

Abstract
INTRODUCTION:
Eukaryotes contain a highly conserved signaling pathway that becomes rapidly activated when adenosine triphosphate (ATP) levels decrease, as happens during conditions of nutrient shortage or mitochondrial dysfunction. The adenosine monophosphate (AMP)–activated protein kinase (AMPK) is activated within minutes of energetic stress and phosphorylates a limited number of substrates to biochemically rewire metabolism from an anabolic state to a catabolic state to restore metabolic homeostasis. AMPK also promotes prolonged metabolic adaptation through transcriptional changes, decreasing biosynthetic genes while increasing expression of genes promoting lysosomal and mitochondrial biogenesis. The transcription factor EB (TFEB) is a well-appreciated effector of AMPK-dependent signals, but many of the molecular details of how AMPK controls these processes remain unknown.
RATIONALE:
The requirement of AMPK and its specific downstream targets that control aspects of the transcriptional adaptation of metabolism remain largely undefined. We performed time courses examining gene expression changes after various mitochondrial stresses in wild-type (WT) or AMPK knockout cells. We hypothesized that a previously described interacting protein of AMPK, folliculin-interacting protein 1 (FNIP1), may be involved in how AMPK promotes increases in gene expression after metabolic stress. FNIP1 forms a complex with the protein folliculin (FLCN), together acting as a guanosine triphosphate (GTP)–activating protein (GAP) for RagC.
The FNIP1-FLCN complex has emerged as an amino acid sensor to the mechanistic target of rapamycin complex 1 (mTORC1), involved in how amino acids control TFEB activation. We therefore examined whether AMPK may regulate FNIP1 to dominantly control TFEB independently of amino acids.
RESULTS:
AMPK was found to govern expression of a core set of genes after various mitochondrial stresses. Hallmark features of this response were activation of TFEB and increases in the transcription of genes specifying lysosomal and mitochondrial biogenesis. AMPK directly phosphorylated five conserved serine residues in FNIP1, suppressing the function of the FLCN-FNIP1 GAP complex, which resulted in dissociation of RagC and mTOR from the lysosome, promoting nuclear translocation of TFEB even in the presence of amino acids. FNIP1 phosphorylation was required for AMPK to activate TFEB and for subsequent increases inperoxisome proliferation–activated receptor gamma coactivator 1-alpha (PGC1α) and estrogen-related receptor alpha (ERRα) mRNAs. Cells in which the five serines in FNIP1 were mutated to alanine were unable to increase lysosomal and mitochondrial gene expression programs after treatment with mitochondrial poisons or AMPK activators despite the presence and normal regulation of all other substrates of AMPK. By contrast, neither AMPK nor its control of FNIP1 were needed for activation of TFEB after amino acid withdrawal, illustrating the specificity to energy-limited conditions.
CONCLUSION:
Our data establish FNIP1 as the long-sought substrate of AMPK that controls TFEB translocation to the nucleus, defining AMPK phosphorylation of FNIP1 as a singular event required for increased lysosomal and mitochondrial gene expression programs after metabolic stresses. This study also illuminates the larger biological question of how mitochondrial damage triggers a temporal response of repair and replacement of damaged mitochondria: Within early hours, AMPK-FNIP1–activated TFEB induces a wave of lysosome and autophagy genes to promote degradation of damaged mitochondria, and a few hours later, TFEB–up-regulated PGC1⍺ and ERR⍺ promote expression of a second wave of genes specifying mitochondrial biogenesis. These insights open therapeutic avenues for several common diseases associated with mitochondrial dysfunction, ranging from neurodegeneration to type 2 diabetes to cancer.
Cells respond to mitochondrial poisons with rapid activation of the adenosine monophosphate–activated protein kinase (AMPK), causing acute metabolic changes through phosphorylation and prolonged adaptation of metabolism through transcriptional effects. Transcription factor EB (TFEB) is a major effector of AMPK that increases expression of lysosome genes in response to energetic stress, but how AMPK activates TFEB remains unresolved. We demonstrate that AMPK directly phosphorylates five conserved serine residues in folliculin-interacting protein 1 (FNIP1), suppressing the function of the folliculin (FLCN)–FNIP1 complex. FNIP1 phosphorylation is required for AMPK to induce nuclear translocation of TFEB and TFEB-dependent increases of peroxisome proliferator–activated receptor gamma coactivator 1-alpha (PGC1α) and estrogen-related receptor alpha (ERRα) messenger RNAs. Thus, mitochondrial damage triggers AMPK-FNIP1–dependent nuclear translocation of TFEB, inducing sequential waves of lysosomal and mitochondrial biogenesis.
Graphical Abstract
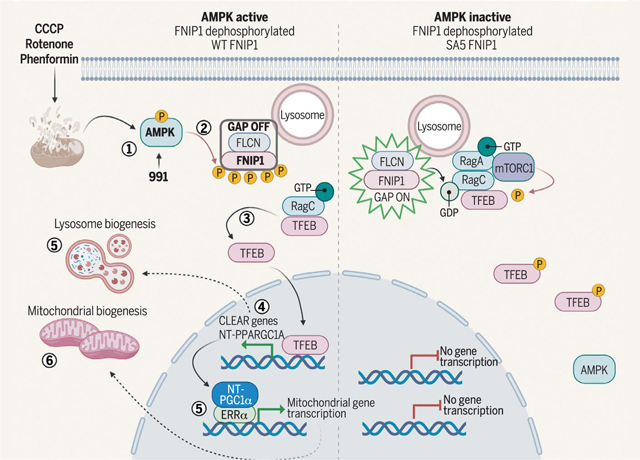
Mitochondrial damage activates AMPK to phosphorylate FNIP1, stimulating TFEB translocation to the nucleus and sequential waves of lysosomal and mitochondrial biogenesis. After mitochondrial damage, activated AMPK phosphorylates FNIP1 (1), causing inhibition of FLCN-FNIP1 GAP activity (2). This leads to accumulation of RagC in its GTP-bound form, causing dissociation of RagC, mTORC1, and TFEB from the lysosome (3). TFEB is therefore not phosphorylated and translocates to the nucleus, inducing transcription of lysosomal or autophagy genes, with parallel increases in NT-PGC1α mRNA (4), which, in concert with ERRα (5), subsequently induces mitochondrial biogenesis (6). CCCP, carbonyl cyanide m-chlorophenylhydrazone; CLEAR, coordinated lysosomal expression and regulation; GDP, guanosine diphosphate; P, phosphorylation. [Figure created using BioRender]
The ability to adapt to prolonged nutrient deprivation is an essential characteristic for survival of all organisms. Eukaryotes contain a highly conserved signaling pathway that becomes rapidly activated when adenosine triphosphate (ATP) levels in the cell decrease, most often as a result of loss of mitochondrial ATP production caused by decreased oxygen or glucose concentrations or in response to mitochondrial poisons that directly interfere with oxidative phosphorylation (OXPHOS) (1). The adenosine monophosphate (AMP)–activated protein kinase (AMPK) becomes activated fully within minutes of OXPHOS inhibition and rapidly phosphorylates a limited number of direct substrates that regulate lipid and glucose metabolism, autophagy, and mechanistic target of rapamycin complex 1 (mTORC1) signaling (2, 3). If energetic stress is prolonged, metabolism is further modified by transcriptional changes to gene expression programs governing distinct metabolic processes (4). The transcription factor EB (TFEB) and related TFE3 are activated in response to nutrient deprivation and energetic stress, and both are suppressed by mTORC1 signaling (5–9) and activated by AMPK signaling (10, 11). mTORC1 directly phosphorylates TFEB on Ser122, Ser142, and Ser211, resulting in its exclusion from the nucleus (6).
Amino acids (AAs) regulate the ability of mTORC1 to phosphorylate TFEB through a guanosine triphosphate (GTP)–activating protein (GAP) complex composed of the folliculin (FLCN) and FLCN-interacting protein 1 (FNIP1) proteins (12–15). The FLCN-FNIP1 complex dictates GTP loading of the guanosine triphosphatase (GTPase) RagC, which results in the release of TFEB and TFE3 from the lysosome and away from mTORC1, causing their nuclear translocation (14). In the nucleus, TFEB and TFE3 directly bind to a well-defined DNA binding element [coordinated lysosomal expression and regulation (CLEAR) motif] found conserved in the proximal promoters of >50 components of the lysosome and a number of autophagy genes (16, 17). In this way, TFEB and TFE3 are held inactive under nutrient-replete conditions, but in response to specific cellular stresses, they translocate into the nucleus and promote lysosomal biogenesis and autophagy (18). AMPK is required for TFEB translocation to the nucleus during energetic stress (10, 11, 19). How AMPK activates TFEB remains unknown, but it is presumed to rely on AMPK-dependent suppression of mTORC1 through its established phosphorylation of the mTORC1 component Raptor and upstream regulator TSC2. We examined the possibility that TFEB and TFE3 were direct substrates of AMPK, but we did not find any evidence supporting this in vivo. Recently, AMPK has been reported to phosphorylate TFEB at three C-terminal residues, and these phosphorylation events may have a role in lysosomal gene transcription (20). However, all three of these tightly clustered sites poorly match the AMPK substrate consensus motif, and mutation of these sites does not disrupt the ability of AMPK to induce nuclear translocation of TFEB and TFE3 (20). Thus, an unknown AMPK-dependent event may govern the translocation of TFEB and TFE3 to the nucleus, without which CLEAR gene transcription cannot occur, even if TFEB and TFE3 are phosphorylated at their C terminus by AMPK-dependent signals.
The precise mechanism of how AMPK activates the transcriptional program of mitochondrial biogenesis is also unresolved. AMPK controls mitochondrial biogenesis and synthesis of peroxisome proliferator–activated receptor gamma coactivator 1-alpha (PGC1α) mRNA in response to energetic stress (21–24). Several mechanisms for how AMPK may promote accumulation of PGC1α mRNA and PGC1α function have been proposed, including control of PGC1α phosphorylation or acetylation (25, 26), but the precise mechanism or mechanisms remain poorly understood. We performed time course analysis of the transcriptional response to mitochondrial OXPHOS inhibitors, revealing a temporal cascade of organellar biogenesis genetically dependent on AMPK. We identified FNIP1 as a direct AMPK substrate whose phosphorylation is critical for TFEB activation and nuclear translocation, which in turn leads to the production of PGC1α and estrogen-related receptor alpha (ERRα) mRNAs, resulting in a wave of lysosomal biogenesis followed by mitochondrial biogenesis.
Electron transport chain inhibitors require AMPK to induce mitochondrial gene transcription
To delineate the role of AMPK in the transcriptional response to mitochondrial energetic stress, we disrupted AMPKα1 and AMPKα2 in human embryonic kidney 293T (HEK293T) cells [AMPK knockout (KO)] by CRISPR-Cas9 and subjected wild-type (WT) control and AMPK KO cells to electron transport chain (ETC) inhibitors, including 100 ng/ml rotenone (complex I), 2 mM phenformin (complex I), and 5 μM CCCP (carbonyl cyanide m-chlorophenyl-hydrazone; a protonophore that disrupts the electrochemical gradient required for ATP production). We performed a time course of RNA sequencing (RNA-seq) of up to 16 hours. Differential expression analysis of all genes in the RNA-seq dataset revealed the gene expression patterns induced by each of the drugs. Differential expression defined by fold change (FC) ≥ 1.3 and P ≤ 0.05 (P values generated from t tests were corrected using the Benjamini-Hochberg method) followed by hierarchical clustering uncovered a set of common genes, whose expression was induced by CCCP, rotenone, and phenformin in WT controls but not in cells lacking AMPK (fig. S1 and Fig. 1A). Approximately one-third of those genes with transcription increased similarly by the three ETC inhibitors in the first 16 hours required AMPK for full gene induction (Fig. 1, A and B). Analysis of this common, AMPK-dependent, and mitochondrial energetic stress–induced gene set at 2- and 4-hour time points—using the Gene Transcription Regulatory Database (GTRD) of chromatin immunoprecipitation sequencing (ChIP-seq) datasets—revealed TFEB to be the most enriched transcription factor (Fig. 1C) based on the binding of transcription factors to the −1000 to +100 bases around the transcription start site of each differentially expressed gene (27).
Fig. 1. Dominant role of AMPK in the transcriptional response to mitochondrial poisons through the MiT-TFE family of transcription factors.

RNA-seq analysis of WT and CRISPR-Cas9–mediated AMPK KO HEK293T cells upon 0- to 16-hour treatment with the mitochondrial poisons CCCP (5 μM), rotenone (100 ng/ml), phenformin (2 mM), and the AMPK-specific activation drug 991 (50 μM). (A) Unbiased heatmap displaying gene expression pattern of all AMPK-dependent, differentially expressed (DE) genes (FC ≥ 1.3, P ≤ 0.05) commonly regulated by all three mitochondrial poisons and 991. (B) Stacked Venn diagram showing the proportion of DE CCCP-induced genes that require AMPK. (C) GSEA analysis shows significantly up-regulated GTRD (ChIP-seq–based Gene Transcription Regulation Database) transcription factor targets upon CCCP treatment. (D) Gene clustering analysis and heat-map displaying the expression pattern of all mitochondria-specific genes as defined by the Mitocarta 3.0 inventory. Right heatmap is a zoomed-in view of the AMPK-dependent mitochondrial genes induced by the four drugs. (E) Overlap in regulation of AMPK-dependent mitochondrial genes in (D) by CCCP, rotenone, phenformin, and 991. (F) Volcano plot depicting DE mitochondrial genes from (D) after 991 compared with DMSO. Red dots represent genes significantly induced by 991 compared with DMSO. The y axis denotes −log10 P values, and the x axis shows log2 FC values. (G) Volcano plot denoting differential expression of mitochondrial genes between WT 16-hour 991-treated cells compared with AMPK KO 16-hour 991-treated cells. Blue dots represent genes significantly down-regulated by AMPK deletion compared with WT AMPK condition. The y axis denotes −log10 P values, and the x axis shows log2 FC values. (H to J) Quantitative RT-PCR (qRT-PCR) for lysosomal gene Lamp2 (H), mitochondrial genes IDH2 (I), and Cox6A1 (J) in WT and AMPK KO HEK293T cells after CCCP. (K to M) qRT-PCR for lysosomal gene Lamp2 (K) and mitochondrial genes IDH2 (L) and ACO2 (M) in WT and AMPK KO HEK293T cells after 991 treatment. All qRT-PCR graphs are shown as the means ± SEMs. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; unpaired t test. (N) Analysis of AMPK signaling and TFEB protein immunoblotting of WT and AMPK KO HEK293T cells treated with DMSO, 991, or phenformin for 1 hour. (O) Analysis of AMPK, TFEB, and mitochondrial protein immunoblotting of WT and AMPK KO HEK293T cells treated with a rotenone (100 ng/ml) time course, ranging from 0 to 24 hours. (P) Analysis of AMPK, TFEB, and mitochondrial protein immunoblotting of WT and AMPK KO HEK293T cells treated with a 991 (50 μM) time course, ranging from 0 to 24 hours. (Q) Analysis of TFEB and TFE3 protein cytoplasm-to-nuclear shuttling by fractionation of WT and AMPK KO HEK293T cells with or without 1-hour 991 treatment, followed by immunoblotting. (R) Quantification of TFEB colocalization with DAPI-stained nuclei. Data are shown as the means ± SEMs of three independent experiments. *P < 0.05; **P < 0.01; unpaired t test. (S) Representative images from immunofluorescence microscopy of endogenous TFEB stained in WT and AMPK KO HEK293T cells, pretreated with 1 hour of DMSO or 991. Nuclei are stained with DAPI. (T) Model. After ETC poisons or the direct small-molecule activator 991, AMPK becomes activated and, through an unknown mechanism, triggers TFEB translocation to the nucleus, where TFEB induces expression of lysosomal and mitochondrial genes.
