

Conspectus

The atmosphere is a key part of the earth system comprising myriad chemical species in all basic forms of matter. Ubiquitous nano- and microscopic aerosol particles and cloud droplets suspended in the air play crucial roles in earth’s climate and the formation of air pollution. Surfaces are a prominent part of aerosols and droplets, due to the high surface area to bulk volume ratios, but very little is known about their specific properties. Many atmospheric compounds are surface-active, leading to enhanced surface concentrations in aqueous solutions. Their distribution between the surface and bulk may determine heterogeneous chemistry and many other properties of aerosol and cloud droplets, but has not been directly observed.
We used X-ray photoelectron spectroscopy (XPS) to obtain direct molecular-level information on the surface composition and structure of aqueous solutions of surface-active organics as model systems for atmospheric aerosol and cloud droplets. XPS is a vacuum-based technique enabled for volatile aqueous organic samples by the application of a high-speed liquid microjet. In combination with brilliant synchrotron X-rays, the chemical specificity of XPS allows distinction between elements in different chemical states and positions within molecular structures. We used core-level C 1s and N 1s signals to identify the alkyl and hydrophilic groups of atmospheric carboxylic acids, alkyl-amines, and their conjugate acids and bases. From this, we infer changes in the orientation of surface-adsorbed species and quantify their relative abundances in the surface. XPS-derived surface enrichments of the organics follow trends expected from their surface activities and we observed a preferential orientation at the surface with the hydrophobic alkyl chains pointing increasingly outward from the solution at higher concentrations. This provides a first direct experimental observation of well-established concepts of surface adsorption and confirms the soundness of the method.
We mapped relative abundances of conjugate acid−base pairs in the aqueous solution surfaces from the respective intensities of distinctive XPS signals. For each pair, the protonation equilibrium was significantly shifted toward the neutral form in the surface, compared to the bulk solution, across the full pH range. This represents an apparent shift of the pKa in the surface, which may be toward either higher or lower pH, depending on whether the acid or base form of the pair is the neutral species. The surface shifts are broadly consistent with the relative differences in surface enrichment of the individual acid and base conjugates in binary aqueous solutions, with additional contributions from nonideal interactions in the surface. In aqueous mixtures of surface-active carboxylate anions with ammonium salts at near-neutral pH, we found that the conjugate carboxylic acids were further strongly enhanced. This occurs as the coadsorption of weakly basic carboxylate anions and weakly acidic ammonium cations forms ion-pair surface layers with strongly enhanced local abundances, increasing the probability of net proton transfer according to Le Chatelier’s principle. The effect is stronger when the evaporation of ammonia from the surface further contributes to irreversibly perturb the protonation equilibrium, leaving a surplus of carboxylic acid. These surface-specific effects may profoundly influence atmospheric chemistry mediated by aqueous aerosols and cloud droplets but are currently not taken into account in atmospheric models.
Key References
Prisle N. L.; Ottosson N.; Öhrwall G.; Söderström J.; Maso M. D.; Björneholm O.. Surface/bulk partitioning and acid/base speciation of aqueous decanoate: direct observations and atmospheric implications. Atmos. Chem. Phys. 2012, 12, 12227–12242. 10.5194/acp-12-12227-2012(1)Surface-sensitive X-ray photoelectron spectroscopy in combination with synchrotron radiation showed that the formation of a decanoate–ammonium ion-pair surface layer with enhanced local concentrations leads to a strong enhancement of decanoic acid from proton transfer in accordance with Le Chatelier’s principle.
Öhrwall G.; Prisle N. L.; Ottosson N.; Werner J.; Ekholm V.; Walz M.-M.; Björneholm O.. Acid–Base Speciation of Carboxylate Ions in the Surface Region of Aqueous Solutions in the Presence of Ammonium and Aminium Ions. J. Phys. Chem. B 2015, 119, 4033–4040. 10.1021/jp509945g (2)Surface-specific carboxylic acid enhancement was observed for different mixtures of carboxylate anions with ammonium or alkyl-ammonium cations and depends on the surface activity (alkyl chain length) of the carboxylate anion with an additional contribution from the possible evaporation of the corresponding amine.
Werner J.; Persson I.; Björneholm O.; Kawecki D.; Saak C.-M.; Walz M.-M.; Ekholm V.; Unger I.; Valtl C.; Caleman C.; Öhrwall G.; Prisle N. L.. Shifted equilibria of organic acids and bases in the aqueous surface region. Phys. Chem. Chem. Phys. 2018, 20, 23281–23293. 10.1039/C8CP01898G (3)The protonation equilibria of carboxylic acid and alkyl-amine conjugate acid−base pairs are significantly shifted toward the neutral form in the aqueous solution surface across the full bulk solution titration curves and consistent with relative differences in individual surface activities.
1. Aerosols and Cloud Droplets in the Atmosphere
The atmosphere is a key part of the earth system. It contains the air we breathe, regulates weather and climate, and supports critical infrastructure. Earth’s atmosphere comprises myriad chemical species in all basic forms of matter. Water is found as vapor, cloud and fog droplets, snow, or ice crystals. Condensed atmospheric phases are present as ubiquitous nano- and microscopic aerosol particles and droplets suspended in the air. These aerosols originate from numerous natural and anthropogenic processes and may comprise a wide range of both organic and inorganic chemical species.4,5
Interactions with water govern many effects of aerosols in the atmosphere. Depending on ambient conditions and aerosol composition and phase state, water can condense onto the aerosol surfaces, forming droplet solutions, which grow into large cloud droplets that eventually precipitate.6,7 Every cloud droplet in the atmosphere contains within it the aerosol seed from which it formed. Aerosols and clouds play crucial roles in earth’s climate, but the underlying mechanisms remain the most poorly constrained, severely affecting projections of future climate change.8 Aerosols and cloud droplets provide media for condensed-phase and heterogeneous chemistry throughout the atmosphere, with significant contributions to the evolution of atmospheric composition.9 Air pollution in particular causes millions of premature deaths and estimated welfare losses of trillions of euros every year.10 A detailed understanding of the atmosphere and its key constituents and processes is vital for robust projections to support the mitigation of both air pollution and climate change.
1.1. Role of Droplet Surfaces
Surfaces are a prominent part of atmospheric aerosols due to their high surface area (A) to bulk volume (V) ratios. The total surface area of cloud droplets in the atmosphere may be similar to that of all of the oceans (Figure 1d). In nano- and microscopic phases, both the surface and bulk are finite and A/V may be orders of magnitude greater than for macroscopic systems.7,11−13 With a finite thickness (δ), for example given by the dimensions of a molecular monolayer,1,14 the surface constitutes an increasing fraction of the total condensed-phase volume as dimensions decrease in the submicrometer range (Figure 1a). For aerosol populations similar to those observed in the atmosphere,15,16 a significant fraction of the total volume can therefore be comprised by their surfaces (Figure 1b).
Figure 1.

Importance of droplet surfaces. (a) Variation of the surface volume fraction with droplet diameter, assuming a surface thickness of δ = 1 nm. (b) Total and surface volume (left axis) calculated for common atmospheric aerosol populations from refs (15) and (16) (right axis). (c) Fraction of surface-adsorbed solute (left axis), assuming a surface of thickness δ = 1 nm with enrichment factor Eσ = 100, compared to the solution bulk, and surface tension resulting from concurrent bulk depletion (right axis), calculated using parametrizations from refs (11) and (17). (d) Total surface area of clouds, calculated with number concentrations for stratus and all clouds from ref (18) and a fixed droplet distribution from ref (19) and, for comparison, of earth’s oceans, calculated with data from ref (20).
Many atmospheric compounds, in particular amphiphilic organics, are surface-active in aqueous solutions, including aerosol and cloud droplets.6,13,21−23 Surface-active components preferentially adsorb at the surface, resulting in enhanced surface concentrations (activity) with respect to the interior (bulk) and a concentration gradient between the surface and bulk phases of a solution. Organic compounds often have the ability to lower the surface tension of an aqueous solution, compared to that of pure water. With enhanced surface activity, such compounds may reduce the surface tension even more efficiently than if homogeneously mixed in the solution. Inorganic ions can also display nonisotropic distributions at the aqueous surface,24,25 but their surface propensities are often not well-constrained in atmospheric mixtures.
