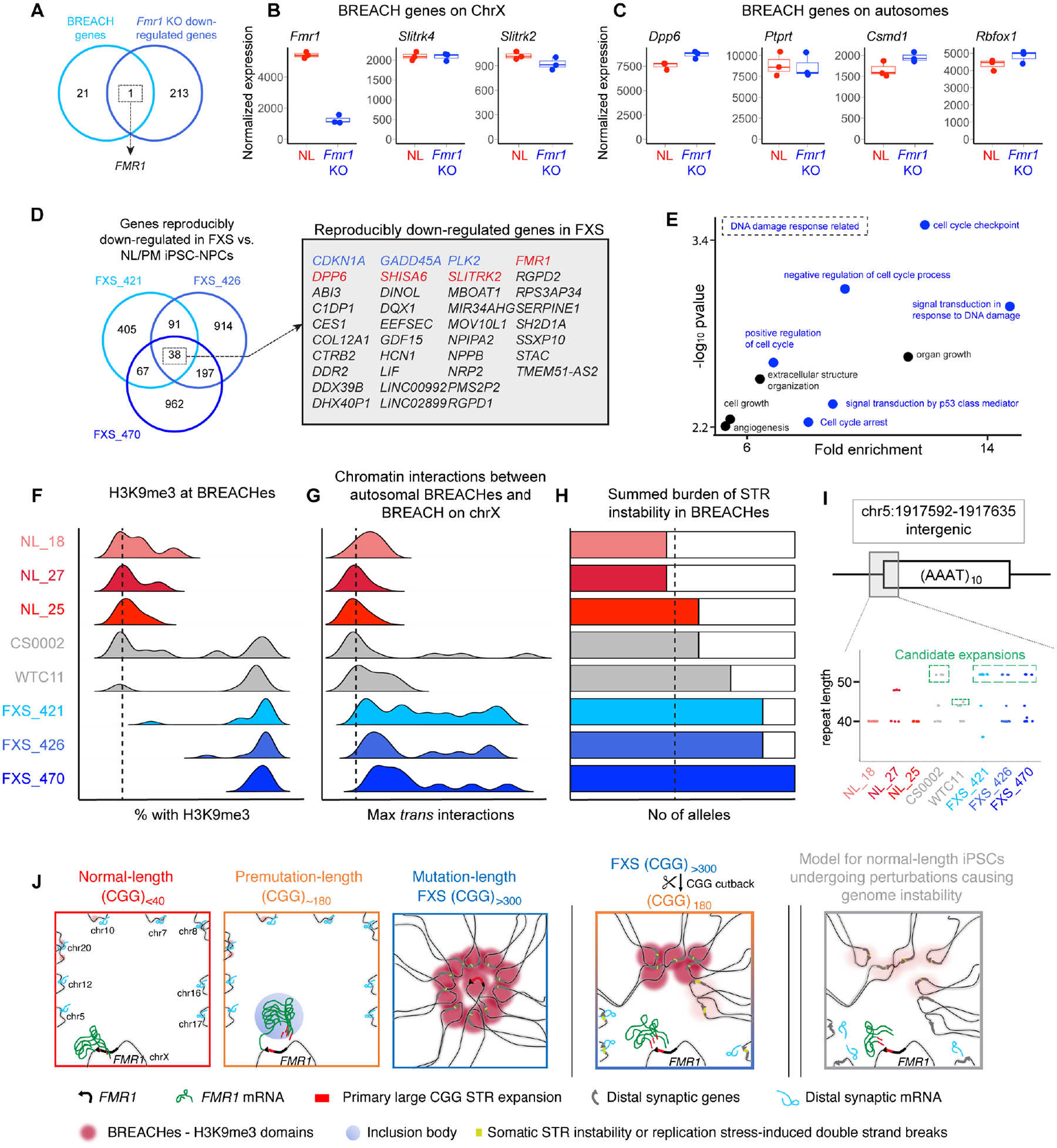
(A) Venn diagram showing the overlap between the genes localized with BREACHes from this study and down-regulated genes in Fmr1 knock-out mouse cortical neurons. (B-C) RNA-seq17 comparing expression of BREACH-localized genes in normal-length versus Fmr1 knock-out neurons. (D) Venn diagram showing reproducibly down-regulated genes (n=38) in mutation-length FXS compared to normal-length and premutation iPSC-NPCs. Red genes localize with BREACHes. Blue genes are linked to the DNA damage response. (E) Gene ontology for reproducibly down-regulated genes (n=34) not present in BREACHes. (F-H) Genomic features at BREACHes in normal-length iPSCs (red) and FXS iPSCs from this study derived without p53 shRNA (blue), as well as two prototypic iPSC lines derived with p53 shRNA (grey). (F) H3K9me3, (G) trans interaction frequency, and (H) summed burden of STR instability. (I) STR length computed directly from reads via the CIGAR string for an AAAT tract on chr5. (J) Schematic model of BREACHEs – Beacons of Repeat Expansion Anchored by Contacting Heterochromatin.
DNA damage and p53-mediated cell cycle arrest pathways are disrupted in human FXS iPSC-NPCs with mutation-length CGG expansion
To shed light on possible signaling pathways linked to genome instability in FXS, we examined RNA-seq in our NL, PM, and ML FXS iPSC-NPC lines. We identified 38 genes genome-wide that were reproducibly downregulated in all 3 FXS iPSC-NPC lines compared to our 3 NL and 1 PM iPSC-NPC lines (Figure 7D). While genes in BREACHes exhibited synaptic ontology, non-BREACH silenced genes (N=34) were enriched in the pathways of the DNA damage response, DNA integrity checkpoints, and p53-mediated cell cycle arrest (Figure 7D–E). It is particularly noteworthy that three tumor suppressor genes were reproducibly silenced, including: (1) a kinase inhibitor, CDKN1A, linked to cell viability during DNA damage40, (2) a kinase, PLK2, involved in cell cycle regulation due to stress-induced DNA damage41,42, and (3) a chromatin regulatory factor, GADD45A, implicated in cell cycle arrest in response to environmental stress43–45. These data suggest that signaling pathways linked to the DNA damage response are reproducibly dysregulated in human iPSC-NPC lines with mutation-length CGG expansion.
Intermediate levels of H3K9me3 signal can occur at BREACHes in normal-length iPSCs exposed to molecular perturbations linked to general genome instability
Given the co-localization of autosomal BREACHes with double-strand breaks and somatic STR instability (Figures 5–6), and the reproducibly dysregulated DNA damage response pathways in FXS cell lines (Figure 7D–E), we hypothesized that Mb-scale heterochromatin domains might have broader relevance beyond FXS in other genetic and pharmacological perturbations linked to genome instability.
We examined publicly available H3K9me3 data from NL iPSCs outside of our lab’s lines which have been subjected to perturbations linked to genome instability (Figure S8). We selected p53 perturbation as a proof-of-principle because it is a well-studied guardian of the genome in which knock-down is reported to increase genome instability and lead to global accumulation of ectopic H3K9me3 in cancer46,47. In the present study, we curated and studied NL, PM, and ML iPSC lines that were matched by ancestry, sex, somatic cell type, and derived without p53 perturbation (Figures 1–5, Table S1). However, more generally, we posited that treatment with p53 shRNA or p53 dominant-negative overexpression during the reprogramming process, which is known to cause karyotype instability in iPSC genomes48, might correlate with H3K9me3 signal in normal-length iPSC lines cultured outside our laboratory’s cohort.
We downloaded and re-analyzed publicly available H3K9me3 ChIP-seq for 11 normal-length male and female human iPSC lines, and also created H3K9me3 ChIP-seq or CUT&RUN data for 5 additional normal-length male human iPSC lines, across a range of ancestries, parent cell types, and reprogramming methods (Figure S8, Tables S3+S4). We stratified N=16 normal-length iPSC lines into those reprogrammed with and without the use of p53 shRNA or p53 dominant-negative overexpression. We observed that the subset of iPSC lines reprogrammed using p53 perturbations showed H3K9me3 signal at several autosomal locations of FXS-recurrent BREACHes (Figure S8A–C). Similarly, on the X-chromosome BREACH, we observed H3K9me3 domain signal upstream of FMR1 in the normal-length male iPSC lines created with p53 perturbation (Figure S8D-F). By contrast, there was negligible or sporadically placed H3K9me3 signal at autosomal and X-chromosomal BREACH locations across most normal-length iPSC lines derived without the use of p53 perturbations, including our own study’s lines (Figure S8A–F). Together, these initial observations suggest that genomic loci spanned by BREACHes in FXS iPSCs might also be susceptible to heterochromatinization in normal-length iPSCs subjected to perturbations which cause genome instability.
We focused on ascertaining if there was evidence for an elevated H3K9me3 signal or burden of genome instability at BREACHes in 2 specific normal-length iPSC lines, WTC11 and CS0002, made with p53 shRNA (Figure S8, Table S1). Consistent with Figure S8, these 2 iPSC lines exhibit a bimodal, intermediate level of H3K9me3 signal at some but not all BREACHes – higher H3K9me3 than this study’s normal-length iPSCs and lower H3K9me3 than this study’s FXS iPSCs (Figure 7F). We created Hi-C data in CS0002 and mined published WTC11 Hi-C data from the 4DN consortium. As expected, both CS0002 and WTC11 iPSCs showed an intermediate level of trans interactions (Figure 7G). Using PCR-free whole genome sequencing, GangSTR, and our custom STR allele length quantification methods, we assayed stepwise somatic STR instability on autosomes in these two iPSC lines derived with p53 shRNA. We observed an increased burden of somatic instability in FXS-recurrent H3K9me3 domains in CS0002 and WTC11– more than this study’s normal-length iPSCs without H3K9me3 and less than this study’s FXS iPSC lines with strong H3K9me3 signal (Figure 7H–I). These observations suggest that normal-length iPSC lines reprogrammed with p53 shRNA can exhibit elevated H3K9me3 signal and increased burden of STR instability at BREACHes.
Altogether, our work highlights a link between BREACHes and genome instability in FXS iPSCs specifically, and also in a subset of reprogrammed iPSCs exposed to perturbations leading to STR instability generally, suggesting that BREACHes might have broad relevance to genome stability beyond the disease of FXS (Figure 7J).
Discussion
Classic models of FXS assert that it is a monogenic disorder in which CGG STR expansion causes local DNA methylation of the FMR1 promoter, leading to transcriptional silencing of FMR1 and loss of FMRP11,12,49. Our data in FXS patient-derived human cell lines and post-mortem brain tissue support a model of spatially coordinated transcriptional silencing via acquisition of Megabase-sized domains of the repressive histone modification H3K9me3 on autosomes and the X-chromosome (Figure 7J). When the CGG STR is normal-length, the FMR1 locus does not connect in trans with distal autosomes (Figure 7J, panel 1). FMR1 mRNA levels increase as the CGG tract expands to premutation-length and genome folding remains intact (Figure 7J, panel 2). Upon mutation-length expansion, we see local promoter DNA methylation and FMR1 silencing as in traditional models. We also observe BREACHes – Beacons of Repeat Expansion Attenuated by Contacting Heterochromatin – including ten Mb-sized H3K9me3 domains on autosomes and a 5 Mb block encompassing FMR1 on the X-chromosome. BREACHes cluster together spatially in trans and demarcate severe Mb-scale misfolding of TADs, subTADs, and loops in cis in many FXS patient-derived samples with mutation-length CGG expansion (Figure 7J, panel 3).
It is particularly noteworthy that BREACH-silenced genes are not ubiquitously and reproducibly silenced in Fmr1 knock-out cell lines and mouse models, suggesting that the CGG STR expansion event itself or a genetic background specific to FXS patients might be an important contributor to the range and severity of genome-wide transcriptional silencing in FXS beyond FMRP loss. Genes encompassed by autosomal BREACHes encode synaptic plasticity, neural adhesion, testis development, and epithelial integrity, which are known systems with clinical presentations in FXS28,29,50. Although preclinical studies are beyond the scope of the current work, we demonstrate the utility of Mb-scale trans interactions in guiding the identification of several FXS genes of interest for follow-on experiments using clinical endpoints.
A critical question arising from our work is whether engineering the length of the CGG STR could reverse BREACHes. Upon CGG cutback from mutation-length to premutation-length, we unexpectedly observe that BREACHes on the X-chromosome and a subset of autosomes lose H3K9me3 signal and spatially disconnect from FMR1 (Figure 7J, panel 4). Our observations of Mb-scale removal of heterochromatin and refolding of the genome extend substantially upon previous studies reporting that excision of the CGG tract results in local removal of H3K9me3 only at the FMR1 promoter31,32. Together, these data are consistent with a model in which mutation-length CGG STR is necessary for H3K9me3 maintenance of at a subset of BREACHes.
Our findings open questions regarding the mechanism(s) by which the mutation-length and premutation-length CGG STR tract or CGG-containing RNA contributes to the establishment, maintenance, and reversal of H3K9me3 at BREACHes. Mutation-length CGG-containing RNA has been implicated in the establishment of local FMR1 silencing via R loop formation during a critical window in early neural differentiation12. By contrast, the mechanisms governing maintenance of FMR1 silencing have not been identified. Here we hypothesize that BREACHes may be required for the long-term maintenance of gene silencing on the X-chromosome and on autosomes in at least some FXS patients. Our work also opens up future lines of inquiry for the exploration of the mechanistic interplay between long-range heterochromatin-mediated silencing and other known molecular phenotypes in FXS, including CGG-RNA-DNA R loops12,51,52, sequestration of specific proteins and the CGG-containing RNA in inclusion bodies53, repeat-associated non-AUG (RAN) translation of the toxic protein FMRpolyG54, alternative splicing defects55, and the downstream effects of FMRP loss17.
The FMR1 CGG STR on the X-chromosome is considered the only genetic mutation in FXS. Unexpectedly, we identified STR tracts on autosomes which exhibit potential for stepwise somatic instability in FXS patient-derived iPSCs in culture. Such stepwise events are significantly smaller in length than the severe CGG expansion event at FMR1, and thus would have been undetectable until now due to the recent availability of single-molecule long-read sequencing and computational technologies to glean STR length information from short-read sequencing. Human iPSCs can exhibit elevated genome instability48, therefore this raised the possibility that specific iPSC lines with a normal-length CGG STR might also exhibit BREACHes due to genetic instability caused by other non-FXS pathways. During preliminary inquiry into our hypothesis, we observed that iPSC lines created with methods involving p53 knock-down or p53 dominant-negative overexpression can show partial BREACH heterochromatinization and possibly an elevated burden of STR instability. These data raise a working model for future testing in which BREACHes might be a generalized phenomenon linked to multiple pathways of genome instability beyond FXS.
Limitations of the Current Study
Here, we find that the mutation-length CGG STR is necessary for the maintenance of H3K9me3 levels in BREACHes on the X-chromosome and multiple autosomes. Another critical open question is if knock-in of a mutation-length CGG (>200 triplets) in a normal-length iPSC line is sufficient for the establishment of H3K9me3 domains and/or trans interactions. Engineering 100% CG-content repetitive tracts is particularly technically challenging because they cannot be synthesized and are susceptible to contraction in E. Coli during cloning. Therefore, studies testing the sufficiency of a mutation-length CGG tract for BREACH establishment will be enabled by future technological advances. Experiments of importance for future work also include dissecting the relative role for premutation-length RNA versus DNA in the removal of H3K9me3 signal at BREACHes. Given recent reports of chromatin folding disruption in cancer and in Huntington’s disease, we hypothesize that heterochromatin-linked trans interactions and long-range TAD/loop dissolution will emerge as generalized principles in diseases and perturbative conditions associated with genome instability56,57. Furthermore, our analysis of BREACHes could be augmented by acquiring a broader range of FXS patient-derived samples allowing for the exploration of sex, age, STR length, brain region, and disease severity on BREACH formation. Although we demonstrate that BREACHes can occur in the caudate nucleus of FXS patients, our data do not suggest every tissue and every FXS patient will have BREACHes as our study is limited by sample size. Given the heterogeneity of brain tissue, examining BREACHes using multi-omic single-cell technology will shed light on the likely heterogenous nature of BREACHes within each brain region.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources, reagents, or other materials should be directed to the Lead Contact, Dr. Jennifer E. Phillips-Cremins (jcremins@seas.upenn.edu).
Materials availability
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement upon reasonable request.
Data and code availability
Raw sequencing files and key intermediate files generated in this study are deposited and freely available from Gene Expression Omnibus (GEO: GSE218680). A complete list of sequencing datasets generated in this study is provided in Table S3. A complete list of genomics datasets reanalyzed from various public repositories and publications is provided in Table S4. Accession numbers are also listed in the key resources table. DNA FISH images and Nanopore long-read sequencing raw files (i.e., fast5) reported in this study are not compatible with GEO but can be shared by the Lead Contact author upon request.
All original code is deposited at Zenodo and is made publicly available as of the date of publication. The DOI (10.5281/zenodo.6558223) is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact author upon request.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CTCF | Millipore | Cat# 07–729; RRID: AB_441965 |
| H3K9me3 | Abcam | Cat# ab8898; RRID: AB_306848 |
| IgG | Sigma-Aldrich | Cat# I8140; RRID: AB_1163661 |
| IgG Alexa Fluor 488 | Invitrogen | Cat# A-11034; RRID: AB_2576217 |
| IgG Alexa Fluor 594 | Invitrogen | Cat# A-21203; RRID: AB_141633 |
| NESTIN | R&D Systems | Cat# MAB1259; RRID: AB_2251304 |
| OCT4 | Cell Signaling Technologies | Cat# 2750; RRID: AB_823583 |
| Bacterial and virus strains | ||
| DH5α-T1R | Invitrogen | Cat# 12297016 |
| Biological samples | ||
| Healthy human caudate nucleus brain tissue from NIH donor 5533 (designated as NL_CN_1) | NIH NeuroBioBank | https://neurobiobank.nih.gov |
| Healthy human caudate nucleus brain tissue from NIH donor 5577 (designated as NL_CN_2) | NIH NeuroBioBank | https://neurobiobank.nih.gov |
| FXS human caudate nucleus brain tissue from NIH donor 5319 (designated as FXS_CN_1) | NIH NeuroBioBank | https://neurobiobank.nih.gov |
| FXS human caudate nucleus brain tissue from NIH donor 5746 (designated as FXS_CN_2) | NIH NeuroBioBank | https://neurobiobank.nih.gov |
| Chemicals, peptides, and recombinant proteins | ||
| 10% Triton X-100 solution | Sigma-Aldrich | Cat# 93443 |
| 100% Ethanol | Decon Labs | Cat# 2716 |
| 20% SDS solution | Fisher Scientific | Cat# BP1311 |
| Accutase | Gibco | Cat# A1110501 |
| AgentCourt Ampure XP beads | Beckman Coulter | Cat# A63881 |
| Alt-R S.p. HiFi Cas9 Nuclease V3 | Integrated DNA Technologies | Cat# 1081060 |
| Aminoallyl-dUTP Solution | Thermo Scientific | Cat# FERR1101 |
| Ammonium Acetate | Invitrogen | Cat# AM9070G |
| BbsI-HF | New England Biolabs | Cat# R3539S |
| Betaine | Sigma-Aldrich | Cat# 61962 |
| Blunt/TA Ligase Master Mix | New England Biolabs | Cat# M0367L |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat# A7906 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat# A7906–50G |
| Calcium chloride (CaCl2) | Fisher Scientific | Cat# BP510 |
| Calcium chloride (CaCl2) | Thermo Fisher | Cat# J63122-AD |
| Concanavalin A magnetic beads | BioMag | Cat# 86057 |
| CUTANA pAG-MNase | EpiCypher | Cat# 15–1016 |
| DAPI | Sigma-Aldrich | Cat# MBD0015–1ML |
| dATP | Thermo Scientific | Cat# R0141 |
| Dextran sulfate | Sigma-Aldrich | Cat# D8906 |
| Digitonin | Millipore | Cat# 300410 |
| Dimethyl sulfoxide | Sigma-Aldrich | Cat# D2650 |
| DMEM/F-12 | Gibco | Cat# 11320033 |
| Duplex buffer | Integrated DNA Technologies | Cat# 11–01-03–01 |
| EDTA, pH 8.0 | Invitrogen | Cat# 15575020 |
| EGTA, pH 8.0 | Bioworld | Cat# 40520008–1 |
| Elution buffer | Qiagen | Cat# 19086 |
| Fetal Bovine Serum | Gibco | Cat# 16000044 |
| Formaldehyde solution | Sigma-Aldrich | Cat# F8775 |
| Formaldehyde solution | Pierce | Cat# 28908 |
| Formamide | Calbiochem | Cat# 344206 |
| Glycine | Sigma-Aldrich | Cat# 50046 |
| Glycogen | Thermo Scientific | Cat# R0561 |
| Hank’s Balanced Salt Solution | Gibco | Cat# 14025092 |
| HEPES-KOH, pH 7.5 | Boston BioProducts | Cat# BBH-75-K |
| High-Vacuum Grease | Dow Corning | Cat# 1658832 |
| Hoechst 33342 Solution | Thermo Scientific | Cat# 62249 |
| Holo-transferrin | Sigma-Aldrich | Cat# T0665 |
| Igepal CA-630 | Sigma-Aldrich | Cat# I8896 |
| Insulin | Sigma-Aldrich | Cat# I1882 |
| Isopropanol | Thermo Fisher | Cat# T036181000 |
| KAPA HiFi HotStart ReadyMix | Roche | Cat# 7958927001 |
| L-ascorbic acid | Sigma-Aldrich | Cat# A8960 |
| LiCl | Sigma-Aldrich | Cat# L9650 |
| Lipofectamine Stem Transfection Reagent | Invitrogen | Cat# STEM00008 |
| Magnesium Acetate (MgAc2) | Sigma-Aldrich | Cat# 63052–100ML |
| Manganese chloride (MnCl2) | Fisher Scientific | Cat# BP541 |
| Matrigel hESC-Qualified Matrix | Corning | Cat# 354277 |
| Maxima H Minus Reverse Transcriptase | Thermo Scientific | Cat# EP0751 |
| mTeSR Plus media | STEMCELL Technology | Cat# 05825 |
| NaCl | Invitrogen | Cat# AM9760G |
| NEBNext Quick Ligation Module | New England Biolabs | Cat# E6056S |
| Noggin | R&D Systems | Cat# 6057-NG |
| Nuclease-free water | Sigma-Aldrich | Cat# W4502 |
| PBS | Corning | Cat# 21–040-CV |
| Penicillin-streptomycin | Gibco | Cat# 15140122 |
| Phenylmethanesulfonyl fluoride (PMSF) solution | Sigma-Aldrich | Cat# 93482 |
| Phusion polymerase | New England Biolabs | Cat# M0530L |
| Polyvinylsulfonic acid (PVSA) | Sigma-Aldrich | Cat# 278424 |
| Potassium chloride (KCl) | Sigma-Aldrich | Cat# P3911 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4368706 |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# P8340 |
| Protease Inhibitor Cocktail (EDTA-free) | Roche | Cat# 11873580001 |
| Protein A Agarose beads | Invitrogen | Cat# 15918014 |
| Protein G Agarose beads | Invitrogen | Cat# 15920010 |
| Proteinase K | New England Biolabs | Cat# P8107S |
| Proteinase K | Qiagen | Cat# 158918 |
| QuickCIP | New England Biolabs | Cat# M0525S |
| rCutSmart buffer | New England Biolabs | Cat# B6004 |
| RevitaCell™ Supplement (100X) | Gibco | Cat# A2644501 |
| RNase A | Roche | Cat# 10109142001 |
| RNase A | Thermo Fisher | Cat# EN0531 |
| RPMI 1640 media | Sigma-Aldrich | Cat# R8758 |
| Saline-Sodium Citrate (SSC) buffer | Corning | Cat# 46–020-CM |
| SB431542 | STEMCELL Technology | Cat# 72234 |
| SlowFade Diamond Antifade Mountant | Invitrogen | Cat# S36967 |
| Sodium bicarbonate | Sigma-Aldrich | Cat# S5761 |
| Sodium deoxycholate | Sigma-Aldrich | Cat# D6750 |
| Sodium Hydroxide (NaOH) | Macron | Cat# 7680 |
| Sodium selenite | Sigma-Aldrich | Cat# S5261 |
| Spermidine | Sigma-Aldrich | Cat# S2501 |
| Sucrose | Sigma-Aldrich | Cat# S0389–500G |
| SuperScript II Reverse Transcriptase | Invitrogen | Cat# 18064014 |
| Synth-a-Freeze | Gibco | Cat# A1254201 |
| T4 DNA ligase | New England Biolabs | Cat# M0202S |
| Taq ligase | New England Biolabs | Cat# M0208L |
| Taq polymerase | New England Biolabs | Cat# M0273 |
| TE buffer, pH 8.0 | Invitrogen | Cat# AM9858 |
| tracrRNA | Integrated DNA Technologies | Cat# 1072532 |
| Tris-HCl, pH 8.0 | Invitrogen | Cat# 15568025 |
| Triton X-100 | Sigma-Aldrich | Cat# T8787–100ML |
| TrypLE | Gibco | Cat# 12605010 |
| Tween 20 | Sigma-Aldrich | Cat# P9416 |
| Ultrapure Phenol/Chloroform/Isoamyl Alcohol | Fisher Scientific | Cat# BP1752I100 |
| VECTASHIELD Antifade Mounting Medium | Vector Laboratories | Cat# H-1200 |
| Versene Solution | Gibco | Cat# 15040066 |
| Critical commercial assays | ||
| Arima-HiC kit | Arima Genomics | Cat# A510008 |
| Bioanalyzer High Sensitivity DNA Analysis Kit | Agilent | Cat# 5067–4626 |
| DNA-free DNA removal kit | Ambion | Cat# AM1906 |
| GeneJet Genomic DNA purification kit | Thermo Scientific | Cat# K0721 |
| Gentra Puregene Cell Kit | Qiagen | Cat# 158767 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368813 |
| Kapa Library Quantification Kit | KAPA Biosystems | Cat# KK4835 |
| Ligation Sequencing Kit | Oxford Nanopore Technologies | Cat# SQK-LSK109 |
| mirVana miRNA Isolation Kit | Invitrogen | Cat# AM1560 |
| Native Barcoding Expansion (PCR-free) kit | Oxford Nanopore Technologies | Cat# EXP-NBD104 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7645S |
| Plasmid Purification Kit | Clontech | Cat# 740588.250 |
