

Abstract
Background
Cystic fibrosis (CF) is an autosomal recessive disorder rarely found in Asian populations. Most males with CF are infertile because of obstructive azoospermia (OA) caused by congenital bilateral absence of the vas deferens (CBAVD). Compound heterozygous mutations of cystic fibrosis transmembrane conductance regulator (CFTR) are among the most common pathogenic factors in CBAVD. However, few genealogical analyses have been performed.
Methods
In this study, whole‐exome sequencing and cosegregation analysis were performed in a Chinese pedigree involving two siblings with CBAVD. Moreover, in vitro gene expressions were used to analyze the pathogenicity of a novel CFTR mutation.
Results
We identified compound heterozygous mutations of CFTR comprising the known disease‐causing variant c.1210‐11T>G (also known as IVS9‐5 T) and c.2144delA;p.q715fs in two siblings with CBAVD. To verify the effects in vitro, we transfected vectors expressing wild‐type and mutated CFTR into 293T cells. The results showed that the CFTR protein containing the frameshift mutation (c.2144delA) was 60 kD smaller. With testicular sperm aspiration/intracytoplasmic sperm injection‐embryo transfer (TESA/ICSI‐ET), both CBAVD patients fathered healthy offspring.
Conclusion
Our study revealed that compound heterozygous mutations of CFTR are involved in CBAVD, expanding the known CFTR gene mutation spectrum of CBAVD patients and providing more evidence that compound heterozygous mutations can cause familial CBAVD.
Keywords: assisted reproductive technology (ART), CFTR gene, compound heterozygous, congenital bilateral absence of the vas deferens (CBAVD), male infertility
We identified compound heterozygous mutations of CFTR in two siblings with CBAVD in a Chinese pedigree. Whole‐exome sequencing and cosegregation analysis were performed; moreover, in vitro gene expressions were used to analyze the pathogenicity of a novel CFTR mutation. With testicular sperm aspiration/intracytoplasmic sperm injection‐embryo transfer (TESA/ICSI‐ET), both CBAVD patients fathered healthy offspring.
1. INTRODUCTION
Cystic fibrosis (CF) is a rare autosomal recessive disease among Asian populations, with an incidence rate of less than 1/100,000. However, in Caucasians, the incidence rate is approximately 1/2500, and the frequency of mutation in the population is approximately 1/25 (Delatycki et al., 2014; Griesenbach et al., 2015). Clinically, it is very typical for CF to be characterized by chronic obstructive pulmonary disease, pancreatic insufficiency, and high electrolyte concentration in sweat. However, it should be noted that CF can also present with atypical genital phenotypes, such as congenital bilateral absence of the vas deferens (CBAVD), which is found in 1% to 2% of cases of male infertility and 6% of cases of obstructive azoospermia among Caucasians (Bombieri et al., 2011). It has been reported that the incidence of CBAVD among infertility patients in China was 1.15%, similar to the rate in Caucasians (Yang et al., 2015).
CBAVD is mainly caused by gene mutations, and mutation in the CFTR (OMIM ID: 602421) gene is one of the most common causes. CFTR is a chloride channel protein that is widely expressed in the respiratory, digestive, reproductive, and endocrine systems and sweat glands to maintain electrolyte homeostasis. Some studies have shown that when the level of CFTR protein decreases to approximately 10% of the normal value, bronchiectasis or pancreatitis might occur, whereas a decrease to a lesser extent can lead to CBAVD (Claustres, 2005; Timmreck et al., 2003). CFTR mutations can cause dysfunction of cell membrane chloride channels, causing the cells to be unable to regulate the flow of chloride ions and water molecules, which can lead to the discharge of sticky secretions from the reproductive tract, blocking and degrading the vas deferens and ultimately leading to the development of CBAVD (de Souza et al., 2018; Gaillard et al., 1997). Studies have shown that the frequency and hot spots of mutations in the CFTR gene are significantly higher in Caucasians than in Asians. Approximately 63% to 83% of patients in Caucasians carry two different mutations in the CFTR gene (i.e., exhibit compound heterozygosity) (Claustres, 2005; Jézéquel et al., 2000; Taulan et al., 2007). However, few results of CFTR gene screening in CBAVD patients in China have been reported, and the distribution of CFTR mutations remains unclear.
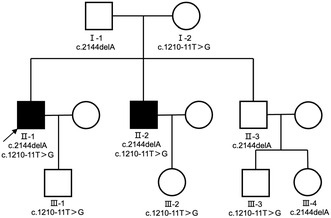
Here, we report a Chinese pedigree in which two patients with CBAVD carried a novel frameshift mutation (nm_000492: c.2144delA;p.q715fs;Het) from their father and a splice site mutation (nm_000492: c.1210‐11T>G; Het) from their mother (Figure 1). The c.1210‐11T>G mutation is located in the poly(T) sequence at the intron 9 (in HGVS name) splice acceptor site and may result in reduced splicing efficiency in exon 10 (in HGVS name), which in turn leads to reduced expression of functional CFTR protein. This mutation is also known as the 5T mutation (IVS9), and it has been shown to be a mild mutation with incomplete epistasis that may lead to an increased risk of CBAVD (Chu et al., 1993; Teng et al., 1997). Our study confirms that the novel compound heterozygous mutation identified is associated with CBAVD, expanding the spectrum of CFTR gene mutations in CBAVD patients in the Chinese population and explaining its pathogenicity.
