

Abstract
Carbometallation of alkenes and alkynes are powerful carbon-carbon bond-forming reactions. The use of compounds containing bonds between carbon and group 13 elements, particularly boron and aluminum, are particularly attractive because of the versatility of subsequent transformations. Uncatalyzed carboboration and carboalumination represent less common classes of reactions. This Short Review discusses uncatalyzed carboboration and carboalumination reactions of alkenes and alkynes, including the reaction design and mechanism.
Keywords: uncatalyzed, carboboration, carboalumination, alkenes, alkynes
Graphical Abstract

1. Introduction
Carbometallation reactions, which involve the addition of a carbon-metal bond to an unsaturated carbon-carbon bond, are commonly used transformations in organic synthesis.1–3 These difunctionalization reactions produce organometallic intermediates possessing a new carbon–carbon bond and a carbon–metal bond that can be functionalized to form a variety of products.4–6 Many organometallic reagents, such as organolithium reagents, Grignard reagents, and organozinc reagents, have been utilized in carbometallation reactions (for examples, Scheme 1).7–9
Scheme 1.
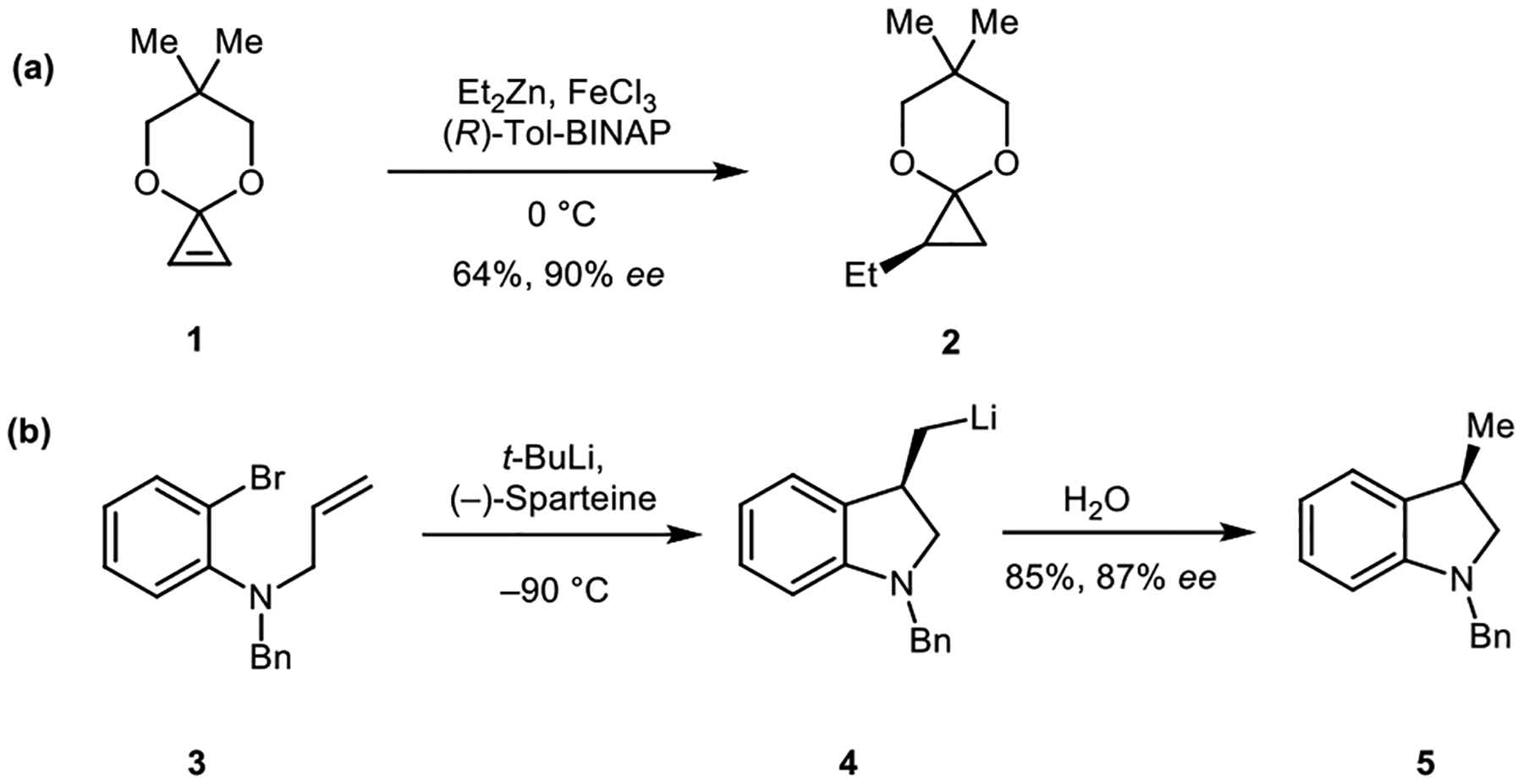
Examples of carbometallation (a) carbozincation and (b) Carbolithiation
Group-13 elements such as boron and aluminum can also be employed in carbometallation reactions. Metal-catalyzed carbometallation reactions involving these reagents have been used in a number of contexts.3, 7 For example, the zirconium-catalyzed carboalumination of alkynes has been developed as a useful reaction in organic synthesis (Scheme 2a).10 Transition-metal-catalyzed carboboration reactions have also emerged as a powerful alkene difunctionalization method (Scheme 2b).11
Scheme 2.
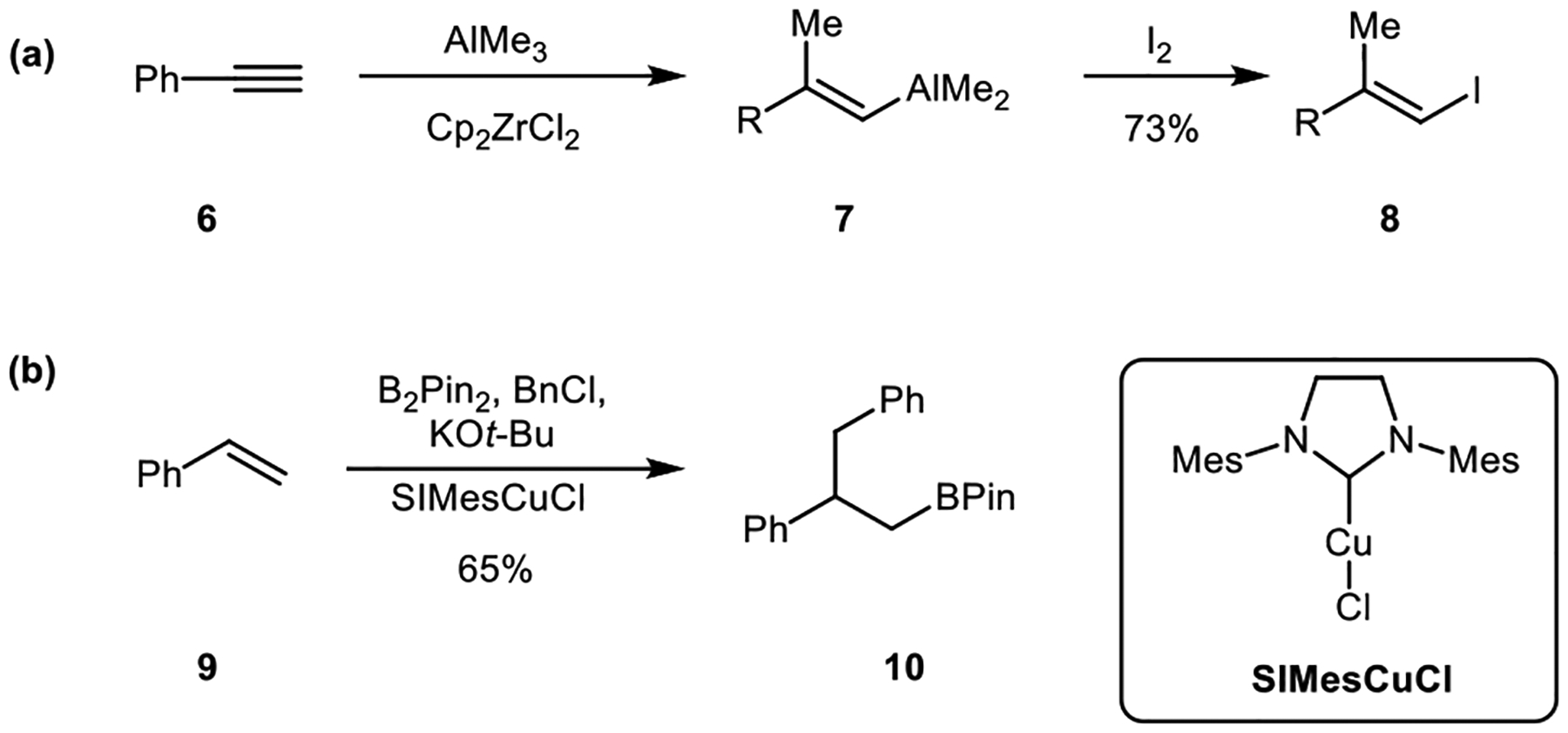
Metal catalyzed carbometallation (a) carboalumination and (b) carboboration
In contrast to metal-catalyzed reactions, uncatalyzed carbometallation reactions have received less attention. Among the methods for performing uncatalyzed carbometallation reactions, group-13 organometallic reagents have emerged as the most general. Organogallium and organoindium reagents are found commonly in uncatalyzed carbometallation reactions because of the moderate Lewis acidity and high π-electron affinity of these reagents.12–13 These transformations have been reviewed.14
Uncatalyzed carboboration and carboalumination reactions represent another class of important transformations. Compared to their transition-metal-catalyzed variants, the reactions are sustainable due to the absence of the expensive and toxic heavy metals. Additionally, the unique reactivity of organoboron and organoaluminum reagents often enable reactions to exhibit unique regio- and stereoselectivity, and reactions of the products provide insights about other reactive intermediates.15–16 Nevertheless, activated unsaturated systems such as strained alkenes are generally required.
This review will summarize the status of uncatalyzed carboboration and carboalumination reactions of alkenes and alkynes. Historical developments and recent examples are documented. Mechanistic insights are also provided. The present review builds upon the last detailed review, which appeared forty years ago.17–18
2. Uncatalyzed carboboration of alkenes
In contrast to the hydroboration reaction, which represents a general method for regioselective functionalization of alkenes, the carboboration reaction, which forms carbon–carbon and carbon–boron bonds in one synthetic operation, is much less common. Just as with hydroboration,19–21 transition metals can facilitate carboborations of alkenes.11 The need for a catalyst underscores the fact that the rate of the background, uncatalyzed reaction is slow.
Reactions of trialkylboranes with alkenes require elevated temperatures.22 Alkenes exchanges are often observed under these conditions (Scheme 3a).22–23 At high temperatures, dec-1-ene underwent carboboration with triethylborane to give the corresponding addition products in low yield (Scheme 3b).23
Scheme 3.