| QIAquick Gel Extraction Kit | Qiagen | Cat# 28706X4 |
| Qubit dsDNA HS assay kit | Invitrogen | Cat# Q32851 |
| Qubit RNA HS assay | Invitrogen | Cat# Q32852 |
| RNA 6000 kit | Agilent | Cat# 5067–1511 |
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| SuperScript First-Strand synthesis system for RT-PCR | Invitrogen | Cat# 11904018 |
| T7 HiScribe Kit | New England Biolabs | Cat# E2040S |
| TruSeq Stranded Total RNA Library Prep Gold kit | Illumina | Cat# 20020598 |
| Deposited data | ||
| ChIP-seq, RNA-seq in B-lymphocytes | This study | GEO: GSE218680 |
| CTCF ChIP-seq in iPSC, iPSC-NPC | This study | GEO: GSE218680 |
| Double stranded DNA breaks in mouse neural progenitor cells | (Wei et al., 2016) | doi: 10.1016/j.cell.2015.12.039. |
| Genome-wide long-read sequencing in iPSC | This study | GEO: GSE218680 |
| H3K9me3 ChIP-seq in 6718 | This study | GEO: GSE218680 |
| H3K9me3 ChIP-seq in CS0002 | This study | GEO: GSE218680 |
| H3K9me3 ChIP-seq in DF19.11 | (Inoue et al., 2017; Kazachenka et al., 2018) | Encode Project identifier: ENCSR704BRU |
| H3K9me3 ChIP-seq in DF6.9 | (Inoue et al., 2017; Kazachenka et al., 2018) | Encode Project identifier: ENCSR681AIW |
| H3K9me3 ChIP-seq in F1, F2, F3, F4, M1, and M2 | (Yokobayashi et al., 2021) | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE165867 |
| H3K9me3 ChIP-seq in iPSC, iPSC-NPC | This study | GEO: GSE218680 |
| H3K9me3 ChIP-seq in iPSC-15b | (Inoue et al., 2017; Kazachenka et al., 2018) | Encode Project identifier: ENCSR558XOU |
| H3K9me3 ChIP-seq in iPSC-18a | (Inoue et al., 2017; Kazachenka et al., 2018) | Encode Project identifier: ENCSR979TIC |
| H3K9me3 ChIP-seq in iPSC-18c | (Inoue et al., 2017; Kazachenka et al., 2018) | Encode Project identifier: ENCSR034LMV |
| H3K9me3 ChIP-seq in SA3.5 | This study | GEO: GSE218680 |
| H3K9me3 CUT&RUN in iPSC | This study | GEO: GSE218680 |
| H3K9me3 CUT&RUN in Kolf2 | This study | GEO: GSE218680 |
| H3K9me3 CUT&RUN in WTC-11 | This study | GEO: GSE218680 |
| H3K9me3 CUT&RUN in port-mortem brain tissue (caudate nucleus) | This study | GEO: GSE218680 |
| Hi-C in iPSC, iPSC-NPC | This study | GEO: GSE218680 |
| Human fetal cortex RNA-seq | (Kang et al., 2021) | GEO: GSE146878 |
| Murine cortical neuron RNA-seq (Fmr1 KO) | (Korb et al., 2017) | GEO: GSE81912 |
| PCR-free WGS from iPSC (a complete list is provided in Table S4) | HipSci | https://www.hipsci.org |
| PCR-free whole genome sequencing in iPSC | This study | GEO: GSE218680 |
| Repli-seq in iPSC | (Emerson et al., 2022) | 4DN: 4DNFI5WEY784 |
| RNA-seq in iPSC, iPSC-NPC | This study | GEO: GSE218680 |
| Targeted long-read sequencing in iPSC | This study | GEO: GSE218680 |
| Original codes | This study | 10.5281/zenodo.6558223 |
| Experimental models: cell lines | ||
| Human healthy iPS cell line - 176 (designated as NL_18 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human healthy iPS cell line – 158.1 (designated as NL_25 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human healthy iPS cell line - 20b (designated as NL_27 in this study) | Harvard Stem Cell Institute iPS Core Facility | https://divvly.com/reagent-3289 |
| Human pre-mutation iPS cell line - 111 (designated as PM_137 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - 135.3 (designated as FXS_421 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - 1H2 (designated as FXS_425 in this study and clonal from FXS_421) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - FXS_SW (designated as FXS_426 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line – GM07730 (designated as FXS_470 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - 135.3_CGG_034 (designated as ML_scClone1 in this study) | This study | N/A |
| Human FXS iPS cell line - 4H2 (designated as ML_scClone2 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - 6D12 (designated as ML_scClone3 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - 135.3_CGG_116 (designated as ML_scClone4 in this study) | This study | N/A |
| Human FXS iPS cell line - 135.3_CGG_125 (designated as ML_scClone5 in this study) | This study | N/A |
| Human FXS iPS cell line - 135.3_CGG_128 (designated as ML_scClone6 in this study) | This study | N/A |
| Human FXS iPS cell line - 135.3_CGG_131 (designated as ML_scClone7 in this study) | This study | N/A |
| Human FXS iPS cell line - 4D3 (designated as ML_CUT_PM_scClone1 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_117 (designated as ML_CUT_PM_scClone2 in this study) | This study | N/A |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_187 (designated as ML_CUT_PM_scClone3 in this study) | This study | N/A |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_275 (designated as ML_CUT_PM_scClone4 in this study) | This study | N/A |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_278 (designated as ML_CUT_PM_scClone5 in this study) | This study | N/A |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_030 (designated as ML_CUT_PM_scClone6 in this study) | This study | N/A |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_313 (designated as ML_CUT_PM_scClone7 in this study) | This study | N/A |
| Human healthy EBV-transformed B-lymphocyte GM09236 (designated as NL_B in this study) | Coriell Institute | https://www.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM09236&Product=CC |
| Human FXS EBV-transformed B-lymphocyte GM04025 (designated as FXS_B_650 in this study) | Coriell Institute | https://www.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM04025&Product=CC |
| Human FXS EBV-transformed B-lymphocyte GM09237 (designated as FXS_B_900 in this study) | Coriell Institute | https://www.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM09237&Product=CC |
| Oligonucleotides | ||
| Primers for DNA-FISH, FMR1 CGG PCR, and qRT-PCR are provided in Table S2 | This study | N/A |
| FMR1 5’UTR targeted gRNA provided in Table S2 | This study | N/A |
| TruSeq RNA Single Indexes Set A | Illumina | Cat# 20020492 |
| TruSeq RNA Single Indexes Set B | Illumina | Cat# 20020493 |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Addgene | #62988 |
| pWPT-GFP | Addgene | #12255 |
| pEFS.Cas9.GFP.Ctrl-B | This study / Addgene | To be uploaded to AddGene upon publication |
| pEFS.Cas9.GFP.CGG.cut | This study / Addgene | To be uploaded to AddGene upon publication |
| Software and algorithms | ||
| OligoMiner (version 1.0.4) | (Passaro et al., 2020) | http://oligominerapp.org |
| TANGO (v0.94) | (Ollion et al., 2013) | https://tango.mnhn.fr/tiki-index.php |
| Minimap2 (version 2.22-r1101) | (Gilbert et al., 2021) | https://github.com/lh3/minimap2 |
| nanopolish (version 0.13.2) | (Simpson et al., 2017) | https://github.com/jts/nanopolish |
| FastQC (v0.11.9) | Andrews, 2010 | https://github.com/s-andrews/FastQC |
| STRique (version 0.4.2) | (Giesselmann et al., 2019) | https://github.com/giesselmann/STRique |
| bwa-mem (v0.7.10-r789) | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net/bwa.shtml |
| deeptools (v3.3.0) | (Ramirez et al., 2016) | https://deeptools.readthedocs.io/en/develop/ |
| Samtools (version 1.11) | (Li et al., 2009) | https://www.htslib.org |
| goleft indexcov (version 0.2.3) | (Pedersen et al., 2017) | https://github.com/brentp/goleft |
| MACS2 (v 2.1.1.20160309) | (Zhang et al., 2008) | https://pypi.org/project/MACS2/ |
| Bowtie (v 0.12.7) | (Langmead et al., 2009) | http://bowtie-bio.sourceforge.net/index.shtml |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Guppy (version 6.2.1) | Oxford Nanopore Technologies | https://community.nanoporetech.com/downloads |
| Bedtools | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| HiC-Pro (version 2.7.7) | (Servant et al., 2015) | https://github.com/nservant/HiC-Pro |
| RSEG program (version 0.4.9) | (Song and Smith, 2011) | http://smithlabresearch.org/software/rseg/ |
| kat (v 2.4.1) | (Mapleson et al., 2017) | https://kat.readthedocs.io/en/latest/index.html |
| DESEQ2 (v1.34.0) | (Love et al., 2014) | doi: 10.1186/s13059-014-0550-8 |
| Kallisto | (Bray et al., 2016) | https://pachterlab.github.io/kallisto/about |
| tximport | (Soneson et al., 2015) | https://bioconductor.org/packages/release/bioc/html/tximport.html |
| WebGestalt (v 0.4.4) | (Liao et al., 2019) | https://github.com/bzhanglab/WebGestaltR |
| W2rapContigger (v 0.1) | (Clavijo et al., 2017) | https://github.com/bioinfologics/w2rap-contigger |
| cutadapt (v 1.18) | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/ |
| Juicer (v 1.5) | (Durand et al., 2016) | https://github.com/aidenlab/juicer |
| 3D-DNA (v180922) | (Dudchenko et al., 2017) | https://github.com/aidenlab/3d-dna |
| Juicebox (v 1.11.08) | (Robinson et al., 2018) | https://aidenlab.org/juicebox/ |
| JupiterPlots (v 3.8.2) | (Chu, 2018) | https://github.com/JustinChu/JupiterPlot |
| GangSTR (version 2.5.0) | (Mousavi et al., 2019) | https://github.com/gymreklab/GangSTR |
| DumpSTR (version 4.0.0) | (Mousavi et al., 2021) | https://github.com/gymreklab/TRTools |
| ExpansionHunter | (Dolzhenko et al., 2019; Dolzhenko et al., 2017) | https://github.com/Illumina/ExpansionHunter |
| ChopChop online tool (version 3.0.0) | (Labun et al., 2019) | https://chopchop.cbu.uib.no |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Huygens Essential deconvolution software v20.04 | Scientific Volume Imaging | https://svi.nl/Huygens-Essential |
| Other | ||
| Dounce Tissue Grinder | Wheaton | Cat# 357544 |
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
EBV-transformed lymphoblastoid culture
We cultured our male EBV-transformed lymphoblastoid B-cell lines as previously described58. We cultured cells in RPMI 1640 media with L-glutamine (Sigma-Aldrich, R8758) supplemented with 15% (v/v) Fetal Bovine Serum (Gibco, 16000044), and 1% (v/v) penicillin-streptomycin (Gibco, 15140122) at 37°C and 5% CO2. We passaged cells every 2–4 days. All information regarding age, developmental stage, sex, ancestry, ethnicity, and race are provided in Table S1.
Induced pluripotent stem cell (iPSC) culture
Fulcrum Therapeutics expanded, curated, and characterized all iPSC lines from this study before shipment to our lab at matched passage (sex: males). iPSCs were routinely tested for karyotype instability, FMR1 expression, CGG length, morphology, and pluripotency markers by Fulcrum Therapeutics. Upon receipt of all clones, all clones were expanded and were frozen down at low passage number. We cultured all iPSC lines in mTeSR Plus media (STEMCELL Technology, 05825) supplemented with 1% (v/v) penicillin-streptomycin (Gibco, 15140122) at 37°C and 5% CO2 on Matrigel hESC-Qualified Matrix (Corning, 354277) coated plates. We passaged all iPSC lines at 60–70% confluency every 2–5 days to ensure that single colonies remained independent without physical merging. We dissociated iPSC by incubating in Versene Solution (Gibco, 15040066) at 37°C for 3 minutes and then deactivated Versene with equal volume of mTeSR Plus media before replating. All iPSC culture plates were coated with 1.2% (v/v) Matrigel hESC-Qualified Matrix in DMEM/F-12 (Gibco, 11320033) for at least 1 hour at room temperature.
We verified the pluripotency state of our cell line clones via visual verification of colony morphology as well as via immunofluorescence staining for the pluripotency marker OCT4 (detailed in “Immunofluorescence staining”). We used whole genome PCR-free sequencing to confirm that all iPSC lines were karyotypically normal after routine passaging in our laboratory (Figure S1) (detailed in “Genomic coverage/mappability plot” and “de novo Genome Assembly”). We identified a small heterozygous deletion (~6.5 mb) on chr18 in FXS_426, covering n=54 refseq genes. The genes were removed from further analyses in Supplementary Figure S3M. The list of genes: BCL2, CCBE1, CDH20, HMSD, KDSR, LINC00305, LINC01538, LINC01544, LINC01916, LINC01924, LOC101927404, LOC105372151, LOC105372152, LOC105372155, LOC105372156, LOC105372157, LOC105372159, LOC105372160, LOC105372161, LOC105372165, LOC105372166, LOC105372167, LOC105372168, LOC105372169, LOC107985156, LOC107985178, LOC112268209, LOC124904313, LOC124904314, LOC124904315, LOC124904316, LOC124904317, LOC124904318, LOC124904356, LOC124904357, MC4R, PHLPP1, PIGN, PMAIP1, RELCH, RNF152, SERPINB10, SERPINB11, SERPINB12, SERPINB13, SERPINB2, SERPINB3, SERPINB4, SERPINB5, SERPINB7, SERPINB8, TNFRSF11A, VPS4B, & ZCCHC2. All information regarding age, developmental stage, sex, ancestry, ethnicity, and race are provided in Table S1.
Generation of iPSC-derived neural progenitor cells (NPCs)
We differentiated human iPSC into NPCs using a well-established protocol59. Briefly, we expanded undifferentiated cells in mTeSR Plus (STEMCELL Technology, 05825) on Matrigel-coated plates as described above. We seeded iPSCs onto freshly coated Matrigel plates in NPC differentiation media at a density of 16,000 cells/cm2. NPC differentiation media consisted of DMEM/F-12 (Gibco, 11320033) with 5 μg/mL insulin (Sigma-Aldrich, I1882), 64 μg/mL L-ascorbic acid (Sigma-Aldrich, A8960), 14 ng/mL sodium selenite (Sigma-Aldrich, S5261), 10.7 ug/mL Holo-transferrin (Sigma-Aldrich, T0665), 543 μg/mL sodium bicarbonate (Sigma-Aldrich, S5761), 10 μM SB431542 (STEMCELL Technology, 72234), and 100 ng/mL Noggin (R&D Systems, 6057-NG). We changed NPC media every day and harvested cells at the end of day 8. Only NPC preparations with the expected rosette morphology and expressing the NPC-specific marker NESTIN (detailed in “Immunofluorescence staining”) were used for downstream genomics and imaging.
FMR1 CGG cut-out isogenic iPSC engineering
We CRISPR-Cas9-mediated CGG tract editing to generate N=7 mutation-length and N=7 premutation-length single-cell subclones from the ML FXS iPSC parent line FXS_421. We created a custom plasmid, pEFS.Cas9.GFP.CGG.cut, expressing Cas9, GFP, and a gRNA targeting the FMR1 5’UTR (sgRNA sequence: 5’- TGACGGAGGCGCCGCTGCCA-3’). We generated pEFS.Cas9.GFP.CGG.cut by modifying pSpCas9(BB)-2A-Puro (PX459) V2.0 (Addgene #62988) as follows: (1) replacing the CMV promoter with an EF1alpha core promoter from Addgene plasmid #12255, (2) adding a GFP sequence from Addgene plasmid #12255, (3) inserting the gRNA targeting the FMR1 CGG STR using BbsI (New England Biolabs, R3539S) restriction digest. We verified the final plasmid sequence via Plasmidsaurus whole-plasmid sequencing service.
We transfected iPSCs in Matrigel coated 6-well plates with 6 μg pEFS.Cas9.GFP.CGG.cut using Lipofectamine Stem Transfection Reagent (Invitrogen, STEM00008) according to the manufacturer’s protocol. Four days post transfection we dissociated the transfected iPSC colonies into single cells using 0.75x TrypLE (Gibco, 12605010), resuspended in HBSS (Gibco, 14025092), and subjected cells to fluorescence activated cell sorting (FACS) to select for the GFP+ population. Fluorescence Activated Cell Sorting (FACS) GFP+ cells were single cell plated into 96-well plates coated with Matrigel hESC-Qualified Matrix (Corning, 354277), containing mTeSR Plus media (STEMCELL Technology, 05825) with 1x RevitaCell (Gibco, A2644501). Media was swapped to mTeSTR Plus media without RevitaCell 3 days post FACS. We then passaged iPSC single cell clones into first 24-well and then 6-well coated tissue culture plates in duplicate, one for freezing down and storage and one for genetic screening.
We first screened iPSC clones for successful FMR1 CGG editing by measuring FMR1 RNA expression. We prepared cell pellets and extracted RNA using the Qiagen RNeasy Mini Kit (Qiagen, 74106) per manufacturer’s protocol. We quantified RNA using a Nanodrop and performed cDNA conversion using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368813) per manufacturer’s protocol with either 100 or 200 ng of RNA input. We performed quantitative real-time polymerase chain reaction (qRT-PCR) for GAPDH and FMR1 (primers listed in Table S2) in duplicate using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, 4368706) on a StepOnePlus Real-Time PCR System (Applied Biosystems). We selected iPSC clones of interest based on recovery or continued repression of FMR1 RNA. We next revived, expanded, and re-screened selected iPSC clones using the same FMR1 CGG qRT-PCR assay in technical triplicate to confirm FMR1 expression followed by targeted Nanopore sequencing (detailed in “Targeted long-read sequencing of CGG at FMR1”) to determine length of the FMR1 CGG sequence. We verified the pluripotency state of all cell line clones via visual verification of colony morphology.
Frozen human brain tissue acquisition
We acquired post-mortem human caudate nucleus brain tissue from healthy male donors and male donors clinically diagnosed with fragile X syndrome from the NIH NeuroBioBank. We stored the tissue at −80°C upon receipt. All information regarding age, developmental stage, sex, ancestry, ethnicity, and race are provided in Table S1.
METHOD DETAILS
Immunofluorescence staining
We performed immunofluorescence staining by fixing iPSCs and iPSC-derived NPCs using 4% formaldehyde (Pierce, 28908) for 12 minutes at room temperature (25°C). We blocked and permeabilized samples in 0.3% Triton X-100 (Sigma-Aldrich, 93443) with 5% BSA (Sigma-Aldrich, A7906) in PBS (Corning, 21–040-CV) at room temperature. We then incubated fixed cells with primary antibodies overnight at 4°C in 0.3% Triton X-100 with 1% BSA in PBS followed by incubation with secondary antibodies for 2 hours at room temperature in 0.3% Triton X-100 with 1% BSA in PBS. Cells were mounted with VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories, H-1200). The following antibodies were used in this study: goat anti-rabbit IgG Alexa Fluor 488 (1:250, Thermo Fisher Scientific, A-11034), donkey anti-mouse IgG Alexa Fluor 594 (1:250, Thermo Fisher Scientific, A-21203), mouse NESTIN (1:100, R&D Systems, MAB1259), rabbit OCT4 (1:200, Cell Signaling, 2750).
Nuclei purification from post-mortem brain tissue
We sectioned tissue from the caudate nucleus into aliquots of ~100 mg. We performed sectioning on dry ice with sterile forceps and a sterile, single-use razor using a petri dish as a platform after all equipment had been pre-chilled on dry ice. Prior to douncing and homogenization, we pre-chilled all buffers, reagents, and equipment on wet ice. We performed the entire procedure on wet ice. We placed tissue in 10 mL of ice-cold Homogenization Buffer (0.32 M sucrose (Sigma-Aldrich, S0389–500G), 5 mM CaCl2 (Thermo Fisher, J63122-AD), 10 mM Tris-HCl pH 8.0 (Invitrogen, 15568025), 3 mM MgAc2 (Sigma-Aldrich, 63052–100ML, 0.1% Triton X-100 (Sigma-Aldrich, T8787–100ML), 0.1 mM EDTA (Invitrogen, 15575020), 1X Protease Inhibitor Cocktail (Roche, 11873580001)) and dounced the tissue with 20 strokes of the loose pestle and 7 strokes of the tight pestle using a 15 mL Dounce Tissue Grinder (Wheaton, 357544). We performed douncing very slowly and gently to avoid unnecessary mechanical stress. We laid 10 mL of homogenized tissue over 14 mL of ice-cold Sucrose Cushion (1.8 M sucrose, 10 mM Tris-HCl pH 8.0, 3 mM MgAc2, 1X Protease Inhibitor Cocktail). We laid an additional 12 mL of ice-cold Homogenization Buffer on top of the homogenized tissue and centrifuged for 2 hours at 4°C at 25,700 RPM (~81,150xg) in a SW Ti 32 swinging bucket rotor. We removed the supernatant and added FANS Buffer (1X PBS (Corning, 21–040-CV), 1% Bovine Serum Albumin (Sigma-Aldrich, A7906–50G), 1X Protease Inhibitor Cocktail), to the pellet. We incubated the pellet on ice for 20 mins before resuspending. We counted the nuclei and centrifuged the solution for 6 mins at 4°C at 600xg. We resuspended nuclei in FANS buffer at a concentration of 3 million nuclei per mL. We blocked nuclei in FANS buffer for 15 mins at 4°C while rotating. We stained nuclei with anti-NeuN (1:1000, Sigma-Aldrich, MAB377X) for 90 mins and added DAPI (1:2000, Sigma-Aldrich, MBD0015–1ML) with 5 mins left on the staining timer. Staining was performed with end-over-end rotation at 4°C. Next, we centrifuged the nuclei for 6 mins at 4°C at 600xg. We resuspended nuclei in FANS buffer at a concentration of 6 million nuclei per mL, filtered the solution using a 5 mL FACS sorting tube (Corning, 352235), and sorted using the MoFlo Astrios (Beckman Coulter). We performed CUT&RUN on sorted nuclei as described below with minor modifications. We sorted nuclei into CUT&RUN Wash Buffer and immediately bound nuclei to Concanavalin A beads after returning from sorting. Additionally, we substituted 0.1%, 0.1%, and 0.05% digitonin in the Antibody Buffer, Digi-Wash Buffer, and 2X Stop Buffer with 0.1%, 0.1%, and 0.04% Triton X-100. All other steps were the same.
Oligopaint DNA FISH probes
We designed Oligopaint probes with OligoMiner (version 1.0.4) to visualize domains that acquired H3K9me3 heterochromatin in FXS (10 loci on autosomes and one locus on the X chromosome)60. We designed primary probes across each of 12 total H3K9me3 domains consistently gained across all three FXS iPSC lines (FXS-recurrent H3K9me3 domains). Although 11 (10 autosomal, 1 X chromosome) FXS-recurrent H3K9me3 domains were reported in Figures 1–2, we divided one autosomal domain on chr8 (chr-8R2) into two (chr-8R2a and chr-8R2b) for imaging experiments due to a gap caused by a highly repetitive part of the genome. We designed primary probes with the following design features: (i) 80 bases of homology to a DNA sequence unique to a H3K9me3 domain, (ii) a 20 bp fiducial sequence, and (iii) a 20 bp barcode sequence unique to one specific H3K9me3 domain (hereafter referred to as a H3K9me3-locus-specific-barcode, one per each of n=12 domains). Primary probe sequences are provided in Table S5. Primary probe densities per H3K9me3 domain are curated in Table S5. We used previously published sequences61 for our fiducial sequence, 5’-AGTCCCGCGCAAACATTATT-3’, and H3K9me3-locus-specific-barcode sequences, provided in Table S5. We ordered primary probes from Twist Biosciences.
