

Abstract
This symposium provided a forum for presentations by the relevant groups on ligand design and ligand binding on the adenosine A1, receptor. Agreement appears to exist that the “N6–C8” model of ligand binding to the receptor is the preferred mode. A consensus has not yet been reached on the actual placement of the ligand in the receptor and the exact amino acids which interact in its binding. Two viable models exist at present. Both can be tested with selective site-directed mutagenic studies on the A1 receptor as well as with additional designed ligands.
Keywords: adenosine; theophylline; MDL 100,991; N6–C8 model; R-PIA
INTRODUCTION
Success in designing selective receptor ligands can be greatly enhanced by combining the concepts of active analog probing to build a receptor pharmacophore with three dimensional computer modeling of the receptor and the proposed ligand binding site. This symposium, entitled “Molecular Modeling of Adenosine Receptor Ligands,” brought together members of some of the most active groups in computer-assisted design of selective ligands for the adenosine A1 receptor (Fig. 1).
Figure 1.

Symposium presented at “Purines ’92: Pharmacology and Clinical Applications” meeting, University of Milan, Milan, Italy, June 24, 1992.
LIGAND MODELING
An investigation of the properties of the adenosine receptor through modeling of the ligands (Fig. 2) that interact with the receptor was presented by van Galen. His modeling of the ligands has led to a better understanding of the interactions between ligand and receptor, and how qualitative and sometimes quantitative predictions can be made in the design of novel agents.
Figure 2.

Prototypical agonist (adenosine) and antagonist (theophylline) ligands for adenosine receptors.
It had been suggested that in the case of the potent xanthine antagonist 8-(2-amino-4-chlorophenyl)-1,3-dipropylxanthine (PACPX), the phenyl substituent lies in the same plane as the xanthine ring because of stabilization by an intramolecular hydrogen bond [Bruns et al., 1983]. However, MOPAC calculations on PACPX and a number of other potent 8-phenyl substituted xanthines clearly showed that there was a considerable energy barrier with dihedral angles of 0° and 180°, i.e., when the phenyl substituent was coplanar with the xanthine ring. The optimal dihedral angle was found to be ca. 220°. This observation provided an explanation for the unexpectedly low affinities of 8-substituted 7-methylxanthines: because of steric hindrance by the 7-methyl group, the 8-substituent cannot attain the dihedral angle required for optimal interaction with the receptor [van de Wenden et al., 1991]. The optimal dihedral angle calculation was also consistent with the inactivity found for a conformationally restricted 8-phenylxanthine, wherein a 2-atom bridge connected the 2-position of the phenyl ring with the 9-position of the xanthine.
van Galen described an active analog approach to explore the N6-region of the A1 receptor in detail. On the basis of a set of 26 N6-substituted adenosine analogs ranging in affinity from subnanomolar to micromolar, a model was developed to map a number of subregions of the N6-domain, as well as some so-called forbidden regions. Comparison of the affinities of compounds that differ in the occupation of one subregion only allowed the accurate prediction of the affinities of compounds that were not used to develop the model [van Galen et al., 1989]. A similar approach was taken by Ortwine et al. [1990], to map the N6-region of the A2 receptor, which assisted in the development of the first potent A2-selective adenosine agonists.
Comparisons of the steric, electrostatic and hydrophobic properties of a number of potent A1 antagonists were used to develop a model of the antagonist binding site of the A1 receptor. Potent antagonists were shown to be flat, aromatic heterocycles that shared common areas of negative and positive electrostatic potential in the plane of the heterocycle, as well as some domains where hydrophobic substituents enhanced affinity [van Galen et al., 1990a]. In the original paper, it was hypothesized that the free electron pair on the nitrogen atom corresponding to N9 in xanthines might act as a hydrogen bond acceptor. However, it was recently shown that 9-deazaxanthines are at least as potent at adenosine receptors as the parent xanthines [Grahner et al., 1992]. On the basis of this antagonist model a new class of non-xanthine antagonists, the imidazo[4,5-c]quinolin-4-amines, was developed. As expected, some representatives were shown to have affinities for the A1 receptor in the lower nanomolar range [van Galen et al., 1991]. Independently, a very similar modeling approach by James et al. [1992] led to the development of imidazo[1,5-a]triazin-4-ones as potent antagonists at the guinea-pig aorta receptor, an A2b-like receptor [Kennedy et al., 1992]. Thus, these compounds might become useful tools in unraveling the physiological functions of A2b receptors.
