

SUMMARY
Patients with early-onset lysosomal storage diseases are ideal candidates for prenatal therapy because organ damage starts in utero. We report the safety and efficacy results of in utero enzyme-replacement therapy (ERT) in a fetus with CRIM (cross-reactive immunologic material)–negative infantile-onset Pompe’s disease. The family history was positive for infantile-onset Pompe’s disease with cardiomyopathy in two previously affected deceased siblings. After receiving in utero ERT and standard postnatal therapy, the current patient had normal cardiac and age-appropriate motor function postnatally, was meeting developmental milestones, had normal biomarker levels, and was feeding and growing well at 13 months of age.
Several early-onset lysosomal storage diseases have multiorgan involvement,1 sometimes even manifesting prenatally. Newborn screening has led to early initiation of enzyme-replacement therapy (ERT) with recombinant enzymes, but this strategy does not completely prevent irreversible organ damage. On the basis of the results of in utero ERT in a mouse model of mucopolysaccharidosis type VII,2 we obtained approval to perform a phase 1 clinical trial of in utero ERT in eight lysosomal storage diseases with antenatal onset for which a recombinant ERT is available (In Utero Enzyme Replacement Therapy for Lysosomal Storage Diseases [IUERT]; ClinicalTrials.gov number, NCT04532047).
Infantile-onset Pompe’s disease, a rapidly progressive lysosomal storage disease caused by acid α-glucosidase (GAA) enzyme deficiency, is a compelling disease to treat in utero because patients have hypertrophic cardiomyopathy prenatally3 and hypotonia at birth; if the disease is untreated, patients typically die by 2 years of age. Infantile-onset Pompe’s disease affects 1 in 138,000 to 226,600 live births worldwide (with a higher incidence in some populations, such as 1 in 50,000 in Taiwan).4–6 Initiating ERT with alglucosidase alfa early after positive newborn screening improves clinical outcomes,7,8 but residual myopathy persists.7,9 Persons with infantile-onset Pompe’s disease have a high degree of sibling concordance.10 Those with a genotype that results in the absence of cross-reactive immunologic material (CRIM-negative) are at the most severe end of the disease spectrum,11,12 and the development of high and sustained levels of IgG antidrug antibodies results in a lack of efficacy of ERT. The induction of immune tolerance concurrent with the initiation of ERT decreases or prevents the formation of antidrug antibodies and can be lifesaving.13 Antidrug antibodies occur in other lysosomal storage diseases, but they are particularly deleterious and well characterized in CRIM-negative infantile-onset Pompe’s disease.14 We treated a fetus with CRIM-negative infantile-onset Pompe’s disease in the context of the early postnatal demise of two previous siblings.
METHODS
CASE PRESENTATION
A 37-year-old woman (gravida 12, para 4) with a known history of three previous pregnancies that were affected with CRIM-negative infantile-onset Pompe’s disease presented for antenatal care in the first trimester. Infantile-onset Pompe’s disease was diagnosed prenatally in the current patient (also referred to as “Sibling 3”) by means of chorionic villus sampling. The fetus was homozygous for the familial known pathogenic variant NM_000152.5(GAA):c.525_526del (p.Asn177fs).
FAMILY HISTORY AND CLINICAL COURSE OF AFFECTED SIBLINGS
The parents are of Pakistani descent and consanguineous (Fig. 1A). Their first child with infantile-onset Pompe’s disease (Sibling 1) received a diagnosis at 5.5 months of age and received immune tolerance induction (four doses of rituximab, nine doses of methotrexate, and monthly intravenous immune globulin)13 concurrent with ERT initiation at 6.6 months of age and again between 17 and 21 months. Treatment with ERT (alglucosidase alfa, 20 mg per kilogram of body weight per dose every other week) was stopped at 24 months, and she died of progressive disease at 29 months. A second pregnancy with an affected fetus was electively terminated. A third affected child (Sibling 2) received a diagnosis prenatally by means of amniocentesis (cardiac hypertrophy was subsequently noted on prenatal echocardiograms), was treated palliatively, and died at 8 months of age from cardiorespiratory failure.
Figure 1. Case Presentation and Skeletal-Muscle and Cardiac Outcomes.