Nano- and microscopic droplets are mesoscale bulk solutions, where strong mutual impacts of surface/bulk equilibria must be taken explicitly into account.13 For example, enhanced surface activity can significantly deplete the finite amount of solute from the bulk phase.11,13,17 The resulting distribution of substance between the surface and bulk phases is referred to as surface/bulk partitioning.6,7 The composition of both phases and composition-dependent solution properties, including surface tension, density, conductivity, and chemical reactivity, may significantly change with surface/bulk partitioning.7,13,26,27 Due to the strong variation of A/V with size, surface/bulk partitioning and composition-dependent properties will depend on the dimensions of nano- and microscopic solutions (Figure 1c). Bzdek et al.12 presented the first direct measurements of size-dependent surface tensions for picoliter aqueous surfactant droplets suspended in air, which were significantly higher than those of macroscopic solutions with identical total compositions.
Both the surface tension and amount of solute in the bulk affect the potential of aqueous droplets to grow by water condensation into cloud droplets.7,13 Two opposing mechanisms together define a threshold droplet size and ambient water saturation, which must be exceeded to activate the droplet for diffusion-limited growth.6 The finite curvature radii of microscopic solutions lead to elevated pressures (Kelvin effect), which scale with the surface tension. Dissolved solutes reduce the water vapor pressure (Raoult effect). Both of these mechanisms can be dramatically impacted by size-dependent surface/bulk partitioning.6,7,11,13,17,26−29 A significant climate effect has been estimated from altered cloud properties by surface-active droplet components21,30,31 but is not well-constrained. A major reason is insufficient knowledge on the variations of the droplet state and surface tension with environmental conditions.
The surface activity of atmospheric aerosol components may also have important implications for aqueous chemistry occurring as droplets grow and shrink during multiple cycles of cloud processing. Similarly as for water vapor, the Kelvin effect and surface tension will influence the condensation–evaporation of other volatile atmospheric species across the droplet surface. This will affect the formation and composition of so-called secondary aerosol mass, which remains a major source of uncertainty in assessing overall atmospheric aerosol and cloud effects.32 The surface tension of cloud droplets may also impact rates of pressure-sensitive chemical reactions in the droplet phase.13,33,34 Surface/bulk partitioning can dramatically alter rate-determining concentrations in both bulk and surface phases and may thereby directly impact chemical reactions in submicrometer droplets.1,13,35 Heterogeneous chemistry on the surfaces of aerosol and cloud droplets, with unique reaction pathways, rates, and products, compared to gas and condensed bulk-phase reactions, is increasingly recognized as potentially significant in the global atmospheric budget.36,37 However, such reactions are currently not well-constrained and rarely explicitly accounted for in atmospheric models.
1.2. Atmospheric Aerosol and Cloud Droplet Models
Very little is known about the specific properties of atmospheric surfaces. Information on the distinct surface composition is obscured in aerosol samples characterized as bulk aggregates21 or with bulk-sensitive methods, such as aerosol mass spectrometry.38 Quantitative surface-sensitive methods generally require macroscopic samples and experimental conditions, such as high vacuum, which do not readily accommodate volatile aerosol and cloud droplet constituents.1,39,40
Complex atmospheric aerosol mixtures introduce exponentially growing composition-dependent parameter spaces. Process-level studies therefore typically focus on simple model mixtures of a single, or few, key atmospheric species with well-defined and stable sample properties. Even when compositions or suitable proxies are identified, direct characterization of size-dependent composition–property relations is extremely challenging.12,41 Models are therefore needed to connect bulk measurements to surface compositions and macroscopic measurements to microscopic properties.13,14,17
Major classes of atmospheric organics comprise carboxylic acids, amines, organosulfates, and their salts, and alcohols. The most common inorganic ions are Na+, NH4+, Cl–, and SO42–. Since liquid water is an integral part of atmospheric aerosols and cloud droplets, it is important to establish the properties of both organic and inorganic components and their mixtures in aqueous solutions. Concentrations of individual compounds vary by many orders of magnitude under ambient conditions.7,13,26,27 Organic compounds, in particular, may be highly dilute due to limited aqueous solubilities and the formation of many different species in atmospheric chemical reactions.
Molecular dynamics (MD) simulations allow a molecular-level investigation of the surface adsorption of solutes and the structure of the surface region.3,42 However, the system size required to represent dilute aqueous solutions and unknown interaction parameters pose challenges for atmospheric mixtures. Concentration gradients between the bulk and surface are represented by a very small number of solute entities, with related uncertainty. The A/V of systems treated in MD simulations may also not represent either atmospheric droplets or macroscopic solutions to validate concentration-dependent properties.12,13,26,27,43
Several thermodynamic surface/bulk partitioning models have been used to evaluate cloud droplet formation by surface-active aerosols.14,26,27 A few are predictive, which is necessary to describe droplet states not accessed in experiments. These models are often based on Gibbs thermodynamics, where surface adsorption is described as an excess with respect to an idealized two-dimensional surface.7,13 The surface excess has no physical volume and does not represent the full composition or structure of the surface. Typically, only the adsorption of a single component is treated, whereas many surface-active species have been identified to coexist in atmospheric aerosol samples.22,23 Our Monolayer surface model11,14,26,27 predicts the full composition of both surface and bulk phases. In principle, the model can be constrained by independent experimental (surface tension and density) data that captures all interactions in the solution. However, such data is often not available for atmospheric aqueous solutions and is approximated with simplified equations of state.13,14
2. Surface-Sensitive XPS on a Liquid Microjet
X-ray photoelectron spectroscopy (XPS) is a
surface-sensitive technique capable of providing direct molecular-level
information with high chemical specificity. Photoelectron spectroscopy
utilizes the photoelectric effect to ionize a sample from inelastic
collisions with photons. The emitted photoelectrons are characterized
in terms of kinetic energy (Ek), from
which their binding energy (Eb) within
the sample is determined as the difference from the known ionizing
photon energy (hν), Eb = hν – Ek. By using X-ray photons, atomic-like core-level orbitals
can be ionized and their electron binding energies reflect the chemical
composition and local environment of the probed sample region. In
combination with a tunable synchrotron light source, a wide range
of chemical species and states can be accessed with high selectivity.
Furthermore, the probing depth into the sample can be varied to optimize
the surface sensitivity for each target species1−3,39,44 via the effective attenuation
length (EAL), which depends on Ek.45 The photoelectron signal intensity (I) originating at a given depth (z) below
the surface decays exponentially as 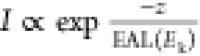 (Figure 2).
(Figure 2).
Figure 2.

XPS on a liquid microjet. Measurement principle, example C 1s spectrum for an octanoate (left fitted peak)–octanoic acid (right fitted peak) aqueous mixture (data from ref (2)), and dependence of signal attenuation (probing depth) on EAL.
Due to strong attenuation of the photoelectron signal also from gas-phase collisions, XPS is a vacuum-based technique. Both water and many other atmospheric species have finite vapor pressures and are either volatile or semivolatile under ambient conditions.38 By probing the sample as a high-speed liquid microjet,46 in combination with differential pumping of the experimental region, XPS can be applied to aqueous solutions of compounds with immediate atmospheric relevance.1−3,42,47,48 Minimum aqueous concentrations that can be studied depend on experimental conditions, including the surface propensity and ionization cross section of the target species and intensity of the ionizing light source. Ionization cross sections vary by several orders of magnitude among different elements, for orbitals within a given element, and with respect to photon energies.49 With high-brilliance synchrotron X-rays, XPS can distinguish elements in chemical states corresponding to different positions within molecular structures, oxidation or protonation states, and solvation environments (Figure 2).
2.1. XPS Experiments for Cloud Droplet Model Solutions
In a series of experiments, we studied the surface adsorption and speciation of surface-active atmospheric organics with Brønsted acid–base character in aqueous solution. Results for conjugate pairs of monocarboxylic acids and alkyl-amines are highlighted here. We focused on the effects of varying surface activity,2 chemical functionality of the hydrophilic group,3 and aqueous bulk solution state in terms of pH3 and mixing with common atmospheric inorganic salts comprising Na+, NH4+, Cl–, and SO42– ions.1,2
XPS experiments were performed at the Swedish National Synchrotron Radiation Laboratory MAX-lab, beamline I411. Aqueous sample stock solutions were prepared with varying pH and solute concentrations between a few millimolar and a few molar, representative of growing and activating cloud droplets.6,7,11,13,17,26,27 These concentrations were below the solubility limit or a possible critical micelle concentration (CMC) of all solutes, to ensure jet stability and avoid phase transitions which would decouple the observed properties from the concentration. The aqueous samples were introduced into the experimental region as a jet with a velocity of ∼25 m s–1, formed by a nozzle with a diameter of ∼20 μm. The microjet intersected with X-rays from the beamline a few millimeters downstream from the nozzle, in the region of stable laminar flow. The temperature of the microjet at the intersection point was estimated to be 5 ± 5 °C. Afterward, the sample was frozen in a liquid-nitrogen-cooled trap to minimize pressure building from evaporation.