Finally, the conformational properties of xanthine-7-ribosides were studied to try to determine whether adenosine interacts with the receptor in the syn or in the anti conformation [van Galen et al., 1990b]. Unlike adenosine which, in solution, does not show a distinct preference for either conformation, theophylline-7-riboside clearly favors the anti conformation. This was shown in both theoretical calculations and in NMR measurements, and is in full agreement with the X-ray crystallographic data. Taken together with recent data which show that theophylline-7-ribosidemay act as a partial agonist at adenosine receptors [Borea et al., 1992], these findings support the notion that adenosine interacts with its receptor in the anti conformation.
Peet presented a new model of ligand binding for xanthines with respect to adenosine at adenosine receptors. As opposed to a standard method of comparing these structures, wherein the four nitrogen atoms of the purine and xanthine rings are directly overlaid, the xanthine ring in the new binding mode is flipped and rotated with respect to adenosine [Peet et al., 1990] (Fig. 3). In this model, N1, N3 and N9 of adenosine overlay with N9, N3 and N1 of theophylline, respectively. This superimposition places the acidic hydrogen atoms of both ligands in the same proximity; thus, the N7–H of theophylline is close to one of the C6 amino hydrogen atoms of adenosine. Also, N7 and the C6 carbonyl oxygen atoms of theophylline are in close proximity to the C6 amino and N7 atoms of adenosine, respectively. This model is referred to as the “N6–C8” model by van der Wenden et al. [1992], since the N6 atom of adenosine derivatives is placed close to the C8 atom of xanthines.
Figure 3.

Three models for the agonist/antagonist binding site on the adenosine A1 receptor: (a) standard; (b) flipped; (c) N6–C8. Template molecules are N6-cyclopentyladenosine (CPA) and 1,3-dipropyl-8-cyclopentylxanthine (DPCPX). From the presentation by A. Ijzerman.
The close placement of the acidic NH groups in the agonist and antagonist ligands is an important feature of the N6–C8 model described by Peet. It is generally known that these groups are necessary for good adenosine A1 receptor affinity.
The new N6–C8 binding mode of theophylline relative to adenosine was corroborated in several ways. Charge localization studies (three methods) were used to show that point charges arising from five pairs of heteroatoms in the N6–C8 mode were similar to those calculated for the standard mode. The program GRID [Goodford, 1985] was used to develop three-dimensional potential energy contour surfaces for a water probe at different energy levels approaching adenosine and theophylline superimposed in the N6–C8 and standard modes. Contour overlap was shown to be better for the N6–C8 model. In addition, Peet described a new synthesis of xanthines which allowed the introduction of chiral substituents at position 8 to test the N6–C8 ligand binding model. Thus, phenylisopropyl groups were incorporated and it was shown that the R-isomer had significantly better affinity that the S-isomer at adenosine A1 receptors, which is analogous to the better affinity of (R)-N6-phenylisopropyladenosine (R-PIA) than the (S)-isomer at adenosine A1 receptors. These data support the hypothesis that the chiral recognition units at N6 on adenosine and C8 on the xanthine nucleus are interacting with the same area of the receptor.
A recent electrotopological study by Joshi and Kier [1992] on 8-substituted xanthines indicated that the first atom attached to C8 was important in the receptor interaction event. This conclusion correlates with the experimental finding of Peet et al. [1990] that the chirality of the 8-phenylisopropyl group on the xanthine nucleus significantly affected the binding affinity. The chiral atom of the 8-phenylisopropyl group is the first atom attached to the C8 position.
The greater importance of the xanthine N7 relative to N9 and the emphasis on the N7-H tautomer in the N6–C8 model were supported with recent synthetic work by Grahner et al. [1992]. 9-Deazaxanthines were shown to be as potent or more potent than the corresponding xanthines at adenosine A1 and A2 receptors. Conversely, the 7-deazaxanthines were decidedly less potent. Two routes to 9-deazaxanthines were described.
A “flipped” ligand binding model of theophylline with respect to adenosine (Fig. 3) was recently described by van Galen et al. [1990b]. This model was very recently compared with the standard and N6–C8 models by van der Wenden et al. [1992], and it was concluded on the basis of steric and electrostatic comparisons that the N6–C8 model was the most probable.