Panel A shows the family pedigree. Squares indicate male family members, circles female family members, triangles pregnancies not carried to term, double bars consanguinity, open symbols unaffected, filled black symbols affected, and diagonal slashes deceased. CRIM denotes cross-reactive immunologic material, IOPD infantile-onset Pompe’s disease, and neg negative. Panel B shows creatine kinase levels at 4 days or more of life in patients with CRIM-negative IOPD (gray dashed curves) treated after newborn screening (NBS) (treated at ≤4 weeks of age)7 as compared with the levels in Sibling 3 (green). IUERT denotes in utero enzyme-replacement therapy. Panel C shows fetal echocardiograms, four-chamber (a and c) and short-axis (b and d), of Sibling 2, who was untreated (top row), and Sibling 3, who received IUERT (bottom row) performed at 34 weeks of gestation. The ventricular-wall thickness, quantified as diastolic measurement of the interventricular septum (IVSd) (asterisk), was 5.7 mm (z score, 7.0) in Sibling 2, whereas it remained normal in S ibling 3 (3.4 mm; z score, 0.6) during fetal therapy. The z scores were calculated according to the methods of Firpo et al.18 LV denotes left ventricle, and RV right ventricle. Panel D shows the left ventricular mass index in Sibling 3 (green) as compared with patients with CRIM-negative IOPD treated after NBS (gray dashed curves)7 and the patient’s two previous affected siblings (Siblings 1 [orange] and 2 [purple]). The first data point for Sibling 1 is just before the initiation of enzyme-replacement therapy (ERT), and the plot shows prolonged time to improvement; Sibling 2 never received ERT.
ETHICS APPROVAL FOR PROVISION OF CLINICAL CARE
The current patient received a diagnosis prenatally and was referred for inclusion in the newly launched phase 1 clinical trial of in utero ERT. Nondirective counseling regarding all options for the pregnancy and the rationale for prenatal therapy was provided by the physician teams at the Ottawa Hospital, Children’s Hospital of Eastern Ontario, the University of California, San Francisco, and Duke University. The family chose to proceed with fetal therapy. Given that challenges with travel during the coronavirus disease 2019 pandemic prevented participation in the trial, a clinical ethics approval was obtained to provide in utero ERT locally at the Ottawa Hospital in the same manner as in the clinical trial.
IN UTERO ERT PROTOCOL
Alglucosidase alfa (20 mg per kilogram of estimated fetal weight) was administered under ultrasonic guidance through the umbilical vein beginning at 24 weeks 5 days of gestation and continued at 2-week intervals through 34 weeks 5 days of gestation (six infusions total). The 2-week dosing interval is consistent with routine practice for in utero blood transfusions and the recommended package-insert administration interval for alglucosidase alfa.15 At each infusion, a small volume of blood was drawn back to confirm successful entry into the umbilical vein; this blood was used to measure plasma trough levels of GAA enzyme activity and serum antidrug antibody titers. (Details of sample collection and analysis are provided in the Methods section in the Supplementary Appendix, available with the full text of this article at NEJM.org.)
After birth, the infant was treated with immune tolerance induction on day 1,13 consistent with standard-of-care treatment for CRIM-negative infantile-onset Pompe’s disease and the Food and Drug Administration–approved investigational new drug protocol of the IUERT trial. On day 4 and every other week thereafter, the patient received an infusion of alglucosidase alfa (20 mg per kilogram per dose). In light of recent studies,16 the patient was transitioned to a dose of 40 mg per kilogram every 2 weeks at 9.6 months of age and a weekly dose of 40 mg per kilogram at 11.3 months of age.
RESULTS
PRENATAL SAFETY AND INFANT DELIVERY
There were no adverse reactions to any of the six infusions in the mother or the fetus. Labor was induced at 37 weeks 3 days of gestation (3 weeks after the last infusion), and the patient was delivered vaginally at 37 weeks 4 days of gestation.
SKELETAL-MUSCLE OUTCOMES
Creatine kinase levels can be a marker of long-term muscle damage, and elevated levels often help in the initial diagnosis of infantile-onset Pompe’s disease.17 Creatine kinase levels in the current patient were normal from day 4 through the most current assessment (13 months), in contrast to those of a newborn cohort of four CRIM-negative patients,7 all of whom had elevated creatine kinase levels before receiving ERT (Fig. 1B).
