

Abstract
A series of tricyclic imidazo[2,1-i]purinones and ring-enlarged analogues derived from xanthine derivatives have been prepared as adenosine receptor (AR) antagonists. In comparison with xanthines, the tricyclic compounds exhibit increased water solubility due to a basic nitrogen atom, which can be protonated under physiological conditions. Substituents were introduced that confer high affinity for A2A or A3 ARs, respectively. A new capillary electrophoresis method was developed for the determination of the enantiomeric purity of selected chiral products using native and modified β-cyclodextrins as chiral discriminators. The compounds were investigated in radioligand binding assays at rat brain A1 and A2A ARs. Selected compounds were additionally investigated in radioligand binding assays at human recombinant A3 ARs and in functional studies (adenylate cyclase assays) at A1 ARs of rat fat cell membranes, A2A ARs of rat PC 12 cell membranes, and mouse A2B ARs of NIH 3T3 cell membranes. Structure–activity relationships were similar to those of corresponding xanthine derivatives. The 2-styrylimidazopurinones were less potent at A2A ARs as compared to 8-styrylxanthine derivatives. The most potent compound at A2A ARs was (S)-1,4-dimethyl-8-ethyl-2-styrylimidazo[2,1-i]purinone (S-25) exhibiting a Ki value of 424 nM at rat A2A ARs. The compound was highly selective for A2A receptors vs A1 and A3 ARs. Selectivity vs A2B ARs, however, was low. Among the 1-unsubstituted 2-phenyl-imidazo[2,1-i]purin-5-one derivatives, very potent and highly selective antagonists for human A3 ARs were identified. The most potent A3 antagonist of the present series was (R)-4-methyl-8-ethyl-2-phenyl-imidazo[2,1-i]purin-5-one (R-24) exhibiting a Ki value of 2.3 nM and high selectivity for A3 receptors vs all other AR subtypes.
Introduction
Adenosine receptors (ARs) are a class of G-protein-coupled receptors (GPCRs) that comprise four subtypes, designated A1, A2A, A2B, and A3.2 All ARs may be coupled to adenylate cyclase (AC) as a second messenger system. A1 and A3 AR stimulation leads to an inhibition of the enzyme, while A2A and A2B AR activation causes a stimulation of AC.2
Selective antagonists at AR subtypes are of considerable interest as novel therapeutics;3,4 for example, A1 AR antagonists are being developed as kidney protective diuretics, for the prevention of renal failure, and as cognition enhancers;5 A2A selective AR antagonists are promising new drugs for the treatment of Parkinson’s disease.6 A3 AR antagonists have been postulated as novel antiinflammatory and antiallergic agents,7 while A2B AR antagonists may possess antiasthmatic properties.8 All AR subtypes have been cloned from various species, including rat and humans. While differences between the two species are moderate for A1 (amino acid sequence identity: 95%), A2A (84%), and A2B (86%) ARs, there are considerable differences between the rat and the human A3 ARs (only 74% sequence identity).2-4 The A3 AR is also unique with regard to tissue distribution in different species: High densities of A3 ARs are found in the testis of rats and only low levels in most other rat tissues. In humans, the highest A3 receptor densities are found in lung and liver and only moderate levels in most other tissues.9
A number of potent and subtype selective AR antagonists have been developed during the past decades.3-7,9,10 However, a major problem is that most of the potent AR antagonists are highly lipophilic and therefore exhibit low water solubility, which may limit their in vivo applicability. A1 AR antagonists with increased water solubility have been developed5 including tricyclic imidazo[2,1-i]purinone derivatives, such as KF 20274 (2) (Chart 1).11 Compound 2 is structurally derived from an A1 selective xanthine derivative, 1,3-dipropyl-8-(3-noradamantyl)xanthine (KW 3902, 1, Chart 1). Increased water solubility was obtained by introduction of a third ring, an imidazoline ring, which contains a basic nitrogen atom subject to protonation at physiological pH. This compound shows a high degree of stereoselectivity at guinea pig A1 ARs, the R enantiomer (Ki = 2.7 nM), being about 40-fold more potent than the S enantiomer (Ki = 120 nM). At rat A2A ARs, both enantiomers are nearly equipotent (R, Ki = 290 nM; S, Ki = 250 nM).11
Chart 1.

A1 Selective AR Antagonists: Xanthine and Derived Imidazo[2,1-i]purin-5-one Derivative
In the present study, we tried to combine substituents that are known to enhance affinity in xanthine derivatives for A2A ARs (e.g., 8-styryl, 7-methyl) or A3 ARs (e.g., 8-phenyl) with the imidazo[2,1-i]purin-5-one nucleus. Furthermore, we investigated the influence of variations of the imidazoline ring, including substitutions, chiral derivatives, and ring enlargement. Our goal was to develop novel AR antagonists with increased water solubility and high selectivity for either A2A or A3 ARs.
Results and Discussion
Chemistry.
The syntheses of the tricyclic purinone derivatives can be subdivided into two parts: First, appropriate xanthine derivatives were synthesized (Scheme 1 and Table 1), which were subsequently converted to the tricyclic purinone derivatives (Scheme 2).1 The 1-substituted uracil derivatives 3a,b required as starting compounds were synthesized according to Papesch and Schröder.12 Starting from these compounds (3a,b), the xanthine derivatives could be obtained by different pathways (Scheme 1).1 One method was their conversion to the nitroso uracils (4a,b). The nitroso compound 4b was reacted with a mixture of benzylamine and benzylamine hydrochloride at high temperatures to yield xanthine derivative 11d. The diamino uracils 7a,b were obtained by reduction of the nitroso uracils 4a,b using sodium dithionite. Because these compounds are unstable due to oxidative dimerization in the presence of oxygen,13 they were used for the next reaction step without further purification. The diaminouracil derivative 7a was reacted with triethyl orthoformate to yield the xanthine derivative 11b. Alternatively, the diaminouracil derivative 7b was condensed with cinnamic aldehyde to yield the imine 8, which was oxidatively cyclized with thionyl chloride14 to afford xanthine 11g. For the synthesis of 3,7,8-tri- or 7,8-disubstituted xanthine derivatives, the 1-substituted 5-N-methylamino-6-aminouracil derivatives 5a,b were prepared by bromination of 3a,b and subsequent substitution with methylamine.15-17 The obtained compound 5a was converted to xanthine 10 using triethyl orthoformate. Debenzylation to obtain xanthine derivative 11c, unsubstituted in the 3-position, was achieved by the Lewis acid aluminum trichloride in toluene. Compound 5b was condensed with cinnamic acid to 6a. Condensation of 5a,b with benzoyl chloride afforded the amides 6b,c. The acylated compounds 6a–c were cyclized in sodium hydroxide solution in a mixture of water and ethanol to yield the xanthine derivatives 9 and 11f,g. To obtain xanthine derivative 11e without substitution in position 3 (R4), the benzyl group of 9 was removed by reaction with boron tribromide yielding the xanthine derivative 11e.
Scheme 1.

Preparation of Intermediate Xanthines Used as Starting Compounds for the Synthesis of Tricyclic Purinone Derivativesa
a Reagents: (i) NaNO2, H2O/CH3COOH. (ii) C6H5CH2NH2•HCl, C6H5CH2NH2, 3 h, 170 °C. (iii) Na2S2O4, H2O/NH3. (iv) (C2H5O)3CH, 12 h Δ. (v) Cinnamic aldehyde, MeOH/CH3COOH, 1 h, room temperature. (vi) SOCl2, 8 h, room temperature. (vii) 1, Br2, 0 °C, 30 min, room temperature; 2, CH3NH2, 70 °C, 4 h. (viii) AlCl3, toluene, 1 h, 70 °C. (ix) 6a: cinnamic acid, EDC, 10 h, room temperature; 6b,c: PhCOCl, pyridine, 12 h, room temperature. (x) NaOH/EtOH, 1 h, Δ. (xi) BBr3, toluene, 2 h, 80 °C.
Table 1.
Intermediate Xanthines Used for the Preparation of Tricyclic Purinone Derivatives
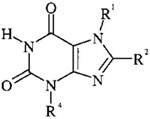
| |||||
|---|---|---|---|---|---|
| compd | R1 | R2 | R4 | mp1 (°C) (lit. mp) | yield1 (%) |
| 11a | CH3 | H | CH3 | 290–295 | a |
| 11b | H | H | benzyl | >250 (>300)34 | 90 |
| 11c | CH3 | H | H | >270 (>330)16 | 75 |
| 11d | H | phenyl | CH3 | >250 (>360)35 | 46 |
| 11e | CH3 | phenyl | H | >250 | 80 |
| 11f | CH3 | phenyl | CH3 | >250 (300)36 | 85 |
| 11g | H | styryl | CH3 | >270 | 80 |
| 11h | CH3 | styryl | CH3 | >270 | 88 |
Theobromine (11a) was commercially available.
Scheme 2.

Synthesis of Imidazo[2,1-i]purinone Derivatives and Ring-Enlarged Analoguesa
a Reagents: (i) P2S5, pyridine, 8 h, Δ. (ii) MeI, NaOH/EtOH, 1 h, room temperature. (iii) Amino alcohol, DMSO, 1 h, 150 °C. (iv) SOCl2, 1 h Δ.
In the second part of the synthesis1 (Scheme 2 and Table 2), the xanthine derivatives 11a–h were converted to the tricyclic purine derivatives according to the route described by Shimada et al.18 The xanthine derivatives 11a–h were thionated with phosphorus pentasulfide in dry pyridine under reflux to yield 6-thioxanthines 12a–h. After methylation with methyl iodide, the compounds 13a–h were treated with amino alcohols in dimethyl sulfoxide (DMSO) under reflux conditions to obtain the amino-substituted purines 14a–o. The final ring closure to the tricyclic purine derivatives 15–30 was performed by cyclization of 14a–o with thionyl chloride at high temperature under reflux conditions.1 As expected, yields for cyclization of un-branched hydroxyalkyl derivatives (e.g., 17, 27, 29, and 30) depended on the ring size of the formed product: formation of five and six ring derivatives proceeded in high yields, while reduced yields were observed in preparation of the diazepine and the diazocine derivatives 29 and 30.