Finally, the synthesis of a multifunctional probe compound, (−)-N-(2-aminoethyl)-2-[4-[2-(2,3,6,9-tetrahydro-1,3-dipropyl-2,6-dioxo-1H-purin-8-yl)propyl] phenoxy]acetamide (MDL 100,991) (Fig. 4), was presented by Peet. MDL 100,991 was designed using the functionalized congener approach of Jacobson et al. [1986], and has subnanomolar (Ki) potency at the adenosine A1 receptor. Placement of MDL 100,991 in the modeled adenosine A1 receptor led to hypothesized sites of interaction, which can be tested by synthetic modifications of the probe molecule or receptor mutagenesis studies.
Figure 4.

Structure of MDL 100,991, a ligand binding site receptor probe.
MODELING OF LIGAND-RECEPTOR INTERACTIONS
The adenosine A1 receptor has been shown to be coupled to its various effector systems through a family of pertussis toxin-sensitive G-proteins [Munshi et al., 1991]. Therefore, as a member of the superfamily of G-protein linked receptors, its three dimensional structure was built on the atomic coordinates of bacteriorhodopsin as outlined by Dr. G. Nordvall.
The structure of bacteriorhodopsin was previously known only at low resolution from the analysis of three independent crystal structures which were averaged [Tsygannik and Baldwin, 1987]. More recently, however, the structure has been resolved to 3.5 Å by Henderson et al. [1990], using high resolution electron cryo-microscopy. This new structural information has been useful for the development of G-protein coupled receptor models [Stiles, 1992; Hibert et al., 1991]. The 3.5 Å model does allow the loop areas of bacteriorhodopsin to be distinguished from the transmembrane helices.
General strategic considerations were discussed for using the structural information for modeling the adenosine A1 receptor (Fig. 5) and related G-protein coupled receptors. Rotation of the helices and sequence alignment or helix fitting are important refinements. Conserved amino acids are generally placed toward the inside of the chimney formed by the helices, as are the polar amino acids. The latter are thought to be important for ligand binding and/or receptor function; an example in the A1 receptor is the aspartic acid in helix 2. Optimization of side chain placement is important to ligand positioning in proposed models.
Figure 5.

Primary sequence of the dog adenosine A1 receptor.
The amino acid sequence of the A1 receptor has been reported for clones from three different cDNA libraries, i.e., canine thyroid [Libert et al., 1989], rat striatum [Mahan et al., 1991] and bovine brain [Olah et al., 1992]. Analyses of the sequences shows a high degree of homology (>90%) in the seven transmembrane α-helices comprising the putative ligand binding site.
Site directed mutagenesis and chemical modification studies have implicated at least one and possibly two histidine residues in the binding of adenosine agonists and possible antagonists to the receptor [Klotz et al., 1988; Olah et al., 1992], These could potentially be His251 in helix VI and His278 in helix VII.
Analysis of the three dimensional structure led Ijzerman to propose that since His278 is present in a largely hydrophilic environment, it was close to the ribose moiety in agonists, particularly, the 2′- and 3′-OH groups which are required for receptor activation. His251 is at the interface between a hydrophobic and hydrophilic region and, consequently, it may be close to the C6-amino group of agonists, which is critical for enhanced receptor affinity (Fig. 6).
Figure 6.

Points of interaction between cyclopentyladenosine and the adenosine A1 receptor as proposed by Ijzerman.
Thus, CPA (N6-cyclopentyladenosine), a highly potent and selective A1 receptor agonist, was docked in several orientations into the A1 receptor pore, all of which were defined by the proximity of the two histidine residues. Two orientations of CPA with the cyclopentyl substituents between the helices led to an overall distortion of receptor architecture, and were not considered further. Of the other possibilities, with CPA entirely within the pore, the most favorable orientation was with the cyclopentyl substituent pointing to the extracellular side of the protein, close to helices IV, V and VI. The ligand binding site on the A1 receptor (arbitrarily defined as the amino acid residues within 4.5 Å from CPA) shows more hydrophilic residues in close proximity to the hydrophilic purine and ribose moieties of CPA. The two histidine residues (251 and 278) may form hydrogen bonds with CPA, via N6-H, and 2’- and 3’-OH, respectively, as does Ser281 with 5’-OH. In this orientation the purine-cyclopentyl torsion angle in CPA is −60°, whereas the ribose moiety adopts a definite anti conformation, fully in line with earlier modeling studies on ligands only. The hydrogen bond formed between His251 and N6-H might well explain the fact that N6-disubstitution of adenosine derivatives is detrimental to affinity, since two substituents would eliminate an energetically favorable hydrogen bond and add steric bulk. It was stressed that a slightly different orientation of CPA probably would allow other hydrogen bonds, e.g., from the ligand to other alcoholic amino acid residues (Thr91, Ser94, and Ser248). Nevertheless, all of these hydrophilic amino acid residues remain an integral part of the ligand binding site. The hydrophobic cyclopentyl substituent, in contrast, is surrounded by two hydrophobic amino acid side chains, viz., Val138 and Phe185, together with Cys255. Upon binding, both the receptor and CPA would change their conformations in order to maximize the interaction strength. Since the docking procedure started with the receptor and the ligand in their energetically optimized conformations, these changes would lead to increases in intramolecular energy content. A comparison between the CPA binding site of the A1 receptor and the retinal binding site of bacteriorhodopsin revealed that both ligands in their bound conformations occupy approximately the same space within the pore. Thus, the lack of overall homology between the two proteins and the entirely different chemical characteristics of both proteins and ligands appear to somehow compensate for each other, allowing a similar location of the binding site.