Volcano plots illustrating genes with the greatest FC and highest degree of statistical significance revealed core components of lysosomes and mitochondria as some of the most increased genes in an AMPK-dependent manner in cells exposed to CCCP for 16 hours (fig. S2, A and B); for example, EPDR1 (ependymin related 1) is a lysosomal protein that binds gangliosides, OXCT1 [3-oxoacid coenzyme A (CoA)–transferase 1] encodes a mitochondrial enzyme in ketone body catabolism, and ACSS3 (acyl-CoA synthetase short chain family member 3) encodes a mitochondrial enzyme in fatty acid oxidation. Gene ontology (GO) enrichment analysis on AMPK-responsive genes in cells treated with CCCP revealed that “lysosomal lumen” and “secretory granule lumen” were among the most enriched GO organelle terms of the AMPK-dependent, CCCP-induced gene set, and “Electron Transport Chain (OXPHOS)” was the second-most enriched WikiPathways term (fig. S2C). Gene set enrichment analysis (GSEA) of the CCCP-induced RNA-seq dataset confirmed enrichment of “KEGG Lysosome” and “Hallmarks OXPHOS” gene sets in WT cells but not in cells lacking AMPK treated with CCCP for 16 hours (fig. S2D). Parallel analysis of the genes differentially expressed in response to rotenone in an AMPK-dependent manner revealed similar enrichment in GO for lysosomes and enrichment for mitochondrial processes in WikiPathways analyses, again with statistically significant enrichment for the “KEGG Lysosome” and “Hallmarks OXPHOS” gene sets (fig. S2, E and F). In addition to observing a common transcriptional response between CCCP, rotenone, and phenformin, we also found that the synthetic small-molecule, direct AMPK activator 991 (28) similarly enriches for the same lysosomal genes and mitochondrial targets (fig. S2, G and H). Transcription factor enrichment analysis of 991-induced genes in the WT condition identified ESRRA as a transcription factor, whose targets are overrepresented in the up-regulated genes (fig. S2I). The estrogen-related receptor alpha gene, ESRRA, which encodes ERRα, is a known key mediator of mitochondrial biogenesis (29, 30).
Having observed a strong lysosomal and mitochondrial component to the common differentially expressed genes upon treatment with all four drugs, we next examined mitochondrial genes more comprehensively. We reanalyzed our data, using the MitoCarta version 3.0, a curated catalog of ~1000 genes encoding the mammalian mitochondrial proteome (31), to assess mitochondrial biogenesis. There was significant overlap in the mitochondrial genes increased after CCCP, rotenone, and CCCP with those increased by 991 (Fig. 1, D and E). The analysis revealed increased transcription of ~300 mitochondrial genes in WT cells treated with ETC poisons or small-molecule, AMPK activator 991 but not cells lacking AMPK (Fig. 1, D, F, and G). Despite the differences in the mechanisms by which these compounds inhibit the ETC, this analysis indicates that a large proportion of the transcriptional response mediated by ETC poisons requires AMPK activation. We validated the increased expression of core lysosomal and mitochondrial genes, including LAMP2 (Fig. 1H), IDH2 (Fig. 1I), Cox6A1, or ACO2 (Fig. 1J), in response to CCCP and 991 (Fig. 1, K to M), all of which showed increased expression in WT cells but not those lacking AMPK when assessed by quantitative polymerase chain reaction (qPCR).
Given the prominent role for TFEB implied by the regulation of genes induced by mitochondrial poisons (Fig. 1C) and TFEB’s major role in AMPK-dependent effects on transcription (9, 10, 17), we examined the regulation of TFEB protein in WT or AMPK KO cells treated with mitochondrial poisons. TFEB isolated from 991-(1 hour) or phenformin-(1 hour) treated WT cells showed a reduced mobility band-shift of endogenous TFEB, which did not occur in TFEB from AMPK KO cells (Fig. 1N). We examined TFEB electrophoretic mobility over longer time courses of treatment with rotenone and 991 and again observed a persistent downward mobility shift in WT cells between 2 and 24 hours after treatment, which was barely detectable in the AMPK KO cells (Fig. 1, O and P). We examined protein abundance of some mitochondrial targets implicated by RNA-seq or qPCR and observed up-regulation of PDHA1 and IDH2 after 991 or rotenone, which was not observed in AMPK KO cells (Fig. 1, O and P).
We assessed the localization of TFEB and TFE3 through nucleocytoplasmic fractionation. In WT cells, 991 treatment led to increased abundance of TFEB and TFE3 in the nuclear fraction with almost none remaining in the cytoplasmic compartment (Fig. 1Q). In cells lacking AMPK, 991 treatment did not lead to translocation of TFEB or TFE3 to the nuclear fractions. Immunofluorescence microscopy of intracellular TFEB with an antibody that detects endogenous TFEB confirmed these results. In WT cells under basal conditions, TFEB was mostly in the cytoplasm with low amounts in the nucleus (Fig. 1, R and S). Upon AMPK activation by 991, conditions under which TFEB is fully dephosphorylated, almost all TFEB translocated to the nucleus. Conversely, in AMPK KO cells, with or without 991, most TFEB remained in the cytoplasm (Fig. 1, R and S). These results indicate that TFEB is a major effector of AMPK after mitochondrial energetic stress (Fig. 1T).
FNIP1 is a conserved AMPK substrate that governs TFEB phosphorylation status and localization
A screen we performed looking for AMPK substrates that could mediate cell growth and metabolism (32) identified FNIP1. FNIP1 is an established interacting partner of FLCN and has been reported to coimmunoprecipitate with AMPK and be regulated by AMPK through unknown details (33, 34). The FNIP1-FLCN complex has emerged as an AA sensor to mTORC1 (11–14), involved in how AAs control TFEB activation (14). We therefore examined whether AMPK may regulate FNIP1 to control TFEB independently of AAs. Analysis of the FNIP1 protein sequence revealed four sites that match the optimal AMPK substrate motif (Ser230, Ser232, Ser261, and Ser593) (Fig. 2A). We used mass spectrometry (MS) to examine the phosphorylation of FNIP1 in vivo. We transfected HEK293T cells with an epitope-tagged FNIP1 cDNA, and the cells treated with dimethyl sulfoxide (DMSO) or phenformin. We detected peptides spanning all four (Ser230, Ser232, Ser261, and Ser593) of these FNIP1 candidate sites (Fig. 2B), phosphorylation of which had increased in the phenformin-treated samples. MS analysis detected a fifth site, Ser220. This also appeared to be highly phosphorylated after AMPK activation and largely conforms to the optimal motif (Fig. 2A), indicating that it too may be a site of AMPK phosphorylation (Fig. 2B).
Fig. 2. FNIP1 as a conserved AMPK substrate that regulates TFEB and TFE3.

(A) ClustalW alignment of five conserved AMPK phosphorylation sites on FNIP1 matching the AMPK substrate consensus motif. (B) AMPK phosphorylation sites on FNIP1 identified by MS in HEK293T cells, expressing FLAG-tagged FNIP1 cDNA, after treatment with vehicle or 1 hour of phenformin. (C) In vitro AMPK kinase assay. WT FNIP1 or FNIP1 mutants (SA2, SA3, SA4, SA5) were immunoprecipitated and incubated with recombinant active AMPK and [γ32P]-ATP. Kinase reactions were separated by SDS-PAGE and incorporation of [γ32P]-ATP was detected by autoradiography. (D) Immunoblot showing endogenous TFEB and FNIP1 (Ser220) phosphorylation status in WT (+/+) or AMPK DKO (−/−) MEFs after DMSO or a 991 time course, as detected by an antibody to FNIP1 P-Ser220. (E) Immunoblots of murine liver lysates from inducible KO of AMPKα1 and α2 (Alb-CreERT2; Prkaa1fl/fl; Prkaa2fl/fl) mice (AMPK KO) or control mice treated with vehicle or AMPK activator compound MK8722 for 2 hours, showing endogenous FNIP1 and TFEB phosphorylation status. (F) Immunoblots of lysates from primary mouse hepatocytes Prkaa2fl/fl) mice (AMPK liver DKO), treated with vehicle or metformin for 5 hours. (G) Immunoblots of endogenous pFNIP1 Ser220 and TFEB in in FNIP1 KO HEK293T cells stably reconstituted with WT, SA4, or SA5 FNIP1, treated with a 991 (50 μM) time course. (H) Nucleocytoplasmic fractionation of WT or SA5 FNIP1 HEK293T cells after 1-hour DMSO or 991 treatment, followed by immunoblotting. (I) Quantitation of TFEB colocalization with DAPI-stained nuclei in (J). Data are shown as the means ± SEMs of three independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; unpaired t test. (J) Representative immunofluorescence images of endogenous TFEB stained in WT FNIP1 and SA5-FNIP1 HEK293T cells, treated with 1-hour DMSO or 991. Nuclei are stained with DAPI.
To test whether FNIP1 is a direct substrate of AMPK, we undertook an in vitro 32P-γ-MgATP phosphorylation assay. We immunopurified FLAG epitope–tagged WT FNIP1 or FNIP1 S-A mutant proteins (Fig. 2C) after transient transfection of cDNA into HEK293T cells. Recombinant AMPK phosphorylated WT FNIP1 and mutation of the five candidate AMPK-site serines (Ser230, Ser232, Ser261, Ser593, and Ser220) showed the greatest effect, seen when all five sites were mutated (hereafter referred to as the SA5 mutant) (Fig. 2C). Notably, variability in the behavior and lack of effect on overall 32P incorporation of some mutants in the in vitro phosphorylation assay leaves the possibility open that some of the sites are not directly regulated by AMPK or are redundantly phosphorylated, masking the impact of loss of one or two sites as judged by overall 32P incorporation in vitro. To verify FNIP1 as an in vivo substrate of AMPK, we generated a phosphor-specific antibody, targeting the pFNIP1 Ser220 site. To validate the Ser220 antibody, we used HEK293T cells lacking FNIP1 generated by CRISPR-Cas9 methodology (fig. S3A) and then transiently transfected those cells with cDNA for WT FNIP1, S220A, SA3 (S-A mutations of Ser230, Ser232, and Ser261), SA4 (S-A mutations of Ser230, Ser232, Ser261, and Ser593), and SA5 FNIP1 mutants. These cells were treated with either DMSO or 991, and then phosphorylation status of FNIP1 was assessed with the pFNIP1 Ser220 antibody. The antibody detected phosphorylation of the S220 site in WT FNIP1, SA3, and SA4 FNIP1 conditions but not in the S220A or SA5 condition, in which the Ser220 site had been mutated to alanine (fig. S3B). A phospho-specific antibody was also generated to the Ser261 site, which was only effective after FNIP1 immunoprecipitation; nonetheless, we observed increased Ser261 phosphorylation in cells overexpressing WT FNIP1 but not SA5 FNIP1, in which Ser261 is mutated to alanine (fig. S3C).
To assess phosphorylation of endogenous FNIP1, we treated mouse embryonic fibroblasts (MEFs) with 991. Endogenous FNIP1 was robustly phosphorylated at the Ser220 site within 30 min, whereas TFEB and TFE3 were dephosphorylated under the same conditions in WT but not AMPK KO MEFs (Fig. 2D). We also examined phosphorylation of endogenous FNIP1 in liver lysates of mice after treatment of mice with MK-8722, an orally available 991 analog that also activates AMPK (35). We detected Ser220 phosphorylation of FNIP1 in livers from WT mice treated with MK-8722 but not in livers from AMPKa1/a2 (Prkaa1l/l; Prkaa2l/l) liver-specific KO mice (Fig. 2E). Other AMPK substrates, such as pRaptor Ser792, were also not phosphorylated in these mice. TFEB and TFE3 were dephosphorylated in livers from WT animals treated with MK-8722 but not in livers lacking AMPK (Fig. 2E). Treatment of primary hepatocytes with metformin induced phosphorylation of endogenous FNIP1 at Ser220 in WT hepatocytes but not hepatocytes lacking AMPK (Fig. 2F). TFEB and TFE3 became dephosphorylated in WT hepatocytes treated with metformin but remained phosphorylated in hepatocytes lacking AMPK, even after metformin administration (Fig. 2F). Thus, FNIP1 appears to be a bona fide, in vitro and in vivo substrate of AMPK, phosphorylated upon direct AMPK stimulation and in response to mitochondrial energetic stress.
Given that both mTOR and AMPK antagonistically regulate TFEB and TFE3 transcription factors and that both pathways converge on the FNIP1-FLCN complex, phosphorylation of FNIP1 by AMPK could represent the dominant mechanism through which AMPK controls these transcription factors. To test for such a role for FNIP1, we used lentivirus to stably reconstitute cells depleted of FNIP1 with full-length cDNA encoding either WT FNIP1, SA4 FNIP1, or SA5 FNIP1 and treated these cells with 991 (Fig. 2G) or phenformin (fig. S3D). In WT FNIP1 cells, AMPK activation led to dephosphorylation of TFEB and TFE3 within 30 to 60 min of 991 or phenformin addition (Fig. 2G and fig. S3D). However, in the SA4 and SA5 FNIP1 cells, TFEB and TFE3 remained in the slow-mobility hyperphosphorylated form whether or not cells were treated with 991 or phenformin (Fig. 2G and fig. S3D). This effect was further enhanced when all five AMPK sites on FNIP1 were mutated. TFEB was fully phosphorylated in FNIP1 SA5 cells, even in the presence of phenformin or 991 (Fig. 2G and fig. S3D). In our prior studies, AMPK promoted activation of TFEB in cells deprived of glucose, but the mechanism was unknown (11). To test whether AMPK control of FNIP1 was involved in the regulation of TFEB after glucose starvation, we examined TFEB in WT FNIP1 and SA5 FNIP1 cells deprived of glucose. Six hours of glucose deprivation activated AMPK, leading to FNIP1 Ser220 phosphorylation in WT FNIP1 cells and TFEB dephosphorylation, as did treatment with 991 (fig. S3E). Conversely in SA5 cells, mutation of AMPK phosphosites on FNIP1 prevented TFEB dephosphorylation induced by glucose starvation (fig. S3E). Furthermore, given the importance of Ser220 as the one serine different between SA4 and SA5 and the apparent in vitro phosphorylation of Ser593 (Fig. 2C), we examined the effect of a Ser220-Ser593 AA mutant (SA2), which revealed incomplete regulation of TFEB, again implying a critical role for all five serines, including 230, 232, and 261 (fig. S3F).
If AMPK phosphorylation of FNIP1 is necessary for TFEB dephosphorylation, localization of TFEB and TFE3 might be expected to change upon FNIP1 phosphorylation. To test this, we isolated nuclear and cytoplasmic fractions from WT FNIP1 cells treated with 991. In contrast to the control, after 991, TFEB was enriched in the nucleus with little cytoplasmic TFEB detected (Fig. 2H). However, after 991 treatment in SA5 FNIP1 cells, most TFEB remained in the cytoplasm (Fig. 2H). Immunofluorescence microscopy to visualize endogenous TFEB and TFE3 confirmed this observation. In WT FNIP1 cells in fresh media, both endogenous TFEB (Fig. 2, I and J) and endogenous TFE3 (fig. S3, G and H) displayed a fully cytoplasmic localization, but upon 991 treatment, both translocated to the nucleus despite the presence of full AAs. Conversely, in SA5 FNIP1 cells, AMPK activation by 991 did not cause translocation of TFEB or TFE3 to the nucleus, and both were primarily cytoplasmic (Fig. 2, I and J, and fig. S3, G and H). Thus, phosphorylation of FNIP1 by AMPK appears to dominantly govern localization of TFEB (fig. S3I).