Table 2.
Synthesized Tricyclic Purinone Derivatives: Substitution Pattern, Method of Preparation, Yields, and Analytical Data1
| compda | stereo- chemistry |
n | R1 | R2 | R4 | R7 | R8 | method | yield (%) |
formula | analyses | mp (°C) |
b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 15 | 0 | CH3 | H | H | H | H | A | 49 | C8H9N5O•HCl•0.7H2O | C, H, N | >250 | ||
| 16 | 0 | CH3 | H | benzyl | H | H | A | 41 | C15H15N5O•2HCl | C, H, Nc | 235 | ||
| 17 | 0 | CH3 | H | CH3 | H | H | A | 75 | C9H11N5O•2HCl•0.2H2O | C, H, N | >250 | ||
| R-18 | R | 0 | CH3 | H | CH3 | H | CH3 | C | 35 | C10H13N5O•2HCl•0.3H2O | C, H, N | >250 | 2.3 |
| S-18 | S | 0 | CH3 | H | CH3 | H | CH3 | C | 39 | C10H13N5O•2HCl•0.5H2O | C, H, N | >250 | −2.8 |
| RS-19 | rac | 0 | CH3 | H | CH3 | H | C2H5 | B | 45 | C11H15N5O•2HCl•0.25H2O | C, H, N | >250 | 0 |
| R-19 | R | 0 | CH3 | H | CH3 | H | C2H5 | B | 42 | C11H15N5O•2HCl•0.1H2O | C, H, N | >250 | 4.2 |
| S-19 | S | 0 | CH3 | H | CH3 | H | C2H5 | B | 32 | C11H15N5O•2HCl•0.1H2O | C, H, N | >250 | −3.9 |
| RS-20 | rac | 0 | CH3 | H | CH3 | CH3 | H | B | 48 | C10H13N5O•2HCl•0.1H2O | C, H, N | 175 | 0 |
| R-20 | R | 0 | CH3 | H | CH3 | CH3 | H | B | 52 | C10H13N5O•2HCl•0.5H2O | C, H, N | 200 | −3.4 |
| S-20 | S | 0 | CH3 | H | CH3 | CH3 | H | B | 54 | C10H13N5O•2HCl•H2O | C, H, N | 200 | 3.3 |
| 21 | 0 | CH3 | phenyl | H | H | H | A | 38 | C14H13N5O•HCl•0.6H2O | Cd, H, N | >250 | ||
| 22 | 0 | CH3 | phenyl | CH3 | H | H | A | 45 | C15H15N5O•HCl•H2O | C, H, N | >250 | ||
| RS-23 | rac | 0 | CH3 | phenyl | CH3 | H | C2H5 | D | 29 | C17H19N5O•HCl•0.3H2O | C, H, N | >250 | 0 |
| R-23 | R | 0 | CH3 | phenyl | CH3 | H | C2H5 | D | 27 | C17H19N5O•HCl•0.5H2O | C, H, N | >250 | 2.4 |
| S-23 | S | 0 | CH3 | phenyl | CH3 | H | C2H5 | D | 28 | C17H19N5O•HCl•0.3H2O | C, H, N | >250 | −2.6 |
| RS-24 | rac | 0 | H | phenyl | CH3 | H | C2H5 | D | 36 | C16H17N5O•HCl•3H2O | C, H, N | >250 | 0 |
| R-24 | R | 0 | H | phenyl | CH3 | H | C2H5 | D | 35 | C16H17N5O•HCl•2H2O | C, H, Ne | >250 | 4.5 |
| S-24 | S | 0 | H | phenyl | CH3 | H | C2H5 | D | 35 | C16H17N5O•HCl•2H2O | C, H, N | >250 | −4.8 |
| R-25 | R | 0 | CH3 | styryl | CH3 | H | C2H5 | D | 16 | C19H21N5O | 335.174f | >250 | 3.3 |
| S-25 | S | 0 | CH3 | styryl | CH3 | H | C2H5 | D | 14 | C19H21N5O | 335.175f | >250 | −3.2 |
| R-26 | R | 0 | H | styryl | CH3 | H | C2H5 | D | 32 | C18H19N5O•HCl•0.6H2O | C, H, N | >250 | 3.2 |
| S-26 | S | 0 | H | styryl | CH3 | H | C2H5 | D | 28 | C18H19N5O•HCl•0.7H2O | C, H, N | >250 | −3.1 |
| 27 | 1 | CH3 | H | CH3 | - | - | A | 52 | C10H13N5O•2HCl•0.2H2O | C, H, N | >250 | ||
| 28 | 1 | CH3 | Cl | CH3 | - | - | A | 30 | C10H12N5OCl•HCl•0.1H2O | C, H, N | 261 | ||
| 29 | 2 | CH3 | H | CH3 | - | - | A | 27 | C11H15N5O•2HCl•0.1H2O | C, H, Ng | 198 | ||
| 30 | 3 | CH3 | H | CH3 | - | - | A | 14 | C12H17N5O•2HCl | C, H, N | 210 |
Hydrochlorides of imidazopurinones.
Solvent, methanol; concentration, 1.00 g/100 mL.
N: calcd, 18.81; found, 18.35.
C: calcd, 53.46; found, 53.04.
N: calcd, 19.04; found, 18.52.
Determined by high-resolution mass spectroscopy.
N: calcd, 22.74; found, 22.24.
Cyclization of chiral compounds with a methyl or ethyl substituent in the α-position with regard to the amino group (18, 19, and 23–26) yielded products with retained stereochemistry in good yields, as described for similar cyclization reactions.11 On the other hand, cyclization of chiral compounds with a methyl substituent β to the amino group (20) resulted in inversion of the configuration. As shown for analogous cyclization reactions,19 the initial chlorination of the alcohol with thionyl chloride without the addition of pyridine follows an SNi mechanism with retention of configuration, while the subsequent step, the actual ring closure, resulted in inversion of the stereocenter.19
An alternative method for the amination of purinone derivatives, such as hypoxanthine, inosine, and xanthosine, via silylation and subsequent nucleophilic substitution of the trimethylsilyloxy group has been described in the literature.20 This would allow for the preparation of hydroxyalkylaminopurinones, such as 14a–o, in a one pot procedure starting from the xanthine derivatives. We treated 3,7-dimethylxanthine (theobromine), 3-methylxanthine, and the corresponding 8-phenyl derivatives with hexamethyldisilazane and ethanolamine in the presence of catalyst (p-toluene-sulfonic acid hydrate, ammonium sulfate, or trimethylsilyltriflate, respectively) without or in the presence of solvent (toluene or pyridine) at 120–160 °C for up to 5 days.1 However, no amination was observed. Yields and analytical data for intermediate and final products are collected in Tables 1 and 2.
Determination of Enantiomeric Purity.
Enantiomeric purity has been investigated exemplarily for compounds R-19 and S-19 with 1H nuclear magnetic resonance (NMR) spectroscopy using tris[3-(heptafluorpropylhydroxymethylen)-d-camphorato]europium(III) as a shift reagent.1,21,22 In contrast to the solution of racemate RS-19, only a single enantiomer could be detected in solutions of R-19 and S-19, respectively.1
Because NMR methods may not be sensitive enough for the quantitative determination of enantiomeric purity, we developed a novel capillary electrophoresis (CE) method using cyclodextrins (CDs) as chiral selectors added to the running buffer. The compounds were detected and identified by using a diode array detector scanning their UV spectra from 190 to 400 nm. Race-mates RS-19 and RS-24 were selected for the development of a method for chiral separation. For all separations, a constant current of 90 μA was applied. Two untreated fused silica capillaries, 60 (50 cm effective length) and 50 cm (40 cm effective length), were used for the investigations. Crucial to the new method was the use of two different buffer solutions simultaneously, the chiral selector added to the inlet buffer differing from that in the outlet buffer. While the inlet buffer contained native β-CD, the outlet buffer contained sulfated β-CD (s-β-CD). If native β-CD was used only, a baseline chiral separation could not be achieved. The separations were performed at pH 4.5 (phosphate buffer). The negatively charged s-β-CD molecules are believed to strongly interact with the positively charged chiral compounds. Low concentrations of s-β-CD ranging from 1.5 to 10 mg/mL added only to the outlet buffer were sufficient to obtain excellent baseline separations as shown in Figure 1. This can be explained by the large countercurrent mobility of s-β-CDs.23,24 The optimal concentration for β-CD in the inlet buffer was found to be 6 mM. Higher concentrations of β-CD led to a decrease in resolution. The use of carboxymethyl-β-cyclodextrin (CMCD) instead of s-β-CD resulted in low chiral resolution. A combination of hydroxypropyl-β-CD (HPCD) with s-β-CD in the inlet buffer and s-β-CD in the outlet buffer also gave good results as shown for compound R-24 in Table 3. The investigated enantiomers generally exhibited high purity (98–100%) (Table 3). The chiral separations were powerful enough for injecting very high amounts of concentrated samples. We were able to detect contaminations by the other enantiomer of below 1%, e.g., R-24 contained only 0.32 and 0.29% of the corresponding S enantiomer (Table 3, Figure 1). However, the racemate RS-20 could not be resolved by the new method, possibly due to a methyl group at the chiral center (vs the larger ethyl group in 19 and 24), resulting in smaller sterical differences. Another explanation could be the larger distance of the chiral center (C7 in 20 vs C8 in 19 and 24) from the basic nitrogen atom (N9), which is believed to interact electrostatically with the negatively charged s-β-CD.
Figure 1.

Electropherograms of chiral separation of two chiral compounds. Fused silica capillary 40 cm effective length × 75 μm ID × 375 OD: constant current, 90 μA; injection time, 5 s with 0.5 psi. (A) Racemate RS-19. Inlet buffer: phosphate 50 mM, β-CD 6 mM, pH 4.5; outlet buffer: phosphate 50 mM, sulfated β-CD 10 mg/mL, pH 4.5. (B) Compound S-24. Inlet buffer: phosphate 50 mM, β-CD 6 mM, pH 4.5; outlet buffer: phosphate 50 mM, sulfated β-CD 1.5 mg/mL, pH 4.5.