Peet presented a different binding model for adenosine A1 agonists. In this model, His278 is not directly involved in any interaction with the agonists but serves to stabilize the location of helix VII through hydrogen bonds with Glu16 and Ser23 on helix I (Fig. 7). This would fix the position of Thr277 on helix VII to selectively interact with the C6-amino group of adenosine derivatives. Receptor activation would occur upon agonist binding through the interaction of Asp55 on helix II with the 2’- and 3’-OH of the ribose sugar (Fig. 8). Additional interactions which occur upon docking the A1 selective agonist R-PIA (N6-R-phenylisopropyladenosine), again in the anti conformation, are Ser281 with the 5’-OH and a hydrophobic region composed of Leu250, Leu253, Phe136, Leu88, and Val87 around the phenylisopropyl substituent. As in the previous model, disubstitution of the N6-amino group would result in decreased binding affinity due to the loss of an energetically favorable hydrogen bond and the addition of steric hindrance.
Figure 7.

Hydrogen bonding relationships for His278 with Glu16 and Ser23, which could stabilize the positioning of Thr277. Partial structure of adenosine A1 receptor displayed as alpha-carbon trace.
Figure 8.

Points of interaction between R-phenylisopropyladenosine and the adenosine A1 receptor as proposed by Peet.
For the placement of an A1 antagonist in the respective binding schemes, both groups utilized the N6–C8 superimposition of agonist and antagonist [Peet et al., 1990]. In Ijzerman’s model, the two n-propyl substituents on the xanthine ring system in both DPCPX and XAC can be accommodated by the A1 receptor upon docking. The propyl group on N3 has significant van der Waals interactions with Val87 and Leu90, whereas the N1-substituent is close to Trp247, Leu250, and Ser281, which is a somewhat less hydrophobic environment. In the model presented by Peet, the N7-H and the C6 carbonyl group can hydrogen bond with the Thr277 on helix VII (Fig. 9). With both models, the extended substituents at the C6-amino position of adenosine derivatives and the C8 position of xanthines fit into a chimney formed by helices III, IV, V, and VI.
Figure 9.