AMPK-dependent phosphorylation of FNIP1 controls mTORC1 binding to TFEB and TFE3
Although TFEB is phosphorylated by extracellular signal–regulated kinase 1 (ERK), glycogen synthase kinase-3 (GSK3), and protein kinase B (PKB or Akt), much regulation of TFEB is thought to be through phosphorylation of Ser122 and Ser211 by mTORC1 (6). To delineate TFEB regulation by AMPK versus mTORC1, we treated WT HEK293T, AMPK KO HEK293T, WT FNIP1, and SA5 FNIP1 HEK293T cells with 991. TFEB and TFE3 became dephosphorylated in WT cells and WT FNIP1 cells within 30 min but remained highly phosphorylated in AMPK KO and SA5 FNIP1 cells (Fig. 3, A and B). mTORC1 signaling was attenuated by 991 in WT HEK293T and WT FNIP1 HEK293T cells within 30 min of treatment, as reflected by the decreased phosphorylation of the direct mTORC1 substrates eukaryotic translation initiation factor 4E (eIF4E)–binding protein 1 (4EBP1) and ribosomal protein S6 kinase beta-1 (P70S6K), and in turn, the P70S6K substrate ribosomal protein S6 (S6). By contrast, in AMPK KO HEK293T cells, mTORC1 signaling did not change with 991 treatment, because of the absence of AMPK (Fig. 3A). However, in both WT and SA5 FNIP1 cells, canonical mTORC1 signaling to P-S6K and 4EBP1 was suppressed upon AMPK activation with 991, whereas TFEB and TFE3 remained fully phosphorylated in the SA5 cells (Fig. 3B). This revealed a disconnect where FNIP1 modification affected mTORC1-dependent phosphorylation of TFEB but not mTORC1 phosphorylation of S6K1 and 4EBP1, consistent with multiple recent studies (15, 36, 37).
Fig. 3. AMPK phosphorylation of FNIP1 blocks FLCN-FNIP1 GAP activity to control RagC and thereby TFEB and TFE3 activity.

(A) WT and AMPK KO HEK293T cells were subjected to a short 991 (50 μM) time course, and lysates were immunoblotted with the indicated antibodies to probe for mTORC1 signaling. (B) WT FNIP1 and SA5 FNIP1 HEK293T cells were treated as in (A), and lysates were immunoblotted to probe for mTORC1 signaling. (C) WT parental or AMPK-null HEK29T cells were AA starved for the indicated times, and lysates were immunoblotted to examine mTORC1 signaling. (D) WT FNIP1 or SA5 FNIP1 HEK293T cells were administered with the mTOR inhibitors AZD8055 or Torin1 either individually or in combination with 991, as indicated. Lysates were subsequently immunoblotted to examine TFEB phosphorylation status. (E) WT or SA5 FNIP1 cells, stably expressing GFP-TFEB cDNA were treated with or without 50-μM 991 for 1 hour. GFP-TFEB was immunoprecipitated from the lysates, and immunoprecipitates were analyzed by Western blotting. (F) Immunoprecipitates of GFP-TFEB, stably expressed in WT FNIP1 and SA5 FNIP1 HEK293T cells, were subjected to immunoblotting to probe interactions with the Rag GTPases. (G) Quantitation of RagC colocalization with Lamp2 in (H). Data are shown as the means ± SEMs of three independent experiments. *P < 0.05; **P < 0.01; unpaired t test. (H) Representative immunofluorescence images of endogenous RagC costained with Lamp2 in WT FNIP1 and SA5 FNIP1 HEK293T cells treated with 1 hour of DMSO or 991 (50 μM). (I) WT FNIP1 or SA5 FNIP1 cells were transiently transfected with HA-RagC mutants locked in either the GTP-bound state (Q120L) or the GDP-bound state (S75N) and subsequently treated with or without 1-hour 991. Lysates were immunoblotted with the indicated antibodies. (J) HA-RagC mutants from (I) were immunoprecipitated from lysates using HA magnetic beads and immunoprecipitates analyzed by Western blotting.
We examined the effect of AA abundance on TFEB phosphorylation status and mTORC1 activity in the context of our FNIP1 phosphorylation site mutants. mTORC1 signaling, as reflected by phosphorylation of p70S6K, S6, and 4EBP1, was decreased in both WT and AMPK KO HEK293T cells deprived of AAs (Fig. 3C). In cells deprived of AAs, TFEB and TFE3 became dephosphorylated in both WT and AMPK KO cells (Fig. 3C), contrasting with cells treated with 991, where TFEB and TFE3 became dephosphorylated in WT but not AMPK KO HEK293T cells (Fig. 3A). Similarly, mTORC1 signaling was decreased after AA deprivation in both WT FNIP1 and SA5 FNIP1 cells. TFEB and TFE3 also became dephosphorylated when both WT FNIP1 and SA5 FNIP1 cells were deprived of AAs for 1 hour (fig. S4A), whereas activation of AMPK with 991 caused TFEB to be dephosphorylated in WT FNIP1 cells but not SA5 FNIP1 cells. These findings reveal that AMPK-FNIP1–mediated control of the micropthalmia–transcription factor E (MiT-TFE) family of transcription factors through mTORC1 is distinct from that caused by a shortage of AAs. However, dephosphorylation of TFEB and TFE3 dephosphorylation in the SA5 FNIP1 cells deprived of AAs was slower than that observed in WT FNIP1 cells, which demonstrates the inherent control that FNIP1 exerts on TFEB and TFE3 in general (fig. S4A).
To clarify whether TFEB phosphorylation remaining in SA5 cells after AMPK activation was from uninhibited mTORC1-dependent phosphorylation or an unknown kinase, we treated WT and SA5 FNIP1 cells with two potent and selective inhibitors of mTOR, AZD8055 and Torin1, with or without 991. Both AZD8055 and Torin1 decreased TFEB phosphorylation in cells expressing SA5 FNIP1, demonstrating that the TFEB hyperphosphorylation observed in these cells was indeed a result of mTORC1 (Fig. 3D). Although phosphorylation of other mTOR substrates, such as S6K1 or 4EBP1, were not affected by mutation of AMPK sites on FNIP1, TFEB specifically was highly regulated by the AMPK-dependent phosphorylation of FNIP1. To further understand the regulation of TFEB in this context, we generated WT FNIP1 and SA5 FNIP1 HEK293T cells that stably express TFEB tagged with green fluorescent protein (GFP) and subjected these cells to 991 treatment (Fig. 3E). GFP-TFEB immunoprecipitates from WT FNIP1 cells revealed that AMPK activation by 991 caused TFEB dissociation from mTOR and Raptor. Conversely in SA5-FNIP1 cells, 991 did not have this effect, and mTOR and Raptor remained associated to GFP-TFEB (Fig. 3E). Although mTORC1 signaling to S6K1 and 4EBP1 is decreased by AMPK activation in SA5-FNIP1 cells, it appears that TFEB is constitutively phosphorylated in cells expressing SA5 FNIP1 treated with 991 because of mTORC1 remaining associated with TFEB.
AMPK phosphorylation of FNIP1 inhibits FNIP1-FLCN GAP activity to control TFE transcription factors through RagC
In cells stimulated with AAs, the Rag GTPases recruit mTORC1 to the lysosomal surface, where it is activated. The Rag proteins function as heterodimers, in which the active complex consists of GTP-bound RagA or RagB in complex with guanosine diphosphate (GDP)–bound RagC or RagD (38, 39). Activation of mTORC1 by intracellular AAs occurs because AAs stimulate GTP binding to RagA and RagB, promoting binding to Raptor and assembly of the activated mTORC1 complex (40). In the absence of AAs, the Rags take up an inactive conformation (GDP-bound RagA or B and GTP-bound RagC or D), causing inactivation and relocalization of mTORC1 to the cytosol. Concordantly, active Rag heterodimers interact with and promote recruitment of TFEB to the lysosomes, leading to mTORC1-dependent phosphorylation and retention of TFEB in the cytosol. Depletion or inactivation of Rags prevents recruitment of TFEB to lysosomes (5, 7, 9). Furthermore, a catalytic arginine in FLCN is required for AA-dependent translocation of TFEB and TFE3, connecting control of FLCN-FNIP1 GAP activity to AA regulation of RagC (15). To further delineate how FNIP1 phosphorylation by AMPK controls TFEB, we treated the WT FNIP1 and SA5 FNIP1 cells stably expressing GFP-TFEB with 991 and immunoprecipitated GFP-TFEB. Within 10 min, interaction of endogenous RagA and especially endogenous RagC interaction with GFP-TFEB was enhanced and maintained for the duration of the treatment (Fig. 3F). Conversely in SA5 FNIP1 cells, RagA and RagC interactions with TFEB did not change after treatment with 991 (Fig. 3F). RagB binding to TFEB was minimal and did not change between WT and SA5 FNIP1 cells or with 991 treatment.
To determine how the FLCN-FNIP1–RagA-C complex controls TFEB after AMPK activation, we tested whether components of the TFEB regulatory machinery changed localization at the lysosome in cells exposed to 991. We used Lyso-IP, a method for the rapid isolation of mammalian lysosomes (41), which uses expression of lysosomal transmembrane protein 192 fused to three tandem hemagglutinin (HA) tags (HA-TMEM192) in WT FNIP1 and SA5 FNIP1 HEK293T cells (fig. S4B). As controls, we also expressed FLAG-TMEM192 in WT FNIP1 and SA5 FNIP1 HEK293T cells. All cell lines were treated with 991, and HA-TMEM192 was immunoprecipitated. The lysosome surface marker, lysosomal-associated membrane protein 1 (Lamp1), was highly enriched in the immunopurified lysosomal fraction with little remaining in the supernatant, which indicates that most of the lysosomes had been extracted with the Lyso-IP technique (fig. S4B). Organelle markers, such as Golgin97 for Golgi, were not detected in the immuneprecipitates, demonstrating that organelle contamination had not occurred in the lysosome purifications. We observed a rapid de crease in association of endogenous RagC and RagA with the lysosome in WT FNIP1-HA-TMEM192 cells within 20 min of treatment with 991 (fig. S4B). By contrast, both RagC and RagA remained in the lysosome fractions in SA5 FNIP1-HA-TMEM192 cells with or without 991. FLCN, FNIP1, and mTOR followed a similar pattern. Depletion of AAs reduced RagA and RagC in lysosome fractions in WT FNIP1 but also SA5 FNIP1 cells, reiterating that AMPK-FNIP1–mediated control of mTOR and TFEB is not required in these conditions and is distinct from TFEB regulation by AAs and mTOR (fig. S4C).
To visualize the changes in RagC and mTOR localization after AMPK activation in cells, we used immunofluorescence imaging of endogenous RagC or mTOR, with antibodies to RagC or mTOR. RagC localized with the lysosomal marker Lamp2 in WT FNIP1 cells in fresh medium (Fig. 3, G and H). If AMPK was activated with 991, RagC localization with Lamp2—measured by Pearson’s correlation—was one-third of that in WT FNIP1 cells but remained localized with Lamp2 in SA5 FNIP1 cells with or without 991 (Fig. 3, G and H), confirming our biochemical observations. mTOR similarly showed decreased lysosomal localization in WT FNIP1 cells treated with 991 but not in cells expressing SA5 FNIP1 (fig. S4, D and E). Thus, AMPK activation appears to displace RagC and mTOR from lysosomes, preventing TFEB phosphorylation (fig. S4F). The enhanced interaction of RagC and TFEB in cells in which AMPK is activated with 991 indicates that RagC is not only required for recruitment of TFEB to the lysosome but also that increased binding to RagC may promote TFEB removal from the lysosome, perhaps by physically chaperoning TFEB into the cytosol.
We explored how FNIP1 phosphorylation by AMPK triggers the disassembly of the lysosomal machinery controlling TFEB. The FLCN-FNIP1 complex functions as a GAP for RagC and RagD, promoting the GDP-bound state of RagC and RagD, which is required for mTOR recruitment to the lysosome (11–13). Thus, phosphorylation of FNIP1 by AMPK might alter the GAP activity of the FLCN-FNIP1 complex. To test this, we transiently transfected either an HA-tagged, GTP-locked mutant of RagC (Q120L) or an HA-tagged, GDP-locked RagC mutant (S75N) in WT FNIP1 and SA5 FNIP1 or WT and AMPK KO cells and treated them with 991 (Fig. 3I and fig. S4G). If phosphorylation of FNIP1 by AMPK blocks the GAP activity of the FLCN-FNIP1 complex toward RagC, then GDP-loaded RagC should accumulate in cells lacking AMPK or SA5 cells. One prediction of this model is that overexpression of the GTP-locked mutant of RagC (Q120L) but not the GDP-locked RagC mutant (S75N) should restore TFEB and TFE3 dephosphorylation in AMPK KO cells and SA5 FNIP1 cells. Overexpression of GTP-locked RagC overrode the effect of SA5 FNIP1, allowing TFEB and TFE3 to be dephosphorylated in SA5 cells with or without 991 (Fig. 3I). We observed the opposite effect in WT FNIP1 cells, in which less TFEB dephosphorylation occurred after treatment with 991, when the GDP-locked RagC mutant was overexpressed compared with when GTP-locked RagC was present (Fig. 3I). Similar results were observed in the AMPK KO cells, where GTP-locked RagC overcame the absence of AMPK, enabling dephosphorylation of TFEB, whereas GDP-locked RagC prevented complete TFEB dephosphorylation in WT cells treated with 991 (fig. S4G).
Thus, AMPK phosphorylation of FNIP1 appears to inhibit FLCN-FNIP1 GAP activity, driving RagC to accumulate in its inactive GTP-bound form, which not only falls off the lysosome (Fig. 3, G and H, and fig. S4B) but also binds more tightly to TFEB (Fig. 3F). Immunoprecipitation experiments using cells expressing GTP- or GDP-locked RagC mutants, under the same conditions as shown in Fig. 3I, showed that in WT FNIP1 cells, more TFEB was bound to RagC in the GTP-locked state, bypassing the need for AMPK activation, in contrast to the GDP-locked state in which binding only occurred if cells were treated with 991 (presumably because of the presence of endogenous RagC, which would also bind TFEB in 991-treated cells) (Fig. 3J). Furthermore, in SA5 FNIP1 cells, exogenous GTP-locked RagC was associated with TFEB, overriding the effect of the SA5 mutations, whereas GDP-locked RagC did not bind TFEB to the same extent, regardless of whether cells were treated with 991 (Fig. 3J). A similar pattern was observed in the WT cells compared with cells lacking AMPK (fig. S4H). These results further corroborate and explain the results shown in Fig. 3F: Increased HA-RagC-TFEB interaction occurs in cells with active AMPK because of inactivation of the FLCN-FNIP1 GAP complex, propelling RagC to its GTP-bound state, which binds to TFEB more strongly than the GDP-bound form of RagC. Although GDP-RagC recruits TFEB to the lysosome for mTOR-dependent phosphorylation, GTP-RagC appears to be required to chaperone TFEB off the lysosome, preventing its phosphorylation.
