

Abstract

Sulfanylbenzamide thioesters are molecules with anti-HIV activity that disrupt zinc coordination in the viral protein NCp7. These molecules are useful as topical microbicides; however, they are too unstable to be used systemically. In this article, a nitroimidazole prodrug was used to protect the sulfanylbenzamide to convey blood stability and oral bioavailability to the molecule. Studies on the molecule called nipamovir were performed to assess the rate of prodrug cleavage, antiviral activity, mechanism of metabolism, and in vivo pharmacokinetics in several different species. An efficient and inexpensive synthesis of nipamovir is also described. The results indicate that nipamovir could be further developed as a new type of drug to treat HIV infection.
Keywords: HIV, antiviral, microbicide, metabolism, pharmacokinetics, synthesis
1.
Human immunodeficiency virus (HIV) is a major global health issue, having claimed about 40 million lives since the beginning of the epidemic. As of 2021, 38.4 million people were living with HIV according to the World Health Organization (WHO). Progress in the reduction of new HIV infections and AIDS-related deaths continues to be led by the availability of new drugs to treat or prevent infections by the virus.1,2 In the past decade, 20 new drugs or combinations targeting the virus have gained FDA approval. Multiple anti-HIV drugs are administered simultaneously to infected patients in combinations called highly active antiretroviral therapy (HAART). In 2018, HAART was administered to 62% of adults and 54% of children living with HIV in low- and middle-income countries.3 The continuous use of HAART over an infected person’s lifetime can be problematic. In some cases, HIV can develop multidrug resistance to the drugs. There are also adverse side-effects that may develop with long-term use of HAART, including bone and renal toxicity, hepatotoxicity, neuropsychiatric events, and cardiovascular diseases.4 Furthermore, access to therapy remains limited in developing countries, with almost half of the global infected population having no access to treatment.2,5 In this context, it is important to develop novel anti-HIV therapeutics that are affordable and have low toxicity and a high barrier to viral resistance.
Nucleocapsid protein 7 (NCp7, Figure 1) is a 55-amino acid protein characterized by the presence of two highly conserved zinc fingers featuring a Cys-Xaa2-Cys-Xaa4-His-Xaa4-Cys (CCHC) motif. Due to their small size, the zinc fingers of NCp7 are often called zinc knuckles. NCp7 is part of the group-specific antigen (Gag) polyprotein that also contains the viral protein matrix (MA) and capsid (CA). NCp7 is responsible for multiple functions, which include binding to viral RNA, ribonucleoprotein formation, chaperoning, and protection of viral RNA from degradation.6
Figure 1.

Primary sequence of NCp7 (1–55) with Zn coordinating residues specified with spherical shapes.
Within the HIV virus life cycle, NCp7 plays a role in reverse transcription, integration, autocatalysis of Gag-Pol, and promotion of Gag assembly.7−10 NCp7 has been investigated as a target for small molecules against HIV for a long time,11−13 and both covalent and noncovalent inhibitors have been reported with antiviral activities in the low micromolar range.14−17 While NCp7 inhibitors are less potent compared with other classes of anti-HIV drugs, the highly conserved sequence of NCp7 in HIV makes this an attractive target for the development of antiviral drugs that would be less susceptible to viral resistance.
Sulfanylbenzamide thioesters (SAMTs), such as SAMT-247 (1, Figure 2), represent an attractive category of HIV inhibitors because of their structural and synthetic simplicity,18−20 high barrier to viral resistance,21 and lack of toxicity in different animal models.21−24 The proposed mechanism by which 1 inhibits HIV involves the disruption of zinc coordination in NCp7 by the transfer of the acetyl group of 1 to the thiol of cysteine side chains of NCp7. This acetyl transfer reaction forms thiolate 2 that is subsequently reacetylated by acetyl-CoA, leading to multiple acetyl transfers from 1 to cysteine side chains.21 Within NCp7, Cys36 is in equilibrium between zinc-coordinated and uncoordinated states.25 Even though the uncoordinated state of Cys36 is present in very small amounts, it provides an opportunity for 1 to react with the free cysteine thiol by transferring its acetyl group. After cysteine acetylation, the acetyl group is then transferred intramolecularly to adjacent lysine residues, resulting in the loss of zinc coordination in NCp7 and the subsequent destabilization of the protein structure. Destabilization of NCp7 triggers the exposure of cysteine sulfhydryls, which undergo oxidative disulfide formation, resulting in Gag/Gag-Pol cross-linking. The cross-linked Gag/Gag-Pol blocks Gag-Pol dimerization and prevents the activation of HIV protease, ultimately producing noninfectious viral particles.26 The formation of disulfide cross-links between different Gag/Gag-Pol proteins may happen spontaneously but could also be promoted by mixed disulfides 3 and benzisothiazolinone 4 which are formed in situ by oxidation of thiol 2.19
Figure 2.

Proposed mechanism of action of sulfanylbenzamide prodrugs.
Previously, our group investigated the pharmacokinetic and antiviral activities of the sulfanylbenzamide esterase-sensitive prodrug 5 (Figure 2),24 as well as substitutions on both the aromatic ring and side chain of 5 to examine the effects on antiviral activity.19,20 While important features of activity/toxicity profiles were observed with different substitutions, the antiviral activity did not improve. Due to the complicated mechanism of action, antiviral activity may be limited by an intracellular thermodynamic equilibrium that limits the extent of acetylation of 2 by acetyl CoA to reform 1.19 Despite these limitations, several preclinical studies have shown that 1 is an effective microbicide that can be used to prevent HIV infection in animal models.27−29 Furthermore, 5 may be used in combination with existing HIV drugs to achieve either additive or synergistic antiviral activity.24
In this report, we investigated whether a nitroimidazole prodrug based on azathioprine (6) could improve the pharmacokinetic profile of 2. Azathioprine (6) is a prodrug of the immunosuppressant drug 6-mercaptopurine (7) which is used in transplant patients (Figure 3).30 We report the design and synthesis of N-(3-amino-3-oxopropyl)-2-((1-methyl-4-nitro-1H-imidazole-5-yl)thio)benzamide 8, carrying a 1-methyl-4-nitro-imidazole moiety as the protecting group on the sulfur (Figure 3). We call molecule 8 nipamovir. The nitroimidazole prodrug 8 may be cleaved in vivo by glutathione and other reactive thiols in the walls of the intestine and in red blood cells without the need for an enzyme. The antiviral activity of 8 has been evaluated both in cellulo and ex vivo in human-derived models in addition to an evaluation of nonspecific toxicity in these systems. The kinetics of prodrug activation were studied by monitoring a reaction with a model thiol by NMR. The pharmacokinetic profile of 8 was evaluated in animal models, and the main metabolic pathway of 2 was studied.
Figure 3.

(a) Prodrug azathioprine 6 and active metabolite 6-mercaptopurine 7. (b) Novel prodrug N-(3-amino-3-oxopropyl)-2-((1-methyl-4-nitro-1H-imidazole-5-yl)thio)benzamide 8 (nipamovir) and free thiol 2 that forms after removal of the prodrug group.
2. Results and Discussion
2.1. Chemistry
The synthesis of 8 is outlined in Scheme 1. The first step involved the protection of the amino group of β-alanine ethyl ester (9) with di-tert-butyl dicarbonate (Boc2O) in the presence of triethylamine as a base in dichloromethane to form derivative 10, followed by reaction with aqueous NH4OH in methanol (MeOH) to form the amide 11. Subsequent deprotection of the amino group by hydrochloric acid in dioxane provided the hydrochloride salt of β-alaninamide 12 with a 61% yield over 3 steps. The salt was coupled with thiosalicylic acid 13 using 1,1′-carbonyldiimidazole in the presence of diisopropylethylamine (DIPEA), then oxidized with hydrogen peroxide to obtain the disulfide 14. Reduction of the disulfide was accomplished using sodium borohydride in methanol/tetrahydrofuran to give thiol 2, which was subsequently protected using 5-chloro-1-methyl-4-nitro-1H-imidazole (CMNI) in ethanol with sodium acetate acting as a base. The final product, 8, was obtained in an overall yield of 48%. The absence of any purification steps and the relatively low production cost ($9.46/g) makes the synthesis of nipamovir scalable and reproducible in industrial settings.
Scheme 1. (a) Boc2O, Et3N, DCM, 0 °C to RT, 22 h; (b) aq. NH4OH 28–30% w/w, MeOH, 40 °C, 4 Days; (c) 4.0 M HCl/Dioxane, RT, 24 h; (d) (i) CDI, DIPEA, DMF, 0 °C to RT, After 2 h, 12 was Added, RT, 4 Days; (ii) H2O2 30% w/w, H2O/Ice, 1 h 45 min (e) NaBH4, MeOH/THF (3:1), RT, 1 h; (f) CMNI, AcONa, EtOH, Reflux, 3 h.

2.2. Anti-HIV Activity
In vitro antiviral activity of nipamovir 8 was tested against HIV-1RF in CEM-SS cells measuring the inhibition of virus-induced cytopathic effects (CPEs) and cell viability following HIV replication, showing an EC50 value of 3.64 ± 3.28 μM and toxicity (TC50) > 100 μM. Then, the activity of the molecule was assessed in human PBMCs against HIV-192HT599 (subtype B, CXCR4) where 8 exhibited an EC50 value of 3.23 ± 2.81 μM and TC50 > 100 μM. Data are reported in Table 1 together with esterase-sensitive prodrug NS-1040 (5) and the thioester SAMT-247 (1). Azidothymidine (AZT) was used as a reference compound. The novel prodrug shows activity similar to that of the previously developed molecules, without noticeable increased toxicity.