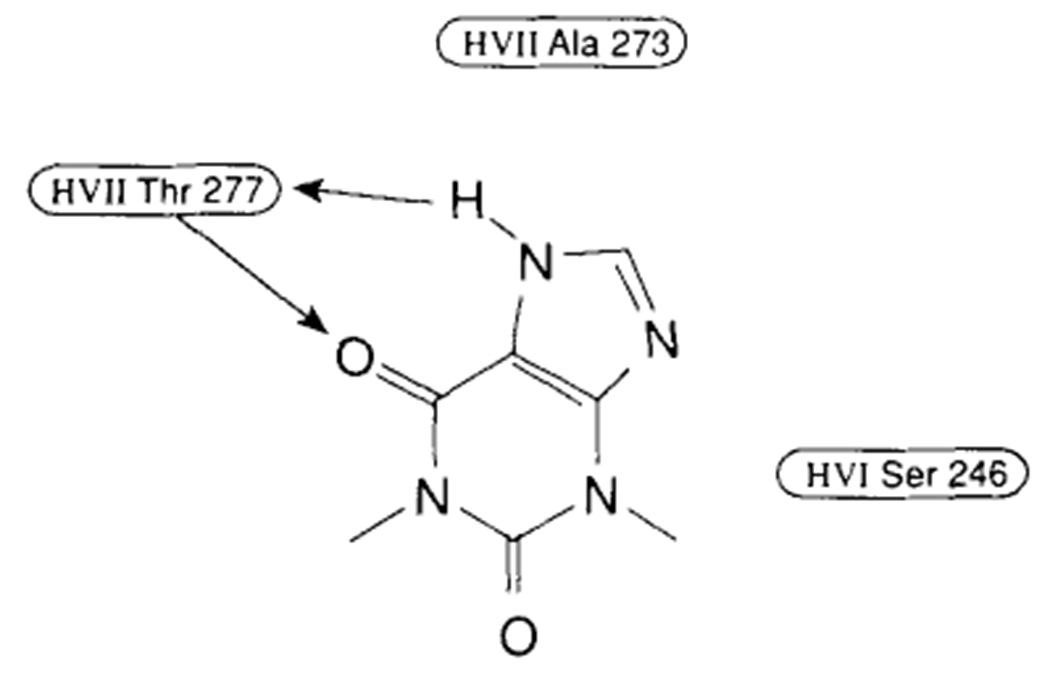
Points of interaction between theophylline and the adenosine A1 receptor as proposed by Peet.
Conclusions drawn from ligand modeling studies are presently more valid than those from receptor modeling studies with our current knowledge base. Ligand modeling is less speculative and involves fewer assumptions, since it is largely based on experimental data. Greater variation exists in receptor models since there are numerous ways of packing helices around the ligands. However, as we learn more about the receptor through mutagenesis studies, specifically designed ligand probes and more sophisticated molecular modeling techniques, valid receptor models will emerge.
ACKNOWLEDGMENTS
The authors wish to thank Linda Austin and Debbra Wagner for their assistance in the preparation of this manuscript.
Contributor Information
Mark W. Dudley, Marion Merrell Dow Research Institute, Cincinnati, Ohio
Norton P. Peet, Marion Merrell Dow Research Institute, Cincinnati, Ohio
David A. Demeter, Marion Merrell Dow Research Institute, Cincinnati, Ohio
Herschel J.R. Weintraub, Marion Merrell Dow Research Institute, Cincinnati, Ohio
Ad P. Ijzerman, Center for Bio-Pharmaceutical Sciences, Leiden, The Netherlands
Gunnar Nordvall, Karolinska Institute, Stockholm, Sweden.
Philip J.M. van Galen, National Institutes of Health, Bethesda, Maryland
Kenneth A. Jacobson, National Institutes of Health, Bethesda, Maryland
REFERENCES
- Borea PA, Varani K, Gardenghi A, Bertolasi V, van Galen PJM, and Ijzerman AP (1992): Theophylline-7-riboside: A partial agonist for adenosine A1 receptors. Int J Purine Pyrimidine Res 3:65. [Google Scholar]
- Bruns RF, Daly JW, Snyder SH (1983): Adenosine receptor binding: Structure activity analysis generates extremely potent xanthine antagonists. Proc Natl Acad Sci USA 80:2077–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodford PJ (1985): A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J Med Chem 28:849–857. [DOI] [PubMed] [Google Scholar]
- Grahner B, Deters D, Miller CE (1992): Xanthine analogues modified at the 9-position: Affinities for A1 and A2 adenosine receptors. Int J Purine Pyrimidine Res 3:92. [Google Scholar]
- Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckmann E, Downing KH (1990): Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J Mol Biol 213:899–929. [DOI] [PubMed] [Google Scholar]
- Hibert MF, Trumpp-Kallmeyer S, Bruinvels A, Hoflak J (1991): Three-dimensional models of neurotransmitter G protein-coupled receptors. Mol Pharmacol 40:8–15. [PubMed] [Google Scholar]
- Jacobson KA, Kirk KL, Padgett WL, Daly JW (1986): A functionalized congener approach to adenosine receptor antagonists: Amino acid conjugates of 1,3-dipropylxanthine. Mol Pharmacol 29:126–133. [PMC free article] [PubMed] [Google Scholar]
- James R, Stutchbury NCJ (1992): Imidazo[l,5-a]triazin-4-ones: The use of electrostatic potentials in the design of adenosine, antagonists. Abstracts of the 203rd American Chemical Society Meeting, Medicinal Chemistry Division, San Francisco, April 5–10, Abstract 166. [Google Scholar]
- Joshi N, Kier LB (1992): An electrotopological state analysis of adenosine A1 inhibitors. Med Chem Res 1:409–416. [Google Scholar]