FNIP1 phosphorylation by AMPK is required for lysosomal biogenesis
The MiT-TFE family of transcription factors, including TFEB and TFE3, are oncogenes and master regulators of lysosome biogenesis and autophagy (16, 17). Given that AMPK activation leads to nuclear translocation of TFEB and TFE3, an increase in their transcriptional activity would be expected. Our RNA-seq data in WT and AMPK KO HEK293T cells displayed an AMPK-dependent lysosomal gene signature. To assess the functional role of FNIP1 phosphorylation by AMPK, we analyzed global transcription in WT and SA5 FNIP1 cells treated with 991 for 0 to 16 hours by RNA-seq. Unsupervised hierarchical clustering of differentially expressed genes (P ≤ 0.05; FC ≥ 1.3) revealed that genes clustered according to 991 treatment or FNIP1 mutation status (WT or SA5) or both, demonstrating that FNIP1 phosphorylation by AMPK governed ~20% of the AMPK-responsive genes, whose transcription increased in cells treated with 991 (fig. S5, A and B). Enrichment analysis of differentially expressed genes found “clathrin-coated endocytic vesicle membrane” and “autolysosome” to be the most overrepresented terms in the GO cellular components category for transcripts less abundant in SA5 FNIP1 cells than in WT FNIP1 cells treated with 991 (fig. S5, C and D); GSEA analysis revealed “KEGG Lysosome” as one of the most enriched gene sets in WT FNIP1 cells treated with 991 for 16 hours compared with SA5 FNIP1 cells with the same treatment (Fig. 4A).
Fig. 4. Lysosomal biogenesis mediated by the MiT-TFE family of transcription factors is dependent on AMPK phosphorylation of FNIP1.

(A) GSEA plot for the “KEGG Lysosome” gene set, which was enriched in WT FNIP1 16-hour 991-treated but not SA5 conditions. (B) RNA-seq analysis of WT FNIP1 and SA5 FNIP1 cells subjected to a 0- to 16-hour 991 (50 μM) time course. Clusteringanalysis and heatmap displaysexpression patterns of AMPK-FNIP1–dependent CLEAR network genes that have been previously validated or GSEA defined. (C) Volcano plot depicting differential expression of CLEAR network genes after 4-hour 991 in WT FNIP1 versus SA5 FNIP1 conditions. Blue dots represent genes significantly down-regulated by mutation of AMPK sites on FNIP1. The y axis denotes −log10 P values, and the x axis shows log2 FC values. (D to I) qRT-PCR of CLEAR network genes SESN (C), Hex A (D), Neu1 (E), Lamp1 (F), FNIP2 (G), and ULK1 (H) in WT FNIP1 and SA5 FNIP1 HEK293T cells subjected to a 0- to 30-hour 991 (50 μM) time course. Graphs are shown as means ± SEMs. n = 3. *P < 0.05; **P < 0.01; ***P < 0.001; unpaired t test. (J) RNA-seq analysis of WT parental and CRISPR-Cas9–mediated TFEB-TFE3 DKO HEK293T cells treated with a 0- to 24-hour 991 time course. Heatmap shows AMPK-FNIP1–dependent genes whose expression is reduced by loss of TFEB-TFE3. (K) Volcano plot denoting CLEAR network DE genes after 16 hours of 991 in WT versus TFEB-TFE3 DKO FNIP1 cells. Blue dots represent genes significantly reduced by deletion of TFEB-TFE3. The y axis denotes −log10 P values, and the x axis shows log2 FC values. (L) Immunoblotting of lysosomal proteins in WT FNIP1 and SA5 FNIP1 HEK 293 cells after a 0- to 30-hour 991 time course. (M) Representative immunofluorescence images of lysosome structures stained with Lamp2 antibody after DMSO or 4-hour 991 treatment of WT FNIP1 and SA5 FNIP1 cells. (N) Quantitation of Lamp2 lysosomal structures from (M) and at the time points indicated, showing percentage of lysosome structures with volume greater than 0.1 μm3 after a 0- to 30-hour 991 time course in WT FNIP1 and SA5 FNIP1 cells. (O) Quantitation of Lamp2 sum intensity per lysosome in (M) and other time points from the same experiment. (P) Model. AMPK phosphorylation of FNIP1, induced by 991 or energetic stress, triggers TFEB entry into the nucleus, where it binds to CLEAR elements on lysosomal gene promoters, inducing lysosomal gene transcription, enhancing lysosomal protein expression and thereby lysosome biogenesis.
To more deeply examine how AMPK and FNIP1 control lysosome biology, we manually curated a list of ~1500 genes, which included experimentally validated CLEAR targets (42) in addition to the Molecular Signatures Database (MsigDB)–GSEA defined TFEB targets. Gene expression pattern analysis showed that ~75% of these CLEAR target genes showed increased transcription after AMPK activation in WT FNIP1 conditions (Fig. 4B and fig. S5, E and F). The genes clustered into four main groups (Fig. 4B)—one group showed early transcriptional activation between 2 and 4 hours only, the second group showed two waves of transcription with one wave at an early time point (2 hours) and the next at a later time point (16 hours), the third group was activated early from 2 hours onward but remained steadily high up to 16 hours, and the fourth group responded mainly at later time points between 8 and 16 hours. A large proportion of these genes, whose expression was increased by 991 in WT FNIP1 cells, did not respond to AMPK activation in cells overexpressing SA5 FNIP1 (~600 genes) (Fig. 4, B and C, and fig. S5C).
To validate some of the targets from our RNA-seq analysis, we subjected another batch of WT FNIP1 and SA5 FNIP1 cells to prolonged treatment with 991 for 0 to 30 hours and performed qPCR with primers targeting several canonical CLEAR network members. Expression patterns were similar to those in the RNA-seq data. A rapid increase in expression of SESN, Hex A, Neu1, Lamp1, FNIP2, and ULK1 mRNA, ranging from ~1.2- to 6-fold, was detected within 2 to 16 hours of 991 administration in WT FNIP1 cells (Fig. 4, D to I). With several lysosomal CLEAR target genes, such as Neu1, SESN, HEXA, and Lamp1, two separate waves of transcription were observed in WT FNIP1 cells—one at early time points between 1 and 2 hours and a second larger wave occurring at 16 to 24 hours. However, when AMPK phosphosites on FNIP1 were mutated, no increased transcription of the CLEAR genes was detected (Fig. 4, D to I). Glucose deprivation showed similar effects with increased transcription of the CLEAR network gene GLA in WT FNIP1 but not in SA5 FNIP1 cells (fig. S5G).
To demonstrate that the differential expression of lysosomal genes between WT and SA5 FNIP1 cells is regulated by TFEB and TFE3, we used CRISPR methods to make HEK293T cells lacking both TFEB and TFE3 (fig. S5, H and I). A cell line lacking only TFEB had minimal changes in gene expression, suggesting redundancy between TFEB and TFE3 (11, 43, 44). We treated parental WT and cells lacking both TFEB and TFE3 [TFEB-TFE3 double knockout (DKO)] with 991 for 24 hours and performed RNA-seq analysis. Specifically focusing on our manually curated list of lysosomal CLEAR genes, this analysis revealed loss of expression in a subset of AMPK-FNIP1– dependent lysosomal genes in the TFEB-TFE3 DKO condition compared with the control, including GLA and LAMTOR4 (Fig. 4, J and K).
To further study the effect of AMPK and FNIP1 on lysosomal proteins, we examined protein expression of lysosomal components including Lamp1, Lamtor 1, and cathepsin B (45). After 991 treatment, a strong correlation in the amounts of nascent, nonglycosylated Lamp1 protein reflected the expression levels of Lamp1 mRNA, showing two separate waves of increased expression for both mRNA and protein in WT FNIP1 cells (Fig. 4L). By contrast, no change in abundance of the Lamp1 protein was detected in the cells overexpressing SA5 FNIP1, which had low amounts like those in WT FNIP cells before AMPK activation; Lamtor1 and cathepsin B followed similar expression patterns to that of Lamp1 (Fig. 4L).
We used immunofluorescence imaging to analyze lysosomal structures by staining for intracellular Lamp2. Lysosomal structures were quantitated by measurement of lysosome volume and Lamp2 fluorescence intensities. The percentage of lysosomal structures above the threshold volume of 0.1 μm3 in WT FNIP1 cells treated with 991 displayed the same biphasic up-regulation pattern observed for Lamp1 mRNA and protein (Fig. 4, G and L to N). In 991-treated SA5 FNIP1 cells, the percentage of lysosomes varied but did not increase beyond the starting time point (Fig. 4, M and N). We also quantitated the Lamp2 sum intensity per lysosome, which did not display a biphasic pattern but did increase at 16 and 24 hours of 991 treatment in WT FNIP1 but not SA5 FNIP1 cells (Fig. 4O). Thus, FNIP1 phosphorylation by AMPK appears to contribute to lysosomal biogenesis (Fig. 4P).
AMPK regulation of PGC1α gene induction is governed by FNIP1 phosphorylation
Phenformin, rotenone, and CCCP all induced expression of PGC1α (PPARGC1A) mRNA in WT cells but not cells lacking AMPK in our RNA-seq datasets. The RNA-seq analysis also showed 991-induced transcription of PPARGC1A mRNA in WT FNIP1 but not SA5 FNIP1 cells (Fig. 4C). The PPARGC1A proximal promoter is a direct target of TFEB and TFE3 (46–48). Therefore, FNIP1 phosphorylation by AMPK, through control of TFEB-TFE3, might be one of the long-sought mechanisms underpinning the transcriptional regulation of mitochondrial biogenesis by AMPK.
The PPARGC1A gene undergoes extensive alternative splicing (49). qPCR assessment of total PPARGC1A using primers against exon 2, which is found in all PGC1α isoforms, showed an increase in transcription in WT FNIP1 but not SA5 FNIP1 cells treated with 991 (Total PPARGC1A, Fig. 5A). In humans, PGC1α transcription has been reported from three distinct promoters: a proximal promoter located just upstream of the canonical exon 1a; a distal alternate promoter followed by an alternative exon 1b, located ~13.7 kb upstream from exon 1a (50); or a much further upstream promoter termed the brain-specific promoter, ~500 kb upstream of the canonical proximal promoter (51, 52). PPARGC1A contains two potential CLEAR elements to direct binding of TFEB and TFE3 in the proximal promoter adjacent to the canonical exon 1a (46, 48). In addition to distinct promoters, alternative splicing between exons 6 and 7 of the PPARGC1A gene produces a transcript encoding the N-terminal isoform of PGC1α, which contains 267 AAs of classical PGC1α and three AAs from the splicing insert (53) (Fig. 5B). NT-PGC1α is a constitutive transcriptional coactivator because it retains the transcription activation and nuclear receptor interaction domains of full-length PGC1α but is no longer subject to phosphorylation-mediated turnover associated with the full-length protein (53, 54), thus leading to a truncated and transcriptionally active form of PGC1α with a longer protein half-life.
Fig. 5. FNIP1 phosphorylation by AMPK is critical for induction of the PGC1α- and ERRα-mediated mitochondrial biogenesis program through MiT-TFE transcription factors.

(A) qRT-PCR showing expression of total PPARGC1A compared with expression of the shorter NT-PPARGC1A splice isoform. Graphs are shown as means ± SEMs. n = 3. *P < 0.05; **P < 0.01; unpaired t test. (B) Schematic of the two predominant splice isoforms of PPARGC1A induced in this cell type in our conditions. (C) Immunoblots reflecting changes in expression of NT-PGC1α protein levels in WT FNIP1 versus SA5 FNIP1 cells in the presence or absence of a 991 time course. Molecular weights are indicated on the right. (D) Densitometry analysis of NT-PGC1α immunoblots. (E) qRT-PCR comparing expression of total PPARGC1A compared with that of shorter NT-PPARGC1A splice isoform in WT and AMPK KO HEK293T after a 0- to 16-hour CCCP time course. (F) qRT-PCR comparing expression of NT-PPARGC1A splice isoform in WT and AMPK KO HEK293T after a 0- to 16-hour 991 time course. For (D) to (F), all values are shown as means ± SEMs. n = 3. *P < 0.05; **P < 0.01; ***P < 0.001; unpaired t test. (G) Gene clustering analysis of RNA-seq data from WT FNIP1 and SA5 FNIP1 cells subjected to 991 time course treatments. Heatmap displays expression pattern of AMPK- and FNIP1-dependent mitochondria specific genes, as defined by the Mitocarta 3.0 inventory. (H) RNA-seq analysis of HEK293T cells transfected with control siRNA (siCont) or siRNA targeting PGC1α (siPGC1α) and treated with 0 to 24 hours of 991, as indicated. Heatmap displays AMPK-FNIP1–dependent Mitocarta 3.0 genes whose expression is lost with PGC1α knockdown. (I) RNA-seq analysis of WT and TFEB-TFE3 DKO HEK293T cells treated with 0 to 24 hours of 991. Heatmap displays expression pattern of AMPK-FNIP1–dependent mitochondrial genes, as defined by Mitocarta 3.0, that are lost upon TFEB-TFE3 deletion. (J) RNA-seq analysis of WT versus ERRα KO HEK293T cells subjected to a 991 time course, as indicated. Heatmap visualizes the expression pattern of AMPK-FNIP1–dependent mitochondrial genes in WT and ERRα KO HEK293T cells. (K) Volcano plot displaying Mitocarta 3.0 DE genes after 16-hour 991 in WT versus SA5 FNIP1 cells. Blue dots represent genes significantly down-regulated by mutation of AMPK sites on FNIP1. (L) Volcano plot depicting DE Mitocarta 3.0 genes in 16-hour 991-treated WT versus 16-hour 991-treated TFEB-TFE3 DKO RNA-seq dataset. Blue dots represent genes significantly down-regulated by CRISPR deletion of TFEB-TFE3. (M) Volcano plot depicting DE Mitocarta 3.0 genes in 16-hour 991 siCont versus 16-hour 991 siPGC1α RNA-seq dataset. Blue dots represent genes significantly down-regulated by knockdown of PGC1α. (N) RNA-seq analysis of WT versus ERRα KO HEK293T cells. Volcano plot displaying DE Mitocarta 3.0 genes after 16-hour 991 in WT versus ERRα null cells. Blue dots represent genes significantly down-regulated by deletion of ERRα. (O) Four-way Venn diagram showing overlap of gene sets controlled by AMPK-FNIP1, TFEB-TFE3, PGC1α, and ERRα. (P) qRT-PCR of mitochondrial genes including IDH2, Cox IV, CytoC, UCP2, and SOD2 in WT FNIP1 and SA5 FNIP1 HEK293T cells subjected to a 0- to 30-hour 991 time course. All data are shown as means ± SEMs. n = 3. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; unpaired t test. (Q) Model. AMPK phosphorylation of FNIP1 after energy stress or 991 facilitates TFEB nuclear entry where it binds to CLEAR network gene promoters, including the PPARGC1A promoter, which induces expression of the short ~35-kDa transcriptional coactivator NT-PGC1α isoform. In turn NT-PGC1α transactivates the ERRα transcription factor for induction of mitochondrial genes.
To further investigate regulation of PPARGC1A in 991-treated cells, we examined endogenous PGC1α protein expression by Western blotting (Fig. 5C). We did not detect the canonical, full-length ~100-kDa isoform of PGC1α in 991-treated WT or SA5 FNIP1 cells. We did, however, detect a time-dependent increase in the expression of the much smaller ~35-kDa N-terminal isoform of the PGC1α protein in WT FNIP1 cells after 991 administration, under the same conditions in which FNIP1 is phosphorylated and TFEB and TFE3 are activated (Fig. 5C). These changes in the 35-kDa PGC1α protein were quantitated by densitometry, which showed 8- to 16-fold increases in PGC1α expression after 991 treatment in WT FNIP1 cells (Fig. 5D). Abundance of the shorter PGC1α protein remained relatively low in SA5 FNIP1 cells (Fig. 5, C and D). We detected similar changes using two commercial antibodies (Santa Cruz and EMD Millipore), both of which have been validated for detection of NT-PGC1α isoforms (53, 55).