Table 1. Antiviral and Toxicological Profiles of SAMT-247 1, NS-1040 5, and Nipamovir 8a.
| CEM-SS/HIV-1RF |
hPBMC/HIV-192HT599 |
|||||
|---|---|---|---|---|---|---|
| compound | EC50 [μM] | TC50 [μM] | TI | EC50 [μM] | TC50 [μM] | TI |
| SAMT-247b | 1.13 ± 0.19 | >100 | >88.5 | 5.74 ± 1.26 | >100 | >17.4 |
| NS-1040c | 1.03 ± 0.54 | >100 | >97.1 | 2.0 ± 1.04 | >100 | >50.0 |
| Nipamovir | 3.64 ± 3.28 | >100 | >27.5 | 3.23 ± 2.81 | >100 | >31.0 |
| AZT | 0.004 ± 0.0003 | >0.5 | >125 | 0.006 ± 0.001 | >1.0 | >182 |
2.3. Ex Vivo Analysis
To investigate the anti-HIV activity of 8 in an in vivo-like system, we tested different concentrations of the molecule in human lymphoid tissue explants infected with HIV-1LAI.04. This ex vivo tissue culture system is known to retain tissue cytoarchitecture and to support HIV-1 replication without exogenous activation.31 Tonsillar tissue blocks were infected with HIV-1LAI.04 and cultured for 12 days in tissue culture medium containing different concentrations of 8, with replacement every 3 days of tissue culture medium containing the corresponding drug treatment. Replication of HIV-1 was evaluated from measurements of the capsid protein p24gag in tissue culture medium and represented as a percentage of HIV-1 replication in untreated control. In the drug-free tissue cultures used as controls, the cumulative HIV-1LAI.04 replication [p24gag] levels in tissues from different donors varied from 5.71 to 150.71 ng/mL. The novel prodrug significantly reduced HIV-1LAI.04 replication in a concentration-dependent manner starting at the concentration of 1, 10, and 100 μM, by 23.68 ± 9.57 (p = 0.0265, n = 5), 55.5 ± 5.35 (p < 0.0001, n = 4), and 100.00 ± 0.00% (p < 0.0001, n = 5), respectively, compared with tonsillar tissue blocks cultured in the absence of drug (Figure 4a). Fitting the data points to a sigmoidal dose–response curve approximates an IC50 value of 4.54 μM (95% confidence interval: 2.61–7.74 μM, Figure 4b) for compound 8. To exclude whether the anti-HIV-1 effect of the molecule was due to cell cytotoxicity, we evaluated the possible cytotoxic effect of nipamovir in human T cells, MT-4 cells.
Figure 4.

(a) Concentration dependence of antiviral activity of 8. (b) IC50 value of 8. Presented are means ± SEM from tissues of at least four donors. Asterisks indicate statistical significance by one-way ANOVA multiple comparison with Dunnett’s correction (*p < 0.05, ****p < 0.0001). (c) TC50 value calculated in MT-4 cells. Results are expressed as percentages of viable cells in drug-free or drug-treated cells in a sigmoidal curve. Results presented are means ± SEM from three independent measurements.
MT-4 cells were cultured in a medium containing different concentrations of 8. After 3 days of cell treatment, cell viability was evaluated from counts of the total and dead cells in control and nipamovir-treated cells using an orange-acridine/propidium-iodide-based assay. No decrease in the number of viable cells was observed after treatment with different amounts of 8 in comparison with the untreated control, even at the highest concentration 100 μM. Indeed, the TC50 value was around 312.30 μM (Figure 4c).
2.4. NMR Kinetics
Azathioprine 6 is cleaved in vivo by reactive thiols, such as glutathione and cysteine residues, to generate 6-mercaptopurine 7, which undergoes a complex pathway of activation/inactivation by different enzymes leading to the final immunosuppressant activity.32 We performed kinetics experiments using NMR spectroscopy to monitor the prodrug’s cleavage by reaction with the reactive thiol Mesna 15 with the subsequent formation of active thiol 2 and derivative 16 (Figure 5).
Figure 5.

Thiol-thioether exchange between Nipamovir 8 and Mesna 15 to produce thiol 2 and derivative 16.
Mesna was chosen as the reactive thiol to simplify the NMR analysis and to facilitate a direct comparison with the previously investigated reaction kinetics between SAMT-247 and the same thiol.18 Thiol-thioether exchange experiments were conducted in a deoxygenated solution (93:7) of PBS 1× buffer/D2O (pH adjusted to 7.39 ± 0.01 with 0.1 M NaOH), allowing for deuterium-frequency locking and shimming while avoiding significant deviation from a native aqueous environment. Tracking the increasing signal of the methyl group on the nitroimidazole ring relative to a dimethylmalonic acid internal standard, a rate constant for the second order reaction was obtained from appropriate integrated rate law plots, in accordance with the protocol described by Whitesides et al.(33) Such derived rate constant (k = 6.34 ± 0.44 M–1 s–1) is 1 order of magnitude smaller than the constant obtained for SAMT-247 (k = 84 ± 11 M–1 s–1), resulting in a considerably slower reaction rate compared to the equimolar ratio previously described for SAMT-247.18 These data suggested an improved stability to nucleophilic attack of the novel prodrug, which encouraged us to further characterize this new derivative.
2.5. Blood Stability, Drug Metabolism, and Pharmacokinetics
Blood stability and metabolite formation studies were initially performed in vitro using blood samples from four different species (mouse, dog, monkey, and human). Nipamovir was incubated in blood samples at 37 °C, and aliquots were withdrawn at predetermined time points (Figure 6, see also Table S3 in Supporting Information) to be analyzed by a LC/MS/MS system. Incubation of nipamovir at 37 °C for 120 min revealed prodrug cleavage and generation of the active thiol 2 with formation of two metabolites: S-methyl derivative 17 and benzisothiazolinone 4. The former is inactive against HIV and nontoxic (hPBMCs EC50 > 100 μM, TC50 > 100 μM), the latter shows anti-HIV activity and moderate cytotoxicity (hPBMCs EC50 5.85 ± 0.63 μM, TC50 76.8 ± 1.03 μM). It can be observed that there is a trend toward slower cleavage from mouse to human with half-life (t1/2) values shifting from 30 to over 60 min (Figure 6).
Figure 6.

Structures of prodrug 8 and metabolites (2, 4, and 17) found in blood. Graphics showing percentage of remaining 8 and formation of metabolites 2, 4, and 17 following incubation of 8 for 120 min in the whole blood of mice, dogs, monkeys, and humans. Formation of metabolites is reported as (AUCmetabolite/AUCinternal standard)*100.
Experiments performed on 4 showed rapid metabolism to the inactive S-methyl-derivative 17 in all of the species, along with the presence of active thiol 2. Studies on the active thiol confirmed the formation of the two metabolites 17 and 4. These data indicate there is a complex equilibrium between thiol 2 and benzisothiazolinone 4 while the formation of 17 is irreversible (see Supporting Information, Figures S8, S9).
The metabolic profile of nipamovir was then evaluated in liver microsomes and hepatocytes from mice, rats, dogs, monkeys, and humans by an LC/UV/MS system. Following incubation for 60 min in NADPH-fortified liver microsomes at 10 μM, no metabolites were found across all five species, suggesting good stability in the presence of CYP enzymes. Instead, after incubation for 4 h in hepatocyte cell cultures of the same species at the same concentration of 10 μM, nipamovir was extensively metabolized to 17 and 4, with no detectable levels of the active thiol 2.
To further investigate the metabolism in blood, kinetic experiments were performed by incubating 8 and 2 in Cynomolgus monkey whole blood. Using a LC/MS/MS method, linear conditions for the formation of methylated derivative 17 were determined with an incubation time of 120 min and blood volume of 0.5 mL (Figure S10).
Incubation of 8 and 2 at different concentrations in blood using the aforementioned conditions enabled us to calculate the kinetic parameters reported in Table 2. The formation of 17 from both prodrug and active thiol showed saturable kinetics (Figure S11) enabling the calculation of Vmax and Km (according to the Michaelis–Menten equation) and the intrinsic clearance. The ability to fit the metabolism data to the Michaelis–Menten equation indicated that there is one main enzyme responsible for the conversion of thiol 2 to methylated derivative 17. The Km value calculated when incubating active thiol 2 in blood indicated that enzymatic saturation could be used to overcome the methylation of the active molecule.
Table 2. Kinetic Parameters of the Formation of 17 From Active Thiol 2 and Prodrug 8.
| incubated compound | Vmax [nmol/min/mL blood] | Km [μM] | intrinsic clearance [mL/min/mL blood] |
|---|---|---|---|
| 2 | 5.56 ± 0.09 | 6.05 ± 0.40 | 0.92 |
| 8 | 9.96 ± 0.14 | 15.3 ± 0.85 | 0.65 |
Pharmacokinetic (PK) profiles of nipamovir 8 and its metabolites 2 and 4, along with the inactive metabolite 17, were initially assessed in male Sprague–Dawley (SD) rats after intravenous (IV) and oral (PO) administration (n = 3 per dose group). Data are listed in Table 3. Following a single IV bolus dose of 5 mg/kg 8 in SD rats, the pharmacokinetics of the prodrug were characterized by a high clearance (169 mL/min/kg), moderate volume of distribution (1.3 L/kg), and a short elimination half-life (0.14 h). Short elimination half-life also characterized the PK profiles of 2 (0.35 h) and 17 (0.84 h), while no measurable levels of 4 were found. AUC0→∞ values were 499, 509, and 1070 h*ng/mL, respectively, for 8, 2, and 17.
Table 3. Pharmacokinetic Parameters of 8, 2, and 17 After Administration of 8 in SD Ratsa.
| 5 mg/kg IV |
300 mg/kg PO |
|||||
|---|---|---|---|---|---|---|
| PK parameter | 8 | 2 | 17 | 8 | 2 | 17 |
| AUC0–inf [ng*h/mL] | 499 ± 30.4 | 509 ± 36.6 | 1070 ± 36.06 | 577 ± 116 | 9560b | 13,700b |
| AUC0–t [ng*h/mL] | 493 ± 31.5 | 502 ± 34.9 | 1052 ± 41.74 | 513 ± 119 | 8023 ± 991 | 12,330 ± 3001 |
| t1/2 [h] | 0.14 ± 0.02 | 0.35 ± 0.02 | 0.84 ± 0.18 | 1.54 ± 0.28 | 3.66b | 3.03b |
| tmax [h] | 0.08 | 0.25 | 0.25 | 0.25 | 2.0 ± 1.0 | |
| Cmax [ng/mL] | 1180 ± 37.86 | 640 ± 48.7 | 219 ± 39.4 | 2950 ± 241 | 1290 ± 86.6 | |
| C0 [ng/mL] | 3537 ± 231 | |||||
| CL [mL/min/kg] | 169 ± 8.41 | |||||
| MRTinf [h] | 0.13 ± 0.01 | 0.36 ± 0.03 | 1.2 ± 0.2 | 2.85 ± 0.86 | 4.41b | 5.76b |
| Vdss | 1.3 ± 0.1 | |||||
| F [%] | 31.4b | |||||
Each value in the table is the mean of three experiments (n = 3), error is reported as SEM.