We designed bridge oligonucleotides with the following features: (i) a 20 bp sequence as the reverse complement to the H3K9me3-locus-specific-barcode in the primary Oligopaint probes and (ii) an adjacent 20 bp sequence which can hybridize to the secondary imaging probe. Finally, we designed a secondary fluorescent dye conjugated oligonucleotide imaging probe with a 20 bp sequence representing the reverse complement to the bridge probe62. We ordered bridge oligonucleotides and dye-conjugated secondary imaging probes from Integrated DNA Technologies (IDT). Bridge and secondary imaging probe sequences are provided in Table S5.
We synthesized primary DNA FISH probes using the oligonucleotide library from Twist Biosciences as the template using two rounds of PCR as previously described63. For the first PCR amplification, we used KAPA HiFi HotStart ReadyMix (Roche, 7958927001), an initial template concentration of 0.04 ng/μL and primers at a concentration of 0.6 μM targeting complementary sequences designed for PCR amplification universal to all DNA FISH probes (“First probe PCR” primers listed in Table S5). We performed PCR starting with a 3-minute 98°C initial denaturation step followed by 20 cycles of denaturation for 20 seconds at 98°C, annealing for 15 seconds at 60°C, and extension for 15 seconds at 72°C, and concluding with a final extension step for 1 minute at 72°C. We next performed a second round of PCR to add (i) the 20 bp fiducial sequence via the forward primer and (ii) a T7 promoter sequence via the reverse primer for subsequent in vitro transcription. We used the purified PCR product from the first PCR at a concentration of 0.004 ng/μL and 0.6 μM primers (“Second probe PCR” primers listed in Table S5) targeting the complementary sequences designed for PCR amplification universal to all DNA FISH probes with the addition of the fiducial and T7 promoter sequence. We performed PCR with KAPA HiFi HotStart ReadyMix and PCR settings from the first PCR as previously described.
We further amplified the primary probe pool using the T7 HiScribe Kit (New England Biolabs, E2040S) for in vitro transcription of the amplified primary probe pool (0.75 ng) per manufacturer’s protocol. We next performed reverse transcription using the entirety of the T7 reaction, 2U of Maxima H Minus Reverse Transcriptase (Thermo Scientific, EP0751) per 75 μL of reaction, and a custom mix of dNTPs (12.5 mM of dATP, dCTP and dGTP and 6.25 mM of dTTP and amino allyl UTP (Thermo Scientific, FERR1101). After incubation for 2 hour at 50°C, we degraded the RNA:DNA hybrids and excess RNA not converted to cDNA with an alkaline hydrolysis mix (0.25 M EDTA (Invitrogen, 15575020), 0.5 M NaOH (Marcon, 7680), and 0.625 μg/μl RNase A (Thermo Scientific, EN0531), followed by purifying the single-stranded cDNA using Plasmid Purification Kit (Clontech, 740588.250) per manufacturer’s protocol. The single-stranded cDNA probe pool was quantifed using a Nanodrop and resuspended in water for a stock concentration of 1.2 μg/μl for hybridization and stored at −20°C.
DNA FISH
We performed Oligopaint DNA FISH as previously described64 with some modifications for iPSCs. We disassociated iPSCs into single cells using TrypLE (Gibco, 12605010) and plated 3 million cells onto Matrigel hESC-Qualified Matrix (Corning, 354277) coated 40 mm glass coverslips (Bioptechs, 40–1313-0319) to maintain the same matrix condition from cell culture. We allowed cells to adhere by placing the cells and coverslips into the incubator at 37°C and 5% CO2 for 4 hours. We performed the fixation and subsequent washes of the coverslips in 60 mm cell culture dishes with 4mL of solution. We fixed the cells by incubating the coverslips in 4% formaldehyde (Thermo Scientific, 28908) and 0.1% Triton X-100 (Sigma-Aldrich, 93443) in PBS (Corning, 21–040-CV) at room temperature (20–25°C) for ten minutes. We washed coverslips three times in PBS for 5 minutes at room temperature. We stored the fixed coverslips at 4°C until staining.
On the first day of the FISH protocol, we added PBS to the coverslips at room temperature for 5 minutes and then performed a series of washes at room temperature to prepare the sample for denaturation: (1) a 10 min wash with 0.5% Triton X-100 in PBS, (2) a 2 minute wash in 70% ethanol (Decon Labs, 2716), (3) a 2 minute wash in 90% ethanol, (4) a 2 minute wash in 100% ethanol followed by 2 minute of drying, (5) a 5 min wash in 2X SSCT buffer (SSC buffer (Corning, 46–020-CM), 0.1% Tween-20 (Sigma-Aldrich, P9416) in nuclease-free water (Sigma, W4502)), and (6) 5 min wash in a 1:1 mixture of 4X SSCT buffer and 100% formamide (Calbiochem, 344206). We then incubated coverslips in a 1:1 mixture of 4X SSCT buffer and 100% formamide at 37°C. We next diluted 175 pmol of the stock single-stranded Oligopaint probe pool into a final volume of 55 μl of primary hybridization buffer (50% formamide, 10% dextran sulfate (Sigma-Aldrich, D8906), 4% polyvinylsulfonic acid (PVSA) (Sigma-Aldrich, 278424) and 0.4 μg/μl RNaseA (Thermo Scientific, EN0531) in nuclease-free water) for a final concentration of 3.2 μM of Oligopaint probe. We pipetted the Oligopaint probe hybridization mix onto 2” x 3” glass slides, placed the coverslips, and sealed with rubber cement. We heat-denatured the samples by placing the slides on a heat block in a water bath set to 80°C for 30 minutes and then incubated slides in a humidified chamber overnight at 37°C.
The following day, we removed the coverslips from the slides and washed the slides in (1) 2X SSCT buffer at 60°C for 15 minutes, (2) 2X SSCT at room temperature for 10 minutes, and (3) 0.2X SSC (SSC buffer in water) at room temperature for 10 minutes. We used secondary hybridization buffer (50% formamide, 10% dextran sulfate, and 4% PVSA in nuclease-free water) to dilute the bridge oligonucleotides and secondary fluorescent dye conjugated imaging probes to final working concentrations of 0.1 μM of each bridge oligonucleotide and 0.2 μM of each secondary dye conjugated imaging probe. We used the bridge probe corresponding to the H3K9me3-locus-specific-barcodes of the domains on chromosomes 12 and X. Our imaging probes included a Cy3 conjugated probe, Cy5 conjugated probe to label the chromosome 12 and X domains, respectively, and a AF488 conjugated probe to label all twelve domains. We pipetted secondary imaging hybridization mix onto 2” x 3” glass slides, placed the coverslips on top, and sealed with rubber cement. Slides were incubated in a dark humidified chamber for 2 hours at room temperature. Following this incubation, we removed the coverslips from the slides and washed them in multiple steps: (1) 2X SSCT at 60°C for 15 minutes, (2) 2X SSCT at room temperature for 10 minutes, and (3) 0.2X SSC (SSC buffer in water) at room temperature for 10 minutes. To stain nuclei, we incubated coverslips in Hoechst 33342 (1:10,000 in 2X SSC, Thermo Scientific, 62249) for 5 minutes at room temperature, and subsequently mounted coverslips on 2” x 3” glass slides using SlowFade Diamond Antifade Mountant (Invitrogen, S36967).
Immunofluorescence and DNA FISH Imaging
We imaged our immunofluorescence and DNA FISH samples on a Leica DMi8 microscope. We used the 20X objective with a 1.6X magnifier for phase contrast and OCT4/NESTIN IF images and the 63X oil-immersion objective (NA 1.4) for DNA FISH images.
Cell fixation for ChIP-seq and Hi-C
We fixed cells as previously described for all downstream ChIP-seq and Hi-C experiments19,65–70. For EBV-transformed lymphoblastoid cells in suspension, we pelleted the appropriate number of cells, resuspended in serum-free RPMI 1640 (Sigma-Aldrich, R8758), and added 1mL of formaldehydes fixation solution for a final concentration of 1% (v/v) formaldehyde (Sigma, F8775). For adherent iPSC and iPSC-derived NPC, we replaced growth media with 10 mL DMEM/F-12 (Gibco, 11320033) and added 1mL of formaldehyde fixation solution for a final concentration of 1% (v/v). The stock formaldehyde fixation solution consisted of 50 mM HEPES-KOH (pH 7.5) (Boston BioProducts, BBH-75-K), 100 mM NaCl (Invitrogen, AM9760G), 1 mM EDTA (Invitrogen, 15575020), 0.5 mM EGTA (Bioworld, 40520008–1), and 11% formaldehyde (Sigma, F8775). We quenched the fixation reaction in 125 mM glycine (Sigma-Aldrich, 50046) for 5 minutes at room temperature and 15 minutes at 4°C and pelleted the cells before storing. For EBV-transformed lymphoblastoid cells in suspension, we pelleted the crosslinked cells. For adherent iPSC and iPSC-derived NPC, we used a cell scraper (Corning, 353089) to remove crosslinked cells from the dish and then pelleted the cells. For all cell lines, we washed pelleted cells in pre-chilled PBS (Corning, 21–040-CV), froze the cell pellets in liquid nitrogen, and stored at −80°C.
ChIP-seq
We performed ChIP-seq as previously described with modifications58,65–67,69–71. Briefly, we lysed crosslinked pellets (consisting of 10 million cells for CTCF ChIP-seq or 3 million cells histone modifications ChIP-seq) in cell lysis buffer (10 mM Tris-HCl, pH 8.0 (Invitrogen, 15568025), 10 mM NaCl (Invitrogen, AM9760G), 0.2% NP-40/Igepal CA-630 (Sigma-Aldrich, I8896), 1X Protease Inhibitor Cocktail (Roche, 11873580001), 1X PMSF (Sigma-Aldrich, 93482) on ice for 10 minutes. We then homogenized the suspension with pestle 30 times. We pelleted nuclei by spinning samples at 2,500xg and 4°C and subsequently lysed the nuclei in 500 μl of Nuclear Lysis Buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA (Invitrogen, 15575020), 1% SDS (Fisher Scientific, BP1311), 1X Protease Inhibitor Cocktail, 1X PMSF) on ice for 20 minutes.
We sonicated lysed nuclei in 300 μl IP Dilution Buffer (20 mM Tris pH 8.0, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100 (Sigma-Aldrich, 93443), 0.01% SDS, 1X Protease Inhibitor Cocktail, 1X PMSF using a QSonica Q800R2 sonicator (settings: 1 hour set, 100% amplitude, 30 seconds pulse, 30 seconds off). We pelleted the nuclear membranes at 18,800xg and 4°C and then resuspended the supernatant-containing chromatin in 800 μl of a pre-clearing solution consisting of 3.7 mL IP Dilution Buffer, 500 μl Nuclear Lysis Buffer, 175 μl of a 1:1 ratio of Protein A:Protein G bead slurry (Invitrogen, 15918014 and 15920010 respectively) and 50 μg of rabbit IgG (Sigma-Aldrich, I8140). This step is to remove the nuclear membrane debris after nuclei lysis and sonication, not for pelleting the nuclei. We incubated this solution at 4°C for 2 hours.
Antibodies used in this study include: CTCF (Millipore, 07–729), H3K9me3 (Abcam, ab8898), and IgG (Sigma-Aldrich, I8140). After pre-clearing, we saved 200 μl as the “input” control and added the remaining solution to an immunoprecipitation (IP) reaction consisting of 1 mL cold PBS (Corning, 21–040-CV), 20 μl Protein A, 20 μl Protein G, and 1 μl/million cells of either CTCF or H3K9me3 antibody and rotated overnight at 4°C. The IP solution was pre-incubated overnight at 4°C before incubating with chromatin. The next day, we pelleted the IP reactions and discarded the supernatant. We washed the remaining pellet once with IP Wash Buffer 1 (20 mM Tris-HCl, pH 8, 2 mM EDTA, 50 mM NaCl, 1% Triton X-100, 0.1% SDS), twice with High Salt Buffer (20 mM Tris-HCl, pH 8, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.01% SDS), once with IP Wash Buffer 2 (10 mM Tris-HCl, pH 8, 1 mM EDTA, 0.25 M LiCl (Sigma-Aldrich, L9650), 1% NP-40/Igepal CA-630, 1% sodium deoxycholate (Sigma-Aldrich, D6750)), and twice with TE buffer (Invitrogen, AM9858). We eluted the IP DNA from the washed beads in 200 μL Elution Buffer (100 mM NaHCO3 (Sigma-Aldrich, S5761) and 1% SDS prepared fresh) by resuspending and spinning at 5,400xg and harvesting the supernatant.
We next degraded RNA with 60 μg RNase A (Sigma-Aldrich, 10109142001) at 65°C for 1 hour and then degraded residual protein by incubating the 200 μl solution with 60 μg proteinase K (New England Biolabs, P8107S) overnight at 65°C. After extracting DNA using phenol:chloroform and ethanol precipitation as previously described72, we prepared ChIP-seq libraries for sequencing using the NEBNext Ultra II DNA Library Prep Kit (New England Biolabs, E7645S) according to the manufacturer’s protocol. We performed size selection of adaptor-ligated libraries using AgentCourt Ampure XP beads (Beckman Coulter, A63881), selecting from fragments under 1 kb, according to the manufacturer’s protocol.
Hi-C
We prepared Hi-C libraries using the Arima Genomics Hi-C kit (Arima Genomics, A510008) according to the manufacturer’s protocol. Briefly, we crosslinked 2 million cells with 1% formaldehyde as described above. We first lysed the cells and permeabilized nuclei before we enzymatically digested chromatin within nuclei of crosslinked cell pellets and created biotinylated ligation junctions between the digested ends according to the manufacturer’s protocols. We extracted DNA and sheared to an average size of ~400 bp using a sonicator (Covaris, S220) at 140 W peak incident power, 10% duty factor, and 200 cycles per burst for 55 seconds. We further selected 200–600 bp DNA fragments using AgenCourt Ampure XP beads (Beckman Coulter, A63881). We then pulled down biotin-tagged ligation junctions using streptavidin beads from the Arima Hi-C kit according to the manufacturer’s protocol. Streptavidin beads containing Hi-C libraries were stored at −20°C for no more than 3 days before library preparation for sequencing was performed. We prepared Hi-C libraries for sequencing by eluting DNA from streptavidin beads by boiling at 98°C for 10 minutes in 15 μl of Elution Buffer. Subsequently, we amplified the libraries using NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs, E7645S) with 8 PCR cycles according to the manufacturer’s protocol.
Total RNA-seq
We isolated total RNA from iPSCs and iPSC-derived NPCs using the mirVana miRNA Isolation Kit (Invitrogen, AM1560) according to the manufacturer’s protocol. All RNA samples had an RNA Integrity Number >9 as assessed by Agilent BioAnalyzer using the RNA 6000 Nano kit (Agilent, 5067–1511). We treated RNA samples with rDNAse I (Ambion, AM1906) according to the manufacturer’s protocol to remove residual genomic DNA. We used 100 ng of DNAse-treated total RNA for RNA-seq library preparation using the TruSeq Stranded Total RNA Library Prep Gold kit (Illumina, 20020598) according to the manufacturer’s instructions. Briefly, we removed rRNA from the input RNA, generated double stranded cDNA using 0.8 U of SuperScript II Reverse Transcriptase (Invitrogen, 18064014), and performed A-tailing and end repair. We ligated the resulting cDNA to TruSeq RNA Single Indexes Set A (Illumina, 20020492) and Set B (Illumina, 20020493) to enable multiplex sequencing. We performed size selection (selecting for 300 bp) and two rounds of bead clean-up (1:1 ratio of sample to Agencourt AMPure XP beads (Beckman Coulter, A63881)) before amplifying the purified samples with 15 PCR cycles.
CUT&RUN
We performed CUT&RUN as previously described on fresh and frozen cells72. We harvested 1×106 iPSCs using either Accutase (Gibco, A1110501) or Versene (Gibco, 15040066) and washed iPSC pellets in PBS (Corning, 21–040-CV). We then washed cell pellets 3x in Wash Buffer (20 mM HEPES-KOH pH 7.5 (Boston BioProducts, BBH-75-K), 150 mM NaCl (Invitrogen, AM9760G), 0.5 mM Spermidine (Sigma-Aldrich, S2501), 1X Protease Inhibitor Cocktail (Roche, 11873580001)) and bound them to activated Concanavalin A beads (BioMag, 86057). We activated Concanavalin A beads by washing 2x and then rotating in binding buffer (20 mM HEPES-KOH pH 7.5, 10 mM KCl (Sigma-Aldrich, P3911), 1 mM CaCl2 (Fisher Scientific, BP510), 1 mM MnCl2 (Fisher Scientific, BP541)) for 10 minutes at room temperature (20–25°C). We incubated the bead bound cells in 100 μl antibody buffer (Wash buffer with 0.1 % digitonin (Millipore, 300410) and 2 mM EDTA (Invitrogen, 15575020)) with a final concentration of 1:100 of antibody (IgG (Sigma-Aldrich, I8140) or H3K9me3 (Abcam, ab8898)) overnight with rotation at 4°C.
We washed cells 3x in Digi-Wash Buffer (Wash Buffer with 0.1% digitonin), resuspending cells in 50 μl Digi-Wash Buffer. We incubated cells with 2.5 μl of CUTANA pAG-MNase (EpiCypher, 15–1016) for 10 minutes at room temperature before we washed the samples 2x in Digi-Wash Buffer, resuspended in 100 μl Digi-Wash Buffer, and placed on ice for 5 minutes. We then performed pAG-MNase chromatin digestion by adding 2 μl of 100 μM CaCl2 and incubated at 4°C with rotation. We stopped the digestion at 2 hours with the addition of 100 μl of 2X Stop Buffer (340 mM NaCl, 20 mM EDTA, 4 mM EGTA (BioWorld, 40520008–1), 0.05% Digitonin, 50 μg/mL mL RNase A (Thermo Scientific, EN0531), 50 μg/mL Glycogen (Thermo Scientific, R0561)) and incubated samples at 37°C for 30 minutes. Finally, we collected the supernatant containing the cleaved chromatin fragments after magnetic removal of immobilized beads. We extracted DNA from the supernatant using phenol:chloroform and ethanol precipitation and performed library preparation using the NEBNext Ultra II Library Prep Kit (New England Biolabs, E7645S) per manufacturer’s instructions.
Illumina Sequencing
We sequenced all libraries on an Illumina NextSeq 500 or NovaSeq 6000 unless specified otherwise. Prior to sequencing, we analyzed library quality and size distribution with Agilent Bioanalyzer High Sensitivity DNA Analysis Kits (Agilent, 5067–4626). We quantified library concentration using the Qubit high sensitivity DNA assay kit (Invitrogen, Q32852) and the Kapa Library Quantification Kit (KAPA Biosystems, KK4835). We sequenced ChIP-seq libraries with 75 bp single-end reads, CUT&RUN and Hi-C libraries with 37 bp paired-end reads, and RNA-seq libraries with 75 bp paired-end reads. The total number of reads sequenced for all datasets generated in this study are listed in Table S3.
qRT-PCR
We quantified gene expression as previously described58. Briefly, we harvested iPSCs and flash froze pellets, storing at −80°C until RNA extraction. We thawed frozen cell pellets on ice and extracted total RNA using either the mirVana miRNA Isolation Kit (Invitrogen, AM1560) or Qiagen RNeasy Mini Kit (Qiagen, 74106) according to the manufacturer’s protocol. We digested any remaining genomic DNA using rDNAse I (Ambion, AM1906). We quantified RNA using the Qubit RNA HS assay (Invitrogen, Q32852) and normalized input into the cDNA conversion reaction. We converted RNA to cDNA by using either the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen, 11904018) with final concentrations of 500 μM dNTPs, 5 mM MgCl2, 10 mM DTT, and 2.5 ng/μl of random hexamers in the first stranding reaction or the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, 4368813) per manufacturer’s instructions.
To perform qRT-PCR reactions, we mixed 2 μl of cDNA with 10 uM forward and 10 uM reverse primers for a final concentration of 400 nM, in 1X Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, 4368706) for a final volume of 20 μl per reaction. Cycle conditions were 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 45 seconds. For all mRNA levels quantified using qRT-PCR (FMR1, SLITRK2, SHISA6, DPP6, and GAPDH), we generated a standard curve by amplifying cDNA with gene-specific primers listed in Table S2. We created standards with serial 10-fold dilutions of cDNA starting at 2 pM. We used the resulting CT values to generate a standard curve and computed the concentration of mRNA transcripts per condition using 100 ng of RNA in the cDNA reaction. We validated the specificity of our amplicons by running the PCR reaction on a gel to verify a single band and confirming a single peak while running a melting curve at the end of each qRT-PCR run.
Genome-wide long read sequencing
We isolated high molecular weight (HMW) DNA for genome-wide long-read sequencing using the Gentra Puregene Cell Kit (Qiagen, 158767) with some minor modifications. Briefly, we lysed cells using 1.5 mL of Cell Lysis Solution per 5 million cells, followed by incubation at 37°C for 1 hour. We then added 10 μl of Proteinase K (Qiagen, 158918) and incubated at 55°C for 1 hour. We removed RNA by adding 10 μl of RNase A and incubating at 37°C for 1 hour. 500 μl of protein precipitation solution (provided in the kit) was added to each tube and vortexed for 10 seconds. Samples were centrifuged at 12,000xg for 5 minutes. The supernatant from each tube was added to a new tube containing 1.5 mL of isopropanol (Thermo Fisher, T036181000) and inverted 50 times. We extracted HMW DNA using a disposable inoculation loop, and washed by dipping into ice-cold 70% ethanol. We resuspended the DNA precipitate in 100 μl of Qiagen elution buffer (Qiagen, 19086) and incubated at 50°C for 30 minutes and then at room temperature overnight to allow full resuspension of the DNA. We quantified DNA using Qubit dsDNA HS kit (Invitrogen, Q32851). We submitted the HMW DNA to the Cold Spring Harbor Laboratory core facility for genome-wide PCR-free long-read sequencing on a PromethION (Oxford Nanopore Technologies).
Targeted long-read sequencing of CGG at FMR1
We performed targeted sequencing of the 5’UTR CGG short tandem repeat expansion at the FMR1 locus by CRISPR-Cas9 targeted genomic digestion of the locus, targeted DNA long reads library preparation, and long read sequencing on the MinION sequencer (Oxford Nanopore Technologies). We designed four CRISPR-Cas9 crRNAs specific to PAM sequences upstream and downstream of the 5’UTR CGG STR in FMR1 (Table S2) using the CHOPCHOP online tool (version 3.0.0 using parameters: Target: FMR1, in: Homo sapiens hg38/GRCh38, using: CRISPR-Cas9, for: nanopore enrichment). We ordered 2 nmol of lyophilized customized single-stranded crRNAs (Integrated DNA Technologies, Table S2) and 2 nmol of single-stranded tracrRNA (Integrated DNA Technologies, 1072532). We resuspended all RNA to 100 μM in 10 mM Tris-EDTA (pH 7.5) (Invitrogen, AM9858) and created a crRNA-tracrRNA pool consisting of 2.5 μM of each crRNA and 10 μM of the tracrRNA in Duplex Buffer (Integrated DNA Technologies, 11–01-03–01). We annealed the crRNA and tracrRNAs to create a crRNA•tracrRNA pool by incubating at 95° C for 5 minutes and cooling to room temperature.
We prepared DNA based on previously published targeted Cas9 targeted sequencing protocols73,74 with modifications. Briefly, we lysed 5 million iPSCs by resuspending in 100 μl of PBS (Corning, 21–040-CV) and adding 10 mL of Tris-Lysis-Buffer solution (10 mM Tris-Cl (pH 8) (Invitrogen, 15568025), 25 mM EDTA (pH 8) (Invitrogen, 15575020), 0.5% SDS (w/v) (Fisher Scientific, BP1311), and 20 μg/mL RNase A (Sigma-Aldrich, 10109142001)) for 1 hour at 37°C followed by proteinase K (New England Biolabs, P8107S) digestion at 50°C for 3 hours. We then performed two phase separations by mixing the sample and 10 mL of ultrapure Phenol/Chloroform/Isoamyl Alcohol (Fisher Scientific, BP1752I100) in Falcon tubes containing phase-lock gel (5g of Corning High Vacuum Grease (Dow Corning, 1658832) autoclaved in 50 mL Falcon tube) and centrifuging at 2800xg for 10 minutes. Next, we performed DNA precipitation by mixing the aqueous phase with 4 mL of 5 M ammonium acetate (Invitrogen, AM9070G) and 30 mL of cold 100% ethanol (Decon Labs, 2716), centrifuging at 12,000xg for 5 minutes, two washes with 70% ethanol, and dried the DNA pellet at room temperature for 5 minutes. We resuspended the DNA in 100 μl of 10 mM Tris-EDTA (pH 8.0) on a rotator at room temperature overnight before storing at 4°C for up to 2 days before use.