Because PPARGC1A is extensively alternatively spliced, we sought to further examine which PPARGC1A mRNA splice isoforms might accumulate in an AMPK-FNIP1–dependent manner. Analysis of the specific PPARGC1A mRNA splice isoforms in our RNA-seq data in WT and AMPK KO cells treated with mitochondrial poisons revealed accumulation of the NT-PPARGC1A short isoform (ENST no. 506055.5) that encodes the 271-AA truncated form but not full-length PGC1α isoforms (full-length, ENST no. 264867.7) after these stresses (fig. S6A). To directly compare with our initial observations of total PPARGC1A mRNA accumulation (targeting common exon 2, present in all splice isoforms), we carried out qPCR with primers targeting exons 5 and 7a to specifically detect expression of the NT-PPARGC1A alternative splice form (Fig. 5A). NT-PPARGC1A appeared to be more abundant in WT FNIP1 cells treated with 991 than total PPARGC1A mRNA; no effect was detected in cells expressing SA5 FNIP1 (Fig. 5A). qPCR from CCCP-treated WT and AMPK KO cells showed a similar pattern to what was observed with 991 treatment in the WT FNIP1 and SA5 FNIP1 cells. After treatment with CCCP, the NT-PPARGC1A short isoform accumulated in WT cells but not cells lacking AMPK (Fig. 5E). Similar results were also seen with the NT-PPARGC1A short isoform after 991 treatment in WT cells but not in cells lacking AMPK (Fig. 5F). Glucose deprivation also induced abundance of NT-PPARGC1A mRNA in WT FNIP1 but not SA5 FNIP1 cells (fig. S6B). Thus, the NT-PGC1α isoform is the predominant splice isoform of PGC1α expressed in HEK293T cells in response to mitochondrial poisons, glucose starvation, or 991 activation of AMPK.
To discern whether FNIP1 is a critical link between AMPK and PGC1α to promote mitochondrial biogenesis, we performed hierarchical clustering and differential expression analyses of our RNA-seq datasets. Again, using the Mitocarta 3.0 catalog, we specifically examined mitochondrial gene expression in WT and SA5 cells treated with 991. This revealed a subset of ~200 mitochondrial genes whose mRNA expression was increased by 991 at time points ranging from 2 to 16 hours, although most of these transcripts accumulated at the longest time point studied (16 hours) (Fig. 5G and fig. S6C). Notably, these transcripts did not accumulate when AMPK phosphosites in FNIP1 were mutated in the SA5 FNIP1 samples, even if cells were treated with 991 (Fig. 5, G and K). To investigate whether phosphorylation of FNIP1 by AMPK triggers mitochondrial biogenesis through PGC1α and to define the mitochondrial gene signature regulated by AMPK and FNIP1 in a PGC1α-dependent manner, we used small interfering RNA (siRNA) to deplete HEK293T cells of PGC1α (90 to 95% efficiency) (fig. S6D) and performed RNA-seq after treating cells with 991. PGC1α depletion led to decreased expression of 291 mitochondrial genes from Mitocarta 3.0 (fig. S6E); a subset of ~80 of these genes was dependent on AMPK phosphorylation of FNIP1 (Fig. 5, H and M). To test whether changes in mitochondrial gene expression were mediated by AMPK-FNIP1 control of TFEB, we also analyzed the expression of Mitocarta 3.0 genes in our WT and TFEB-TFE3 DKO RNA-seq dataset. We specifically examined the AMPK- and FNIP1-dependent mitochondrial genes and made an observation similar to that observed with the CLEAR network genes: Expression of ~50% of AMPK-FNIP1–dependent mitochondrial genes increased by 991 treatment in WT but not TFEB-TFE3 DKO samples (Fig. 5, I and L, and fig. S6F). We investigated mitochondria gene expression in cells expressing WT FNIP1 or SA5 FNIP1 after treatment with 991, validating transcription of several PGC1α mitochondrial targets by qPCR. mRNA levels of IDH2, Cox IV, Cyto C, SOD2, and UCP2 remained low at early time points but accumulated between 16 and 24 hours after 991 administration (Fig. 5P). Cells deprived of glucose for 6 hours also showed increased transcription of reported PGC1α target genes such as PDHA1 in WT FNIP1 cells (fig. S6G). In cells expressing SA5 FNIP1, transcription of all mitochondrial genes tested did not increase despite prolonged stimulation of cells with 991 or glucose deprivation (Fig. 5P and fig. S6G). Although many lysosomal genes showed two waves of expression, mitochondrial gene expression generally occurred at later time points, consistent with regulation by PGC1α, mRNA of which was itself transcribed in sync with the first wave of lysosomal biogenesis (model in Fig. 5Q).
AMPK-FNIP1 induction of mitochondrial biogenesis requires ERRα
Our RNA-seq analysis detected an AMPK-dependent increase in transcripts for the nuclear receptors ESRRA and ESSRG, an effect that was abolished by deleting AMPK or PPARGC1A siRNA knockdown. In WT cells, mRNAs of genes encoding nuclear receptors, including ESRRA and ESRRG, were increased from 2 to 16 hours after AMPK activation, and depletion of PPARGC1A suppressed this effect (fig. S7A). PGC1α initiates mitochondrial biogenesis through interaction with ERRα (30, 56, 57). Transcription of the ERRα gene is also increased by PGC1α (29). The requirement of PGC1α for 991 to promote ESRRA expression (fig. S7A) suggests that at least some of the AMPK- and FNIP1-dependent mitochondrial biogenesis program mediated by PGC1α might be through transcriptional coactivation of ERRα. Furthermore, transcription factor enrichment analysis of the global transcriptome of WT cells and those lacking AMPK revealed that ESRRA targets were overrepresented upon 991 treatment in the WT condition and underrepresented in cells lacking AMPK (fig. S2J). To test this, we genetically deleted ESRRA by CRISPR (fig. S7C). RNA-seq analysis of 991-treated WT cells or cells lacking ERRα showed that many Mitocarta 3.0 genes required ERRα for basal expression or increased expression in response to 991 (fig. S7D). We focused on the AMPK- and FNIP1-dependent Mitocarta genes to assess which of these were up-regulated by 991 in WT controls but suppressed when ERRα was absent (Fig. 5, J and N). We validated the expression of several of these genes by qPCR, including CPT1A, COX6A1, IDH2, and PDHA1 (fig. S7F), and observed increased transcription in cells treated with 991, with maximal increase at later times of 16 to 24 hours. 991 did not induce transcription of these genes in cells lacking ERRα.
We next performed a four-way comparison of the 991-regulated Mitocarta genes that varied between (i) WT and SA5 FNIP1 cells, (ii) WT and TFEB-TFE3 DKO cells, (iii) control and PGC1α siRNA cells, and (iv) WT and cells lacking ERRα (Fig. 5O). We observed an overlap between all groups, with 131 out of 190 AMPK- and FNIP1-dependent genes being coregulated by one or multiple of the other factors. Twenty Mitocarta genes were regulated by all four factors, and 61 genes were regulated by three of the four conditions (in an SA5-FNIP1–dependent manner), which we propose as a core minimal AMPK-FNIP1-TFEB-PGC1α-ERRα–dependent gene set involved in mitochondrial biogenesis (Fig. 5O and fig. S7E).
AMPK-FNIP1 is required for mitochondrial and lysosomal biogenesis
We performed immunoblotting of mitochondrial proteins as a measure of mitochondrial biogenesis. We detected increased expression of key mitochondrial proteins, including isocitrate dehydrogenase type 2 (IDH2), ubiquinolcytochrome Creductase binding protein (UQCRB), and NADH:ubiquinone oxidoreductase subunit B3 (NDUFB3), after 991 treatment in WT FNIP1 cells but not cells expressing SA5 FNIP1 (Fig. 6A); these were all detected in our RNA-seq data showing similar expression patterns. In agreement, expression of mitochondrial proteins, such as UQCRB, PDHA1, and NDUFB3, was increased by 991 treatment of WT but not TFEB-TFE3 DKO cells, indicating that these proteins also require TFEB and TFE3 (Fig. 6B). Finally, we also immunoblotted for these mitochondrial proteins after siRNA knockdown of PGC1α or CRISPR deletion of ERRα, observing that PGC1α (fig. S7B) and ERRα (Fig. 6C) were both required for these mitochondrial proteins to accumulate in 991-treated cells.
Fig. 6. AMPK-FNIP1–mediated mitochondrial biogenesis affects mitochondrial function and behavior.

(A to C) Western blots probing mitochondrial protein expression after a 991 time course ranging from 0 to 30 hours in WT FNIP1 and SA5 FNIP1 HEK293T cells (A), WT and TFEB-TFE3 DKO HEK293T cells (B), and WT and ERRα KO HEK293T cells (C). (D) Mitochondrial DNA content analysis. The ratio of mitochondrial (16S) to nuclear (actin) DNA was determined by qRT-PCR after treatment for 24 hours with 991 or DMSO (vehicle), as indicated. (E) Quantitation of IDH2 staining in (F). (F) Representative Airyscan microscopy images of mitochondrial IDH2 staining in WT FNIP1 and SA5 FNIP1 HEK293T cells treated for 24 hours with 991 or DMSO, as indicated. (G) Representative Airyscan images of Lamp2-stained lysosomes and Cox IV–stained mitochondria in WT FNIP1 and SA5 FNIP1 HEK293T cells treated for 24 hours with DMSO or 991, as indicated. (H) Quantitation of mitochondrial volume in (G). (I) Quantitation of lysosomal volume in (G). (J) Seahorse assay to measure OCR in WT compared with AMPK KO HEK293T cells. (K) Seahorse assays displaying OCR in WT FNIP1 compared with SA5 FNIP1 HEK293T cells. (L) Seahorse assays measuring OCR in WT compared with ERRα KO HEK293T cells. Graphs are shown as the means ± SEMs. n = 3. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; unpaired t test.
Although most of the genes required for mitochondrial biogenesis are encoded by the nucleus, the mitochondrial genome encodes 13 proteins, which are components of the OXPHOS system. As another parameter to measure mitochondrial biogenesis, we quantitated relative mitochondrial DNA (mtDNA) copy number by qPCR (58) (i.e., the ratio between a mitochondrial gene to a reference nuclear gene) in our cell lines at the 24-hour time point when mitochondrial gene transcription and protein expression was maximal. In cells treated with 991 for 24 hours, we observed an ~30% increase in mtDNA abundance in WT FNIP1 cells but not in SA5 FNIP1 cells (Fig. 6D).
To visualize mitochondria in WT and SA5 FNIP1 cells, we performed Airyscan microscopy after staining cells with IDH2 or CoxIV antibodies. Exposure of cells to 991 for 24 hours increased endogenous IDH2 staining inmitochondria in WT but not SA5 FNIP1 cells (Fig. 6, E and F). Again using Airyscan microscopy, we observed an increase in both mitochondrial (CoxIV) and lysosomal (LAMP2) volumes after 24 hours of 991 treatment in cells expressing WT but not SA5 FNIP1 (Fig. 6, G to I, and fig. S8A). We also observed an apparent increase in mitochondria-lysosome colocalization in 991-treated WT FNIP1 cells (Fig. 6G and fig. S8B), which we further examined using serial section EM. In EM data, an increase in mitochondria to lysosome contacts was observed (as defined by <20-nm distances between mitochondrial outer membrane and lysosome) only in the WT 991-treated condition (fig. S8, C to E).
Finally, we conducted Seahorse experiments to measure oxygen consumption rate (OCR) as a measure of mitochondrial function in WT cells or cells lacking AMPK (Fig. 6J), WT FNIP1 and SA5 FNIP1 cells (Fig. 6K), and WT and cells lacking ERRα (Fig. 6L). OCR was decreased in cells lacking AMPK compared with that in WT cells. Concordantly, OCR was also reduced in the SA5 FNIP1 cell line compared with WT FNIP1 cells. Moreover, a comparison of WT cells with cells lacking ERRα revealed that OCR was also decreased when ERRα was deleted. This further supports the common role of AMPK, FNIP1, and ERRα in controlling mitochondrial biogenesis and function.
Discussion
FNIP1 was originally identified as a binding partner for the FLCN hamartoma suppressor (34) and in the same study was found to coimmunoprecipitate with endogenous AMPK subunits, although a functional role for FNIP1 mediating aspects of AMPK function has never been examined in the decades since. Subsequent studies have focused on how AMPK signaling is hyperactivated in FLCN-deficient states (59–62). Our findings indicate that the physiological relationship between AMPK and FLCN-FNIP1 is that after metabolic stress, AMPK lies upstream of FLCN-FNIP1, wherein AMPK-dependent phosphorylation of FNIP1 acutely inhibits FLCN-FNIP1 GAP function, leading to TFEB activation.
Our results are consistent with the emerging model that the FNIP1-FLCN GAP complex controls RagC to retain TFEB and TFE3 at the lysosome and that TFEB and TFE3 are specific and selective substrates of mTORC1 in this regulation (15, 36, 37). A critical arginine required for GAP activity of FLCN in the FLCN-FNIP1 complex is required for AA-induced control of translocation of TFEB and TFE3 (15). Our data indicate that AMPK activation inhibits FLCN-FNIP1 GAP activity and promotes the accumulation of GTP-loaded RagC, the inactive state of the Rag complex. This mechanism allows activation of TFEB and TFE3 under low-energy conditions, even if AAs are plentiful. AMPK phosphorylation of FNIP1 promotes dissociation of RagC and mTOR from the lysosome as well as separation of mTOR from TFEB itself, consistent with loss of mTORC1-mediated TFEB phosphorylation. In cells that are AMPK deficient or just mutated in the five AMPK sites in FNIP1, even in the face of energy stress, the FNIP1-FLCN complex cannot be regulated by AMPK, and mTOR remains bound to TFEB at the lysosomal surface, rendering TFEB resistant to activation by mitochondrial poisons, glucose deprivation, or direct AMPK activators. By contrast, these cells still exhibit normal regulation of TFEB in response to AA withdrawal. Thus, AMPK maintains specific control of TFEB and TFE3 through its phosphorylation of FNIP1, independent of AA regulation of the FLCN-FNIP1 complex. Collectively, FNIP1 phosphorylation by AMPK at Ser220, Ser230, Ser232, Ser261, and Ser593 is a key mechanism by which AMPK controls the MiT-TFE family of transcription factors to increase lysosomal biogenesis and in parallel to increase PGC1α mRNA, which induces mitochondrial biogenesis, placing FNIP1 at the center of multiple AMPK-dependent processes for which the direct biochemical substrate of AMPK had remained elusive.