Patients with infantile-onset Pompe’s disease can have motor deficits7,9 as early as baseline assessment after newborn screening ascertainment. These deficits were not present in our patient, in contrast to those seen in two of four patients among the CRIM-negative early-treated newborn screening cohort7 (Table S1 in the Supplementary Appendix). The current patient consistently demonstrated age-appropriate gross and fine motor development, muscle tone, and power. Specifically, the score on the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (a standardized measure of infant strength; range, 0 to 64) was 42 at 3 days and appropriately increased to the maximum score of 64 by 5 months 24 days. She was at the 50th percentile for the Alberta Infant Motor Scale score at a chronologic age of 7 months 5 days. At this age, she was able to roll, reach and pivot from prone forearm support, sit (un-sustained without arm support), and stand with support. At 10 months 13 days of age, she was at the 50th to 75th percentile and was able to actively resist or oppose testing with more vigor than previously; she was able to transition to and from the floor through a half-kneel with hands on furniture, cruise, and creep reciprocally (Table S2). She walked independently at 11.5 months of age. These findings contrast with the development of Sibling 1, who had (according to the parents’ report) poor head control shortly after birth and who presented with clinically notable hypotonia and head lag at 2 months of age, and with the development of Sibling 2, in whom hypotonia and increased work of breathing first manifested in the second month of life.
CARDIAC OUTCOMES
Each of the patient’s echocardiograms (including prenatal) was normal. Sibling 2 had a fetal echocardiogram that showed severe left ventricular hypertrophy (z score, 7.0) at 34 weeks of gestation; a fetal echocardiogram for Sibling 3 at 34 weeks of gestation showed an absence of left ventricular hypertrophy (z score, 0.6) (Fig. 1C). On day 1, the current patient had a normal left ventricular mass index (39.47 g per square meter of body-surface area; z score, −2.54), in contrast to both previous siblings’ first echocardiograms and to a CRIM-negative newborn screening cohort treated with postnatal ERT initiation at 4 weeks of age or younger7 (Fig. 1D). Figure S1A and S1B and Video 1 compare the echocardiograms of all three siblings at similar ages. In addition, electrocardiograms showed severe biventricular hypertrophy with T-wave inversion in the inferolateral leads in Siblings 1 and 2. The electrocardiograms of the current patient were normal (Fig. S2).
PLACENTAL PATHOLOGICAL ANALYSIS
Glycogen deposits are evident in the placentas of patients with infantile-onset Pompe’s disease, supporting the early onset of storage disease and providing a readily accessible tissue for monitoring the potential benefit of in utero ERT. Two previous reports of placental pathological features in fetuses with infantile-onset Pompe’s disease showed membrane-bound glycogen, unbound cytoplasmic glycogen rosettes, and glycogen-filled lysosomes on electron microscopy.19,20 These features were absent in the placenta of the current patient (Fig. 2A). On light microscopy, periodic acid–Schiff–positive granules were notable in the villi of an untreated infantile-onset Pompe’s disease placenta and absent in the current patient (Fig. 2B).
Figure 2. Placental Pathological Analysis.