We performed CRISPR-Cas9 targeted genomic digestion by first dephosphorylating genomic DNA. We incubated 5 μg of high molecular weight DNA, 3 μl NEB rCutSmart Buffer (New England Biolabs, B6004), and 3 μl of QuickCIP enzyme (New England Biolabs, M0525S) at 37°C for 20 min followed by 80°C for 2 min, and 20°C for 15 minutes. Next, we assembled Cas9 ribonucleoproteins (RNPs) in vitro in a 100 μl reaction by incubating 10 μM crRNA•tracrRNA pool, 1X NEB CutSmart buffer, nuclease-free water (Sigma-Aldrich, W4502), and 62 μM HiFi Cas9 (Integrated DNA Technologies, 1081060) on ice for 30 minutes. We then digested the DNA by incubating 10 μL of RNPs with 5 μg of dephosphorylated high molecular weight DNA, 10 mM dATP (Thermo Scientific, R0141), and 1 μL Taq polymerase (New England Biolabs, M0273) at 37°C for 60 minutes, followed dA-tailing of blunt ends by incubation the sample at 72°C for 5 minutes. We purified our Cas9-cut genomic DNA by adding 16 μl of 5 M ammonium acetate (Invitrogen, AM9070G) and 126 μl of cold 100% ethanol, spinning down at 16,000xg for 5 minutes. We then washed the DNA pellet twice with 70% ethanol to remove excessive salts. We dried the DNA pellet at room temperature for 5 minutes before resuspending the DNA in 10 mM Tris-HCl (pH 8.0) at 50°C for 1 hour followed by rotation at 4°C overnight. We performed size selection for Cas9-cut DNA with the Blue Pippin (Sage Science) using the “0.75DF 3–10 kb Marker S1” cassette definition and size range mode at 5–12 kb.
To prepare the library for sequencing, we barcoded each sample by adding 3 μl of barcode (Oxford Nanopore Technologies, EXP-NBD104) and 50 μl of Blunt/TA Ligase Master Mix (New England Biolabs, M0367) to each sample. We incubated the samples at room temperature for 10 minutes and then performed a cleanup using 50 μl of Agencourt AMPure XP beads (Beckman Coulter, A63881), eluting the library in a final volume of 16 μl nuclease-free water. We quantified samples using a Qubit fluorometer and Qubit dsDNA HS assay kit (Invitrogen, Q32851) and then ligated the barcoded DNA to the Nanopore adapters for MinION flowcell sequencing using the NEBNext Quick Ligation Module (New England Biolabs, E6056S) and Ligation Sequencing Kit (Oxford Nanopore Technologies, SQK-LSK109). In short, we prepared an Adapter Ligation Solution consisting of 20 μl NEBNext Quick Ligation Buffer, 10 μl NEBNext Quick T4 DNA ligase, and 5 μl Nanopore Adapter Mix (AMII) (Oxford Nanopore Technologies, EXP-NBD104). We then mixed 20 μl of Adapter Ligation Solution with 65 μl barcode-ligated DNA. Immediately after mixing, we added the remaining 15 μl of the Adapter Ligation Solution and incubated for 10 minutes at room temperature. We next purified our DNA libraries by first bringing the total volume to 100 μl using nuclease-free water and then adding 100 μl of TE (pH 8.0) and 80 μl of AMPure XP Beads. We incubated the sample for 10 minutes at room temperature before separating the beads using a magnet and discarding the supernatant. We washed the beads with 250 μl Nanopore Long Fragment Buffer twice and then air-dried the DNA pellet for ~30 seconds. We eluted the library in 14 μl Nanopore Elution Buffer. Finally, we mixed 13 μl of the library with 37.5 μl Nanopore Sequencing Buffer and 25.5 μl loading beads and loaded the library onto the MinION flowcell for sequencing. We sequenced the libraries for 48 hours.
PCR-free Whole Genome Sequencing
We extracted genomic DNA from all iPSC lines using the GeneJet Genomic DNA purification kit (Thermo Scientific, K0721) per manufacturer’s protocol. Genewiz performed library prep and sequencing on the HiSeqX platform with 150 bp paired-end reads.
QUANTIFICATION AND STATISTICAL ANALYSIS
Targeted Nanopore long-read sequencing
We performed base-calling of raw nanopore fast5 using Guppy (version 6.2.1) and aligned the output fasta files to hg38 using minimap2 (version 2.22-r1101). We performed several quality-control steps to ensure only high-quality reads were used in downstream analysis: (1) removing reads that did not align to the FMR1 gene, (2) using only reads that mapped to the reverse strand due to cast errors for the ultra-high-GC content CGG STR in the forward strand, (3) filtering out truncated reads that did not contain an upstream sequence to the CGG tract “ACCAAACCAA” and at least four consecutive CGGs, and (4) removing reads that contain more than nine consecutive “TA” nucleotides within the CGG repeats, as these reflect base calling errors. We created a custom script to count the number of CGGs in the remaining high-quality reads by finding the first and last instances of the string “CGGCGGCGG”, counting the number of CGGs between them and subtracting five CGGs from the total sum. These five CGGs were excluded because they reflect CGGs located within the FMR1 5’UTR but upstream and external to the continuous CGG tract. We plotted the CGG counts of the reads that also had corresponding methylation scores from Nanopolish and STRique (See ‘DNA methylation‘)
DNA methylation
We called DNA methylation from the long-reads using two different methods. We used nanopolish (version 0.13.2) to call methylation in the 19 CpG dinucleotides in the 500 bp FMR1 promoter (hg38, chrX:147911419–147911919). Because nanopolish cannot call DNA methylation over a variable number of CGG triplets, we used STRique (version 0.4.2) to call methylation over the CGG tract itself in our normal-length, pre-mutation, and FXS iPSCs.
For the FMR1 promoter, we first indexed the fast5 files using the nanopolish command ‘index’. We called CpG methylation using the command ‘call-methylation’ in the window ‘chrX:147,902,117–147,960,927’. We considered Log2 likelihood >0.1 as methylated and <−0.1 as un-methylated. For every single-molecule read in every iPSC line, we computed the proportion of 19 CpGs that were methylated. We removed the reads that didn’t have CGG counts from our custom code and didn’t pass STRique filtering (see below). We plotted the proportion as Kernel Distribution Estimation (KDE) using the function ‘density’ in R.
To determine CpG methylation specifically at the CGG STR in the 5’UTR of FMR1, we first indexed the fast5 files using the STRique command ‘index’. We then computed methylation status and CGG counts using the STRique command ‘count’ with the respective models ‘r9_4_450bps_mCpG.model’ and ‘r9_4_450bps.model’. We only used reads with prefix and suffix scores greater than 4 for further analyses as the reads with <4 were of low-quality mapping scores to the upstream and downstream regions of the CGG tract. We removed reads that didn’t have CGG counts from our custom code and promoter methylation values from nanopolish. We then calculated the total methylated CpGs over CGG and plotted as jitter plots. We also plotted methylated (1) and unmethylated (0) nucleotides as red and black stripes along the repeats, respectively.
Hi-C data processing
We processed Hi-C reads using Hi-C Pro (version 2.7.7). Briefly, we aligned paired-end reads independently to the hg38 human genome using Bowtie2 (v2.2.9) (global parameters: --verysensitive –L 30 –score-min L,−0.6,−0.2 –end-to-end --reorder; local parameters: --very-sensitive –L 20 –score-min L,−0.6,−0.2 –end-to-end --reorder). We then filtered out unmapped reads, non-uniquely mapped reads, and PCR duplicates, and then paired the remaining uniquely aligned reads. We assembled raw cis contact matrices for all samples into 20kb, 40kb, and 100kb non-overlapping bins and balanced using the Knight-Ruiz algorithm. We normalized the balanced cis matrices across all iPSC-NPC lines using distance-dependent median-of-ratios size factors to normalize for sequencing depth75,76. We assembled trans m x n contact matrices by binning hg38 aligned, in situ Hi-C paired-end reads into uniform 1 Mb-sized non-overlapping bins and balancing using the Knight-Ruiz algorithm with default parameters. We quantile normalized trans matrices across samples to facilitate direct comparison.
A/B compartment identification
To determine A/B compartment status genome-wide, we calculated the eigenvector of 100 kb Knight-Ruiz-balanced cis Hi-C matrices for each chromosome as previously described77,78. Briefly, we first normalized the balanced matrix by the expected distance dependence mean counts value, followed by removal of rows and columns that were composed of less than 2% non-zero counts. We then calculated the z-score of the off-diagonal counts and calculated a Pearson correlation matrix for the cis-interaction matrixes. We selected the largest eigenvalue of the Pearson correlation matrix computed from the Hi-C matrix as the eigenvector. Coordinates corresponding to transitions between positive and negative eigenvector values demarcate boundaries of compartments. Using the established pattern of gene density in A/B compartments, we assigned positive eigenvector values to the gene-dense A compartment, and negative values to the gene-poor B compartment.
Hi-C contact matrix difference maps
To directly compare Hi-C contact matrices between two iPSC-NPC lines, difference heatmaps were created by taking the log2 ratio of the two contact matrices for the region of interest. Any values in either contact matrix that were less than 5 were dropped before normalizing.
Quantifying long-range interaction frequency
To determine the interaction frequency between FMR1 and SLITRK2, we used Knight-Ruiz normalized Hi-C data binned at 20 kb and summed the normalized counts in bins corresponding to interactions between the hg38 coordinates of the two genes in the cis X chromosome interaction matrix. To determine the interaction frequency between FMR1 and SLITRK4, we used Knight-Ruiz normalized Hi-C data binned at 40 kb and summed the normalized counts in bins corresponding to interactions between the hg38 coordinates of the two genes in the cis X chromosome interaction matrix.
Insulation score and boundary strength
To calculate insulation score, we tiled a 200 kb square window (10 × 10 bins on 20 kb binned data) with one bin offset from the diagonal across the genome on Knight-Ruiz-balanced cis Hi-C maps79,80. We then summed, normalized by the chromosome-wide mean, and log transformed counts in the 20 × 20 bin window to obtain the Insulation Score (IS) of that window. We characterize “boundary strength” within a domain by calculating the difference between the window with the lowest insulation score in the domain and the average insulation score across a 200 kb neighboring region.
ChIP-seq mapping for libraries generated in this study
We processed ChIP-seq data as previously described58,65–67,69–71. Briefly, we mapped 75 bp single-end reads to the hg38 reference genome using Bowtie (v 0.12.7) with parameters: “--tryhard --time --sam -S -m2”. We removed optical and PCR duplicates using Samtools commands “sort” and “markdup -r” (version 1.11). We filtered the bam files keeping only reads that were properly mapped and then indexed the files with Samtools functions “view -F 4” and “index”, respectively. Using the Samtools function “view –hbs”, we downsampled reads to achieve equal read numbers across samples using a seed value of 42. We created index files for each downsampled file. We called CTCF peaks using MACS2 (v 2.1.1.20160309) with a cutoff of p-value < 1×10−8 using input samples as control files. For CTCF visualization, we produced bigwigs using deepTools (v3.3.0) bamCoverage with default parameters. For H3K9me3 bigwig visualization, we performed input subtract using deepTools bamCompare with the flag “-operation subtract”. We called H3K9me3 domains using the RSEG program (See ‘H3K9me3 domain calling’ for more information).
Re-analyzing published H3K9me3 ChIP-seq data used in Supplementary Figure 8
We analyzed previously published sequencing data (Table S4) by soft trimming reads with a quality score less than 20 and removing reads smaller than 15 bp using cutadapt v1.18. We mapped reads using Bowtie2 (version 2.2.5) with default parameters for single end datasets and with the parameters “--local --very-sensitive-local --no-mixed --no-discordant -I 10 -X 700” for paired end datasets. We removed duplicates and unmapped reads and then converted the file to bam format using Samtools (version 1.11) fixmate, sort, markdup “-r”, and view “-F 4” commands. We downsampled mapped reads for inputs and H3K9me3 samples to the lowest number of mapped fragments using Samtools view with parameters “-hbs” and a seed of 42. Indices were created for each file using Samtools “index”. We then input normalized bam files using BamCompare from deeptools (version 3.3.0) using the “--binSize 10 --smoothLength 30 --extendReads 200 –operation subtract” parameters for single end datasets and with parameters “--binSize 10 --smoothLength 30 --extendReads 200 --samFlagInclude 64 –operation subtract” for paired end datasets. We included “--samFlagInclude 64” to ensure properly paired reads were only counted once in order to compare signal with single end datasets. iPSC-18c was downsampled to a lower sequencing depth because the sequencing depth was significantly lower than other previously published ChIP-seq datasets.
Binning ChIP-seq
We plotted H3K9me3 signal in heatmap form by binning ChIP-seq signal in each domain into 100 equally sized bins and calculating the average H3K9me3 ChIP-seq signal in each bin. The flanking 50 kb regions around each domain were also binned into 100 equally sized bins, and the average H3K9me3 ChIP-seq signal in each bin was calculated and plotted.
CUT&RUN Data Processing
We analyzed CUT&RUN sequencing data using Bowtie2 (version 2.2.5) with parameters “--local --very-sensitive-local --no-mixed --no-discordant --phred33 -I 10 -X 700”. We removed duplicates and unmapped reads and then converted the file to bam format using Samtools (version 1.11) fixmate, sort, markdup “-r”, and view “-F 4” commands. We downsampled mapped reads for IgG and H3K9me3 samples to the lowest number of mapped reads for each comparison group using Samtools view with parameters “-hbs” and a seed of 42. Indices were created for each file using Samtools “index”. We then input normalized bam files using BamCompare from deeptools (version 3.3.0) using the “–extendReads –binSize 10 –smoothLength 30 –operation subtract” parameters.
CUT&RUN data processing for brain tissue
We performed CUT&RUN data processing as earlier described with minor modifications. After mapping, we kept duplicates instead of removing them. Unlike with ChIP-seq, this is an acceptable method of data processing. In CUT&RUN, targeted DNA fragmentation is performed using a pA/G-MNase fusion protein tethered to an antibody which is bound to the target. As a result, duplicates are expected based on MNase cutting DNA in a non-random pattern. This is unacceptable in ChIP-seq as DNA is randomly sheared and therefore, duplicates are expected to be primarily from PCR over-cycling. Lastly, we converted downsampled bam files to bigwigs with log2 input normalization using BamCompare from Deeptools (v3.3.0) with parameters “--extendReads –binSize 10 –smoothLength 30 --operation log2”
iPSC and iPSC-NPC H3K9me3 domain calling from ChIP-seq
We computationally identified H3K9me3 domains using the RSEG package (version 0.4.9)81. First, we converted downsampled, filtered bam files into bed files using BedTools (v2.92.2) bamtobed and sorted as described in RSEG documentation. We ran RSEG-Diff on the H3K9me3 ChIP-seq samples against their inputs with parameters “-mode 2 -s 800000 -bin-size 100 -P -posterior-cutoff 0.9995” and the deadzone flag (-d). We generated hg38 deadzones using the RSEG deadzone command with default parameters using kmer sizes of 37 and 75 bp.
We filtered and filled gaps iteratively from the full list of domain calls for iPSC and iPSC-NPC H3K9me3 ChIP-seq. We removed domains less than 10 kb in size and within 2.5 Mb of centromeres and 1 Mb of telomeres. Next, gaps within 50 kb of domains were merged if the average H3K9me3 signal was at least 40% of the mean signal in the flanking regions. If the gap consisted of 70% of dead zones and was at least 50 kb in size, gaps between domains were merged. Next, we merged domains within 7.5 kb of each other to fill small, local gaps using BedTools merge. Then, we excluded domains less than 47.5 kb in order to remove small domains. Next, domains within 65 kb were merged, and domains less than 47.5 kb were removed to fill mediumsized gaps and remove medium-sized domains, respectively. Finally, to fill large gaps, gaps within 750 kb of domains were merged if the average H3K9me3 signal was at least 25% of the mean signal in either flanking regions, and the flanking regions were at least 400 kb in size. If the gap consisted of 70% of dead zones and was at least 50 kb in size, gaps between domains were merged.
To focus our analysis on Mb-scale H3K9me3 domains specific to our FXS iPSC and iPSC-NPC cell lines, we performed additional domain filtering. First, we concatenated all domain calls in NL_18, NL_27, NL_25, and PM_137 as “control domains”. We merged domains within 100 kb of each other and kept domains that were present in at least 2 of 4 genotypes if there was reciprocal overlap of at least 15%. Only “control domains” that were larger than 100 kb were kept. Similarly, we concatenated all domain calls in FXS_421, FXS_426, and FXS_470 as “FXS domains” and kept domains that were present in all three FXS samples if there was reciprocal overlap of at least 15%. Only “FXS domains” larger than 500 kb were kept. To generate “FXS-recurrent domains”, we subtracted “control domains” from “FXS domains” only keeping the resulting domains if they were larger than 300 kb, and the result was merged if domains were within 600 kb. Lastly, resulting domains were required to overlap with the pre-concatenated domain calls for each line.
To generate “FXS-variable domains”, we filtered out domains less than 250 kb in size for each individual cell line. Then we subtracted domains from “FXS-recurrent domains” and “control domains”. Next domains from each individual FXS cell line were subtracted from each other if there was at least 60% reciprocal overlap. Domains in each cell line were merged if they were within 200 kb of each other, and domains less than 350 kb were removed. Finally, domains from each cell line were concatenated together and domains within 500 kb were merged to form Mb-scale “FXS-variable domains”. We defined “Genotype-invariant H3K9me3 domains” as domains present in at least 6 of 7 of FXS iPSC-NPCs with at least 50% reciprocal overlap.
B-lymphoblastoid H3K9me3 domain calling from ChIP-seq
We computationally identified H3K9me3 domains using the RSEG package (version 0.4.9)81. First, we converted downsampled, filtered bam files into bed files using BedTools (v2.92.2) bamtobed and sorted as described in RSEG documentation. We ran RSEG-Diff on the H3K9me3 ChIP-seq samples against their inputs with parameters “-mode 2 -s 800000 -bin-size 100 -P posterior-cutoff 0.9995” and the deadzone flag (-d). We generated hg38 deadzones using the RSEG deadzone command with default parameters using kmer sizes of 37 and 75 bp.
We filtered and filled gaps iteratively from the full list of domain calls for B-lymphoblastoid H3K9me3 ChIP-seq. We removed domains less than 2.5 kb in size and within 2.5 Mb of centromeres and 1 Mb of telomeres. Next, gaps within 250 kb of domains were merged if the average H3K9me3 signal was at least 20% of the mean signal in the flanking regions. If the gap consisted of 70% of deadzones and was at least 50 kb in size, gaps between domains were merged. Next, we merged domains within 27.5 kb of each other to fill small, local gaps using BedTools merge. Then, we excluded domains less than 55 kb in order to remove small domains. Next, domains within 50 kb were merged, and domains less than 75 kb were removed to fill medium-sized gaps and remove medium-sized domains, respectively. Finally, to fill large gaps, gaps within 2 Mb of domains were merged if the average H3K9me3 signal was at least 40% of the mean signal in either flanking regions, and the flanking regions were at least 150 kb in size. If the gap consisted of 70% of deadzones and was at least 50 kb in size, gaps between domains were merged.
To focus our analysis on Mb-scale H3K9me3 domains that spread or are acquired de novo in our FXS lymphoblastoid B-cells lines, we performed additional domain filtering to generate large domains present in both FXS B lymphoblastoid B-cells lines but not the normal-length cell line. First, we concatenated all domain calls in FXS_B_650 and FXS_B_900 as “FXS domains”. We merged domains within 100 kb of each other and kept domains that were present in both genotypes if there was reciprocal overlap of at least 25%. Only “FXS domains” that were larger than 100 kb were kept. To generate domains consistently present in both FXS lymphoblastoid B-cells lines, “FXS-recurrent domains”, we subtracted domains greater than 100 kb in the NL_B cell line from “FXS domains” and only kept the resulting domains if they were larger than 100 kb. Finally, the result was merged if domains were within 200 kb. Resulting “FXS-recurrent domains” were required to overlap with the pre-concatenated domain calls for each FXS lymphoblastoid B-cells line.
“FXS-recurrent domains” were used to identify spreading and de novo domains in FXS patient-derived EBV-transformed lymphoblastoid B-cells. Spreading domains were generated by intersecting 10 bp flanking regions of filtered NL_B domains greater than 250 kb with “FXS-recurrent domains” greater than 500 kb. This identified “FXS-recurrent SPREAD domains” that were immediately adjacent to NL_B domains. These spreading domains were then inverse intersected with “FXS-recurrent domains” to identify domains which were not adjacent to NL_B domains which represent “FXS-recurrent DE NOVO domains”.
Brain tissue H3K9me3 domain calling from CUT&RUN
We computationally identified H3K9me3 domains using the RSEG package (version 0.4.9)81. First, we converted downsampled, filtered bam files into bed files using BedTools (v2.92.2) bamtobed and sorted as described in RSEG documentation. We ran RSEG on the H3K9me3 CUT&RUN samples with parameters “-s 800000 -bin-size 100 -P -posterior-cutoff 0.5 -duplicates” and the deadzone flag (-d). We generated hg38 deadzones using the RSEG deadzone command with default parameters using a kmer size of 37.
We filtered and filled gaps iteratively from the full list of domain calls for brain tissue H3K9me3 CUT&RUN. We removed domains less than 2 kb in size and within 2.5 Mb of centromeres and 1 Mb of telomeres. Next, gaps within 150 kb of domains were merged if the average H3K9me3 signal was at least 40% of the mean signal in the flanking regions. If the gap consisted of 70% of deadzones and was at least 50 kb in size, gaps between domains were merged. Next, we merged domains within 5 kb of each other to fill small, local gaps using BedTools merge. Then, we excluded domains less than 5 kb in order to remove small domains. Next, domains within 10 kb were merged, and domains less than 45 kb were removed to fill medium-sized gaps and remove medium-sized domains, respectively. Finally, to fill large gaps, gaps within 2 Mb of domains were merged if the average H3K9me3 signal was at least 70% of the mean signal in the flanking regions, and the flanking regions were at least 105 kb in size. If the gap consisted of 70% of deadzones and was at least 50 kb in size, gaps between domains were merged. Brain tissue H3K9me3 CUT&RUN signal was floored at zero to only consider signal from CUT&RUN where H3K9me3 was more enriched than the input.
To focus our analysis on Mb-scale H3K9me3 domains that spread or are acquired de novo in our FXS brain tissue, we performed additional domain filtering to generate large domains present in both FXS caudate nucleus but not the control tissue samples. First, we concatenated all domain calls in FXS_CN_1 and FXS_CN_2 as “FXS domains”. We concatenated all domain calls in NL_CN_1 and NL_CN_2 as “control domains”. We merged domains within 200 kb of each other and kept domains that were present in both genotypes if there was reciprocal overlap of at least 25%. Only “FXS domains” and “control domains” that were larger than 200 kb and 150kb, respectively, were kept. To generate domains consistently present in both FXS_CN_1/2, “FXS-recurrent domains”, we subtracted domains greater than 150 kb in the NL_CN_1/2 from “FXS domains” and only kept the resulting domains if they were larger than 250 kb. Finally, the result was merged if domains were within 300 kb. Resulting “FXS-recurrent domains” were required to overlap with the pre-concatenated domain calls for each FXS_CN_1/2.
“FXS-recurrent domains” were used to identify spreading and de novo domains in FXS patient-derived caudate nucleus brain tissue. Spreading domains were generated by intersecting 10 bp flanking regions from “control domains” with “FXS-recurrent domains” greater than 250 kb. This identified “FXS-recurrent SPREAD domains” that were immediately adjacent to NL_CN_1/2 domains. These spreading domains were then inverse intersected with “FXS-recurrent domains” to identify domains which were not adjacent to NL_B domains which represent “FXS-recurrent DE NOVO domains”.
iPSC H3K9me3 domain calling from CUT&RUN
We computationally identified H3K9me3 domains using the RSEG package (version 0.4.9)81. First, we converted downsampled, filtered bam files into bed files using BedTools (v2.92.2) bamtobed and sorted as described in RSEG documentation. We ran RSEG-Diff on the H3K9me3 ChIP-seq samples against their inputs with parameters “-mode 2 -s 800000 -bin-size 100 -P -posterior-cutoff 0.9995” and the deadzone flag (-d). We generated hg38 deadzones using the RSEG deadzone command with default parameters using kmer sizes of 37 and 75 bp.
We filtered and filled gaps iteratively from the full list of domain calls for iPSC H3K9me3 CUT&RUN. We removed domains less than 15 kb in size and within 2.5 Mb of centromeres and 1 Mb of telomeres. Next, gaps within 15 kb of domains were merged if the average H3K9me3 signal was at least 30% of the mean signal in the flanking regions. If the gap consisted of 70% of deadzones and was at least 50 kb in size, gaps between domains were merged. Next, we merged domains within 2.5 kb of each other to fill small, local gaps using BedTools merge. Then, we excluded domains less than 40 kb in order to remove small domains. Next, domains within 150 kb were merged, and domains less than 75 kb to fill medium-sized gaps and remove medium-sized domains, respectively. Finally, to fill large gaps, gaps within 200 kb of domains were merged if the average H3K9me3 signal was at least 30% of the mean signal in either flanking region. If the gap consisted of 70% of deadzones and was at least 50 kb in size, gaps between domains were merged.
Identification of genes in H3K9me3 domains
We identified genes as co-localized to H3K9me3 domains if the promoter (TSS +/− 1 kb) of the gene was contained within the domain or the gene overlapped with the domain by 50%. We performed the intersections using the BedTools (v2.30.0) function ‘intersect’.