Materials and methods
Antibodies and reagents
Abcam antibodies used include Total OXPHOS Rodent WB Antibody Cocktail [catalog no. (Cat#) ab110413], monoclonal anti-UQCRB (Cat# ab190360 [EPR15591]), monoclonal anti-NDUFB3 (Cat# ab202585 [EPR15571]), monoclonal Tomm20 (Cat# ab56783), and monoclonal LAMP2 (Cat# ab25631). Cell Signaling Technology (CST) antibodies used in the study were as follows: TFEB (Cat# 4240), monoclonal TFEB (D2O7D, Cat# 37785), monoclonal Phospho-TFEB S122 (Cat# 86843), Tfe3 (Cat# 14779), FNIP1 (Cat# 36892), Phospho-FNIP1 S220 (Cat# 40812, in development), FLCN (Cat# 3697 (D14G9), RagA (D8B5, Cat# 4357), RagB (D18F3 Cat# 8150), RagC (D8H5, Cat# 9480), IDH2 (D8E3B, Cat# 56439), PDHA1 (C54G1, Cat# 3205), Tricarboxylic Acid Cycle Antibody Sampler Kit (Cat# 47767), monoclonal anti-LAMP2 (H4B4, Abcam Cat# ab25631), monoclonal anti-LAMP1 (D2D11, Cat# 9091), ERRα (Cat# 13826), AMPKα (Cat# 2532), Phospho AMPKα T172 (40H9, Cat# 2535), ACC (Cat# 3662), Phospho-ACC S79 (Cat# 3661), Raptor (Cat# 2280), Phospho-Raptor S792 (Cat# 2083), anti-Ulk1 (D8H5 Cat# 8054), Phospho-S6K T389 (Cat# 9205), 4EBP1(Cat# 9452), S6 (5G10, Cat# 2217), Phospho-S6 S235/236 (Cat# 4858), mTOR (Cat# 2972), LAMTOR1/C11orf59 (D11H6, Cat# 8975), monoclonal Hdac3 (7G6C5 Cat# 3949), Golgin-97 (D8P2K Cat# 13192), monocloncal GAPDH (D16H11, Cat# 8884), GFP (D5.1, Cat# 2956), and DYKDDDDK (FLAG) tag (Cat# 2368). Α-Tubulin antibody was from Sigma-Aldrich (B-5–1-2, Cat# T5168). Monoclonal anti-PGC1α antibodies were from Santa Cruz Biotechnology (D-5, Cat# sc-518025) and Millipore (4C1.3, Cat# ST1202). Polyclonal Tfeb antibody was from Bethyl Laboratories (Cat# A303–673A). GFP-Trap Agarose was from ChromoTek (Cat# gta-20) and Pierce HA Magnetic Beads were from Thermo Fisher Scientific (Cat# 88837). 991 was purchased from Glixx Laboratories (Cat# GLXC-09267), and phenformin hydrochloride (Cat# P7045), rotenone (Cat# R8875), CCCP (Cat# C2759), and metformin (Cat# PHR1084) were from Sigma-Aldrich.
Plasmids
The cDNA encoding human FNIP1 (Uniprot Q8TF40) was generated by reverse transcription polymerase chain reaction (RT-PCR) from RNA obtained from IMR90 cells. Fusion tag FLAG-CHERRY was added to the N-terminal end of FNIP1 and subcloned into pDONR221 with BP Clonase (Invitrogen). The cDNA for human FLCN was obtained from Invitrogen (IOH12359). Mammalian expression vectors were generated, by recombining ENTR clones into DEST vectors using LR Clonase (Invitrogen). Destination vectors include: pLentiCMV/TO (Addgene no. 17293), pcDNA3 N-term FLAG DEST, pcDNA3 N-Term MYC DEST, and pQCXIB CMV/TO (Addgene no. 17400). Site-directed mutagenesis for FNIP1 was performed using QuikChange II XL (Stratagene) according to the manufacturer’s instructions. Untagged human AMPKα1 cDNA was cloned into pLentiCMV/TO puro Gateway destination vector. Other plasmids used include pLJC5-Tmem192–2xFlag (41) (Addgene no. 102929), pLJC5-Tmem192–3xHA (41) (Addgene no. 102930), pEGFP-N1-TFEB (5) (Addgene no. 38119), pRK5-HA GST RagC 120L (Addgene no. 19306), and pRK5-HA GST RagC 75L (Addgene no. 19305).
Cell culture and cell lines
All cells were cultured in Dulbecco’s modified essential medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) (Hyclone, Thermo Fisher Scientific), 2 mM l-glutamine, penicillin/streptomycin (Gibco) at 37°C in 5% CO2 and maintained under antibiotic selection for stable cell lines. Stably reexpressing WT FNIP1 and SA5 FNIP1 cells were generated by using lentivirus-mediated transduction of FNIP1 KO HEK293T cells with human Myc– tagged FLCN cDNA, in combination with either FLAG-Cherry–tagged WT FNIP1, SA4 FNIP1, or SA5 FNIP1 cDNA under double puromycin (Sigma) and hygromycin (Invitrogen) selection. GFP-TFEB cell lines were generated by stable infection of WT FNIP1 and SA5 FNIP1 HEK293T cells with lentivirus expressing GFP-TFEB cDNA and blasticidin resistance. Lyso-IP cells were generated by stable infection of WT FNIP1 or SA5 FNIP1 with lentivirus expressing pLJC5-Tmem192–3xHA or pLJC5-Tmem192–2xFlag cDNA. Mitochondrial poisons and AMPK-activating drugs were used at the following concentrations: 991 (50 μM), CCCP (5 μM), phenformin (2.5 mM), and rotenone (1 mM). For AA starvation experiments, cells were first washed with RPMI AA-free medium supplemented with 10% dialysed FBS, then the same media added for the times indicated in figures. For AA-replete conditions, RPMI AA-free medium was supplemented with 10% dialysed FBS in addition to 1x L-glutamine, 1x essential AAs, and 1x nonessential AAs for the times indicated in the figures. For glucose deprivation experiments, glucose-free DMEM media (Invitrogen) was supplemented with 10% dialysed FBS and a range of glucose concentrations including 1 mM, 2.5 mM, and 25 mM. For transient expression of proteins and packaging of virus, HEK293T cells were transfected with the plasmid of interest using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) following the manufacturer’s protocol.
Mouse studies
All procedures using animals were approved by the Salk Institute Institutional Animal Care and Use Committee (IACUC) protocol 11–000029. AMPKa1 and AMPKa2 floxed allele (Prkaa1fl/fl; Prkaa2fl/fl) mice bearing Albumin-creERT2 were treated with tamoxifen (1 mg per day) or vehicle (control) for 5 consecutive days. Eight weeks after tamoxifen injection, mice were fasted overnight, refed for 1 hour, then for 2 hours with vehicle or MK-8722 (30 mg/kg) before euthanizing as previously described (63). Livers were collected, and lysates were prepared in lysis buffer. In Fig. 2F, primary hepatocytes were made and treated with metformin as previously described (64).
Western blots
For biochemical analysis of cells, cells were washed with ice-cold phosphate-buffered saline (PBS) and lysed in buffer containing 20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM pyrophosphate, 50 mM NaF, 5 mM β-glycero-phosphate, 50 nM calyculin A, 1 mM Na3VO4, and protease inhibitors (Roche). Lysates were clarified by centrifugation at 16,000 ×g for 10 min at 4°C. Protein concentration was calculated using the BCA protein kit (Pierce). Lysates were resolved on 10 to 12% SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gels, depending on molecular weight of proteins assessed and immunoblotted. Nuclear and cytoplasmic fractions were isolated using a NE-PER nuclear and cytoplasmic extraction kit (Thermo Fisher Scientific) according to manufacturer’s instructions.
Immunoprecipitation
Cells were lysed in standard lysis buffer as described above and samples equilibrated. GFP-TFEB was immunoprecipitated from 2 mg of lysates using GFP-Trap beads (Chromotek) at 4°C for 2 hours under rotation. Subsequently, the beads were washed three times with lysis buffer. Protein complexes were eluted from beads using Laemmli sample buffer with 2% (v/v) beta-mercaptoethanol. Coimmunoprecipitating proteins were detected by immunoblot analysis.
Immunopurification of lysosomes (Lyso-IPs)
Lyso-IP cells were generated by infection with viruses containing Tmem192–3xHA (HA-Lyso) or Tmem192–2xFlag (Cont-Lyso). The Lyso-IP protocol was carried out as described in Abu-Remaileh et al. (41). Briefly, WT FNIP1/HA-Lyso or SA5 FNIP1/HA-Lyso cells in addition to control cells were treated with short 991 time courses and washed with PBS then scraped into 1-ml KPBS (136 mM KCl, 10 mM KH2PO4, pH 7.25 adjusted with KOH). Cells were gently homogenized with 20 strokes of a 2-ml homogenizer. The homogenate was then centrifuged at 1000 ×g for 2 min and the supernatant containing the cellular organelles including lysosomes was incubated with 100 μl beads of anti-HA magnetic beads, prewashed in KPBS, on a gentle rotator for 15 min at 4°C. Immunoprecipitates were then gently washed three times with KPBS and the lysosome fraction was eluted from the beads through incubation in lysis buffer for 10 min.
CRISPR-Cas9 techniques
Small guide RNAs (sgRNAs) targeting human TFEB, TFE3, and ERRα were selected using the CRISPR design tool Benchling program https://www.benchling.com/crispr/. Guides with high targeting scores and low probability of off-target effects were chosen (table S1). At least three independent sgRNA sequences were tested for each gene. Oligonucleotides for sgRNAs were synthesized by IDT, annealed in vitro, and subcloned into BsmbI-digested plentiCRISPRv.2-puro (Addgene no. 52961) or lentiCRISPRv.2-blast (Addgene no. 98293). Validation of guide specificity was assessed by Western blotting of low-passage cells after puromycin selection.
HEK293T AMPK KO cells were generated using the Cas9 nickase strategy. Briefly, a pair of guide RNAs (gRNAs) targeting exon 1 was designed for both human PRKAA1 and PRKAA2 genes using the online design tool at http://crispr.mit.edu (AMPKα1 A/B and AMPKα2 A/B, respectively) (table S1). Each gRNA duplex was cloned into pX462 vector encoding SpCas9n-2A-Puro (Addgene no. 48141). HEK293T cells were transfected with the gRNA pair to generate single AMPKα1 or AMPKα2 KO or transfected with both pairs together to generate double AMPK α1/α2 KO (DKO). After puromycin selection, single-cell cloning was performed by cell sorting into 96-well plates. Individual clones were screened by Western blot and a clone lacking both AMPK α1 and α2 protein expression was selected. HEK293T FNIP1 KO cells were made using a single gRNA (table S1) targeting exon 2 and subsequently cloned into pX459 encoding SpCas9(BB)-2A-Puro (Addgene no. 48139). Single-cell clones were isolated after selection with puromycin and screened by immunoblotting.
RNA interference studies
All siRNAs were purchased from Horizon Discovery. siRNAs against a nontargeting sequence was used as a negative control (ON-TARGET-plus nontargeting control siRNA, Cat# D-001810–01-05) or ON-plus SMARTpool siRNAs targeting PPARGC1A (L-005111–00-0005) or targeting TFEB (Cat# L-009798–00-00050) or TFE3 (Cat# L-009363–00-0005) were used. HEK293T cells were plated in a 6-well plate and allowed to adhere overnight. Cells were transfected with the nontargeting control siRNA pool (20 nM), or PPARGC1A siRNA pool (20 nM). Transfection was carried out using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer’s protocol. 48 hours after transfection, cells were treated with 991 as indicated in the figures and then harvested for downstream applications.
Lentivirus production
Lentiviruses were produced by transfecting HEK293T cells with the cDNA construct of interest including 3xHA-Tmem192, 2xFLAG-Tmem192, FLAG-WT FNIP1, FLAG-SA4 FNIP1, FLAG-SA5 FNIP1, MYC-FLCN, or GFP-TFEB constructs, in combination with VSV-G and CMV-ΔVPR packaging plasmids. Sixteen hours later, the media was changed to DMEM with 10% FBS. The virus containing supernatant was collected the next day and 0.45-μm filtered, then frozen at −80°C or supplemented with 8 μg/ml polybrene and applied to destination cells for 24 hours. Sixteen hours later, the media was refreshed and the appropriate antibiotic was added for selection.
In vitro kinase assays
HEK293T cells transiently transfected with FLAG-FNIP1 or FLAG-tagged FNIP1 mutants (SA2, SA3, SA4, SA5) were lysed and subjected to FLAG immunoprecipitation. Immunoprecipitates were washed three times in lysis buffer, followed by three times in kinases assay buffer (50 mM Tris pH 7.5 10 mM MgCl2). Subsequently, immunoprecipitates were subjected to a kinase reaction containing 0.1 mM [γ32P]-ATP (PerkinElmer) and 2 mM dithiothreitol (DTT) with or without 0.1 μg of active recombinant 50 ng of active recombinant AMPK (Millipore no. 14–840) in the presence of kinase assay buffer. The reaction was incubated at 30°C for 30 min. Reactions were terminated with LDS sample buffer and resolved by SDS-PAGE electrophoresis. Proteins were detected with Coomassie staining. Dried gels were exposed to UltraCruz autoradiography film overnight, in an autoradiography cassette and the films were later developed using an auto-developer.
Immunofluorescence
Cells were seeded on coverslips (precoated with poly-L-lysine). Following treatments described in figure legends, cells were fixed with 4% (v/v) paraformaldehyde and permeabilized with 1% (v/v) NP-40. Cells were blocked using 5% bovine serum albumin (BSA) in PBS, then incubated for 1 hour with primary antibodies, followed by three washes in 0.2% BSA/PBS. Coverslips were incubated for 1 hour with secondary antibodies and counterstained with 4′,6-diamidino-2-phenylindole (DAPI) for 5 min, then washed three times in 0.2% BSA/PBS and once in water. Coverslips were mounted with Fluoromount-G (Southern Biotech).
Confocal imaging of TFEB, TFE3, and RagC
For visualization of TFEB, TFE3, and RagC, the following primary rabbit antibodies were used: TFEB (CST no. 37785), TFE3 (CST no.14779), and RagC (CST no. 9480) followed by anti-rabbit secondary antibodies conjugated to Alexa Fluor (AF) 594. Cells were imaged on a Zeiss LSM 700 confocal microscope using the 63X objective. Quantification was performed using imageJ software.
Airyscan confocal imaging of IDH2, Cox IV, and Lamp2
For imaging IDH2, coverslips were incubated in anti-IDH2 (CST no. 56439) antibody, followed by secondary anti-rabbit antibody conjugated to AF-568. IDH2 was costained with phalloidin 647 and Tomm20 using anti-Tomm20 (Abcam no. ab56783) antibody, followed by secondary antibody conjugated to AF-488. Cox IV and Lamp2 were costained with anti-Cox IV (CST no. 11967) and anti-Lamp2 (Abcam no. ab18528) antibodies, followed by anti-rabbit secondary conjugated to AF-594 and anti-mouse secondary conjugated to AF-488, respectively. Cells were imaged with a 63× 1.4 NA oil objective on a ZEISS 880 LSM Airyscan confocal system with an inverted stage. High-resolution Airyscan images were acquired using a pixel dwell time of 0.66 μs and 2x Nyquist pixel size of 43 nm per pixel in SR mode (i.e., a virtual pinhole size of 0.2 Airy units), then processed using ZEISS Zen software with the Airyscan parameter determined by auto-filter settings. The zoom factor was set to 2 to obtain a large field of view. Alexa Fluor 488 was imaged with a 488-nm laser with a laser power of ~64 μW, Alexa Fluor 568 and 594 were imaged with the 561-nm laser with a laser power of ~268 μW, and Alexa Fluor 647 was imaged with the 633-nm laser with a laser power of ~98 μW.
Fluorescence image quantification
IDH2 fluorescence intensity quantification was performed with Imaris (version 9.6.0, Bitplane, Zurich, Switzerland). Mitochondrion regions were masked by surfaces generated TOM20 fluorescence using automatic threshold settings in Imaris. Mitochondrial associated IDH2 was quantified by summing up the IDH2 voxel intensity values inside the segmented TOM20 regions and normalizing to the mitochondria volume. Statistical significance was determined using an unpaired t test. Quantification was performed with Imaris software 9.6.0 (Bitplane). COX IV fluorescence image channels were preprocessed with a Gaussian smoothing filter with a filter size of 0.0707 μm. Background subtraction was performed with an estimation of the diameter of the largest sphere that fits into the object as 0.265 μm. Mitochondrial clusters were then defined by surfaces generated with automatic threshold settings, which makes use of an iterative selection method. Clusters smaller than 0.003 μm3 were removed. Clusters for LAMP 2 fluorescence signal were defined similarly but without smoothing or background subtraction. Touching objects were separated using region growing with a seed point diameter of 0.25 μm. The Imaris quality filter with a lower threshold of 86.5 was then used to select positive signals. Clusters smaller than 0.065 μm3 were removed. Cell volume was defined by first smoothing the LAMP2 signal with a Gaussian filter with a diameter of 1 μm. Surfaces generated with manual threshold of 41.5 absolute intensity. Lysosomes colocalized with mitochondria were defined as lysosomes located within 0.2 μm from its closest mitochondria cluster. The lysosome colocalized volume, total lysosome volume, and mitochondria volume were normalized to the cell volume. Statistical significance was determined using an unpaired t test.