FIGURE 1.
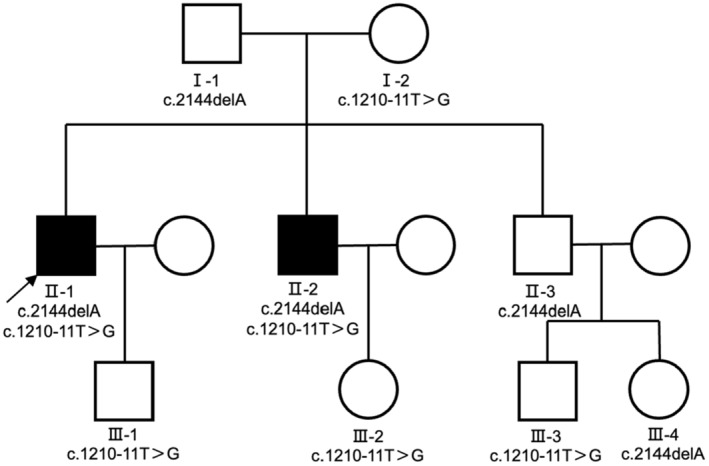
Pedigree structure of the CBAVD family. Black squares indicate CBAVD patients. Arrow indicates the proband.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical compliance
This study was approved by the Ethics Committee of the Henan Provincial People's Hospital. All subjects provided written informed consent for the study, which was conducted in accordance with the Declaration of Helsinki.
2.2. Study participants
We recruited a Chinese pedigree with two siblings with CBAVD from Henan Provincial People's Hospital. The proband was 31 years old and infertile for 2 years after marriage. Physical examination found bilateral absence of the vas deferens, and he was diagnosed with CBAVD at Henan Provincial People's Hospital (Henan University People's Hospital) in combination with auxiliary examinations. Written informed consent was obtained from all participants. This study was approved by the Ethics Committee of the Henan Provincial People's Hospital.
2.3. Whole‐exome sequencing
Genomic DNA was extracted from blood samples using the DNeasy Blood & Tissue Kit (Qiagen). IDT xGen Exome Research Panel V1.0 (Integrated DNA Technologies) was used for whole‐exome sequencing of samples. A Qubit 2.0 fluorometer (Thermo Fisher Scientific) and 2100 Bioanalyzer high‐sensitivity DNA assay (Agilent Technologies) were used to evaluate and measure the quantity and quality of sequencing libraries.
Qualified libraries were sequenced on the Illumina NovaSeq platform (Illumina, San Diego, USA) for 2 × 150‐bp paired‐end next‐generation sequencing. BWA v0.7.13 was used to compare the FASTQ files with the human reference genome (hg19/GRCh37) (Li & Durbin, 2009). Variants (single‐nucleotide variants and indels) were genotyped from recalibrated BAM files using GATK 4.0 and annotated with ANNOVAR based on data such as HGVS variant description, population frequency, disease or phenotype, and variant function prediction (McKenna et al., 2010; Wang et al., 2010). The American College of Medical Genetics (ACMG) guidelines classify variant types as pathogenic, probably pathogenic, variants of unknown significance (VUS), probably benign, or benign (Richards et al., 2015). Copy number variants were called by the DNA copy R package and manually examined using the Integrative Genomics Viewer after filtering and classification according to the ACMG guidelines (Brandt et al., 2020; Thorvaldsdóttir et al., 2013; Venkatraman & Olshen, 2007).
2.4. Sanger sequencing
Sanger sequencing was used to confirm mutations and analyze cosegregation using the following primers: forward 5′‐F‐TTCCTGCATCCATGGCATCT‐3′ and reverse 5′‐GCCAACTCCACCGTCAACAG‐3′ for c.2144delA, and forward 5′‐ATGTCCTCTAGAAACCGTATGC‐3′ and reverse 5′‐CCAAAAATACCTTCCAGCACTA‐3′ for c.1210‐11T>G.
2.5. Plasmid construction
RNA was extracted from healthy human blood samples using the RNAsimple Total RNA Kit (DP419, TIANGEN), and reverse transcription was performed using First‐Strand cDNA Synthesis SuperMix (AT301, TransGen Biotech) to synthesize cDNA. The target fragment was amplified with DNA polymerase (TaKaRa Premix Taq Version 2.0 plus dye) using the following primers (designed by Primer 5.0 software): 5′‐ATGCAGAGGTCGCCTCTGG‐3′ and 5′‐CAACCAAAGAAGCAGCCACC‐3′. The amplification products were cloned into the polyclonal site of the pEGFP‐C1 vector using the restriction endonucleases BamHI and EcoRI (New England Biolabs). The wild‐type plasmid was then extracted using the TIANprep Mini Plasmid Kit (DP103, TIANGEN) and sent to Zhengzhou Sunya Biotechnology Co., Ltd. for Sanger sequencing verification.