Additions of trialkylboranes to alkenes through (a) alkenes exchanges and (b) carboboration
The most reactive boranes for uncatalyzed carboboration reactions of alkenes are allylboranes. These reagents add to alkenes by ene reactions through six-membered-ring transition states. For example, cyclopropenes underwent ene reactions with allyboranes in a net syn-addition (Scheme 4a).24 A second product, alkene 21, was isolated that involved addition across the sterically more accessible carbon–carbon bond of the cyclopropene ring. A related transformation was observed for additions of trialkylboranes with 1-methylcyclopropene (Scheme 4b).25 The reaction proceeded through a ring-opening reaction related to the formation of 21 (Scheme 4a) followed by an ene reaction of the resulting allylborane with the excess cyclopropene.
Scheme 4.

Carboboration of cyclopropene with (a) allylboranes (b) Trialkylboranes
Other allylboranes react with alkenes in a net carboboration reaction. Allyldichloroborane underwent carboboration with allylic silane 24 to give alcohol 26 as a single diastereomer after oxidation of the intermediate borane (Scheme 5).26–27 The synaddition across the double bond is consistent with the ene mechanism. The regioselectivity reflects the polarization of the carbon–carbon double bond of the alkene and the relative steric interactions that would develop.28 As with other reactions of cyclic allylic silanes, reactions occur from the face opposite to the silyl group.29–31.
Scheme 5.