SEM values were not calculated since AUC extrapolated was greater than 25% in one subject.
After administration of a single oral (PO) dose of 300 mg/kg of 8 in SD rats, a moderate elimination half-life was found for 8 (1.54 h), 2 (3.66 h), and 17 (3.03 h). AUC0→∞ were 577, 9560, and 13,700 h*ng/mL, respectively. To calculate the oral bioavailability of the active thiol, the dose was estimated using the following equation: (actual dose of 8) × (MW of 2 ÷ MW of 8), resulting in an oral bioavailability of 31.4% for 2. Derivative 4 was not detected. The PK profiles of prodrug, free thiol, and S-methyl derivative after 5 mg/kg IV and 300 mg/kg PO administration of 8 in SD rats are depicted in Figure 7 and values are reported in Table 3.
Figure 7.

Mean plasma concentrations of 8, 2, and 17 after 5 mg/kg IV (left) and 300 mg/kg PO (right) administration of 8 in SD rats. Each data point in the graphs is the mean of three experiments (n = 3), error is reported as SEM.
PK profiles were then evaluated in male Cynomolgus monkeys after a single intravenous (IV) and oral (PO) administration (n = 3 per dose group). Following 1.1 mg/kg IV bolus injection of 8, data revealed high clearance (60 mL/min/kg), moderate volume of distribution (1.2 L/kg), and a short elimination half-life (0.48 h) of the prodrug. A short elimination half-life characterized 2 (1.20 h) and 17 (1.77 h), while the concentration of 4 was below the quantification limit of 10 ng/mL. AUC0→∞ values for 8, 2, and 17 were 316, 22, and 119 h*ng/mL, respectively. After administration of a single oral dose of 300 mg/kg nipamovir, the prodrug showed a moderate elimination half-life (2.53 h), while the pharmacokinetics of the thiol and its methylated metabolite were characterized by a long half-life (7.55 and 8.79 h, respectively). AUC0→∞ were 94, 5555, and 14,342 h*ng/mL for 8, 2, and 17 with an oral bioavailability for the active thiol of 93.4%. The PK profiles of the three compounds after 1.1 mg/kg IV and 300 mg/kg PO administration of 8 in Cynomolgus monkeys are depicted in Figure 8 and values are reported in Table 4.
Figure 8.

Mean plasma concentrations of nipamovir 8, amovir 2, and S-methyl 17 after 1.1 mg/kg IV (left) and 300 mg/kg PO (right) administration of 8 in Cynomolgus monkeys. Each data point in the graphs is the mean of three experiments (n = 3), error is reported as SEM.
Table 4. Pharmacokinetic Parameters of 8, 2, and 17 Following the Administration of 8 in Cynomolgus Monkeys.
| 1.1 mg/kg IV |
300 mg/kg PO |
|||||
|---|---|---|---|---|---|---|
| PK parameter | 8 | 2 | 17 | 8 | 2 | 17 |
| AUC0–inf [ng*h/mL] | 316 ± 39.3 | 22.0 ± 1.53 | 119 ± 18.1 | 94.0a | 5555a | 14,342a |
| AUC0–t [ng*h/mL] | 315 ± 40.1 | 15.3 ± 1.45 | 116 ± 17.9 | 87.7 ± 1.86 | 5779 ± 898 | 15,416 ± 3564 |
| t1/2 [h] | 0.48 ± 0.06 | 1.20 ± 0.14 | 1.77 ± 0.15 | 2.53a | 7.55a | 8.79a |
| tmax [h] | 0.08 | 0.14 ± 0.06 | 2.0 | 1.00 ± 0.50 | 3.17 ± 2.42 | 2.33 ± 0.88 |
| Cmax [ng/mL] | 16.7 ± 0.88 | 25.0 ± 3.60 | 26.0 ± 3.61 | 631 ± 62.4 | 1443 ± 163 | |
| C0 [ng/mL] | 1659 ± 349 | |||||
| CL [mL/min/kg] | 60 ± 8.5 | |||||
| MRTinf [h] | 0.34 ± 0.01 | 1.61 ± 0.15 | 3.45 ± 0.06 | 4.01a | 9.53a | 11.9a |
| Vdss | 1.2 ± 0.2 | |||||
| F [%] | 93.4a | |||||
SEM values were not calculated since AUC extrapolated was greater than 25% in one subject.
PK studies showed a better profile in nonhuman primates compared to rats with high oral bioavailability and higher plasma concentration levels after 24 h for the active molecule 2. PK studies also showed a high rate of metabolism to inactive methylated derivative 17 in both species.
We hypothesized that methylation of active thiol could be likely caused by the activity of thiopurine methyltransferase (TPMT), which is responsible for inactivation of thiopurine drugs.30 We, therefore, performed blood stability experiments incubating the prodrug 8 alone and together with sulfasalazine, olsalazine, 5-amino-salicylic acid (5-ASA), and N-acetyl-5-amino-salicylic acid (N-Ac-5-ASA), which are known inhibitors of the enzyme in vitro.34 Sulfasalazine and olsalazine are two commercially available prodrugs used for the treatment of ulcerative colitis and rheumatoid arthritis. These prodrugs release 5-ASA, which is acetylated to the active metabolite N-Ac-5-ASA. The exact mechanism of action for these drugs is not clear, but it revolves around the anti-inflammatory properties of 5-ASA.34 Prodrug 8 was incubated at 10 μM concentration at 37 °C for 30 min in blood samples from rats and monkeys, alone and in combination with 0.2 and 1 mM of Sulfasalazine, 5-ASA, and N-Ac-5-ASA, while for olsalazine, concentrations of 0.05 and 0.5 mM were used. Blood samples were then analyzed in a LC/UV/MS system for metabolite ID and profiling, and the results showed a decreased formation of the inactive methylated derivative 17, supporting the hypothesis that TPMT is the main enzyme responsible for the metabolism of active molecule 2. Inhibition results are summarized in Table 5.
Table 5. Inhibition Rate of Formation of S-Methyl Derivative 17 After Incubation of 8 With Different Salicylate Derivatives in Rat’s and Monkey’s Blood.
| co-incubated compound | concentration [mM] | inhibition percentage in rat’s blood [%] | inhibition percentage in monkey’s blood [%] |
|---|---|---|---|
| sulfasalazine | 0.2 | 8 | 12 |
| 1.0 | 42 | 80 | |
| olsalazine | 0.05 | 15 | a |
| 0.5 | 42 | 37 | |
| 5-ASA (mesalamine) | 0.2 | 28 | 51 |
| 1.0 | 91 | 81 | |
| N-Ac-5-ASA | 0.2 | a | 45 |
| 1.0 | 32 | 94 |
Reduction of formation was not relevant. Result presented were obtained with one single incubation in the two different species.
Following these preliminary blood stability studies, we performed PK studies in male SD-rats (Figure 9, Table 6) and male hamsters (Figure 10, Table 7) administering a single oral dose of 300 mg/kg of 8 after either vehicle or mesalamine (1000 mg/kg) pretreatment. From the data obtained in the rat study, we observed an increased absorption of prodrug 8 in addition to an increased concentration of active drug 2 and metabolized derivative 17. Interestingly, the study performed on male hamsters showed increased plasma concentration for all three species but with a more pronounced increase in the concentration of the thiol 2 compared to the methylated metabolite 17 for the second to last (8 h) and last (24 h) data points. These observations suggest a reduction of the methylation rate in this species. The increased absorption of 8 in the two studies could be attributed to the salicylates’ ability to enhance drug permeation and absorption, a well-documented feature of this class of molecules.35−37
Figure 9.

Mean plasma concentrations of 8, 2, and 17 following a single PO dose of 300 mg/kg 8 with and without PO pretreatment with 1000 mg/kg mesalamine in male SD-rats. Each data point in the graph is the mean of three experiments (n = 3), and error is reported as SEM.
Table 6. Pharmacokinetic Parameters of 8 and Its Metabolites 2 and 17 Following a Single PO Dose of 300 mg/kg 8 With and Without PO Pre-treatment With 1000 mg/kg Mesalamine in Male SD-Ratsa.
| 300 mg/kg PO
(vehicle) |
300 mg/kg PO
(mesalamine) |
|||||
|---|---|---|---|---|---|---|
| PK parameter | 8 | 2 | 17 | 8 | 2 | 17 |
| AUC0–inf [ng*h/mL] | 516 ± 138 | 1240b | 10,800 ± 1582 | 804b | 2130b | 18,900b |
| AUC0–t [ng*h/mL] | 495 ± 142 | 956 ± 161 | 10,400 ± 1722 | 612 ± 143 | 1740 ± 91.6 | 16,400 ± 3051 |
| t1/2 [h] | 1.78 ± 0.42 | 5.95b | 5.53 ± 1.39 | 3.18b | 3.01b | 11.0b |
| tmax [h] | 0.33 ± 0.08 | 0.25 | 2.00 ± 1.00 | 0.42 ± 0.08 | 0.42 ± 0.08 | 4.33 ± 2.03 |
| Cmax [ng/mL] | 369 ± 184 | 606 ± 111 | 1230 ± 65.6 | 226 ± 24.8 | 662 ± 54 | 1400 ± 215 |
| MRTinf [h] | 2.30 ± 0.26 | 4.78b | 7.12 ± 0.96 | 4.41b | 4.43b | 16.0b |
Each value in the table is the mean of three experiments (n = 3), error is reported as SEM.
