

Summary
Immune checkpoint blockade (ICB) therapy targeting cytotoxic T-lymphocyte-associated protein 4, programmed death 1, and programmed death ligand 1 has shown durable remission and clinical success across different cancer types. However, patient outcomes vary among disease indications. Studies have identified prognostic biomarkers associated with immunotherapy response and patient outcomes derived from diverse data types, including next-generation bulk and single-cell DNA, RNA, T cell and B cell receptor sequencing data, liquid biopsies, and clinical imaging. Owing to inter- and intra-tumor heterogeneity and the immune system’s complexity, these biomarkers have diverse efficacy in clinical trials of ICB. Here, we review the genetic and genomic signatures and image features of ICB studies for pan-cancer applications and specific indications. We discuss the advantages and disadvantages of computational approaches for predicting immunotherapy effectiveness and patient outcomes. We also elucidate the challenges of immunotherapy prognostication and the discovery of novel immunotherapy targets.
Graphical abstract
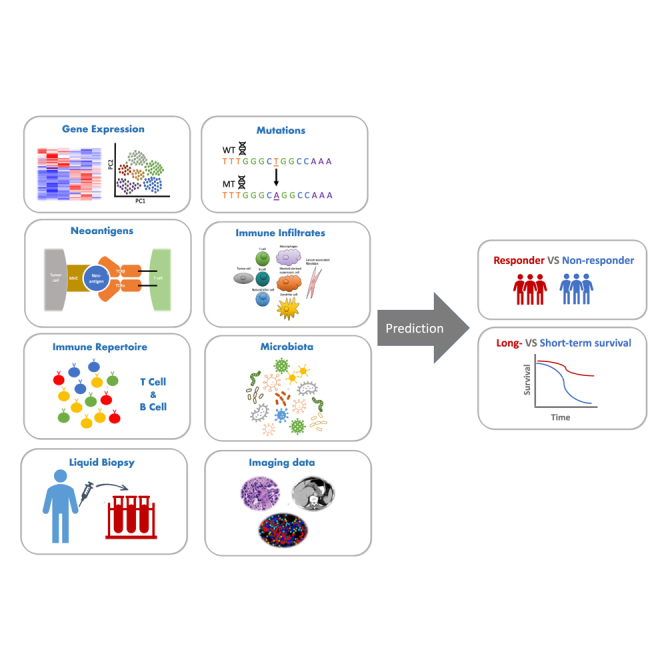
Liu et al. reviewed recently developed biomarkers derived from multi-omics and multi-modality data resources for predicting cancer patient outcomes treated with immune checkpoint blockade therapy. These biomarkers play an important role in patient selection for clinical decision-making, offer insights into mechanisms driving tumor-cell-intrinsic and -extrinsic heterogeneity, and contribute to discovering novel therapeutic targets. As immunotherapy advances, the integration of advanced computational approaches with ever-expanding data resources promises continued progress and substantial benefits for cancer patients.
Introduction
The United States Food and Drug Administration (FDA) approved the first immune checkpoint blockade (ICB) therapy for treating melanoma 12 years ago, ushering in the era of cancer immunotherapy. The mechanism of ICB therapy is based on insights into the interactions between the human immune system and tumor cells. In brief, neoantigens can bind to a cell’s major histocompatibility complex (MHC) class I and II molecules, be presented on the cell’s surface, and elicit an immune response by interacting with T cell receptors (TCRs). This interaction helps T cells recognize non-self peptides, which allows the immune system to distinguish tumor from normal cells,1 thus enabling the process of immune surveillance. The ability to specifically target neoantigens has been documented across diverse cancer immunotherapies, including immune checkpoint blockade2 and cancer vaccines.3,4 Immune cells secrete cytokines when activated, which contribute to the killing of tumor cells. Tumor cells, however, can escape immune surveillance and interact with tumor-killing immune cells to suppress their activity. ICB agents block such suppressive interactions to enhance the activation of immune cells and allow the latter to kill tumor cells effectively.5 Cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) was the first FDA-approved ICB target; it is an immune-suppressive cell ligand expressed on the surface of regulatory T cells (Tregs), as well as other T cell subsets, which indirectly increases the threshold of immune cell activation through competing for the binding ligands with CD28, a co-stimulatory signal ligand for tumor-killing cells (T cells).6
The FDA has now given the green light to several antibodies targeting negative regulators of the immune response, such as programmed death 1 (PD-1) and its ligand programmed death ligand 1 (PD-L1), for the treatment of multiple cancer types, including melanoma, non-small cell lung cancer (NSCLC), and renal cell carcinoma (RCC). PD-1 is a homolog of CD28 that can be expressed on tumor-infiltrating T cells and other immune cells. Targeting PD-1/PD-L1 is direct, unlike CTLA-4, enhancing the activation of T cells and killing tumor cells expressing PD-L1.6 Combination therapy targeting both lymphocyte-activation gene 3 (LAG-3) and PD-1 is now available to treat unresectable or metastatic melanoma. Currently, the United States has nine FDA-approved drugs targeting immune checkpoint interactions: ipilimumab, pembrolizumab, nivolumab, atezolizumab, durvalumab, avelumab, cemiplimab, dostarlimab, and relatlimab.
Despite the success of these agents, patient outcomes are heterogeneous. The biological basis of this heterogeneity requires molecular information to target patient populations for these therapies and to optimize their response. Clinical studies have utilized advanced computing methods to identify molecular biomarkers associated with response and survival. Currently validated and proposed biomarkers include gene expression signatures, mutational burden and related signatures, antigen specificity, measures of immune cell infiltration, immune repertoire characteristics, microbiome species, features derived from imaging modalities, and multi-modal data integrations. Although many prognostic biomarkers have been proposed, few have been approved for clinical use, including PD-L1 expression levels, microsatellite instability and defective mismatch repair, and tumor mutation burden. Here, we describe published biomarkers, discuss the computational and clinical implications of those insights, and explore challenges and potential future developments for identifying biomarkers of ICB response.
Gene expression signatures
Transcriptomic sequencing quantifies RNA expression across a genome. RNA quantification and biological functions elucidated with transcriptomics in precision cancer medicine have been reviewed elsewhere.7 For ICB outcome prediction, the expression levels of genes associated with immune response mechanisms, from prior knowledge of cancer immunology, are commonly used. Those genes include known ICB targets (CTLA4, PD-1, and PD-L1), cytokines and chemokines,8,9,10 and genes involved in neoantigen presentation and immune-related biological pathways.11,12 However, those genes have varied predictive effectiveness due to the large extent of inter- and intra-tumor heterogeneity and the complexity of the immune system. It is therefore necessary to measure the tumor microenvironment more precisely for effective ICB-response prediction.
Cytotoxic T lymphocytes (CTLs) are the most direct and powerful tumor-killing cells in the innate immune system.13 Two genes, granzyme A (GZMA) and perforin (PRF1), were identified to be strongly upregulated by CD8+ T cell activation.14 The geometric mean of transcriptomic levels of these two genes can be used to quantify the extent of cytolytic activity, which is also positively correlated with the expression levels of antigen presentation genes.14 Based on the level of cytolytic activity, tumors can be characterized as “hot” and “cold.” Hot tumors are immune inflamed with high levels of T cell infiltrates, which may be in a dysfunctional status, and cold tumors can be further characterized as “immune desert” (absence of T cells) or “immune excluded” (only exhibiting peripheral invasion of T cells).15 Patients have also been stratified into hot and cold tumors by the average expression of CTL-related genes16 such as CD8A, CD8B, GZMA, GZMB, and PRF1. A T cell dysfunction score was then developed to predict patient response for hot tumors and a T cell exclusion score for cold tumors (Figure 1A and Table 1).
Figure 1.

Gene signatures are usually derived from a study cohort and validated through other independent cohorts
(A) Gene set signatures are discovered from clustering or differential gene expression analysis. A gene set score can be calculated as the weighted sum of normalized gene expression levels.
(B) Gene pairs are derived from the differential expression analysis between responders and non-responders or between long-term and short-term survivors. The MAPK4 and TBX3 gene pair, for instance, was selected through testing the predictive efficacy of each pair using a logistic regression and Cox-PH model.
(C) The IMPRES score was derived from calculating the logical relations of the 15 curated gene pairs list.
Table 1.
Signatures derived from gene expression data
| Reference | Cancer | Signatures | Notes |
|---|---|---|---|
| Ayers et al.17 | pan-cancer | TIGIT, CD27, CD8A, PDCD1LG2, LAG3, CD274, CXCR6, CMKLR1, NKG7, CCL5, PSMB10, IDO1, CXCL9, HLA-DQA1, CD276, STAT1, HLA-DRB1, HLA-E | T cell inflammation |
| Rooney et al.14 | pan-cancer | GZMA, PRF1 | cytolytic activity |
| Rooney et al.14 | pan-cancer | B2M, HLA-A, HlA-B, HLA-C, CASP8 | antigen presentation |
| Jiang et al.16 | melanoma and NSCLC | TIDE score | – |
| Lemvigh et al.18 | AML | ZNF683 | expressed in CE8+ effector and memory T cells |
| Helmink et al.20 | melanoma | CD79B, CD1D, CCR6, LAT, SKAP1, CETP, EIF1AY, RBP8, PTGDS | TLS-related genes |
| Hugo et al.25 | melanoma | IPRES: GSVA score from 21 curated gene sets | – |
| Freeman et al.21 | melanoma | MAP4K, TBX3 | gene pair expressed in immune and tumor cell |
| Auslander et al.22 | melanoma | IMPRES: score derived from the logical relationship of 15 gene pairs | – |
| Hwang et al.142 | NSCLC | CBLB, CCR7, CD27, CD48, FOXO1, FYB, HLA-B, HLA-G, IFIH1, IKZF4, LAMP3, NFKBIA, SAMHD1 | M1 Signatures |
| – | – | HLA-DOA, GPR18, STAT1 | peripheral T cell |
| – | – | CD137, PSMB9 | – |
| van Galen et al.143 | AML | NPTX2, H1F0, EMP1, MEIS1, CALCRL, TPSD1, TPT1, CRHBP, CLNK, TSC22D1, DST, NRIP1, ABCB1, GABRA4, ZBTB20, ABCA9, TPSB2, KMT2A, FAM30A, MEF2C, TMEM74, PDZRN4, ST3GAL1, XIRP2, RBPMS, TMEM25, C20orf203, GNG11, SLC6A13, HOPX | HSC-like |
| – | – | CDK6, HSP90AB1, SPINK2, EEF1B2, PCNP, TAPT1-AS1, HINT1, LRRC75A-AS1, DSE, PEBP1, LOC107984974, H2AFY, EEF1A1, SMIM24, PSME1, SOX4, LINC01623, EEF1G, EBPL, EIF4B, PARP1, MEST, TMEM70, TFDP2, ATP5G2, NAP1L1, MSI2, TPM4, SPN, SELL | progenitor-like |
| Wang et al.144 | melanoma and GBM | CXCL9, CXCL10, CXCL11, CXCR3, CD3, CD4, CD8a, CD8b, CD274, PDCD1, CXCR4, CCL5 | hot tumor-related gene |
| – | – | CXCL1, CXCL2, CCL20 | cold tumor-related genes |
Through differential gene expression analysis at bulk and single-cell levels, researchers have identified genes highly expressed in tumor-killing cells. For instance, an 18-gene signature of anti-PD1 treatment was identified according to differentially expressed genes between responders and non-responders of metastatic melanoma; this signature was later validated in head and neck squamous cell carcinoma (HNSCC) and gastric cancer.17 Those 18 genes, which show higher expression in responders, were associated with interferon-γ (IFN-γ) signaling, representing the mechanism of the PD-1/PD-L1 pathway that kills tumor cells. Recently, the ZNF683 gene was found to regulate T cell cytotoxicity and activation and to be highly expressed in CD8+ effector/memory T cells based on single-cell RNA sequencing (scRNA-seq) data of Richter syndrome patients; this finding was further validated in melanoma and HNSCC.18 In addition to T cells, tertiary lymphoid structures (TLSs), organized aggregates of immune cells that form in chronically inflamed environments, have been shown to correlate with clinical outcomes in NSCLC, colorectal cancer (CRC), melanoma, and breast cancer.19 A nine-gene signature was used to describe the TLS phenotype of melanoma after anti-CTLA4 or anti-PD1 treatment, as patients with higher average expression of these nine genes have significantly longer survival than those with relatively lower expression20 (Figure 1A and Table 1).
A paired-gene approach has also been used to derive predictive immune scores of ICB. For example, by using a multi-variate Cox hazard model for survival and logistic regression model for response screening for all gene pairs in patients with long and short survival, the gene pair MAP4K1 & TBX3 was the most significantly associated with survival21 (Figure 1B and Table 1). The immuno-predictive score (IMPRES), which was derived by selecting 15 gene pairs created from 28 immune checkpoint genes, predicts the risk of tumor progression in a neuroblastoma cohort of 108 patients.22 The IMPRES score ranges from 0 to 15 and is determined by counting the number of gene pairs in a patient’s transcriptomic profile that have a similar logical relation to the selected gene pairs (Figure 1C and Table 1). Similarly, gene partners inferred from the protein expression data from The Cancer Genome Atlas (TCGA), which show a synthetic rescue (SR) relationship with PD-1/PD-L1 or CTLA-4, were used to predict patient response.23,24 The SR score was defined as the fraction of inferred gene partners whose expression is below the tertile across the patient cohort and is associated with positive responses.