Table 3.
Determination of Enantiomeric Purity of Selected Compounds by CE Using CDs as Chiral Selectors
| compd | CDa added to inlet bufferb |
CDa added to outlet bufferb |
capillary effective length (cm) |
determined purity ± RSDc(%) |
determined impurityd ± RSD (%) |
|---|---|---|---|---|---|
| R-19 | β-CD (6 mM) | s-β-CD (10 mg/mL) | 40 | 99.23 ± 0.03 (n = 3) | 0.77 ± 0.03 |
| R-24e | β-CD (6 mM) | s-β-CD (1.5 mg/mL) | 40 | 99.68 ± 0.01 (n = 2) | 0.32 + 0.01 |
| R-24e | HPCD (6 mM) + s-β-CD (1.5 mg/mL) | s-β-CD (1.5 mg/mL) | 50 | 99.71 ± 0.04 (n = 5) | 0.29 ± 0.04 |
| S-24 | β-CD (6 mM) | s-β-CD (1.5 mg/mL) | 40 | 97.69 ± 0.04 (n = 3) | 2.31 ± 0.04 |
β-CD, β-cyclodextrin; s-β-CD, sulfated β-cyclodextrin; HPCD, 2-hydroxypropyl-cyclodextrin.
Phosphate buffer, 50 mM, pH 4.5.
RSD = relative standard deviation.
By the other enantiomer.
Two different batches.
Water Solubility and Estimation of pKa Values.
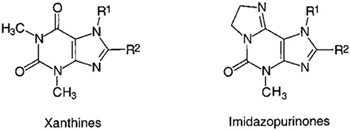
The solubility of two representative imidazo[2,1-i]purin-5-ones was determined by UV spectroscopy.1 In Table 4, solubilities of compounds 17 and 22 are listed. For comparison, solubilities of corresponding xanthine derivatives are included. The introduction of an 8-phenyl group into theophylline results in a drastic reduction of solubility. The effect of an 8-styryl residue is even more dramatic. On the other hand, 7-methyl-substituted xanthines generally display higher water solubility than the corresponding 7-unsubstituted compounds (compare theophylline and caffeine derivatives). The two imidazopurinone derivatives 17 and 22 possess significantly higher water solubility than caffeine and 8-phenylcaffeine, respectively.1 It is expected that the other imidazopurinones also will possess higher water solubility than the corresponding substituted xanthines.
Table 4.
Solubilities of Imidazo[2,1-i]purin-5-one Derivatives in Comparison with Xanthines1
| |||
|---|---|---|---|
| compd | R1 | R2 | water solubility (μM)a |
| Xanthines | |||
| theophylline | H | H | 44 400b,37 |
| 8-phenyltheophylline | H | phenyl | 1038 |
| 8-styryltheophylline | H | styryl | 0.22 |
| caffeine | methyl | H | 108 000b,37 |
| 8-phenylcaffeine | methyl | phenyl | 487 |
| 8-styrylcaffeine | methyl | styryl | 2.8 |
| Imidazopurinonesc | |||
| 17 | methyl | H | 1 210 000 |
| 22 | methyl | phenyl | 1450 |
At 20 °C unless otherwise noted.
At 25 °C.
Hydrochlorides.
To assess whether imidazo[2,1-i]purinone derivatives are protonated at physiologic pH value, UV spectra of 1,4-dimethylimidazo[2,1-i]purin-5-one (17) were recorded at various pH values in steps of one pH unit from pH 2 to pH 13.1 At pH equal to or greater to 8, the compound is present as the free base (UVmax = 287 nm), while at pH values of 7 or lower a bathochromic shift of 8 nm to a UVmax of 295 nm along with a hyperchromic effect is observed (data not shown).1 Therefore, the pKa value can be expected to be around physiologic pH of 7.4, and the compound should be at least partially protonated under physiological conditions.
Biological Evaluation.
All imidazopurinones were investigated in radioligand binding studies at rat brain A1 and A2A ARs using [3H]N6-cyclohexyladenosine (CHA), and [3H]-2-[[[4-(carboxyethyl)phenyl]ethyl]amino]-5′-N-(ethylcarboxamido)]adenosine (CGS21680), respectively, as radioligands.1 Selected compounds were evaluated in AC assays at rat A1 and A2A ARs: Antagonism of R-PIA-elicited inhibition of AC via A1 ARs in rat fat cell membranes and antagonism of NECA-induced stimulation of AC via A2A ARs in rat pheochromocytoma (PC12) cell membranes was measured. A3 AR affinity was determined at recombinant human A3 ARs stably expressed in HEK-293 cells using [125I]N2-methyl-3,4-dihydroxy-5-[6-[3-iodobenzyl)amino]-9H-9-purinyl)-3-tetrahydro-2-furancarboxamide ([125I]AB-MECA) as a radioligand. Activity at A2B ARs was determined by AC assays in NIH 3T3 fibroblast cells (derived from Swiss mouse embryo cells): the inhibition of the NECA-induced stimulation of adenosine cyclic 3′,5′-phosphate (cAMP) accumulation by the imidazopurinones was determined.
Because data from different species (rat, mouse, and human) were compared, species differences have to be taken into account. However, for A1, A2A, and A2B ARs, differences between human and rodent receptors have been shown to be moderate due to high amino acid sequence homologies.2 In contrast, large species differences have been reported between human and rodent A3 ARs, most known A3 antagonists being inactive or only weakly active at rat A3 while exhibiting high affinity for human A3 receptors.2,7
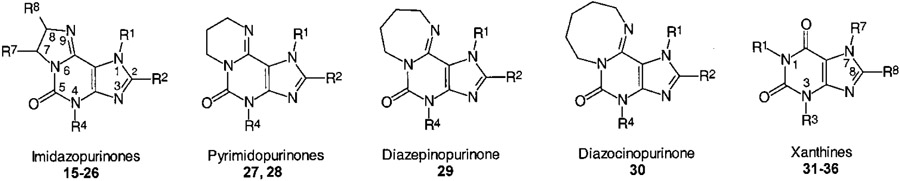
Structure–Activity Relationships (SAR). 2-Unsubstituted Tricyclic Purinone Derivatives.
2-Unsubstituted 1,4-dimethyl-imidazo[2,1-i]purin-5-one (17), corresponding to caffeine (31), was selected as a lead structure.1 Ring enlargement of the dihydroimidazole ring in 17 to the corresponding pyrimido- (27), diazepino- (29), and diazocino- (30) purinone derivatives led to an increase in A1 AR affinity; the optimum was reached with the seven ring compound 29. At A2A ARs, the five, six, and seven ring derivatives (17, 27, and 29) exhibited similar affinities, while the eight ring derivative 30 was considerably weaker. Thus, the imidazopurinone 17 showed the highest degree of A2A selectivity (ca. 4-fold) as compared with the higher homologues and was selected for further modifications.1 If the methyl group in the 4-position was lacking (corresponding to the 3-substituent in xanthines), the A2A selectivity of 17 was lost shown in the marked reduction in A2A affinity in 15. A larger substituent in the 4-position, a benzyl group (compound 16), was well-tolerated by the A2A but less by the A1 ARs. The imidazoline ring positions seven and eight of the lead structure 17 were substituted with methyl or ethyl groups (compounds 18–20) yielding chiral molecules. An R-methyl group in the 7-position (R-20) increased A1 affinity (4-fold) and decreased A2A affinity (2-fold), while the S enantiomer (S-20) showed increased A2A affinity (2-fold) with no change in A1 affinity in comparison with the unsubstituted compound 17. Thus, the R enantiomer (R-20) was somewhat A1 selective and the S enantiomer (S-20) was slightly A2A selective; however, the degree of stereoselectivity was low. A methyl group in the 8-position did not increase but rather decreased AR affinity (compound 18), while an (S)-configurated ethyl group (S-19) increased A2A affinity and thus was favorable for high A2A selectivity.
At human A3 ARs, the lead compound 17 exhibited 40% displacement of radioligand binding at a concentration of 10 μM. Modifications as in compounds 15 (4-unsubstituted), RS-20 (7-methyl), 27 (six ring analogue), 29 (seven ring analogue), and 30 (eight ring analogue) reduced A3 affinity yielding compounds without significant A3 receptor binding at a concentration of 10 μM. Introduction of an ethyl group in the 8-position (RS-19) yielded a compound with low A3 affinity (28% displacement at 100 μM).
2-Substituted Tricyclic Purinone Derivatives.
An increase in AR affinity could be obtained by the introduction of substituents in the 2-position of the tricyclic purinone derivatives (corresponding to the 8-position in xanthines).1 Chlorination of the pyrimidopurinone 27 to its 2-chloro derivative 28 slightly increased both A1 (4-fold) and A2A affinity (2-fold) (Table 5). A phenyl group in the 2-position (compounds 21–23) resulted in a large increase in A1 AR affinity with no major effect on A2A affinity (compare 21 to 15 and 22,23 to 19). Compound 24 lacking a 1-methyl group was particularly potent. None of the compounds were highly selective for A1 receptors. A 2-styryl had about the same effect as a phenyl group at A1 ARs; however, in the case of compound 25, it led to a dramatic increase in affinity for A2A ARs yielding potent A2A antagonists R-25 and S-25 with up to 40-fold selectivity for A2A vs A1 ARs. 2-Styryl-substituted imidazopurinones that were lacking a methyl group in the 1-position (compounds R-26 and S-26) were 20-fold more potent at A1 ARs but exhibited somewhat reduced affinity for A2A ARs as compared to the 1-methyl analogues R-25 and S-25. A similar effect had been observed with 8-styrylxanthine derivatives with regard to methylation in the 7-position.25 It was postulated that a neighboring methyl group might force the styryl residue into the putatively bioactive cisoid conformation.26 For binding to the A1 AR, however, the unsubstituted NH is an important hydrogen bond donor.5,27 For the 2-phenyl imidazopurinones, 1-methyl substitution was highly unfavorable. 2-Phenyl derivatives lacking the 1-methyl group were ca. 40-fold more potent at A1 ARs than their methylated analogues (compare R-23/R-24, S-23/S-24). At the A2A ARs, this effect was less pronounced.