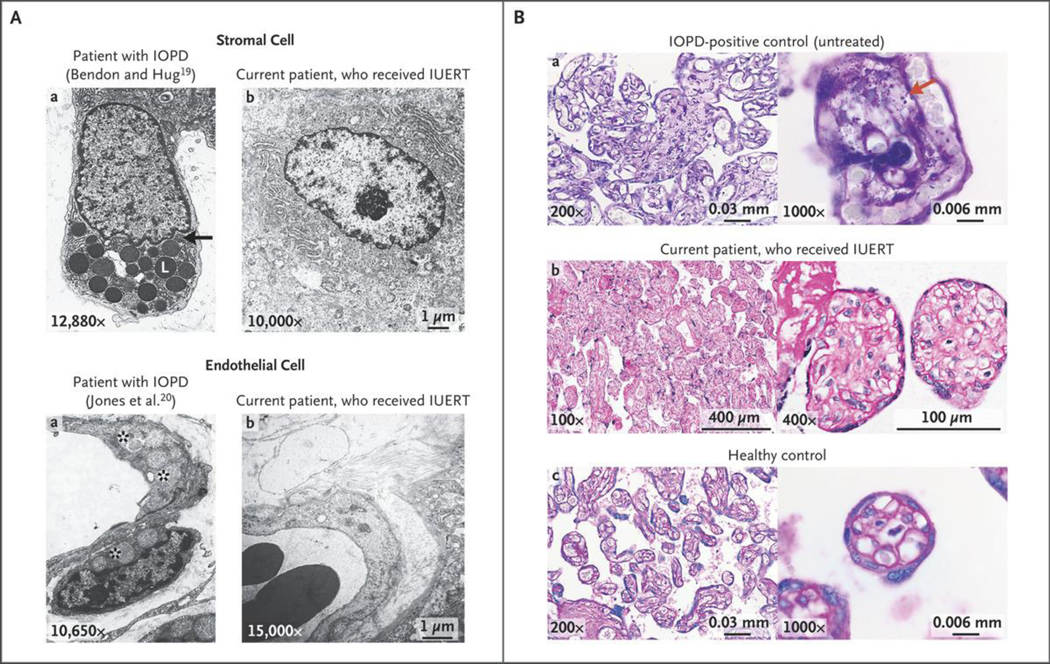
Panel A shows electron microscopic images from previously studied patients with IOPD who did not receive IUERT19,20 (left column) and from the current patient, who received IUERT (right column). The top row shows numerous membrane-bound glycogen lobules (L) in a stromal cell as well as unbound cytoplasmic glycogen rosettes (black arrow) in the untreated patient, which are not present in the patient who received IUERT. The bottom row shows that the capillary endothelium and pericytes contain glycogen-filled lysosomes (asterisks) in a fetus with IOPD, which are not present in the current patient. Panel B shows periodic acid–Schiff (PAS) staining (without diastase digestion) of placental villi. The top row shows photomicrographs from an untreated IOPD placenta at term with PAS-positive granules throughout, which are not present in the current patient (middle row) or the healthy unaffected placenta (bottom row) at a gestational age similar to that of the current patient at term. The right column of images shows the same at higher magnifications; the red arrow highlights PAS-positive granules.
IGG ANTIDRUG ANTIBODY LEVELS
We detected antidrug antibodies after the third in utero infusion; these peaked at 1:3200 at 34 weeks 5 days of gestation (Fig. 3A). Postnatally, antibody levels peaked at 1:6400 and decreased to 1:3200 by 8 weeks. Titers remained lower than 1:12,800, a level at or above which, if persistent, is considered to be clinically significant.12 Monthly rituximab was given as a precaution at 11 weeks of age and then subsequently every 2 months beginning at 27 weeks of age, with the most recent antibody titers ranging from negative to 1:400. Given the persistently low titers, the administration of rituximab was changed to once every 3 months at 43 weeks of age. Monthly intravenous immune globulin was continued to provide passive immunity.13 In contrast to the current patient, the proband (Sibling 1, who started immune tolerance induction and ERT at 6.6 months of age) had antibody titers of 1:6400, which persisted for a longer duration and decreased only after a second full course of immune tolerance induction. Assays of maternal IgG with reactivity against ERT that were performed during pregnancy, 2 days after delivery, and 7 months after delivery yielded negative results.
Figure 3. (facing page). Laboratory Monitoring.

Panel A shows the time course for antidrug antibody levels and immune tolerance induction in the current patient (Sibling 3, green; left graph) and the proband (Sibling 1, red; right graph). The antidrug antibody levels of Siblings 1 and 3 reached the same peak, with Sibling 1 having a longer duration of titers at this level. Sibling 3 received immune tolerance induction as previously published,13 followed by monthly rituximab alone, then rituximab alone every other month, and later every 3 months; Sibling 1 received immune tolerance induction as previously published,13 followed by a repetition of a full course of immune tolerance induction (three medications), owing to her persistent titers at 1:6400. Pharmacokinetic and clinical concerns are present when titers reach a level of 1:12,800 or greater.12,15 IVIG denotes intravenous immune globulin. Panel B shows plasma trough levels of acid α-glucosidase (GAA) enzyme activity (obtained before each ERT infusion) in Sibling 3’s fetal plasma over the course of IUERT. Panel C shows glucose tetrasaccharide (Glc4) levels in amniotic fluid samples for Sibling 3 (green) over the course of IUERT infusions as compared with the levels for gestational age–matched unaffected controls (gray). These findings show concordance with unaffected controls and a rise in the levels of this biomarker with older gestational age. Panel D shows the Glc4 levels in neonatal urine samples obtained postnatally from Sibling 3 (green), as compared with baseline values in a previously studied CRIM-positive cohort in the neonatal period at less than 1 month of age (light blue)23 and with values in patients with CRIM-negative IOPD (gray dashed curves) treated after NBS (treated at ≤4 weeks of age).7 Cr denotes creatinine, and pos positive.