Gene set enrichment analysis has also been applied to calculate predictive immune scores. For instance, an innate anti-PD-1 resistant score (IPRES) was developed using data from 28 metastatic melanoma patients (Table 1). The IPRES score is determined from the averaged single-sample gene set variation analysis (GSVA) scores obtained from 26 curated transcriptomic gene sets; these gene sets were determined from public chemical and genetic perturbations, molecular signature databases and published signatures of MAPK inhibitor treatment, post-operation wound and melanoma invasion, and proliferation datasets.25
Mutation burden and mutational signatures
Tumor mutation burden (TMB) is a widely used biomarker for stratifying cancer patients who may benefit from ICB, based on the assumption that more mutations may increase the number of neoantigens, thus allowing for enhanced immunogenicity.26 Quantifying the number of mutations per megabase (Mb) of DNA is an FDA-approved biomarker for predicting patient outcomes in certain cancer types.27,28,29,30 However, TMB is not predictive across all cancer types, and experimental measures of TMB are often inconsistent, which can limit comparisons across trials and datasets.26,31 Importantly, the dissimilar underlying biology and mutation levels of different cancer types make it difficult to derive a single pan-cancer threshold for patient selection. In addition, statistical analyses for quantifying TMB and assessing its accuracy for response prediction can be sensitive to method and parameter choices as well as the specific assay used to determine mutations.
A genomics meta-analysis aggregating data from over 10,000 patients across 31 tumor types in TCGA showed that a classification of tumors into TMB-high (TMB-H) and TMB-low (TMB-L) types was cancer-type specific and that no single cutoff was universally valid across cancer types in terms of clinical relevance.26 This analysis found that a more relevant split of cancer types regarding their ICB response was whether CD8+ T cell infiltration positively correlated with neoantigen load. For tumors for which infiltrating CD8+ T cell numbers positively correlated with neoantigen load, TMB-H predicted immune response, and its use as a biomarker was justified.
However, the predictive accuracy of the FDA-approved TMB test varies across cancer types. For some of the most common cancer types (glioma, prostate, and breast cancers), TMB-H is not predictive. The FDA granted accelerated approval to pembrolizumab for treating unresectable or metastatic TMB-H solid tumors that progressed following prior treatment and without satisfactory alternative treatment options, for which TMB-H was determined by the 309-gene FoundationOne CDx assay with ≥10 mutations per Mb of DNA.32
Other TMB-derived biomarkers may also be effective in predicting immune response. A meta-analysis of whole-exome and transcriptomic data for >1,000 ICB-treated patients across seven tumor types identified that clonal TMB (i.e., the number of non-synonymous mutations estimated to be present in every tumor cell) was the strongest independent predictor of ICB response.33 Specific copy-number alterations can also be significant ICB predictors. For instance, 9q34 loss is associated with response and CCND1 amplification with resistance to ICB therapy.33 Similarly, 9p21 loss induces a cold tumor microenvironment and immune resistance.34 Persistent TMB (pTMB), defined as mutations that exist in haploid or polyploid regions of the genome, represents another potential mutational biomarker; these mutations are less likely to be edited by the immune system compared to mutations in diploid regions of the genome.35 This metric was evaluated across TCGA datasets and eight ICB cohorts consisting of NSCLC, melanoma, mesothelioma, and HNSCC patients, demonstrating that pTMB is weakly correlated with TMB or clonal TMB in most low-mutation-burden cancers and positively correlated with infiltrates of CD8, CD4 T cells, and M1 macrophages.35 Compared to TMB, pTMB was better at distinguishing TCGA early-stage (I, II, and III) patient survival and ICB response.35 The observation that pTMB from the simulated copy-number variations of the target gene regions captured by the FDA-approved FoundationOne CDx panel was also more significantly able than TMB to differentiate responders from non-responders of ICB-treated melanoma and NSCLC cohorts further suggests a potential for clinical applications of pTMB.35
Mutation types have also been analyzed for their ability to predict ICB response through aggregation into mutational processes. Classification of mutational signatures into 20 mutational process definitions showed that five were significantly associated with ICB response—signature 1A (aging), signature 4 (tobacco), signature 7 (ultraviolet; UV), signature 10 (DNA polymerase epsilon; POLE), and signature 2 + 13 (APOBEC cytidine deaminases)—even after correcting for total mutation count.36 This finding suggests that the mechanism driving mutation acquisition is important for ICB outcomes and that the predictive capacity of mutation-based scores is not solely a consequence of the number of mutations. These signatures (except 1A, aging) were associated with a significantly improved chance of ICB response. Additionally, the prognostic value of tumor aneuploidy among tumors with low TMB (<80th percentile) showed a less predictive capacity than the standard TMB measure.37 A higher tumor aneuploidy score (AS), defined as the fraction of chromosome arms with arm-level copy-number alterations in a sample, was associated with poorer prognosis following ICB treatment among tumors with low TMB but not among those with high TMB. However, a reanalysis of the same dataset separately for each cancer type found that the original AS only significantly predicted survival in one out of ten cancer types.38 In addition, the AS was not significantly associated with any individual cancer type. By changing the cutoff based on which a copy-number change is defined (from 0.1 to 0.2), this study found that the fraction of the genome encompassed by copy-number alterations had a stronger predictive value than the AS.
The FDA approved the anti-PD1 treatment pembrolizumab for the first-line treatment of CRC patients with microsatellite instability-high (MSI-H) or mismatch-repair-deficient (dMMR)39,40 characteristics. dMMR status is determined by immunohistochemical analysis when MMR proteins (MLH1, MSH2, MSH6, and PMS2) are absent. MSI status is determined by PCR-based analysis, and MSI-H is defined as detecting more than two out of five tumor microsatellite loci (BAT-25, BAT-26, D2S123, D5S346, and D17S250).41 In a clinical trial of 307 CRC patients, anti-PD1 treatment improved progression-free survival (PFS) and overall survival (OS) compared to the chemotherapy arm.42 In another advanced rectal cancer cohort of 12 dMMR patients, complete responses were observed in all patients treated with anti-PD-1 monotherapy with no reported progression or adverse events.43
Computational methods have also been applied to portray the MSI landscape across 39 cancer types from whole-exome sequencing data from TCGA and Therapeutically Applicable Research to Generate Effective Treatments (TARGET) projects.44 MSI was detected in 27 cancer types, among which only 12 were found to have more than a single MSI-H tumor represented in the cohort. Uterine corpus endometrial carcinoma has the largest MSI-H prevalence among all cancers, suggesting that ICB therapy may be effective for this cancer type. In a clinical trial with a mix of 697 MMR-proficient (pMMR) and 130 dMMR endometrial cancer patients designed to compare the effectiveness of combined target and anti-PD1 therapy versus chemotherapy alone,45 more positive outcomes were observed in the combined treatment group than in the chemotherapy group; however, the trial was not designed for dMMR patients. Furthermore, among nine metastatic prostate cancer patients with MSI-H status detected from circulating tumor DNA in the blood, there was one complete response to ICB treatment and two partial responses while treatment was ongoing.46 These findings suggest that MSI-H can be a potential indication for ICB treatment not only for CRC but also for other MSI-H cancers.
Antigen specificity
Tumor mutations can alter the amino acid sequence and increase the abundance and diversity of neoantigen peptides. However, only limited mutation-derived peptides are presented on the surface of an individual’s MHCs, which may explain why TMB is not the optimal biomarker. Neoantigen load or tumor neoantigen burden, defined as the number of neoantigens per Mb in the genome, may be more effective for predicting ICB response.47,48
Neoantigen load alone is not sufficient for predicting ICB response, because a patient may not have T cells that recognize the novel antigen even if presented on an MHC. Neoantigen load calculated from whole-exome sequencing (WES) data from 337 ICB-treated melanoma patients did not demonstrate a significant association with ICB response.49 However, the survival rate was proportional to neoantigen quality, defined as the probability of a peptide being recognized by a TCR sequence amplified by inferred wild-type and mutant peptide MHC class I affinities. Several computational models have been developed to describe neoantigen quality. A neoantigen quality fitness model, describing fitness as the product of the TCR recognition probability and the binding affinity ratio between mutant and wild-type neoantigens, was applied to two ICB melanoma cohorts and one NSCLC cohort.50 The results demonstrated that a higher fitness score was significantly associated with longer survival. This fitness model was extended by adding a cross-reactivity distance, which estimates the antigenic distance required for T cells to discriminate between a mutant and wild-type peptide.50,51 This extended model revealed that cancer immunoediting, a hypothesis that T cells selectively and dynamically edit tumor cell clones to promote less immunogenic tumor cell escape from immunosurveillance, takes place in pancreatic ductal carcinomas, which have a low mutation or neoantigen load; this model was able to describe the clonal evolution of tumors.
Immune infiltration
Tumors comprise cancer and non-cancer cells, including immune cells, stromal cells, and endothelial cells, which collectively form the tumor microenvironment (TME). Among immune cells, tumor-infiltrating lymphocytes (TILs) have been the focus of attention as predictive biomarkers of ICB response due to their role in clearing tumor cells.52 TILs are a heterogeneous population of TCR-expressing T cells, with the CD8+ CTLs and CD4+ T cells among the best-characterized subsets.
TIL quantifications are based on the transcriptomic level of IFN-γ signaling and are mostly done through manual histopathological assessment of hematoxylin and eosin (H&E)-stained tissue sections produced during a patient’s clinical diagnostic workup.53 Immunohistochemical markers such as CD4, CD8, FOXP3, and CD25 provide some, albeit limited and static, characterization of TIL subsets. Multi-color flow cytometry allows for a wider array of phenotypic markers with quantitative outputs but loses spatial information and has limited clinical application because it requires fresh tumor samples.54 Beyond evaluating phenotypic markers, diverse molecular and computational techniques are available for profiling TILs, including scRNA-seq,55 multi-omics profiling in combination with computational deconvolution of immune cell populations,56 and multiplex imaging57 capturing the spatial and functional resolution of TILs. Given that the distribution of TILs within the TME and the spatial relationships between tumor and immune cells are important factors when assessing mechanisms of resistance to ICB agents, advanced spatial transcriptomics profiling allows for the molecular characterization of TILs within their microenvironment. This approach then offers a means of interrogating functional TIL states and their interactions with other cell types.
The presence of TILs is a favorable overall prognostic indicator in several tumor types,58,59,60,61 but TIL quantification has been an inconsistent predictor of response to ICB in clinical studies.62,63 The predictive efficacy of TILs varies by different ICB agents; for instance, a study of melanoma treated with CTLA-4 blockade did not find the pre-treated TIL abundance to be a predictor for post-treatment response.64 However, in various cancers treated with PD-L1 blockade, a higher density of CD8+ T cells in the tumor core and invasive margins in pre-treatment biopsies was correlated with response.62,65 The difference between these findings may be explained by the observation that Tregs express CTLA-4 while PD-1 is expressed on tumor-infiltrating T cells.6 In addition, examining the ratio of CD8+ CTLs in different compartments or with other T cell subsets could be a useful proxy measurement of the TME composition. Indeed, a CD8+/CD4+ ratio of less than 2, inferring relatively fewer CTLs relative to helper T cells, is associated with a negative response to PD-1 blockade in melanoma.66 This ratio also predicted ICB response in NSCLC patients, whereas CD8+ CTLs alone did not.
TIL quantification is highly variable across cancer types and patients67 and can be improved by incorporating heterogeneity in tumor specificity, phenotype, and functional states of TILs. Phenotypic analysis of clinical tumor samples has shown a large proportion of TILs similar to tissue-resident memory (TRM) cells based on canonical markers of tissue residency such as CD69, CD103, and/or CD49,68,69,70 as well as a higher enrichment of memory/residency programs relative to other T cell states.71 This observation suggests the importance of TRM cell populations in tumor control. Indeed, several tumor types have shown correlations between intra-tumoral CD103+CD8+ cells and improved patient prognosis. For instance, oral squamous cell carcinoma patients who received neoadjuvant ICB treatment were found to harbor clonally expanded TILs enriched in tissue-resident memory and cytotoxicity programs.72 Notably, PD-1 expression in tumors is most prominent in cells with TRM characteristics,73 suggesting that TRM cells may be an essential determinant of ICB response, particularly with PD-1 blockade.
TILs are not all individually scattered in a tumor and sometimes aggregate or form TLSs.19 Patients with soft-tissue sarcomas exhibited the highest overall response (OS) and improved PFS to PD-1 treatment with high levels of immune infiltration, if prior to treatment markers of TLS and B cell lineage gene expression signatures were present as compared to patients without these markers.74 In melanoma patients, the density of TLS, CD20+ B cells, and the ratio of TLS to tumor area in the treated samples were higher in responders than non-responders to neoadjuvant ICB.20 Furthermore, responders had a correlated higher expression of B cell markers in pre-treatment and treatment samples. Overall, TLS shows promise as a biomarker; however, the assessment of TLS lacks standardized criteria, T and B cell markers, and consensus on distinguishing TLS from a TIL aggregate.75,76
Incorporating information on spatial TIL distribution may improve predictive models, as CTLs need close contact with tumor cells to function. Indeed, the ratio of CD8+ T cells, independent of CD8+ density, at the invasive margin relative to the tumor core was significantly higher in melanoma biopsies of responders to PD-1 blockade as compared to non-responders.77 Although spatial transcriptomics approaches improve the resolution of TILs, current inconsistencies between study findings highlight gaps in our understanding, a need for standardized criteria for TIL assessment, and the need for validation studies to translate these findings into clinically helpful information.78
Immune repertoire
Assessment of the adaptive immune receptor repertoire, an individual’s sum of unique T and B cell receptors, provides a snapshot of an individual’s adaptive immune status.79 Over 90% of human T cells have TCRs consisting of paired α and β chains, with the remainder having TCRs consisting of paired γ and δ chains.80 The complementarity-determining region 3 (CDR3) of the β chain is essentially unique and the primary contributor to TCR diversity, due to the incredible diversity produced through the rearrangement of the variable (V), diverse (D), joining (J), and constant (C) gene segments, in addition to genomic insertions and deletions.80,81 Advances in single-cell analysis have enabled paired chain resolution and improved the accuracy of clonotype analysis and modeling of antigen specificity.82,83
Both the Shannon and Simpson indices can be used to measure the extent of diversity of the immune repertoire, taking into consideration the number of distinct clonotypes (richness) and the relative abundance of these clonotypes (evenness) to extract biological insights and the degree of overlap between repertoires (Figures 2A–2C). Meaningful comparisons can only be made if repertoires have sufficient sampling and similar repertoire sizes.84 This lack of standardization in measuring clonality and reporting TCR repertoires makes comparing studies difficult, with the diversity value affected by the sequencing depth.