Regioselective and stereoselective allylboration of allylic silanes
Intramolecular allylboration can also be achieved. Aryldiallylborane 28, synthesized from dimethyl (2-allylphenyl)boronate 27, underwent endo-cyclization with the terminal alkene unit to yield benzoborinane 29 (Scheme 6).32
Scheme 6.

Intramolecular carboboration with allylboranes
Activated alkenes, such as strained alkenes, also participate in uncatalyzed carboboration reactions. Norbornadiene underwent an addition reaction with phenyldichlorborane to form dichlolorborane 32 (Scheme 7).33 The formation of the tricyclic product is likely through a transannular π-participation process, as has been observed for reactions between norbornadiene and bromine.34–37 The electronic nature of the boron atom was critical to the success of the reaction: neither diphenylchloroborane nor triphenylborane reacted with norbornadiene.
Scheme 7.

Thermal carboboration of norbornadiene with arylborane
An early report of 1,2-carboboration was provided for highly strained trans-cyclohexenes (Scheme 8). Cyclohexenes underwent photosensitized isomerization to form strained and reactive E-isomers 34, which were trapped by reaction with trialkylboranes.38–39 The authors proposed a structure for the carboboration products after oxidation of the carbon–boron bond of the product 36 with hydrogen peroxide. This compound would have been formed by syn-addition across the E-double bond. It was proposed that this product was formed by a stepwise, zwitterionic mechanism that begins with initial nucleophilic attack of the alkene on the trialkylborane was proposed.
Scheme 8.

Carboboration of trans-cyclohexenes with triethylboranes
More recent studies, however, have indicated that the structure of alcohol was not correct. Instead, the product of the photoinduced carboboration was a ring-contracted product. For example, trans-cyclohexene 39, formed by irradiation of cyclohexene 38, underwent addition of the electrophilic boron species to form a zwitterionic species followed by a 1,2-alkyl shift and migration of an alkyl group from the boron atom to form five-membered ring 41 (Scheme 9).40 Alcohol 42 was isolated after oxidation. The mechanism was examined computationally, which indicated that the two migrations are not concerted, but instead occur sequentially through a short-lived intermediate.41
Scheme 9.

Carboboration of trans-cyclohexenes via a ring contraction
Seven-membered-ring trans-alkenes also undergo strain-promoted carboboration (Scheme 10).16 Uncatalyzed syn-addition of trialkylboranes to trans-alkene 43 occurred at room temperature to yield borane 44 as a single diastereomer. The resulting boranes were air-stable, which contrasts with the general pyrophoric reactivity of trialkylboranes.42 The stability of the product may be derived from the steric protection of the boron atom by the substituents on the seven-membered ring and hyperconjugation involving adjacent bonds.
Scheme 10.

Carboboration of seven-membered-ring trans-alkenes
Vinylboronate complexes reacted with electrophiles in a 1,2-carboboration pathway (Scheme 11).43 The vinylboronate intermediate 46 was formed from boronic ester 45 upon addition of n-butyllithium. Carboboration occurred in the presence of an electrophile, which caused the 1,2-migration of n-butyl group, resulting in intermediate 47. The product 48 was formed through a second 1,2-migration.
Scheme 11.

1,2-Carboboration of vinylboron ate complexes
Net uncatalyzed carboboration can be achieved using boron alkylidenes and unactivated alkenes (Scheme 12).44 Bisboronate 49 underwent deborylation to form boron alkylidene 50, which underwent intramolecular addition of the tethered alkene to form boracyclobutane 52. This intermediate could be trapped by electrophiles to form adducts 53.
Scheme 12.

Net carboboration of nonstrained alkenes
Net carboboration of alkenes can also be accomplished using radical intermediates. Alkene 54 reacted with diboron reagent 55 and perfluorobutyl iodide to yield 1,2-carboboration product 56 (Scheme 13a).45 A radical cascade mechanism was proposed involving perfluoroalkyl radicals that were generated by irradiation with blue light. The perfluoroalkyl radicals added to alkene 54 and the resulting alkyl radicals then abstracted a dialkoxyboryl group from the diboron reagent 55 to afford the 1,2-carboboration product 56. Boron-centered radicals can also be used to achieve net carboboration of alkenes. This reaction likely involves addition of a boron-centered radical to a styrene to make a benzylic radical, which can add to another alkene to form the carbon–carbon bond (Scheme 13b).46
Scheme 13.

Radical triggered carboboration of alkenes
Allenes are reactive towards Lewis acidic boron reagents. 1,2-Addition of tris(pentafluorophenyl)borane to the terminal double bond of allenyl ketone 60 was reported to yield cyclic product 61 (Scheme 14a).47 The transformation gave alkenes with complete E-selectivity. Another example of carboboration on allenes involved alkenylborane 62 (Scheme 14b).48 Phenylacetylene underwent hydroboration, and the resulting alkenylborane 62 reacted with phenylallene to give 1,4-diene 63.
Scheme 14.

Uncatalyzed carboboration of allenes
3. Uncatalyzed carboboration of alkynes
Uncatalyzed carboboration of alkynes represent another useful reaction in organic synthesis because the vinyl borane products can undergo diverse transformations.1, 7–8 In general, a more electrophilic boron reagent or an activated alkyne is required to enable the electrophilic attack on the carbon–carbon triple bond.
Just as observed with alkenes, alkynes undergo ene-type reactions with allyboranes (Scheme 15).32, 49–50 Propyne underwent intermolecular allylboration of triallylborane, followed by intramolecular allylboration to form borocyclohexene 66. Another intramolecular carboboration occurred to give bicyclic boron derivative 68. This reaction sequence has been employed to synthesize a variety of 1-bora- and 1-azaadamantanes.51
Scheme 15.

Allylboration of propyne
Alkenylboranes can also undergo carboboration with alkynes. As demonstrated previously (Scheme 14b), alkenylborane 62 underwent a series of carboboration reactions with an excess of phenylacetylene to form tetrahydropentalene 70 (Scheme 16).52 Computational studies supported a sequene of carboboration reactions to form octatetraene 69 followed by a sequence of rearrangements and cycloaddition reactions to form the tetrahydropentalene 70.
Scheme 16.