PHARMACOKINETICS
Plasma trough levels of GAA enzyme activity were batch-analyzed after the course of in utero ERT. Trough GAA levels were higher in the first 6 weeks after the initial in utero ERT infusion (Fig. 3B and Fig. S3A) but decreased to near zero before the final two in utero ERT infusions and remained at this level postnatally.
GLUCOSE TETRASACCHARIDE LEVELS
Glucose tetrasaccharide (Glc4) is a biomarker of accumulated glycogen and can be measured in amniotic fluid prenatally or urine postnatally.21 In healthy fetuses, liver glycogen accumulation begins during the first trimester and continues throughout gestation.22 Figure 3C and Figure S3B show the patient’s Glc4 trajectory in utero as compared with the levels for gestational age–matched unaffected controls, findings that indicate concordance with unaffected controls. The initial postnatal level of urine Glc4 for Sibling 3 (obtained 3 weeks after the last in utero infusion) was similar to that of a cohort of patients with CRIM-positive infantile-onset Pompe’s disease23 and two patients with CRIM-negative infantile-onset Pompe’s disease,7 with all cases diagnosed by means of newborn screening. The patient’s level decreased rapidly after postnatal ERT and subsequently remained normal (Fig. 3D).
DISCUSSION
We describe a protocol for in utero ERT that showed safety for both the mother and fetus, as well as efficacy in decreasing placental glycogen deposits. Although cardiomyopathy was present in the patient’s two previous affected siblings, cardiac manifestations did not develop in this patient, who was thriving at 13 months of age. Our results are consistent with in utero ERT attenuating or even halting the disease process in the fetal period. Furthermore, although it is accepted that starting treatment as early as possible improves outcomes in patients with lysosomal storage diseases (e.g., when the diagnosis is established after newborn screening), our results suggest that moving the window for therapeutic intervention into the prenatal period may further improve postnatal outcomes.
Glycogen is known to accumulate in utero and leads to the clinical sequelae that are present by the time of birth in patients with infantile-onset Pompe’s disease. The placental pathological features in our patient, as compared with those in untreated patients, suggest that in utero ERT prevented glycogen buildup. The improved cardiac results in our in utero ERT–treated patient in the third trimester and at birth (as compared with those in the untreated sibling and in previously studied cohorts3), together with normal creatine kinase levels and age-appropriate motor function, are consistent with an overall cardiac and musculoskeletal benefit of in utero ERT. Although cardiomyocytes tend to be most responsive to postnatal ERT owing to their robust expression of mannose-6-phosphate (M6P) receptors24 (to which the normalization of left ventricular mass index in patients with infantile-onset Pompe’s disease with clinically significant hypertrophy7 has been attributed), cellular glycogen accumulation persists in cardiac conduction cells,25 and patients may still be at risk for life-threatening arrhythmias.26
It is important to accurately predict a severe phenotype for potential candidates for in utero ERT with the use of clinical information on previous affected family members or published cases with the same genotype. The availability of prenatal treatment options may affect parental decisions about prenatal carrier screening and prenatal genetic diagnosis. Although newborn screening programs have improved outcomes for infants receiving a diagnosis at birth, newborn screening is not available for all conditions, and we think that there is a strong rationale for prenatal therapy for conditions in which organ pathologic processes start before birth and in which immune responses to replaced proteins complicate therapy.