Figure 2.

Clonal diversity is used to describe the evenness and richness of immune repertoires and microbiota
(A) CDR3 sequences can be clustered into multiple unique clonotypes, which contribute to the diversity of TCR and BCR repertoires.
(B) Visualization of Shannon and inverse Simpson diversity indices used to characterize immune repertoires into low and moderate populations and high clonality of equal clonotype richness. Diversity indices take into consideration richness and evenness. High index values indicate an equal distribution of the CDR3 sequences and low diversity. Clonality is inversely proportional to evenness.
(C) Diversity indices are frequently used to describe the diversity of the TCR/BCR repertoire and the gut microbiome.
Given that ICB is believed to work by activation of previously primed T cells,85 patients with higher intra-tumoral TCR clonality are expected to respond better than those who have relatively low clonality. However, high intra-tumoral TCR clonality in the pre-treatment sample was inconsistently correlated with clinical response to PD1 blockade in melanoma and lung cancer patients.28,62,84,86,87 However, in the post-treatment biopsies of responders, there was 10-fold more TCR clonal expansion than in non-responders, while the tumor specificity of these clones was not assessed.62 Additionally, prior or combination therapy, compared to monotherapy, may enhance TCR clonality and lead to better immune response. For instance, melanoma patients previously treated with anti-CTLA4 blockade who responded to subsequent anti-PD1 therapy had higher TCR richness, whereas naive responders had higher TCR evenness.28 Furthermore, higher clonality was observed in responders to anti-PD1 as a monotherapy or in combination with anti-CTLA4 therapy.86 Additionally, anti-PD1/CTLA4 combination therapy patients had an expansion of intra-tumoral clones in the peripheral blood but not anti-PD1 monotherapy patients. In addition to melanoma, increased TCR clonality has also been associated with responders in NSCLC, glioblastoma, metastatic bladder cancer, and pancreatic cancer.88,89 These findings suggest that expansion of tumoral TCR clonotypes is a feature of response to PD1 blockade.
The intra-tumoral B cell receptor (BCR) repertoire typically has very high clonality due to the presence of clonal plasma cells, with most of the current work based on transcriptomes derived from bulk tumors or sorted B cell subsets.90 In recent reviews of the limited number of studies examining the BCR repertoire, high clonality and large amounts of somatic hypermutations were associated with a better prognosis in melanoma but not with response in lung adenocarcinoma.90,91 Furthermore, spatial BCR profiling in renal RCC demonstrated that in situ somatic hypermutation occurred within the TLS, and resulting plasma cells disseminated into the tumor.92,93 Additionally, the presence of tumor cells coated with high levels of immunoglobulin was correlated with a positive response to a combination of PD-1 and CTLA-4 blockade therapy. These findings implicate that B cells play an essential role within the TME and particularly in the TLS. Given the importance and ubiquity of intra-tumor clonal heterogeneity, the use of spatial transcriptomic technologies will be a key addition to more accurately interpreting immune repertoire results in the future.
Microbiota signatures
The intestinal microbiome extensively interacts with the host’s innate and adaptive immune system; details of this interaction and the role of the gut microbiome in cancer have been reviewed elsewhere.94 The microbiota affects the efficacy of immunotherapy agents, and several studies have explored the association of microbiomes with clinical response (Table 2). Clinical trial studies demonstrated that fecal microbiota transplantation (FMT), a transfer of gut microbiota from a healthy donor to a recipient, may promote anti-PD1 treatment efficacy and result in a better response in refractory melanoma patients who have progressed on previous ICB treatment.95,96 Studies that explored the gut microbiome’s effect on ICB efficacy in germ-free mice and patients found that Bacteroides thetaiotaomicron, Bacteroides fragilis, and members of the order Clostridiales were associated with anti-CTLA4 efficacy in melanoma and NSCLC, and the commensal genus Bifidobacterium enhanced anti-PD-L1 immunotherapy in melanoma.97,98 Similarly, recent findings ascertained that the relative abundance of the Bifidobacteriaceae family was enriched in responders to anti-PD1/PD-L1 treatment of melanoma and NSCLC.99,100 In addition, the Ruminococcus genus is associated with better response and survival in melanoma and NSCLC.101,102 Akkermansia muciniphila has also been associated with positive outcomes in NSCLC and RCC.100,103,104
Table 2.
Signatures derived from microbiome data
| Microbiota | Cancer | Cohorts | Reference |
|---|---|---|---|
| Bacteroides | melanoma | 15 patients with FMT and anti-PD1 | Davar et al.95 |
| – | melanoma | 10 prognostic patients with FMT and anti-PD1. FMT was received from 2 donors; significance was only found within the donor 1 group but not found when comparing donor 1 and donor 2 groups together | Baruch et al.96 |
| – | RCC | 26 patients with anti-PD1 | Sivan et al.98 |
| – | melanoma | 27 patients with immunotherapy | Peters et al.101 |
| – | melanoma | 94 patients with anti-PD1 treatment from multiple cohorts | McCulloch et al.106 |
| – | RCC | 31 patients with anti-PD1 or combined anti-CTLA4 and anti-PD1 treatment | Salgia et al.103 |
| – | melanoma | 54 patients with combined anti-CTLA4 and anti-PD1 treatment | Andrews et al.107 |
| Bifidobacterium | melanoma | 42 patients with anti-PD1 treatment; score calculating the ratio of 43 OTUs between different clinical-beneficial patients | Matson et al.99 |
| – | RCC | 31 patients with anti-PD1 or combined anti-CTLA4 and anti-PD1 treatment | Salgia et al.103 |
| – | melanoma | 147 patients from multiple cohorts | Lee et al.105 |
| Akkermansia muciniphila | RCC | 31 patients with anti-PD1 or combined anti-CTLA4 and anti-PD1 treatment | Salgia et al.103 |
| – | NSCLC | 338 patients with anti-PD1 treatment; patients were stratified by Akkermansia muciniphila levels, and worst survival was observed in the overabundance group, followed by low abundance group and no-abundance group | Derosa et al.100 |
| – | RCC | 69 patients with anti-PD treatment | Derosa et al.104 |
| Ruminococcus genus | melanoma | 27 patients with immunotherapy | Peters et al.101 |
| – | melanoma | 112 patients with anti-PD1 treatment | Gopalakrishnan et al.108 |
| – | NSCLC | 70 patients with anti-PD1/PD-L1 treatment | Hakozaki et al.102 |
In melanoma, a cross-cohort analysis integrating 147 metagenomics samples demonstrated that no consistent single microbiota species could be used as a biomarker for clinical response, and inconsistent microbiomes were present in different cohorts.105 Regarding microbiomes enriched in responders, one meta-study identified Actinobacteria phylum and the Lachnospiraceae family,106 and another study found Bacteroides stercoris, Parabacteroides distasonis, and Fournierella massiliensis.107 Additionally, the abundance of the Faecalibacterium genus was positively correlated with T cell infiltrates and antigen presentation and processing.108 Regarding microbiomes present in non-responders, Bacteroidetes, Proteobacteria,106 Klebsiella aerogenes, and Lactobacillus rogosae were reported.107
A recent study found that NSCLC patients with Akkermansia muciniphila accompanied by Eubacterium hallii and Bifidobacterium adolescentis have better OS and response.100 This study also identified two subgroups of patients who present as Akkermansia muciniphila-positive (Akk+), and stratified patients into Akk-high (overabundance) and Akk-low (normal abundance) groups in addition to patients with no presentation of this genus (Akk−). The Akk-low patients had a significant increase in median survival compared to overabundant Akk-high and Akk− patients, while the overabundance group had the worst survival. Ruminococcus and Agathobacter were also found to be associated with response and PFS in Japanese patients.102
For RCC patients, a relatively higher abundance of Akkermansia muciniphila was observed in clinical-benefit patients compared to non-clinical-benefit patients.103 The relative abundance of Bifidobacterium adolescentis, Barnesiella intestinihominis, and Bacteroides eggerthii differed between patients with and without clinical benefits. Similarly, Akkermansia muciniphila, Bacteroides salyersiae, and Eubacterium siraeum were observed to be more enriched in responders.104
Besides considering the abundance of a single microbiome species, a compound score was recently developed that calculates the ratio of 43 differential operational taxonomy units between clinical-benefit and non-clinical-benefit samples; this score is correlated with tumor size and clinical outcomes.99 In addition, the Shannon and Simpson indices, often used for quantifying the immune repertoire, are other commonly used biomarkers that measure the within-sample diversity of the intestinal microbiome109 (Figure 2C). Such diversity indices have shown a positive correlation with patient response,96,103,108 and survival102 in some studies but are not always significant.101,106,107,110
Compared to gut and stool, tissues that harbor the majority of microbiomes in the human body, the blood microbiome remains incompletely understood. This limitation is due to concerns regarding potential contamination and false-positive results of identifying microbiota in this tissue.111,112 No consistent microbiome results were obtained in a large-scale study of 9,770 healthy individuals.113 Although the blood microbiome is challenging for the prediction of ICB response, it is informative for distinguishing different cancer types and healthy donors.114 Overall the major microbiota (Bacteroides, Bifidobacterium, Akkermansia muciniphila, and Ruminococcus) from gut or stool are commonly identified as differentially enriched among patients115,116; however, as the different species identified may be affected by the tissue collected, environment, or antibiotics usage of individual patients, care is warranted in the interpretation of these findings.
Liquid biopsy
Liquid biopsy has several advantages over tissue-based assays, including lower cost, ease of access, the ability to perform longitudinal assessment, and the potential to better assess tumor heterogeneity. Liquid biopsy can assay measures of tumor circulating nucleic acids, circulating tumor cells (CTCs), exosomes, and nucleosomes, as well as other analytes found in bodily fluids.117,118 Cell-free DNA, of which circulating tumor DNA (ctDNA) is a subset, has been shown to correlate with tumor burden and stage. However, this observation may not hold for all cancer types, and the utility may be limited to certain cancers as a result.117 In addition, early-stage cancers may have significantly lower levels of ctDNA than advanced-stage cancers, which may limit its usefulness as a diagnostic marker in early-stage disease.118
Blood-based TMB (bTMB) derived from ctDNA has been correlated with tumor-tissue-derived TMB, and several retrospective studies found bTMB to be predictive of response.119 However, studies also indicate a need for assay optimization, standard measures of bTMB, and further exploration into optimal cutoffs or the use of bTMB as a continuous metric. Normalization of bTMB measurements to total ctDNA was superior to bTMB or ctDNA alone in predicting NSCLC patient clinical outcomes.120 Cancer driver genes may lead to overestimating bTMB and are excluded in some bTMB measures.121 In one study, a bTMB of ≥20 mutations/Mb was predictive of clinical benefit, PFS, and OS but only in patients who received a combination of ICB treatment. Another study122 used a pre-defined bTMB cutoff of ≥16 mutations/1.1 Mb to assess response in locally advanced or metastatic NSCLC and did not find a statistically significant improvement in PFS. This study did, however, observe an association between higher cutoffs and better PFS outcomes whereby a cutoff of ≥20 was significantly associated with PFS, and follow-up showed a significant difference in OS in the bTMB ≥16 groups. The ctDNA assay used for the study did not include insertion/deletion mutations, which might be more antigenic than single-nucleotide variants and lead to a more informative bTMB score. However, a randomized cohort of NSCLC patients receiving atezolizumab or platinum-based chemotherapy did not have a statistically significant difference in PFS at bTMB ≥16.123
Multiple studies have explored ctDNA’s association with prognosis and its use as a surrogate for minimum residual disease. For instance, early levels of ctDNA during treatment as well as changes in ctDNA have been found to be predictive of response. These findings can help guide treatment decisions and provide complementary information to radiographic assessments of response. NSCLC patients with detectable ctDNA following chemoradiation had significantly better outcomes when treated with ICB than patients who did not receive ICB.124 In contrast, patients with undetectable ctDNA levels showed no difference in outcomes whether they received ICB or not.124 Similarly, among urothelial carcinoma patients with detectable ctDNA, ICB treatment led to better disease-free survival and OS than chemotherapy. In contrast, no significant difference was found between treatment groups in patients with undetectable ctDNA.125 When ctDNA levels across 16 ICB-treated advanced-stage tumor types were assessed, high pre-treatment ctDNA variant allele frequencies (VAFs) were prognostic for poor OS, while on-treatment decreases in VAFs and lower on-treatment VAFs were predictive of ICB response and better survival.126 Early changes in ctDNA levels correlated with clinical outcomes in ICB-treated NSCLC patients.127,128
Assessments of ctDNA levels and bTMB must consider mutations due to clonal hematopoiesis of indeterminate potential (CHIP).117,119 Several studies have approached this issue differently, such as eliminating pre-reported CHIP genes,121 excluding variants with low allele frequency,122,123 and personalized filtering using white blood cell controls.129
In addition to bTMB and ctDNA levels, blood-based measures of MSI may help predict the outcome, and ctDNA methylation patterns or fragmentation profiles may provide epigenomic windows into the tumor landscape which reveal mechanisms of resistance or are predictive of response.117,119 Pre-treatment plasma MSI and TMB-high ICB-treated patients with advanced tumors were associated with better PFS.130
The ctDNA-derived marker may be better utilized with other biomarkers for ICB-response prediction. For example, the bTMB is more informative if combined with other factors relevant to neoantigen stimulation of the immune system.123 Two Bayesian models—one which combined pre-treatment PD-L1, normalized bTMB, and circulating CD8 T cell counts and a second which added early on-treatment ctDNA dynamics—predicted durable clinical benefit with areas under the curves (AUCs) of 0.74 and 0.93, respectively, in a stage IV NSCLC validation cohort.120 The neutrophil-to-lymphocyte ratio (NLR) and ctDNA VAF dynamics were similarly related to patient progression in a small sample of NSCLC patients, and the ctDNA and NLR may be complementary in predicting long-term outcomes.131
CTCs represent tumor cells that have escaped the primary tumor environment and entered the bloodstream. CTCs are rare and more technically challenging to detect but offer new information and information potentially complementary to ctDNA for assessment and prediction of ICB response as well as exploration of mechanisms of migration, metastasis, and therapeutic resistance.