Carboboration of alkyne with alkenylboranes
Trialkylboranes can also be used in the uncatalyzed 1,1-carboboration of alkynes. The reaction involves electrophilic addition to the alkyne followed by migration of the substituent M on the alkyne then migration of the substituent R on boron (Scheme 17). This reaction has been summarized in other papers.53–54 Nonetheless, the reaction scope has been limited to alkynes containing main group metal or transition metal substituents.
Scheme 17.

1,1-Carboboration of metal-substituted alkynes
Strong boron Lewis acids extended the scope of this uncatalyzed 1,1-carboboration on alkynes. The use of pentafluoroaryl boranes such as tris(pentafluorophenyl)borane allowed for the 1,2-migration of alkyl groups.55–56 For example, the reaction of 4-octyne with tris(pentafluorophenyl)borane formed the vinylboranate 74, which was subjected to cross-coupling to give compound 75 (Scheme 18).57 The migration of an alkyl group on the alkyne provides unique insights to carbon–carbon σ-bond activation.55
Scheme 18.

1,1-Carboboration with tris(pentafluorophenyl)borane
Borenium ions are also electrophilic enough to participate in the 1,1-carboboration of alkynes. Borenium ions are characterized as cationic boron species with two covalently bonded substituents and a dative ligand.58 1,1-Carboboration was reported with borenium cation 76 and silyl-protected alkyne under mild conditions (Scheme 19).59 The 1,1-carboboration product 77 underwent a ligand exchange with pinacol to form borinate ester 78, which could be used in further transformations.
Scheme 19.

1,1-Carboboration of alkynes with borenium ions
Although 1,2-carboboration of alkynes are relatively rare, the use of borenium ions permit this transformation. A vinyl cation intermediate 80, which is formed upon attack of the electrophilic boron species on the alkyne, would either undergo 1,2-migration of one of the alkyne’s substituents to form a 1,1-carboboration product 81, as observed in Schemes17−19, or the substituent R on boron could migrate to form the 1,2-carboboration product 82 (Scheme 20). It was proposed that aryl groups on the boron atom would have a higher migratory aptitude, which would favor their migration, leading to net 1,2-carboboration.60
Scheme 20.

Stepwise reaction pathyways leading 1,1- or 1,2-carboboration of alkynes with borenium ions
Reactions of a series of quinolato(aryl)borenium cations led to 1,2-carboboration of alkynes. The borenium ion 83, formed by reacting a quinoline substrate with BCl3 followed by halogen abstraction by AlCl3, inserted into 3-hexyne to yield 1,2-carboboration product 85 (Scheme 21a).60 The resulting alkenylborane could be converted to the pinacolboronate 86, which could undergo subsequent transformations. Computational studies supported the higher migratory aptitude of aryl groups versus alkyl groups, which leads to 1,2-carboboration. Phosphine-coordinated borenium ions undergo similar reactions. The borenium ion 87, which was prepared from 1-dihenylphosphino-8-iodonaphthalene, underwent syn-addition to the alkyne more readily than the quinolinestabilized borenium ion 83 (Scheme 21b).61
Scheme 21.

1,2-Carboboration of alkynes with borenium ions
Divalent borinium ions are more electrophilic than trivalent borenium ions, which allows them to react readily with alkynes. Borinium ion 89 underwent two sequential 1,2-carboboration reactions with two equivalents of diphenylacetylene to form borinium ion 91 (Scheme 22).62 X-ray diffraction analysis revealed that the positive charge on the product was delocalized across the entire conjugated π-system.
Scheme 22.

Carboboration of alkynes with borinium ions
Other arylboranes can undergo 1,2-carboboration of electron-rich alkynes. Dichlorophenylborane reacted with ynamide 92 to yield 1,2-carboboration product 93 with complete regio- and stereoselectivity (Scheme 23a).63 The reaction is believed to involve formation of a keteniminium intermediate followed by migration of a phenyl group from the boron atom to the carbon atom.
Scheme 23.

Carboboration with arylborane
Borafluorenes also react with alkynes to form new carbon-carbon and carbon–boron bonds. For example, 9-chloro-9-borafluorene reacted with diphenylacetylene to form the borapin 95, which may be stabilized by orbital interactions within the seven-membered ring (Scheme 23b).64–65 Oxidation of intermediate 95 with excess iron trichloride led to extended π-conjugated molecules through a carbon-carbon coupling process. The computational studies supported a concerted mechanism of carboboration for formation of 95.66
Anti-1,2-carboboration of alkynes has been reported. The reaction between a heteroarylacetylene 97 and alkenylboronic acid 98 in the presence of tartaric acid formed the five-membered boronic acid derivative 99 (Scheme 24a).67 The Brønsted acid enhanced the electrophilicity of the boronic acid by formation of the dioxaborolanone, which promoted the electrophilic attack to the alkyne. The anti-addition was rationalized by formation of an oxaborole, which requires the boron and hydroxyl groups to be added to the same side. Alkynylboration can occur in a similar fashion. An alkynylboronate reacted with propargylic alcohol 100 in the presence of a base to yield oxaborole 101 (Scheme 24b).68–69
Scheme 24.

Anti-1,2-carboboration of alkynes
4. Uncatalyzed carboalumination of alkenes
The additions of organoalanes to alkenes have become powerful methods in synthetic organic chemistry because of the ready availability of organoaluminum reagents and the versatility of subsequent transformations.70 While the metal-catalyzed carboalumination of alkenes are a common transformation, fewer examples of uncatalyzed reactions exist.71 Efforts to develop hydroalumination led to the observation of carboalumination of alkenes. Attempts to add AlH3 to ethene yielded triethylaluminum, which underwent insertion to form higher molecular-weight organoaluminum compounds (Scheme 25).72–73 These uncatalyzed carboalumination required extreme temperatures and pressures, however.
Scheme 25.

General insertion of trialkylaluminums into alkenes
Detailed studies of these transformations provided evidence of the reaction mechanism. Oligomeric organoaluminum compounds dissociate into monomeric species, which are the active species in the reaction.74 The rate-determining step of the subsequent carboalumination is believed to be the electrophilic attack of the tricoordinate monomer on the unsaturated system (Scheme 26).75 The stable π-complex 107 reacts through four-centered transition state 108, leading to the final products.76 This proposed mechanism was supported by experiments showing that the addition of Lewis bases, which would compete for the monomeric organoaluminum species, greatly slowed or inhibited the reaction.77 Kinetic studies also support this mechanism.78
Scheme 26.