Identification of reprogrammed vs resistant domains
We categorized FXS-recurrent H3K9me3 domains as either reprogrammed or resistant to CGG deletion based on if the length of the RSEG domain call in the edited iPSC line was less than half the size of that in the parent disease cell line (reprogrammed) or not (resistant). Domains were considered lowered if the length of the RSEG domain in the edited iPSC lines was greater than 50% of the parent line with less than two-thirds of the H3K9me3 CUT&RUN signal.
RNA-seq gene expression analysis
We mapped RNA-seq reads to the hg38 ensembl reference transcriptome release 107 for both cDNA and ncRNA using kallisto (v 0.44.0) quant with 100 bootstraps of transcript quantification82 as described in the kallisto documentation. We converted the resulting quantifications into DESEQ2 format and mapped transcript level counts to gene level counts in R using the package “tximport” (v1.22.0) according to DESEQ2 documentation recommendations83. We filtered out genes with total counts less than 60 across all samples from analysis and normalized data using the DESEQ2 median of ratios-based method. We determined differentially called genes across the iPSC-NPC lines studied in a pairwise manner using DESEQ2 (v1.34.0) LRT with adjusted p-value < 0.005.
Gene ontology analysis
We performed gene ontology enrichment using the WebGestalt R package (v 0.4.4) with the following settings: Organism of interest = homo sapiens; Method of interest = overrepresentation enrichment, Functional database = geneontology, biological_process_noRedun. We identified gene name identifiers for each set of classified genes and used the genome_protein-coding set as the reference set. We plotted the enrichment ratios and −log10(p-values) for the top 5 gene ontology terms with an p-value < 0.01 and enrichment ratio > 4. All protein-coding genes with TSSs co-localized to “FXS-recurrent H3K9me3 domains” or “FXS-variable H3K9me3 domains” or “genotype-invariant H3K9me3 domains” were input into WebGESTALT. Only protein coding genes were included using the genome protein-coding set as the reference set.
GTEX gene expression data
We obtained gene expression across human tissues from the GTEX consortium. We obtained the data used for the analyses described in this manuscript from https://www.gtexportal.org/home/datasets from the GTEx Portal in 04/2020. To generate the heatmap in Figure 2, we first retrieved the expression of all genes in 11 “FXS-recurrent H3K9me3 domains”. We removed genes with 0 expression across all tissues, resulting in a final list of 54 genes. We calculated the gene expression z-score across tissues to ensure strong expression of a gene in one tissue type does not diminish the expression in all other tissues. Finally, we clustered genes on the gene expression data using scipy.cluster (v1.9.0) KMeans function to cluster into 4 groups labeled by the tissue types dominating each cluster.
RNA-seq analysis of the human fetal cortex
We analyzed publicly available RNA-seq in human male control and FXS fetal cortex to examine the down-regulation genes present in our 11 FXS-recurrent H3K9me3 domains identified in FXS iPSC-NPCs. We downloaded the N=1 male normal-length healthy brain tissue RNA-seq dataset and the N=1 male FXS patient RNA-seq dataset for re-analysis starting from raw fastq files from GEO (GSE146878). We processed the fastq files using the pseudo-alignment tool ‘Kallisto’ with default parameters and hg38 transcriptome. Given the files exhibited marked technical differences in read depth, we performed quantile normalization of Kallisto-calculated TPM in the control and FXS sample using the function ‘normalize.quantiles’ of the R package ‘preprocessorCore’ and used normalized data for further analysis. We extracted the transcripts for each gene co-localized in the N=11 iPSC-NPC FXS-recurrent H3K9me3 domains and calculated the fold change as the log2 ratio of TPM in FXS to non-diseased/normal-length brain tissue. To create a null distribution, we computed the same fold change in 100 iterations of random intervals (10 size-matched random intervals on autosomes and 1 size-matched random interval on the X chromosome) and calculated the median log2 fold change from each draw to create a null distribution. We computed a one-tailed empirical P-value as the proportion of random intervals with log2 fold change less than the value computed for the FXS-recurrent H3K9me3 domains.
RNA-seq analysis of Fmr1 knock-out mouse cortical neurons
We analyzed publicly available RNA-seq data-sets of WT and Fmr1 KO mouse cortical neurons (both male and female embryos from a single pregnant mouse) to examine the presence of BREACHes. We downloaded the N=3 WT and the N=3 Fmr1 KO RNA-seq datasets for re-analysis starting from raw fastq files from GEO (GSE81912). We processed the fastq files using the pseudo-alignment tool ‘Kallisto’ with default parameters and mm10 transcriptome. We converted the resulting quantifications into DESEQ2 format and mapped transcript level counts to gene level counts in R using the package “tximport” (v1.22.0) according to DESEQ2 documentation recommendations (Love et al., 2014). We filtered out genes with total counts less than 50 across all samples from analysis and normalized data using the DESEQ2 median of ratios-based method. We defined genes log2 fold change (KO/WT) < −1 as down-regulated genes.
Measurements of distances between H3K9me3 domains using DNA FISH images
We deconvolved DNA FISH images with Huygens Essential deconvolution software v20.04 (Scientific Volume Imaging) using the Classic MLE algorithm with a signal to noise ratio of 40 and 50 iterations (DNA FISH) or signal to noise ratio of 40 and 2 iterations (DAPI stain). We subsequently analyzed our DNA FISH data with TANGO (v0.94)84. We used TANGO to segment nuclei and perform DNA FISH signal calling using the “Hysteresis” algorithm. We manually curated the segmentation to remove merged multiple nuclei. Processing parameters are curated in Table S5. To measure the distance between the domains on chromosomes X (chrX) and 12 (chr12), we removed nuclei where the number of H3K9me3 domains on chrX and chr12 did not equal one and two respectively, and then took the smallest of the distances between the chrX spot and the two spots representing chr12. For chrX to all domain measurements, we first removed nuclei that that had more than 23 foci (11 autosomal domains * 2 + 1 domain on chrX), and where the domain on chrX did not co-localize with any of these foci. For the remaining nuclei, we measured the edge-to-edge spatial distance between the spot representing chrX and the spots representing all other distal domains using the “Distance” algorithm in TANGO (border-to-border). We performed two-tailed Mann-Whitney-U tests to evaluate the difference between the distributions of each measurement among the iPSC lines.
Enrichment of genomic features in FXS-recurrent H3K9me3 domains
We tested the following genomic features for enrichment in FXS-recurrent H3K9me3 domains: (1) number of genes, (2) length of genes, (3) replicated-stress induced double stranded breaks, and (4) S phase replication timing. We evaluated the null hypothesis that the average of a given feature in our 10 FXS-recurrent H3K9me3 domains would be similar to the average in random genomic intervals. Our alternative hypothesis was that the average of a given feature in our 10 FXS-recurrent H3K9me3 domains would be significantly different from the average in random genomic intervals. We used the following test statistics: (1) gene density (Figure 5A): the average number of genes within each interval divided by the size of the interval in base pairs, (2) gene length (Figure 5B): the average length of genes within each interval, (3) replication timing (Figure 5C): the average log2(Early/Late) signal across the interval using a previously published two-fraction Repli-seq experiment performed in a non-diseased normal-length line (https://data.4dnucleome.org/files-processed/4DNFI5WEY784/), and (4) replication stress-induced double strand breaks (Figure 5D): Percent of the 10 intervals in a given draw of random intervals overlapping replication-stress induced double stranded breaks mapped in mouse neural progenitor cells and lifted over from mouse to hg3835.
We compute the same test statistics across N=1,000 iterations of size-matched random genomic intervals without H3K9me3 (N=10). We computed a one-tailed empirical p-value as the percentage of the null distribution that is either less than (left-tailed) or greater than (right-tailed) the test statistic computed on the 10 FXS-recurrent autosomal H3K9me3 domains.
We further tested the enrichment of the above-mentioned genomic features in the invariant H3K9me3 domains specific to iPSC (Figure 2A) in Figures 5E–H to test if the genomic features observed in Figures 5A–D are specific to FXS-recurrent H3K9me3 domains.
CGGx3 enrichment analysis
We extracted the position of every CGG in the hg38 fasta file using custom code. We merged genomic coordinates to get contiguous CGG tracts using bedtools merge using default parameters (i.e., with -d 0 for no gaps between coordinates). We used CGG tracts of unit length >=3 (i.e., >=CGGx3) and those present in gene TSS + 2kb for further analyses. We evaluated the null hypothesis that the average of >=CGGx3 count in our 10 FXS-recurrent H3K9me3 domains would be similar to the average in random genomic intervals. Our alternative hypothesis was that the average of >=CGGx3 count in our 10 FXS-recurrent H3K9me3 domains would be significantly different from the average in random genomic intervals. We formulated an empirical statistical test in which we randomly sampled N=10 size- and gene TSS density-matched genomic intervals with replacement and computed a test statistic of the total number of STRs present inside the domains. We computed the same test statistic for N=1,000 iterations of random intervals and computed a one-tailed empirical p-value as the percentage of the null distribution that is greater than or equal to the test statistic in our N=10 FXS-recurrent H3K9me3 domains.
Genomic coverage/mappability plot
We checked read quality using FastQC (v0.11.9). We aligned the fastq files to the hg38 reference genome using bowtie2 in the end-to-end method with the default parameters. We sorted the reads and removed reads with mapping quality less than 30 using Samtools functions “sort” and “view -q 30”. We downsampled the samples to match corresponding sequencing depth (Table S3) and we calculated genome coverage for all iPSC lines using the published command line tool “goleft indexcov” (version 0.2.4) on aligned bam files with parameters --sex “X,Y”85.
De novo genome assembly
We constructed de novo assembly using PCR-free Whole Genome Sequencing data as previously described86. Briefly, we removed any adapter sequences and quality trimmed ends of reads using cutadapt (v 1.18) with parameters “-j 16 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCA -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT -q 20,20 --minimum-length 60”. Reads less than 60 bp were removed from further analysis and quality checked using FastQC (v 0.11.9). After filtering reads, we analyzed the k-mer distribution using kat (v 2.4.1). Next, we used W2rapContigger (v 0.1) with parameters “-t 48 -m 600 --min_freq 4 -d 16 -K 136” to create a draft assembly from only raw reads using a 60-mer de bruijn graph and an expanded de bruijn graph up to a k-mer size of 136. Parameters for W2rapContigger were chosen based on our analysis of k-mer distributions and the raw reads. Next, we adapter trimmed, and quality trimmed the ends of our raw Hi-C reads using cutadapt with parameters “-j 16 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCA -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT --nextseq-trim=20 -q 20,20 --minimum-length 10”. We applied Juicer (v 1.5) with parameters “-s Arima -p assembly -S early” to map Hi-C reads onto our W2rapContigger draft assembly. We used the output from Juicer and the W2rapContigger draft assembly as inputs to 3D-DNA (v180922) with default parameters. We viewed the output candidate assembly in Juicebox (v 1.11.08), made manual corrections to address assembly errors, and input the edited assembly into 3D-DNA again to finalize the assembly. All sequences over 500 kb were extracted as the final assembly. We mapped our final assembly to hg38 and visualized syntenic regions using JupiterPlots (v 3.8.2).
STR tract genotyping for HipSci Consortium iPSC lines and iPSC lines from the present study
We performed STR genotyping on the PCR-free whole genome sequencing data from N=120 ancestry-, sex-, sequencing depth, and cell type-matched non-diseased iPSC-lines from the HipSci Consortium87. We obtained PCR-free whole genome sequencing data from public repositories (Table S4) as pre-processed CRAM files aligned to hg19. We first converted cram files into fastq files using ‘samtools fastq’ with default parameters and realigned to hg38 using bwa mem with the parameter -T 0. We then downsampled all reads to ~500 million reads to be comparable to the sequencing depth of the NL and FXS iPSC lines used in this study. Next, we ran GangSTR (version 2.5.0) on all hiPSC bam libraries with the STR input file “hg38_ver13.bed” from GangSTR GitHub page (https://github.com/gymreklab/GangSTR), which consists of >830,000 STRs. Default parameters with one additional parameter declaring sex as males (--samp-sex M) were used. We then filtered out low quality GangSTR predictions using DumpSTR (version 4.0.0) with the following parameters ‘--gangstr-min-call-DP 10 --gangstr-max-call-DP 1000’. Since DumpSTR was limited by the quality score from a haploid X chromosome, we focused only on autosomes. The resulting data consisted of an allele-specific STR tract length estimate for 832,380 STRs genome-wide in N=120 non-diseased iPSC lines. We also ran ExpansionHunter (version 5.0.0) using a custom json file created using the STRs from GangSTR’s “hg38_ver13.bed” file. We created a resulting data consisting of 832,380 STRs in N=120 non-diseased iPSC lines. We ran GangSTR and EH with the same parameters for the NL and FXS iPSC lines used in this study, including: NL_18, NL_27, NL_25, FXS_421, FXS_426, FXS_470, CS0002, & WTC11.
Identification of candidate FXS long STRs in FXS iPSC
The N=120 sex-, sequencing depth-, ancestry-, and cell type-matched PCR-free whole genome sequencing datasets from the HipSci Consortium iPSC lines afforded us the ability to assess the distribution of allele lengths for a given STR tract across a set of non-diseased, normal-length iPSC. We generated more than 830,000 STR length distributions, one per each STR tract, representing the expected null distribution of lengths for non-diseased, normal-length iPSCs (Figure 6A). For each STR on autosomes, we generated an expected null distribution of allele lengths using both alleles per all N=120 normal-length iPSCs (N=240 alleles). We filtered out any STRs that were the same length across the entire hiPSC population and across FXS iPSC – such STRs were classified as “Stable”. All STRs that were not classified as “Stable” were moved forward for statistical testing.
We identified a group of “FXS long candidate expansions” in each of our full-mutation FXS iPSC lines as alleles that are significantly longer than the distribution N=240 alleles from normal-length iPSCs (P-value<0.03). As a cross-check for our “FXS long candidate expansion” STR hits, we conducted the same statistical tests using both GangSTR and Expansion Hunter, requiring the STRs pass the significance threshold using both algorithms (Figure S6C). To report only the most conservative, rigorous results, we also required that the “FXS long candidate expansions” were reproducible in all 3 FXS iPSC lines (Figure 6B). We ultimately finalized a conservative list of N=71 FXS significantly long candidate expansions which are reproducibly longer than expected in all 3 FXS iPSCs compared to null distribution of alleles in N=120 normal-length hiPSC lines using two independent algorithms of GangSTR and Expansion Hunter (Table S6).
Quantifying the extent of stepwise somatic instability per STR in each FXS iPSC
To query the extent to which our candidate unstable STRs display somatic instability, we developed custom algorithms (STAR Methods) to compute the number of unique alleles across reads for each individual STR in each FXS iPSC line. First, for a given allele, we extracted all reads that aligned over that STR from the PCR-free whole genome sequencing mapped bam file for a given iPSC line. We then calculated a per base-pair alignment score from the CIGAR string of all reads against the genome assembly. For each read, we extracted the STR length present in the read by subtracting the number of base pairs that were shown as D in the CIGAR string (i.e., deletions) and adding the number of base pairs that were shown as I (i.e., insertions) to the total STR length. Thus, for each STR, we generated a list composed of precise STR lengths present across all reads that mapped to that STR. We finally calculated the number of alleles for a given STR as the number of unique STR lengths. We stratified “FXS significantly long candidate expansions” (N=71) into those with somatic instability (>= 3 alleles) in at least one line (N=53) and those that are somatically stable (1–2 alleles) in all 3 FXS iPSC lines (N=18).
Contingency table for the association of somatic instability with FXS long STRs
To test the association of FXS long STRs with somatic instability, we formulated a 2×2 contingency table with “FXS significantly long candidate expansions” and sequence-matched stable STRs (e.g. unchanging length across all N=120 hiPSC cell lines) for a given FXS iPSC line in the rows and propensity for somatic instability in the columns (1–2 alleles per STR – column 1; 3+ alleles per STR – column 2). We computed an Odds Ratio test statistic and applied Fisher’s Exact test to compute p-values.
Enrichment of somatically unstable STRs in FXS-recurrent domains
We formulated a statistical test to ascertain if our identified somatically unstable STRs in FXS iPSCs were enriched in FXS-recurrent autosomal H3K9me3 domains as compared to size-matched random genomic intervals without H3K9me3. Our null hypothesis was that FXS-reproducible long STRs with somatic instability would be distributed uniformly across the genome. Our alternative hypothesis was that FXS-reproducible STRs with somatic instability would be significantly enriched in FXS-recurrent H3K9me3 domains. We defined an STR as co-localized if it was located within an FXS-recurrent H3K9me3 domain. We formulated an empirical statistical test in which we randomly sampled N=10 size-matched genomic intervals with replacement and computed a test statistic of the total number of STRs present inside the domains. We computed the same test statistic for N=1,000 iterations of random intervals and computed a one-tailed empirical p-value as the percentage of the null distribution that is greater than or equal to the test statistic in our N=10 FXS-recurrent H3K9me3 domains.
H3K9me3 and Hi-C signal quantification in BREACHes for Figures 7C–D
We examined H3K9me3 and Hi-C signal in BREACHes for the normal-length and FXS iPSC lines used in this study as well as two candidate normal-length iPSC lines made with p53 shRNA. We processed H3K9me3 and input ChIP-seq or CUT&RUN data by downsampling to the same sequencing depth and quantile normalization to allow direct comparison. Similarly, for Hi-C trans interactions, we quantile normalized the 1 Mb-binned trans matrices to allow for direct comparison. Data were analyzed for 8 iPSC lines representing three classes of genotypes and H3K9me3 phenomena, including:
Group 1 – normal-length iPSCs made without p53 knock-down and exhibiting no H3K9me3 signal at BREACHes (NL_18, NL_27, & NL_25)
Group 2 – FXS iPSCs made without p53 knock-down and exhibiting strong reproducible H3K9me3 signal at BREACHes (FXS_421, FXS_426, & FXS_470)
Group 3 – normal-length iPSCs made with a perturbation of p53 via shRNA or dominant negative overexpression and exhibiting sporadic H3K9me3 signal at BREACHes (CS0002 & WTC11)
We calculated the coverage of the input normalized H3K9me3 signal in 100 kb non-overlapping bins across all N=10 autosomal BREACHes in hg38 using ‘bedtools coverage’ using default parameters. For each BREACH, we computed the percentage of 100 kb bins which exhibited H3K9me3 signal. For Hi-C, we computed the interaction frequency of the maximum bin in the trans interaction between each autosomal FXS-recurrent H3K9me3 domain and the domain on the X-chromosome.
STR instability burden in BREACHes for Figures 7E–F
We examined STR allele length in BREACHes for the normal-length and FXS iPSC lines used in this study as well as two candidate normal-length iPSC lines made with p53 shRNA. We used our custom code to compute STR allele estimates across all 8 iPSC lines for only “FXS significantly long candidate STR expansions” co-localized in BREACHes which also exhibited somatic instability (>= 3 alleles) in all 3 FXS iPSC lines. Data were analyzed for 8 iPSC lines representing three classes of genotypes and H3K9me3 phenomena, including:
Group 1 – normal-length iPSCs made without p53 knock-down and exhibiting no H3K9me3 signal at BREACHes (NL_18, NL_27, & NL_25)
Group 2 – FXS iPSCs made without p53 knock-down and exhibiting strong reproducible H3K9me3 signal at BREACHes (FXS_421, FXS_426, & FXS_470)
Group 3 – normal-length iPSCs made with a perturbation of p53 via shRNA or dominant negative overexpression and exhibiting sporadic H3K9me3 signal at BREACHes (CS0002 & WTC11)
We computed the burden as the summed number of unique alleles per STR in each line at the 2 candidate somatically unstable STRs on chromosome 5 and chromosome 16 co-localized with BREACHes and reproducibly somatically unstable in all 3 FXS iPSC lines (Figure S7).
Calculation of a heterochromatin-sink score
We calculated a Heterochromatin-Sink score from a recently published universal annotation of the human genome88 that assigns every 200 bp genome bin to one of 100 different states. The Heterochromatin-Sink score was computed by summing the total number of bins labeled as heterochromatin states (“HET1” – “HET9”) in 5 kb bins tiled across the genome. The resulting data resembles a bedGraph file where each chromosomal interval is associated with a number. This dataset was then transformed into a bigwig file using bedGraphToBigWig. The “Het-Sink” score for a given region is then the average bigwig signal in that region.
Supplementary Material
Figure S1: Morphology, expected homogeneous marker expression, and karyotyping in human induced pluripotent stem cells (iPSCs) and iPSC-derived neural progenitor cells (iPSC-NPCs), related to Figure 1. (A) Phase contrast images of iPSC colony morphology in an independent field of view from the immunofluorescence staining. An independent field of view was used for phase contrast imaging and immunofluorescence. (B) Immunofluorescence staining of human iPSC lines for OCT4 (green) and NESTIN (cyan) co-localized with DAPI (blue) as a nuclear marker. Independent field of view from phase contrast images. (C) de novo genome assemblies for autosomal chromosomes across all 7 iPSC lines used in this study. Genome assembly was constructed from iPSC-NPC Hi-C data and iPSC PCR-free whole genome sequencing using W2rapContigger, Juicer, and 3D-DNA (STAR Methods). Layers connecting rectangles on the Jupiter plots demonstrate mapping between the de novo genome assembly (right half of circle) with the hg38 reference genome (left half of circle). Plots without translocations should have clear chromosome layers with no overlaps or intersections with other chromosome layers. FXS-recurrent H3K9me3 domains are denoted with black stars along the reference chromosomes (left half). Grey stripes denote masked regions. No clear evidence of a translocation was observed. (D) Genome coverage in 16 kb bins across chromosomes containing FXS-recurrent H3K9me3 domains (“grey dashed boxes”) calculated from PCR free whole genome sequencing data generated in iPSCs. Karyotype was largely normal across all seven lines. In one specific case, the FXS_426 line showing a small heterozygous deletion on chr18 (“green dashed box”) which was not a BREACH locus or chromosome examined in this study. Coverage was normalized from 0 to 2 where 0 is a homozygous deletion and 2 is a homozygous duplication. Dashed lines represent BREACHes. (E) Phase contrast images of iPSC-derived NPC rosettes. An independent field of view was used for phase contrast imaging and immunofluorescence. (F) Immunofluorescence staining of human iPSC-derived NPCs for OCT4 (green) and NESTIN (cyan) co-stained with DAPI (blue) as a nuclear marker. Scale bars, 250 μm.
Figure S2: DNA methylation, 3D genome folding disruption, and acquisition of a Mb-sized heterochromatin domain at the FMR1 locus in iPSC-NPCs with normal-length, premutation-length, and mutation-length CGG STR lengths, related to Figure 1. (A) Schematic representation of the FMR1 locus with the CpGs within the CGG tract (analyzed in D) and promoter (analyzed in C) annotated. (B) Visual representation of reverse Nanopore reads (i.e., single alleles) spanning upstream and downstream regions of FMR1 across iPSC lines. CGGs are highlighted in green and other nucleotides are grey. Base calling accomplished with guppy (STAR methods). (C) Visual representation of the DNA methylation status of the 19 CpGs present in the FMR1 promoter called using nanopolish (STAR methods). (D) Visual representation of methylation status of CpGs within the FMR1 CGG tract by the STR-specific tool STRique (STAR methods). In panels (C) and (D), DNA methylation is annotated per read, with read order kept consistent across (B), (C), and (D). (E) Hi-C data across the two iPSC-NPC lines not shown in Figure 1 are shown as a heatmap of interaction frequency for an 8 Mb region around FMR1. A/B compartment score, input normalized H3K9me3 ChIP-seq, and CTCF ChIP-seq in iPSC-derived NPCs is displayed below the heatmaps. Horizontal black lines above H3K9me3 ChIP-seq tracks represent H3K9me3 domain calls. Vertical blue lines above CTCF ChIP-seq tracks represent peak calls using MACS2. FMR1, SLITRK2, and SLITRK4 are highlighted in red, blue, and green, respectively. (F) Hi-C fold-change interaction frequency maps. Gained and lost contacts compared to NL_18 are highlighted in red and blue, respectively. (G) Summed interactions using normalized Hi-C counts between FMR1 and the TAD immediately upstream or downstream is shown as a difference from NL_18. (H) CTCF and input normalized H3K9me3 ChIP-seq for 2 Mb downstream and 6 Mb upstream of FMR1 are overlaid for each iPSC-NPC line. Input normalized H3K9me3 and CTCF ChIP-seq signal ranges from −0.25 to 2.5 and 5 to 30, respectively. (I, J) Zoom-ins to Box 1 and Box 2 from (E) showing (I) FMR1-SLITRK2 or (J) FMR1-SLITRK4 interactions. Arrows point to loops which are disrupted in full mutation-length iPSC-NPC lines. (K,L) Boxplot showing the interaction frequency measured with Hi-C between FMR1 and either (K) SLITRK2 or (L) SLITRK4.