SEM values were not calculated since AUC extrapolated was greater than 25% in one subject.
Figure 10.

Mean plasma concentrations of 8 and its metabolites 2 and 17 following a single PO dose of 300 mg/kg 8 with and without PO pretreatment with 1000 mg/kg mesalamine in male hamsters. Each data point in the graph is the mean of three experiments (n = 3), error is reported as SEM.
Table 7. Pharmacokinetic Parameters of 8 and Its Metabolites 2 and 17 Following a Single PO Dose of 300 mg/kg 8 With and Without PO Pre-treatment With 1000 mg/kg Mesalamine in Male Hamstersa.
| 300 mg/kg PO
(vehicle) |
300 mg/kg PO
(mesalamine) |
|||||
|---|---|---|---|---|---|---|
| PK parameter | 8 | 2 | 17 | 8 | 2 | 17 |
| AUC0–inf [ng*h/mL] | 464 ± 152 | b | 5820 ± 537 | 1090 ± 377 | 5580c | 9370c |
| AUC0–t [ng*h/mL] | 434 ± 158 | 4390 ± 1120 | 5520 ± 239 | 1030 ± 406 | 7030 ± 1556 | 7480 ± 328 |
| t1/2 [h] | 3.07 ± 1.08 | b | 5.24c | 4.97 ± 0.94 | 3.97c | 8.10c |
| tmax [h] | 0.19 ± 0.06 | 6.67 ± 0.67 | 4.67 ± 0.67 | 0.50 | 1.00 ± 0.50 | 6.00 ± 2.00 |
| Cmax [ng/mL] | 171 ± 90.5 | 476 ± 27.4 | 397 ± 41.3 | 372 ± 134 | 586 ± 19.0 | 483 ± 25.4 |
| MRTinf [h] | 4.07 ± 1.20 | b | 8.25c | 5.00 ± 0.65 | 6.73c | 11.7c |
Each value in the table is the mean of three experiments (n = 3), error is reported as SEM.
Not calculated; there was insufficient data to define the elimination phase.
SEM values were not calculated since AUC extrapolated was greater than 25% in one subject.
3. Conclusions
In this study, we reported the synthesis and development of a novel prodrug, nipamovir, that could be further developed to treat HIV-1 infections. Capitalizing on our previous findings on esterase-sensitive prodrug analogs, we envisioned that the utilization of the 5-thio-1-methyl-4-nitroimidazolyl prodrug could be used to mask the reactive thiol and improve the pharmacokinetic profile of the previously investigated thioester SAMT-247 and the esterase-sensitive prodrug NS-1040. The molecule, nipamovir (8), was synthesized at a relatively low cost of less than $10 per gram with minimal purification so that the synthetic procedure could be easily translated to an industrial setting. Nipamovir showed comparable antiviral activity to SAMT-247 and NS-1040 in both cellular assays and a human ex vivo model of HIV infection, along with low toxicity, encouraging further experiments on the 5-thio-1-methyl-4-nitroimidazolyl moiety as a prodrug for this class of molecules. Kinetic studies by nuclear magnetic spectroscopy were conducted and demonstrated a lower susceptibility to nucleophilic attack compared to the parent thioester SAMT-247, indicating good in vivo stability of the novel prodrug. Extensive metabolic profiling studies were conducted to assess the release of the active thiol in different animal species, allowing us to clarify the inactivation pathway. Pharmacokinetic studies in rats and monkeys showed a good profile for the novel prodrug with excellent oral bioavailability and good half-life for the active thiol, also confirming the inactivation pathway by methylation of the reactive sulfur. In order to diminish the metabolism of our active molecule, which we hypothesized was due to the activity of the TPMT enzyme, in vitro blood stability experiments were performed with inhibitors of the enzyme to evaluate whether the methylation rate could be lowered. Successful results obtained with coincubation of active thiol and mesalamine and its active metabolite N-acetyl-5-aminosalicylic acid led us to conduct further pharmacokinetic experiments in rats and hamsters. Data from animals pretreated with mesalamine showed an increased absorption of the prodrug, likely due to the well-documented property of salicylates to enhance permeability. In rats we continued to observe formation of the inactive methylated derivative, while in male hamsters, we were able to overcome some of the methylation metabolism. In conclusion, our results demonstrate that the 5-thio-1-methyl-4-nitroimidazolyl moiety is a useful prodrug for the sulfanylbenzamide derivatives to be used for in vivo evaluation in several different animal species. Further studies are required to better assess therapeutic plasma concentration and establish an appropriate dosage for nipamovir alone and/or in combination with already approved anti-HIV drugs. Altogether, the results pave the way to the further development of sulfanylbenzamide derivatives as therapeutic agents to treat HIV infections.
4. Materials and Methods
4.1. Chemicals, Synthesis, and Molecule Characterization
Unless otherwise stated, all reactions were carried out under an atmosphere of argon. Commercial solvents were obtained from Millipore Sigma. Commercial reagents were obtained from Millipore Sigma with the following exceptions: N,N′-carbonyldiimidazole (Oakwood Chemical); 30% hydrogen peroxide (Fisher Scientific); β-alanine ethyl ester hydrochloride (9; Oakwood Chemical); di-tert-butyl dicarbonate (Advanced Chemtech); 28–30% ammonium hydroxide solution (Oakwood Chemical); 4 M hydrogen chloride in 1,4-dioxane (Oakwood Chemical); PBS 1× buffer solution (K-D Medical); Mesna (Combi-Blocks); 5-Chloro-1-methyl-4-nitroimidazole (Combi-Blocks). Commercial reagents and solvents were used as received. Analytical thin-layered chromatography (TLC) was performed using plates precoated with silica gel (Sigma-Aldrich; 60 Å, 17 mm particle size), impregnated with a fluorescent indicator (254 nm). TLC plates were visualized by exposure to ultraviolet (UV) light. 1H and 13C NMR spectra were recorded on Bruker (400 and 500 MHz) spectrometers. Chemical shifts (δ) are quoted in parts per million and are referenced to residual protium in the NMR solvent (DMSO-d6, δ 2.50). Signals are reported as δ ppm (multiplicity, number of protons); s = singlet, d = doublet, t = triplet, q = quartet, sext = sextet, m = multiplet, dd = doublet of doublets, td = triplet of doublets, bs = broad signal. High-resolution mass spectrometry (HRMS) data were obtained using a Waters Xevo-G2 XS qTOF instrument.
4.1.1. Synthesis of Ethyl 3-((tert-butoxycarbonyl)amino)propanoate 10
β-Alanine ethyl ester hydrochloride 9 (75.0 g, 488 mmol, 1.00 equiv) was charged in a round-bottomed flask and dissolved in dichloromethane (DCM, 735 mL), then cooled to 0 °C with an ice-bath. First, di-tert-butyl dicarbonate (Boc2O, 160 g, 732 mmol, 1.50 equiv) was added to the mixture, then triethylamine (Et3N, 170 mL, 1.22 mol, 2.50 equiv) was added dropwise over the course of 10 min. Ice-bath was removed, and the mixture was stirred at 23 °C for 20 h. Mixture was transferred in a separation funnel and washed with sat. aq. sodium bicarbonate (NaHCO3, 450 mL × 1), water (H2O, 450 mL × 1), and brine (450 mL × 1). Organic layer was dried over sodium sulfate (Na2SO4), filtered, and concentrated to obtain a crude mixture of desired product and Boc2O as a yellow oil (mol ratio 6:1) which was used without further purification. HRMS-ESI+ (m/z): [M + Na]+ calcd for C10H19NNaO4, 240.1206; found, 240.1211.
4.1.2. Synthesis of Tert-butyl (3-Amino-3-oxopropyl)carbamate 11
Ethyl 3-((tert-butoxycarbonyl)amino)propanoate 10 was charged in a round-bottomed flask, and then a 1:1 mixture of methanol (MeOH)/aq. ammonia (NH4OH) 28–30% w/w (952 mL) was added. Mixture was heated at 40 °C for 72 h. Progression of the reaction was monitored by NMR. Once the reaction was completed, mixture was cooled and concentrated under vacuum until an aqueous slurry was obtained. It was filtered and washed with cold water (600 mL), then dried under vacuum overnight to obtain the desired product as white crystalline solid (57.3 g, 304 mmol). Yield: 62% over 2 steps.
1H NMR (500 MHz, DMSO-d6): δ 7.30 (s, 1H), 6.80 (s, 1H), 6.72 (s, 1H), 3.08 (q, 2H, J = 6.7 Hz), 2.18 (t, 2H, J = 7.4 Hz), 1.36 (s, 9H). 13C NMR (125 MHz, DMSO-d6): δ 173.03, 155.96, 78.04, 37.09, 35.87, 28.75. HRMS-ESI+ (m/z): [M + Na]+ calcd for C8H16N2NaO3, 211.1053; found, 211.1055.
4.1.3. Synthesis of 3-Aminopropanamide Hydrochloride 12
A round-bottomed flask was charged with a 4.0 M solution of hydrogen chloride (HCl) in dioxane (903 mL) and then cooled to 0 °C with an ice-bath. Tert-butyl (3-amino-3-oxopropyl)carbamate 11 (57.3 g, 304 mmol) was added over the course of 30 min, the ice-bath was removed, and the mixture was stirred at 23 °C for 22 h. Gaseous HCl was removed, then mixture was concentrated under vacuum. Solid was filtered, washed with diethyl ether (Et2O, 2 × 550 mL), and dried to obtain a white, crystalline solid (37.1 g, 298 mmol). Yield: 98%. 1H NMR (400 MHz, DMSO-d6): δ 7.95 (s, 3H), 7.59 (s, 1H), 7.05 (s, 1H), 2.94 (t, 2H, J = 6.6 Hz), 2.46 (t, 2H, J = 6.6 Hz). HRMS-ESI+ (m/z): [M + H]+ calcd for C3H9N2O, 89.0709; found, 89.0716.