Electron microscopy imaging and quantification
Cells were cultured on 10-cm dishes to reach ~70% confluence before fixation. Materials were sourced from Electron Microscopy Sciences (Hatfield, PA) unless noted otherwise. Culture media was gently poured off and 4 ml of warm 37°C fixative (3% glutaraldehyde in 0.1 M sodium cacodylate buffer with 3 mM CaCl2) was added to the dish before being replaced with ice-cold fixative after 10 s. After an hour of fixation at 4°C, cells were rinsed with 0.1 M sodium cacodylate buffer with 3 mM CaCl2 three times for 10 min and postfixed with reduced osmium tetroxide (0.1 M sodium cacodylate buffer, 0.1 M CaCl2, 1.5% osmium tetroxide, 1.5% potassium ferrocyanide) for 40 min in the dark at room temperature. Dishes were rinsed with ice-cold deionized water three times, with the final rinse of 1 ml of water left in the dish, and scraped with a sharpened piece of Teflon secured in a hemostat. The cell suspension was collected into Eppendorf tubes and left overnight at 4°C. The following day, cells were stained with aqueous 1% uranyl acetate at room temperature for an hour before serial dehydration in ice-cold ethanol solutions. Dehydrated cells were rinsed twice with anhydrous ethanol at room temperature and infiltrated with Eponate 12 resin (hard formulation) for 2 hours at 3:1 and 1:1 ethanol:resin mixtures and left in 1:3 resin to infiltrate overnight. The following day, cells were infiltrated in two changes of pure resin throughout the day and pelleted in a third change of fresh resin at 12,000 rpm in a tabletop centrifuge (Pelco) and left to polymerize in an oven at 60°C for 48 hours.
Polymerized blocks were removed from Eppendorf tubes using razor blades and trimmed for utlrathin serial sectioning as previously described (65) using diamond knives (Diatome) mounted on a Zeiss UC8 ultramicrotome. Series of ultrathin (70-nm) sections were collected onto silicon chips and imaged using a scanning electron microscope (Zeiss Sigma VP) equipped with a backscattered electron detector (Gatan) using Atlas5 (Fibics) control software and scan generator. Ribbons of serial sections from each condition were screened to identify a cluster of cells suitable for further analysis. For each series, the region of interest in each section was identified and captured at an overview resolution (200 nm per pixel) and a midlevel resolution (8 nm per pixel) to minimize drift during high-resolution (2 nm per pixel) acquisition. High-resolution stacks of images were aligned. Approximately 50 mitochondria from two randomly sampled subvolumes of registered 3DEM image stacks were segmented using Vast Lite (66). The two volumes (total 105.28 μm3, average = 52.64 μm3) of densely labeled mitochondria (n = 53, average = 26.5) from the WT-DMSO condition were proofread. Because of the prevalence of touching mitochondria, all labels were first eroded to ensure separated boundaries. A three-dimensional (3D) U-Net was used to detect boundaries and LSDs in a multitask learning framework. The network had an input shape of [48,284,284] and output shape of [16,196,196] (voxels, zyx). It consisted of three layers and was down-sampled by a factor of [1,2,2] in the first two layers and [2,2,2] in the final layer. The reverse was done for the up-sampling path. Twelve initial feature maps were used and multiplied by a factor of 5 between layers. The number of feature maps in the last layer was increased to 14 to account for the 13 feature maps generated by the boundaries (3) and LSDs (10). A mean squared error loss and Adam optimizer were used to train the network. A single voxel neighborhood [1,1,1] was used for the boundaries. The LSDs used a sigma of 140 and were down-sampled by a factor of 2. Training was done using Tensorflow and Gunpowder. Inference, seeded watershed, and percentile agglomeration were performed in a block-wise fashion for each condition. Four subvolumes from the non–WT-DMSO conditions (1 WT-991, 1 SA5-DMSO, 2 SA5–991, 116.73 μm3, average = 29.2 μm3) were proofread (n = 110, average = 27.5) and used to refine training along with the original two volumes. Additionally, 11 negative samples which did not contain any mitochondria (i.e., resin, cytosol, background) were used for retraining (~450 μm3, average = 40.9 μm3). Each batch randomly sampled from one of the 17 total subvolumes such that the probability of choosing either a positive or negative sample was 50%. After training, segmentations were created for each condition, and small objects (i.e., oversegmented debris) were filtered out. Mitochondria were then randomly sampled from each volume (n = 100) and proofread for subsequent analysis. Iterative cycles of training and manual proofreading were used until a fairly accurate but excessively permissive segmentation of mitochondria was achieved for all conditions. One hundred mitochondria were randomly sampled from the machine-generated segments. Segmentations were meticulously proofread by human experts before inclusion in analysis. Contingency tables of sampled mitochondria with and without lysosomal contacts were assembled for pairs of experimental conditions (i.e., combinations of WT/SA5, DMSO/991 treatment). Chi square tests were performed in Python using the chi2_contingency function in the scipy.stats library.
Mass spectrometry
HEK293T cells were transfected with an epitope-tagged FNIP1 cDNA, and cells were treated with DMSO or phenformin. After immunopurification of FNIP1 protein, FNIP1 was isolated from an SDS-polyacrylamide gel. Bands on the gels were cut out and subjected to reduction with dithiothreitol, alkylation with iodoacetamide, and in-gel digestion with chymotrypsin overnight at pH 8.3, followed by reversed-phase microcapillary or liquid chromatography with tandem mass spectrometry (LC-MS/MS). LC-MS/MS was performed using an Easy-nLC nanoflow HPLC (Proxeon Biosciences) with a self-packed 75 μm id × 15 cm C18 column coupled to a LTQ-Orbitrap XL mass spectrometer (Thermo Fisher Scientific) in the data-dependent acquisition and positive ion mode at 300 nL/min. Peptide ions from predicted phosphorylation sites were also targeted in MS/MS mode for quantitative analyses. MS/MS spectra collected through collision-induced dissociation in the ion trap were searched against the concatenated target and decoy (reversed) single entry and full Swiss-Prot protein databases using Sequest (Proteomics Browser Software, Thermo Fisher Scientific) with differential modifications for Ser/Thr/Tyr phosphorylation (+79.97) and the sample processing artifacts Met oxidation (+15.99), deamidation of Asn and Gln (+0.984) and Cys alkylation (+57.02). Phosphorylated and unphosphorylated peptide sequences were identified if they initially passed the following Sequest scoring thresholds against the target dtabase: 1+7 ions, Xcorr ≥ 2.0 Sf ≥ 0.4, P ≥ 5; 2+ ions, Xcorr ≥ 2.0, Sf ≥ 0.4, P ≥ 5; 3+ ions, Xcorr ≥ 2.60, Sf ≥ 0.4, P ≥ 5 against the target protein database. Passing MS/MS spectra were manually inspected to ensure that all b- and y-fragment ions aligned with the assigned sequence and modification sites. Determination of the exact phosphorylation sites was aided using FuzzyIons and GraphMod and phosphorylation site maps were created using ProteinReport software (Proteomics Browser Software suite, Thermo Fisher Scientific). False discovery rates (FDRs) of peptide hits (phosphorylated and unphosphorylated) were estimated below 1.5% based on reversed database hits.
Seahorse assays
OCR of cells were measured using the Seahorse XF96 Cell Mito Stress Test Kit (Seahorse Biosciences) with an XF96 Extracellular Flux Analyzer (Seahorse Bioscience) in accordance with the manufacturer’s instructions. Seahorse XF Cell Mito Stress Test Kit (Cat# 103015–100) and Seahorse xFe96 FluxPak (Cat# 102416–100) were purchased from Agilent Technologies. The XF96 cell culture microplates were polylysine coated before seeding HEK293T cells in XF base media supplemented with 1 mM pyruvate, 2 mM glutamine, and 10 mM glucose. Each condition was seeded in six replicate wells. The Seahorse sensor cartridge was hydrated overnight in a non-CO2 incubator at 37°C. On the day of the assay, cells were incubated in a CO2-free incubator at 37°C for 1 hour to allow for temperature and pH equilibration before loading into the XF96 apparatus. Mitochondrial stress tests were conducted following Seahorse guidelines (Agilent Technologies). Inhibitors were used at the following concentrations: 1 μM oligomycin, 1 μM FCCP, and 0.5 μM antimycin A + 0.5 μM rotenone. Analyses were conducted using Wave software (Agilent Technologies).
mRNA extraction and qPCR
RNA extracted from cells using QIAGEN rNeasy Plus mini kit (Cat# 74134) and 1 μg of RNA was reverse transcribed using Iscript cDNA Synthesis Kit (Bio-Rad, Cat# 1708891). qPCR used diluted cDNA, relevant primers, and SYBR Green PCR master mix (Thermo Fisher Scientific, Cat# 4309155) in a C1000 Thermal Cycler (BioRad). Relative mRNA levels calculated using the ΔΔCT method, with β-actin serving as the internal control. All mRNA measurements performed in triplicate. All primers are listed in table S1.
Mitochondrial DNA content analysis
Mitochondrial DNA was quantified by determining the mtDNA/nDNA ratio. Total genomic DNA was extracted from cells using the Qiagen dNeasy Blood & Tissue Kit (Cat# 69504). Extracted DNA was diluted to a final concentration of 10 ng of DNA per microliter, and mitochondrial DNA was quantified relative to the nuclear DNA specific gene β-actin by qPCR. Primers used for β-actin and mitochondrial 16S rRNA genes are listed in table S1. Reactions used SYBR Green PCR Master Mix (Thermo Fisher Scientific), using a C1000 Thermal Cycler (BioRad). The relative mtDNA copy number was calculated using the ΔΔCT method.
RNA-seq
Cells were administered with 991, CCCP, rotenone, or phenformin time courses, ranging from 0 to 24 hours. Each time point and condition was prepared in triplicate. Total RNA was isolated using the QIAGEN rNeasy Plus mini kit. The quality of the isolated total RNA was assessed using Agilent TapeStation 4200 and RNA-seq libraries were prepared with 500 ng of total RNA using the TruSeq stranded mRNA Sample Preparation Kit according to the manufacturer’s protocol (Illumina). Libraries were quantified, multiplexed, and pooled for sequencing at paired-end 75 base pairs using the Illumina NextSeq500 or NovsSeq6000 platform at the Salk Next-Generation Sequencing Core. Raw sequencing data were demultiplexed and converted in the FASTQ files using CASAVA (version 1.8.2). Libraries were sequenced at an average depth of 12 to 40 million reads per sample.
Bioinformatic analysis of RNA-seq data
Raw RNA-seq reads in FASTQ files were quality-tested using FASTQC (v0.11.8) (Andrews 2010) and mapped to the human reference genome (GRCh38) with STAR (v2.5.3a) aligner with default parameters (67). Raw or TPM (transcripts per million) gene expression levels were quantified across all the exons of the top isoform in RefSeq with analyzeRepeats.pl in HOMER (v4.11.1) (68). Differential expression analysis was performed on the Biojupies platform (https://amp.pharm.mssm.edu/biojupies/) (69) with raw gene counts, comparing gene expression levels between control and experimental groups using the limma R package (70). Replicates were used to compute within-group dispersion and correction for batch effects. Volcano plots were generated to display the results of differential gene expression analysis using VolcanoseR (71). Gene fold changes were log2 transformed and displayed on the x axis; P values were corrected using the Benjamini-Hochberg method, transformed using −log10 and displayed on the y axis. Differentially expressed genes were defined as having a P value ≤ 0.05; FC ≥ 1.3. Red points indicate significantly up-regulated genes, and blue points indicate significantly down-regulated genes. Raw gene counts were first normalized to TPM (transcripts per million). TPM counts were log transformed, filtered, scaled, and centered before gene clustering and heatmap generation using ClustVis (72), Heatmapper (73), or the R package (pheatmap). ClustVis uses code from BoxPlotR; several R packages are used internally, including shiny, ggplot2, pheatmap, gridSVG, rColorBrewer, FactoMineR, pcaMethods, gProfileR, shinyBS, shinyjs, and others. The source code of ClustVis is available in GitHub. For enrichment analyses, the up-regulated and down-regulated gene sets were generated by extracting genes with the respectively highest and lowest values from the gene expression signature. The gene sets were subsequently submitted to Enrichr (74), which is freely available at http://amp.pharm.mssm.edu/ Enrichr/. The following libraries were used for the analysis: GO_Cellular_Component_2018, WikiPathways_2016, and ChEA_2016. Significant terms were determined by using a cut-off of P < 0.1 after applying Benjamini-Hochberg correction. GSEA was carried out with GSEA desktop v4.0.3 using preranked lists generated from FDR values, setting gene set permutations to 1000, using the Hallmark or the c2 collections in mSigDB v7.2 or “KEGG Lysosome” gene sets. Four-way Venn diagrams were generated using Venny 2.1 (https://bioinfogp.cnb.csic.es/tools/venny/).
Statistical analysis
Statistical parameters including the exact value of n, measures (means ± SEMs), and statistical significance are reported in the figures and figure legends. Data are judged to be statistically significant when P < 0.05 by two-tailed Student’s t test. In figures, asterisks denote statistical significance as calculated by Student’s t test (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). Statistical analyses were performed using Graph Pad Prism 7. Analysis of RNA-seq data has been described in the section “Bioinformatic analysis of RNA-seq data.”
Supplementary Material
ACKNOWLEDGMENTS
Funding:
This study was supported by grants to R.J.S. from the National Institutes of Health (NIH) (R35CA220538, P01CA120964, and R01DK080425) and the Leona M. and Harry B. Helmsley Charitable Trust grant no. 2012-PG-MED002. The study received support to R.J.S. and G.S.S. by an AHA-Allen Initiative in Brain Health and Cognitive Impairment award made jointly through the American Heart Association and the Paul G. Allen Frontiers Group (19PABH134610000). The Salk NCI Cancer Center CCSG P30 CA014195 supported the Functional Genomics Core, the Bioinformatics Core, the Mass Spectrometry Core, the Flow Cytometry Core, and the Biophotonics Core. This research was also supported by the Nathan Shock Center for Aging Research at the Salk Institute P30 AG068635. U.M., L.F., R.G., A.S., and S.W.N. are supported by the Waitt Foundation and NCI CCSG P30 CA014195. U.M. is a Chan-Zuckerberg Initiative Imaging Scientist and is supported by NSF NeuroNex award 2014862 and NIH R21 DC018237. N.M. was supported by a Glenn Foundation Fellowship. We thank G. Kasof at Cell Signaling Technology for partnership in developing the pSer220 FNIP1 antibodies. We thank J. Moresco and J. Deitrich for their assistance in the Mass Spectrometry Core for Fig. 2B. All RNA-seq datasets in this study available through the Gene Expression Omnibus (GEO) under the accession no. GSE218827.
Footnotes
Competing interests: All authors declare that they have no competing interests.
Data and materials availability:
All RNA-seq datasets generated for this study are available on GEO under the accession no. GSE218827.