Table 5.
AR Affinities and Selectivities of Tricyclic Purinone Derivatives
|
||||||||
|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R4 | R7 | R8 |
Ki (μM) ± SEM or % displacement at indicated concentration (in brackets) |
||
| A1 affinity rat brain cortical membranes1 [3H]CHA |
A2A affinity rat brain striatal membranes1 [3H]CGS21680 |
A3 affinity human recombinant receptors [125I]AB-MECA |
||||||
| 2-Unsubstituted Imidazopurinones | ||||||||
| 15 | methyl | H | H | H | H | 65.5 ± 7.6 | 40.9 ± 6.3 | >10a |
| 16 | methyl | H | benzyl | H | H | 24% (30 μM) | 16.1 ± 3.8 | ndb |
| 17 | methyl | H | methyl | H | H | 79.6 ± 0.6 | 20.7 ± 3.0 | 40% (10 μM) |
| R-18 | methyl | H | methyl | H | (R)-methyl | 14% (3 μM) | 12% (30 μM) | nd |
| S-18 | methyl | H | methyl | H | (S)-methyl | 10% (10 μM) | 46% (30 μM) | nd |
| RS-19 | methyl | H | methyl | H | (R,S)-ethyl | 46% (250 μM) | 16.2 ± 0.6 | 28% (100 μM) |
| R-19 | methyl | H | methyl | H | (R)-ethyl | 75.5 ± 4.5 | 28.3 ± 5.4 | nd |
| S-19 | methyl | H | methyl | H | (S)-ethyl | 50% (250 μM) | 10.9 ± 1.2 | nd |
| RS-20 | methyl | H | methyl | (R/S)-methyl | H | 59 ± 8 | 64.6 ± 9.8 | >10a |
| R-20 | methyl | H | methyl | (R)-methyl | H | 23 ± 4 | 50.4 ± 13.4 | nd |
| S-20 | methyl | H | methyl | (S)-methyl | H | 70 ± 6 | 12.0 ± 1.9 | nd |
| 2-Substituted Imidazopurinones | ||||||||
| 21 | methyl | phenyl | H | H | H | 1.83 ± 0.9 | 11.9 ± 1.6 | 0.047 ± 0.0048 |
| 22 | methyl | phenyl | methyl | H | H | 9.7 ± 2.6 | 20.3 ± 2.6 | 3.33 ± 0.72 |
| RS-23 | methyl | phenyl | methyl | H | (R,S)-ethyl | 6.1 + 0.8 | 19.2 ± 3.9 | 17% (10 μM) |
| R-23 | methyl | phenyl | methyl | H | (R)-ethyl | 17.5 ± 5.0 | 36.3 ± 1.2 | nd |
| S-23 | methyl | phenyl | methyl | H | (S)-ethyl | 4.7 ± 0.3 | 16.2 ± 4.6 | nd |
| RS-24 | H | phenyl | methyl | H | (R,S)-ethyl | 0.265 ± 0.1 | 3.1 ± 0.5 | nd |
| R-24 (PSB-11) | H | phenyl | methyl | H | (R)-ethyl | 0.44 ± 0.1 | 2.1 ± 0.14 | 0.0023 ± 0.0011 |
| S-24 | H | phenyl | methyl | H | (S)-ethyl | 0.115 + 0.001 | 3.33 ± 0.93 | 0.0098 ± 0.0038 |
| R-25 | methyl | styryl | methyl | H | (R)-ethyl | 21.7 ± 3.8 | 0.547 ± 0.035 | 1.70 ± 0.48 |
| S-25 | methyl | styryl | methyl | H | (S)-ethyl | 14.9 ± 2.6 | 0.424 ± 0.007 | 30.6 ± 8.3 |
| R-26 | H | styryl | methyl | H | (R)-ethyl | 0.95 ± 0.15 | 0.89 ± 0.01 | 0.64 ± 0.199 |
| S-26 | H | styryl | methyl | H | (S)-ethyl | 0.73 ± 0.13 | 0.67 ± 1.73 | >10a |
| Ring-Enlarged Analogues of Imidazopurinones | ||||||||
| 27 | methyl | H | methyl | 43.2 ± 13.4 | 28.5 ± 5.2 | >10a | ||
| 28 | methyl | Cl | methyl | 10.5 ± 6.1 | 11.9 ± 0.87 | nd | ||
| 29 | methyl | H | methyl | 19.8 ± 9.5 | 18.5 ± 1.3 | >10a | ||
| 30 | methyl | H | methyl | 60.3 ± 8.1 | 51 ± 4 | >10a | ||
| Xanthine Derivatives (for Comparison) | ||||||||
| R7 | R8 | R3 | R1 | name | ||||
| 31 | methyl | H | methyl | methyl | caffeine | 4139 | 4339 | 13.37 |
| 32 | methyl | phenyl | methyl | methyl | 8-phenylcaffeine | 1540 | 2540 | nac |
| 33 | H | phenyl | methyl | methyl | 8-phenyltheophylline | 0.08939 | 0.8339 | na |
| 34 | methyl | styryl | methyl | methyl | 8-styrylcaffeine | 3.941 | 0.09441 | na |
| 35 | H | styryl | methyl | methyl | 8-styryltheophylline | 0.6541 | 0.2941 | na |
nd = not determined.
Less than 10% displacement at 10 μM.
Data not available.
The introduction of a 2-phenyl substituent had an even more dramatic effect on A3 affinity than it had on A1 and A2A affinity of imidazopurinone derivatives. The A3 affinity of 1-methylimidazopurinone 15 was increased in the 2-phenyl derivative 21 by greater than 200-fold (Ki A3 = 47 nM, >38-fold selective vs A1, A2A, and A2B ARs). An additional methyl group in the 4-Position of 21 (compound 22) led to a 70-fold reduction in A3 afinity, while A1 affinity was only reduced by 5-fold, and A2A affinity was reduced by 2-fold. An additional ethyl group in the 8-position of the imidazoline ring (compound RS-23) resulted in a further decrease in A3 affinity without much influence on A1 and A2A affinity. However, the corresponding 1-unmethylated 2-phenyl-substituted imidazopurinones (R-24, S-24) were very potent ligands for the human A3 AR exhibiting affinities in the low nanomolar concentration range (R-24/S-24). Compound R-24 was highly selective for A3 ARs vs all other AR subtypes and very potent showing a Ki value of 2.3 nM at human A3 ARs. 2-Styryl substitution (compounds 25 and 26) also increased A3 affinity, however, to a much smaller extent than 2-phenyl substitution.
Stereoselectivity.
Chiral 2-unsubstituted (18–20) and 2-phenyl-substituted imidazopurinones (23 and 24) bearing substituents at the imidazoline ring showed low degrees of enantioselectivity at ARs. High affinity compounds displayed the same low degree of enantioselectivity as compounds with lower AR affinity. 2-Styryl-substituted imidazopurinones (25 and 26) also showed low enantioselectivities at A1 and A2A ARs, but larger differences were observed at A3 receptors (R-25 > S-25, 18-fold; R-26 > S-26, ≫16-fold). Thus, the degree of enantioselectivity (i) depended on the receptor subtype and (ii) was affected by substitution in the 1-position (methyl or hydrogen) and the 2-position (styryl, phenyl, or hydrogen). While the A3 AR preferred the R-configurated enantiomers of 8-substituted derivatives, the A1 AR showed a preference for the S enantiomers in the case of 2-phenyl and 2-styryl derivatives and for the R enantiomers in the case of 2-unsubstituted imidazopurinones. Enantioselectivity of 8-substituted imidazopurinones was lowest at the A2A AR; in most cases, the S enantiomer was slightly more potent than the R enantiomer. One exception was seen with the 2-phenyl derivatives R-24/S-24; in this case, the R enantiomer was only slightly more potent than the S enantiomer. At the A1 ARs, higher (4-fold) and opposite stereoselectivity was observed for the 2-phenyl-substituted imidazopurinone R-24/S-24 (S > R) in comparison with the enantioselectivity at A2A ARs. A2B and A3 ARs both exhibited ca. 4-fold stereoselectivity for the R enantiomer R-24 over the S enantiomer S-24, which was opposite to the results at the A1 AR. Thus, despite only moderate degrees of stereoselectivity of the compounds vs one particular AR subtype, certain pure enantiomers may exhibit a much higher degree of receptor subtype selectivity than their counterparts as shown for the enantiomeric pair R-24/S-24. The S enantiomer was only 12-fold A3 selective vs A1, while the R enantiomer was 190-fold selective for the A3 AR vs A1.
Comparison with Xanthines.
SARs for the various imidazopurinones roughly paralleled those of corresponding xanthine derivates at A1 and A2A ARs. Published data of corresponding xanthine derivatives are included in Table 5 for comparison. The lead structure 17 can be compared with caffeine (31). Larger alkyl substituents (propyl, butyl) in the 1-position of xanthine derivatives may lead to an increase in A1 and A2A affinity,4,5 similarly as observed with increasing ring size in tricyclic compounds (17, 27, and 29). 8-Pheny (33) and 8-styryl (35) substitution in xanthines and 7-methylation of those 8-substituted xanthine derivatives (32 and 34) result in similar effects at A1 and A2A ARs as analogous substitution in the corresponding positions of imidazopurinones. Therefore, it is very likely that the tricyclic purinone derivatives interact with the same binding site of the receptors and in a similar binding mode as the xanthines. A recently published molecular modeling study, in which the A1 selective antagonists KW 3902 (1) and KF 20274 (2) were compared, came to the same conclusion.28 In general, tricyclic imidazopurinone derivatives appeared to be somewhat less potent at A1 and A2A ARs than corresponding xanthine derivatives. Limited data on SARs of xanthine derivatives at A3 ARs preclude a similar conclusion for the A3 receptors at present.
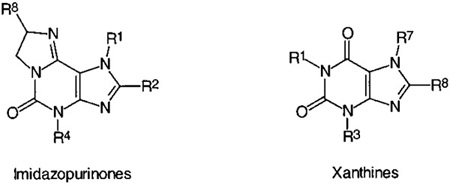
Functional Assays.