4.1.4. Synthesis of 2,2′-Disulfanediylbis(N-(3-amino-3-oxopropyl)benzamide) 14
Thiosalicyclic acid 13 (32.6 g, 211 mmol, 1.00 equiv) was charged in a round-bottomed flask and dissolved in anhydrous dimethyl formamide (DMF, 403 mL). Mixture was cooled to 0 °C over 30 min. 1,1′-Carbonyldiimidazole (CDI, 35.9 g, 221 mmol, 1.05 equiv) was added and the mixture was stirred at 0 °C for further 30 min. N,N-Diisopropylethylamine (DIPEA, 121 mL, 696 mmol, 3.30 equiv) was added to the flask, which was removed from the ice-bath to reach room temperature and stirred for 2 h. 3-Aminopropanamide hydrochloride 12 (28.9 g, 232 mmol, 1.10 equiv) was added, and mixture stirred at room temperature for 72 h, then solvents were evaporated under vacuum to obtain an orange slurry. This was diluted with a water/ice 50:50 mixture (3.00 L) and transferred in a beaker. A 30% w/w hydrogen peroxide (H2O2) solution (8.62 mL, 84.4 mmol, 0.40 mmol) was added and mixture stirred for 2 h, then filtered and washed with H2O (2 × 1.50 L) to obtain the desired product as a white solid (43.6 g, 97.7 mmol). Yield: 93%.
1H NMR (500 MHz, DMSO-d6): δ 8.68 (t, 1H, J = 5.4 Hz), 7.63–7.61 (m, 2H), 7.46–7.43 (m, 2H), 7.41 (s, 1H), 7.30–7.27 (m, 2H), 3.45 (q, 2H, J = 6.5 Hz), 2.38 (t, 2H, J = 7.3 Hz). 13C NMR (125 MHz, DMSO-d6): δ 172.93, 167.35, 137.21, 134.17, 131.65, 128.50, 126.40, 126.11, 36.53, 35.36. HRMS-ESI+ (m/z): [M + H]+ calcd for C20C23N4O432S2, 447.1155; found, 447.1168.
4.1.5. Synthesis of N-(3-Amino-3-oxopropyl)-2-mercaptobenzamide 2
Compound 14 (21.0 g, 47.0 mmol) was dissolved in a MeOH/tetrahydrofuran (THF) 3:1 mixture (336 mL/112 mL) in a beaker under N2 flux. Mixture was cooled to 5 °C with an ice-bath, and then sodium borohydride (NaBH4, 12.6 g, 333 mmol, 7.08 equiv) was added carefully over 6 times, waiting for the mixture to cool down after each addition. Once the reducing agent was all added, ice-bath was removed and mixture stirred at room temperature for 1 h. Mixture was cooled to 0 °C with an ice-bath and an aqueous solution of HCl was added, forming a precipitate which was filtered out. Filtrate was concentrated under a vacuum to obtain a solid that was subsequently sonicated, diluted with H2O (400 mL), and sonicated again. Mesna 15 (2.08 g, 12.7 mmol, 0.27 equiv) was added, and mixture was sonicated again, then put under stirring and acidified with conc. HCl to pH 1. Precipitate formed was filtered and dried to yield the desired product as a golden crystalline solid (19.3 g, 86.2 mmol). Yield: 92%.
1H NMR (500 MHz, DMSO-d6): δ 8.46 (t, 1H, J = 5.0 Hz), 7.46 (dd, 1H, J = 7.7, 1.2 Hz), 7.40 (dd, 1H, J = 7.9, 0.6 Hz), 7.38 (s, 1H), 7.28 (td, 1H, J = 7.6, 1.4 Hz), 7.16 (td, 1H, J = 7.6, 1.0 Hz), 5.36 (s, 1H), 3.39 (q, 2H, J = 7.3 Hz). 13C NMR (125 MHz, DMSO-d6): δ 172.93, 167.35, 137.21, 134.17, 131.65, 128.50, 126.40, 126.11, 36.53, 35.36. HRMS-ESI+ (m/z): [M + Na]+ calcd for C10H12N2NaO232S, 247.0512; found, 247.0519.
4.1.6. Synthesis of N-(3-Amino-3-oxopropyl)-2-((1-methyl-4-nitro-1H-imidazole-5-yl)thio)benzamide 8
Compound 2 (33.2 g, 148 mmol) was suspended in ethanol (EtOH, 950 mL) in a round-bottomed flask, and then 5-chloro-1-methyl-4-nitro-1H-imidazole (19.1 g, 118 mmol, 0.8 equiv) and sodium acetate (24.3 g, 296 mmol, 2 equiv) were added. Mixture was heated to reflux for 3 h. After cooling, the solid was filtered, washed with EtOH (500 mL) and H2O (500 mL), and then dried to obtain the final product as a bright yellow solid (38.2 g, 109 mmol). Yield: 92%.
1H NMR (500 MHz, DMSO-d6): δ 8.69 (t, 1H, J = 5.0 Hz), 8.14 (s, 1H), 7.63 (d, 1H, J = 7.4 Hz), 7.40 (s, 1H), 7.31 (app t, 1H, J = 7.4 Hz), 7.28 (t, 1H, J = 7.2 Hz), 6.87 (s, 1H), 6.70 (d, 1H, J = 7.8 Hz), 3.57 (s, 3H), 3.43 (q, 2H, J = 6.6 Hz), 2.38 (t, 2H, J = 7.3 Hz). 13C NMR (125 MHz, DMSO-d6): δ 172.92, 167.25, 149.59, 139.81, 135.10, 133.88, 131.82, 128.84, 127.39, 126.51, 123.68, 36.52, 35.36, 33.26. HRMS-ESI+ (m/z): [M]+ calcd for C14H16N5O432S, 350.0923; found, 350.0921.
4.1.7. Synthesis of N-(3-Amino-3-oxopropyl)-2-(methylthio)benzamide 17
Compound 2 (1.00 g, 4.46 mmol) was dissolved in anhydrous THF (0.2 M), then methyl iodide (MeI, 0.56 mL, 8.92 mmol, 2 equiv) and Et3N (1.55 mL, 11.1 mmol, 2.5 equiv) were added. Reaction mixture was stirred at room temperature for 1.5 h. After concentration, crude was washed with water and then purified by flash chromatography with linear gradient 0 to 10% MeOH in ethyl acetate (EtOAc) to obtain the desired product as a white solid (256 mg, 1.07 mmol). Yield: 24%. 1H NMR (500 MHz, DMSO-d6): δ 8.28 (t, 1H, J = 5.4 Hz), 7.41 (dt, 1H, J = 7.6, 1.4 Hz), 7.36–7.32 (m, 3H), 7.17 (dt, 1H, J = 7.4, 1.1 Hz), 6.83 (s, 1H), 3.40–3.36 (m, 2H), 2.39 (s, 3H), 2.33 (t, 2H, J = 7.3 Hz). 13C NMR (125 MHz, DMSO-d6): δ 172.90, 167.97, 138.05, 135.77, 130.56, 127.88, 125.98, 124.51, 36.23, 35.38, 15.66.
4.1.8. Synthesis of 3-(3-Oxobenzo[d]isothiazol-2(3H)-yl)propenamide 4
Compound 4 was prepared according to modified published procedure.16
4.2. Biological Assays
4.2.1. Anti-HIV Evaluation in Human PBMCs
PHA-P stimulated PBMCs from three donors were pooled together and resuspended in fresh tissue culture medium at 1 × 106 cells/ml and plated in the interior wells of a 96 well round-bottom microplate at 50 μL/well. A 100 μL volume of 9 concentrations of compound serially diluted was transferred to the round-bottom 96-well plate containing the cells in triplicate. Fifty microliters (50 μL) of HIV-1 at a predetermined dilution was added. Each plate contained cell control wells and virus control wells in parallel with the experimental wells. After 7 days in culture, efficacy was evaluated by measuring the reverse transcriptase in the culture supernatants, and the cells were stained with the tetrazolium dye XTT to evaluate cytotoxicity.
4.2.2. Cell and Human Tissue Cultures
Human T-lymphocyte MT-4 cell line (obtained through the NIH AIDS Reagent Program, Germantown, MD) was cultured in Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco BRL, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS). Human tonsillar tissues were obtained from routine tonsillectomies (Children’s Hospital, Washington, DC). Tissues were dissected into 2 mm3 blocks, placed onto collagen sponge gel (Gelfoam, Pfizer, New York, NY) at the air–liquid interface, and cultured in RPMI 1640 medium supplemented with FBS at 15%, 1 mM nonessential amino acids, 1 mM sodium pyruvate, amphotericin B at 2.5 μg/mL, and gentamycin sulfate at 50 μg/mL.
4.2.3. Antiviral Assay in Human Tissue
The anti-HIV-1 activity of nipamovir 8 was investigated in human tonsillar tissues ex vivo. A prototypic X4 HIV-1 isolate, LAI.04 (HIV-1LAI.04; Rush University Virology Quality Assurance Laboratory, Chicago, IL), was used. For HIV-1 infection in human tonsillar tissues ex vivo, nine blocks per well were placed on collagen sponge gel and infected with 7.5 μL of HIV-1LAI.04 on top of each block. HIV-1LAI.04 infected tonsillar tissues were cultured in tissue culture medium containing or not containing different concentrations of drug test (0.01, 0.1, 1, 10, 100 μM). All tissue cultures ex vivo were kept for 12 days at 37 °C, replaced with the medium containing or not containing different concentrations of drug every 3 days. We used 18 tissue blocks per each condition (nine tissue blocks per well). Levels of HIV-1 infection were monitored from measurements of the release of HIV-1 gag protein p24 into the tissue culture medium, using an immunofluorescent cytometric bead-based assay by Luminex. The anti-HIV-1 activity of each compound was expressed as percentage of untreated drug-free control. The concentration required to inhibit viral replication by 50% (IC50) was calculated by fitting the data points to a sigmoidal dose–response curve with Prism software (version 7.0; GraphPad).