Figure S3: FXS-recurrent H3K9me3 domains on autosomes and the X-chromosome in iPSCs, EBV-transformed B-lymphoblasts, and iPSC-NPCs derived from FXS patients with mutation-length CGG tracts, related to Figure 1 & 2. (A-D) Characterization of the FXS_421 iPSC parent line and a subclone, FXS_425, derived from the same parent line to assess clonal variation in heterochromatin domains. (A) Number of CGG triplets in the FMR1 5’UTR based on Nanopore long read sequencing. (B) Percent of CGGs in the FMR1 5’UTR which are methylated based on STRique analysis of single-molecule Nanopore long-reads. (C) Input normalized H3K9me3 CUT&RUN is shown for a 6 Mb locus around FMR1. Tracks for input normalized H3K9me3 and CTCF CUT&RUN and ChIP-seq are plotted underneath heatmaps. (D) FMR1 DPP6, and SHISA6 expression using qRT-PCR with n=2 biological replicates averaged across n=2 technical replicates. (E-G) Characterization of the H3K9me3 domain on the X-chromosome in the iPSC state. (E) Input normalized H3K9me3 ChIP-seq profile in seven iPSC lines in an 8 Mb region around FMR1. FMR1, SLITRK2, and SLITRK4 genes are highlighted in red, blue, and green respectively. (F) Zoom-in on data from (A) is shown in a 150 kb window around the FMR1 gene. (G) Input normalized H3K9me3 ChIP-seq from (A) is overlaid for all seven iPSC lines. (H-J) Characterization of the H3K9me3 domain on the X-chromosome in wild type normal-length (NL_B) and FXS patient-derived (FXS_B_900, FXS_B_650) EBV-transformed B-lymphoblastoid cell lines (hereafter referred to as B-lymphoblastoid cells). (H) FMR1, SLITRK2, and SLITRK4 gene expression via RNA-seq. (I, J) Zoom in to (I) 1.5 Mb and (J) 150 kb around FMR1 (location marked in grey rectangle. (K) N=10 FXS-recurrent H3K9me3 domains on autosomes in iPSC-NPC cell type. Input normalized H3K9me3 ChIP-seq signal shown for all 7 iPSC-NPC lines. H3K9me3 domain calls from RSEG are shown above H3K9me3 ChIP-seq track as horizontal lines. Of N=11 total H3K9me3 domains gained in FXS, one is at FMR1 on the X chromosome, and the remaining 10 are shown here. (L) Gene ontology analysis for n=368 protein-coding genes co-localized with genotype-invariant H3K9me3 domains. (M) Total number of up- and down-regulated genes for pre-mutation (PM_137), and three FXS mutation-length iPS-NPCs (FXS_421, FXS_426, FXS_470) compared to normal-length (NL_18) as determined by DESeq2. (N) Gene ontology analysis for n=24 protein-coding genes co-localized with FXS-variable H3K9me3 domains present in only 1 of 3 FXS iPSC-NPC lines. For L+N, Gene ontology analysis using WebGESTALT with settings Over-Representation Analysis, geneontology, Biological Process, Cellular Component, Molecular function, with “genome-protein coding” as the reference. We only examine protein-coding genes, and used a P-value cutoff of p < 0.01 and enrichment > 4.
Figure S4: FXS iPSC-NPC and cell type-specific H3K9me3 domains arise in FXS patient-derived EBV-transformed lymphoblastoid B-cells and patient-derived caudate nucleus tissue and correlate with gene silencing, related to Figure 2. (A,B,C) Previously published human fetal cortex RNA-seq (GEO: GSE146878) showing isoform expression of key genes (A) FMR1, (B) DPP6, and (C) RBFOX1 co-localized in FXS-recurrent H3K9me3 domains in iPSC-NPCs. Each dot represents as different isoform for a given gene. To our knowledge, only one sex-matched RNA-seq data set from healthy and FXS patient brain tissue was publicly available for re-analysis. (D) RNA-seq fold change for FXS fetal cortex compared to healthy control fetal cortex. Genes localized in FXS-recurrent H3K9me3 domains are significantly lower in expression in FXS vs. healthy brain tissue compared to genes in size-matched random intervals. (E) Input normalized H3K9me3 ChIP-seq in one normal-length and two FXS EBV-transformed lymphoblastoid B-cells are plotted in n=10 autosomal FXS-recurrent H3K9me3 domains (shown as a blue line above ChIP-seq tracks) identified in iPSC-NPCs. A subset of FXS-recurrent H3K9me3 domains from iPSC-NPCs are not developmentally silenced in normal-length lymphoblastoid B-cells but acquire H3K9me3 de novo (n=2) or through domain spreading (n=2) in FXS lymphoblastoid B-cells (highlighted in red). (F) Percent of FXS-recurrent H3K9me3 domains identified in iPSC-NPCs present or absent in normal-length and mutation-length lymphoblastoid B-cells. (G) Two representative B-cell specific H3K9me3 domains out of n=24 that spread in mutation-length B-cells relative to normal-length B-cells. H3K9me3 domain calls are shown as red horizontal lines above the ChIP-seq signal. (H) Two representative FXS B-cell specific H3K9me3 domains out of n=36 that are acquired de novo in mutation-length FXS relative to normal-length lymphoblastoid B-cells. H3K9me3 domain calls are shown as red horizontal lines above the ChIP-seq signal. (I) Gene expression assayed by RNA-seq for n=25 genes identified as co-localized with H3K9me3 spreading and de novo domains in FXS lymphoblastoid B-cells. Each dot is the expression of a given gene which is the average of two biological replicates after DESeq2 normalization. (J) Input normalized H3K9me3 CUT&RUN in two normal-length and two FXS patient-derived caudate nucleus tissue samples are plotted in n=10 autosomal FXS-recurrent H3K9me3 domains. Black horizontal lines above the CUT&RUN tracks are H3K9me3 domain calls. Domains in the tissue samples are classified as de novo or spreading relative to the control tissue H3K9me3 signal. One locus shows enriched signal at the BREACH but did not have a domain called. (K) One representative de novo (n=161) and spreading (n=398) H3K9me3 domain in FXS caudate nucleus tissue. H3K9me3 domain calls are shown as red horizontal lines above the CUT&RUN signal. (L) Percent of FXS-recurrent iPSC-NPC H3K9me3 domains, de novo brain tissue domains, and spreading domains identified in FXS caudate nucleus tissue.
Figure S5: Engineering the FMR1 CGG STR tract from mutation-length to permutation-length removes H3K9me3 at a subset of FXS-recurrent autosomal domains, related to Figure 3. (A-B) Schematic of N=7 premutation-length single-cell-derived CGG cut-back iPSC clones and N=7 mutation-length single-cell-derived iPSC clones generated with CRISPR-Cas9 genome editing from ML FXS_421 iPSCs. (C,D) Input normalized H3K9me3 CUT&RUN profiles encompassing the FMR1 locus on chromosome X shown for 8 Mb (left) and zoomed-in to 150 kb (right) around FMR1 across all iPSC single-cell clones. (C) N=7 mutation-length single-cell iPSC clones. (D) N=7 premutation-length cutback single-cell iPSC clones. FMR1, SLITRK2, and SLITRK4 are highlighted in red, blue, and green, respectively. (E,F) H3K9me3 CUT&RUN signal for n=11 FXS-recurrent autosomal and X-chromosome H3K9me3 domains. One domain per row. (E) N=7 mutation-length single-cell iPSC clones. (F) N=7 premutation-length cutback single-cell iPSC clones. The top five rows (outlined by a red box) represent the loci most amenable to reprogramming upon CGG tract reduction. The chromosome and key gene at each loci are shown to the right of the row. (G, H) Input normalized H3K9me3 CUT&RUN profiles encompassing three autosomal loci (chr5, chr17, chr16) amenable to reprogramming after premutation-length CGG cutback are shown. (G) N=7 mutation-length single-cell iPSC clones. (H) N=7 premutation-length cutback single-cell iPSC clones. (I,J) H3K9me3 CUT&RUN signal for n=43 genotype-invariant H3K9me3 domains. One domain per row. (I) N=7 mutation-length single-cell iPSC clones. (J) N=7 premutation-length cutback single-cell iPSC clones.
Figure S6: Somatic STR instability is significantly associated with STRs which are significantly longer in FXS iPSCs than the expected null distribution from N=120 normal-length human iPSCs as assessed reproducibly with Expansion Hunter and GangSTR and is observed at FXS iPSC-NPC BREACHes and cell type-specific H3K9me3 domains in FXS patient-derived B-lymphoblastoid cells, related to Figure 4 & 6. (A-B) Pairwise Hi-C trans interactions between FXS-recurrent H3K9me3 domain loci on autosomes (N=10) and the X-chromosome are compared for between NL_27 (lower triangle) and FXS_470 (upper triangle) in (A) and NL_25 (lower triangle) and PM_137 (upper triangle) in (B). H3K9me3 domains +/− 3 Mb annotated by chromosome. Input normalized H3K9me3 Chip-seq signal for all domains in all iPSC-NPC cell lines shown are plotted alongside Hi-C heatmaps. Cyan boxes highlight FXS-gained trans interactions. (C) Venn diagrams depicting the reproducible overlap between FXS significantly long STRs identified by GangSTR and Expansion Hunter (EH). (D) STRs are categorized into expanded or stable across the population of N=120 normal-length hiPSC lines (N=240 alleles) and stratified with or without somatic instability (<= 2 unique STR lengths; >= 3 unique STR lengths) across reads within each line for each STR (STAR Methods). The Odds Ratio and p-values are computed from the 2×2 contingency table with a Fisher’s exact test. (E) Stratification of “FXS-reproducible long STRs” into those exhibiting stepwise somatic instability (green: >=1 FXS iPSC line with somatic instability) and those that do not (orange: somatically stable).
Figure S7: Validation of significantly expanded STRs co-localized with FXS-recurrent autosomal H3K9me3 domains using short- and long-read sequencing, related to Figure 6. We identified 3 STR tracts with significant stepwise somatic instability in all 3 FXS iPSC lines and co-localized in FXS-recurrent autosomal H3K9me3 domains. For each locus, PCR free whole genome sequencing short-reads are shown for N=3 NL iPSC lines and N=3 ML FXS iPSC lines. PCR free Nanopore long read sequencing is also shown as validation of the results in the FXS_412 iPSC line (designated as FXS_421 LR). Insertions (in bp) are highlighted as purple-filled boxes with total inserted base-pairs written in white. Deletions (in bp) are shown as purple text against a white background. Under the mapped reads, strip plots demonstrate STR lengths computed directly from each individual short read per iPSC line (STAR Methods). Each dot represents one read. More than two alleles is indicative of possible somatic instability. Bottom: Distribution of STR tract length (bp) across n=240 alleles of ancestry-, sex-, sequencing depth-, and cell type-matched n=120 normal-length HipSci iPSC lines compared to the maximum allele of each FXS iPSC line. Distributions for a reproducibly FXS long STR in RBFOX1 (left panel), PTPRT (middle panel), and an intergenic region on chr5 (right panel) are shown. Overlayed blue dashed lines indicating the GangSTR-computed length of the STR in the three FXS iPSC lines. Empirical one-tailed P-value.
Figure S8: H3K9me3 domains can be detected on autosomes and upstream of FMR1 on the X-chromosome in a small subset of normal-length human iPSC lines derived with shRNA against p53 or dominant negative p53 overexpression, related to Figure 7. (A) Frequency of autosomal H3K9me3 domains in normal-length (NL) or premutation-length (PM) iPSC lines reprogrammed (i) without p53 perturbation (left), (ii) reprogrammed with p53 shRNA or p53 dominant negative expression (middle), or (iii) this study’s FXS iPSC lines reprogrammed without p53 perturbation (right). (B) Input normalized H3K9me3 ChIP-Seq or CUT&RUN data previously published or created in this study and re-analyzed in a cohort for Supplementary Figure 8. Data is shown for N=16 normal-length iPSC lines external to our lab (N=11 published data sets and N=5 libraries generated from iPSC line external to our lab) as well as the main iPSC lines for the present study (N=3 normal-length, N=1 premutation-length, and N=3 mutation-length FXS). The iPSC lines were stratified into those reprogrammed without p53 shRNA (grey) or with p53 shRNA or p53 dominant negative expression (orange). All of the present study’s iPSC lines were reprogrammed without p53 perturbation. FXS lines highlighted in orange. Het-Sink score shown in blue, is derived from a recently published universal genome annotation (Vu and Ernst, 2022). (C) Randomization test comparing Het-Sink score, a measure of heterochromatin tendency, in FXS-recurrent H3K9me3 domains compared to n=1,000 draws of random genomic intervals matched by size and chromosome arm. (D) The frequency of observing an H3K9me3 domain over either a 5 Mb region upstream of FMR1 or the FMR1 gene body in N=13 normal-length or premutation-length male iPSC lines and in N=3 FXS iPSC lines used in this study. (E-F) Input normalized H3K9me3 or CUT&RUN ChIP-seq tracks in a (E) 7 Mb window or a (F) 150 kb window upstream of FMR1 in N=13 normal-length or premutation-length male iPSC lines reprogrammed without p53 shRNA (grey) or with p53 shRNA or p53 dominant negative expression (orange). For full information about iPSCs in this figure, see Table S1.
Table S2; Related to Figure 1, 3: Primers and sgRNAs used for qRT-PCR and Cas9 targeted Nanopore sequencing of FMR1.
Table S3; Related to Figure 1–7: Sequencing depth and mappability for sequencing datasets generated in this study.
Table S4; Related to Figure 5, 6, 7: Sequencing depth and mappability for sequencing datasets re-analyzed in this study.
Table S5; Related to Figure 4: Oligopaint primary, bridge, and secondary imaging probes used for DNA FISH imaging of trans interactions and TANGO processing chain parameters for DNA FISH analysis.
Table S6; Related to Figure 6: Short tandem repeats exhibiting reproducibly longer tracts in all three FXS iPSC lines compared to N=120 normal-length human IPSC lines using both GangSTR and Expansion Hunter
Highlights.
We find BREACHes: Beacons of Repeat Expansion Anchored by Contacting Heterochromatin.
BREACHes are Mb-scale H3K9me3 domains co-localized via trans interactions.
BREACHes harbor long, late replicating synaptic genes and STRs prone to instability.
Select BREACHes in FXS are reversible via CGG engineering to premutation-length.
Acknowledgments
We acknowledge a generous gift from Dr. & Mrs. Paul Bianco in support of this work.
Funding:
The New York Stem Cell Foundation (JEPC); NIH NIMH (1R01MH120269; 1DP1MH129957; JEPC); 4D Nucleome Common Fund (1U01DK127405, 1U01DA052715; JEPC); NSF CAREER Award (CBE-1943945; JEPC); CZI Neurodegenerative Disease Pairs Awards (2020–221479-5022; DAF2022–250430; JEPC); NIH F30 (F300HD098015; LZ); Blavatnik Family Fellowship (LZ); NIH F31 Fellowship (F31NS129317; TM); NYSCF Druckenmiller Postdoctoral Fellowship (SAH).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests:
LZ, CG, and JEPC are inventors on patent US20220323553A1 related to this work (https://patents.google.com/patent/US20220323553A1/en).
Data and materials availability:
All data is provided at GEO GSE218680.
REFERENCES
- 1.Santoro MR, Bray SM, and Warren ST (2012). Molecular mechanisms of fragile X syndrome: a twenty-year perspective. Annu Rev Pathol 7, 219–245. 10.1146/annurev-pathol-011811-132457. [DOI] [PubMed] [Google Scholar]
- 2.Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP, and et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65, 905–914. 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 3.Yu S, Pritchard M, Kremer E, Lynch M, Nancarrow J, Baker E, Holman K, Mulley JC, Warren ST, Schlessinger D, and et al. (1991). Fragile X genotype characterized by an unstable region of DNA. Science 252, 1179–1181. 10.1126/science.252.5009.1179. [DOI] [PubMed] [Google Scholar]
- 4.Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas MF, and Mandel JL (1991). Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science 252, 1097–1102. 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- 5.Mirkin SM (2007). Expandable DNA repeats and human disease. Nature 447, 932–940. 10.1038/nature05977. [DOI] [PubMed] [Google Scholar]
- 6.La Spada AR, and Taylor JP (2010). Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet 11, 247–258. 10.1038/nrg2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nelson DL, Orr HT, and Warren ST (2013). The unstable repeats--three evolving faces of neurological disease. Neuron 77, 825–843. 10.1016/j.neuron.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McMurray CT (2010). Mechanisms of trinucleotide repeat instability during human development. Nat Rev Genet 11, 786–799. 10.1038/nrg2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hagerman RJ, and Hagerman P (2016). Fragile X-associated tremor/ataxia syndrome - features, mechanisms and management. Nat Rev Neurol 12, 403–412. 10.1038/nrneurol.2016.82. [DOI] [PubMed] [Google Scholar]
- 10.Orr HT, and Zoghbi HY (2007). Trinucleotide repeat disorders. Annual review of neuroscience 30, 575–621. 10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 11.Sutcliffe JS, Nelson DL, Zhang F, Pieretti M, Caskey CT, Saxe D, and Warren ST (1992). DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum Mol Genet 1, 397–400. 10.1093/hmg/1.6.397. [DOI] [PubMed] [Google Scholar]
- 12.Colak D, Zaninovic N, Cohen MS, Rosenwaks Z, Yang WY, Gerhardt J, Disney MD, and Jaffrey SR (2014). Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science 343, 1002–1005. 10.1126/science.1245831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Avitzour M, Mor-Shaked H, Yanovsky-Dagan S, Aharoni S, Altarescu G, Renbaum P, Eldar-Geva T, Schonberger O, Levy-Lahad E, Epsztejn-Litman S, and Eiges R (2014). FMR1 epigenetic silencing commonly occurs in undifferentiated fragile X-affected embryonic stem cells. Stem Cell Reports 3, 699–706. 10.1016/j.stemcr.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Esch CE, Ghazvini M, Loos F, Schelling-Kazaryan N, Widagdo W, Munshi ST, van der Wal E, Douben H, Gunhanlar N, Kushner SA, et al. (2014). Epigenetic characterization of the FMR1 promoter in induced pluripotent stem cells from human fibroblasts carrying an unmethylated full mutation. Stem Cell Reports 3, 548–555. 10.1016/j.stemcr.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urbach A, Bar-Nur O, Daley GQ, and Benvenisty N (2010). Differential modeling of fragile X syndrome by human embryonic stem cells and induced pluripotent stem cells. Cell Stem Cell 6, 407–411. 10.1016/j.stem.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alisch RS, Wang T, Chopra P, Visootsak J, Conneely KN, and Warren ST (2013). Genome-wide analysis validates aberrant methylation in fragile X syndrome is specific to the FMR1 locus. BMC Med Genet 14, 18. 10.1186/1471-2350-14-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Korb E, Herre M, Zucker-Scharff I, Gresack J, Allis CD, and Darnell RB (2017). Excess Translation of Epigenetic Regulators Contributes to Fragile X Syndrome and Is Alleviated by Brd4 Inhibition. Cell 170, 1209–1223 e1220. 10.1016/j.cell.2017.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dahlhaus R (2018). Of Men and Mice: Modeling the Fragile X Syndrome. Front Mol Neurosci 11, 41. 10.3389/fnmol.2018.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sun JH, Zhou L, Emerson DJ, Phyo SA, Titus KR, Gong W, Gilgenast TG, Beagan JA, Davidson BL, Tassone F, and Phillips-Cremins JE (2018). Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 175, 224–238 e215. 10.1016/j.cell.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coffee B, Zhang F, Warren ST, and Reines D (1999). Acetylated histones are associated with FMR1 in normal but not fragile X-syndrome cells. Nat Genet 22, 98–101. 10.1038/8807. [DOI] [PubMed] [Google Scholar]
- 21.Coffee B, Zhang F, Ceman S, Warren ST, and Reines D (2002). Histone modifications depict an aberrantly heterochromatinized FMR1 gene in fragile x syndrome. Am J Hum Genet 71, 923–932. 10.1086/342931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu XS, Wu H, Krzisch M, Wu X, Graef J, Muffat J, Hnisz D, Li CH, Yuan B, Xu C, et al. (2018). Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 172, 979–992 e976. 10.1016/j.cell.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haenfler JM, Skariah G, Rodriguez CM, Monteiro da Rocha A, Parent JM, Smith GD, and Todd PK (2018). Targeted Reactivation of FMR1 Transcription in Fragile X Syndrome Embryonic Stem Cells. Front Mol Neurosci 11, 282. 10.3389/fnmol.2018.00282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kumari D, and Usdin K (2010). The distribution of repressive histone modifications on silenced FMR1 alleles provides clues to the mechanism of gene silencing in fragile X syndrome. Hum Mol Genet 19, 4634–4642. 10.1093/hmg/ddq394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Telias M (2019). Molecular Mechanisms of Synaptic Dysregulation in Fragile X Syndrome and Autism Spectrum Disorders. Front Mol Neurosci 12, 51. 10.3389/fnmol.2019.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gothelf D, Furfaro JA, Hoeft F, Eckert MA, Hall SS, O’Hara R, Erba HW, Ringel J, Hayashi KM, Patnaik S, et al. (2008). Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP). Ann Neurol 63, 40–51. 10.1002/ana.21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin L, Sun W, Throesch B, Kung F, Decoster JT, Berner CJ, Cheney RE, Rudy B, and Hoffman DA (2013). DPP6 regulation of dendritic morphogenesis impacts hippocampal synaptic development. Nature communications 4, 2270. 10.1038/ncomms3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pfeiffer BE, and Huber KM (2009). The state of synapses in fragile X syndrome. Neuroscientist 15, 549–567. 10.1177/1073858409333075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Atkin JF, Flaitz K, Patil S, and Smith W (1985). A new X-linked mental retardation syndrome. Am J Med Genet 21, 697–705. 10.1002/ajmg.1320210411. [DOI] [PubMed] [Google Scholar]
- 30.Kang Y, Zhou Y, Li Y, Han Y, Xu J, Niu W, Li Z, Liu S, Feng H, Huang W, et al. (2021). A human forebrain organoid model of fragile X syndrome exhibits altered neurogenesis and highlights new treatment strategies. Nat Neurosci 24, 1377–1391. 10.1038/s41593-021-00913-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie N, Gong H, Suhl JA, Chopra P, Wang T, and Warren ST (2016). Reactivation of FMR1 by CRISPR/Cas9-Mediated Deletion of the Expanded CGG-Repeat of the Fragile X Chromosome. PloS one 11, e0165499. 10.1371/journal.pone.0165499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Park CY, Halevy T, Lee DR, Sung JJ, Lee JS, Yanuka O, Benvenisty N, and Kim DW (2015). Reversion of FMR1 Methylation and Silencing by Editing the Triplet Repeats in Fragile X iPSC-Derived Neurons. Cell Rep 13, 234–241. 10.1016/j.celrep.2015.08.084. [DOI] [PubMed] [Google Scholar]
- 33.Haws SA, Simandi Z, Barnett RJ, and Phillips-Cremins JE (2022). 3D genome, on repeat: Higher-order folding principles of the heterochromatinized repetitive genome. Cell 185, 2690–2707. 10.1016/j.cell.2022.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Subramanian PS, Nelson DL, and Chinault AC (1996). Large domains of apparent delayed replication timing associated with triplet repeat expansion at FRAXA and FRAXE. Am J Hum Genet 59, 407–416. [PMC free article] [PubMed] [Google Scholar]
- 35.Wei PC, Chang AN, Kao J, Du Z, Meyers RM, Alt FW, and Schwer B (2016). Long Neural Genes Harbor Recurrent DNA Break Clusters in Neural Stem/Progenitor Cells. Cell 164, 644–655. 10.1016/j.cell.2015.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitra I, Huang B, Mousavi N, Ma N, Lamkin M, Yanicky R, Shleizer-Burko S, Lohmueller KE, and Gymrek M (2021). Patterns of de novo tandem repeat mutations and their role in autism. Nature 589, 246–250. 10.1038/s41586-020-03078-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Trost B, Engchuan W, Nguyen CM, Thiruvahindrapuram B, Dolzhenko E, Backstrom I, Mirceta M, Mojarad BA, Yin Y, Dov A, et al. (2020). Genome-wide detection of tandem DNA repeats that are expanded in autism. Nature 586, 80–86. 10.1038/s41586-020-2579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Allen-Brady K, Miller J, Matsunami N, Stevens J, Block H, Farley M, Krasny L, Pingree C, Lainhart J, Leppert M, et al. (2009). A high-density SNP genome-wide linkage scan in a large autism extended pedigree. Molecular psychiatry 14, 590–600. 10.1038/mp.2008.14. [DOI] [PubMed] [Google Scholar]
- 39.Griswold AJ, Dueker ND, Van Booven D, Rantus JA, Jaworski JM, Slifer SH, Schmidt MA, Hulme W, Konidari I, Whitehead PL, et al. (2015). Targeted massively parallel sequencing of autism spectrum disorder-associated genes in a case control cohort reveals rare loss-of-function risk variants. Mol Autism 6, 43. 10.1186/s13229-015-0034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cazzalini O, Scovassi AI, Savio M, Stivala LA, and Prosperi E (2010). Multiple roles of the cell cycle inhibitor p21(CDKN1A) in the DNA damage response. Mutat Res 704, 12–20. 10.1016/j.mrrev.2010.01.009. [DOI] [PubMed] [Google Scholar]
- 41.Matthew EM, Yen TJ, Dicker DT, Dorsey JF, Yang W, Navaraj A, and El-Deiry WS (2007). Replication stress, defective S-phase checkpoint and increased death in Plk2-deficient human cancer cells. Cell Cycle 6, 2571–2578. 10.4161/cc.6.20.5079. [DOI] [PubMed] [Google Scholar]