A selection of four compounds (21, R-24, S-24, and S-25) was investigated in functional studies at A1, A2A, and A2B ARs. KB values obtained from AC assays are listed in Table 6 in comparison with literature data for selected xanthine derivatives. The imidazopurinones were clearly shown to act as antagonists at ARs. KB values obtained in AC assays correlated well with Ki values from binding studies at A1 and A2A ARs. The investigated 2-phenyl- and 2-styrylimidazopurinones showed moderate antagonist potency at A2B ARs with KB values in micromolar concentrations. Activities at A2A and A2B receptors were similar. Thus, the styryl derivative S-25, which was 35-fold selective for A2A receptors vs A1 and 72-fold selective vs A3 ARs, exhibited only low selectivity vs A2B ARs (4–9-fold). However, compounds 21, R-24, and S-24, that were selective for A3 ARs vs A1 and A2A, were also A3 selective vs A2B ARs.
Table 6.
Functional Antagonist Activity of Selected Imidazopurinones at A1, A2A, and A2B ARs in Comparison with Standard Xanthine Derivatives
|
|||||||
|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R4 | R8 | KB ± SEM (μM) | ||
| A1 | A2A | A2B | |||||
| Imidazopurinones | |||||||
| 21 | methyl | phenyl | H | H | 0.50 ± 0.11 | 17 ± 2 | 7.0 ± 0.3 |
| R-24 | H | phenyl | methyl | (R)-ethyl | 0.43 ± 0.10 | 2.5 ± 0.2 | 2.1 ± 0.3 |
| S-24 | H | phenyl | methyl | (S)-ethyl | 0.063 ± 0.021 | 4.3 ± 0.1 | 7.6 ± 7.3 |
| S-25 | methyl | styryl | methyl | (S)-ethyl | 33 ± 10 | 1.0 ± 0.1 | 3.7 ± 0.3 |
| Xanthines | |||||||
| R7 | R8 | R3 | R1 | ||||
| 11f | methyl | phenyl | H | methyl | 0.32 ± 0.03 | 5.7 ± 0.7 | 5.2 ± 0.9 |
| caffeine | methyl | H | methyl | methyl | 60a | 36 ± 4a | 25 ± 2 |
| theophylline | H | H | methyl | methyl | 8.7a | 13.7 ± 0.4a | 16 ± 8 |
| enprofylline | H | H | propyl | H | 32 ± 2b | 120 ± 17a | 18 ± 4 |
Data from Choi; et al. Life Sci. 1988, 43, 387–398; Hide; et al. Mol. Pharmacol. 1992, 41, 352–359; Ukena; et al. Life Sci. 1986, 39, 743–750; and Brackett; Daly. Biochem. Pharmacol. 1994, 47, 801–804.
Binding data: Rat cerebral cortical membranes, [3H]R-PIA. Müller; et al. J. Med. Chem. 1993, 36, 3341–3349; also, 81 μM. Shamim; et al. J. Med. Chem. 1989, 32, 1231.
Conclusion
The investigation of the SARs of imidazo[2,1-i]purinones and ring-enlarged analogues derived from xanthine derivatives resulted in the development of novel A2A and A3 AR antagonists exhibiting enhanced water solubility in comparison with corresponding xanthine derivatives. 2-Phenyl-substituted R-24 exhibited the highest A3 affinity of the present series with a Ki value of 2.3 nM and 190-fold selectivity vs A1 and greater than 900-fold selectivity vs A2A and A2B ARs. Thus, R-24 will be a useful research tool for future studies at human A3 ARs. 1-Methyl-2-styryl-substituted S-25 was the most potent A2A antagonist with a Ki value of 424 nM, 35- and 72-fold selectivity vs A1 and A3 ARs, respectively, and 4–9-fold selectivity vs A2B receptors. This study provides novel lead compounds for the develoment of A2A and A3 AR selective antagonists, respectively, exhibiting good water solubility, which is an important prerequisite for in vivo activity.
Experimental Section
Synthetic Procedures.1
NMR spectra were performed on a Bruker AC-80 (1H: 80 MHz, 13C: 20 MHz), a Bruker AC200 (1H: 200 MHz, 13C: 50 MHz), a Bruker AC250 (1H: 250, 13C: 60 MHz), or a Bruker DMX 600 (1H: 600 MHz, 13C: 150 MHz) spectrometer. DMSO-d6 was used as solvent. The chemical shifts are reported in parts per million (δ). Signals of the deuterated solvent served as internal standard: δ 1H: 2.50 ppm; 13C: 39.1 ppm. Coupling constants are in Hertz (Hz). UV spectra were recorded on an HP8452A spectrometer equipped with a diode array detector (Hewlett-Packard). All compounds were checked for purity by thin-layer chromatography (TLC) using aluminum sheets with silica gel 60 F254 (Merck). For column chromatography, silica gel (0.05–0.2 mm, Merck) was used. Dry column chromatography was performed using silica gel H according to Stahl (Merck). Enantiomeric purity of compounds R-19 and S-19 was exemplarily determined by 1H NMR spectroscopy in the presence of tris[3-(heptafluorpropylhydroxymethylen)-d-camphorato]europium-(III) as a shift reagent as described.21,22 Only one enantiomer could be detected by this method. Optical rotation was measured with a Perkin-Elmer 241 polarimeter using solutions in methanol (1.00%). The melting points were determined with a Büchi 510 or a Büchi 530 melting point apparatus and are uncorrected. Elemental analyses were performed by the Institute of Chemistry, University of Tübingen, the Institute of Inorganic Chemistry, University of Würzburg, or the Pharmaceutical Institute, Department of Pharmaceutical Chemistry, University of Bonn, respectively. The mass spectra were obtained on a 8200 Finnigan-MAT mass spectrometer.
Only selected 1H NMR data for one representative member of each class of compounds are given. Complete 1H NMR data and selected 13C NMR data are available as Supporting Information. Theobromine (11a) was commercially available. Chiral amino alcohols were purchased from Merck, Aldrich, or Fluka in the highest available purity grade (typically at least 96% ee, usually ca. 99% ee).
6-Amino-1-benzyl-5-nitrosouracil (4a).
6-Amino-1-benzyluracil (3a, 10.85 g, 50 mmol) was suspended in 125 mL of water. Acetic acid (125 mL) was added, and the suspension was heated to 70 °C. Slowly, 6.9 g (100 mmol) of NaNO2 in 22 mL of H2O was added. The suspension was stirred for 1 h at room temperature. After it was cooled to 4 °C, the precipitate was collected by filtration and purified by recystallization from H2O:EtOH (50:50). Yield, 90%. 1H NMR: δ 5.09 (s, 2H, CH2), 7.20–7.40 (m, 5H, ar), 9.13 (s, 1H, N─O─H), 11.68 (s, 1H, N─H), 13.35 (s, 1H, =NH).
6-Amino-1-methyl-5-nitrosouracil (4b).
6-Amino-1-methyluracil (3b, 5.0 g, 36 mmol) was suspended in 150 mL of 50% aqueous acetic acid. The suspension was kept at a temperature of 60 °C until all solid had dissolved. After it was cooled to room temperature, 5 g of NaNO2 (72 mmol) in 20 mL of H2O was added over a period of 30 min. The solution was stirred for 1 h. After it was cooled to 4 °C, the precipitate was collected by filtration, subsequently suspended in H2O, filtered off, and dried. An analytical sample was recrystallized from H2O. Yield, 93%. 1H NMR: δ 3.20 (s, 2H, CH3), 8.94, 11.16 ((2*s, 1H, NH, NOH), 13.11 (s, 1H, N3─H).
6-Amino-1-benzyl-5-N-methylaminouracil (5a) and 6-Amino-5-N-methylamino-1-methyluracil (5b).
To a mixture of 41 mmol of 3a (8.9 g) or 3b (5.78 g), respectively, and 3.44 g (41 mmol) of NaHCO3 in 40 mL of MeOH, 6.55 g (41 mmol) of Br2 was added at 0 °C over a period of 30 min. After it was stirred at room temperature for 30 min, the solution was cooled to 4 °C, and the white precipitate was collected by filtration. The solid was dissolved in 17 mL of aqueous methylamine solution (30%) and stirred at a temperature of 70 °C for a period of 4 h. The methylamine was subsequently removed by distillation. The residue was taken up in EtOH and brought to a pH value of 7 by adding concentrated HCl solution and kept at 4 °C for a period of 5 h. The product was then collected by filtration and purified by recrystallization from H2O (5a) (yield, 75%) or EtOH (5b) (yield, 70%). 1H NMR: 5a δ 2.37 (s, 3H, CH3), 2.82 (s, 1H, NH), 5.08 (s, 2H, CH2), 6.46 (s, 2H, NH2), 7.10–7.40 (m, 5H, ar), 10.47 (s, 1H, NH). 5b: δ 2.36 (s, 3H, CH3), 2.78 (s, 1H, NH), 3.21 (s, 3H, CH3), 6.38 (s, 2H, NH2), 10.48 (s, 1H, NH).
1 N-(6-Amino-1-methyl-2,4-dioxo-1,2,3,4-tetrahydro-5-pyrimidinyl)-1N-methyl-3-phenyl-(E)-2-propenamide (6a).
To a solution of 5a in 40 mL of MeOH, (E)-cinnamic acid (0.72 g, 5.05 mmol) and N-(diethylaminopropyl-N′-ethylcarbodiimide-HCl (EDC) (0.98 g, 5.1 mmol) were added. The mixture was stirred for 10 h. Then, 20 mL of H2O was added, and the mixture was stirred for 3 h. The precipitated product was collected by filtration and purified by dissolving it in 11 mL of dimethylformamide (DMF) and subsequent precipitation by the addition of 30 mL of H2O. Yield, 1.04 g (69%). 1H NMR: δ 2.95 (s, 3H, CH3), 3.27 (s, 3H, CH3), 6.74 (d, 1H, ═CH), 7.06 (s, 2H, NH2), 7.30–7.65 (m, 6H, ar + ═CH), 10.66 (s, 1H, NH).