General mechanism of uncatalyzed carboalumination of alkenes
The ease of hydroalumination compared to carboalumination can be understood by considering several factors. The hydridic hydrogen atom can bridge more easily between the β-carbon atom and the aluminum atom, causing 110 to be a lower energy transition state than 111 (Figure 1).74 Steric destabilization between the alkyl group being transferred and the substituent on the alkene also raises the energy of the transition state for carboalumination.74
Figure 1.

Transition states between hydroalumination and carboalumination on alkenes
Some side reactions prevent the synthetic utility of uncatalyzed carboalumination. Cis-trans isomerization of alkenes can be problematic when reaction mixtures are held at the elevated temperatures required for these reactions.17 This isomerization may occur because carboalumination can be reversible. That reverse reaction, decarboalumination, is observed with some reactive aluminum reagents, leading to a net transmetallation process (Scheme 27).75
Scheme 27.

decarboalumination of 3,3,3-triphenyl-propen
Despite the forcing conditions typically required to achieve uncatalyzed carboalumination of alkenes, additions to strained and conjugated alkenes occur at lower temperatures. Norbornadiene underwent uncatalyzed carboalumination at 80 °C (Scheme 28).75, 79 This reaction yielded both alkene 114 and diphenyl alkane 115. These products were likely formed by carboalumination of the double bonds through concerted four-membered-ring transition states followed by protonation of the resulting organoalane intermediates.
Scheme 28.

Carboalumination of nordondiene with triphenylaluminum
Cyclopropenes also reacted with organoaluminum reagents in the absence of a catalyst. As with carboboration of cyclopropenes (Scheme 4), carboalumination of 3,3-dimethylcyclopropene with triethylaluminum formed compound 117 by ring-opening of the cyclopropene and subsequent carboalumination (Scheme 29a).80 By contrast, reaction of 1,2,3-triphenylcyclopropene gave only alkene 119, the product obtained by carboalumination of a carbon-carbon single bond (Scheme 29b).81
Scheme 29.

Carboalumination of cyclopropenes with triethylaluminum
Highly strained alkenes, such as seven-membered-ring trans-alkenes, reacted with trialkylaluminum reagents at room temperature (Scheme 30).15 A concerted, syn-addition pathway was proposed. The intermediate that possessed a carbon–aluminum bond could undergo subsequent reactions, such as protonation and oxidation, providing functionalized products as single diastereomers.
Scheme 30.

Carboalumination on seven-membered ring trans-alkenes
Intramolecular uncatalyzed carboalumination occurred with alkenylaluminum compounds. A regioselective hydroalumination of silyl-substituted alkyne 123 formed alkenylaluminum intermediate 124 (Scheme 31a). Isomerization of the double bond was followed by intramolecular 1,2-carboalumination with the second double bond, which formed intermediate 126.82 Addition of water to the reaction mixture yielded 127. Mechanistic experiments showed that the intramolecular carboalumination reaction proceeded through a syn-addition.82–83 At higher temperature, the related enyne 128 underwent skeletal rearrangement to form 1,2-dihydronaphthalen-1-ylaluminum reagent 131 via a cyclopropylcarbinylaluminum species 130 (Scheme 31b).84 Subsequent dehydroalumination formed the naphthalene 132.
Scheme 31.

Intramolecular uncatalyzed carboalumination of alkenes
5. Uncatalyzed carboalumination of alkynes
Uncatalyzed carboalumination of carbon–carbon triple bonds occurs under mild conditions. For example, acetylene reacted with trimethylaluminum at 40–60 °C, compared to the reaction of ethylene, which required heating to 150 °C.17
The carboalumination of alkynes generally occurs with syn-stereoselectivity. The mechanism of the addition resembles the mechanism proposed for carboalumination of alkenes (Scheme 26). The electrophilic addition of the tricoordinate monomer to the unsaturated substrate is believed to be the rate-determining step. The higher reactivity of alkynes compared to alkenes is ascribed to the less steric hinderance developed in the transition state for carbometallation of alkynes (Figure 2).17
Figure 2.