- 42.Burns TF, Fei P, Scata KA, Dicker DT, and El-Deiry WS (2003). Silencing of the novel p53 target gene Snk/Plk2 leads to mitotic catastrophe in paclitaxel (taxol)-exposed cells. Mol Cell Biol 23, 5556–5571. 10.1128/MCB.23.16.5556-5571.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, Doderlein G, Maltry N, Wu W, Lyko F, and Niehrs C (2007). Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 445, 671–675. 10.1038/nature05515. [DOI] [PubMed] [Google Scholar]
- 44.Jin S, Mazzacurati L, Zhu X, Tong T, Song Y, Shujuan S, Petrik KL, Rajasekaran B, Wu M, and Zhan Q (2003). Gadd45a contributes to p53 stabilization in response to DNA damage. Oncogene 22, 8536–8540. 10.1038/sj.onc.1206907. [DOI] [PubMed] [Google Scholar]
- 45.Niehrs C, and Schafer A (2012). Active DNA demethylation by Gadd45 and DNA repair. Trends Cell Biol 22, 220–227. 10.1016/j.tcb.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 46.Mungamuri SK, Benson EK, Wang S, Gu W, Lee SW, and Aaronson SA (2012). p53-mediated heterochromatin reorganization regulates its cell fate decisions. Nat Struct Mol Biol 19, 478–484, S471. 10.1038/nsmb.2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zheng H, Chen L, Pledger WJ, Fang J, and Chen J (2014). p53 promotes repair of heterochromatin DNA by regulating JMJD2b and SUV39H1 expression. Oncogene 33, 734–744. 10.1038/onc.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, et al. (2017). Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545, 229–233. 10.1038/nature22312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou Y, Kumari D, Sciascia N, and Usdin K (2016). CGG-repeat dynamics and FMR1 gene silencing in fragile X syndrome stem cells and stem cell-derived neurons. Mol Autism 7, 42. 10.1186/s13229-016-0105-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tan H, Li H, and Jin P (2009). RNA-mediated pathogenesis in fragile X-associated disorders. Neurosci Lett 466, 103–108. 10.1016/j.neulet.2009.07.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Groh M, Lufino MM, Wade-Martins R, and Gromak N (2014). R-loops associated with triplet repeat expansions promote gene silencing in Friedreich ataxia and fragile X syndrome. PLoS Genet 10, e1004318. 10.1371/journal.pgen.1004318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loomis EW, Sanz LA, Chedin F, and Hagerman PJ (2014). Transcription-associated R-loop formation across the human FMR1 CGG-repeat region. PLoS Genet 10, e1004294. 10.1371/journal.pgen.1004294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tassone F, Iwahashi C, and Hagerman PJ (2004). FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS). RNA Biol 1, 103–105. 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- 54.Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V, et al. (2013). CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron 78, 440–455. 10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ, et al. (2010). Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. The EMBO journal 29, 1248–1261. 10.1038/emboj.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alcala-Vida R, Seguin J, Lotz C, Molitor AM, Irastorza-Azcarate I, Awada A, Karasu N, Bombardier A, Cosquer B, Skarmeta JLG, et al. (2021). Age-related and disease locus-specific mechanisms contribute to early remodelling of chromatin structure in Huntington’s disease mice. Nature communications 12, 364. 10.1038/s41467-020-20605-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Griffin GK, Wu J, Iracheta-Vellve A, Patti JC, Hsu J, Davis T, Dele-Oni D, Du PP, Halawi AG, Ishizuka JJ, et al. (2021). Epigenetic silencing by SETDB1 suppresses tumour intrinsic immunogenicity. Nature 595, 309–314. 10.1038/s41586-021-03520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sun JH, Zhou L, Emerson DJ, Phyo SA, Titus KR, Gong W, Gilgenast TG, Beagan JA, Davidson BL, Tassone F, and Phillips-Cremins JE (2018). Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell. 10.1016/j.cell.2018.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, et al. (2013). Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148. 10.1016/j.cell.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beliveau BJ, Kishi JY, Nir G, Sasaki HM, Saka SK, Nguyen SC, Wu CT, and Yin P (2018). OligoMiner provides a rapid, flexible environment for the design of genome-scale oligonucleotide in situ hybridization probes. Proc Natl Acad Sci U S A 115, E2183–E2192. 10.1073/pnas.1714530115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Su JH, Zheng P, Kinrot SS, Bintu B, and Zhuang X (2020). Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell 182, 1641–1659 e1626. 10.1016/j.cell.2020.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nir G, Farabella I, Perez Estrada C, Ebeling CG, Beliveau BJ, Sasaki HM, Lee SD, Nguyen SC, McCole RB, Chattoraj S, et al. (2018). Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLoS Genet 14, e1007872. 10.1371/journal.pgen.1007872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moffitt JR, and Zhuang X (2016). RNA Imaging with Multiplexed Error-Robust Fluorescence In Situ Hybridization (MERFISH). Methods Enzymol 572, 1–49. 10.1016/bs.mie.2016.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosin LF, Nguyen SC, and Joyce EF (2018). Condensin II drives large-scale folding and spatial partitioning of interphase chromosomes in Drosophila nuclei. PLoS Genet 14, e1007393. 10.1371/journal.pgen.1007393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beagan JA, Pastuzyn ED, Fernandez LR, Guo MH, Feng K, Titus KR, Chandrashekar H, Shepherd JD, and Phillips-Cremins JE (2020). Three-dimensional genome restructuring across timescales of activity-induced neuronal gene expression. Nat Neurosci 23, 707–717. 10.1038/s41593-020-0634-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim JH, Rege M, Valeri J, Dunagin MC, Metzger A, Titus KR, Gilgenast TG, Gong W, Beagan JA, Raj A, and Phillips-Cremins JE (2019). LADL: light-activated dynamic looping for endogenous gene expression control. Nat Methods 16, 633–639. 10.1038/s41592-019-0436-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim JH, Titus KR, Gong W, Beagan JA, Cao Z, and Phillips-Cremins JE (2018). 5C-ID: Increased resolution Chromosome-Conformation-Capture-Carbon-Copy with in situ 3C and double alternating primer design. Methods 142, 39–46. 10.1016/j.ymeth.2018.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beagan JA, Duong MT, Titus KR, Zhou L, Cao Z, Ma J, Lachanski CV, Gillis DR, and Phillips-Cremins JE (2017). YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res 27, 1139–1152. 10.1101/gr.215160.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beagan JA, Gilgenast TG, Kim J, Plona Z, Norton HK, Hu G, Hsu SC, Shields EJ, Lyu X, Apostolou E, et al. (2016). Local Genome Topology Can Exhibit an Incompletely Rewired 3D-Folding State during Somatic Cell Reprogramming. Cell Stem Cell 18, 611–624. 10.1016/j.stem.2016.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, et al. (2013). Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295. 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Beagan JA, Duong MT, Titus KR, Zhou L, Cao Z, Ma J, Lachanski CV, Gillis DR, and Phillips-Cremins JE (2017). YY1 and CTCF orchestrate a 3D chromatin looping switch during early neural lineage commitment. Genome Res. 10.1101/gr.215160.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meers MP, Bryson TD, Henikoff JG, and Henikoff S (2019). Improved CUT&RUN chromatin profiling tools. Elife 8. 10.7554/eLife.46314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Giesselmann P, Brandl B, Raimondeau E, Bowen R, Rohrandt C, Tandon R, Kretzmer H, Assum G, Galonska C, Siebert R, et al. (2019). Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nature biotechnology 37, 1478–1481. 10.1038/s41587-019-0293-x. [DOI] [PubMed] [Google Scholar]
- 74.Gilpatrick T, Lee I, Graham JE, Raimondeau E, Bowen R, Heron A, Downs B, Sukumar S, Sedlazeck FJ, and Timp W (2020). Targeted nanopore sequencing with Cas9-guided adapter ligation. Nature biotechnology 38, 433–438. 10.1038/s41587-020-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang H, Emerson DJ, Gilgenast TG, Titus KR, Lan Y, Huang P, Zhang D, Wang H, Keller CA, Giardine B, et al. (2019). Chromatin structure dynamics during the mitosis-to-G1 phase transition. Nature 576, 158–162. 10.1038/s41586-019-1778-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fernandez LR, Gilgenast TG, and Phillips-Cremins JE (2020). 3DeFDR: statistical methods for identifying cell type-specific looping interactions in 5C and Hi-C data. Genome Biol 21, 219. 10.1186/s13059-020-02061-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rowley MJ, and Corces VG (2018). Organizational principles of 3D genome architecture. Nat Rev Genet 19, 789–800. 10.1038/s41576-018-0060-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rowley MJ, Nichols MH, Lyu X, Ando-Kuri M, Rivera ISM, Hermetz K, Wang P, Ruan Y, and Corces VG (2017). Evolutionarily Conserved Principles Predict 3D Chromatin Organization. Mol Cell 67, 837–852 e837. 10.1016/j.molcel.2017.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Beagan JA, and Phillips-Cremins JE (2020). On the existence and functionality of topologically associating domains. Nat Genet 52, 8–16. 10.1038/s41588-019-0561-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Norton HK, Emerson DJ, Huang H, Kim J, Titus KR, Gu S, Bassett DS, and Phillips-Cremins JE (2018). Detecting hierarchical genome folding with network modularity. Nat Methods 15, 119–122. 10.1038/nmeth.4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Song Q, and Smith AD (2011). Identifying dispersed epigenomic domains from ChIP-Seq data. Bioinformatics 27, 870–871. 10.1093/bioinformatics/btr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nature biotechnology 34, 525–527. 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 83.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ollion J, Cochennec J, Loll F, Escude C, and Boudier T (2013). TANGO: a generic tool for high-throughput 3D image analysis for studying nuclear organization. Bioinformatics 29, 1840–1841. 10.1093/bioinformatics/btt276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pedersen BS, Collins RL, Talkowski ME, and Quinlan AR (2017). Indexcov: fast coverage quality control for whole-genome sequencing. Gigascience 6, 1–6. 10.1093/gigascience/gix090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dudchenko O, Batra SS, Omer AD, Nyquist SK, Hoeger M, Durand NC, Shamim MS, Machol I, Lander ES, Aiden AP, and Aiden EL (2017). De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 356, 92–95. 10.1126/science.aal3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Streeter I, Harrison PW, Faulconbridge A, The HipSci C, Flicek P, Parkinson H, and Clarke L (2017). The human-induced pluripotent stem cell initiative-data resources for cellular genetics. Nucleic Acids Res 45, D691–D697. 10.1093/nar/gkw928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vu H, and Ernst J (2022). Universal annotation of the human genome through integration of over a thousand epigenomic datasets. Genome Biol 23, 9. 10.1186/s13059-021-02572-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Morphology, expected homogeneous marker expression, and karyotyping in human induced pluripotent stem cells (iPSCs) and iPSC-derived neural progenitor cells (iPSC-NPCs), related to Figure 1. (A) Phase contrast images of iPSC colony morphology in an independent field of view from the immunofluorescence staining. An independent field of view was used for phase contrast imaging and immunofluorescence. (B) Immunofluorescence staining of human iPSC lines for OCT4 (green) and NESTIN (cyan) co-localized with DAPI (blue) as a nuclear marker. Independent field of view from phase contrast images. (C) de novo genome assemblies for autosomal chromosomes across all 7 iPSC lines used in this study. Genome assembly was constructed from iPSC-NPC Hi-C data and iPSC PCR-free whole genome sequencing using W2rapContigger, Juicer, and 3D-DNA (STAR Methods). Layers connecting rectangles on the Jupiter plots demonstrate mapping between the de novo genome assembly (right half of circle) with the hg38 reference genome (left half of circle). Plots without translocations should have clear chromosome layers with no overlaps or intersections with other chromosome layers. FXS-recurrent H3K9me3 domains are denoted with black stars along the reference chromosomes (left half). Grey stripes denote masked regions. No clear evidence of a translocation was observed. (D) Genome coverage in 16 kb bins across chromosomes containing FXS-recurrent H3K9me3 domains (“grey dashed boxes”) calculated from PCR free whole genome sequencing data generated in iPSCs. Karyotype was largely normal across all seven lines. In one specific case, the FXS_426 line showing a small heterozygous deletion on chr18 (“green dashed box”) which was not a BREACH locus or chromosome examined in this study. Coverage was normalized from 0 to 2 where 0 is a homozygous deletion and 2 is a homozygous duplication. Dashed lines represent BREACHes. (E) Phase contrast images of iPSC-derived NPC rosettes. An independent field of view was used for phase contrast imaging and immunofluorescence. (F) Immunofluorescence staining of human iPSC-derived NPCs for OCT4 (green) and NESTIN (cyan) co-stained with DAPI (blue) as a nuclear marker. Scale bars, 250 μm.
Figure S2: DNA methylation, 3D genome folding disruption, and acquisition of a Mb-sized heterochromatin domain at the FMR1 locus in iPSC-NPCs with normal-length, premutation-length, and mutation-length CGG STR lengths, related to Figure 1. (A) Schematic representation of the FMR1 locus with the CpGs within the CGG tract (analyzed in D) and promoter (analyzed in C) annotated. (B) Visual representation of reverse Nanopore reads (i.e., single alleles) spanning upstream and downstream regions of FMR1 across iPSC lines. CGGs are highlighted in green and other nucleotides are grey. Base calling accomplished with guppy (STAR methods). (C) Visual representation of the DNA methylation status of the 19 CpGs present in the FMR1 promoter called using nanopolish (STAR methods). (D) Visual representation of methylation status of CpGs within the FMR1 CGG tract by the STR-specific tool STRique (STAR methods). In panels (C) and (D), DNA methylation is annotated per read, with read order kept consistent across (B), (C), and (D). (E) Hi-C data across the two iPSC-NPC lines not shown in Figure 1 are shown as a heatmap of interaction frequency for an 8 Mb region around FMR1. A/B compartment score, input normalized H3K9me3 ChIP-seq, and CTCF ChIP-seq in iPSC-derived NPCs is displayed below the heatmaps. Horizontal black lines above H3K9me3 ChIP-seq tracks represent H3K9me3 domain calls. Vertical blue lines above CTCF ChIP-seq tracks represent peak calls using MACS2. FMR1, SLITRK2, and SLITRK4 are highlighted in red, blue, and green, respectively. (F) Hi-C fold-change interaction frequency maps. Gained and lost contacts compared to NL_18 are highlighted in red and blue, respectively. (G) Summed interactions using normalized Hi-C counts between FMR1 and the TAD immediately upstream or downstream is shown as a difference from NL_18. (H) CTCF and input normalized H3K9me3 ChIP-seq for 2 Mb downstream and 6 Mb upstream of FMR1 are overlaid for each iPSC-NPC line. Input normalized H3K9me3 and CTCF ChIP-seq signal ranges from −0.25 to 2.5 and 5 to 30, respectively. (I, J) Zoom-ins to Box 1 and Box 2 from (E) showing (I) FMR1-SLITRK2 or (J) FMR1-SLITRK4 interactions. Arrows point to loops which are disrupted in full mutation-length iPSC-NPC lines. (K,L) Boxplot showing the interaction frequency measured with Hi-C between FMR1 and either (K) SLITRK2 or (L) SLITRK4.
Figure S3: FXS-recurrent H3K9me3 domains on autosomes and the X-chromosome in iPSCs, EBV-transformed B-lymphoblasts, and iPSC-NPCs derived from FXS patients with mutation-length CGG tracts, related to Figure 1 & 2. (A-D) Characterization of the FXS_421 iPSC parent line and a subclone, FXS_425, derived from the same parent line to assess clonal variation in heterochromatin domains. (A) Number of CGG triplets in the FMR1 5’UTR based on Nanopore long read sequencing. (B) Percent of CGGs in the FMR1 5’UTR which are methylated based on STRique analysis of single-molecule Nanopore long-reads. (C) Input normalized H3K9me3 CUT&RUN is shown for a 6 Mb locus around FMR1. Tracks for input normalized H3K9me3 and CTCF CUT&RUN and ChIP-seq are plotted underneath heatmaps. (D) FMR1 DPP6, and SHISA6 expression using qRT-PCR with n=2 biological replicates averaged across n=2 technical replicates. (E-G) Characterization of the H3K9me3 domain on the X-chromosome in the iPSC state. (E) Input normalized H3K9me3 ChIP-seq profile in seven iPSC lines in an 8 Mb region around FMR1. FMR1, SLITRK2, and SLITRK4 genes are highlighted in red, blue, and green respectively. (F) Zoom-in on data from (A) is shown in a 150 kb window around the FMR1 gene. (G) Input normalized H3K9me3 ChIP-seq from (A) is overlaid for all seven iPSC lines. (H-J) Characterization of the H3K9me3 domain on the X-chromosome in wild type normal-length (NL_B) and FXS patient-derived (FXS_B_900, FXS_B_650) EBV-transformed B-lymphoblastoid cell lines (hereafter referred to as B-lymphoblastoid cells). (H) FMR1, SLITRK2, and SLITRK4 gene expression via RNA-seq. (I, J) Zoom in to (I) 1.5 Mb and (J) 150 kb around FMR1 (location marked in grey rectangle. (K) N=10 FXS-recurrent H3K9me3 domains on autosomes in iPSC-NPC cell type. Input normalized H3K9me3 ChIP-seq signal shown for all 7 iPSC-NPC lines. H3K9me3 domain calls from RSEG are shown above H3K9me3 ChIP-seq track as horizontal lines. Of N=11 total H3K9me3 domains gained in FXS, one is at FMR1 on the X chromosome, and the remaining 10 are shown here. (L) Gene ontology analysis for n=368 protein-coding genes co-localized with genotype-invariant H3K9me3 domains. (M) Total number of up- and down-regulated genes for pre-mutation (PM_137), and three FXS mutation-length iPS-NPCs (FXS_421, FXS_426, FXS_470) compared to normal-length (NL_18) as determined by DESeq2. (N) Gene ontology analysis for n=24 protein-coding genes co-localized with FXS-variable H3K9me3 domains present in only 1 of 3 FXS iPSC-NPC lines. For L+N, Gene ontology analysis using WebGESTALT with settings Over-Representation Analysis, geneontology, Biological Process, Cellular Component, Molecular function, with “genome-protein coding” as the reference. We only examine protein-coding genes, and used a P-value cutoff of p < 0.01 and enrichment > 4.
Figure S4: FXS iPSC-NPC and cell type-specific H3K9me3 domains arise in FXS patient-derived EBV-transformed lymphoblastoid B-cells and patient-derived caudate nucleus tissue and correlate with gene silencing, related to Figure 2. (A,B,C) Previously published human fetal cortex RNA-seq (GEO: GSE146878) showing isoform expression of key genes (A) FMR1, (B) DPP6, and (C) RBFOX1 co-localized in FXS-recurrent H3K9me3 domains in iPSC-NPCs. Each dot represents as different isoform for a given gene. To our knowledge, only one sex-matched RNA-seq data set from healthy and FXS patient brain tissue was publicly available for re-analysis. (D) RNA-seq fold change for FXS fetal cortex compared to healthy control fetal cortex. Genes localized in FXS-recurrent H3K9me3 domains are significantly lower in expression in FXS vs. healthy brain tissue compared to genes in size-matched random intervals. (E) Input normalized H3K9me3 ChIP-seq in one normal-length and two FXS EBV-transformed lymphoblastoid B-cells are plotted in n=10 autosomal FXS-recurrent H3K9me3 domains (shown as a blue line above ChIP-seq tracks) identified in iPSC-NPCs. A subset of FXS-recurrent H3K9me3 domains from iPSC-NPCs are not developmentally silenced in normal-length lymphoblastoid B-cells but acquire H3K9me3 de novo (n=2) or through domain spreading (n=2) in FXS lymphoblastoid B-cells (highlighted in red). (F) Percent of FXS-recurrent H3K9me3 domains identified in iPSC-NPCs present or absent in normal-length and mutation-length lymphoblastoid B-cells. (G) Two representative B-cell specific H3K9me3 domains out of n=24 that spread in mutation-length B-cells relative to normal-length B-cells. H3K9me3 domain calls are shown as red horizontal lines above the ChIP-seq signal. (H) Two representative FXS B-cell specific H3K9me3 domains out of n=36 that are acquired de novo in mutation-length FXS relative to normal-length lymphoblastoid B-cells. H3K9me3 domain calls are shown as red horizontal lines above the ChIP-seq signal. (I) Gene expression assayed by RNA-seq for n=25 genes identified as co-localized with H3K9me3 spreading and de novo domains in FXS lymphoblastoid B-cells. Each dot is the expression of a given gene which is the average of two biological replicates after DESeq2 normalization. (J) Input normalized H3K9me3 CUT&RUN in two normal-length and two FXS patient-derived caudate nucleus tissue samples are plotted in n=10 autosomal FXS-recurrent H3K9me3 domains. Black horizontal lines above the CUT&RUN tracks are H3K9me3 domain calls. Domains in the tissue samples are classified as de novo or spreading relative to the control tissue H3K9me3 signal. One locus shows enriched signal at the BREACH but did not have a domain called. (K) One representative de novo (n=161) and spreading (n=398) H3K9me3 domain in FXS caudate nucleus tissue. H3K9me3 domain calls are shown as red horizontal lines above the CUT&RUN signal. (L) Percent of FXS-recurrent iPSC-NPC H3K9me3 domains, de novo brain tissue domains, and spreading domains identified in FXS caudate nucleus tissue.
Figure S5: Engineering the FMR1 CGG STR tract from mutation-length to permutation-length removes H3K9me3 at a subset of FXS-recurrent autosomal domains, related to Figure 3. (A-B) Schematic of N=7 premutation-length single-cell-derived CGG cut-back iPSC clones and N=7 mutation-length single-cell-derived iPSC clones generated with CRISPR-Cas9 genome editing from ML FXS_421 iPSCs. (C,D) Input normalized H3K9me3 CUT&RUN profiles encompassing the FMR1 locus on chromosome X shown for 8 Mb (left) and zoomed-in to 150 kb (right) around FMR1 across all iPSC single-cell clones. (C) N=7 mutation-length single-cell iPSC clones. (D) N=7 premutation-length cutback single-cell iPSC clones. FMR1, SLITRK2, and SLITRK4 are highlighted in red, blue, and green, respectively. (E,F) H3K9me3 CUT&RUN signal for n=11 FXS-recurrent autosomal and X-chromosome H3K9me3 domains. One domain per row. (E) N=7 mutation-length single-cell iPSC clones. (F) N=7 premutation-length cutback single-cell iPSC clones. The top five rows (outlined by a red box) represent the loci most amenable to reprogramming upon CGG tract reduction. The chromosome and key gene at each loci are shown to the right of the row. (G, H) Input normalized H3K9me3 CUT&RUN profiles encompassing three autosomal loci (chr5, chr17, chr16) amenable to reprogramming after premutation-length CGG cutback are shown. (G) N=7 mutation-length single-cell iPSC clones. (H) N=7 premutation-length cutback single-cell iPSC clones. (I,J) H3K9me3 CUT&RUN signal for n=43 genotype-invariant H3K9me3 domains. One domain per row. (I) N=7 mutation-length single-cell iPSC clones. (J) N=7 premutation-length cutback single-cell iPSC clones.
Figure S6: Somatic STR instability is significantly associated with STRs which are significantly longer in FXS iPSCs than the expected null distribution from N=120 normal-length human iPSCs as assessed reproducibly with Expansion Hunter and GangSTR and is observed at FXS iPSC-NPC BREACHes and cell type-specific H3K9me3 domains in FXS patient-derived B-lymphoblastoid cells, related to Figure 4 & 6. (A-B) Pairwise Hi-C trans interactions between FXS-recurrent H3K9me3 domain loci on autosomes (N=10) and the X-chromosome are compared for between NL_27 (lower triangle) and FXS_470 (upper triangle) in (A) and NL_25 (lower triangle) and PM_137 (upper triangle) in (B). H3K9me3 domains +/− 3 Mb annotated by chromosome. Input normalized H3K9me3 Chip-seq signal for all domains in all iPSC-NPC cell lines shown are plotted alongside Hi-C heatmaps. Cyan boxes highlight FXS-gained trans interactions. (C) Venn diagrams depicting the reproducible overlap between FXS significantly long STRs identified by GangSTR and Expansion Hunter (EH). (D) STRs are categorized into expanded or stable across the population of N=120 normal-length hiPSC lines (N=240 alleles) and stratified with or without somatic instability (<= 2 unique STR lengths; >= 3 unique STR lengths) across reads within each line for each STR (STAR Methods). The Odds Ratio and p-values are computed from the 2×2 contingency table with a Fisher’s exact test. (E) Stratification of “FXS-reproducible long STRs” into those exhibiting stepwise somatic instability (green: >=1 FXS iPSC line with somatic instability) and those that do not (orange: somatically stable).