4.2.4. Cell Viability in MT-4 Cells
Cell viability assays were performed in MT-4 cells using the automated cell counting system Cellometer Auto 2000 (Nexcelom bioscience, San Diego, CA). We determined the numbers of total and dead cells in control cultures and in each concentration of drug tested after 3 days of culture using a dual-fluorescence acridine orange/propidium iodide method. Acridine orange stained all nucleated cells to generate green fluorescence. Propidium iodide stained all dead nucleated cells to generate red fluorescence. Cell toxicity for each concentration of each tested compound was expressed as a percentage of cells in the untreated control. Cellular toxicity (TC50) was calculated by fitting the data points to a sigmoidal dose–response curve with Prism software (version 7.0; GraphPad).
4.3. NMR-Based Kinetic Experiments
4.3.1. Thioether–thiol (8 + 15) Exchange Rate Constant Determination
A solution of dimethyl malonic acid (DMA; 0.3 mg, 2.27 μmol, 0.18
equiv) in D2O (10 μL) and Mesna (15; 111 mg, 0.68 mmol, 50
equiv) were sequentially added to a deoxygenated (30 min purge with nitrogen) solution
of PBS 1× buffer–D2O (93:7, 4.9 mL) in a 20 mL vial. Monitoring
with a calibrated Orion Star A111 pH benchtop meter, the pH of the resulting solution
was adjusted to 7.39 ± 0.01 with an aqueous sodium hydroxide solution (0.1 M in
H2O). An aliquot (∼750 μL) of the pH-adjusted solution was
transferred to an NMR tube, and the resulting sample was used to lock (onto
D2O) and shim the instrument. The NMR sample and a solution of Nipamovir
(8; 4.5 mg, 12.9 μmol, 1 equiv) in
DMSO-d6 (90 μL) were added to the reaction mixture,
at which point a timer was started. The vial was capped and shaken, and then an aliquot
(∼750 μL) of the resulting solution was transferred to an NMR tube. A
water-suppressed 1H NMR spectrum at 25 °C was obtained every 15 s over
the course of 5 min, with gradually increasing intervals between data collection from
30–60 s for a total of 41 data points. After referencing to the residual protium
of D2O, integration of the methyl group (s, 3H) relative to the integration
of the dimethyl malonic acid internal standard, 1.27 (s, 6H) was used to calculate
reactant concentrations. Being our case of thiol-thioether exchange a second-order
reaction with one of the reactants (Mesna) in excess compared to Nipamovir (50:1 ratio),
data was fit to the integrated form of the pseudo-first order rate equation:
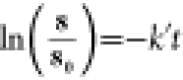 ,
derived from the pseudo-first order equation: [8] = [8]0e–k″t,
where 80 is the concentration of 8 at the beginning
of the experiment and k′ the pseudo-first order rate constant.
The rate constant was interpreted as the slope of the resulting linear plot. In order to
calculate the second-order rate constant k, we can express
k′ = k[Mesna–]0
= kθ[Mesna]0, where [Mesna–] and
[Mesna–]0 are the concentration of Mesna thiolate and
Mesna at the start of the experiment, respectively, and θ is the fraction of Mesna
thiolate at the experimental pH, calculated as
,
derived from the pseudo-first order equation: [8] = [8]0e–k″t,
where 80 is the concentration of 8 at the beginning
of the experiment and k′ the pseudo-first order rate constant.
The rate constant was interpreted as the slope of the resulting linear plot. In order to
calculate the second-order rate constant k, we can express
k′ = k[Mesna–]0
= kθ[Mesna]0, where [Mesna–] and
[Mesna–]0 are the concentration of Mesna thiolate and
Mesna at the start of the experiment, respectively, and θ is the fraction of Mesna
thiolate at the experimental pH, calculated as  . The reported rate constant is the
average of three trials (Tables S1, S2).
. The reported rate constant is the
average of three trials (Tables S1, S2).
4.4. Blood Stability Assays
4.4.1. Blood Stability Assay of 8, 2 and 4 in Blood Samples from Mice, Dogs, Monkeys and Humans
Aliquots of 792 μL of blood from CD-1 mice, Beagle dogs, Cynomolgus monkeys, and humans were mixed with 100 μM solution of 8, 4, and 2 to reach a final concentration of 1 μM for each compound and incubated in water bath at 37 °C for 0, 15, 30, 60, 90, and 120 min. At each time point, aliquots of 50 μL were drawn in duplicate and vortexed, followed by an addition of 200 μL of a solution of Warfarin (100 ng/mL in acetonitrile) as internal standard, vortexed again, and centrifuged at 14,000 rpm for 10 min at 4 °C. Aliquots of 100 μL of the supernatant were transferred to a 96-well plate containing 100 μL of water in each well and vortexed for 10 min before being analyzed using a validated LC–MS/MS method developed on a Shimadzu HPLC system interfaced to a Sciex API4000 mass spectrometer. Eluents were gradient mixtures of water and acetonitrile with 0.1% formic acid. HPLC column was ACE Excel5 C18, 50 × 2.1 mm, 5 μM.
4.4.2. Blood Stability Assay of 8 in the Presence of TPMT Inhibitors
Nipamovir 8 was incubated at a 10 μM concentration in whole blood from SD-rats and Cynomolgus Monkeys. The compound was incubated in absence and presence of sulfasalazine (0.2 and 1 mM), olsalazine (0.05 and 0.5 mM), 5-aminosalicylic acid (0.2 and 1 mM), and N-acetyl-5-aminosalicylic acid (0.2 and 1 mM). After incubation at 37 °C for 60 min, reaction was stopped by adding AcCN to the mixture. Sample was vortexed and centrifuged to precipitate proteins, then the supernatant was transferred in clean tubes for drying under N2 stream. Dried residues were reconstituted with AcCN and aliquots were submitted to LC/UV/MS analysis.
4.5. Metabolic Profiling
4.5.1. Metabolite Profiles of nipamovir 8 in Liver Microsomes from Mouse, Rat, Dog, Monkey, and Human
Metabolic stability was determined by using pooled hepatic microsomal fractions from five different species: mice, rats, dogs, monkeys, and humans. Final microsomal protein concentration was 1 mg/mL and compound concentration 10 μM. After incubation at 37 °C for 60 min, reaction was stopped by adding AcCN to the mixture. Sample was vortexed and centrifuged to precipitate proteins, then the supernatant was transferred in clean tubes for drying under N2 stream. Dried residues were reconstituted with AcCN and aliquots submitted to analysis using a LC/UV/MS method developed on a Agilent 1200 HPLC system interfaced to a LTQ/Orbitrap (Thermo Finnigan) mass spectrometer. Eluents were gradient mixtures of water with 0.05% TFA and acetonitrile with 0.1% TFA. HPLC column was Luna C18(2), 250 × 2.0 mm, 5 μM.
4.5.2. Metabolite Profiles of nipamovir 8 in Liver Hepatocytes from Mouse, Rat, Dog, Monkey, and Human
Metabolic stability was assessed using mouse, rat, dog, monkey, and human hepatocytes (2 × 106 cells/mL) at a compound concentration of 10 μM. After incubation at 37 °C for 4 h under 5% CO2, reaction was stopped by adding AcCN to the mixture. Sample was vortexed and centrifuged to precipitate proteins, then the supernatant was transferred in clean tubes for drying under N2 stream. Dried residues were reconstituted with AcCN and aliquots submitted to analysis using an LC/UV/MS method developed on an Agilent 1200 HPLC system interfaced to an LTQ/Orbitrap (Thermo Finnigan) mass spectrometer. Eluents were gradient mixtures of water with 0.05% TFA and acetonitrile with 0.1% TFA. HPLC column was Luna C18(2), 250 × 2.0 mm, 5 μM.
4.6. Determination of the Kinetics of Metabolism of 8 and 2 to 17 in Monkey Whole Blood
Nipamovir 8 and thiol 2 were incubated at 0.5 μM concentration in four different samples of monkey whole blood of volumes 0.1, 0.25, 0.5, and 1 mL, respectively. Before starting incubating, PBS was added to each sample in order to reach a final volume of 1 mL.
Once linear conditions for 17 formation had been established, 8 and 2 were incubated at different concentrations (0.1, 0.25, 0.5, 1.0, 1.5, 2.0, 2.5, 5.0, 10.0, 25.0, 50.0, 100, 150, and 200 μM) in 0.5 mL of monkey whole blood for 120 min. 5.0 μL of a solution of each concentration under study was added to aliquots of blood (495 μL) and incubated at 37 °C for 120 min. At the end of the incubation, 50 μL of sample were withdrawn in duplicate and treated with AcCN (200 μL) containing the internal standard, vortexed, and centrifuged. 100 μL aliquots of supernatant were transferred into a 96-well plate containing 100 μL of water in each well, then analyzed using a validated LC–MS/MS method developed on a Shimadzu HPLC system interfaced to a Sciex API4000 mass spectrometer. Eluents were gradient mixtures of water and acetonitrile with 0.1% formic acid. HPLC column was ACE Excel5 C18, 50 × 2.1 mm, 5 μM. Data were analyzed with GraphPad Prism software to calculate Vmax and Km.
4.7. Pharmacokinetics
All animal studies were conducted in facilities accredited by AAALAC International using protocols that were approved by the Institutional Animal Care and Use Committee (IACUC) of Frontage Laboratories, Inc.