The kinetic energies (Ek) of photoelectrons emitted from ionization by the X-rays are recorded with a spectrometer. The photoelectrons enter the spectrometer through a skimmer placed a few millimeters above the surface of the microjet. This makes it possible to operate the spectrometer at much lower pressure than the experimental region. The liquid jet was perpendicular to the propagation axis of the X-rays and the spectrometer detection axis. The latter was at a 54.7° angle, also known as the ”magic angle”, relative to the polarization plane of the X-rays to minimize angular distribution effects in the recorded photoelectron signal.50
An XPS spectrum consists of photoelectron (PE) signal intensities recorded at a fixed X-ray energy across a well-defined range of Ek, corresponding to relevant Eb for a given target core-level orbital (Figure 2). In each spectrum, the majority of photoelectrons cluster around one or more Ek values, forming spectral peaks representative of different chemical species or molecular environments. Recorded spectra for each target orbital are fitted to obtain peak positions Eb and areas (intensities) I, which are used to identify distinct chemical species or states and quantify their relative abundances in the probed sample region.
Carboxylic acids and their conjugate bases (carboxylate anions) were probed via the C 1s PE signals from the alkyl chain (−CH2−) and the carboxylic acid group (−COOH) and its deprotonated carboxylate form (−COO–). Alkyl amines and their conjugate acids were observed via both C 1s and N 1s signals from the alkyl chain (−CH2−) and the amine (−NH2) and ammonium (−NH3+) groups, respectively. Binding energies are calibrated with a well-known reference, such as the liquid water 1b1 peak at 11.16 eV.51 XPS allows a clear distinction between the conjugates of each acid−base pair from the chemical shifts of Eb peaks in the recorded spectra (Figure 2).
For a given Ek, PE signal intensities I are directly proportional to the abundances (c) of ionized chemical species or states in the probed sample region. Additionally, PE intensities are affected by the photoionization cross section of the target orbital, the density profile ρ(z) of the species with respect to the surface (z = 0), and the depth of the probed region determined by the photoelectron EAL(Ek).44,52 To the extent that these factors can be accounted for, PE intensities can be compared between different samples when measured during unchanged conditions. However, in practice this is nontrivial. We monitor the stability of the measurements, including sample injection, synchrotron radiation, and alignment of the liquid jet, X-ray beam, and skimmer, across the acquisition time via an internal reference. The EAL is not exactly known for aqueous solutions, and quantitative comparison of spectra for very different Ek values has significant uncertainty. The recorded PE signals originate from both the surface and bulk (near-surface) regions in the sample. By using photon energies to yield C 1s and N 1s photoelectrons with Ek ≈ 70 eV and EAL ≈ 10 Å, the relative contribution from species in the surface to the recorded PE intensities is close to being maximized.53
Absolute surface abundances (cσ) are estimated in terms of surface enrichment factors (Eσ = cσ/caq) for each species with respect to their known bulk concentrations (caq). We impose a simple two-layer model of the surface,1,3,42 resolving observed PE signals into contributions from the surface (Iσ) and bulk (Iaq)
| 1 |
where nσ and naq represent the sensitivity of measurements to the surface and bulk layers, respectively. Surface enrichments are then obtained from1,3
| 2 |
With given EAL and surface thickness δ, Iσ and Iaq are
estimated3 for an isotropic species, where cσ = caq, to
determine the relative sensitivity as nσ/naq = Iσisotropic/Iaqisotropic via eq 1. δ
is obtained from MD density profiles3,42 or approximated
with a molecular monolayer.1,11,14 For highly surface sensitive measurements, the two-layer model typically
yields  . By comparing to PE signals from similar
chemical states in species with little or no surface activity (surface
depleted), where cσ = 0, eq 1 further yields naq = Idepleted/caqdepleted. For example, we have compared carboxylic C 1s intensities
from surface-active carboxylate anions to those of formate.
. By comparing to PE signals from similar
chemical states in species with little or no surface activity (surface
depleted), where cσ = 0, eq 1 further yields naq = Idepleted/caqdepleted. For example, we have compared carboxylic C 1s intensities
from surface-active carboxylate anions to those of formate.
Although peak shapes and absolute chemical shifts may be affected by solvent effects54 for both neutral and ionic solutes, these were not explicitly investigated. Observed Eb values agree well with previous references, and conjugates are readily identified in each spectrum from relative shifts of their corresponding peaks. PE intensities are obtained as integral areas of fitted spectral peaks, which are assumed to be unaffected by peak shape due to the proportionality of signal and abundance.
3. Key Results
3.1. Direct Observations of Surface Adsorption
We observed clear PE signals from our samples, confirming sufficient surface concentrations and photoionization cross sections of the target species.1−3 XPS spectra of surface-active atmospheric organics showed significant enrichment in the aqueous surface, in accordance with expectations from adsorption theory.7,11,14,17,26,27,42,55 The two-layer model yielded surface enrichment factors between a few multiples and several orders of magnitude for the investigated monocarboxylic acids, alkyl-amines, and their conjugates.1−3 Greater Eσ values were observed for species expected to be more surface-active in aqueous solution (Figure 3a). For each conjugate pair, the neutral species was more strongly enriched in the surface than the charged form. We observed stronger surface enrichments of alkyl-ammonium ions compared to carboxylate ions with similar alkyl chain lengths.3 XPS-derived surface abundances increased with bulk concentration in solution and eventually saturated, akin to Langmuir surface-adsorption curves.3 Surface saturation occurred at lower bulk concentrations and the fractional surface saturation at a given bulk concentration was higher for species expected to be more surface-active. No significant impact on organic surface adsorption, in terms of salting-out and common-ion effects from mixing with inorganic cosolutes,56,57 was observed for the investigated concentrations and relative mixing states.1,2
Figure 3.

Direct observation of surface adsorption. (a) Langmuir-like adsorption curves in terms of surface abundances from XPS spectra for bytyric acid and butyrate anions (data from ref (3)). (b) Orientation with respect to the surface observed for octanoate and propionate anions via relative alkyl/carboxylic group C 1s peak areas (data from ref (2)).
Surface enrichments obtained from XPS spectra depend on the nonisotropic density profiles of the surface-active species in the probed sample region and the surface sensitivity of the measurements. The surface activity of the investigated amphiphilic species stems from a sensitive balance between hydrophobic interactions of the alkyl chain and hydrophilic interactions of the polar or charged groups in the solution.3 A higher relative permittivity is required to stabilize highly polar or charged species than less polar molecular species. The relative permittivity is likely lower in the outermost surface region than in the bulk aqueous solution due to the enrichment of less polar organic species and reduced overall density. This explains the relatively higher propensity of molecular conjugates for the immediate surface region. Charged species can be stabilized in the surface region through the formation of ion pairs reducing the overall local charge. The local charge screening is more effective when ions are closer and could therefore be less efficient for carboxylate than alkyl-ammonium ions due to stronger aqueous-phase hydration.
Surface adsorption can be seen as full or partial separation into organic surface (σ) and aqueous bulk (aq) phases with distinct compositions.14,28,38 Here, the activity coefficients of each species i
| 3 |
where ai and [i] are the corresponding activity (with respect to a given reference state) and concentration of i, will vary significantly.57 For more surface-active species, γi is reduced more in the surface, compared to the bulk, than for the less-surface-active species. Therefore, greater Eσ = [i]σ/[i]aq values are necessary to yield the same activity aσi = aaqi at equilibrium with a given [i]aq. Activity coefficients are expected to decrease with H-bond-forming ability and increase with the length of the alkyl chain in the dilute aqueous bulk and vice versa for the organic surface.57
For all surface-adsorbed
organics, we observed a preferential orientation
with the hydrophobic alkyl chains pointing away from and the hydrophilic
groups embedded into the solution. This is evident as an enhancement
of the alkyl/hydrophilic group PE intensity ratio, relative to the
stoichiometric ratio expected for free, randomly oriented solutes.