Transition states between carboalumination on alkynes and alkenes
The addition of alkylaluminum reagents to alkynes occurs with retention of configuration of the alkyl group that is transferred. Alkylalane 135, in which the relative configuration was established in a hydroalumination step, reacted with diphenylacetylene to form vinylalane 136 with the illustrated stereochemistry (Scheme 32).85
Scheme 32.
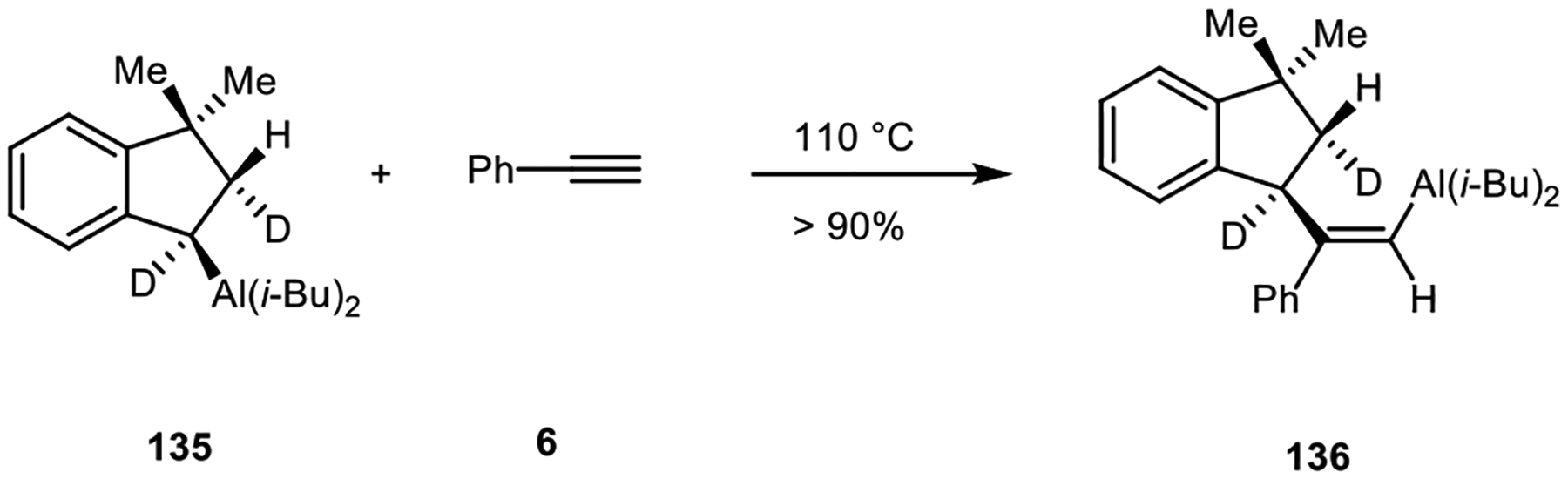
Carboalumination of alkyne with retention configuration
The lack of regioselectivity of carboalumination of alkynes limits the synthetic uses of the reaction. In the case of alkynes where the two ends of the alkyne were sterically distinct, the reactions can be regioselective, however. Reactions of triphenylaluminum with internal alkynes 137 and 139 resulted in products where the aluminum atom was placed at the more sterically congested carbon atom (Scheme 33).86
Scheme 33.
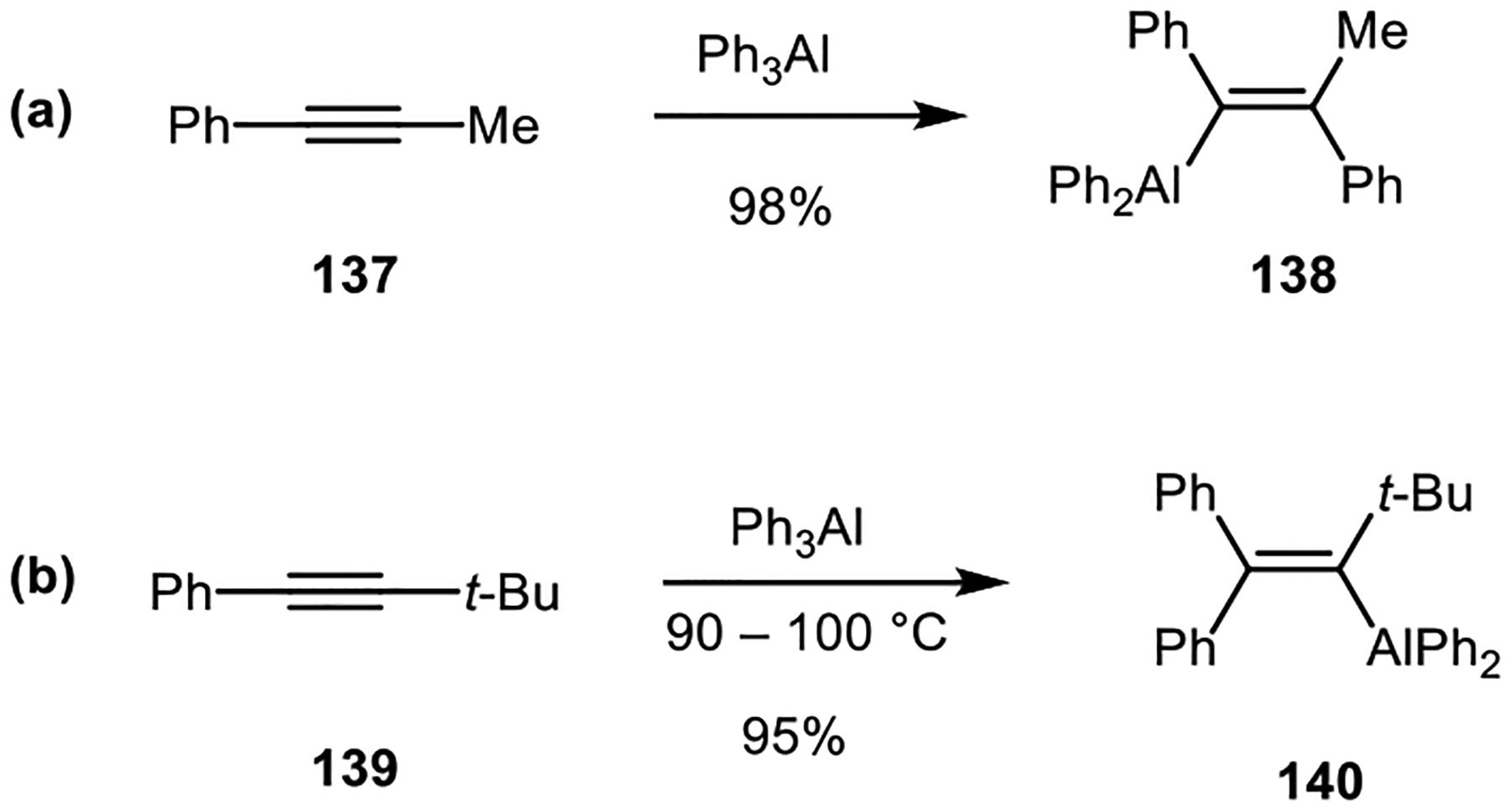
Carboalumination of alkynes with opposite regioselectivity
Another limitation of the uncatalyzed carboalumination of alkynes is the considerable amount of products formed from β-H elimination side reactions. When tricoordinate aluminum reagents have C–H bonds β to the aluminum atom, as is the case for triisobutylaluminum, dialkylaluminum hydrides are formed in situ, and these compounds underwent hydroalumination instead of carboalumination (Scheme 34a).18, 87 Carboalumination also does not occur with terminal alkynes, which instead undergo metalation at the terminal C–H bond (Scheme 34b).88
Scheme 34.