4.7.1. Pharmacokinetic Studies in SD-Rats
This study was conducted in accordance with the applicable Frontage Standard Operating Procedures (SOPs) and IACUC protocols. Male SD rats weighing between 200 and 350 g were obtained from Charles River Laboratories. Rats in group 1 (n = 3) were cannulated with a dual jugular vein and femoral artery catheter for IV injection and blood collection, respectively. Group 2 rats (n = 3) were cannulated with a femoral artery catheter for blood collection. Rats had access to food and water ad libitum; group 1 rats (IV) were not fasted, while group 2 rats (PO) were fasted and food made available 4 h postdose. IV dosing solutions were prepared in DMSO/PEG400/Saline (5:35:60, v/v/v), while PO dosing solutions were prepared in 1% aqueous methylcellulose with 0.1% Tween 80. Group 1 rats were administered a single IV bolus at 5 mg/kg (4 mL/kg of a 1.25 mg/mL solution of 8), while group 2 rats were administered a single oral dose at 300 mg/kg (10 mL/kg of a 30 mg/mL solution of Nipamovir) by oral gavage. Blood samples (200 μL each time point) were obtained via Culex at predetermined time points (0, 5, 15, 30, 60, 120, 240, 360, 480, and 1440 min) after dosing. Plasma was obtained by centrifugation within 30 min after collection and aliquots of dosing solutions were taken and analyzed at the same time with plasma samples using a validated LC–MS/MS method developed on a Shimadzu HPLC system interfaced to a Sciex API 5000 mass spectrometer. Eluents were gradient mixtures of water and acetonitrile with 0.1% formic acid. HPLC column was ACE PFP C18, 50 × 2.1 mm, 5 μM. PK parameters of the analytes were obtained using Phoenix WinNonlin software (version 8.1).
4.7.2. Pharmacokinetic Studies in Cynomolgus Monkeys
This study was conducted in accordance with the applicable Frontage Standard Operating Procedures (SOPs) and IACUC protocols. Male Cynomolgus monkeys weighing between 3 and 5 kg were divided into two groups. Group 1 (n = 3) consisted of nonfasted male monkeys dosed intravenously with 1 mg/kg of 8via a cephalic vein. Group 2 animals (n = 3) fasted overnight prior to oral administration of 300 mg/kg of 8via oral gavage. IV dosing solutions were prepared in ethanol/PEG400/saline (10:60:30, v/v/v), while PO dosing suspensions were prepared in 0.5% aqueous methylcellulose with 0.1% Tween 80. Group 1 monkeys were administered a single IV bolus (1 mL/kg of a 1 mg/mL solution of 8), while group 2 monkeys were administered a single oral dose (4 mL/kg of a 60 mg/mL solution of 8) by oral gavage. Blood samples (1 mL each time point) were obtained at predetermined time points: 5, 15, and 30 min predose, and 60, 120, 240, 360, 600, and 1440 min postdose for the IV group; 15 and 30 min predose, and 60, 120, 240, 360, 600, and 1440 min postdose for the PO group. Plasma was harvested by centrifugation (0.5 mL) and mixed with 1 M NaF (50 μL) and 1 M citric acid (25 μL), and then 20 μL of each treated plasma sample was crashed with 200 μL of acetonitrile (containing 1% formic acid) immediately after collection. The rest of the plasma samples were stored at −75 ± 15 °C until shipped for analysis using a validated LC–MS/MS method developed on a Shimadzu HPLC system interfaced to a Sciex API 5000 mass spectrometer. Eluents were gradient mixtures of water and acetonitrile with 0.1% formic acid. HPLC column was ACE Excel5 C8, 50 × 2.1 mm, 5 μM. PK parameters of the analytes were obtained using Phoenix WinNonlin software (version 8.1).
4.7.3. Pharmacokinetic Studies in Male SD-Rats with or without Pretreatment with Mesalamine
This study was conducted in accordance with the applicable Frontage Standard Operating Procedures (SOPs) and IACUC protocols. Male SD rats weighing between 200 and 350 g were obtained from Charles River Laboratories. Rats were cannulated with a femoral artery catheter for blood collection. PO dosing solutions were prepared in 1% aqueous methylcellulose with 0.1% Tween 80 and administered by oral gavage. Group 1 animals (n = 3) were dosed orally with vehicle (1% aqueous methylcellulose with 0.1% Tween 80) first, followed by PO dose at 300 mg/kg of 8 (10 mL/kg of a 30 mg/mL solution), 30 min after the vehicle dose; while group 2 animals (n = 3) were dosed orally with mesalamine at 1000 mg/kg first (10 mL/kg of a 100 mg/mL solution), followed by PO dose at 300 mg/kg of 8, 30 min after administration of mesalamine. Rats had access to food and water ad libitum, fasted before PO dosing, and food made available 4 h post dosing. Blood samples (250 μL each time point) were obtained via Culex at predetermined time points (5, 15, 30, 60, 120, 240, 360, 480, and 1440 min) after dosing. Plasma (100 μL) was obtained by centrifugation within 30 min after collection, and aliquots of dosing solutions were taken and analyzed at the same time with plasma samples using a validated LC–MS/MS method developed on a Shimadzu HPLC system interfaced to a Sciex API 5000 mass spectrometer. Eluents were gradient mixtures of water and acetonitrile with 0.1% formic acid. HPLC column was ACE PFP C18, 50 × 2.1 mm, 5 μM. PK parameters of the analytes were obtained using Phoenix WinNonlin software (version 8.1).
4.7.4. Pharmacokinetic Studies in Male Hamsters with or without Pretreatment with Mesalamine
This study was conducted in accordance with the applicable Frontage Standard Operating Procedures (SOPs) and IACUC protocols. Golden Syrian male hamsters weighing between 110 and 150 g were obtained from either Charles River Laboratories. PO dosing solutions were prepared in 1% aqueous methylcellulose with 0.1% Tween 80 and administered by oral gavage. Group 1 animals (n = 3) were dosed orally with vehicle (1% aqueous methylcellulose with 0.1% Tween 80) first, followed by PO dose at 300 mg/kg of 8 (10 mL/kg of a 30 mg/mL solution), 30 min after the vehicle dose; while group 2 animals (n = 3) were dosed orally with mesalamine at 1000 mg/kg first (10 mL/kg of a 100 mg/mL solution), followed by PO dose at 300 mg/kg of 8, 30 min after administration of mesalamine. Hamsters had access to food and water ad libitum, fasted overnight before PO dosing, and food was made available 4 h post dosing. Blood samples (90 μL per time point via retro-orbital or jugular vein) were obtained via Culex at predetermined time points (5, 15, 30, 60, 120, 240, 360, 480, and 1440 min) after dosing. Plasma (40 μL) was obtained by centrifugation within 30 min after collection, and aliquots of dosing solutions were taken and analyzed at the same time with plasma samples using a validated LC–MS/MS method developed on a Shimadzu HPLC system interfaced to a Sciex API 6500 mass spectrometer. Eluents were gradient mixtures of water and acetonitrile with 0.1% formic acid. HPLC column was a Cortecs T3, 50 × 4.6 mm, 2.7 μM. PK parameters of the analytes were obtained using Phoenix WinNonlin software (version 8.1).
Acknowledgments
Support for this work comes from the Intramural Research Program of NIDDK, NIH, and the Office of AIDS Research at NIH. We gratefully acknowledge Dr. Robert O’Connor for his assistance in NMR analysis and Dr. John Lloyd together with the mass spectrometry core facility of NIDDK for analysis of all compounds. We also acknowledge ImQuest and Frontage for their efforts to conduct some of the reported work under contract from NIH.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.3c00260.
NMR spectra for 2, 8, 11, 12, 14, 17; kinetic data and NMR spectra for thioester exchange rate constant; blood stability data for 2, 4, 8; and rate of 17 formation in blood (PDF)
Author Contributions
M.R. participated in research design and execution (chemistry) and contributed to the writing of the manuscript; H.N. and M.T.S. participated in research design and execution (chemistry); R.A.N.P. participated in the research design and execution (biology) and contributed to the writing of the manuscript; V.M. participated in the research design and execution (biology); D.H.A. participated in research design (chemistry), acquired funding, and contributed to the writing of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- https://www.who.int/news-room/fact-sheets/detail/hiv-aidsWorld Health Organization—HIV/AIDS fact sheet, 2019.
- https://www.unaids.org/en/resources/fact-sheetGlobal HIV & AIDS statistics - fact sheet, 2019.
- https://aidsinfo.nih.gov/understanding-hiv-aids/fact-sheets. U.S. Department of Health and Human Services - HIV/AIDS fact sheets, 2020.
- http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services, 2020.