For example, stoichiometric C 1s peak
ratios between the carboxylate/carboxylic and alkyl carbons would
be 1:7 for octanoate and 1:2 for propionoate (Figure 3b). With preferential orientation, PE signals
from alkyl and hydrophilic groups originate at different depths z and are attenuated to different degrees as 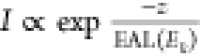 . The alkyl signal is increasingly enhanced
compared to the carboxylic signal for higher bulk concentrations,
indicating that the alkyl chain is increasingly perpendicular to the
surface closer to saturation. This confirms, by direct observation,
the expected behavior for a surface state above the dilute (Traube’s
law) limit.58
. The alkyl signal is increasingly enhanced
compared to the carboxylic signal for higher bulk concentrations,
indicating that the alkyl chain is increasingly perpendicular to the
surface closer to saturation. This confirms, by direct observation,
the expected behavior for a surface state above the dilute (Traube’s
law) limit.58
3.2. Apparent Shift in Surface Acidity
Carboxylic acids and alkyl-ammonium cations act as Brønsted acids (proton donors) in aqueous solution, and their conjugate carboxylate anions and alkyl-amines act as Brønsted bases (proton acceptors). The hydrolysis of monoprotic acid HA to form its conjugate base B is given by
| 4 |
with the acid dissociation equilibrium constant
| 5 |
where aH3O+, aH2O, aB, and aHA are the activities of H3O+, H2O, B, and HA, respectively. In dilute aqueous bulk solutions, approximating aH2O ≈ 1 and other activities by the corresponding concentrations yields the Henderson–Hasselbalch equation
| 6 |
We mapped the relative abundances 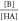 of conjugate acid−base pairs in
the surface of dilute aqueous solutions from the respective intensities
of distinctive XPS spectral peaks. For each pair, observed
of conjugate acid−base pairs in
the surface of dilute aqueous solutions from the respective intensities
of distinctive XPS spectral peaks. For each pair, observed 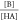 values were significantly shifted toward
the neutral form in the surface compared to the aqueous bulk solution
at a given pH. The shifts were observed for both proton donors (butyric
and pentanoic acid) and acceptors (n-butyl and n-hexyl amine) as neutral species and across a wide range
of bulk pH (2–13). The relative intensities of XPS peaks corresponding
to acid and base forms of each conjugate pair were fitted as a function
of bulk solution pH to a reformulation of eq 6 in terms of the acid fraction
values were significantly shifted toward
the neutral form in the surface compared to the aqueous bulk solution
at a given pH. The shifts were observed for both proton donors (butyric
and pentanoic acid) and acceptors (n-butyl and n-hexyl amine) as neutral species and across a wide range
of bulk pH (2–13). The relative intensities of XPS peaks corresponding
to acid and base forms of each conjugate pair were fitted as a function
of bulk solution pH to a reformulation of eq 6 in terms of the acid fraction  (Figure 4a). For each acid−base pair, the fitted titration
curves represent an apparent shift of the pKa (as the pH where
(Figure 4a). For each acid−base pair, the fitted titration
curves represent an apparent shift of the pKa (as the pH where  ) in the surface, compared to a dilute bulk
aqueous solution. The shift may be toward either higher or lower pH,
depending on whether the acid or base form of the pair is the neutral
species.
) in the surface, compared to a dilute bulk
aqueous solution. The shift may be toward either higher or lower pH,
depending on whether the acid or base form of the pair is the neutral
species.
Figure 4.

Surface-specific protonation. (a) Shifted equilibria toward the neutral species of conjugate acid−base pairs and apparent pKa in the surface of aqueous solutions (data from ref (3)). (b) Enhanced protonation of octanoate in aqueous mixtures with ammonium and diethanolammonium cations (data from ref (2)).
The magnitude of the shift varies with bulk concentration
and surface
activities of the conjugates. For the investigated samples, apparent
pKa shifts are roughly between 0.5 and
>1 pH unit.3 Considerable intensity
of
the protonated acidic form was previously observed in XPS spectra
from aqueous formic acid59 and sodium propionate,2 octanoate,2 and decanoate1 salts, at pH up to five units higher than the
bulk pKa. This corresponds to a shift
in 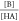 of up to roughly 4 orders of magnitude.
Suppressed dissociation at the aqueous surface was also observed with
XPS for highly concentrated nitric60 and
sulfuric54 acid, involving similar preferential
stabilization of the neutral species. Our measurements for dilute
aqueous solutions suggest strong contributions from the surface activity
of both organic acids and bases across the full titration curve pH
range and further influence of ion pairing with cosolutes.
of up to roughly 4 orders of magnitude.
Suppressed dissociation at the aqueous surface was also observed with
XPS for highly concentrated nitric60 and
sulfuric54 acid, involving similar preferential
stabilization of the neutral species. Our measurements for dilute
aqueous solutions suggest strong contributions from the surface activity
of both organic acids and bases across the full titration curve pH
range and further influence of ion pairing with cosolutes.
These
observations do not per se imply that the
surface itself is more acidic or basic than bulk water.61 First, we directly observe the abundances of
the surface-active conjugate acid−base pairs but not water
or its conjugate acid H3O+ and base OH–. Second, contrary to the dilute aqueous bulk, the probed surfaces
are predominantly organic phases with water present as a solute.11,17,26−28,48 The degree of acid protonation in the aqueous surface
is expected to vary due to different hydration enthalpies of protonated
and deprotonated species.62 These thermodynamic
properties will further change for a predominantly organic surface
phase. The observed shifts in 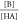 and apparent pKa in the surface can be attributed to differences in the surface propensities
and/or radial density profiles of the acid and base forms. If the
nonisotropic density profile ρ(z) of the charged
form of the pair peaks deeper below the surface, then this will bias
the observed PE signal to reflect a lower relative abundance in the
surface than for the neutral form. However, comprehensive angle-resolved
XPS experiments for environmental organosulfates in aqueous solutions
demonstrated that relative surface abundances were due to differences
in peak intensity (ρ), rather than peak depth (z), of the density profiles with respect to the surface.63
and apparent pKa in the surface can be attributed to differences in the surface propensities
and/or radial density profiles of the acid and base forms. If the
nonisotropic density profile ρ(z) of the charged
form of the pair peaks deeper below the surface, then this will bias
the observed PE signal to reflect a lower relative abundance in the
surface than for the neutral form. However, comprehensive angle-resolved
XPS experiments for environmental organosulfates in aqueous solutions
demonstrated that relative surface abundances were due to differences
in peak intensity (ρ), rather than peak depth (z), of the density profiles with respect to the surface.63
Given that the observed 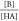 values indeed reflect the relative composition
of the surface, the apparent shifts in pKa can be rationalized in terms of the relative change in activity
coefficients of the conjugates between the organic surface and aqueous
bulk. With respect to the dilute aqueous phase (reference state),
activity coefficients for the charged form of each conjugate pair
are expected to increase more in the organic surface compared to the
neutral species. This must be compensated for by a corresponding decrease
in their relative abundances in the surface, leading to the apparent
shift in pKa as
values indeed reflect the relative composition
of the surface, the apparent shifts in pKa can be rationalized in terms of the relative change in activity
coefficients of the conjugates between the organic surface and aqueous
bulk. With respect to the dilute aqueous phase (reference state),
activity coefficients for the charged form of each conjugate pair
are expected to increase more in the organic surface compared to the
neutral species. This must be compensated for by a corresponding decrease
in their relative abundances in the surface, leading to the apparent
shift in pKa as
| 7 |
When the charged species is the base, the shift is in the direction of higher pKa and vice versa when it is the acid (Figure 4a).
In our experiments,
surface shifts in 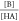 are broadly consistent with the relative
differences in the surface enrichment of the individual acid and base
conjugates obtained for binary aqueous solutions (Figure 3). For example, with pKa = 4.9 for octanoic acid, a 0.1 M aqueous sodium
octanoate bulk solution at neutral pH will comprise about 0.001 M
octanoic acid, whereas we observe XPS peak ratios of
are broadly consistent with the relative
differences in the surface enrichment of the individual acid and base
conjugates obtained for binary aqueous solutions (Figure 3). For example, with pKa = 4.9 for octanoic acid, a 0.1 M aqueous sodium
octanoate bulk solution at neutral pH will comprise about 0.001 M
octanoic acid, whereas we observe XPS peak ratios of 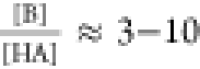 in the surface.2 From binary aqueous surface tension isotherms, the Monolayer model14 predicts individual Eσ values of 4990 and 371 for 0.001 M octanoic acid and 0.1 M sodium
octanoate, respectively, corresponding to
in the surface.2 From binary aqueous surface tension isotherms, the Monolayer model14 predicts individual Eσ values of 4990 and 371 for 0.001 M octanoic acid and 0.1 M sodium
octanoate, respectively, corresponding to  for the surface.
for the surface.