6-Amino-1-benzyl-5-methyl(phenyl)carboxamido-2,3-dioxo-1,2,3,4-tetrahydropyrimidine (6b) and 6-Amino-1-methyl-5-methyl(phenyl)carboxamido-2,3-dioxo-1,2,3,4-tetrahydro-pyrimidine (6c).
A suspension of 14 mmol of 5a (4.90 g) or 5b (3.84 g) in 30 mL of dry pyridine was cooled to 0 °C. Benzoyl chloride (2.23 mL, 16 mmol) was added upon stirring. The mixture was stirred for 12 h, and then, the solvent was removed by distillation. After 30 mL of H2O was added, the precipitate was filtered off and recrystallized from EtOH/H2O (50/50). The yields amounted to 73% for both compounds. 1H NMR: 6b δ 3.01 (s, 3H, CH3), 4.94 (s, 2H, CH2), 6.79 (s, 2H, NH2), 7.15–7.45 (m, 10H, ar), 10.62 (s, 1H, NH). 6c: δ 2.96 (s, 3H, CH3), 3.33 (s, 3H, CH3), 7.00–7.40 (m, 5H, ar), 10.46 (s, 1H, NH).
5,6-Diamino-1-benzyluracil (7a) and 5,6-Diamino-1-methyluracil (7b).
6-Amino-5-nitrosouracil 4a (2.5 g, 10 mmol) or 4b (1.7 g, 10 mmol) was dissolved in a mixture of 50 mL of 12.5% aqueous ammonia solution. Na2S2O4 (3.5 g, 20 mmol) in 15 mL of H2O was slowly added. The solution was reduced to half of its volume by rotary evaporation and then cooled to 4 °C. The precipitate was filtered off and used for the next reaction without any further purification due to instability.
6-Amino-1-methyl-5-[3-phenyl-(E,2E)-2-propenylidenamino]-1,2,3,4-tetrahydro-2,4-pyrimidindione (8).
1-Methyl-5,6-diaminouracil 7b (3.9 mmol) was dissolved in 30 mL of MeOH and 200 μL (3.5 mmol) of acetic acid. The cinnamic aldehyde (0.48 mL, 3.8 mmol) was added, and the mixture was stirred for 1 h. The product precipitated after the addition of 30 mL of H2O, was filtered off, and was dried over P2O5. Yield, 91%. 1H NMR: 3.34 (s, 3H, CH3), 6.97 (s, 2H, NH2), 6.98 (m, 1H, C═CH), 7.20—7.55 (m, 6H, ar + C═CH), 9.47 (1H, N═CH), 10.71 (s, 1H, NH).
3-Benzyl-7-methyl-8-phenylxanthine (9).
Pyrimidine derivative 6b (5.26 g, 15 mmol) was suspended in a mixture of 26 mL of 2 N NaOH and 8 mL of EtOH. The suspension was refluxed for 1 h, then cooled to 4 °C, and diluted with 34 mL of H2O. The solution was acidified to pH 5 by the addition of acetic acid. The precipitated product was collected by filtration and purified by recrystallization from H2O:EtOH (50:50). Yield, 85%. 1H NMR: δ 3.97 (s, 3H, CH3), 7.20–7.40 (m, 5H, ar), 7.45–7.60 (m, 3H, ar), 7.70—7.9 (m, 2H, ar).
3-Benzyl-7-methylxanthine (10).
6-Amino-1-benzyl-5-methylaminouracil 5a (1.0 g, 4 mmol) was suspended in triethylorthoformate (20 mL) and refluxed for 12 h. After the mixture was cooled to room temperature, the formed precipitate was filtered off and purified by recrystallization from H2O: EtOH (50:50). Yield, 91%. 1H NMR: δ 3.85 (s, 3H, CH3), 5.09 (s, 2H, CH2), 7.10–7.35 (m, 5H, ar), 7.97 (s, 1H, CH), 11.22 (s, 1H, NH).
3-Benzylxanthine (11b).
1-Benzyl-5,6-diaminouracil 7a (6.5 g, 28 mmol) was suspended in 130 mL of triethylorthoformate and refluxed for 8 h. After the mixture was cooled to room temperature, the precipitate was collected by filtration and subsequently recrystallized from H2O:MeOH (50:50). Yield, 90%. 1H NMR: δ 5.14 (s, 2H, CH2), 7.20–7.40 (m, 5H, ar), 8.03 (s, 1H, CH), 11.21 (s, 1H, NH), 13.45 (s, 1H, NH).
7-Methylxanthine (11c).
3-Benzyl-7-methylxanthine (10, 1.54 g, 6 mmol) and dry AlCl3 (1.6 g, 12 mmol) were suspended in 6 mL of dry toluene. The mixture was stirred for 1 h at 70 °C. After it was cooled to room temperature, 30 mL of ice-cold H2O was added over a period of 3 h. After it was stirred for 2 h, the precipitate was collected by filtration and dried at 80 °C. Yield, 75%. 1H NMR: δ 3.83 (s, 3H, CH3), 7.90 (s, 1H, CH), 10.86 (s, 1H, NH), 11.53 (s, 1H, NH).
3-Methyl-8-phenylxanthine (11d).
A suspension of 6-amino-1-methyl-5-nitrosouracil (4b, 8.5 g, 35.1 mmol) and benzylamine hydrochloride (8 g, 55.7 mmol) in 20 mL of benzylamine was heated at 170 °C for 3 h. After it was cooled to room temperature, the suspension was diluted with 40 mL of EtOH. The precipitate was filtered off, suspended in 40 mL of H2O, and stirred for 2 h. The white precipitate was collected by filtration and subsequently recrystallized from acetic acid. The collected product was washed with a large amount of H2O (ca. 200 mL) and dried at 110 °C. Yield, 46%. 1H NMR: δ 3.48 (s, 3H, CH3), 7.40–8.20 (m, 5H, ar).
7-Methyl-8-phenylxanthine (11e).
3-Benzyl-7-methyl-8-phenylxanthine (9, 3.32 g, 10 mmol) was dissolved in 100 mL of dry toluene, BBr3 (10 g, 40 mmol) was added, and the mixture was stirred at 80 °C for 2 h. After the mixture was cooled to room temperature, 30 mL of ice-cold H2O was added over a period of 1 h. The mixture was stirred for 2 h. The product was collected by filtration and purified by recrystallization from H2O. Yield, 80%. 1H NMR: δ 3.94 (s, 3H, CH3), 7.40–7.60 (m, 3H, ar), 7.70–7.90 (m, 2H, ar), 10.90 (s, 1H, NH), 11.60 (s, 1H, NH).
3,7-Dimethyl-8-phenylxanthine (11f).
Compound 6c (1.65 g, 6 mmol) was suspended in 13 mL of 2 N NaOH and 4 mL of EtOH and refluxed for 1 h. The mixture was cooled to 4 °C and diluted with 17 mL of H2O. Acetic acid was added until the product precipitated. The solid was collected by filtration and washed with 20 mL of H2O. The product could be recrystallized from acetic acid. Yield, 85%. 1H NMR: δ 3.39 (s, 3H, CH3), 3.97 (s, 3H, CH3), 7.44–7.83 (m, 5H, ar), 11.16 (s, 1H, NH).
3-Methyl-8-styrylxanthine (11g).
Compound 8 (9.46 g, 35 mmol) was dissolved in 50 mL of thionyl chloride. The solution was stirred for 8 h at room temperature. Excess SOCl2 was removed by vacuum distillation. To the residue, saturated aqueous NaHCO3 solution was added. The product was collected by filtration. Yield, 80%. 1H NMR: δ 3.41 (s, 3H, CH3), 7.03 (d, 1H, C═CH), (7.30—7.70 (m, 6H, ar + C═CH), 11.12 (s, 1H, NH), 13.55 (s, 1H, NH).
3,7-Dimethyl-8-styrylxanthine (11h).
Compound 6a (0.54 g, 1.8 mmol) was dissolved in a mixture of 4 mL of EtOH and 2 mL of 2 N NaOH. The solution was refluxed for 2 h. After it was cooled to room temperature, concentrated HCl solution was added until a pH value of 4 was achieved. The precipitate was collected by filtration and subsequently purified by recrystallization from H2O:EtOH (50:50). Yield, 88%. 1H NMR: δ 3.35 (s, 3H, CH3), 3.85 (s, 3H, CH3), 6.74 (d, 1H, C═CH), 7.35–7.85 (m, 6H, ar + C═CH), 11.10 (s, 1H, NH).
6-Thioxanthines (12a–h). General Procedure.
A mixture of xanthine 10a–h (55.4 mmol) and phosphorus pentasulfide (20 g, 90 mmol) in 200 mL of dry pyridine was refluxed for 8 h. After the mixture was cooled to room temperature, 400 mL of H2O was added over a period of 2 h. The solution was reduced in vacuo to one-third of its volume. The precipitate was collected by filtration and suspended in 100 mL of 2 N NaOH. The solution was filtered, and the product was precipitated by adding dilute HCl solution to achieve a pH value of 4 and collected by filtration. The product can be purified by recrystallization from EtOH in the presence of charcoal. 1H NMR: 12d δ 3.49 (s, 3H, CH3), 7.40–7.60 (m, 3H, ar), 8.10–8.30 (m, 2H, ar).
6-Methylsulfanylxanthines (13a–h). General Procedure.
6-Thioxanthine 12a–h (5 mmol) was suspended in 12.5 mL of 0.5 N NaOH. EtOH (ca. 5 mL) was added until a clear solution was obtained. At room temperature, 7.1—20 mmol (0.44—1.25 mL) of CH3I was added slowly. After it was stirred for 1 h, the product was collected by filtration, washed with H2O (10 mL), and recrystallized from H2O or a mixture of H2O: EtOH (50:50). 1H NMR: 13d δ 2.66 (s, 3H, CH3), 3.55 (s, 3H, CH3), 7.50–7.60 (m, 3H, ar), 8.10–8.30 (m, 2H, ar).