Biomarkers derived from imaging modalities
Immunohistochemistry (IHC) and newer technologies, such as multiplex immunohistochemistry (mIHC), multiplex immunofluorescence (mIF), co-detection by indexing (CODEX), and multiplexed ion beam imaging (MIBI), are approaches to measure the level of different cell components from the TME (Table 3). PD-L1 expression measured by IHC was one of the first biomarkers explored in relation to ICB treatment stratification and response.132 For cancers such as NSCLC, HNSCC, triple-negative breast cancer (TNBC), and cervical cancer, PD-L1 expression levels above a particular threshold may be required for immunotherapy use. For other cancer types, PD-L1 levels can be used as a complementary test to guide treatment decisions. For some indications, such as NSCLC, treatment decisions are based on tumor cell expression only (tumor proportion score), while tumor and immune cell expression are assessed for other indications (combined proportion score).133
Table 3.
Signatures derived from imaging data
| Reference | Cancer | Signatures | Notes |
|---|---|---|---|
| Nishino et al.132 | melanoma, NSCLC, advanced-stage solid tumors | PD-L1 | – |
| Berry et al.135 | melanoma | CD8+FoxP3+PD-1 | – |
| – | CD163+PD-L1− | – | |
| Chen et al.136 | gastric cancer | TIIC signature | TIIC signature: CD4+FoxP3−PD-L1+, CD8+PD-1−LAG3−, and CD68+STING+ cell density + the effective score of CD8+PD-1+LAG3− T cells; effective score = proportion of immune cells within a specific distance from the tumor cells |
| Lopez de Rodas et al.137 | NSCLC | spatial heterogeneity score | spatial heterogeneity score uses Rao's Q index to measure diversity based on pairwise distances between cell types (tumor, CD4+, CD8+, CD20+, and stromal cells) and relative abundance of each cell type |
| Wu et al.138 | head and neck cancer, colorectal cancer | graphical embeddings of spatial relationships between cells | delineates graph neural network spatial motifs associated with cancer recurrence and response |
| Patwa et al.139 | TNBC | scoring of interactions between cells expressing PD-1, PD-L1, IDO, and LAG-3 | – |
| Zugazagoitia et al.140 | NSCLC | immune-stromal CD56 and CD4 protein expression | – |
| tumor VISTA and CD127 protein expression | – | ||
| Larroquette et al.141 | NSCLC | high CD163+ cell infiltration (possibly high expression of ITGAM, CD27, and CCL5) | – |
| high CSF1R expression in tumor cells | higher CSF1R in tumors of responders | ||
| Johannet et al.148 | metastatic melanoma | DCNN-derived response classification + clinical variables | ECOG performance score and immunotherapy category |
| Khorrami et al.149 | NSCLC | changes in a machine-learning-derived radiomic feature set | – |
| Vaidya et al.150 | advanced NSCLC | CT-scan radiomic features | – |
| Trebeschi et al.151 | advanced melanoma and NSCLC | CT-scan radiomic features | only borderline significant for melanoma |
The evidence and reliability of PD-L1 as a biomarker have been reviewed elsewhere.132,134 In brief, the evaluation of PD-L1 measurements as a biomarker is clouded by technical variability, the choice of scoring method, and threshold variation among clinical trials as well as the timing of specimen collection relative to ICB treatment initiation. This variation impairs the ability to compare PD-L1 levels among different trials. PD-L1 measurements alone or in combination with TMB are generally not sufficient to delineate responders from non-responders. The utility of PD-L1 in guiding ICB treatment choice, as well as which type of PD-L1 measure is most informative, currently appears to vary based on tumor type.134 Standardization of PD-L1 IHC measurements will help improve the reliability of its use as a biomarker across tumor types.
Imaging data can measure PD-L1 levels and also assess immune cell infiltration levels, tissue features, and other protein markers to enhance the utility of histological specimens. A six-marker mIF panel (PD-1, PD-L1, CD8, FoxP3, CD163, and Sox10/S100) identified a rare subpopulation of CD8+ T cells associated with response to anti-PD1 therapy in melanoma patients.135 In the same study, CD163+PD-L1− myeloid cells were associated with non-response. Similarly, an mIHC tumor-infiltrating immune cell (TIIC) signature was associated with gastric cancer anti-PD1/PD-L1 response and patient survival.136 The TIIC signature incorporates both immune cell densities and measures of distance between tumor cells and particular immune cell populations.136
Spatial information, in combination with the ability to measure expression levels of multiple proteins, also opens up the possibility of extracting higher-level information, such as cell-cell interactions and network and neighborhood analyses, which may be associated with therapeutic responses. A continuous measure of spatial heterogeneity developed using Rao’s Q Index identified an association between a large extent of spatial immune heterogeneity and reduced survival.137 A graph deep-learning model of the spatial architecture of tumors predicted patient outcomes when applied to CODEX datasets, including an anti-PD1-treated HNSCC dataset.138 Profiling interactions between immunoregulatory proteins such as PD-1, PD-L1, IDO, and LAG-3 in TNBC with MIBI resulted in statistically significant associations with recurrence and survival, which were not found when profiling single-cell levels of each marker.139
Newer spatial profiling techniques can combine the power of spatial information, proteomic markers, and mRNA expression. Digital spatial profiling of proteomics data identified immune cell/stromal compartment CD56 and CD4 levels as positively associated with durable clinical benefit, longer PFS, and better OS, and tumor compartment V-domain immunoglobulin suppressor of T cell activation (VISTA) and CD127 were associated with no clinical benefit in an anti-PD1-treated NSCLC cohort.140 Furthermore, the differential expression profile of high and low CD163+-cell-infiltrated NSCLC tumors identified ICB-response markers and potential targets to enhance ICB response.141
Machine-learning approaches for discovering biomarkers
Supervised machine-learning (ML) algorithms, such as logistic regression (LR), random forest (RF), and extreme gradient boosting (XGBoosting), are widely used to classify patients into response groups and identify genes that predict ICB treatment success. LR is popular for binary classification and determining linear relationships between given inputs and prediction outcomes. RF and XGBoosting are non-linear tree-ensemble algorithms. RF trains multiple decision trees for prediction in parallel and aggregates the results from those trees using a majority or probability vote. XGBoosting sequentially derives boosted trees through gradient descent optimizations of selected loss objective functions. XGBoosting has more hyperparameters to tune during training and is more adaptable to different data types but may also be more prone to overfitting as compared to RF.
To identify transcriptomics signatures, an RF model applied to gene expression data identified macrophage M1 and peripheral T cell gene signatures predictive of durable clinical benefit in 34 NSCLC cancer patients.142 Additionally, an RF classifier for cell-type classification from scRNA-seq data from patients with acute myeloid leukemia identified gene expression patterns that correlated with the prediction score.143 In addition to discovering gene signatures from RNA-seq data, an ML-based text-mining approach applied to literature containing immune-related keywords identified a 15-gene signature associated with immune-supportive/immunosuppressive tumors. This 15-gene signature was later used to identify aurora kinase inhibitors as promoters of T cell infiltration in mouse models of TNBC.144 Additionally, an XGBoosting model using whole-exome and transcriptomics-derived features as well as RNA-derived immune repertoire features from melanoma patients was used to estimate neoantigen quality.145
Deep learning, an advanced ML technique with particularly dense or convolution kernels embedded in a flexible network structure, can efficiently learn hidden data patterns directly from the raw sequence and clinical image data. For example, a DeepTCR model trained a variational autoencoder with both raw CDR3 sequences and V/D/J gene usage to represent a TCR sequence in a more comprehensive manner.146 A follow-up study further incorporating the human leukocyte antigen background together with TCR sequence information improved the ability to characterize the extent of TCR diversity.147 In addition to the application of these methods to sequencing data, a proof-of-principle response classifier integrating clinical variables and H&E data of clinical specimens accurately predicted PFS for ICB-treated advanced melanoma.148 Radiographic data from computed tomography (CT) scans or other imaging modalities also provide valuable data without needing a biopsy or other invasive techniques. For instance, features derived from CT scans before and early during ICB treatment predicted the response and OS in advanced NSCLC patients.149,150,151
Signatures derived from multi-modal and machine-learning approaches
The integration of multiple data modalities has the potential to improve the prediction accuracy of ICB outcomes. Flexible data processing using different deep-learning structures has been popular to integrate multi-modal data. For example, a deep-learning multi-modal model called DyAM integrates radiology, histology, and genetic mutation data to predict patient response.152 The multi-modal model has shown improved predictive performance compared to single-modal models, TMB, and PD-L1 tumor proportion on an NSCLC anti-PD-1/PD-L1 cohort of 247 patients. The DyAM model applies attention-based multiple-instance learning153 algorithms and outputs a DyAM score as a predictive biomarker, which is a weighted sum of the logistic regression of each data modality where weights are dynamically optimized by the attention-based score that learns the most contributed features to the predictive outcome through model training (Figure 3A). In addition, the multi-variable approach can also be used to aggregate multi-modal data with the predictive outputs feature using RF or XGBoosting methods, which output features importance for predicting response to ICB treatment (Figure 3B).
Figure 3.

Multi-modal data integrated using deep-learning and multi-variate models
(A) Multi-modal data are concatenated and used as inputs for a deep-learning model. Attention-based multiple instances learning during model training outputs normalized attention weights and patient risk scores. DyAM score, which aggregates features, weights, and risk scores, is used as a predictive marker.
(B) High-importance features can be selected using the non-linear random forest or XGBoosting methods. Top features are used to build a multi-variable logistic model to predict patient response.
In a large study, a multi-variable approach applied to WES and RNA-seq data from more than 1,000 patient samples from 12 ICB cohorts was used to derive 55 unique biomarkers across mutational, neoantigen, gene expression, and immune infiltration signatures.33 The total variance explained by all tested biomarkers was ∼0.6, suggesting that 40% of the variance remained unexplained by the model. Among all features, clonal TMB, expression of CXCL9 (a critical chemokine that binds CXCR3 on T cells, enhancing recruitment of cytotoxic CD8+ T cells into the tumor), and three mutational signatures (UV, APOBEC, and tobacco signatures) were the most predictive at the pan-cancer level. This study also applied the XGBoosting ML method to develop a multi-variable model with 11 selected features, which are top-importance features at both individual and pan-cancer levels. On three independent testing cohorts, the AUC score from the 11-feature multi-variable model outperformed prediction results using TMB only as a baseline comparison and performed comparably with a much simpler, two-marker model including clonal TMB and CXCL9 expression. Furthermore, analysis of whole-genome sequencing, RNA-seq, IHC, and methylation data from 77 anti-PD1/PD-L1 melanoma patients indicated that TMB, neoantigen load, PD-L1 expression, gene signatures associated with IFN-γ response, a score to describe T cell dysfunction and exclusion (TIDE), cytolytic activity and chemokines, and immune infiltrates of macrophage M1 and CD8 T cells were able to distinguish responders and non-responders, while TMB was the measure most significantly correlated with outcome. A bivariable model (TMB + IFN-γ score) performed better than TMB-only or IFN-γ-only univariate models.
Recently, single-cell sequencing techniques have been developed to generate multi-modal data simultaneously, such as CITE-seq integrating gene expression and epitopes,154 SHARE-seq and sci-CAR characterizing chromatin accessibility and gene expression,155,156 TAE-seq integrating gene expression, epitopes, and chromatin accessibility,157 and Slide-seq combining RNA and spatial profiling.158 These multi-modal assays have been applied to patient samples to help better understand the immune system-tumor interface. For example, multi-modal single-cell TCR repertoire sequencing overcomes a critical deficiency of TCR repertoire assessment, which currently does not characterize which T cell subsets express the TCR clones.159 To this end, integrating spatial transcriptomics and TCR receptor sequencing demonstrated that the localization of infiltrating CD8+ T cell subsets was linked to their phenotypic state in brain metastases.160,161 Recently, Slide-TCR-seq, which integrates gene expression, TCR clonality, and spatial-resolution data, was used to characterize the TCR repertoire in the mouse spleen.162 Perturb-CITE-seq, which measures the clustered regularly interspaced short palindromic repeat (CRISPR)-Cas9 perturbations with single-cell transcriptome and protein readouts, was used to uncover tumor immune evasion mechanisms from patient-derived melanoma cell and tumor-infiltrating lymphocyte co-cultures.163 However, studies applying these advanced multi-modal sequencing techniques have primarily been performed in pre-clinical models, and patient data from ICB clinical cohorts to discover novel biomarkers are limited to date.