REFERENCES AND NOTES
- 1.González A, Hall MN, Lin SC, Hardie DG, AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 31, 472–492 (2020). doi: 10.1016/j.cmet.2020.01.015; pmid: 32130880 [DOI] [PubMed] [Google Scholar]
- 2.Hardie DG, Keeping the home fires burning: AMP-activated protein kinase. J. R. Soc. Interface 15, 20170774 (2018). doi: 10.1098/rsif.2017.0774; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Herzig S, Shaw RJ, AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol 19, 121–135 (2018). doi: 10.1038/nrm.2017.95; pmid: 28974774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kelly DP, Scarpulla RC, Transcriptional regulatory circuits controlling mitochondrial biogenesis and function. Genes Dev. 18, 357–368 (2004). doi: 10.1101/gad.1177604; pmid: 15004004 [DOI] [PubMed] [Google Scholar]
- 5.Roczniak-Ferguson A. et al. , The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal 5, ra42 (2012). doi: 10.1126/scisignal.2002790; pmid: 22692423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puertollano R, Ferguson SM, Brugarolas J, Ballabio A, The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 37, e98804 (2018). doi: 10.15252/embj.201798804; pmid: 29764979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Settembre C. et al. , A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 (2012). doi: 10.1038/emboj.2012.32; pmid: 22343943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vega-Rubin-de-Celis S, Peña-Llopis S, Konda M, Brugarolas J , Multistep regulation of TFEB by MTORC1. Autophagy 13, 464–472 (2017). doi: 10.1080/15548627.2016.1271514; pmid: 28055300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martina JA, Chen Y, Gucek M, Puertollano R, MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 (2012). doi: 10.4161/auto.19653; pmid: 22576015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young NP et al. , AMPK governs lineage specification through Tfeb-dependent regulation of lysosomes. Genes Dev. 30, 535–552 (2016). doi: 10.1101/gad.274142.115; pmid: 26944679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Eichner LJ et al. , Genetic Analysis Reveals AMPK Is Required to Support Tumor Growth in Murine Kras-Dependent Lung Cancer Models. Cell Metab. 29, 285–302.e7 (2019). doi: 10.1016/j.cmet.2018.10.005; pmid: 30415923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tsun ZY et al. , The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 52, 495–505 (2013). doi: 10.1016/j.molcel.2013.09.016; pmid: 24095279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen K. et al. , Cryo-EM Structure of the Human FLCN-FNIP2-Rag-Ragulator Complex. Cell 179, 1319–1329.e8 (2019). doi: 10.1016/j.cell.2019.10.036; pmid: 31704029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petit CS, Roczniak-Ferguson A, Ferguson SM, Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J. Cell Biol 202, 1107–1122 (2013). doi: 10.1083/jcb.201307084; pmid: 24081491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lawrence RE et al. , Structural mechanism of a Rag GTPase activation checkpoint by the lysosomal folliculin complex. Science 366, 971–977 (2019). doi: 10.1126/science.aax0364; pmid: 31672913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Settembre C. et al. , TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433 (2011). doi: 10.1126/science.1204592; pmid: 21617040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sardiello M. et al. , A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009). doi: 10.1126/science.1174447; pmid: 19556463 [DOI] [PubMed] [Google Scholar]
- 18.Perera RM, Di Malta C, Ballabio A, MiT/TFE Family of Transcription Factors, Lysosomes, and Cancer. Annu. Rev. Cancer Biol 3, 203–222 (2019). doi: 10.1146/annurev-cancerbio-030518-055835; pmid: 31650096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Collodet C. et al. , AMPK promotes induction of the tumor suppressor FLCN through activation of TFEB independently of mTOR. FASEB J. 33, 12374–12391 (2019). doi: 10.1096/fj.201900841R; pmid: 31404503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paquette M. et al. , AMPK-dependent phosphorylation is required for transcriptional activation of TFEB and TFE3. Autophagy 17, 3957–3975 (2021). doi: 10.1080/15548627.2021.1898748; pmid: 33734022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winder WW et al. , Activation of AMP-activated protein kinase increases mitochondrial enzymes in skeletal muscle. J. Appl. Physiol 88, 2219–2226 (2000). doi: 10.1152/jappl.2000.88.6.2219; pmid: 10846039 [DOI] [PubMed] [Google Scholar]
- 22.Bergeron R. et al. , Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab 281, E1340–E1346 (2001). doi: 10.1152/ajpendo.2001.281.6.E1340; pmid: 11701451 [DOI] [PubMed] [Google Scholar]
- 23.Zong H. et al. , AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc. Natl. Acad. Sci. U.S.A 99, 15983–15987 (2002). doi: 10.1073/pnas.252625599; pmid: 12444247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reznick RM, Shulman GI, The role of AMP-activated protein kinase in mitochondrial biogenesis. J. Physiol 574, 33–39 (2006). doi: 10.1113/jphysiol.2006.109512; pmid: 16709637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jäger S, Handschin C, St.-Pierre J, Spiegelman BM, AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc. Natl. Acad. Sci. U.S.A 104, 12017–12022 (2007). doi: 10.1073/pnas.0705070104; pmid: 17609368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cantó C, Auwerx J, PGC-1α, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol 20, 98–105 (2009). doi: 10.1097/MOL.0b013e328328d0a4; pmid: 19276888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kolmykov S. et al. , GTRD: An integrated view of transcription regulation. Nucleic Acids Res. 49, D104–D111 (2021). doi: 10.1093/nar/gkaa1057; pmid: 33231677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinberg GR, Carling D, AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov 18, 527–551 (2019). doi: 10.1038/s41573-019-0019-2; pmid: 30867601 [DOI] [PubMed] [Google Scholar]
- 29.Mootha VK et al. , Errα and Gabpa/b specify PGC-1α-dependent oxidative phosphorylation gene expression that is altered in diabetic muscle. Proc. Natl. Acad. Sci. U.S.A 101, 6570–6575 (2004). doi: 10.1073/pnas.0401401101; pmid: 15100410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schreiber SN et al. , The estrogen-related receptor α (ERRα) functions in PPARg coactivator 1α (PGC-1α)-induced mitochondrial biogenesis. Proc. Natl. Acad. Sci. U.S.A 101, 6472–6477 (2004). doi: 10.1073/pnas.0308686101; pmid: 15087503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rath S. et al. , MitoCarta3.0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 49, D1541–D1547 (2021). doi: 10.1093/nar/gkaa1011; pmid: 33174596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gwinn DM et al. , AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 (2008). doi: 10.1016/j.molcel.2008.03.003; pmid: 18439900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Behrends C, Sowa ME, Gygi SP, Harper JW, Network organization of the human autophagy system. Nature 466, 68–76 (2010). doi: 10.1038/nature09204; pmid: 20562859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baba M. et al. , Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc. Natl. Acad. Sci. U.S.A 103, 15552–15557 (2006). doi: 10.1073/pnas.0603781103; pmid: 17028174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Myers RW et al. , Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 357, 507–511 (2017). doi: 10.1126/science.aah5582; pmid: 28705990 [DOI] [PubMed] [Google Scholar]
- 36.Napolitano G. et al. , A substrate-specific mTORC1 pathway underlies Birt-Hogg-Dubé syndrome. Nature 585, 597–602 (2020). doi: 10.1038/s41586-020-2444-0; pmid: 32612235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gosis BS et al. , Inhibition of nonalcoholic fatty liver disease in mice by selective inhibition of mTORC1. Science 376, eabf8271 (2022). doi: 10.1126/science.abf8271; pmid: 35420934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sekiguchi T, Hirose E, Nakashima N, Ii M, Nishimoto T, proteins Novel G, Rag C and Rag D, interact with GTP-binding proteins, Rag A and Rag B. J. Biol. Chem 276, 7246–7257 (2001). doi: 10.1074/jbc.M004389200; pmid: 11073942 [DOI] [PubMed] [Google Scholar]
- 39.Gao M, Kaiser CA, A conserved GTPase-containing complex is required for intracellular sorting of the general amino-acid permease in yeast. Nat. Cell Biol 8, 657–667 (2006). doi: 10.1038/ncb1419; pmid: 16732272 [DOI] [PubMed] [Google Scholar]
- 40.Sancak Y. et al. , The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320, 1496–1501 (2008). doi: 10.1126/science.1157535; pmid: 18497260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abu-Remaileh M. et al. , Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813 (2017). doi: 10.1126/science.aan6298; pmid: 29074583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palmieri M. et al. , Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet 20, 3852–3866 (2011). doi: 10.1093/hmg/ddr306; pmid: 21752829 [DOI] [PubMed] [Google Scholar]
- 43.Pastore N. et al. , TFE3 regulates whole-body energy metabolism in cooperation with TFEB. EMBO Mol. Med 9, 605–621 (2017). doi: 10.15252/emmm.201607204; pmid: 28283651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pastore N. et al. , Nutrient-sensitive transcription factors TFEB and TFE3 couple autophagy and metabolism to the peripheral clock. EMBO J. 38, e101347 (2019). doi: 10.15252/embj.2018101347; pmid: 31126958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonam SR, Wang F, Muller S, Lysosomes as a therapeutic target. Nat. Rev. Drug Discov 18, 923–948 (2019). doi: 10.1038/s41573-019-0036-1; pmid: 31477883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Settembre C. et al. , TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol 15, 647–658 (2013). doi: 10.1038/ncb2718; pmid: 23604321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mansueto G. et al. , Transcription Factor EB Controls Metabolic Flexibility during Exercise. Cell Metab. 25, 182–196 (2017). doi: 10.1016/j.cmet.2016.11.003; pmid: 28011087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salma N, Song JS, Arany Z, Fisher DE, Transcription Factor Tfe3 Directly Regulates Pgc-1alpha in Muscle. J. Cell. Physiol 230, 2330–2336 (2015). doi: 10.1002/jcp.24978; pmid: 25736533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jannig PR, Dumesic PA, Spiegelman BM, Ruas JL, SnapShot: Regulation and biology of PGC-1α. Cell 185, 1444–1444.e1 (2022). doi: 10.1016/j.cell.2022.03.027; pmid: 35427500 [DOI] [PubMed] [Google Scholar]
- 50.Miura S, Kai Y, Kamei Y, Ezaki O, Isoform-specific increases in murine skeletal muscle peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) mRNA in response to β2- adrenergic receptor activation and exercise. Endocrinology 149, 4527–4533 (2008). doi: 10.1210/en.2008-0466; pmid: 18511502 [DOI] [PubMed] [Google Scholar]
- 51.Martínez-Redondo V, Pettersson AT, Ruas JL, The hitchhiker’s guide to PGC-1α isoform structure and biological functions. Diabetologia 58, 1969–1977 (2015). doi: 10.1007/s00125-015-3671-z; pmid: 26109214 [DOI] [PubMed] [Google Scholar]
- 52.Soyal SM et al. , A greatly extended PPARGC1A genomic locus encodes several new brain-specific isoforms and influences Huntington disease age of onset. Hum. Mol. Genet 21, 3461–3473 (2012). doi: 10.1093/hmg/dds177; pmid: 22589246 [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y. et al. , Alternative mRNA splicing produces a novel biologically active short isoform of PGC-1α. J. Biol. Chem 284, 32813–32826 (2009). doi: 10.1074/jbc.M109.037556; pmid: 19773550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olson BL et al. , SCFCdc4 acts antagonistically to the PGC-1α transcriptional coactivator by targeting it for ubiquitin-mediated proteolysis. Genes Dev. 22, 252–264 (2008). doi: 10.1101/gad.1624208; pmid: 18198341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chang JS, Ghosh S, Newman S, Salbaum JM, A map of the PGC-1α- and NT-PGC-1α-regulated transcriptional network in brown adipose tissue. Sci. Rep 8, 7876 (2018). doi: 10.1038/s41598-018-26244-4; pmid: 29777200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huss JM, Torra IP, Staels B, Giguère V, Kelly DP, Estrogen-related receptor a directs peroxisome proliferator-activated receptor a signaling in the transcriptional control of energy metabolism in cardiac and skeletal muscle. Mol. Cell. Biol 24, 9079–9091 (2004). doi: 10.1128/MCB.24.20.9079-9091.2004; pmid: 15456881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Luo C. et al. , ERRα Maintains Mitochondrial Oxidative Metabolism and Constitutes an Actionable Target in PGC1α-Elevated Melanomas. Mol. Cancer Res 15, 1366–1375 (2017). doi: 10.1158/1541-7786.MCR-17-0143; pmid: 28596418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rooney JP et al. , in Mitochondrial Regulation, Palmeira C, Rolo A, eds., vol. 1241 of Methods in Molecular Biology (Humana Press, 2015), pp. 23–38.25308485 [Google Scholar]
- 59.Siggs OM et al. , Mutation of Fnip1 is associated with B-cell deficiency, cardiomyopathy, and elevated AMPK activity. Proc. Natl. Acad. Sci. U.S.A 113, E3706–E3715 (2016). doi: 10.1073/pnas.1607592113; pmid: 27303042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reyes NL et al. , Fnip1 regulates skeletal muscle fiber type specification, fatigue resistance, and susceptibility to muscular dystrophy. Proc. Natl. Acad. Sci. U.S.A 112, 424–429 (2015). doi: 10.1073/pnas.1413021112; pmid: 25548157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan M. et al. , The tumor suppressor folliculin regulates AMPK-dependent metabolic transformation. J. Clin. Invest 124, 2640–2650 (2014). doi: 10.1172/JCI71749; pmid: 24762438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Possik E. et al. , Folliculin regulates Ampk-dependent autophagy and metabolic stress survival. PLOS Genet. 10, e1004273 (2014). doi: 10.1371/journal.pgen.1004273; pmid: 24763318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hung C-M et al. , AMPK/ULK1-mediated phosphorylation of Parkin ACT domain mediates an early step in mitophagy. Sci. Adv 7, eabg4544 (2021). doi: 10.1073/pnas.1413021112; pmid: 25548157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Howell JJ et al. , Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 25, 463–471 (2017). doi: 10.1172/JCI71749; pmid: 24762438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Harris KM et al. , Uniform serial sectioning for transmission electron microscopy. J. Neurosci 26, 12101–12103 (2006). doi: 10.1523/JNEUROSCI.3994-06.2006; pmid: 17122034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berger DR, Seung HS, Lichtman JW, VAST (Volume Annotation and Segmentation Tool): Efficient Manual and Semi-Automatic Labeling of Large 3D Image Stacks. Front. Neural Circuits 12, 88 (2018). doi: 10.3389/fncir.2018.00088; pmid: 30386216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dobin A. et al. , STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). doi: 10.1093/bioinformatics/bts635; pmid: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heinz S. et al. , Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). doi: 10.1016/j.molcel.2010.05.004; pmid: 20513432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Torre D, Lachmann A, Ma’ayan A, BioJupies: Automated Generation of Interactive Notebooks for RNA-Seq Data Analysis in the Cloud. Cell Syst. 7, 556–561.e3 (2018). doi: 10.1016/j.cels.2018.10.007; pmid: 30447998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ritchie ME et al. , limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). doi: 10.1093/nar/gkv007; pmid: 25605792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goedhart J, Luijsterburg MS, VolcaNoseR is a web app for creating, exploring, labeling and sharing volcano plots. Sci. Rep 10, 20560 (2020). doi: 10.1038/s41598-020-76603-3; pmid: 33239692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Metsalu T, Vilo J, ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, W566–W570 (2015). doi: 10.1093/nar/gkv468; pmid: 25969447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Babicki S. et al. , Heatmapper: Web-enabled heat mapping for all. Nucleic Acids Res. 44, W147–W153 (2016). doi: 10.1093/nar/gkw419; pmid: 27190236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuleshov MV et al. , Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). doi: 10.1093/nar/gkw377; pmid: 27141961 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All RNA-seq datasets generated for this study are available on GEO under the accession no. GSE218827.