Although there is evidence in preclinical models that in utero administration of a new antigen will induce tolerance,2 one theoretical risk of in utero ERT in CRIM-negative infantile-onset Pompe’s disease (arguably the subset of disease with the highest risk of the development of antidrug antibodies14) is sensitization to the enzyme in utero. Accordingly, we monitored IgG antidrug antibodies with each prenatal infusion. The patient received six ERT infusions in utero without having a clinically significant antibody response. Of note, without immune tolerance induction, a CRIM-negative patient receiving six doses (12 weeks) of postnatal ERT could expect the development of high sustained antibody titers.11,12
Our administration schedule was extrapolated from postnatal experience with ERT, given the lack of knowledge of fetal pharmacokinetics. We observed elevated GAA trough levels early in gestation, possibly owing to the diminished expression of M6P receptors in fetal tissue early in gestation.27 Later in gestation, when M6P receptors are known to be more abundant,27 GAA enzyme activity reached a trough close to zero.
Direct umbilical-vein administration of therapeutic compounds can be accomplished safely by physicians experienced with in utero blood transfusions. Like any fetal intervention, this procedure carries a risk of preterm delivery. Nondirective counseling for parents of affected pregnancies is critical to ensure informed choices regarding the risk–benefit profile of this new therapy. Our phase 1 clinical trial of in utero ERT for fetuses with genetically confirmed early-onset lysosomal storage diseases will enable the collection of additional safety and efficacy data in a larger cohort.
Supplementary Material
Acknowledgments
The protocol development and the collection, shipping, and analysis of samples were supported by funds from the Center for Maternal–Fetal Precision Medicine (University of California, San Francisco [UCSF]), the Y.T. and Alice Chen Pediatric Genetics and Genomics Research Center (Duke University), Newborn Screening Ontario, CHEO Research Institute, and a grant (R01 DK067859) from the National Institutes of Health. Sanofi Genzyme provided the enzyme for this patient.
We thank the patient and her family for their participation; Ankit Desai, Akos Herzeg, Eleanor Rodriguez-Rassi, Seung-Hye Jung, Catherine Rehder, and Erika Bariciak for helpful discussions regarding this work; Nathan McIntosh, Deidre Kelly, Wendy Mears, and Alan Mears for helpful discussions and for preparing all the fetal samples for analysis; and Pam Derish (UCSF Department of Surgery) for critical reading of an earlier version of the manuscript.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
REFERENCES
- 1.Platt FM. Emptying the stores: lysosomal diseases and therapeutic strategies. Nat Rev Drug Discov 2018; 17: 133–50. [DOI] [PubMed] [Google Scholar]
- 2.Nguyen Q-H, Witt RG, Wang B, et al. Tolerance induction and microglial engraftment after fetal therapy without conditioning in mice with mucopolysaccharidosis type VII. Sci Transl Med 2020; 12(532): eaay8980. [DOI] [PubMed] [Google Scholar]
- 3.Hamdan MA, El-Zoabi BA, Begam MA, Mirghani HM, Almalik MH. Antenatal diagnosis of Pompe disease by fetal echocardiography: impact on outcome after early initiation of enzyme replacement therapy. J Inherit Metab Dis 2010; 33: Suppl 3: S333–S339. [DOI] [PubMed] [Google Scholar]
- 4.Ausems MG, Verbiest J, Hermans MP, et al. Frequency of glycogen storage disease type II in the Netherlands: implications for diagnosis and genetic counselling. Eur J Hum Genet 1999; 7: 713–6. [DOI] [PubMed] [Google Scholar]
- 5.Tang H, Feuchtbaum L, Sciortino S, et al. The first year experience of newborn screening for Pompe disease in California. Int J Neonatal Screen 2020; 6: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chiang S-C, Hwu W-L, Lee N-C, Hsu L-W, Chien Y-H. Algorithm for Pompe disease newborn screening: results from the Taiwan screening program. Mol Genet Metab 2012; 106: 281–6. [DOI] [PubMed] [Google Scholar]
- 7.Li C, Desai AK, Gupta P, et al. Transforming the clinical outcome in CRIM-negative infantile Pompe disease identified via newborn screening: the benefits of early treatment with enzyme replacement therapy and immune tolerance induction. Genet Med 2021; 23: 845–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chien Y-H, Lee N-C, Thurberg BL, et al. Pompe disease in infants: improving the prognosis by newborn screening and early treatment. Pediatrics 2009; 124(6): e1116–e1125. [DOI] [PubMed] [Google Scholar]