- Kennedy I, Gurden MF, Strong P, Hornby E, Foster M, Vardey CJ (1992): Pharmacological classification of adenosine receptors in isolated tissues. Int J Purine Pyrimidine Res 3:83. [Google Scholar]
- Klotz K-N, Lohse MJ, Schwabe U (1988): Chemical modification of A1 adenosine receptors in rat brain membranes. J Biol Chem 263:17522–17526. [PubMed] [Google Scholar]
- Libert FM, Parmentier M, Lefort A, Dinsart C, Van Sande J, Maenhout C, Simons MJ, Dumont JE, Vassart G (1989): Selective amplification and cloning of four new members of the G protein-coupled receptor family. Science 244:569–572. [DOI] [PubMed] [Google Scholar]
- Mahan LC, McVittie LD, Smyk-Randall EM, Nakata FJ, Gerfen CR, Sibley DR (1991): Cloning and expression of an A1 adenosine receptor from rat brain. Mol Pharmacol 40:1–7. [PubMed] [Google Scholar]
- Munshi R, Pang I, Sternweis PC, Linden J (1991): A1 receptors of bovine brain coupled to quanine nucleotide-binding proteins Gi1, Gi2 and G0. J Biol Chem 266:22285–22289. [PubMed] [Google Scholar]
- Olah ME, Ren H, Ostrowski J, Jacobson KA, Stiles GL (1992): Cloning, expression, and characterization of the unique bovine A1 adenosine receptor. J Biol Chem 267:10764–10770. [PMC free article] [PubMed] [Google Scholar]
- Ortwine DF, Bridges AJ, Humblet C, Trivedi BK (1990): In Jacobson KA, Daly JW, Manganiello V (eds): Purines in Cellular Signalling: Targets for New Drugs. New York: Springer, pp 152–157. [Google Scholar]
- Peet NP, Lentz NL, Meng EC, Dudley MW, Ogden AML, Demeter DA, Weintraub HJR, Bey P (1990): A novel synthesis of xanthines: Support for a new binding mode for xanthine with respect to adenosine at adenosine receptors. J Med Chem 33:3127–3130. [DOI] [PubMed] [Google Scholar]
- Stiles GL (1992): Adenosine receptors. J Biol Chem 267:6451–6454. [PubMed] [Google Scholar]
- Tsygannik IN, Baldwin JM (1987): Three-dimensional structure of deoxy-cholate-treated purple membrane at 6 Å resolution and molecular averaging of three crystal forms of bacteriorhodopsin. Eur J Biophys 14:263–272. [Google Scholar]
- van der Wenden EM, van Galen PJM, Ijzerman AP, Soudijn W (1991): Mapping the xanthine C8-region of the adenosine A1 receptor with computer graphics. Eur J Pharmacol Mol Pharmacol Sect. 206:315–323. [DOI] [PubMed] [Google Scholar]
- van der Wenden EM, Ijzerman AP, Soudijn W (1992): A steric and electrostatic comparison of three models for the agonist/antagonist binding site on the adenosine A1 receptor. J Med Chem 35:629–635. [DOI] [PubMed] [Google Scholar]
- van Galen PJM, Leusen FJJ, Ijzerman AP, Soudijn W (1989): Mapping the N6-region of the adenosine A1 receptor with computer graphics. Eur J Pharmacol Mol Pharmacol Sect 172:19–27. [DOI] [PubMed] [Google Scholar]
- van Galen PJM, van Vlijmen HWT, Ijzerman AP, Soudijn W (1990a): A model for the antagonist binding site on the adenosine A1 receptor, based on steric, electrostatic, and hydrophobic properties. J Med Chem 33:1708–1713. [DOI] [PubMed] [Google Scholar]
- van Galen PJM, Ijzerman AP, Soudijn W (1990b): Xanthine-7-ribosides as adenosine A1 receptor antagonists: Further evidence for adenosine’s anti mode of binding. Nucleosid Nucleotid 9:275–291. [Google Scholar]
- van Galen PJM, Nissen P, van Wijngaarden I, Ijzerman AP, Soudijn W (1991): 1H-Imidazo[4,5-c]quinolin-4-amines: Novel nonxanthine adenosine antagonists. J Med Chem 34:1202–1206. [DOI] [PubMed] [Google Scholar]