Outlook
Here, we have provided examples of biomarkers discovered from various data resources and have described different computational methodologies developed to analyze those data types and extract features associated with ICB therapy response. These methods have demonstrated an association between clinical outcome and individual data-derived features, including FDA-approved mutational markers (TMB and MSI-H), neoantigen load and neoantigen quality (MHC binding and TCR recognition), gene signatures, relationships between gene pairs or among gene sets, immune cell infiltrates (TIL, CD8, CD4, tissue-memory T cells, CD8/CD4 ratio, and TLS), immune repertoire diversity, and gut microbiota. Moreover, image-derived PD-L1 expression and spatial transcriptomic profiling, as well as integrative multi-modal features, have been developed from multi-variable regression or ML approaches. Despite the tremendous progress in checkpoint therapy, as well as acknowledgment of the risk of adverse events from ICB treatment, a single reliable biomarker that accurately predicts ICB response for all cancer types or its toxicities does not currently exist. Biomarkers can also enhance our understanding of ICB resistance and provide the basis for enhanced or novel treatments. The complex and continually evolving nature of tumor-immune cell interactions remains challenging to capture with a single biomarker or modality.
The rapid progress in novel multi-omics sequencing technologies and the diverse data types generated (next-generation sequencing, clinical, and imaging data) invites the development of computational methods to efficiently integrate and analyze these datasets. ML approaches have the advantage of being able to integrate, process, and explore those large-scale diverse data types in order to develop novel ICB biomarkers. A reliable ML model requires high-quality training and testing data, which is challenging given the non-standardized patient data available in clinical practice. Therefore, data harmonization is critical when processing patient data from multiple cohorts and is necessary before training any prediction model. In addition, data overfitting by ML approaches can result in poor generalizability to new patient cohorts, especially when external validation must be performed across independent clinical trials to develop biomarkers with meaningful clinical predictions.
Considering the data quality and modality at both single-indication and pan-cancer levels, currently there is a limited amount of data for developing and validating ICB signatures. Unfortunately, not all data are publicly available, making it time- and effort-consuming to request access because of varying restrictions by different organizations. The National Cancer Institute Cancer Moonshot project aiming to prevent cancer death and improve the experience of cancer patients launched the Cancer and Immune Monitoring Analysis Center–Cancer Immunological Data Commons (CIMAC-CIDC) Network,164 which standardizes and systematically collects assay and clinical data from many clinical trials and will help fuel this progress. The explosion of new technologies to profile genomic, transcriptomic, and proteomic cell states has coincided with ICB’s revolutionary effect on cancer therapy. We anticipate that ever-more-accurate computational methods will be published to take advantage of diverse data resources and that next-generation sequencing technology at the bulk, single-cell, multi-omics, and multi-modal levels will be applied to clinical trials to help better characterize ICB-response mechanisms.
Acknowledgments
We gratefully acknowledge support from the CIMAC-CIDC network, the Dana-Farber Cancer Institute’s Center for Cancer Evolution, and the National Library of Medicine T15 (T15LM007092) program.
Author contributions
Y.L., J.A., C.J. Wong, S.B., S.C., C.J. Wu, and F.M. drafted the manuscript. Y.L. and C.J. Wong drafted the figures. F.M. supervised the writing of the manuscript. All authors read and approved the final manuscript.
Declaration of interests
C.J. Wu holds equity in BioNTech, Inc., and receives research support from Pharmacyclics. F.M. is a co-founder of and has equity in Harbinger Health, has equity in Zephyr AI, and serves as a consultant for both companies. She is also on the board of directors of Exscientia Plc. F.M. declares that none of these relationships are directly or indirectly related to the content of this manuscript.
References
- 1.Mardis E.R. Neoantigens and genome instability: impact on immunogenomic phenotypes and immunotherapy response. Genome Med. 2019;11:71. doi: 10.1186/s13073-019-0684-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kiyotani K., Toyoshima Y., Nakamura Y. Immunogenomics in personalized cancer treatments. J. Hum. Genet. 2021;66:901–907. doi: 10.1038/s10038-021-00950-w. [DOI] [PubMed] [Google Scholar]
- 3.Ott P.A., Hu Z., Keskin D.B., Shukla S.A., Sun J., Bozym D.J., Zhang W., Luoma A., Giobbie-Hurder A., Peter L., et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 2017;547:217–221. doi: 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keskin D.B., Anandappa A.J., Sun J., Tirosh I., Mathewson N.D., Li S., Oliveira G., Giobbie-Hurder A., Felt K., Gjini E., et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 2019;565:234–239. doi: 10.1038/s41586-018-0792-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ribas A., Wolchok J.D. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Seidel J.A., Otsuka A., Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018;8:86. doi: 10.3389/fonc.2018.00086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsimberidou A.M., Fountzilas E., Bleris L., Kurzrock R. Transcriptomics and solid tumors: The next frontier in precision cancer medicine. Semin. Cancer Biol. 2022;84:50–59. doi: 10.1016/j.semcancer.2020.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berraondo P., Sanmamed M.F., Ochoa M.C., Etxeberria I., Aznar M.A., Pérez-Gracia J.L., Rodríguez-Ruiz M.E., Ponz-Sarvise M., Castañón E., Melero I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer. 2019;120:6–15. doi: 10.1038/s41416-018-0328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagarsheth N., Wicha M.S., Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017;17:559–572. doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Si Z., Hu H. Identification of CXCL13 as an Immune-Related Biomarker Associated with Tumorigenesis and Prognosis in Cutaneous Melanoma Patients. Med. Sci. Mon. Int. Med. J. Exp. Clin. Res. 2021;27:e932052. doi: 10.12659/MSM.932052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kraehenbuehl L., Weng C.-H., Eghbali S., Wolchok J.D., Merghoub T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nat. Rev. Clin. Oncol. 2022;19:37–50. doi: 10.1038/s41571-021-00552-7. [DOI] [PubMed] [Google Scholar]
- 12.Baumeister S.H., Freeman G.J., Dranoff G., Sharpe A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016;34:539–573. doi: 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 13.Farhood B., Najafi M., Mortezaee K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019;234:8509–8521. doi: 10.1002/jcp.27782. [DOI] [PubMed] [Google Scholar]
- 14.Rooney M.S., Shukla S.A., Wu C.J., Getz G., Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell. 2015;160:48–61. doi: 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen D.S., Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–330. doi: 10.1038/nature21349. Preprint at. [DOI] [PubMed] [Google Scholar]
- 16.Jiang P., Gu S., Pan D., Fu J., Sahu A., Hu X., Li Z., Traugh N., Bu X., Li B., et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 2018;24:1550–1558. doi: 10.1038/s41591-018-0136-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ayers M., Lunceford J., Nebozhyn M., Murphy E., Loboda A., Kaufman D.R., Albright A., Cheng J.D., Kang S.P., Shankaran V., et al. IFN-γ–related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017;127:2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lemvigh C.K., Parry E.M., Deng S.L., Dangle N.J., Ruthen N., Knisbacher B.A., Broséus J., Hergalant S., Guieze R., Li S., et al. ZNF683 (Hobit) Marks a CD8+ T Cell Population Associated with Anti-Tumor Immunity Following Anti-PD-1 Therapy for Richter Syndrome. Blood. 2022;140:1807–1808. doi: 10.1016/j.ccell.2023.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schumacher T.N., Thommen D.S. Tertiary lymphoid structures in cancer. Science. 2022;375:eabf9419. doi: 10.1126/science.abf9419. [DOI] [PubMed] [Google Scholar]
- 20.Helmink B.A., Reddy S.M., Gao J., Zhang S., Basar R., Thakur R., Yizhak K., Sade-Feldman M., Blando J., Han G., et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature. 2020;577:549–555. doi: 10.1038/s41586-019-1922-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freeman S.S., Sade-Feldman M., Kim J., Stewart C., Gonye A.L.K., Ravi A., Arniella M.B., Gushterova I., LaSalle T.J., Blaum E.M., et al. Combined tumor and immune signals from genomes or transcriptomes predict outcomes of checkpoint inhibition in melanoma. Cell Rep. Med. 2022;3:100500. doi: 10.1016/j.xcrm.2021.100500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Auslander N., Zhang G., Lee J.S., Frederick D.T., Miao B., Moll T., Tian T., Wei Z., Madan S., Sullivan R.J., et al. Robust prediction of response to immune checkpoint blockade therapy in metastatic melanoma. Nat. Med. 2018;24:1545–1549. doi: 10.1038/s41591-018-0157-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee J.S., Nair N.U., Dinstag G., Chapman L., Chung Y., Wang K., Sinha S., Cha H., Kim D., Schperberg A.V., et al. Synthetic lethality-mediated precision oncology via the tumor transcriptome. Cell. 2021;184:2487–2502.e13. doi: 10.1016/j.cell.2021.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dinstag G., Shulman E.D., Elis E., Ben-Zvi D.S., Tirosh O., Maimon E., Meilijson I., Elalouf E., Temkin B., Vitkovsky P., et al. Clinically oriented prediction of patient response to targeted and immunotherapies from the tumor transcriptome. Méd. 2023;4:15–30.e8. doi: 10.1016/j.medj.2022.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hugo W., Zaretsky J.M., Sun L., Song C., Moreno B.H., Hu-Lieskovan S., Berent-Maoz B., Pang J., Chmielowski B., Cherry G., et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell. 2017;168:542. doi: 10.1016/j.cell.2017.01.010. [DOI] [PubMed] [Google Scholar]
- 26.McGrail D.J., Pilié P.G., Rashid N.U., Voorwerk L., Slagter M., Kok M., Jonasch E., Khasraw M., Heimberger A.B., Lim B., et al. High tumor mutation burden fails to predict immune checkpoint blockade response across all cancer types. Ann. Oncol. 2021;32:661–672. doi: 10.1016/j.annonc.2021.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Van Allen E.M., Miao D., Schilling B., Shukla S.A., Blank C., Zimmer L., Sucker A., Hillen U., Foppen M.H.G., Goldinger S.M., et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science. 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riaz N., Havel J.J., Makarov V., Desrichard A., Urba W.J., Sims J.S., Hodi F.S., Martín-Algarra S., Mandal R., Sharfman W.H., et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell. 2017;171:934–949.e16. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cristescu R., Mogg R., Ayers M., Albright A., Murphy E., Yearley J., Sher X., Liu X.Q., Lu H., Nebozhyn M., et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science. 2018;362:eaar3593. doi: 10.1126/science.aar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jardim D.L., Goodman A., de Melo Gagliato D., Kurzrock R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell. 2021;39:154–173. doi: 10.1016/j.ccell.2020.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gurjao C., Tsukrov D., Imakaev M., Luquette L.J., Mirny L.A. Limited evidence of tumour mutational burden as a biomarker of response to immunotherapy. bioRxiv. 2020:260265. Preprint at. [Google Scholar]
- 32.Food U.S., Administration D., Others . US Food and Drug Administration; 2020. FDA Approves Pembrolizumab for Adults and Children with TMB-H Solid Tumors. News release. [Google Scholar]
- 33.Litchfield K., Reading J.L., Puttick C., Thakkar K., Abbosh C., Bentham R., Watkins T.B.K., Rosenthal R., Biswas D., Rowan A., et al. Meta-analysis of tumor- and T cell-intrinsic mechanisms of sensitization to checkpoint inhibition. Cell. 2021;184:596–614.e14. doi: 10.1016/j.cell.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Han G., Yang G., Hao D., Lu Y., Thein K., Simpson B.S., Chen J., Sun R., Alhalabi O., Wang R., et al. 9p21 loss confers a cold tumor immune microenvironment and primary resistance to immune checkpoint therapy. Nat. Commun. 2021;12:5606. doi: 10.1038/s41467-021-25894-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niknafs N., Balan A., Cherry C., Hummelink K., Monkhorst K., Shao X.M., Belcaid Z., Marrone K.A., Murray J., Smith K.N., et al. Persistent mutation burden drives sustained anti-tumor immune responses. Nat. Med. 2023;29:440–449. doi: 10.1038/s41591-022-02163-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alexandrov L.B., Jones P.H., Wedge D.C., Sale J.E., Campbell P.J., Nik-Zainal S., Stratton M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015;47:1402–1407. doi: 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spurr L.F., Weichselbaum R.R., Pitroda S.P. Tumor aneuploidy predicts survival following immunotherapy across multiple cancers. Nat. Genet. 2022;54:1782–1785. doi: 10.1038/s41588-022-01235-4. [DOI] [PubMed] [Google Scholar]
- 38.Chang T.-G., Cao Y., Shulman E.D., Ben-David U., Schäffer A.A., Ruppin E. Optimizing cancer immunotherapy response prediction by tumor aneuploidy score and fraction of copy number alterations. npj Precis. Oncol. 2023;7:54. doi: 10.1038/s41698-023-00408-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Center for Drug Evaluation & Research. FDA approves pembrolizumab for first-line treatment of MSI-H/dMMR colorectal cancer. US Food and Drug Administration https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-pembrolizumab-first-line-treatment-msi-hdmmr-colorectal-cancer.