Side reactions of carboalumination of alkynes
6. Conclusion
The uncatalyzed carbometallation reactions of alkenes and alkynes with alkylmetal reagents containing group 13 elements are uncommon, but they show potential utility for synthetic organic chemistry. Both carboboration and carboalumination reactions occur in particular with strained alkenes with, in many cases, high regioselectivity and stereoselectivity. Highly electrophilic reagents, such as borenium ions and borinium ions, are the most reactive, suggesting ways of improving both the carboboration and carboalumination reactions by the study of fundamental main-group organometallic chemistry. This field will likely advance as new types of organoboron and organoaluminum reagents are developed and as new types of reactive unsaturated systems are designed.
Funding Information
National Institutes of Health, National Institute of General Medical Sciences (R01GM129286 and R01GM118730).
Biosketches

Professor Keith Woerpel received his B.S. with Highest Distinction from the University of Virginia. He earned A.M. and Ph.D. degrees from Harvard University, studying with Professor David Evans. He then was an NIH postdoctoral fellow at the University of California, Berkeley, with Professor Robert Bergman. He was appointed to the faculty at the University of California, Irvine, where he ascended to the rank of Professor. He is currently the Margaret and Herman Sokol Professor of Medicinal Chemistry at New York University. His research interests involve developing stereochemical models to understand complex reactions, exploring the unusual reactivities of silylenes and strained cyclic alkenes, and devising new methods for the synthesis of biologically active organic peroxides.