2.6. Mutant plasmid construction and identification
Mutant plasmids were constructed using the Mut Express II Fast Mutagenesis Kit V2 (C214, Vazyme). Using the wild‐type plasmid as a template, the following primers were designed and used: 5′‐CCATTGTGCAAAGACTCCCTTACAAATGAATGGC‐3′ and 5′‐GGAGTCTTTGCACAATGGAAAATTTTCGTATAGA‐3′. The primer annealing temperature was set at 63°C. The cyclized recombinant products were transformed into DH5α cells (C502, Vazyme), cultured, and identified by PCR. Next, the positive clones were screened, and the plasmids were extracted using a TIANprep Mini Plasmid Kit (DP103, TIANGEN) to obtain the mutant plasmids, which were sent to Zhengzhou Sunya Biotechnology Co., Ltd. for Sanger sequencing to verify whether the targeted mutation was successfully constructed.
2.7. Cell culture and transient transfection assays
HEK293T cells frozen in liquid nitrogen were resuscitated according to the conventional method, DMEM culture medium with a volume fraction of 10% fetal bovine serum was added, and the cells were incubated in an incubator at 37°C with 5% CO2. Cells were transfected in 6‐well plates when the cell density reached 80%–90% and then transfected using the Lipofectamine 2000 DNA Transfection Reagent Protocol (Life Technologies) when the cell density reached 80%–90% again. Lipofectamine 2000 reagent and plasmids were diluted with OPTI‐MEM (Gibco) and then mixed together. The mixture was incubated for 20 min at room temperature and then added dropwise to HEK293T cell culture medium in a 6‐well plate, which was shaken gently before placing it in the incubator. The expression of green fluorescent protein was observed by inverted fluorescence microscopy after 48 h of transfection.
2.8. Western blot
After transfection for 48 h, cells were washed with PBS and lysed with RIPA Lysis Buffer (P0013B, Beyotime Biotechnology) containing protease inhibitors. SDS‐PAGE Sample Loading Buffer 6X (Beyotime Biotechnology) was added to the cell samples, and proteins were denatured by boiling at high temperature. After the samples were cooled, SDS‐PAGE protein gel electrophoresis was performed. A 6% SDS‐PAGE gel (P0012A, Beyotime Biotechnology) was prepared, and the extracted protein samples were separated by gel electrophoresis, transferred to PVDF membranes, blocked with 10% skimmed milk powder for 2 h at room temperature, washed with TBST, and then incubated overnight at 4°C with GFP primary antibody (50430, Proteintech) diluted 1:2000. The membrane was washed with TBST, and horseradish peroxidase (HRP)‐labeled goat anti‐rabbit secondary antibody (A0208, Beyotime Biotechnology) diluted 1:1000 was added and incubated for 2 h at room temperature. Finally, the membrane was exposed to ultrasensitive ECL reagent (P0018, Beyotime Biotechnology) for color development, and the protein bands were imaged by a high‐sensitivity chemiluminescence ChemiDoc XRS system (Bio‐Rad).
3. RESULTS
3.1. Clinical characteristics
In the present study, twelve subjects from the family were enrolled, including two patients with CBAVD (Figure 1) who did not exhibit typical CF phenotypes. Both were diagnosed with impalpable scrotal vas deferens by physical examination and invisible vas deferens by ultrasound. Sequencing of their peripheral blood samples indicated the CFTR gene mutation. Based on these findings, they were diagnosed with the genital form of CF (Anguiano et al., 1992). The semen examination of the proband (family number II‐1) showed no sperm on centrifugation, decreased volume, and decreased pH, while the liquefaction time was normal. Testicular sperm aspiration (TESA) showed sperm in the testis. The chromosomal karyotype was 46,XY, and the AZF gene was normal. Additionally, sex hormone examination indicated that the proband had normal serum follicle‐stimulating hormone (FSH), luteinizing hormone (LH), and testosterone (T) levels. The results of two sweat tests on the right upper limb were equivocal; the chloride concentrations were 35 and 31 mmol/L, respectively. Physical examination showed no signs such as emphysematous chest and clubbed fingers, except for bilateral absence of vas deferens. He had no history of any chronic respiratory disease. Chest CT scan showed no abnormalities; the color Doppler examination showed that the pancreas, hepar, and bilateral kidneys were normal, but the bilateral vas deferens and seminal vesicles were not detected. His younger brother (family number II‐2) had a similar clinical presentation (Table 1), while his father and elder brother and the offspring within the pedigree had no CBAVD symptoms. The parents of this pedigree did not report any consanguinity.
TABLE 1.