- 40.Center for Drug Evaluation & Research. FDA grants nivolumab accelerated approval for MSI-H or dMMR colorectal cancer. US Food and Drug Administration https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-nivolumab-accelerated-approval-msi-h-or-dmmr-colorectal-cancer.
- 41.Li K., Luo H., Huang L., Luo H., Zhu X. Microsatellite instability: a review of what the oncologist should know. Cancer Cell Int. 2020;20:16. doi: 10.1186/s12935-019-1091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.André T., Shiu K.K., Kim T.W., Jensen B.V., Jensen L.H., Punt C., Smith D., Garcia-Carbonero R., Benavides M., Gibbs P., et al. Pembrolizumab in Microsatellite-Instability–High Advanced Colorectal Cancer. N. Engl. J. Med. 2020;383:2207–2218. doi: 10.1056/NEJMoa2017699. [DOI] [PubMed] [Google Scholar]
- 43.Cercek A., Lumish M., Sinopoli J., Weiss J., Shia J., Lamendola-Essel M., El Dika I.H., Segal N., Shcherba M., Sugarman R., et al. PD-1 Blockade in Mismatch Repair–Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022;386:2363–2376. doi: 10.1056/NEJMoa2201445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonneville R., et al. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO Precision Oncology. 2017:1–15. doi: 10.1200/PO.17.00073. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makker V., Colombo N., Casado Herráez A., Santin A.D., Colomba E., Miller D.S., Fujiwara K., Pignata S., Baron-Hay S., Ray-Coquard I., et al. Lenvatinib plus Pembrolizumab for Advanced Endometrial Cancer. N. Engl. J. Med. 2022;386:437–448. doi: 10.1056/NEJMoa2108330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barata P., Agarwal N., Nussenzveig R., Gerendash B., Jaeger E., Hatton W., Ledet E., Lewis B., Layton J., Babiker H., et al. Clinical activity of pembrolizumab in metastatic prostate cancer with microsatellite instability high (MSI-H) detected by circulating tumor DNA. J. Immunother. Cancer. 2020;8:e001065. doi: 10.1136/jitc-2020-001065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y., Shi T., Song X., Liu B., Wei J. Gene fusion neoantigens: Emerging targets for cancer immunotherapy. Cancer Lett. 2021;506:45–54. doi: 10.1016/j.canlet.2021.02.023. [DOI] [PubMed] [Google Scholar]
- 48.Lazdun Y., Si H., Creasy T., Ranade K., Higgs B.W., Streicher K., Durham N.M. A New Pipeline to Predict and Confirm Tumor Neoantigens Predict Better Response to Immune Checkpoint Blockade. Mol. Cancer Res. 2021;19:498–506. doi: 10.1158/1541-7786.MCR-19-1118. Preprint at. [DOI] [PubMed] [Google Scholar]
- 49.Chen H., Yang M., Wang Q., Song F., Li X., Chen K. The new identified biomarkers determine sensitivity to immune check-point blockade therapies in melanoma. OncoImmunology. 2019;8:1608132. doi: 10.1080/2162402X.2019.1608132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Łuksza M., Riaz N., Makarov V., Balachandran V.P., Hellmann M.D., Solovyov A., Rizvi N.A., Merghoub T., Levine A.J., Chan T.A., et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature. 2017;551:517–520. doi: 10.1038/nature24473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Łuksza M., Sethna Z.M., Rojas L.A., Lihm J., Bravi B., Elhanati Y., Soares K., Amisaki M., Dobrin A., Hoyos D., et al. Neoantigen quality predicts immunoediting in survivors of pancreatic cancer. Nature. 2022;606:389–395. doi: 10.1038/s41586-022-04735-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Linette G.P., Carreno B.M. Tumor-Infiltrating Lymphocytes in the Checkpoint Inhibitor Era. Curr. Hematol. Malig. Rep. 2019;14:286–291. doi: 10.1007/s11899-019-00523-x. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H., van der Merwe P.A., Sivakumar S. Biomarkers of response to PD-1 pathway blockade. Br. J. Cancer. 2022;126:1663–1675. doi: 10.1038/s41416-022-01743-4. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Young Y.K., Bolt A.M., Ahn R., Mann K.K. Analyzing the Tumor Microenvironment by Flow Cytometry. Methods Mol. Biol. 2016;1458:95–110. doi: 10.1007/978-1-4939-3801-8_8. [DOI] [PubMed] [Google Scholar]
- 55.Chung W., Eum H.H., Lee H.O., Lee K.M., Lee H.B., Kim K.T., Ryu H.S., Kim S., Lee J.E., Park Y.H., et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017;8:15081–15112. doi: 10.1038/ncomms15081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Finotello F., Trajanoski Z. Quantifying tumor-infiltrating immune cells from transcriptomics data. Cancer Immun., Immunotherapy. 2018;67:1031–1040. doi: 10.1007/s00262-018-2150-z. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parra E.R., Francisco-Cruz A., Wistuba I.I. State-of-the-Art of Profiling Immune Contexture in the Era of Multiplexed Staining and Digital Analysis to Study Paraffin Tumor Tissues. Cancers. 2019;11:247. doi: 10.3390/cancers11020247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paijens S.T., Vledder A., de Bruyn M., Nijman H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021;18:842–859. doi: 10.1038/s41423-020-00565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clarke B., Tinker A.V., Lee C.H., Subramanian S., van de Rijn M., Turbin D., Kalloger S., Han G., Ceballos K., Cadungog M.G., et al. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod. Pathol. 2009;22:393–402. doi: 10.1038/modpathol.2008.191. [DOI] [PubMed] [Google Scholar]
- 60.Lee H.E., Chae S.W., Lee Y.J., Kim M.A., Lee H.S., Lee B.L., Kim W.H. Prognostic implications of type and density of tumour-infiltrating lymphocytes in gastric cancer. Br. J. Cancer. 2008;99:1704–1711. doi: 10.1038/sj.bjc.6604738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sheu B.-C., Kuo W.H., Chen R.J., Huang S.C., Chang K.J., Chow S.N. Clinical significance of tumor-infiltrating lymphocytes in neoplastic progression and lymph node metastasis of human breast cancer. Breast. 2008;17:604–610. doi: 10.1016/j.breast.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 62.Tumeh P.C., Harview C.L., Yearley J.H., Shintaku I.P., Taylor E.J.M., Robert L., Chmielowski B., Spasic M., Henry G., Ciobanu V., et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Presti D., Dall'Olio F.G., Besse B., Ribeiro J.M., Di Meglio A., Soldato D. Tumor infiltrating lymphocytes (TILs) as a predictive biomarker of response to checkpoint blockers in solid tumors: A systematic review. Crit. Rev. Oncol. Hematol. 2022;177:103773. doi: 10.1016/j.critrevonc.2022.103773. Preprint at. [DOI] [PubMed] [Google Scholar]
- 64.Hamid O., Schmidt H., Nissan A., Ridolfi L., Aamdal S., Hansson J., Guida M., Hyams D.M., Gómez H., Bastholt L., et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J. Transl. Med. 2011;9:204. doi: 10.1186/1479-5876-9-204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., Sosman J.A., McDermott D.F., Powderly J.D., Gettinger S.N., et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Uryvaev A., Passhak M., Hershkovits D., Sabo E., Bar-Sela G. The role of tumor-infiltrating lymphocytes (TILs) as a predictive biomarker of response to anti-PD1 therapy in patients with metastatic non-small cell lung cancer or metastatic melanoma. Med. Oncol. 2018;35:25. doi: 10.1007/s12032-018-1080-0. [DOI] [PubMed] [Google Scholar]
- 67.Simoni Y., Becht E., Fehlings M., Loh C.Y., Koo S.L., Teng K.W.W., Yeong J.P.S., Nahar R., Zhang T., Kared H., et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557:575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 68.Boddupalli C.S., Bar N., Kadaveru K., Krauthammer M., Pornputtapong N., Mai Z., Ariyan S., Narayan D., Kluger H., Deng Y., et al. Interlesional diversity of T cell receptors in melanoma with immune checkpoints enriched in tissue-resident memory T cells. JCI Insight. 2016;1:e88955. doi: 10.1172/jci.insight.88955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edwards J., Wilmott J.S., Madore J., Gide T.N., Quek C., Tasker A., Ferguson A., Chen J., Hewavisenti R., Hersey P., et al. CD103+ Tumor-Resident CD8+ T Cells Are Associated with Improved Survival in Immunotherapy-Naïve Melanoma Patients and Expand Significantly During Anti-PD-1 Treatment. Clin. Cancer Res. 2018;24:3036–3045. doi: 10.1158/1078-0432.CCR-17-2257. [DOI] [PubMed] [Google Scholar]
- 70.Ganesan A.-P., Clarke J., Wood O., Garrido-Martin E.M., Chee S.J., Mellows T., Samaniego-Castruita D., Singh D., Seumois G., Alzetani A., et al. Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol. 2017;18:940–950. doi: 10.1038/ni.3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jaiswal A., Verma A., Dannenfelser R., Melssen M., Tirosh I., Izar B., Kim T.G., Nirschl C.J., Devi K.S.P., Olson W.C., Jr., et al. An activation to memory differentiation trajectory of tumor-infiltrating lymphocytes informs metastatic melanoma outcomes. Cancer Cell. 2022;40:524–544.e5. doi: 10.1016/j.ccell.2022.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Luoma A.M., Suo S., Wang Y., Gunasti L., Porter C.B.M., Nabilsi N., Tadros J., Ferretti A.P., Liao S., Gurer C., et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell. 2022;185:2918–2935.e29. doi: 10.1016/j.cell.2022.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Amsen D., Van Gisbergen K.P.J.M., Hombrink P., Van Lier R.A.W. Tissue-resident memory T cells at the center of immunity to solid tumors. Nat. Immunol. 2018;19:538–546. doi: 10.1038/s41590-018-0114-2. Preprint at. [DOI] [PubMed] [Google Scholar]
- 74.Petitprez F., de Reyniès A., Keung E.Z., Chen T.W.W., Sun C.M., Calderaro J., Jeng Y.M., Hsiao L.P., Lacroix L., Bougoüin A., et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577:556–560. doi: 10.1038/s41586-019-1906-8. [DOI] [PubMed] [Google Scholar]
- 75.Buisseret L., Desmedt C., Garaud S., Fornili M., Wang X., Van den Eyden G., de Wind A., Duquenne S., Boisson A., Naveaux C., et al. Reliability of tumor-infiltrating lymphocyte and tertiary lymphoid structure assessment in human breast cancer. Mod. Pathol. 2017;30:1204–1212. doi: 10.1038/modpathol.2017.43. [DOI] [PubMed] [Google Scholar]
- 76.Barmpoutis P., Di Capite M., Kayhanian H., Waddingham W., Alexander D.C., Jansen M., Kwong F.N.K. Tertiary lymphoid structures (TLS) identification and density assessment on H&E-stained digital slides of lung cancer. PLoS One. 2021;16:e0256907. doi: 10.1371/journal.pone.0256907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chen P.-L., Roh W., Reuben A., Cooper Z.A., Spencer C.N., Prieto P.A., Miller J.P., Bassett R.L., Gopalakrishnan V., Wani K., et al. Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov. 2016;6:827–837. doi: 10.1158/2159-8290.CD-15-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hendry S., Salgado R., Gevaert T., Russell P.A., John T., Thapa B., Christie M., van de Vijver K., Estrada M.V., Gonzalez-Ericsson P.I., et al. Assessing Tumor-infiltrating Lymphocytes in Solid Tumors: A Practical Review for Pathologists and Proposal for a Standardized Method from the International Immunooncology Biomarkers Working Group: Part 1: Assessing the Host Immune Response, TILs in Invasive Breast Carcinoma and Ductal Carcinoma in Situ, Metastatic Tumor Deposits and Areas for Further Research. Adv. Anat. Pathol. 2017;24:235–251. doi: 10.1097/PAP.0000000000000162. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rubelt F., Busse C.E., Bukhari S.A.C., Bürckert J.P., Mariotti-Ferrandiz E., Cowell L.G., Watson C.T., Marthandan N., Faison W.J., Hershberg U., et al. Adaptive Immune Receptor Repertoire Community recommendations for sharing immune-repertoire sequencing data. Nat. Immunol. 2017;18:1274–1278. doi: 10.1038/ni.3873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Laydon D.J., Bangham C.R.M., Asquith B. Estimating T-cell repertoire diversity: Limitations of classical estimators and a new approach. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015;370:20140291. doi: 10.1098/rstb.2014.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aoki H., Shichino S., Matsushima K., Ueha S. Revealing Clonal Responses of Tumor-Reactive T-Cells Through T Cell Receptor Repertoire Analysis. Front. Immunol. 2022;13:807696. doi: 10.3389/fimmu.2022.807696. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pai J.A., Satpathy A.T. High-throughput and single-cell T cell receptor sequencing technologies. Nat. Methods. 2021;18:881–892. doi: 10.1038/s41592-021-01201-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Efremova M., Vento-Tormo R., Park J.-E., Teichmann S.A., James K.R. Vol. 7. 2020. Immunology in the Era of Single-Cell Technologies. [DOI] [PubMed] [Google Scholar]