The Langmuir adsorption model assumes an ideal dilute state with no interactions between adsorbing species in the surface. This is not expected when hydrophobic interactions significantly reduce the surface tension compared to that of pure water. Carboxylic acids and carboxylate anions form strong complexes,64 which could stabilize surface layers with roughly even proportions of the conjugates. At higher concentrations, species close to individual saturation may compete for surface adsorption. This is not seen in XPS spectra from our experiments, where concentrations were well below the reported solubility and/or CMC.3 Nonideal interactions could decouple adsorption into the mixed surface from binary solutions of the same concentrations. A simple relation between the solution composition and apparent shift in surface pKa is therefore not expected.
3.3. Surface-Specific Chemistry
We investigated dilute aqueous mixtures of common atmospheric inorganic ions with surface-active carboxylate anion (Cx–) salts at near-neutral pH.1,2 Specifically in mixtures with NH4+ ions, surface abundances of conjugate carboxylic acids (CxH) were further strongly enhanced, beyond what was observed in aqueous organic mixtures (Figure 4b). For a given bulk concentration, this did not involve a simultaneous decrease in the surface abundance of the carboxylate anions. Therefore, total abundances of the organic pair ([Cx–] + [CxH]) in the surface were enhanced in NH4+ mixtures. This enhancement was stronger for the more surface-active pairs. Bulk-sensitive 1H NMR spectra confirmed the vast majority of the carboxylic acid−carboxylate anion pair to be on the ionic form, in complete agreement with the measured solution pH. The significant enhancement of CxH observed with XPS must therefore be specific to the solution surface.
The ammonium cation may act as a proton donor in aqueous solutions. Surface adsorption of Cx– and coadsorption of electrostatically attracted NH4+ cations form an ion-pair layer with strongly enhanced local abundances, increasing the probability of net proton transfer
| 8 |
according to Le Chatelier’s principle. The evaporation of ammonia from the surface may further contribute to irreversibly perturb the protonation equilibrium between NH4+ and Cx–, leaving a surplus of carboxylic acid. We observed gas-phase NH3 evaporating from the solutions, as clearly distinguishable from solvated aqueous ammonia and ammonium in recorded N 1s XPS spectra. A strong surface enhancement of octanoic acid was also observed in aqueous mixtures of octanoate and diethanolammonium (DEA), the corresponding acid of diethanolamine (NH–(C2H4OH)2), which is much less volatile than ammonia (Henry’s law coefficients 3.87 × 10–8 atm L mol−1 and 1.61 × 10–2 atm L mol−1, respectively, at 25 °C).2 The octanoic acid fractions of the carboxylic C 1s signal were somewhat smaller for solutions with DEA (0.34) than with NH4+(0.46) but smallest (0.23) without either (Figure 4b). The evaporation of ammonia from the solution is therefore not necessary to drive the strong enhancement of the carboxylic acid form in the surface but likely amplifies the effect.
4. Atmospheric Significance
Liquid microjets have enabled surface-sensitive XPS experiments for aqueous solutions of surface-active organics as model systems for atmospheric aerosols and cloud droplets. We have gained direct, quantitative, molecular-level insights into the surface, which are not readily accessible with other methods. Even for these simple systems, we observed complex surface structures and chemistry, which are highly distinct from the solution bulk. These define the chemical and phase state, reactant species, and accessible functional groups governing rates, pathways, and products of heterogeneous and surface-specific reactions.
Surface adsorption can significantly deplete the bulk of submicrometer droplets, leading to increased water activity.7,12,13,26,27 The formation of bilayers between ionic surface-active species and coadsorbed counterions can further promote bulk depletion, decreasing water uptake but increasing the existing potential for aqueous solvation.35 The surface-specific (potentially irreversible) conversion of organic ions to their more surface-active neutral conjugates could significantly decrease the aqueous surface tension. This may promote atmospheric droplet growth by the condensation of water and other semivolatile species, thereby influencing cloud–climate interactions.
As the surface becomes increasingly saturated with adsorbing organic species, the more outward orientation of the alkyl chains presents a more hydrophobic surface to the gas phase. This may decrease droplet growth from the uptake of water and other hydrophilic species and shield hydrophilic groups from gas-phase reactions while making alkyl chains more available. The evaporation of small neutral molecules formed in surface-specific reactions, including NH31,2 but potentially many others such as HCl65 from
| 9 |
could lead to significant mass loss from the aerosol phase. When the evaporating species have (Brønsted) acid−base character, it could also dramatically change the droplet acidity. Such a mechanism may contribute to the strong variation of observed pH for atmospheric aerosols.41
These surface-mediated mechanisms may profoundly affect the evolution of atmospheric chemistry due to the large volume fraction of aerosol and cloud droplets comprising their surfaces but are currently not taken into account in atmospheric models.30,31,66 Thermodynamic frameworks considering only the surface excess or a single adsorbing component are too simple to capture these variations of the droplet state.13,26,27 We found that the chemical environment of the surface changes in highly specific ways with concentration, pH, and cosolutes in the bulk. For aqueous droplets in the atmosphere, these may vary greatly with the ambient environment and air-mass history. By a somewhat fortuitous coincidence, the liquid microjet temperature during our measurements is immediately relevant for droplets in a rising air parcel or a cloud in the atmosphere.
4.1. Outlook
Faint and complex signals may prohibit applications of liquid microjet XPS to actual atmospheric samples (we tried). Immediate applications for atmospheric research involve the characterization of increasingly complex model systems and realistic experimental conditions. This will be crucial to validating and guiding new developments of models connecting surface and bulk composition−property relations.
Applications of liquid microjet XPS to more complex atmospheric model systems would benefit from several methodological developments. It is not known to what extent the surface of the high-speed liquid microjet has equilibrated at the time of measurement. In our experiments, we found no change in the phenomena observed by varying the intersection point of the X-ray beam with the jet,2 supporting similar previous assertions.46 The depth into solution with which the adsorption of surface-active solutes has equilibrated can be estimated from the solute diffusivity as 0.25–1.1 μm, 2 to 3 orders of magnitude greater than the surface thickness (Figure 5). Measurements at elevated, near-ambient pressures are possible with high-brilliance synchrotron lights sources, bringing the jet closer to steady state with respect to the gas phase and allowing the study of gas−solution heterogeneous reactions.67
Figure 5.

Estimated maximum diffusion distance for the surface adsorption of decanoate anions and propionic acid, indicating the depth into the microjet which has equilibrated with the surface at the time of measurement at different positions on the microjet. Diffusion coefficients (D) are from refs (68) and (69). Concentrations C = 0 mol L−1 indicate values extrapolated to infinite dilution.
The ability to extract absolute concentrations from liquid microjet XPS measurements without introducing overly simplifying assumptions will be pivotal for the quantitative analysis of surface-specific chemistry.42,55 In particular, absolute depth profiles for various solution components will reveal the extent of the nonisotropic surface region and constrain the impact of distinct surface properties. For example, surface depth profiles of conjugate acid−base pairs would resolve how much of the observed shifts can be attributed to different surface enrichments.43 Major challenges are the poor constraint on photoelectron EAL in different aqueous solutions and the limited number of depths possible to probe in each experiment. Advanced spectral analysis based on new machine learning methods has great potential for enabling this.44,52
Acknowledgments
The author warmly thanks her team members for help with making the figures [Konstantin Tumashevich (Figures 2, 3, and 4), Jack J. Lin (Figure 5), and Kunal Ghosh (Figure 1)], the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme, Project SURFACE (grant agreement no. 717022), and the Academy of Finland (grant nos. 257411, 308238, 314175, 316743, 335649, and 351476) for funding.