6-(Hydroxyalkylamino)xanthines (14a–o).
6-Methylsulfanylxanthine 13a–h (1 mmol) and 5 mmol of the appropriate amino alcohol in 1 mL of DMSO was heated for 1 h at 150 °C. The solvent and the excess amount of alcohol were removed in vacuo. The workup was done using four different methods: The residue was recrystallized from EtOH (method A); prior to recrystallization, the residue was purified by dry column flash chromatography as previously described29 (method B); or the residue was column chromatographed with CHCl3: methanol (gradient from 9:1 to 3:1) (method D). A fourth method was dissolution of the residue in methanol and acidification to a pH value of ca. 3 by adding concentrated HCl solution. The precipitate was then collected by filtration and recrystallized from EtOH (method C). 1H NMR: R-14i δ 0.95 (t, 3H, CH3), 1.52.1,71 (m, 2H, CH2), 3.35–3.65 (m, 3H, CH2–CH), 3.52 (s, 3H, CH3), 4.2 (vbr, 1H, OH), 7.50–7.70 (m, 3H, ar) 8.05—8.25 (m, 2H, ar), 10.08 (d, 1H, NH).
Tricyclic Purinone Derivatives (15–30).
1-Methyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (15), 4-benzyl-1-methyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-9-ium-5-one chloride (16), 1,4-dimethyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-on chloride (17), 1,4,8-trimethyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (R-18), 1,4,8-trimethyl-(8S)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (S-18), 8-ethyl-1,4-dimethyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (RS-19), 8-ethyl-1,4-dimethyl-(8-R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (R-19), ethyl-1,4-dimethyl-(8-S)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (S-19), 1,4,7-trimethyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (RS-20), 1,4,7-trimethyl-(7R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (R-20), 1,4,7-trimethyl-(7S)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (S-20), 1-methyl-2-phenyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-ium-5-one chloride (21), 1,4-dimethyl-2-phenyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (22), 8-ethyl-1,4-dimethyl-2-phenyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-9-ium-5-on chloride (RS-23), 8-ethyl-1,4-dimethyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-9-ium-5-one chloride (R-23), 8-ethyl-1,4-dimethyl-2-phenyl-(8S)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-9-ium-5-one chloride (S-23), 8-ethyl-4-methyl-2-phenyl-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-9-ium-5-one chloride (RS-24), 8-ethyl-4-methyl-2-phenyl-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-9-ium-5-one chloride (R-24), 8-ethyl-4-methyl-2-phenyl-(8S)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]-purin-9-ium-5-one chloride (S-24), 8-ethyl-1,4-dimethyl-2-[2-phenyl-(E)-1-ethenyl]-(8R)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (R-25), 8-ethyl-1,4-dimethyl-2-[2-phenyl-(E)-1-ethenyl]-(8S)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (S-25), 8-ethyl-4-methyl-2-[2-phenyl-(E)-1-ethenyl]-(8R)-4,5,7,8-tetrahydro1H-imidazo[2,1-i]purin-9-ium-5-one chloride (R-26), 8-ethyl-4-methyl-2-[2-phenyl-(E)-1-ethenyl]-(8S)-4,5,7,8-tetrahydro-1H-imidazo[2,1-i]purin-9-ium-5-one chloride (S-26), 1,4-dimethyl-1,4,5,7,8,9-hexahydropyrimido[2,1-i]-purin-10-ium-5-on chloride (27), 2-chloro-1,4-dimethyl-1,4,5,7,8,9-hexahydropyrimido[2,1-i]purin-10-ium-5-one chloride (28), 1,4-dimethyl-4,5,6,8,9,10-hexahydro-1H-[1,3]diazepino[2,1-i]purin-11-ium-5-One chloride (29), 1,4-dimethyl-1,4,5,7,8,9,10,11-octahydro-[1,3]diazocino[2,1-i]purin-12-ium-5-one chloride (30).
To 1 mL of thionyl chloride (cooled to 0 °C), 0.5 mmol of 6-(hydroxylalkyamino)xanthine (14a–o) was added. The mixture was refluxed for 60 min. Excess thionyl chloride was removed by vacuum distillation. Workup was performed using five different methods.
Method A.
The residue was purified by dry column flash chromatography as previously described29 and subsequently recrystallized from EtOH.
Method B.
To the residue, a saturated solution of NaHCO3 was added and the product was extracted with CHCl3, purified by dry column flash chromatography,29 and subsequently recrystallized from EtOH.
Method C.
The residue was purified by dry column flash chromatography.29 The solvent was removed by rotary evaporation. The residue was suspended in ethyl acetate, and HCl gas was blown through the mixture. The precipitate was collected by filtration and recrystallized from EtOH.
Method D.
The residue was purified by column chromatography using CHCl3:methanol (gradient from 9:1 to 3:1), suspended in CHCl3, filtered off, and recrystallized from EtOH.
Method E.
The residue was purified by column chromatography using CHCl3:methanol (gradient from 9:1 to 3:1) and subsequently recrystallized from EtOH.
The purification method, yield, analytical characterization, mp, and where applicable optical rotation of compounds 15–30 are provided in Table 2. 1H NMR: R-24 δ 0.91 (t, 3H, CH3), 1.20–1.40 (m, 2H, CH2), 3.73 (s, 3H, CH3), 3.70–3.90 (m, 2H, CH2), 4.20–4.40 (m, 1H, CH), 7.30–7.50 (m, 3H, ar), 8.10–8.20 (m, 2H, ar). Additional NMR data are provided in Supporting Information.
Determination of Water Solubility.1
A saturated solution of the test compounds in water was prepared. After the undissolved residue was filtered off, the solution was diluted with water and the UV absorption was determined at λmax. The concentration of the solution was calculated using a previously determined standard calibration curve for each compound.
Chiral Separation and Quantitative Determination of Enantiomeric Purity by CE.
The experiments were performed on a P/ACE CE system MDQ glycoprotein (Beckman Coulter Instruments, Fullerton, CA) equipped with a diode array detection system. The electrophoretic separations were carried out using eCAP untreated fused silica capillaries (60 (50 cm effective length) or 50 cm (40 cm effective length) × 75 μm internal diameter (ID) × 375 μm outside diameter (OD) obtained from Beckman Coulter). The separation was performed using an applied constant current of 90 μA and a data acquisition rate of 8 Hz. Analytes were detected and identified using a diode array detector, which provided UV spectra of the compounds by scanning the absorption from 190 to 400 nm. The CE instrument was fully controlled through a PC, which operated with the analysis software 32 KARAT obtained from Beckman Coulter. The evaluation of the electropherograms was done using the same software. The capillary temperature was kept constant at 25 °C. The temperature of the sample-storing partition was adjusted to 25 °C.
β-CD (28707), sulfated β-CD sodium salt (28248), (2-hydroxypropyl)-β-CD (56332), and carboxymethyl-β-CD (21906) were purchased from Fluka (Buchs, Switzerland). Potassium dihydrogen phosphate for the running buffer was purchased from Kraemer and Martin (Bonn, Germany).
For the preparation of the samples, 10 mM stock solutions in DMSO were diluted 1:100 with distilled water containing 3 mM HCl. Sometimes, higher sample concentrations had to be prepared to make sure that even small amounts of impurities by the undesired enantiomer could be detected.
The capillary was conditioned every day by rinsing it with 0.1 M sodium hydroxide solution for 20 min and with water for 10 min before starting the measurements. Between the runs, the capillary was conditioned by rinsing with 0.1 N HCl solution for 1 min, then with distilled water for 1 min, and subsequently with buffer for 1 min. Sample injections were made hydrodynamically applying 0.5 psi pressure for 5–20 s at the inlet side of the capillary. After the work was completed, the capillary was rinsed with 0.1 M sodium hydroxide solution for 20 min and with water for 10 min followed by drying with air for 3 min before shutting down the instrument. For rinsing procedures, 30 or 40 psi of pressure was applied.
Radioligand Binding Assays.
Inhibition of binding of CHA to A1 ARs of rat cerebral cortical membranes and inhibition of CGS21680 to A2A ARs of rat striatal membranes were assayed as previously described.1,30 2-Chloroadenosine (10 μM for the A1, 20 μM for the A2A assay) was used to define nonspecific binding. Inhibition of the receptor radioligand binding was determined over a range of at least 5—6 concentrations of the compounds in triplicate in at least three separate experiments. The Cheng–Prusoff equation and KD values of 1 nM for [3H]CHA and 14 nM for [3H]CGS21680 were used to calculate Ki values from IC50 values, determined by the nonlinear curve fitting program Graph Pad Prism 2.01 (GraphPad, SanDiego, CA). Binding of [125I]AB-MECA to human A3 ARs stably expressed in HEK-293 cells (Receptor Biology, Inc., Baltimore, MD) was determined as described.31
AC Assays.
Antagonism of R-PIA-elicited inhibition of AC via A1 ARs in rat fat cell membranes and antagonism of NECA-elicited stimulation of AC via A2A ARs in rat PC12 cell membranes were assayed as described.32 Inhibition of NECA-induced stimulation of AC by test compounds was determined at A2B receptors in NIH 3T3 fibroblast cell membranes as described.33 KB values were calculated using the Schild equation, and the ratio of EC50 values for NECA activation or the ratio of IC50 values for R-PIA inhibition were calculated in the presence and absence of antagonist.
Supplementary Material
Acknowledgment.
C.E.M. is grateful for support by the Fonds der Chemischen Industrie.
Footnotes
Supporting Information Available: Additional 1H and 13C NMR data of synthesized compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Significant parts of the present results are based on data from the Ph.D. thesis of J. Hipp1 and are so cited.