Clinical laboratory evaluation of the two CBAVD patients in the study.
| II‐1 | II‐2 | Normal range | |
|---|---|---|---|
| Age (years) | 31 | 27 | – |
| Gender | Male | Male | – |
| Sperm volume (mL) | 0.2 | 1.2 | >1.5 |
| Total sperm count (106/ejaculate) | 0 | 0 | >39 |
| Seminal PH | 6.5 | 6.5 | 7.2–8 |
| Karyotype | 46, XY | 46, XY | – |
| AZF deletion | Undetectable | Undetectable | – |
| Kidney | Bilaterally normal | Bilaterally normal | – |
| Testis | 37 × 25 × 19 mm (right) | 36 × 25 × 18 mm (right) | – |
| 38 × 24 × 20 mm (left) | 36 × 25 × 16 mm (left) | – | |
| Epididymis caput | 12 × 10 mm (right) | 16 × 8 mm (right) | – |
| 11 × 9 mm (left) | 10 × 7 mm (left) | – | |
| Epididymis corpus | Absence | Absence | –‐ |
| Epididymis cauda | Absence | Absence | – |
| Vas deference | Absence | Absence | – |
| Seminal vesicle | Absence | Absence | – |
| FSH (mIU/mL) | 4.73 | 10.29 | 1.5–12.4 |
| LH (mIU/mL) | 2.73 | 5.50 | 1.7–8.6 |
| T (ng/mL) | 4.58 | 5.28 | 2.49–8.36 |
| E2 (pg/mL) | 27.72 | 35.94 | 27.1–52.2 |
| Sweat chloride (mmol/L) | 35 and 31 | 33 and 37 | CF > 60, N < 30 Equivocal 31–59 |
In addition, to develop an assisted reproductive program, the two patients' wives also underwent CFTR gene testing. After excluding the presence of pathogenic CFTR mutations in the patient's wives, the couples were referred to the genetic counseling program of other reproductive centers on CF and fully informed of the risks of treatment; the two couples received TESA/ICSI‐ET. The fertilization rate, available embryo rate, and implantation rate of the two couples were 90.9%/81.3%, 50.0%/76.9%, and 100%/50%, respectively (Table 2). The proband and his wife gave birth to a boy, his younger brother had a girl, and his older brother naturally fathered a boy and a girl. Follow‐up indicated that all the offspring were healthy, as of 2022.
TABLE 2.
Clinical outcomes of the two couples following ICSI.
| II‐1 | II‐2 | |
|---|---|---|
| Male age (years) | 31 | 27 |
| Female age (years) | 28 | 27 |
| No. of ICSI cycles | 1 | 1 |
| No. of oocytes injected | 11 | 16 |
| Fertilization rate | 10/11 (90.9%) | 13/16 (81.3%) |
| Cleavage rate | 10/10 (100%) | 13/13 (100%) |
| Available embryo rate | 5/10 (50%) | 10/13 (76.9%) |
| Number of embryos transferred | 2 | 2 |
| Implantation rate | 2/2 (100%) | 1/2 (50%) |
| Clinical pregnancy | Yes | Yes |
| Live birth | Yes | Yes |
3.2. Mutational analysis
In this study, a novel frameshift mutation (NM_000492: c.2144delA; p.q715fs; Het) and a known splice site mutation (NM_000492: c.1210‐11T>G; Het) were identified in a Chinese pedigree with two CBAVD patients (Figure 2). The frameshift mutation is not found in ExAC (http://exac.broadinstitute.org) or 1000 Genomes (http://www.internationalgenome.org) and has not been reported previously. According to the 2015 ACMG guidelines for the interpretation of genetic variants, this novel frameshift variant is considered a pathogenic variant. Cosegregation analysis confirmed that these two mutations were in trans. Sanger sequencing was used to validate the family analysis, which confirmed the compound heterozygous CFTR mutations in the pedigree (Figure 2). Specifically, the mother of two siblings with CBAVD was found to carry a heterozygous mutation of c.1210‐11T>G in the CFTR gene, while the father carried a heterozygous mutation of c.2144delA. Notably, the other two adult males in the pedigree who harbored the frameshift mutation (c.2144delA) had no clinical features of CF or CBAVD.
FIGURE 2.
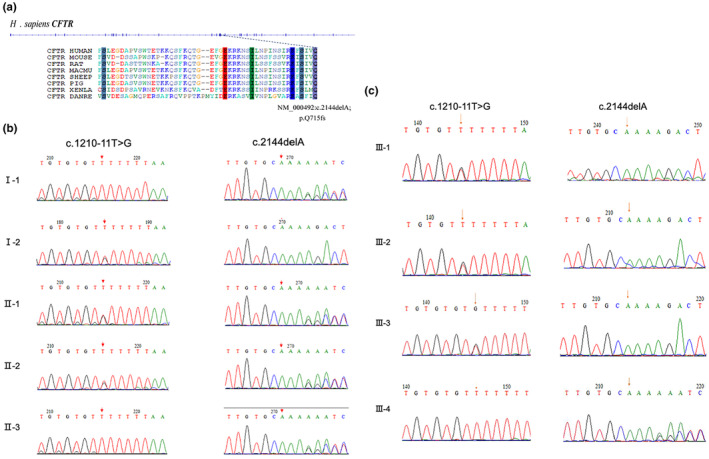
Genetic variations detected in the CBAVD family. Structure of CFTR gene and the novel frameshift mutation in CFTR protein (a). Validation of both variants of CFTR among family members by Sanger sequencing (b, c).