Biography
Nønne L. Prisle is a professor of atmospheric science and director of the Center for Atmospheric Research at the University of Oulu, Finland. Her main research interests include nanoparticles and their atmospheric and climate effects, currently focusing on applications of ultrabrilliant X-ray spectroscopic and imaging and machine learning methods. She obtained her Ph.D. in chemistry (2009) and M.Sc. in physics and chemistry (2006) from the University of Copenhagen and B.Sc. in theoretical physics from the University of Southern Denmark (2004), both in Denmark, and earned the title of docent in atmospheric physics from the University of Helsinki, Finland (2015). She was a postdoctoral researcher at the Finnish Meteorological Institute and Helsinki University’s Institute for Atmospheric Research, Finland, as well as a visiting researcher at Carnegie Mellon University, USA, Uppsala University, Sweden, and Georgia Institute of Technology, USA. She currently serves as vice chair of the board of the Finnish Synchrotron Radiation Users Organization, a board member of the Finnish Association for Aerosol Research, and associate editor of Environmental Science: Atmospheres. She is active in the popularization of aerosol, atmospheric, and climate research, including the TEDx talk “Small steps for us, a great leap for our planet” (2020), podcast “Why having your head in the clouds is a good thing” (2020), and graphic novel “Little Things” (2019).
The author declares no competing financial interest.
Special Issue
Published as part of the Accounts of Chemical Research special issue “Applications of Liquid Microjets in Chemistry”.
References
- Prisle N. L.; Ottosson N.; Öhrwall G.; Söderström J.; Maso M. D.; Björneholm O. Surface/bulk partitioning and acid/base speciation of aqueous decanoate: direct observations and atmospheric implications. Atmos. Chem. Phys. 2012, 12, 12227–12242. 10.5194/acp-12-12227-2012. [DOI] [Google Scholar]
- Öhrwall G.; Prisle N. L.; Ottosson N.; Werner J.; Ekholm V.; Walz M.-M.; Björneholm O. Acid–Base Speciation of Carboxylate Ions in the Surface Region of Aqueous Solutions in the Presence of Ammonium and Aminium Ions. J. Phys. Chem. B 2015, 119, 4033–4040. 10.1021/jp509945g. [DOI] [PubMed] [Google Scholar]
- Werner J.; Persson I.; Björneholm O.; Kawecki D.; Saak C.-M.; Walz M.-M.; Ekholm V.; Unger I.; Valtl C.; Caleman C.; Öhrwall G.; Prisle N. L. Shifted equilibria of organic acids and bases in the aqueous surface region. Phys. Chem. Chem. Phys. 2018, 20, 23281–23293. 10.1039/C8CP01898G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Jimenez J. L.; Canagaratna M. R.; Allan J. D.; Coe H.; Ulbrich I.; Alfarra M. R.; Takami A.; Middlebrook A. M.; Sun Y. L.; Dzepina K.; Dunlea E.; Docherty K.; DeCarlo P. F.; Salcedo D.; Onasch T.; Jayne J. T.; Miyoshi T.; Shimono A.; Hatakeyama S.; Takegawa N.; Kondo Y.; Schneider J.; Drewnick F.; Borrmann S.; Weimer S.; Demerjian K.; Williams P.; Bower K.; Bahreini R.; Cottrell L.; Griffin R. J.; Rautiainen J.; Sun J. Y.; Zhang Y. M.; Worsnop D. R. Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically-influenced Northern Hemisphere midlatitudes. Geophys. Res. Lett. 2007, 34, n/a. 10.1029/2007GL029979. [DOI] [Google Scholar]
- Hallquist M.; Wenger J. C.; Baltensperger U.; Rudich Y.; Simpson D.; Claeys M.; Dommen J.; Donahue N. M.; George C.; Goldstein A. H.; Hamilton J. F.; Herrmann H.; Hoffmann T.; Iinuma Y.; Jang M.; Jenkin M. E.; Jimenez J. L.; Kiendler-Scharr A.; Maenhaut W.; McFiggans G.; Mentel T. F.; Monod A.; Prevot A. S. H.; Seinfeld J. H.; Surratt J. D.; Szmigielski R.; Wildt J. The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. 10.5194/acp-9-5155-2009. [DOI] [Google Scholar]
- Prisle N. L.; Raatikainen T.; Sorjamaa R.; Svenningsson B.; Laaksonen A.; Bilde M. Surfactant partitioning in cloud droplet activation: a study of C8, C10, C12 and C14 normal fatty acid sodium salts. Tellus B 2022, 60B, 416–431. 10.1111/j.1600-0889.2008.00352.x. [DOI] [Google Scholar]
- Prisle N. L.; Raatikainen T.; Laaksonen A.; Bilde M. Surfactants in cloud droplet activation: mixed organic-inorganic particles. Atmos. Chem. Phys. 2010, 10, 5663–5683. 10.5194/acp-10-5663-2010. [DOI] [Google Scholar]
- Intergovernmental Panel on Climate Change (IPCC) . Climate Change 2021 - The Physical Science Basis: Working Group I Contribution to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change; Cambridge University Press, 2023.
- McNeill V. F. Aqueous Organic Chemistry in the Atmosphere: Sources and Chemical Processing of Organic Aerosols. Environ. Sci. Technol. 2015, 49, 1237–1244. 10.1021/es5043707. [DOI] [PubMed] [Google Scholar]
- Watkiss P.; Pye S.; Holland M.. Baseline Scenarios for Service Contract for carrying out cost-benefit analysis of air quality related issues, in particular in the clean air for Europe (CAFE) programme. AEAT/ED51014/ Baseline Issue 5 2005.
- Lin J. J.; Malila J.; Prisle N. L. Cloud droplet activation of organic-salt mixtures predicted from two model treatments of the droplet surface. Environ. Sci. Process. Impacts 2018, 20, 1611–1629. 10.1039/C8EM00345A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzdek B. R.; Reid J. P.; Malila J.; Prisle N. L. The surface tension of surfactant-containing, finite volume droplets. Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 8335–8343. 10.1073/pnas.1915660117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prisle N. L. A predictive thermodynamic framework of cloud droplet activation for chemically unresolved aerosol mixtures, including surface tension, non-ideality, and bulk-surface partitioning. Atmos. Chem. Phys. 2021, 21, 16387–16411. 10.5194/acp-21-16387-2021. [DOI] [Google Scholar]
- Malila J.; Prisle N. L. A monolayer partitioning scheme for droplets of surfactant solutions. J. Adv. Model. Earth Syst. 2018, 10, 3233–3251. 10.1029/2018MS001456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra G.; Ghosh K.; Dwivedi A. K.; Kumar M.; Kumar S.; Chintalapati S.; Tripathi S. An application of probability density function for the analysis of PM2.5 concentration during the COVID-19 lockdown period. Sci. Total Environ. 2021, 782, 146681. 10.1016/j.scitotenv.2021.146681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkola H.; Korhonen H.; Lehtinen K. E. J.; Makkonen R.; Asmi A.; Järvenoja S.; Anttila T.; Partanen A.-I.; Kulmala M.; Järvinen H.; Laaksonen A.; Kerminen V.-M. SALSA – a Sectional Aerosol module for Large Scale Applications. Atmos. Chem. Phys. 2008, 8, 2469–2483. 10.5194/acp-8-2469-2008. [DOI] [Google Scholar]
- Lin J. J.; Kristensen T. B.; Calderón S. M.; Malila J.; Prisle N. L. Effects of surface tension time-evolution for CCN activation of a complex organic surfactant. Environ. Sci. Process. Impacts 2020, 22, 271–284. 10.1039/C9EM00426B. [DOI] [PubMed] [Google Scholar]