- Loubiere S.; Meiners C.; Sloan C.; Freedberg K. A.; Yazdanpanah Y. Economic evaluation of ART in resource-limited countries. Curr. Opin. HIV AIDS 2010, 5 (3), 225–231. 10.1097/COH.0b013e3283384a9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darlix J.-L.; Godet J.; Ivanyi-Nagy R.; Fossé P.; Mauffret O.; Mély Y. Flexible nature and specific functions of the HIV-1 nucleocapsid protein. J. Mol. Biol. 2011, 410 (4), 565–581. 10.1016/j.jmb.2011.03.037. [DOI] [PubMed] [Google Scholar]
- Guo J.; Wu T.; Kane B. F.; Johnson D. G.; Henderson L. E.; Gorelick R. J.; Levin J. G. Subtle Alterations of the Native Zinc Finger Structures Have Dramatic Effects on the Nucleic Acid Chaperone Activity of Human Immunodeficiency Virus Type 1 Nucleocapsid Protein. J. Virol. 2002, 76 (9), 4370–4378. 10.1128/JVI.76.9.4370-4378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J.; Wu T.; Anderson J.; Kane B. F.; Johnson D. G.; Gorelick R. J.; Henderson L. E.; Levin J. G. Zinc finger structures in the human immunodeficiency virus type 1 nucleocapsid protein facilitate efficient minus-and plus-strand transfer. J. Virol. 2000, 74 (19), 8980–8988. 10.1128/JVI.74.19.8980-8988.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckman J. S.; Bosche W. J.; Gorelick R. J. Human immunodeficiency virus type 1 nucleocapsid Zn2+ fingers are required for efficient reverse transcription, initial integration processes, and protection of newly synthesized viral DNA. J. Virol. 2003, 77 (2), 1469–1480. 10.1128/JVI.77.2.1469-1480.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundquist W. I.; Krausslich H. G. HIV-1 assembly, budding, and maturation. Cold Spring Harbor Perspect. Med. 2012, 2 (7), a006924. 10.1101/cshperspect.a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice W. G.; Schaeffer C. A.; Harten B.; Villinger F.; South T. L.; Summers M. F.; Henderson L. E.; Bess J. W.; Arthur L. O.; McDougal J. S.; et al. Inhibition of HIV-1 infectivity by zinc-ejecting aromatic C-nitroso compounds. Nature 1993, 361 (6411), 473–475. 10.1038/361473a0. [DOI] [PubMed] [Google Scholar]
- Rice W. G.; Supko J. G.; Malspeis L.; Buckheit R. W.; Clanton D.; Bu M.; Graham L.; Schaeffer C. A.; Turpin J. A.; Domagala J.; et al. Inhibitors of HIV nucleocapsid protein zinc fingers as candidates for the treatment of AIDS. Science 1995, 270 (5239), 1194–1197. 10.1126/science.270.5239.1194. [DOI] [PubMed] [Google Scholar]
- Zhan P.; Pannecouque C.; De Clercq E.; Liu X. Y. Anti-HIV Drug Discovery and Development: Current Innovations and Future Trends. J. Med. Chem. 2016, 59 (7), 2849–2878. 10.1021/acs.jmedchem.5b00497. [DOI] [PubMed] [Google Scholar]
- Mori M.; Kovalenko L.; Lyonnais S.; Antaki D.; Torbett B. E.; Botta M.; Mirambeau G.; Mély Y.. Nucleocapsid protein: a desirable target for future therapies against HIV-1. The Future of HIV-1 Therapeutics; Springer, 2015; pp 53–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancineto L.; Iraci N.; Tabarrini O.; Santi C. NCp7: targeting a multitasking protein for next-generation anti-HIV drug development part 1: covalent inhibitors. Drug discovery today 2018, 23 (2), 260–271. 10.1016/j.drudis.2017.10.017. [DOI] [PubMed] [Google Scholar]
- Iraci N.; Tabarrini O.; Santi C.; Sancineto L. NCp7: targeting a multitask protein for next-generation anti-HIV drug development part 2. Noncovalent inhibitors and nucleic acid binders. Drug discovery today 2018, 23 (3), 687–695. 10.1016/j.drudis.2018.01.022. [DOI] [PubMed] [Google Scholar]
- Sun S. K.; Huang B. S.; Li Z.; Wang Z.; Sun L.; Gao P.; Kang D. W.; Chen C. H.; Lee K. H.; Daelemans D.; et al. Discovery of potential dual-target prodrugs of HIV-1 reverse transcriptase and nucleocapsid protein 7. Bioorg. Med. Chem. Lett. 2020, 30 (16), 127287. 10.1016/j.bmcl.2020.127287. [DOI] [PubMed] [Google Scholar]
- Nikolayevskiy H.; Scerba M. T.; Deschamps J. R.; Appella D. H. Reaction Kinetics Direct a Rational Synthesis of an HIV-1 Inactivator of Nucleocapsid Protein 7 and Provide Mechanistic Insight into Cellular Metabolism and Antiviral Activity. Chem. Eur J. 2018, 24 (38), 9485–9489. 10.1002/chem.201801253. [DOI] [PubMed] [Google Scholar]
- Nikolayevskiy H.; Robello M.; Scerba M. T.; Pasternak E. H.; Saha M.; Hartman T. L.; Buchholz C. A.; Buckheit R. W. Jr.; Durell S. R.; Appella D. H. The structure-activity profile of mercaptobenzamides’ anti-HIV activity suggests that thermodynamics of metabolism is more important than binding affinity to the target. Eur. J. Med. Chem. 2019, 178, 818–837. 10.1016/j.ejmech.2019.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha M.; Scerba M. T.; Shank N. I.; Hartman T. L.; Buchholz C. A.; Buckheit R. W. Jr.; Durell S. R.; Appella D. H. Probing mercaptobenzamides as HIV inactivators via nucleocapsid protein 7. ChemMedChem 2017, 12 (10), 714–721. 10.1002/cmdc.201700141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins L. M. M.; Ott D. E.; Hayashi R.; Coren L. V.; Wang D.; Xu Q.; Schito M. L.; Inman J. K.; Appella D. H.; Appella E. Small-molecule inactivation of HIV-1 NCp7 by repetitive intracellular acyl transfer. Nat. Chem. Biol. 2010, 6 (12), 887–889. 10.1038/nchembio.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schito M. L.; Goel A.; Song Y.; Inman J. K.; Fattah R. J.; Rice W. G.; Turpin J. A.; Sher A.; Appella E. In vivo antiviral activity of novel human immunodeficiency virus type 1 nucleocapsid p7 zinc finger inhibitors in a transgenic murine model. AIDS Res. Hum. Retrovir. 2003, 19 (2), 91–101. 10.1089/088922203762688595. [DOI] [PubMed] [Google Scholar]
- Schito M. L.; Soloff A. C.; Slovitz D.; Trichel A.; Inman J. K.; Appella E.; Turpin J. A.; Barratt-Boyes S. M. Preclinical evaluation of a zinc finger inhibitor targeting lentivirus nucleocapsid protein in SIV-infected monkeys. Curr. HIV Res. 2006, 4 (3), 379–386. 10.2174/157016206777709492. [DOI] [PubMed] [Google Scholar]
- Hartman T.; Yang L.; Helfrick A.; Hassink M.; Shank N.; George Rosenker K.; Scerba M.; Saha M.; Hughes E.; Wang A.; et al. Preclinical evaluation of a mercaptobenzamide and its prodrug for NCp7-targeted inhibition of human immunodeficiency virus. Antivir. Res. 2016, 134, 216–225. 10.1016/j.antiviral.2016.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh L.; Tugarinov V.; Appella D. H.; Clore G. M. Targeting a Dark Excited State of HIV-1 Nucleocapsid by Antiretroviral Thioesters Revealed by NMR Spectroscopy. Angew. Chem., Int. Ed. 2018, 57 (10), 2687–2691. 10.1002/anie.201713172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller Jenkins L. M.; Paine E. L.; Deshmukh L.; Nikolayevskiy H.; Lyons G. C.; Scerba M. T.; George Rosenker K.; Luecke H. F.; Louis J. M.; Chertova E.; et al. Inhibition of HIV Maturation via Selective Unfolding and Cross-Linking of Gag Polyprotein by a Mercaptobenzamide Acetylator. J. Am. Chem. Soc. 2019, 141 (20), 8327–8338. 10.1021/jacs.9b02743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugaonkar S. R.; Clark J. T.; English L. B.; Johnson T. J.; Buckheit K. W. Jr; Bahde R. J.; Appella D. H.; Buckheit R. W.; Kiser P. F. An intravaginal ring for the simultaneous delivery of an HIV-1 maturation inhibitor and reverse-transcriptase inhibitor for prophylaxis of HIV transmission. J. Pharmaceut. Sci. 2015, 104 (10), 3426–3439. 10.1002/jps.24551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace G. S.; Cheng-Mayer C.; Schito M. L.; Fletcher P.; Miller Jenkins L. M.; Hayashi R.; Neurath A. R.; Appella E.; Shattock R. J. Human immunodeficiency virus type 1 nucleocapsid inhibitors impede trans infection in cellular and explant models and protect nonhuman primates from infection. J. Virol. 2009, 83 (18), 9175–9182. 10.1128/JVI.00820-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y.; Zhu J.; Hassink M.; Jenkins L. M. M.; Wan Y.; Appella D. H.; Xu J.; Appella E.; Zhang X. A novel preventive strategy against HIV-1 infection: combinatorial use of inhibitors targeting the nucleocapsid and fusion proteins: Combination of SAMT10 and SFT against HIV-1. Emerg. Microb. Infect. 2017, 6 (1), 1–8. 10.1038/emi.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran P.; Attard N. Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer 2008, 8 (1), 24–36. 10.1038/nrc2292. [DOI] [PubMed] [Google Scholar]
- Grivel J.-C.; Margolis L. Use of human tissue explants to study human infectious agents. Nat. Protoc. 2009, 4 (2), 256–269. 10.1038/nprot.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennard L. The clinical pharmacology of 6-mercaptopurine. Eur. J. Clin. Pharmacol. 1992, 43 (4), 329–339. 10.1007/BF02220605. [DOI] [PubMed] [Google Scholar]
- Bracher P. J.; Snyder P. W.; Bohall B. R.; Whitesides G. M. The relative rates of thiol-thioester exchange and hydrolysis for alkyl and aryl thioalkanoates in water. Origins Life Evol. Biospheres 2011, 41 (5), 399–412. 10.1007/s11084-011-9243-4. [DOI] [PubMed] [Google Scholar]
- Szumlanski C. L.; Weinshilboum R. M. Sulphasalazine inhibition of thiopurine methyltransferase: possible mechanism for interaction with 6-mercaptopurine and azathioprine. Br. J. Clin. Pharmacol. 1995, 39 (4), 456–459. 10.1111/j.1365-2125.1995.tb04478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosny E. A.; Khan Ghilzai N. M.; Al-Najar T. A.; Elmazar M. M. A. Hypoglycemic Effect of Oral Insulin in Diabetic Rabbits Using pH-Dependent Coated Capsules Containing Sodium Salicylate Without and with Sodium Cholate. Drug Dev. Ind. Pharm. 1998, 24 (3), 307–311. 10.3109/03639049809085625. [DOI] [PubMed] [Google Scholar]
- Hosny E. A.; Al-Shora H. I.; Elmazar M. M. Oral delivery of insulin from enteric-coated capsules containing sodium salicylate: effect on relative hypoglycemia of diabetic beagle dogs. Int. J. Pharm. 2002, 237 (1–2), 71–76. 10.1016/S0378-5173(02)00024-8. [DOI] [PubMed] [Google Scholar]
- Hurni M. A.; Noach A.; Blom-Roosemalen M.; de Boer A. G.; Nagelkerke J. F.; Breimer D. D. Permeability enhancement in Caco-2 cell monolayers by sodium salicylate and sodium taurodihydrofusidate: assessment of effect-reversibility and imaging of transepithelial transport routes by confocal laser scanning microscopy. J. Pharmacol. Exp. Ther. 1993, 267 (2), 942–950. [PubMed] [Google Scholar]; PMID: 7504101
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.