3.3. In vitro gene expression
The wild‐type plasmid constructed by ligating the target fragment of the pEGFP‐C1 vector with the restriction endonucleases BamHI and EcoRI was analyzed by DNA sequencing, and the sequencing results showed that the wild‐type recombinant plasmid (CFTR‐W) was successfully constructed (Figure 3). Then, the wild‐type recombinant plasmid was used as a template to construct the mutant plasmid (CFTR‐M). The pEGFP‐C1 empty vector was used as the control group. The three plasmids (CFTR‐W, CFTR‐M, and pEGFP‐C1) were transfected into HEK293T cells, and then, green fluorescence was observed under an inverted fluorescence microscope after 48 h. The CFTR‐W and CFTR‐M plasmids showed strong green fluorescent protein expression in HEK293T cells, with an infection rate above 90%, while no green fluorescent protein was expressed in pEGFP‐C1 vector cells (Figure 4), suggesting successful transfection and high transfection efficiency. After 72 h of transfection, the three groups of cells were lysed, and the protein expression in the HEK293T cells was detected by western blotting. The protein size of the mutant CFTR was reduced by 60 kD compared to that of the wild‐type group (220 kD), while the protein size of the control pEGFP‐C1 was approximately 27 kD (Figure 5).
FIGURE 3.
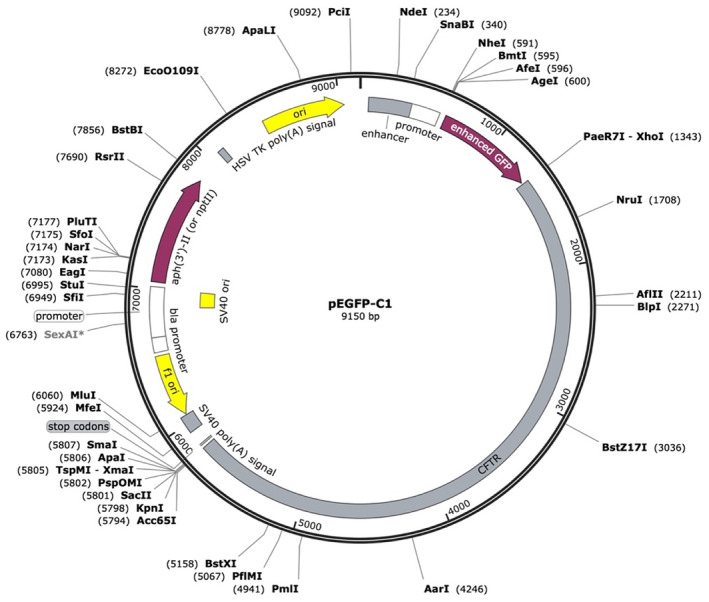
Structure of the wild‐type recombinant plasmid (CFTR‐W).
FIGURE 4.
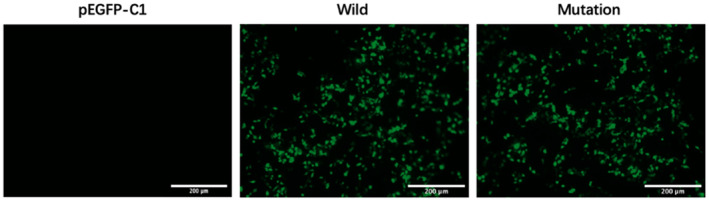
The green fluorescent protein expression at 48 h after transfection (200×).
FIGURE 5.
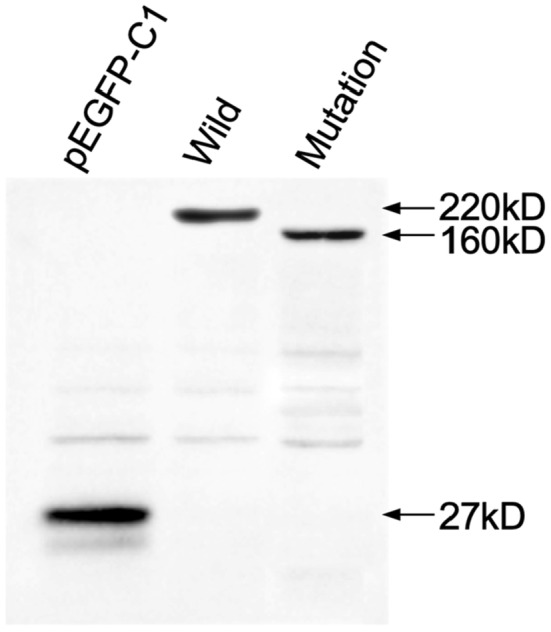
Western blotting (WB) assay detected the size of mutant CFTR protein. The pEGFP‐C1 was used as the control group.