- 84.Kidman J., Principe N., Watson M., Lassmann T., Holt R.A., Nowak A.K., Lesterhuis W.J., Lake R.A., Chee J. Characteristics of TCR Repertoire Associated With Successful Immune Checkpoint Therapy Responses. Front. Immunol. 2020;11:587014. doi: 10.3389/fimmu.2020.587014. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hopkins A.C., Yarchoan M., Durham J.N., Yusko E.C., Rytlewski J.A., Robins H.S., Laheru D.A., Le D.T., Lutz E.R., Jaffee E.M. T cell receptor repertoire features associated with survival in immunotherapy-treated pancreatic ductal adenocarcinoma. JCI insight. 2018;3:e122092. doi: 10.1172/jci.insight.122092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Amaria R.N., Reddy S.M., Tawbi H.A., Davies M.A., Ross M.I., Glitza I.C., Cormier J.N., Lewis C., Hwu W.J., Hanna E., et al. Neoadjuvant immune checkpoint blockade in high-risk resectable melanoma. Nat. Med. 2018;24:1649–1654. doi: 10.1038/s41591-018-0197-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Forde P.M., Chaft J.E., Pardoll D.M. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. N. Engl. J. Med. 2018;379:e14–e1986. doi: 10.1056/NEJMc1808251. [DOI] [PubMed] [Google Scholar]
- 88.Porciello N., Franzese O., D’Ambrosio L., Palermo B., Nisticò P. T-cell repertoire diversity: friend or foe for protective antitumor response? J. Exp. Clin. Cancer Res. 2022;41:356. doi: 10.1186/s13046-022-02566-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Joshi K., de Massy M.R., Ismail M., Reading J.L., Uddin I., Woolston A., Hatipoglu E., Oakes T., Rosenthal R., Peacock T., et al. Spatial heterogeneity of the T cell receptor repertoire reflects the mutational landscape in lung cancer. Nat. Med. 2019;25:1549–1559. doi: 10.1038/s41591-019-0592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sharonov G.V., Serebrovskaya E.O., Yuzhakova D.V., Britanova O.V., Chudakov D.M. B cells, plasma cells and antibody repertoires in the tumour microenvironment. Nat. Rev. Immunol. 2020;20:294–307. doi: 10.1038/s41577-019-0257-x. Preprint at. [DOI] [PubMed] [Google Scholar]
- 91.Fridman W.H., Petitprez F., Meylan M., Chen T.W.W., Sun C.M., Roumenina L.T., Sautès-Fridman C. B cells and cancer: To B or not to B? J. Exp. Med. 2021;218:e20200851. doi: 10.1084/jem.20200851. Preprint at. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Meylan M., Petitprez F., Becht E., Bougoüin A., Pupier G., Calvez A., Giglioli I., Verkarre V., Lacroix G., Verneau J., et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity. 2022;55:527–541.e5. doi: 10.1016/j.immuni.2022.02.001. [DOI] [PubMed] [Google Scholar]
- 93.Liu S., Iorgulescu B., Li S., Morriss J., Borji M., Murray E., Braun D., Livak K., Wu C., Chen F. 76 Spatial mapping of T cell receptors and transcriptomes in renal cell carcinoma following immune checkpoint inhibitor therapy. J. Immunother. Cancer. 2021;9:A84–A85. [Google Scholar]
- 94.Hayase E., Jenq R.R. Role of the intestinal microbiome and microbial-derived metabolites in immune checkpoint blockade immunotherapy of cancer. Genome Med. 2021;13:107. doi: 10.1186/s13073-021-00923-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Davar D., Dzutsev A.K., McCulloch J.A., Rodrigues R.R., Chauvin J.M., Morrison R.M., Deblasio R.N., Menna C., Ding Q., Pagliano O., et al. Fecal microbiota transplant overcomes resistance to anti-PD-1 therapy in melanoma patients. Science. 2021;371:595–602. doi: 10.1126/science.abf3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Baruch E.N., Youngster I., Ben-Betzalel G., Ortenberg R., Lahat A., Katz L., Adler K., Dick-Necula D., Raskin S., Bloch N., et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;371:602–609. doi: 10.1126/science.abb5920. [DOI] [PubMed] [Google Scholar]
- 97.Vétizou M., Pitt J.M., Daillère R., Lepage P., Waldschmitt N., Flament C., Rusakiewicz S., Routy B., Roberti M.P., Duong C.P.M., et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–1084. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sivan A., Corrales L., Hubert N., Williams J.B., Aquino-Michaels K., Earley Z.M., Benyamin F.W., Lei Y.M., Jabri B., Alegre M.L., et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti-PD-L1 efficacy. Science. 2015;350:1084–1089. doi: 10.1126/science.aac4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Matson V., Fessler J., Bao R., Chongsuwat T., Zha Y., Alegre M.L., Luke J.J., Gajewski T.F. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–108. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Derosa L., Routy B., Thomas A.M., Iebba V., Zalcman G., Friard S., Mazieres J., Audigier-Valette C., Moro-Sibilot D., Goldwasser F., et al. Intestinal Akkermansia muciniphila predicts clinical response to PD-1 blockade in patients with advanced non-small-cell lung cancer. Nat. Med. 2022;28:315–324. doi: 10.1038/s41591-021-01655-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Peters B.A., Wilson M., Moran U., Pavlick A., Izsak A., Wechter T., Weber J.S., Osman I., Ahn J. Relating the gut metagenome and metatranscriptome to immunotherapy responses in melanoma patients. Genome Med. 2019;11:61. doi: 10.1186/s13073-019-0672-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hakozaki T., Richard C., Elkrief A., Hosomi Y., Benlaïfaoui M., Mimpen I., Terrisse S., Derosa L., Zitvogel L., Routy B., Okuma Y. The Gut Microbiome Associates with Immune Checkpoint Inhibition Outcomes in Patients with Advanced Non-Small Cell Lung Cancer. Cancer Immunol. Res. 2020;8:1243–1250. doi: 10.1158/2326-6066.CIR-20-0196. [DOI] [PubMed] [Google Scholar]
- 103.Salgia N.J., Bergerot P.G., Maia M.C., Dizman N., Hsu J., Gillece J.D., Folkerts M., Reining L., Trent J., Highlander S.K., Pal S.K. Stool Microbiome Profiling of Patients with Metastatic Renal Cell Carcinoma Receiving Anti-PD-1 Immune Checkpoint Inhibitors. Eur. Urol. 2020;78:498–502. doi: 10.1016/j.eururo.2020.07.011. [DOI] [PubMed] [Google Scholar]
- 104.Derosa L., Routy B., Fidelle M., Iebba V., Alla L., Pasolli E., Segata N., Desnoyer A., Pietrantonio F., Ferrere G., et al. Gut Bacteria Composition Drives Primary Resistance to Cancer Immunotherapy in Renal Cell Carcinoma Patients. Eur. Urol. 2020;78:195–206. doi: 10.1016/j.eururo.2020.04.044. [DOI] [PubMed] [Google Scholar]
- 105.Lee K.A., Thomas A.M., Bolte L.A., Björk J.R., de Ruijter L.K., Armanini F., Asnicar F., Blanco-Miguez A., Board R., Calbet-Llopart N., et al. Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma. Nat. Med. 2022;28:535–544. doi: 10.1038/s41591-022-01695-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McCulloch J.A., Davar D., Rodrigues R.R., Badger J.H., Fang J.R., Cole A.M., Balaji A.K., Vetizou M., Prescott S.M., Fernandes M.R., et al. Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat. Med. 2022;28:545–556. doi: 10.1038/s41591-022-01698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Andrews M.C., Duong C.P.M., Gopalakrishnan V., Iebba V., Chen W.S., Derosa L., Khan M.A.W., Cogdill A.P., White M.G., Wong M.C., et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat. Med. 2021;27:1432–1441. doi: 10.1038/s41591-021-01406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gopalakrishnan V., Spencer C.N., Nezi L., Reuben A., Andrews M.C., Karpinets T.V., Prieto P.A., Vicente D., Hoffman K., Wei S.C., et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reese A.T., Dunn R.R. Drivers of Microbiome Biodiversity: A Review of General Rules, Feces, and Ignorance. mBio. 2018;9:12944–e1318. doi: 10.1128/mBio.01294-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wind T.T., Gacesa R., Vich Vila A., de Haan J.J., Jalving M., Weersma R.K., Hospers G.A.P. Gut microbial species and metabolic pathways associated with response to treatment with immune checkpoint inhibitors in metastatic melanoma. Melanoma Res. 2020;30:235–246. doi: 10.1097/CMR.0000000000000656. [DOI] [PubMed] [Google Scholar]
- 111.Cheng H.S., Tan S.P., Wong D.M.K., Koo W.L.Y., Wong S.H., Tan N.S. The Blood Microbiome and Health: Current Evidence, Controversies, and Challenges. Int. J. Mol. Sci. 2023;24:5633. doi: 10.3390/ijms24065633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Castillo D.J., Rifkin R.F., Cowan D.A., Potgieter M. The Healthy Human Blood Microbiome: Fact or Fiction? Front. Cell. Infect. Microbiol. 2019;9:148. doi: 10.3389/fcimb.2019.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Tan C.C.S., Ko K.K.K., Chen H., Liu J., Loh M., SG10K_Health Consortium. Chia M., Nagarajan N. No evidence for a common blood microbiome based on a population study of 9,770 healthy humans. Nat. Microbiol. 2023;8:973–985. doi: 10.1038/s41564-023-01350-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Poore G.D., Kopylova E., Zhu Q., Carpenter C., Fraraccio S., Wandro S., Kosciolek T., Janssen S., Metcalf J., Song S.J., et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature. 2020;579:567–574. doi: 10.1038/s41586-020-2095-1. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 115.Zhang J., Dai Z., Yan C., Zhang W., Wang D., Tang D. A new biological triangle in cancer: intestinal microbiota, immune checkpoint inhibitors and antibiotics. Clin. Transl. Oncol. 2021;23:2415–2430. doi: 10.1007/s12094-021-02659-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Frankel A.E., Deshmukh S., Reddy A., Lightcap J., Hayes M., McClellan S., Singh S., Rabideau B., Glover T.G., Roberts B., Koh A.Y. Cancer Immune Checkpoint Inhibitor Therapy and the Gut Microbiota. Integr. Cancer Ther. 2019;18 doi: 10.1177/1534735419846379. 1534735419846379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kilgour E., Rothwell D.G., Brady G., Dive C. Liquid Biopsy-Based Biomarkers of Treatment Response and Resistance. Cancer Cell. 2020;37:485–495. doi: 10.1016/j.ccell.2020.03.012. [DOI] [PubMed] [Google Scholar]
- 118.Kustanovich A., Schwartz R., Peretz T., Grinshpun A. Life and death of circulating cell-free DNA. Cancer Biol. Ther. 2019;20:1057–1067. doi: 10.1080/15384047.2019.1598759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sivapalan L., Murray J.C., Canzoniero J.V., Landon B., Jackson J., Scott S., Lam V., Levy B.P., Sausen M., Anagnostou V. Liquid biopsy approaches to capture tumor evolution and clinical outcomes during cancer immunotherapy. J. Immunother. Cancer. 2023;11:e005924. doi: 10.1136/jitc-2022-005924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Nabet B.Y., Esfahani M.S., Moding E.J., Hamilton E.G., Chabon J.J., Rizvi H., Steen C.B., Chaudhuri A.A., Liu C.L., Hui A.B., et al. Noninvasive Early Identification of Therapeutic Benefit from Immune Checkpoint Inhibition. Cell. 2020;183:363–376.e13. doi: 10.1016/j.cell.2020.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Si H., Kuziora M., Quinn K.J., Helman E., Ye J., Liu F., Scheuring U., Peters S., Rizvi N.A., Brohawn P.Z., et al. A Blood-based Assay for Assessment of Tumor Mutational Burden in First-line Metastatic NSCLC Treatment: Results from the MYSTIC Study. Clin. Cancer Res. 2021;27:1631–1640. doi: 10.1158/1078-0432.CCR-20-3771. [DOI] [PubMed] [Google Scholar]
- 122.Kim E.S., Velcheti V., Mekhail T., Yun C., Shagan S.M., Hu S., Chae Y.K., Leal T.A., Dowell J.E., Tsai M.L., et al. Blood-based tumor mutational burden as a biomarker for atezolizumab in non-small cell lung cancer: the phase 2 B-F1RST trial. Nat. Med. 2022;28:939–945. doi: 10.1038/s41591-022-01754-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Peters S., Dziadziuszko R., Morabito A., Felip E., Gadgeel S.M., Cheema P., Cobo M., Andric Z., Barrios C.H., Yamaguchi M., et al. Atezolizumab versus chemotherapy in advanced or metastatic NSCLC with high blood-based tumor mutational burden: primary analysis of BFAST cohort C randomized phase 3 trial. Nat. Med. 2022;28:1831–1839. doi: 10.1038/s41591-022-01933-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Moding E.J., Liu Y., Nabet B.Y., Chabon J.J., Chaudhuri A.A., Hui A.B., Bonilla R.F., Ko R.B., Yoo C.H., Gojenola L., et al. Circulating Tumor DNA Dynamics Predict Benefit from Consolidation Immunotherapy in Locally Advanced Non-Small Cell Lung Cancer. Nat. Can. (Ott.) 2020;1:176–183. doi: 10.1038/s43018-019-0011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Powles T., Assaf Z.J., Davarpanah N., Banchereau R., Szabados B.E., Yuen K.C., Grivas P., Hussain M., Oudard S., Gschwend J.E., et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature. 2021;595:432–437. doi: 10.1038/s41586-021-03642-9. [DOI] [PubMed] [Google Scholar]