- Jallu P. R.; Ghosh K.; Kokkola H.; Prisle N. L.. Climate effects of surface active marine organic aerosol. In preparation. [Google Scholar]
- Igel A. L.; van den Heever S. C. The Importance of the Shape of Cloud Droplet Size Distributions in Shallow Cumulus Clouds. Part I: Bin Microphysics Simulations. J. Atmos. Sci. 2017, 74, 249–258. 10.1175/JAS-D-15-0382.1. [DOI] [Google Scholar]
- Fairbridge R. W. The Changing Level of the Sea. Sci. Am. 1960, 202, 70–79. 10.1038/scientificamerican0560-70. [DOI] [Google Scholar]
- Facchini M.; Mircea M.; Fuzzi S.; Charlson R. Cloud Albedo Enhancement by Surface-Active Organic Solutes in Growing Droplets. Nature 1999, 401, 257–259. 10.1038/45758. [DOI] [Google Scholar]
- Gérard V.; Nozière B.; Baduel C.; Fine L.; Frossard A. A.; Cohen R. C. Anionic, Cationic, and Nonionic Surfactants in Atmospheric Aerosols from the Baltic Coast at Askö, Sweden: Implications for Cloud Droplet Activation. Environ. Sci. Technol. 2016, 50, 2974–2982. 10.1021/acs.est.5b05809. [DOI] [PubMed] [Google Scholar]
- Kroflič A.; Frka S.; Simmel M.; Wex H.; Grgić I. Size-resolved surface active substances of atmospheric aerosol: reconsideration of the impact on cloud droplet formation. Environ. Sci. Technol. 2018, 52, 9179–9187. 10.1021/acs.est.8b02381. [DOI] [PubMed] [Google Scholar]
- Petersen P. B.; Saykally R. J. On the nature of ions at the liquid water surface. Annu. Rev. Phys. Chem. 2006, 57, 333–364. 10.1146/annurev.physchem.57.032905.104609. [DOI] [PubMed] [Google Scholar]
- Piatkowski L.; Zhang Z.; Backus E. H. G.; Bakker H. J.; Bonn M. Extreme surface propensity of halide ions in water. Nat. Commun. 2014, 5, 4083. 10.1038/ncomms5083. [DOI] [PubMed] [Google Scholar]
- Vepsäläinen S.; Calderón S. M.; Malila J.; Prisle N. L. Comparison of six approaches to predicting droplet activation of surface active aerosol - Part 1: moderately surface active organics. Atmos. Chem. Phys. 2022, 22, 2669–2687. 10.5194/acp-22-2669-2022. [DOI] [Google Scholar]
- Vepsäläinen S.; Calderón S. M.; Prisle N. L. Comparison of six approaches to predicting droplet activation of surface active aerosol - Part 2: strong surfactants. EGUsphere 2023, 2023, 1–23. 10.5194/egusphere-2022-1188. [DOI] [Google Scholar]
- Prisle N. L.; Dal Maso M.; Kokkola H. A simple representation of surface active organic aerosol in cloud droplet formation. Atmos. Chem. Phys. 2011, 11, 4073–4083. 10.5194/acp-11-4073-2011. [DOI] [Google Scholar]
- Prisle N. L.; Lin J. J.; Purdue S.; Lin H.; Meredith J. C.; Nenes A. Cloud condensation nuclei activity of six pollenkitts and the influence of their surface activity. Atmos. Chem. Phys. 2019, 19, 4741–4761. 10.5194/acp-19-4741-2019. [DOI] [Google Scholar]
- Prisle N. L.; Asmi A.; Topping D.; Partanen A.-I.; Romakkaniemi S.; Dal Maso M.; Kulmala M.; Laaksonen A.; Lehtinen K. E. J.; McFiggans G.; Kokkola H. Surfactant effects in global simulations of cloud droplet activation. Geophys. Res. Lett. 2012, 39, L05802. 10.1029/2011GL050467. [DOI] [Google Scholar]
- Lowe S. J.; Partridge D. G.; Davies J. F.; Wilson K. R.; Topping D.; Riipinen I. Key drivers of cloud response to surface-active organics. Nat. Commun. 2019, 10, 5214. 10.1038/s41467-019-12982-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsigaridis K.; Daskalakis N.; Kanakidou M.; Adams P. J.; Artaxo P.; Bahadur R.; Balkanski Y.; Bauer S. E.; Bellouin N.; Benedetti A.; Bergman T.; Berntsen T. K.; Beukes J. P.; Bian H.; Carslaw K. S.; Chin M.; Curci G.; Diehl T.; Easter R. C.; Ghan S. J.; Gong S. L.; Hodzic A.; Hoyle C. R.; Iversen T.; Jathar S.; Jimenez J. L.; Kaiser J. W.; Kirkevåg A.; Koch D.; Kokkola H.; Lee Y. H.; Lin G.; Liu X.; Luo G.; Ma X.; Mann G. W.; Mihalopoulos N.; Morcrette J. J.; Müller J. F.; Myhre G.; Myriokefalitakis S.; Ng N. L.; O’Donnell D.; Penner J. E.; Pozzoli L.; Pringle K. J.; Russell L. M.; Schulz M.; Sciare J.; Seland Ø.; Shindell D. T.; Sillman S.; Skeie R. B.; Spracklen D.; Stavrakou T.; Steenrod S. D.; Takemura T.; Tiitta P.; Tilmes S.; Tost H.; van Noije T.; van Zyl P. G.; von Salzen K.; Yu F.; Wang Z.; Zaveri R. A.; Zhang H.; Zhang K.; Zhang Q.; Zhang X. The AeroCom evaluation and intercomparison of organic aerosol in global models. Atmos. Chem. Phys. 2014, 14, 10845–10895. 10.5194/acp-14-10845-2014. [DOI] [Google Scholar]
- Riva M.; Sun J.; McNeill V. F.; Ragon C.; Perrier S.; Rudich Y.; Nizkorodov S. A.; Chen J.; Caupin F.; Hoffmann T.; George C. Particles Influences the Rate and Products of Chemical Reactions. Environ. Sci. Technol. 2021, 55, 7786–7793. 10.1021/acs.est.0c07386. [DOI] [PubMed] [Google Scholar]
- Petters S. S. Constraints on the Role of Laplace Pressure in Multiphase Reactions and Viscosity of Organic Aerosols. Geophys. Res. Lett. 2022, 49, e2022GL098959. 10.1029/2022GL098959. [DOI] [Google Scholar]
- Romakkaniemi S.; Kokkola H.; Smith J. N.; Prisle N. L.; Schwier A. N.; McNeill V. F.; Laaksonen A. Partitioning of semivolatile surface-active compounds between bulk, surface and gas phase. Geophys. Res. Lett. 2011, 38, L03807. 10.1029/2010GL046147. [DOI] [Google Scholar]
- Chapleski R. C.; Zhang Y.; Troya D.; Morris J. R. Heterogeneous chemistry and reaction dynamics of the atmospheric oxidants, O3, NO3, and OH, on organic surfaces. Chem. Soc. Rev. 2016, 45, 3731–3746. 10.1039/C5CS00375J. [DOI] [PubMed] [Google Scholar]
- Tsui W. G.; McNeill V. F. Modeling Secondary Organic Aerosol Production from Photosensitized Humic-like Substances (HULIS). Environ. Sci. Technol. Lett. 2018, 5, 255–259. 10.1021/acs.estlett.8b00101. [DOI] [PubMed] [Google Scholar]
- Prisle N. L.; Engelhart G. J.; Bilde M.; Donahue N. M. Humidity Influence on Gas-Particle Phase Partitioning of α-Pinene + O3 Secondary Organic Aerosol. Geophys. Res. Lett. 2010, 37, L01802. 10.1029/2009GL041402. [DOI] [Google Scholar]
- Lin J. J.; Raj R K.; Wang S.; Kokkonen E.; Mikkelä M.-H.; Urpelainen S.; Prisle N. L. Pre-deliquescent water uptake in deposited nanoparticles observed with in situ ambient pressure X-ray photoelectron spectroscopy. Atmos. Chem. Phys. 2021, 21, 4709–4727. 10.5194/acp-21-4709-2021. [DOI] [Google Scholar]
- Pelimanni E.; Saak C.-M.; Michailoudi G.; Prisle N.; Huttula M.; Patanen M. Solvent and cosolute dependence of Mg surface enrichment in submicron aerosol particles. Phys. Chem. Chem. Phys. 2022, 24, 2934–2943. 10.1039/D1CP04953D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pye H. O. T.; Nenes A.; Alexander B.; Ault A. P.; Barth M. C.; Clegg S. L.; Collett J. L. Jr.; Fahey K. M.; Hennigan C. J.; Herrmann H.; Kanakidou M.; Kelly J. T.; Ku I. T.; McNeill V. F.; Riemer N.; Schaefer T.; Shi G.; Tilgner A.; Walker J. T.; Wang T.; Weber R.; Xing J.; Zaveri R. A.; Zuend A. The acidity of atmospheric particles and clouds. Atmos. Chem. Phys. 2020, 20, 4809–4888. 10.5194/acp-20-4809-2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner J.; Julin J.; Dalirian M.; Prisle N. L.; Öhrwall G.; Persson I.; Björneholm O.; Riipinen I. Succinic acid in aqueous solution: connecting microscopic surface composition and macroscopic surface tension. Phys. Chem. Chem. Phys. 2014, 16, 21486–21495. 10.1039/C4CP02776K. [DOI] [PubMed] [Google Scholar]