4. DISCUSSION
The clinical phenotype of CF is related to the amount of protein transcripts after CFTR gene mutation and the specificity of protein transcripts in different tissues. CBAVD can occur in isolation without any typical clinical manifestations of CF. This may be caused by a differential sensitivity to anomalous mRNA splicing or dysfunctional CFTR protein among various organs (Cuppens & Cassiman, 2004; Mak et al., 1997). Among the different tissues, the growth and development of the vas deferens was susceptible to CFTR gene mutations; in addition, the expression level was significantly higher in respiratory epithelial cells than in epididymal epithelial cells. However, CBAVD patients with IVS9‐TG13‐5T may not have lung symptoms. One study found that a low level of normal CFTR transcripts was sufficient to maintain normal airway function in bronchial epithelial cells, suggesting a high tolerance of lung tissue to dysfunctional CFTR protein (Chu et al., 1992). The poly(T) sequence is located at the splice acceptor site at the end of intron 9 and influences the amount of skipping of exon 10. The 5T mutation may reduce exon 9 splicing efficiency and thereby promote the production of immature proteins with no channel activity (Chu et al., 1993). These proteins cannot contribute to chloride channel activity, and an extreme decrease in the level of normal CFTR chloride channels in the cell membrane may cause CF (Delaney et al., 1993; Gregory et al., 1991).
Portuguese researchers have carried out extensive mutation screening of CFTR exons and exon‐intron splicing sites in European patients with CBAVD and found that 78% of CBAVD patients carry at least one CFTR mutation and 48% of them have two mutation sites. One study has shown that when the CFTR gene is heterozygous for the 5T mutation, its transcript level only reaches 6%–16% of normal, which may be one of the reasons why mild phenotypes, such as CBAVD, can occur even in heterozygous patients (Kronn et al., 1998). Another study showed that among family members carrying the same single mutation, only some of them developed CBAVD symptoms, suggesting that CFTR gene mutation is not an absolute determinant of the occurrence and development of CBAVD and that other factors may be involved (Daudin et al., 2000). Previous studies have suggested that only one copy of a CFTR mutation is not sufficient to cause CBAVD, and the combination of frequent and rare mutations may be the main cause of CBAVD (de Souza et al., 2018; Ge et al., 2019; Lissens et al., 1996). This conclusion is consistent with our study. This pedigree study provides evidence for a CBAVD pedigree and confirms the composite heterozygous status of CFTR variants, which can provide more evidence for investigating CBAVD gene mutations. The results of in vitro functional assays suggest that the truncated protein may not be able to perform the same function as normal, intact protein.
Most CBAVD patients have normal spermatogenesis function, and their spouses can become pregnant by TESA/ICSI‐ET. However, the CFTR gene mutation is likely to be passed on to the next generation, and even more severe CF symptoms may occur in the offspring, so genetic consultation before ART is particularly critical, especially for patients with a family history of CBAVD; patients and their spouses should be screened for CFTR mutations. For patients with spouses who do not carry the mutation, the rate of their child developing CBAVD is very low, potentially less than 1% (Shin et al., 1997). However, when a CFTR gene mutation is found, genetic consultation should be conducted to avoid passing CBAVD to the next generation.
5. CONCLUSIONS
We identified a novel compound heterozygous mutation in CFTR (c.2144delA;p.q715fs and c.1210‐11T>G) in a Chinese pedigree. Through molecular and cellular studies, we elucidated the effect of the novel mutation on the CFTR protein and explained its potential pathogenicity. We also expanded the CFTR gene mutation profile of CBAVD patients in the Chinese population. It was demonstrated that single rare CFTR mutations may not cause clinical symptoms, but compound heterozygous mutations can cause the development and progression of CBAVD. Furthermore, CBAVD patients with CFTR mutations can have healthy offspring by ART.
AUTHOR CONTRIBUTIONS
Lingyi Li, Xiaowei Qu, and Haibin Guo conceived and designed the experiments. Lingyi Li and Chenchen Cui conducted the experiments. Xiaowei Qu and Ke Feng collected the samples and performed clinical assessments. Yinghong Fang collected the clinical data of the patients. Yanqing Xia and Feng Wan analyzed the WES data. Cuilian Zhang and Hengtao Ge performed the ART treatment. Lingyi Li and Xiaowei Qu wrote the manuscript. All authors read and approved the final manuscript.
FUNDING INFORMATION
This study was supported by Henan Provincial Science and Technology Research Project (222102310047), Medical Science and Technology Project of Henan Province (LHGJ20230046 and LHGJ20230005), and Action Plan Project for Innovation and Quality Improvement of Postgraduate Training, School of Clinical Medicine, Henan University, China (SYLYC2022176).
CONFLICT OF INTEREST STATEMENT
The authors declare that there is no conflict of interest regarding the publication of this paper.
ETHICS STATEMENT AND CONSENT TO PARTICIPATE
This study was approved by the Ethics Committee of the Henan Provincial People's Hospital. All subjects provided written informed consent for the study, which was conducted in accordance with the Declaration of Helsinki.
ACKNOWLEDGMENTS
We thank all patients and their family members for their participation.
Li, L. , Qu, X. , Cui, C. , Feng, K. , Xia, Y. , Wan, F. , Ge, H. , Fang, Y. , Zhang, C. , & Guo, H. (2024). Compound heterozygous mutations in CFTR causing congenital bilateral absence of the vas deferens in a Chinese pedigree. Molecular Genetics & Genomic Medicine, 12, e2364. 10.1002/mgg3.2364
Lingyi Li and Xiaowei Qu should be considered joint first author.