References
- (1).Hipp J. Synthese, Eigenschaften, pharmakologische Testung und Struktur-Wirkungsbeziehungen tricyclischer, wasserlöslicher Purinon-Derivate und verwandter Verbindungen als Adenosinrezeptor-Antagonisten (Synthesis, properties, pharmacological testing and structure–activity relationships of tricyclic water-soluble purinone derivatives and related compounds as adenosine receptor antagonists). Dissertation, University of Würzburg, 1997. [Google Scholar]
- (2).Fredholm BB; Arslan G; Halldner L; Kull B; Schulte G; Wasserman W Structure and function of adenosine receptors and their genes. Naunyn-Schmiedberg’s Arch. Pharmacol 2000, 362, 364–374. [DOI] [PubMed] [Google Scholar]
- (3).Poulsen SA; Quinn RJ Adenosine receptors: new opportunities for future drugs. Bioorg. Med. Chem 1998, 6, 619–641. [DOI] [PubMed] [Google Scholar]
- (4).Müller CE; Stein B Adenosine receptor antagonists: structures and potential therapeutic applications. Curr. Pharm. Des 1996, 2, 501–530. [Google Scholar]
- (5).Müller CE A1-Adenosine receptor antagonists. Exp. Opin. Ther. Pat 1997, 7, 419–440. [Google Scholar]
- (6).Müller CE A2A Adenosine receptor antagonists – future drugs for Parkinson’s disease? Drugs Future 2000, 25, 1043–1052. [Google Scholar]
- (7).Müller CE A3 Adenosine receptor antagonists. Mini-Rev. Med. Chem 2001, 1, 433–443. [DOI] [PubMed] [Google Scholar]
- (8).Feoktistov I; Biaggioni I Adenosine A2B receptors. Pharmacol. Rev 1997, 49, 381–402. [PubMed] [Google Scholar]
- (9).Baraldi PG; Cacciari B; Romagnoli R; Merighi S; Varani K; Borea PA; Spalluto G A3 Adenosine receptor ligands: history and perspectives. Med. Res. Rev 2000, 20, 103–128. [DOI] [PubMed] [Google Scholar]
- (10).Klotz K-N Adenosine receptors and their ligands. Naunyn-Schmiedberg’s Arch. Pharmacol 2000, 362, 382–391. [DOI] [PubMed] [Google Scholar]
- (11).Suzuki F; Shimada J; Nonaka H; Ishii A; Shiozaki S; Ichikawa S; Ono E 7,8-Dihydro-8-ethyl-2-(3-noradamantyl)-4-propyl-1H-imidazo[2,1-i]purin-5(4H)-one: a potent and water-soluble adenosine A1 antagonist. J. Med. Chem 1992, 35, 3578–3581. [DOI] [PubMed] [Google Scholar]
- (12).Papesch V; Schroeder EF Synthesis of 1-mono- and 1,3-disubstituted 6-aminouracils. Diuretic activity. J. Org. Chem 1951, 16, 1879–1891. [Google Scholar]
- (13).Tylor EC Jr.; Loux HM; Falco EA; Hitchings GH Pyrimidopteridines by oxidative self-condensation of aminopyrimidines. J. Am. Chem. Soc 1955, 77, 2243–2248. [Google Scholar]
- (14).Senga K; Shimizu K; Nishigaki S Oxidatve cyclization of 6-amino-5-benzylidene-amino-1,3-dimethyluracils with thionyl chloride. A convenient synthesis of 8-substituted theophyllines. Chem. Pharm. Bull 1977, 25, 495–497. [Google Scholar]
- (15).Schroeder EF 5-Halo-6-aminouracils and derivatives. US 1956, 2, 731, 465. [Google Scholar]
- (16).Hutzenlaub W; Pfleiderer W Vereinfachte Synthesen für 7-Methyl-und 1,7-Dimethylxanthin und -harnsäure (Simplified syntheses for 7-methyl- and 1,7-dimethylxanthine and -uric acid). Liebigs Ann. Chem 1979, 1847–1854. [Google Scholar]
- (17).Rybár A; Hesek D; Szemes F; Alföldi J 3,7-Dialkyl-8-alkyl- or -aryl-3,7-dihydropurine-2,6-diones. Collect. Czech. Chem. Commun 1990, 55, 2257–2269. [Google Scholar]
- (18).Shimada J; Kuroda T; Suzuki F A convenient synthesis of tricyclic purine derivatives. J. Heterocycl. Chem 1993, 30, 241–246. [Google Scholar]
- (19).Peet NP; Lentz NL; Sunder S; Dudley MW; Ogden AML Conformationally restrained, chiral (phenylisopropyl)-amino-substituted pyrazolo[3,4-d]pyrimidines and purines with selectivity for adenosine A1 and A2 receptors. J. Med. Chem 1992, 35, 3263–3269. [DOI] [PubMed] [Google Scholar]
- (20).Vorbrüggen H; Krolkiewiecz K Nucleosidsynthesen. XVIII. Aminierung von Heterocyclen. I. Eine neue einfache Synthese von Cytidinen (Nucleoside syntheses. XVIII. Amination of heterocycles. I. A new simple synthesis of cytidines). Liebigs Ann. Chem 1975, 745–761. [Google Scholar]
- (21).Schurig V. Current methods for determination of enantiomeric compositions (part 2): NMR spectroscopy with chiral lanthanide shift reagents. Kontakte 1985, 22–36. [Google Scholar]
- (22).Parker D. NMR determination of enantiomeric purity. Chem. Rev 1991, 91, 1441–1457. [Google Scholar]
- (23).Tait RJ; Thompson DO; Stella VJ; Stobaugh JF Sulfobutyl ether β-cyclodextrin as chiral discriminator for use with capillary electrophoresis. Anal. Chem 1994, 66, 4013–4018. [Google Scholar]
- (24).Christians T; Holzgrabe U Enantioseparation of dihydropyridine derivatives by means of neutral and negatively charged β-cyclodextrin derivatives using capillary electrophoresis. Electrophoresis 2000, 21, 3609–3617. [DOI] [PubMed] [Google Scholar]
- (25).Müller CE; Geis U; Hipp J; Schobert U; Frobenius W; Pawlowski M; Suzuki F; Sandoval-Ramírez J Synthesis and structure–activity relationships of DMPX (3,7-dimethyl-1-propargyl-xanthine) derivatives, A2A-selective adenosine receptor antagonists. J. Med. Chem 1997, 40, 4396–4405. [DOI] [PubMed] [Google Scholar]
- (26).Müller CE; Schobert U; Hipp J; Geis U; Frobenius W; Pawlowski M Configurationally stable analogues of 8-styrylxanthines as A2A-adenosine receptor antagonists. Eur. J. Med. Chem 1997, 32 (7), 709–719. [Google Scholar]
- (27).Müller CE A1 Adenosine receptors and their ligands: overview and recent developments. Farmaco 2001, 56, 77–80. [DOI] [PubMed] [Google Scholar]
- (28).Dooley MJ; Kono M; Suzuki F Conformational search for the N6-substituted adenosine analogues and related adenosine A1 receptor antagonists. Bioorg. Med. Chem 1996, 4, 917–921. [DOI] [PubMed] [Google Scholar]
- (29).Müller CE; Deters D; Dominik A; Pawlowski M Syntheses of Paraxanthine and Isoparaxanthine Analogues (1,7- and 1,9-Substituted Xanthine Derivatives). Synthesis 1998, 1428–1436. [Google Scholar]
- (30).Müller CE; Sauer R; Geis U; Frobenius W; Talik P; Pawlowski M Aza-analogues of 8-styrylxanthines as A2A-adenosine receptor antagonists. Arch. Pharm. Pharm. Med. Chem 1997, 330, 181–189. [DOI] [PubMed] [Google Scholar]
- (31).Kim Y-C; Ji X-D; Jacobson KA Derivatives of the triazoloquinazoline adenosine antagonist (CGS15943) are selective for the human A3 receptor subtype. J. Med. Chem 1996, 39, 4142–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Ukena D; Daly JW; Kirk KL; Jacobson KA Functionalized congeners of 1,3-dipropyl-8-phenylxanthine: potent antagonists for adenosine receptors that modulate membrane adenylate cyclase in pheochormocytoma cells, platelets and fat cells. Life Sci. 1986, 38, 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Brackett LE; Daly JW Functional characterization of the A2b adenosine receptor in NIH 3T3 fibroblasts. Biochem. Pharmacol 1994, 47, 801–814. [DOI] [PubMed] [Google Scholar]
- (34).Neimann Z; Bergmann F Selective removal of the benzyl group from position 3 of purines. Isr. J. Chem 1968, 6, 9–16. [Google Scholar]
- (35).Goldner H; Dietz G; Carstens E Eine neue Xanthin-Synthese (A new xanthine synthesis). Liebigs Ann. Chem 1965, 691, 142–158. [Google Scholar]
- (36).Jacobson KA; Shi D; Gallo-Rodríguez C; Manning M; Müller C; Daly JW; Neumeyer JL; Kiriasis L; Pfleiderer W Effects of trifluoromethyl and other substituents on activity of xanthines at adenosine receptors. J. Med. Chem 1993, 36, 2639–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Paruta AN; Sheth BB Solubility of the xanthines, antipyrine, and several derivatives in syrup vehicles. J. Pharm. Sci 1966, 55, 896. [DOI] [PubMed] [Google Scholar]
- (38).Daly JW; Padgett W; Shamim MT; Butts-Lamb P; Waters J 1,3-Dialkyl-8-(p-sulfophenyl)xanthines: potent water-soluble antagonists for A1- and A2 adenosine receptors. J. Med. Chem 1989, 28, 487–492. [DOI] [PubMed] [Google Scholar]
- (39).Grahner B; Winiwarter S; Lanzner W; Müller CE Synthesis and structure–activity relationships of deazaxanthines: analogues of potent A1- and A2-adenosine receptor antagonists. J. Med. Chem 1994, 37, 1526–1534. [DOI] [PubMed] [Google Scholar]
- (40).Daly JW Analogues of caffeine and theophylline: activity as antagonists at adenosine receptors. In Role of Adenosine and Adenine Nucleotides in the Biological System; Imai S, Nakazawa M, Eds.; Elsevier: Amsterdam, 1991; pp 119–129. [Google Scholar]
- (41).Jacobson KA; Gallo-Rodriguez C; Melman N; Fischer B; Maillard M; van Bergen A; van Galen PJ; Karton Y Structure–activity relationships of 8-styrylxanthines as A2-selective adenosine antagonists. J. Med. Chem 1993, 36, 1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.