- de la Puente M.; David R.; Gomez A.; Laage D. Acids at the Edge: Why Nitric and Formic Acid Dissociations at Air-Water Interfaces Depend on Depth and on Interface Specific Area. J. Am. Chem. Soc. 2022, 144, 10524–10529. 10.1021/jacs.2c03099. [DOI] [PubMed] [Google Scholar]
- Ozon M.; Tumashevich K.; Lin J. J.; Prisle N. L. Inversion model for extracting chemically resolved depth profiles across liquid interfaces of various configurations from XPS data: PROPHESY. J. Synchrotron Radiat. 2023, 30, 941–961. 10.1107/S1600577523006124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski A.; Powell C. J. The electron attenuation length revisited. Surf. Sci. Rep. 2002, 47, 33–91. 10.1016/S0167-5729(02)00031-6. [DOI] [Google Scholar]
- Winter B.; Faubel M. Photoemission from Liquid Aqueous Solutions. Chem. Rev. 2006, 106, 1176–1211. 10.1021/cr040381p. [DOI] [PubMed] [Google Scholar]
- Walz M.-M.; Caleman C.; Werner J.; Ekholm V.; Lundberg D.; Prisle N. L.; Öhrwall G.; Björneholm O. Surface behavior of amphiphiles in aqueous solution: a comparison between different pentanol isomers. Phys. Chem. Chem. Phys. 2015, 17, 14036–14044. 10.1039/C5CP01870F. [DOI] [PubMed] [Google Scholar]
- Walz M.-M.; Werner J.; Ekholm V.; Prisle N. L.; Öhrwall G.; Björneholm O. Alcohols at the aqueous surface: chain length and isomer effects. Phys. Chem. Chem. Phys. 2016, 18, 6648–6656. 10.1039/C5CP06463E. [DOI] [PubMed] [Google Scholar]
- Yeh J. J.; Lindau I. Atomic subshell photoionization cross sections and asymmetry parameters: 1:1 ≤ Z ≤ 103. At. Data Nucl. Data Tables 1985, 32, 1–155. 10.1016/0092-640X(85)90016-6. [DOI] [Google Scholar]
- Cooper J.; Zare R. N. Thermodynamics of Multicomponent, Miscible, Ionic Solutions. 2. Mixtures Including Unsymmetrical Electrolytes. J. Chem. Phys. 1968, 48, 942–943. 10.1063/1.1668742. [DOI] [Google Scholar]
- Winter B.; Weber R.; Widdra W.; Dittmar M.; Faubel M.; Hertel I. V. Full Valence Band Photoemission from Liquid Water Using EUV Synchrotron Radiation. J. Phys. Chem. A 2004, 108 (14), 2625–2632. 10.1021/jp030263q. [DOI] [Google Scholar]
- Ozon M.; Tumashevich K.; Prisle N. L. Quantitative alignment parameter estimation for analyzing X-ray photoelectron spectra. J. Synchrotron Radiat. 2023, 30, 766–779. 10.1107/S1600577523004150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottosson N.; Faubel M.; Bradforth S. E.; Jungwirth P.; Winter B. Photoelectron Spectroscopy of Liquid Water and Aqueous Solution: Electron Effective Attenuation Lengths and Emission-Angle Anisotropy. J. Electron Spectrosc. 2010, 177, 60–70. 10.1016/j.elspec.2009.08.007. [DOI] [Google Scholar]
- Margarella A. M.; Perrine K. A.; Lewis T.; Faubel M.; Winter B.; Hemminger J. C. Dissociation of Sulfuric Acid in Aqueous Solution: Determination of the Photoelectron Spectral Fingerprints of H2SO4, HSO4–, and SO42– in Water. J. Phys. Chem. C 2013, 117, 8131–8137. 10.1021/jp308090k. [DOI] [Google Scholar]
- Toribio A. R.; Prisle N. L.; Wexler A. S. Statistical Mechanics of Multilayer Sorption: Surface Concentration Modeling and XPS Measurement. J. Phys. Chem. Lett. 2018, 9, 1461–1464. 10.1021/acs.jpclett.8b00332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toivola M.; Prisle N. L.; Elm J.; Waxman E. M.; Volkamer R.; Kurtén T. Can COSMOTherm Predict a Salting in Effect?. J. Phys. Chem. A 2017, 121, 6288–6295. 10.1021/acs.jpca.7b04847. [DOI] [PubMed] [Google Scholar]
- Michailoudi G.; Hyttinen N.; Kurtén T.; Prisle N. L. Solubility and Activity Coefficients of Atmospheric Surfactants in Aqueous Solution Evaluated Using COSMOtherm. J. Phys. Chem. A 2020, 124, 430–443. 10.1021/acs.jpca.9b09780. [DOI] [PubMed] [Google Scholar]
- Kjaer K.; Als-Nielsen J.; Helm C. A.; Tippman-Krayer P.; Mobwald H. Synchrotron x-ray diffraction and reflection studies of arachidic acid monolayers at the air-water interface. J. Phys. Chem. 1989, 93, 3200–3206. 10.1021/j100345a063. [DOI] [Google Scholar]
- Brown M. A.; Vila F.; Sterrer M.; Thürmer S.; Winter B.; Ammann M.; Rehr J. J.; van Bokhoven J. A. Electronic Structures of Formic Acid (HCOOH) and Formate (HCOO–) in Aqueous Solutions. J. Phys. Chem. Lett. 2012, 3, 1754–1759. 10.1021/jz300510r. [DOI] [PubMed] [Google Scholar]
- Lewis T.; Winter B.; Stern A. C.; Baer M. D.; Mundy C. J.; Tobias D. J.; Hemminger J. C. Does Nitric Acid Dissociate at the Aqueous Solution Surface?. J. Phys. Chem. C 2011, 115 (43), 21183–21190. 10.1021/jp205842w. [DOI] [Google Scholar]
- Saykally R. J. Two sides of the acid-base story. Nat. Chem. 2013, 5, 82–84. 10.1038/nchem.1556. [DOI] [PubMed] [Google Scholar]
- Tabe Y.; Kikkawa N.; Takahashi H.; Morita A. Surface Acidity of Water Probed by Free Energy Calculation for Trimethylamine Protonation. J. Phys. Chem. C 2014, 118, 977–988. 10.1021/jp4078882. [DOI] [Google Scholar]
- Lewis T.; Winter B.; Thürmer S.; Seidel R.; Stephansen A. B.; Freites J. A.; Tobias D. J.; Hemminger J. C. Molecular Arrangement of a Mixture of Organosulfur Surfactants at the Aqueous Solution–Vapor Interface Studied by Photoelectron Intensity and Angular Distribution Measurements and Molecular Dynamics Simulations. J. Phys. Chem. C 2019, 123, 8160–8170. 10.1021/acs.jpcc.8b08260. [DOI] [Google Scholar]
- Kanicky J. R.; Shah D. O. Effect of Premicellar Aggregation on the pKa of Fatty Acid Soap Solutions. Langmuir 2003, 19, 2034–2038. 10.1021/la020672y. [DOI] [Google Scholar]
- Rissler J.; Preger C.; Eriksson A. C.; Lin J. J.; Prisle N. L.; Svenningsson B. Missed Evaporation from Atmospherically Relevant Inorganic Mixtures Confounds Experimental Aerosol Studies. Environ. Sci. Technol. 2023, 57, 2706–2714. 10.1021/acs.est.2c06545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta G.; Zheng M.; Prisle N. L.. Impact of acidity and surface modulated acid dissociation on cloud response to organic aerosol. EGUsphere 2023, 2023, 10.5194/egusphere-2023-438. [DOI] [Google Scholar]
- Chen S.; Artiglia L.; Orlando F.; Edebeli J.; Kong X.; Yang H.; Boucly A. Impact of Tetrabutylammonium on the Oxidation of Bromide by Ozone. ACS Earth Space Chem. 2021, 5, 3008–3021. 10.1021/acsearthspacechem.1c00233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn L. A.; Stokes R. H. The diffusion of monocarboxylic acids in aqueous solution at 25°. Aust. J. Chem. 1965, 18, 285–296. 10.1071/CH9650285. [DOI] [Google Scholar]
- Deng Z.; Lü H.; Leaist D. G. Mutual Diffusion Coefficients and Resistance Coefficients for Aqueous Solutions of Sodium Alkanoate Surfactants. J. Chem. Eng. Data 1996, 41, 214–217. 10.1021/je950234b. [DOI] [Google Scholar]