DATA AVAILABILITY STATEMENT
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
REFERENCES
- Anguiano, A. , Oates, R. D. , Amos, J. A. , Dean, M. , Gerrard, B. , Stewart, C. , Maher, T. A. , White, M. B. , & Milunsky, A. (1992). Congenital bilateral absence of the vas deferens. A primarily genital form of cystic fibrosis. JAMA, 267(13), 1794–1797. [PubMed] [Google Scholar]
- Bombieri, C. , Claustres, M. , De Boeck, K. , Derichs, N. , Dodge, J. , Girodon, E. , Sermet, I. , Schwarz, M. , Tzetis, M. , Wilschanski, M. , Bareil, C. , Bilton, D. , Castellani, C. , Cuppens, H. , Cutting, G. R. , Drevínek, P. , Farrell, P. , Elborn, J. S. , Jarvi, K. , … Ferec, C. (2011). Recommendations for the classification of diseases as CFTR‐related disorders. Journal of Cystic Fibrosis: Official Journal of the European Cystic Fibrosis Society, 10(Suppl. 2), S86–S102. 10.1016/S1569-1993(11)60014-3 [DOI] [PubMed] [Google Scholar]
- Brandt, T. , Sack, L. M. , Arjona, D. , Tan, D. , Mei, H. , Cui, H. , Gao, H. , Bean, L. J. H. , Ankala, A. , Del Gaudio, D. , Johnson, A. K. , Vincent, L. M. , Reavey, C. , Lai, A. , Richard, G. , & Meck, J. M. (2020). Adapting ACMG/AMP sequence variant classification guidelines for single‐gene copy number variants. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 22(2), 336–344. 10.1038/s41436-019-0655-2 [DOI] [PubMed] [Google Scholar]
- Chu, C. S. , Trapnell, B. C. , Curristin, S. , Cutting, G. R. , & Crystal, R. G. (1993). Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nature Genetics, 3(2), 151–156. 10.1038/ng0293-151 [DOI] [PubMed] [Google Scholar]
- Chu, C. S. , Trapnell, B. C. , Curristin, S. M. , Cutting, G. R. , & Crystal, R. G. (1992). Extensive posttranscriptional deletion of the coding sequences for part of nucleotide‐binding fold 1 in respiratory epithelial mRNA transcripts of the cystic fibrosis transmembrane conductance regulator gene is not associated with the clinical manifestations of cystic fibrosis. The Journal of Clinical Investigation, 90(3), 785–790. 10.1172/JCI115952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claustres, M. (2005). Molecular pathology of the CFTR locus in male infertility. Reproductive Biomedicine Online, 10(1), 14–41. 10.1016/s1472-6483(10)60801-2 [DOI] [PubMed] [Google Scholar]
- Cuppens, H. , & Cassiman, J.‐J. (2004). CFTR mutations and polymorphisms in male infertility. International Journal of Andrology, 27(5), 251–256. 10.1111/j.1365-2605.2004.00485.x [DOI] [PubMed] [Google Scholar]
- Daudin, M. , Bieth, E. , Bujan, L. , Massat, G. , Pontonnier, F. , & Mieusset, R. (2000). Congenital bilateral absence of the vas deferens: Clinical characteristics, biological parameters, cystic fibrosis transmembrane conductance regulator gene mutations, and implications for genetic counseling. Fertility and Sterility, 74(6), 1164–1174. 10.1016/s0015-0282(00)01625-3 [DOI] [PubMed] [Google Scholar]
- de Souza, D. a. S., Faucz, F. R. , Pereira‐Ferrari, L. , Sotomaior, V. S. , & Raskin, S. (2018). Congenital bilateral absence of the vas deferens as an atypical form of cystic fibrosis: Reproductive implications and genetic counseling. Andrology, 6(1), 127–135. 10.1111/andr.12450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney, S. J. , Rich, D. P. , Thomson, S. A. , Hargrave, M. R. , Lovelock, P. K. , Welsh, M. J. , & Wainwright, B. J. (1993). Cystic fibrosis transmembrane conductance regulator splice variants are not conserved and fail to produce chloride channels. Nature Genetics, 4(4), 426–431. 10.1038/ng0893-426 [DOI] [PubMed] [Google Scholar]
- Delatycki, M. B. , Burke, J. , Christie, L. , Collins, F. , Gabbett, M. , George, P. , Haan, E. , Ioannou, L. , Martin, N. , McKenzie, F. , O'Leary, P. , Scoble‐Williams, N. , Turner, G. , Massie, J. , & Human Genetics Society of Australasia . (2014). Human Genetics Society of Australasia position statement: Population‐based carrier screening for cystic fibrosis. Twin Research and Human Genetics: The Official Journal of the International Society for Twin Studies, 17(6), 578–583. 10.1017/thg.2014.65 [DOI] [PubMed] [Google Scholar]
- Gaillard, D. A. , Carré‐Pigeon, F. , & Lallemand, A. (1997). Normal vas deferens in fetuses with cystic fibrosis. The Journal of Urology, 158(4), 1549–1552. [PubMed] [Google Scholar]