- 126.Zhang Q., Luo J., Wu S., Si H., Gao C., Xu W., Abdullah S.E., Higgs B.W., Dennis P.A., van der Heijden M.S., et al. Prognostic and Predictive Impact of Circulating Tumor DNA in Patients with Advanced Cancers Treated with Immune Checkpoint Blockade. Cancer Discov. 2020;10:1842–1853. doi: 10.1158/2159-8290.CD-20-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ricciuti B., Jones G., Severgnini M., Alessi J.V., Recondo G., Lawrence M., Forshew T., Lydon C., Nishino M., Cheng M., Awad M. Early plasma circulating tumor DNA (ctDNA) changes predict response to first-line pembrolizumab-based therapy in non-small cell lung cancer (NSCLC) J. Immunother. Cancer. 2021;9:e001504. doi: 10.1136/jitc-2020-001504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Vega D.M., Nishimura K.K., Zariffa N., Thompson J.C., Hoering A., Cilento V., Rosenthal A., Anagnostou V., Baden J., Beaver J.A., et al. Changes in Circulating Tumor DNA Reflect Clinical Benefit Across Multiple Studies of Patients With Non-Small-Cell Lung Cancer Treated With Immune Checkpoint Inhibitors. JCO Precis. Oncol. 2022;6:e2100372. doi: 10.1200/PO.21.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hellmann M.D., Nabet B.Y., Rizvi H., Chaudhuri A.A., Wells D.K., Dunphy M.P.S., Chabon J.J., Liu C.L., Hui A.B., Arbour K.C., et al. Circulating Tumor DNA Analysis to Assess Risk of Progression after Long-term Response to PD-(L)1 Blockade in NSCLC. Clin. Cancer Res. 2020;26:2849–2858. doi: 10.1158/1078-0432.CCR-19-3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Georgiadis A., Durham J.N., Keefer L.A., Bartlett B.R., Zielonka M., Murphy D., White J.R., Lu S., Verner E.L., Ruan F., et al. Noninvasive Detection of Microsatellite Instability and High Tumor Mutation Burden in Cancer Patients Treated with PD-1 Blockade. Clin. Cancer Res. 2019;25:7024–7034. doi: 10.1158/1078-0432.CCR-19-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hwang M., Canzoniero J.V., Rosner S., Zhang G., White J.R., Belcaid Z., Cherry C., Balan A., Pereira G., Curry A., et al. Peripheral blood immune cell dynamics reflect antitumor immune responses and predict clinical response to immunotherapy. J. Immunother. Cancer. 2022;10:e004688. doi: 10.1136/jitc-2022-004688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nishino M., Ramaiya N.H., Hatabu H., Hodi F.S. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat. Rev. Clin. Oncol. 2017;14:655–668. doi: 10.1038/nrclinonc.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.https://www.accessdata.fda.gov/drugsatfda_docs/label/2022/125554s114lbl.pdf
- 134.Paver E.C., Cooper W.A., Colebatch A.J., Ferguson P.M., Hill S.K., Lum T., Shin J.S., O'Toole S., Anderson L., Scolyer R.A., Gupta R. Programmed death ligand-1 (PD-L1) as a predictive marker for immunotherapy in solid tumours: a guide to immunohistochemistry implementation and interpretation. Pathology. 2021;53:141–156. doi: 10.1016/j.pathol.2020.10.007. [DOI] [PubMed] [Google Scholar]
- 135.Berry, S., Giraldo N.A., Green B.F., Cottrell T.R., Stein J.E., Engle E.L., Xu H., Ogurtsova A., Roberts C., Wang D., et al., . Analysis of multispectral imaging with the AstroPath platform informs efficacy of PD-1 blockade." Science 372.eaba2609. [DOI] [PMC free article] [PubMed]
- 136.Chen Y., Jia K., Sun Y., Zhang C., Li Y., Zhang L., Chen Z., Zhang J., Hu Y., Yuan J., et al. Predicting response to immunotherapy in gastric cancer via multi-dimensional analyses of the tumour immune microenvironment. Nat. Commun. 2022;13:4851–4912. doi: 10.1038/s41467-022-32570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lopez de Rodas M., Nagineni V., Ravi A., Datar I.J., Mino-Kenudson M., Corredor G., Barrera C., Behlman L., Rimm D.L., Herbst R.S., et al. Role of tumor infiltrating lymphocytes and spatial immune heterogeneity in sensitivity to PD-1 axis blockers in non-small cell lung cancer. J. Immunother. Cancer. 2022;10:e004440. doi: 10.1136/jitc-2021-004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wu Z., Trevino A.E., Wu E., Swanson K., Kim H.J., D'Angio H.B., Preska R., Charville G.W., Dalerba P.D., Egloff A.M., et al. Graph deep learning for the characterization of tumour microenvironments from spatial protein profiles in tissue specimens. Nat. Biomed. Eng. 2022;6:1435–1448. doi: 10.1038/s41551-022-00951-w. [DOI] [PubMed] [Google Scholar]
- 139.Patwa A., Yamashita R., Long J., Risom T., Angelo M., Keren L., Rubin D.L. Multiplexed imaging analysis of the tumor-immune microenvironment reveals predictors of outcome in triple-negative breast cancer. Commun. Biol. 2021;4:852. doi: 10.1038/s42003-021-02361-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Zugazagoitia J., Gupta S., Liu Y., Fuhrman K., Gettinger S., Herbst R.S., Schalper K.A., Rimm D.L. Biomarkers Associated with Beneficial PD-1 Checkpoint Blockade in Non-Small Cell Lung Cancer (NSCLC) Identified Using High-Plex Digital Spatial Profiling. Clin. Cancer Res. 2020;26:4360–4368. doi: 10.1158/1078-0432.CCR-20-0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Larroquette M., Guegan J.P., Besse B., Cousin S., Brunet M., Le Moulec S., Le Loarer F., Rey C., Soria J.C., Barlesi F., et al. Spatial transcriptomics of macrophage infiltration in non-small cell lung cancer reveals determinants of sensitivity and resistance to anti-PD1/PD-L1 antibodies. J. Immunother. Cancer. 2022;10:e003890. doi: 10.1136/jitc-2021-003890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hwang S., Kwon A.Y., Jeong J.Y., Kim S., Kang H., Park J., Kim J.H., Han O.J., Lim S.M., An H.J. Immune gene signatures for predicting durable clinical benefit of anti-PD-1 immunotherapy in patients with non-small cell lung cancer. Sci. Rep. 2020;10:643. doi: 10.1038/s41598-019-57218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.van Galen P., Hovestadt V., Wadsworth Ii M.H., Hughes T.K., Griffin G.K., Battaglia S., Verga J.A., Stephansky J., Pastika T.J., Lombardi Story J., et al. Single-Cell RNA-Seq Reveals AML Hierarchies Relevant to Disease Progression and Immunity. Cell. 2019;176:1265–1281.e24. doi: 10.1016/j.cell.2019.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang H., Li S., Wang Q., Jin Z., Shao W., Gao Y., Li L., Lin K., Zhu L., Wang H., et al. Tumor immunological phenotype signature-based high-throughput screening for the discovery of combination immunotherapy compounds. Sci. Adv. 2021;7:eabd7851. doi: 10.1126/sciadv.abd7851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Abbott C.W., Boyle S.M., Pyke R.M., McDaniel L.D., Levy E., Navarro F.C.P., Mellacheruvu D., Zhang S.V., Tan M., Santiago R., et al. Prediction of Immunotherapy Response in Melanoma through Combined Modeling of Neoantigen Burden and Immune-Related Resistance Mechanisms. Clin. Cancer Res. 2021;27:4265–4276. doi: 10.1158/1078-0432.CCR-20-4314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sidhom J.W., Larman H.B., Pardoll D.M., Baras A.S. DeepTCR is a deep learning framework for revealing sequence concepts within T-cell repertoires. Nat. Commun. 2021;12:2309. doi: 10.1038/s41467-021-21879-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sidhom J.-W., et al. Vol. 8. 2022. Deep Learning Reveals Predictive Sequence Concepts within Immune Repertoires to Immunotherapy; p. 5089.https://www.science.org [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Johannet P., Coudray N., Donnelly D.M., Jour G., Illa-Bochaca I., Xia Y., Johnson D.B., Wheless L., Patrinely J.R., Nomikou S., et al. Using Machine Learning Algorithms to Predict Immunotherapy Response in Patients with Advanced Melanoma. Clin. Cancer Res. 2021;27:131–140. doi: 10.1158/1078-0432.CCR-20-2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Khorrami M., Prasanna P., Gupta A., Patil P., Velu P.D., Thawani R., Corredor G., Alilou M., Bera K., Fu P., et al. Changes in CT radiomic features associated with lymphocyte distribution predict overall survival and response to immunotherapy in non-small cell lung cancer. Cancer Immunol. Res. 2020;8:108–119. doi: 10.1158/2326-6066.CIR-19-0476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vaidya P., Bera K., Patil P.D., Gupta A., Jain P., Alilou M., Khorrami M., Velcheti V., Madabhushi A. Novel, non-invasive imaging approach to identify patients with advanced non-small cell lung cancer at risk of hyperprogressive disease with immune checkpoint blockade. J. Immunother. Cancer. 2020;8:e001343. doi: 10.1136/jitc-2020-001343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Trebeschi S., Drago S.G., Birkbak N.J., Kurilova I., Cǎlin A.M., Delli Pizzi A., Lalezari F., Lambregts D.M.J., Rohaan M.W., Parmar C., et al. Predicting response to cancer immunotherapy using noninvasive radiomic biomarkers. Ann. Oncol. 2019;30:998–1004. doi: 10.1093/annonc/mdz108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Vanguri R.S., Luo J., Aukerman A.T., Egger J.V., Fong C.J., Horvat N., Pagano A., Araujo-Filho J.d.A.B., Geneslaw L., Rizvi H., et al. Multimodal integration of radiology, pathology and genomics for prediction of response to PD-(L)1 blockade in patients with non-small cell lung cancer. Nat. Can. (Ott.) 2022;3:1151–1164. doi: 10.1038/s43018-022-00416-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Ilse, M., Tomczak, J. & Welling, M. Attention-based Deep Multiple Instance Learning. in Proceedings of the 35th International Conference on Machine Learning (eds. Dy, J. & Krause, A.) vol. 80 2127–2136 (PMLR, 10--15 Jul 2018).
- 154.Stoeckius M., Hafemeister C., Stephenson W., Houck-Loomis B., Chattopadhyay P.K., Swerdlow H., Satija R., Smibert P. Simultaneous epitope and transcriptome measurement in single cells. Nat. Methods. 2017;14:865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ma S., Zhang B., LaFave L.M., Earl A.S., Chiang Z., Hu Y., Ding J., Brack A., Kartha V.K., Tay T., et al. Chromatin Potential Identified by Shared Single-Cell Profiling of RNA and Chromatin. Cell. 2020;183:1103–1116.e20. doi: 10.1016/j.cell.2020.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cao J., Cusanovich D.A., Ramani V., Aghamirzaie D., Pliner H.A., Hill A.J., Daza R.M., McFaline-Figueroa J.L., Packer J.S., Christiansen L., et al. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science. 2018;361:1380–1385. doi: 10.1126/science.aau0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Swanson E., Lord C., Reading J., Heubeck A.T., Genge P.C., Thomson Z., Weiss M.D., Li X.J., Savage A.K., Green R.R., et al. Simultaneous trimodal single-cell measurement of transcripts, epitopes, and chromatin accessibility using TEA-seq. Elife. 2021;10:e63632. doi: 10.7554/eLife.63632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Rodriques S.G., Stickels R.R., Goeva A., Martin C.A., Murray E., Vanderburg C.R., Welch J., Chen L.M., Chen F., Macosko E.Z. Slide-seq: A scalable technology for measuring genome-wide expression at high spatial resolution. Science. 2019;363:1463–1467. doi: 10.1126/science.aaw1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Briggs, A. W. et al. Tumor-infiltrating immune repertoires captured by single-cell barcoding in emulsion. doi:.
- 160.Sudmeier L.J., Hoang K.B., Nduom E.K., Wieland A., Neill S.G., Schniederjan M.J., Ramalingam S.S., Olson J.J., Ahmed R., Hudson W.H. Distinct phenotypic states and spatial distribution of CD8+ T cell clonotypes in human brain metastases. Cell Rep. Med. 2022;3:100620. doi: 10.1016/j.xcrm.2022.100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hudson W.H., Sudmeier L.J. Localization of T cell clonotypes using the Visium spatial transcriptomics platform. STAR Protoc. 2022;3:101391. doi: 10.1016/j.xpro.2022.101391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Liu S., Iorgulescu J.B., Li S., Borji M., Barrera-Lopez I.A., Shanmugam V., Lyu H., Morriss J.W., Garcia Z.N., Murray E., et al. Spatial maps of T cell receptors and transcriptomes reveal distinct immune niches and interactions in the adaptive immune response. Immunity. 2022;55:1940–1952.e5. doi: 10.1016/j.immuni.2022.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Frangieh C.J., Melms J.C., Thakore P.I., Geiger-Schuller K.R., Ho P., Luoma A.M., Cleary B., Jerby-Arnon L., Malu S., Cuoco M.S., et al. Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nat. Genet. 2021;53:332–341. doi: 10.1038/s41588-021-00779-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chen H.X., Song M., Maecker H.T., Gnjatic S., Patton D., Lee J.J., Adam S.J., Moravec R., Liu X.S., Cerami E., et al. Network for biomarker immunoprofiling for cancer immunotherapy: Cancer Immune Monitoring and Analysis Centers and Cancer Immunologic Data Commons. Clin. Cancer Res. 2021;27:5038–5048. doi: 10.1158/1078-0432.CCR-20-3241. [DOI] [PMC free article] [PubMed] [Google Scholar]