Figure S7: Validation of significantly expanded STRs co-localized with FXS-recurrent autosomal H3K9me3 domains using short- and long-read sequencing, related to Figure 6. We identified 3 STR tracts with significant stepwise somatic instability in all 3 FXS iPSC lines and co-localized in FXS-recurrent autosomal H3K9me3 domains. For each locus, PCR free whole genome sequencing short-reads are shown for N=3 NL iPSC lines and N=3 ML FXS iPSC lines. PCR free Nanopore long read sequencing is also shown as validation of the results in the FXS_412 iPSC line (designated as FXS_421 LR). Insertions (in bp) are highlighted as purple-filled boxes with total inserted base-pairs written in white. Deletions (in bp) are shown as purple text against a white background. Under the mapped reads, strip plots demonstrate STR lengths computed directly from each individual short read per iPSC line (STAR Methods). Each dot represents one read. More than two alleles is indicative of possible somatic instability. Bottom: Distribution of STR tract length (bp) across n=240 alleles of ancestry-, sex-, sequencing depth-, and cell type-matched n=120 normal-length HipSci iPSC lines compared to the maximum allele of each FXS iPSC line. Distributions for a reproducibly FXS long STR in RBFOX1 (left panel), PTPRT (middle panel), and an intergenic region on chr5 (right panel) are shown. Overlayed blue dashed lines indicating the GangSTR-computed length of the STR in the three FXS iPSC lines. Empirical one-tailed P-value.
Figure S8: H3K9me3 domains can be detected on autosomes and upstream of FMR1 on the X-chromosome in a small subset of normal-length human iPSC lines derived with shRNA against p53 or dominant negative p53 overexpression, related to Figure 7. (A) Frequency of autosomal H3K9me3 domains in normal-length (NL) or premutation-length (PM) iPSC lines reprogrammed (i) without p53 perturbation (left), (ii) reprogrammed with p53 shRNA or p53 dominant negative expression (middle), or (iii) this study’s FXS iPSC lines reprogrammed without p53 perturbation (right). (B) Input normalized H3K9me3 ChIP-Seq or CUT&RUN data previously published or created in this study and re-analyzed in a cohort for Supplementary Figure 8. Data is shown for N=16 normal-length iPSC lines external to our lab (N=11 published data sets and N=5 libraries generated from iPSC line external to our lab) as well as the main iPSC lines for the present study (N=3 normal-length, N=1 premutation-length, and N=3 mutation-length FXS). The iPSC lines were stratified into those reprogrammed without p53 shRNA (grey) or with p53 shRNA or p53 dominant negative expression (orange). All of the present study’s iPSC lines were reprogrammed without p53 perturbation. FXS lines highlighted in orange. Het-Sink score shown in blue, is derived from a recently published universal genome annotation (Vu and Ernst, 2022). (C) Randomization test comparing Het-Sink score, a measure of heterochromatin tendency, in FXS-recurrent H3K9me3 domains compared to n=1,000 draws of random genomic intervals matched by size and chromosome arm. (D) The frequency of observing an H3K9me3 domain over either a 5 Mb region upstream of FMR1 or the FMR1 gene body in N=13 normal-length or premutation-length male iPSC lines and in N=3 FXS iPSC lines used in this study. (E-F) Input normalized H3K9me3 or CUT&RUN ChIP-seq tracks in a (E) 7 Mb window or a (F) 150 kb window upstream of FMR1 in N=13 normal-length or premutation-length male iPSC lines reprogrammed without p53 shRNA (grey) or with p53 shRNA or p53 dominant negative expression (orange). For full information about iPSCs in this figure, see Table S1.
Table S2; Related to Figure 1, 3: Primers and sgRNAs used for qRT-PCR and Cas9 targeted Nanopore sequencing of FMR1.
Table S3; Related to Figure 1–7: Sequencing depth and mappability for sequencing datasets generated in this study.
Table S4; Related to Figure 5, 6, 7: Sequencing depth and mappability for sequencing datasets re-analyzed in this study.
Table S5; Related to Figure 4: Oligopaint primary, bridge, and secondary imaging probes used for DNA FISH imaging of trans interactions and TANGO processing chain parameters for DNA FISH analysis.
Table S6; Related to Figure 6: Short tandem repeats exhibiting reproducibly longer tracts in all three FXS iPSC lines compared to N=120 normal-length human IPSC lines using both GangSTR and Expansion Hunter
Data Availability Statement
Raw sequencing files and key intermediate files generated in this study are deposited and freely available from Gene Expression Omnibus (GEO: GSE218680). A complete list of sequencing datasets generated in this study is provided in Table S3. A complete list of genomics datasets reanalyzed from various public repositories and publications is provided in Table S4. Accession numbers are also listed in the key resources table. DNA FISH images and Nanopore long-read sequencing raw files (i.e., fast5) reported in this study are not compatible with GEO but can be shared by the Lead Contact author upon request.
All original code is deposited at Zenodo and is made publicly available as of the date of publication. The DOI (10.5281/zenodo.6558223) is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact author upon request.
KEY RESOURCE TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CTCF | Millipore | Cat# 07–729; RRID: AB_441965 |
| H3K9me3 | Abcam | Cat# ab8898; RRID: AB_306848 |
| IgG | Sigma-Aldrich | Cat# I8140; RRID: AB_1163661 |
| IgG Alexa Fluor 488 | Invitrogen | Cat# A-11034; RRID: AB_2576217 |
| IgG Alexa Fluor 594 | Invitrogen | Cat# A-21203; RRID: AB_141633 |
| NESTIN | R&D Systems | Cat# MAB1259; RRID: AB_2251304 |
| OCT4 | Cell Signaling Technologies | Cat# 2750; RRID: AB_823583 |
| Bacterial and virus strains | ||
| DH5α-T1R | Invitrogen | Cat# 12297016 |
| Biological samples | ||
| Healthy human caudate nucleus brain tissue from NIH donor 5533 (designated as NL_CN_1) | NIH NeuroBioBank | https://neurobiobank.nih.gov |
| Healthy human caudate nucleus brain tissue from NIH donor 5577 (designated as NL_CN_2) | NIH NeuroBioBank | https://neurobiobank.nih.gov |
| FXS human caudate nucleus brain tissue from NIH donor 5319 (designated as FXS_CN_1) | NIH NeuroBioBank | https://neurobiobank.nih.gov |
| FXS human caudate nucleus brain tissue from NIH donor 5746 (designated as FXS_CN_2) | NIH NeuroBioBank | https://neurobiobank.nih.gov |
| Chemicals, peptides, and recombinant proteins | ||
| 10% Triton X-100 solution | Sigma-Aldrich | Cat# 93443 |
| 100% Ethanol | Decon Labs | Cat# 2716 |
| 20% SDS solution | Fisher Scientific | Cat# BP1311 |
| Accutase | Gibco | Cat# A1110501 |
| AgentCourt Ampure XP beads | Beckman Coulter | Cat# A63881 |
| Alt-R S.p. HiFi Cas9 Nuclease V3 | Integrated DNA Technologies | Cat# 1081060 |
| Aminoallyl-dUTP Solution | Thermo Scientific | Cat# FERR1101 |
| Ammonium Acetate | Invitrogen | Cat# AM9070G |
| BbsI-HF | New England Biolabs | Cat# R3539S |
| Betaine | Sigma-Aldrich | Cat# 61962 |
| Blunt/TA Ligase Master Mix | New England Biolabs | Cat# M0367L |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat# A7906 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat# A7906–50G |
| Calcium chloride (CaCl2) | Fisher Scientific | Cat# BP510 |
| Calcium chloride (CaCl2) | Thermo Fisher | Cat# J63122-AD |
| Concanavalin A magnetic beads | BioMag | Cat# 86057 |
| CUTANA pAG-MNase | EpiCypher | Cat# 15–1016 |
| DAPI | Sigma-Aldrich | Cat# MBD0015–1ML |
| dATP | Thermo Scientific | Cat# R0141 |
| Dextran sulfate | Sigma-Aldrich | Cat# D8906 |
| Digitonin | Millipore | Cat# 300410 |
| Dimethyl sulfoxide | Sigma-Aldrich | Cat# D2650 |
| DMEM/F-12 | Gibco | Cat# 11320033 |
| Duplex buffer | Integrated DNA Technologies | Cat# 11–01-03–01 |
| EDTA, pH 8.0 | Invitrogen | Cat# 15575020 |
| EGTA, pH 8.0 | Bioworld | Cat# 40520008–1 |
| Elution buffer | Qiagen | Cat# 19086 |
| Fetal Bovine Serum | Gibco | Cat# 16000044 |
| Formaldehyde solution | Sigma-Aldrich | Cat# F8775 |
| Formaldehyde solution | Pierce | Cat# 28908 |
| Formamide | Calbiochem | Cat# 344206 |
| Glycine | Sigma-Aldrich | Cat# 50046 |
| Glycogen | Thermo Scientific | Cat# R0561 |
| Hank’s Balanced Salt Solution | Gibco | Cat# 14025092 |
| HEPES-KOH, pH 7.5 | Boston BioProducts | Cat# BBH-75-K |
| High-Vacuum Grease | Dow Corning | Cat# 1658832 |
| Hoechst 33342 Solution | Thermo Scientific | Cat# 62249 |
| Holo-transferrin | Sigma-Aldrich | Cat# T0665 |
| Igepal CA-630 | Sigma-Aldrich | Cat# I8896 |
| Insulin | Sigma-Aldrich | Cat# I1882 |
| Isopropanol | Thermo Fisher | Cat# T036181000 |
| KAPA HiFi HotStart ReadyMix | Roche | Cat# 7958927001 |
| L-ascorbic acid | Sigma-Aldrich | Cat# A8960 |
| LiCl | Sigma-Aldrich | Cat# L9650 |
| Lipofectamine Stem Transfection Reagent | Invitrogen | Cat# STEM00008 |
| Magnesium Acetate (MgAc2) | Sigma-Aldrich | Cat# 63052–100ML |
| Manganese chloride (MnCl2) | Fisher Scientific | Cat# BP541 |
| Matrigel hESC-Qualified Matrix | Corning | Cat# 354277 |
| Maxima H Minus Reverse Transcriptase | Thermo Scientific | Cat# EP0751 |
| mTeSR Plus media | STEMCELL Technology | Cat# 05825 |
| NaCl | Invitrogen | Cat# AM9760G |
| NEBNext Quick Ligation Module | New England Biolabs | Cat# E6056S |
| Noggin | R&D Systems | Cat# 6057-NG |
| Nuclease-free water | Sigma-Aldrich | Cat# W4502 |
| PBS | Corning | Cat# 21–040-CV |
| Penicillin-streptomycin | Gibco | Cat# 15140122 |
| Phenylmethanesulfonyl fluoride (PMSF) solution | Sigma-Aldrich | Cat# 93482 |
| Phusion polymerase | New England Biolabs | Cat# M0530L |
| Polyvinylsulfonic acid (PVSA) | Sigma-Aldrich | Cat# 278424 |
| Potassium chloride (KCl) | Sigma-Aldrich | Cat# P3911 |
| Power SYBR Green PCR Master Mix | Applied Biosystems | Cat# 4368706 |
| Protease inhibitor cocktail | Sigma-Aldrich | Cat# P8340 |
| Protease Inhibitor Cocktail (EDTA-free) | Roche | Cat# 11873580001 |
| Protein A Agarose beads | Invitrogen | Cat# 15918014 |
| Protein G Agarose beads | Invitrogen | Cat# 15920010 |
| Proteinase K | New England Biolabs | Cat# P8107S |
| Proteinase K | Qiagen | Cat# 158918 |
| QuickCIP | New England Biolabs | Cat# M0525S |
| rCutSmart buffer | New England Biolabs | Cat# B6004 |
| RevitaCell™ Supplement (100X) | Gibco | Cat# A2644501 |
| RNase A | Roche | Cat# 10109142001 |
| RNase A | Thermo Fisher | Cat# EN0531 |
| RPMI 1640 media | Sigma-Aldrich | Cat# R8758 |
| Saline-Sodium Citrate (SSC) buffer | Corning | Cat# 46–020-CM |
| SB431542 | STEMCELL Technology | Cat# 72234 |
| SlowFade Diamond Antifade Mountant | Invitrogen | Cat# S36967 |
| Sodium bicarbonate | Sigma-Aldrich | Cat# S5761 |
| Sodium deoxycholate | Sigma-Aldrich | Cat# D6750 |
| Sodium Hydroxide (NaOH) | Macron | Cat# 7680 |
| Sodium selenite | Sigma-Aldrich | Cat# S5261 |
| Spermidine | Sigma-Aldrich | Cat# S2501 |
| Sucrose | Sigma-Aldrich | Cat# S0389–500G |
| SuperScript II Reverse Transcriptase | Invitrogen | Cat# 18064014 |
| Synth-a-Freeze | Gibco | Cat# A1254201 |
| T4 DNA ligase | New England Biolabs | Cat# M0202S |
| Taq ligase | New England Biolabs | Cat# M0208L |
| Taq polymerase | New England Biolabs | Cat# M0273 |
| TE buffer, pH 8.0 | Invitrogen | Cat# AM9858 |
| tracrRNA | Integrated DNA Technologies | Cat# 1072532 |
| Tris-HCl, pH 8.0 | Invitrogen | Cat# 15568025 |
| Triton X-100 | Sigma-Aldrich | Cat# T8787–100ML |
| TrypLE | Gibco | Cat# 12605010 |
| Tween 20 | Sigma-Aldrich | Cat# P9416 |
| Ultrapure Phenol/Chloroform/Isoamyl Alcohol | Fisher Scientific | Cat# BP1752I100 |
| VECTASHIELD Antifade Mounting Medium | Vector Laboratories | Cat# H-1200 |
| Versene Solution | Gibco | Cat# 15040066 |
| Critical commercial assays | ||
| Arima-HiC kit | Arima Genomics | Cat# A510008 |
| Bioanalyzer High Sensitivity DNA Analysis Kit | Agilent | Cat# 5067–4626 |
| DNA-free DNA removal kit | Ambion | Cat# AM1906 |
| GeneJet Genomic DNA purification kit | Thermo Scientific | Cat# K0721 |
| Gentra Puregene Cell Kit | Qiagen | Cat# 158767 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat# 4368813 |
| Kapa Library Quantification Kit | KAPA Biosystems | Cat# KK4835 |
| Ligation Sequencing Kit | Oxford Nanopore Technologies | Cat# SQK-LSK109 |
| mirVana miRNA Isolation Kit | Invitrogen | Cat# AM1560 |
| Native Barcoding Expansion (PCR-free) kit | Oxford Nanopore Technologies | Cat# EXP-NBD104 |
| NEBNext Ultra II DNA Library Prep Kit for Illumina | New England Biolabs | Cat# E7645S |
| Plasmid Purification Kit | Clontech | Cat# 740588.250 |
| QIAquick Gel Extraction Kit | Qiagen | Cat# 28706X4 |
| Qubit dsDNA HS assay kit | Invitrogen | Cat# Q32851 |
| Qubit RNA HS assay | Invitrogen | Cat# Q32852 |
| RNA 6000 kit | Agilent | Cat# 5067–1511 |
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| SuperScript First-Strand synthesis system for RT-PCR | Invitrogen | Cat# 11904018 |
| T7 HiScribe Kit | New England Biolabs | Cat# E2040S |
| TruSeq Stranded Total RNA Library Prep Gold kit | Illumina | Cat# 20020598 |
| Deposited data | ||
| ChIP-seq, RNA-seq in B-lymphocytes | This study | GEO: GSE218680 |
| CTCF ChIP-seq in iPSC, iPSC-NPC | This study | GEO: GSE218680 |
| Double stranded DNA breaks in mouse neural progenitor cells | (Wei et al., 2016) | doi: 10.1016/j.cell.2015.12.039. |
| Genome-wide long-read sequencing in iPSC | This study | GEO: GSE218680 |
| H3K9me3 ChIP-seq in 6718 | This study | GEO: GSE218680 |
| H3K9me3 ChIP-seq in CS0002 | This study | GEO: GSE218680 |
| H3K9me3 ChIP-seq in DF19.11 | (Inoue et al., 2017; Kazachenka et al., 2018) | Encode Project identifier: ENCSR704BRU |
| H3K9me3 ChIP-seq in DF6.9 | (Inoue et al., 2017; Kazachenka et al., 2018) | Encode Project identifier: ENCSR681AIW |
| H3K9me3 ChIP-seq in F1, F2, F3, F4, M1, and M2 | (Yokobayashi et al., 2021) | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE165867 |
| H3K9me3 ChIP-seq in iPSC, iPSC-NPC | This study | GEO: GSE218680 |
| H3K9me3 ChIP-seq in iPSC-15b | (Inoue et al., 2017; Kazachenka et al., 2018) | Encode Project identifier: ENCSR558XOU |
| H3K9me3 ChIP-seq in iPSC-18a | (Inoue et al., 2017; Kazachenka et al., 2018) | Encode Project identifier: ENCSR979TIC |
| H3K9me3 ChIP-seq in iPSC-18c | (Inoue et al., 2017; Kazachenka et al., 2018) | Encode Project identifier: ENCSR034LMV |
| H3K9me3 ChIP-seq in SA3.5 | This study | GEO: GSE218680 |
| H3K9me3 CUT&RUN in iPSC | This study | GEO: GSE218680 |
| H3K9me3 CUT&RUN in Kolf2 | This study | GEO: GSE218680 |
| H3K9me3 CUT&RUN in WTC-11 | This study | GEO: GSE218680 |
| H3K9me3 CUT&RUN in port-mortem brain tissue (caudate nucleus) | This study | GEO: GSE218680 |
| Hi-C in iPSC, iPSC-NPC | This study | GEO: GSE218680 |
| Human fetal cortex RNA-seq | (Kang et al., 2021) | GEO: GSE146878 |
| Murine cortical neuron RNA-seq (Fmr1 KO) | (Korb et al., 2017) | GEO: GSE81912 |
| PCR-free WGS from iPSC (a complete list is provided in Table S4) | HipSci | https://www.hipsci.org |
| PCR-free whole genome sequencing in iPSC | This study | GEO: GSE218680 |
| Repli-seq in iPSC | (Emerson et al., 2022) | 4DN: 4DNFI5WEY784 |
| RNA-seq in iPSC, iPSC-NPC | This study | GEO: GSE218680 |
| Targeted long-read sequencing in iPSC | This study | GEO: GSE218680 |
| Original codes | This study | 10.5281/zenodo.6558223 |
| Experimental models: cell lines | ||
| Human healthy iPS cell line - 176 (designated as NL_18 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human healthy iPS cell line – 158.1 (designated as NL_25 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human healthy iPS cell line - 20b (designated as NL_27 in this study) | Harvard Stem Cell Institute iPS Core Facility | https://divvly.com/reagent-3289 |
| Human pre-mutation iPS cell line - 111 (designated as PM_137 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - 135.3 (designated as FXS_421 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - 1H2 (designated as FXS_425 in this study and clonal from FXS_421) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - FXS_SW (designated as FXS_426 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line – GM07730 (designated as FXS_470 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - 135.3_CGG_034 (designated as ML_scClone1 in this study) | This study | N/A |
| Human FXS iPS cell line - 4H2 (designated as ML_scClone2 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - 6D12 (designated as ML_scClone3 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human FXS iPS cell line - 135.3_CGG_116 (designated as ML_scClone4 in this study) | This study | N/A |
| Human FXS iPS cell line - 135.3_CGG_125 (designated as ML_scClone5 in this study) | This study | N/A |
| Human FXS iPS cell line - 135.3_CGG_128 (designated as ML_scClone6 in this study) | This study | N/A |
| Human FXS iPS cell line - 135.3_CGG_131 (designated as ML_scClone7 in this study) | This study | N/A |
| Human FXS iPS cell line - 4D3 (designated as ML_CUT_PM_scClone1 in this study) | Fulcrum Therapeutics | https://www.fulcrumtx.com |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_117 (designated as ML_CUT_PM_scClone2 in this study) | This study | N/A |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_187 (designated as ML_CUT_PM_scClone3 in this study) | This study | N/A |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_275 (designated as ML_CUT_PM_scClone4 in this study) | This study | N/A |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_278 (designated as ML_CUT_PM_scClone5 in this study) | This study | N/A |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_030 (designated as ML_CUT_PM_scClone6 in this study) | This study | N/A |
| Human pre-mutation FXS iPS cell line - 135.3_CGG_313 (designated as ML_CUT_PM_scClone7 in this study) | This study | N/A |
| Human healthy EBV-transformed B-lymphocyte GM09236 (designated as NL_B in this study) | Coriell Institute | https://www.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM09236&Product=CC |
| Human FXS EBV-transformed B-lymphocyte GM04025 (designated as FXS_B_650 in this study) | Coriell Institute | https://www.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM04025&Product=CC |
| Human FXS EBV-transformed B-lymphocyte GM09237 (designated as FXS_B_900 in this study) | Coriell Institute | https://www.coriell.org/0/Sections/Search/Sample_Detail.aspx?Ref=GM09237&Product=CC |
| Oligonucleotides | ||
| Primers for DNA-FISH, FMR1 CGG PCR, and qRT-PCR are provided in Table S2 | This study | N/A |
| FMR1 5’UTR targeted gRNA provided in Table S2 | This study | N/A |
| TruSeq RNA Single Indexes Set A | Illumina | Cat# 20020492 |
| TruSeq RNA Single Indexes Set B | Illumina | Cat# 20020493 |
| Recombinant DNA | ||
| pSpCas9(BB)-2A-Puro (PX459) V2.0 | Addgene | #62988 |
| pWPT-GFP | Addgene | #12255 |
| pEFS.Cas9.GFP.Ctrl-B | This study / Addgene | To be uploaded to AddGene upon publication |
| pEFS.Cas9.GFP.CGG.cut | This study / Addgene | To be uploaded to AddGene upon publication |
| Software and algorithms | ||
| OligoMiner (version 1.0.4) | (Passaro et al., 2020) | http://oligominerapp.org |
| TANGO (v0.94) | (Ollion et al., 2013) | https://tango.mnhn.fr/tiki-index.php |
| Minimap2 (version 2.22-r1101) | (Gilbert et al., 2021) | https://github.com/lh3/minimap2 |
| nanopolish (version 0.13.2) | (Simpson et al., 2017) | https://github.com/jts/nanopolish |
| FastQC (v0.11.9) | Andrews, 2010 | https://github.com/s-andrews/FastQC |
| STRique (version 0.4.2) | (Giesselmann et al., 2019) | https://github.com/giesselmann/STRique |
| bwa-mem (v0.7.10-r789) | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net/bwa.shtml |
| deeptools (v3.3.0) | (Ramirez et al., 2016) | https://deeptools.readthedocs.io/en/develop/ |
| Samtools (version 1.11) | (Li et al., 2009) | https://www.htslib.org |
| goleft indexcov (version 0.2.3) | (Pedersen et al., 2017) | https://github.com/brentp/goleft |
| MACS2 (v 2.1.1.20160309) | (Zhang et al., 2008) | https://pypi.org/project/MACS2/ |
| Bowtie (v 0.12.7) | (Langmead et al., 2009) | http://bowtie-bio.sourceforge.net/index.shtml |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Guppy (version 6.2.1) | Oxford Nanopore Technologies | https://community.nanoporetech.com/downloads |
| Bedtools | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| HiC-Pro (version 2.7.7) | (Servant et al., 2015) | https://github.com/nservant/HiC-Pro |
| RSEG program (version 0.4.9) | (Song and Smith, 2011) | http://smithlabresearch.org/software/rseg/ |
| kat (v 2.4.1) | (Mapleson et al., 2017) | https://kat.readthedocs.io/en/latest/index.html |
| DESEQ2 (v1.34.0) | (Love et al., 2014) | doi: 10.1186/s13059-014-0550-8 |
| Kallisto | (Bray et al., 2016) | https://pachterlab.github.io/kallisto/about |
| tximport | (Soneson et al., 2015) | https://bioconductor.org/packages/release/bioc/html/tximport.html |
| WebGestalt (v 0.4.4) | (Liao et al., 2019) | https://github.com/bzhanglab/WebGestaltR |
| W2rapContigger (v 0.1) | (Clavijo et al., 2017) | https://github.com/bioinfologics/w2rap-contigger |
| cutadapt (v 1.18) | (Martin, 2011) | https://cutadapt.readthedocs.io/en/stable/ |
| Juicer (v 1.5) | (Durand et al., 2016) | https://github.com/aidenlab/juicer |
| 3D-DNA (v180922) | (Dudchenko et al., 2017) | https://github.com/aidenlab/3d-dna |
| Juicebox (v 1.11.08) | (Robinson et al., 2018) | https://aidenlab.org/juicebox/ |
| JupiterPlots (v 3.8.2) | (Chu, 2018) | https://github.com/JustinChu/JupiterPlot |
| GangSTR (version 2.5.0) | (Mousavi et al., 2019) | https://github.com/gymreklab/GangSTR |
| DumpSTR (version 4.0.0) | (Mousavi et al., 2021) | https://github.com/gymreklab/TRTools |
| ExpansionHunter | (Dolzhenko et al., 2019; Dolzhenko et al., 2017) | https://github.com/Illumina/ExpansionHunter |
| ChopChop online tool (version 3.0.0) | (Labun et al., 2019) | https://chopchop.cbu.uib.no |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Huygens Essential deconvolution software v20.04 | Scientific Volume Imaging | https://svi.nl/Huygens-Essential |
| Other | ||
| Dounce Tissue Grinder | Wheaton | Cat# 357544 |
All data is provided at GEO GSE218680.