- Ge, B. , Zhang, M. , Wang, R. , Wang, D. , Li, T. , Li, H. , & Wang, B. (2019). A rare frameshift variant in trans with the IVS9‐5 T allele of CFTR in a Chinese pedigree with congenital aplasia of vas deferens. Journal of Assisted Reproduction and Genetics, 36(12), 2541–2545. 10.1007/s10815-019-01617-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory, R. J. , Rich, D. P. , Cheng, S. H. , Souza, D. W. , Paul, S. , Manavalan, P. , Anderson, M. P. , Welsh, M. J. , & Smith, A. E. (1991). Maturation and function of cystic fibrosis transmembrane conductance regulator variants bearing mutations in putative nucleotide‐binding domains 1 and 2. Molecular and Cellular Biology, 11(8), 3886–3893. 10.1128/mcb.11.8.3886-3893.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesenbach, U. , Pytel, K. M. , & Alton, E. W. F. W. (2015). Cystic fibrosis gene therapy in the UK and elsewhere. Human Gene Therapy, 26(5), 266–275. 10.1089/hum.2015.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jézéquel, P. , Dubourg, C. , Le Lannou, D. , Odent, S. , Le Gall, J. Y. , Blayau, M. , Le Treut, A. , & David, V. (2000). Molecular screening of the CFTR gene in men with anomalies of the vas deferens: Identification of three novel mutations. Molecular Human Reproduction, 6(12), 1063–1067. 10.1093/molehr/6.12.1063 [DOI] [PubMed] [Google Scholar]
- Kronn, D. , Jansen, V. , & Ostrer, H. (1998). Carrier screening for cystic fibrosis, Gaucher disease, and Tay‐Sachs disease in the Ashkenazi Jewish population: The first 1000 cases at New York University Medical Center, New York, NY. Archives of Internal Medicine, 158(7), 777–781. 10.1001/archinte.158.7.777 [DOI] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics, 25(14), 1754–1760. 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissens, W. , Mercier, B. , Tournaye, H. , Bonduelle, M. , Férec, C. , Seneca, S. , Devroey, P. , Silber, S. , Van Steirteghem, A. , & Liebaers, I. (1996). Cystic fibrosis and infertility caused by congenital bilateral absence of the vas deferens and related clinical entities. Human Reproduction (Oxford, England), 11 Suppl. 4, 55–78; discussion 79–80. 10.1093/humrep/11.suppl_4.55 [DOI] [PubMed] [Google Scholar]
- Mak, V. , Jarvi, K. A. , Zielenski, J. , Durie, P. , & Tsui, L. C. (1997). Higher proportion of intact exon 9 CFTR mRNA in nasal epithelium compared with vas deferens. Human Molecular Genetics, 6(12), 2099–2107. 10.1093/hmg/6.12.2099 [DOI] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20(9), 1297–1303. 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine: Official Journal of the American College of Medical Genetics, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, D. , Gilbert, F. , Goldstein, M. , & Schlegel, P. N. (1997). Congenital absence of the vas deferens: Incomplete penetrance of cystic fibrosis gene mutations. The Journal of Urology, 158(5), 1794–1798; discussion 1798–1799. 10.1016/s0022-5347(01)64131-4 [DOI] [PubMed] [Google Scholar]
- Taulan, M. , Girardet, A. , Guittard, C. , Altieri, J.‐P. , Templin, C. , Beroud, C. , Des Georges, M. , & Claustres, M. (2007). Large genomic rearrangements in the CFTR gene contribute to CBAVD. BMC Medical Genetics, 8, 22. 10.1186/1471-2350-8-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng, H. , Jorissen, M. , Van Poppel, H. , Legius, E. , Cassiman, J. J. , & Cuppens, H. (1997). Increased proportion of exon 9 alternatively spliced CFTR transcripts in vas deferens compared with nasal epithelial cells. Human Molecular Genetics, 6(1), 85–90. 10.1093/hmg/6.1.85 [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir, H. , Robinson, J. T. , & Mesirov, J. P. (2013). Integrative genomics viewer (IGV): High‐performance genomics data visualization and exploration. Briefings in Bioinformatics, 14(2), 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmreck, L. S. , Gray, M. R. , Handelin, B. , Allito, B. , Rohlfs, E. , Davis, A. J. , Gidwani, G. , & Reindollar, R. H. (2003). Analysis of cystic fibrosis transmembrane conductance regulator gene mutations in patients with congenital absence of the uterus and vagina. American Journal of Medical Genetics. Part A, 120A(1), 72–76. 10.1002/ajmg.a.20197 [DOI] [PubMed] [Google Scholar]
- Venkatraman, E. S. , & Olshen, A. B. (2007). A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics (Oxford, England), 23(6), 657–663. 10.1093/bioinformatics/btl646 [DOI] [PubMed] [Google Scholar]
- Wang, K. , Li, M. , & Hakonarson, H. (2010). ANNOVAR: Functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Research, 38(16), e164. 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, X. , Sun, Q. , Yuan, P. , Liang, H. , Wu, X. , Lai, L. , & Zhang, Y. (2015). Novel mutations and polymorphisms in the CFTR gene associated with three subtypes of congenital absence of vas deferens. Fertility and Sterility, 104(5), 1268–1275.e1‐2. 10.1016/j.fertnstert.2015.07.1143 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.