- 9.Prater SN, Patel TT, Buckley AF, et al. Skeletal muscle pathology of infantile Pompe disease during long-term enzyme replacement therapy. Orphanet J Rare Dis 2013; 8: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith WE, Sullivan-Saarela JA, Li JS, et al. Sibling phenotype concordance in classical infantile Pompe disease. Am J Med Genet A 2007; 143A: 2493–501. [DOI] [PubMed] [Google Scholar]
- 11.Kishnani PS, Goldenberg PC, DeArmey SL, et al. Cross-reactive immunologic material status affects treatment outcomes in Pompe disease infants. Mol Genet Metab 2010; 99:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Desai AK, Baloh CH, Sleasman JW, Rosenberg AS, Kishnani PS. Benefits of prophylactic short-course immune tolerance induction in patients with infantile Pompe disease: demonstration of long-term safety and efficacy in an expanded cohort. Front Immunol 2020; 11: 1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kazi ZB, Desai AK, Berrier KL, et al. Sustained immune tolerance induction in enzyme replacement therapy-treated CRIM-negative patients with infantile Pompe disease. JCI Insight 2017; 2(16): e94328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kishnani PS, Dickson PI, Muldowney L, et al. Immune response to enzyme replacement therapies in lysosomal storage diseases and the role of immune tolerance induction. Mol Genet Metab 2016; 117: 66–83. [DOI] [PubMed] [Google Scholar]
- 15.Lumizyme (alglucosidase alfa). Cambridge, MA: Genzyme, 2016. (package insert). [Google Scholar]
- 16.Ditters IAM, Huidekoper HH, Kruij-shaar ME, et al. Effect of alglucosidase alfa dosage on survival and walking ability in patients with classic infantile Pompe disease: a multicentre observational cohort study from the European Pompe Consortium. Lancet Child Adolesc Health 2022; 6: 28–37. [DOI] [PubMed] [Google Scholar]
- 17.Yang C-F, Liu H-C, Hsu T-R, et al. A large-scale nationwide newborn screening program for Pompe disease in Taiwan: towards effective diagnosis and treatment. Am J Med Genet A 2014; 164A: 54–61. [DOI] [PubMed] [Google Scholar]
- 18.Firpo C, Hoffman JI, Silverman NH. Evaluation of fetal heart dimensions from 12 weeks to term. Am J Cardiol 2001; 87: 594–600. [DOI] [PubMed] [Google Scholar]
- 19.Bendon RW, Hug G. Morphologic characteristics of the placenta in glycogen storage disease type II (alpha-1,4-glucosidase deficiency). Am J Obstet Gynecol 1985; 152:1021–6. [DOI] [PubMed] [Google Scholar]
- 20.Jones CJ, Lendon M, Chawner LE, Jau-niaux E. Ultrastructure of the human placenta in metabolic storage disease. Placenta 1990;11: 395–411. [DOI] [PubMed] [Google Scholar]
- 21.Young SP, Piraud M, Goldstein JL, et al. Assessing disease severity in Pompe disease: the roles of a urinary glucose tetrasaccharide biomarker and imaging techniques. Am J Med Genet C Semin Med Genet 2012; 160C:50–8. [DOI] [PubMed] [Google Scholar]
- 22.Gruppuso PA, Sanders JA. Regulation of liver development: implications for liver biology across the lifespan. J Mol Endocrinol 2016; 56:R115–R125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chien Y-H, Goldstein JL, Hwu W-L, et al. Baseline urinary glucose tetrasaccharide concentrations in patients with infantile- and late-onset Pompe disease identified by newborn screening. JIMD Rep 2015; 19: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amalfitano A, Bengur AR, Morse RP, et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med 2001; 3: 132–8. [PubMed] [Google Scholar]
- 25.Pena LD, Proia AD, Kishnani PS. Post-mortem findings and clinical correlates in individuals with infantile-onset Pompe disease. JIMD Rep 2015; 23: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McDowell R, Li JS, Benjamin DK Jr, et al. Arrhythmias in patients receiving enzyme replacement therapy for infantile Pompe disease. Genet Med 2008; 10: 758–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Funk B, Kessler U, Eisenmenger W, Hansmann A, Kolb HJ, Kiess W. Expression of the insulin-like growth factor-II/mannose-6-phosphate receptor in multiple human tissues during fetal life and early infancy. J Clin Endocrinol Metab 1992;75: 424–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.