Yudong Liu received his B.S. in Chemical Biology at Xiamen University in 2017, conducting research in late modification of organic functional skeleton through C-H activation under Associate Professor Jianbin Lin and Professor Huijun Zhang before joining the Ph.D. program in Chemistry at NYU.
References
- 1.Normant JF; Alexakis A Synthesis 1981, 1981, 841. [Google Scholar]
- 2.Marek I; Basheer A In Science of Synthesis, Stereoselective Synthesis 1, 1st ed., Vol. 1; de Vries JG; Evans PA; Molander GA, Eds.; Georg Thieme Verlag: Stuttgart, 2011, 325. [Google Scholar]
- 3.Ding A; Guo H In Comprehensive Organic Synthesis II 2nd ed., Vol. 4; Knochel P, Ed. Elsevier: Amsterdam, 2014, 891. [Google Scholar]
- 4.Marek I J. Chem. Soc., Perkin Trans. 1 1999, 535. [Google Scholar]
- 5.Banon-Tenne D; Marek I In Transition Metals for Organic Synthesis, 2nd ed., Beller M; Bolm C, Eds.; Wiley-VCH: New York, 2004, 563. [Google Scholar]
- 6.Hogan A-ML; O’Shea DF Chem. Commun 2008, 3839. [DOI] [PubMed] [Google Scholar]
- 7.Fallis AG; Forgione P Tetrahedron 2001, 57, 5899. [Google Scholar]
- 8.Flynn AB; Ogilvie WW Chem. Rev 2007, 107, 4698. [DOI] [PubMed] [Google Scholar]
- 9.Knochel P In Comprehensive Organic Synthesis, Vol. 4; Trost BM; Fleming I, Eds.; Pergamon: Oxford, 1991, 865. [Google Scholar]
- 10.Negishi E; Tan Z In Metallocenes in Regio- and Stereoselective Synthesis, Vol. 8; Takahashi T, Ed. Springer: Berlin, Heidelberg, 2004, 139. [Google Scholar]
- 11.Liu Z; Gao Y; Zeng T; Engle KM Isr. J. Chem 2020, 60, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jung H-J; Cho Y; Kim D; Mehrkhodavandi P Catal. Sci. Technol 2021, 11, 62. [Google Scholar]
- 13.Augé J; Lubin-Germain N; Uziel J Synthesis 2007, 2007, 1739. [Google Scholar]
- 14.Nishimoto Y; Yasuda M Chem. - Asian J 2020, 15, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greene MA; Liu Y; Sanzone JR; Woerpel KA Org. Lett 2020, 22, 7518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanzone JR; Hu CT; Woerpel KA J. Am. Chem. Soc 2017, 139, 8404. [DOI] [PubMed] [Google Scholar]
- 17.Wardell JL; Paterson ES In The Metal—Carbon Bond (1985), Hartley FR; Patai S, Eds.; Wiley-VCH: New York, 1985, 219. [Google Scholar]
- 18.Zietz JR; Robinson GC; Lindsay KL In Comprehensive Organometallic Chemistry, Wilkinson G; Stone FGA; Abel EW, Eds.; Pergamon: Oxford, 1982, 365. [Google Scholar]
- 19.Männig D; Nöth H Angew. Chem. Int. Ed 1985, 24, 878. [Google Scholar]
- 20.Evans DA; Fu GC; Hoveyda AH J. Am. Chem. Soc 1988, 110, 6917. [Google Scholar]
- 21.Evans DA; Fu GC J. Am. Chem. Soc 1991, 113, 4042. [Google Scholar]
- 22.Koster R Angew. Chem. Int. Ed 1964, 3, 174. [Google Scholar]
- 23.Köster R Justus Liebigs Ann. Chem 1958, 618, 31. [Google Scholar]
- 24.Bubnov YN; Nesmeyanova OA; Rudashevskaya TY; Mikhailov BM; Kazansky BA Tetrahedron Lett 1971, 12, 2153. [Google Scholar]
- 25.Mikhailov BM; Bubnov YN; Nesmeyanova OA; Kiselev VG; Rudashevskaya TY; Kazansky BA Tetrahedron Lett 1972, 13, 4627. [Google Scholar]
- 26.Singleton DA; Waller SC; Zhang Z; Frantz DE; Leung S-WJ Am. Chem. Soc 1996, 118, 9986. [Google Scholar]
- 27.Frantz DE; Singleton DA Org. Lett 1999, 1, 485. [DOI] [PubMed] [Google Scholar]
- 28.Dubac J; Laporterie A Chem. Rev 1987, 87, 319. [Google Scholar]
- 29.Angelaud R; Landais Y J. Org. Chem 1996, 61, 5202. [Google Scholar]
- 30.Roberson CW; Woerpel KA Org. Lett 2000, 2, 621. [DOI] [PubMed] [Google Scholar]
- 31.Roberson CW; Woerpel KA J. Am. Chem. Soc 2002, 124, 11342. [DOI] [PubMed] [Google Scholar]
- 32.Kuznetsov NY; Starikova ZA; Averkiev BB; Bubnov YN Russ. Chem. Bull 2005, 54, 678. [Google Scholar]
- 33.Joy F; Lappert MF; Prokai BJ Organomet. Chem 1966, 5, 506. [Google Scholar]
- 34.Schmerling L; Luvisi JP; Welch RW J. Am. Chem. Soc 1956, 78, 2819. [Google Scholar]
- 35.Youngblood GT; Trivette CD; Wilder PJ Org. Chem 1958, 23, 684. [Google Scholar]
- 36.Winstein SJ Am. Chem. Soc 1961, 83, 1516. [Google Scholar]
- 37.Walkowiak J; Marciniak B; Koroniak HJ Fluorine Chem 2012, 143, 287. [Google Scholar]
- 38.Miyamoto N; Isiyama S; Utimoto K; Nozaki H Tetrahedron Lett 1971, 12, 4597. [Google Scholar]
- 39.Miyamoto N; Isiyama S; Utimoto K; Nozaki H Tetrahedron 1973, 29, 2365. [Google Scholar]
- 40.Jin S; Nguyen VT; Dang HT; Nguyen DP; Arman HD; Larionov OV J. Am. Chem. Soc 2017, 139, 11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roytman VA; Jin S; Nguyen VT; Nguyen VD; Haug GC; Larionov OV; Singleton DA J. Am. Chem. Soc 2020, 142, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brown HC; Midland MM Angew. Chem. Int. Ed 1972, 11, 692. [Google Scholar]
- 43.You C; Studer A Angew. Chem. Int. Ed 2020, 59, 17245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu X; Deaton TM; Haeffner F; Morken JP Angew. Chem. Int. Ed 2017, 56, 11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cheng Y; Mück-Lichtenfeld C; Studer AJ Am. Chem. Soc 2018, 140, 6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ren S-C; Zhang F-L; Qi J; Huang Y-S; Xu A-Q; Yan H-Y; Wang Y-FJ Am. Chem. Soc 2017, 139, 6050. [DOI] [PubMed] [Google Scholar]
- 47.Melen RL; Wilkins LC; Kariuki BM; Wadepohl H; Gade LH; Hashmi ASK; Stephan DW; Hansmann MM Organometallics 2015, 34, 4127. [Google Scholar]
- 48.Averdunk A; Hasenbeck M; Müller T; Becker J; Gellrich U Chem. - Eur. J 2022, n/a, e202200470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mikhailov BM; Bubnov YN; Frolov SI Bull. Acad. Sci. USSR, Div. Chem. Sci 1967, 16, 2193. [Google Scholar]
- 50.Frolov SI; Bufonov YN; Mikhailov BM Bull. Acad. Sci. USSR, Div. Chem. Sci 1969, 18, 1846. [Google Scholar]
- 51.Bubnov YN; Gurskii ME; Grandberg AI; Pershin DG Tetrahedron 1986, 42, 1079. [Google Scholar]
- 52.Hasenbeck M; Müller T; Averdunk A; Becker J; Gellrich U Chem. - Eur. J 2022, 28, e202104254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wrackmeyer B Coord. Chem. Rev 1995, 145, 125. [Google Scholar]
- 54.Wrackmeyer B Heteroat. Chem 2006, 17, 188. [Google Scholar]
- 55.Kehr G; Erker G Chem. Sci 2016, 7, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kehr G; Erker G Chem. Commun 2012, 48, 1839. [DOI] [PubMed] [Google Scholar]
- 57.Chen C; Kehr G; Fröhlich R; Erker GJ Am. Chem. Soc 2010, 132, 13594. [DOI] [PubMed] [Google Scholar]
- 58.Piers WE; Bourke SC; Conroy KD Angew. Chem. Int. Ed 2005, 44, 5016. [DOI] [PubMed] [Google Scholar]
- 59.Lawson JR; Fasano V; Cid J; Vitorica-Yrezabal I; Ingleson MJ Dalton Trans 2016, 45, 6060. [DOI] [PubMed] [Google Scholar]
- 60.Cade IA; Ingleson MJ Chem. - Eur. J 2014, 20, 12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Devillard M; Brousses R; Miqueu K; Bouhadir G; Bourissou D Angew. Chem. Int. Ed 2015, 54, 5722. [DOI] [PubMed] [Google Scholar]
- 62.Tanaka N; Shoji Y; Hashizume D; Sugimoto M; Fukushima T Angew. Chem. Int. Ed 2017, 56, 5312. [DOI] [PubMed] [Google Scholar]
- 63.You C; Sakai M; Daniliuc CG; Bergander K; Yamaguchi S; Studer A Angew. Chem. Int. Ed 2021, 60, 21697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shoji Y; Tanaka N; Muranaka S; Shigeno N; Sugiyama H; Takenouchi K; Hajjaj F; Fukushima T Nat. Commun 2016, 7, 12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schulman JM; Disch RL Organometallics 2000, 19, 2932. [Google Scholar]
- 66.Shoji Y; Shigeno N; Takenouchi K; Sugimoto M; Fukushima T Chem. - Eur. J 2018, 24, 13223. [DOI] [PubMed] [Google Scholar]
- 67.Roscales S; Csákÿ AG Org. Lett 2015, 17, 1605. [DOI] [PubMed] [Google Scholar]
- 68.Nogami M; Hirano K; Kanai M; Wang C; Saito T; Miyamoto K; Muranaka A; Uchiyama MJ Am. Chem. Soc 2017, 139, 12358. [DOI] [PubMed] [Google Scholar]
- 69.Nogami M; Hirano K; Morimoto K; Tanioka M; Miyamoto K; Muranaka A; Uchiyama M Org. Lett 2019, 21, 3392. [DOI] [PubMed] [Google Scholar]
- 70.Huo S In PATAI’S Chemistry of Functional Groups, Rappoport Z, Ed. Wiley-VCH: New York, 2016, 1. [Google Scholar]
- 71.Xu S; Negishi E Acc. Chem. Res 2016, 49, 2158. [DOI] [PubMed] [Google Scholar]
- 72.Ziegler K; Gellert H-G; Martin H; Nagel K; Schneider J Justus Liebigs Ann. Chem 1954, 589, 91. [Google Scholar]
- 73.Ziegler K; Gellert H-G; Zosel K; Holzkamp E; Schneider J; Söll M; Kroll W-R Justus Liebigs Ann. Chem 1960, 629, 121. [Google Scholar]
- 74.Eisch JJ In Comprehensive Organometallic Chemistry II, Vol. 11; Stone FGA; Wilkinson G, Eds.; Elsevier: Oxford, 1995, 277. [Google Scholar]
- 75.Eisch JJ; Burlinson NE; Boleslawski M J. Organomet. Chem 1976, 111, 137. [Google Scholar]
- 76.Bundens JW; Yudenfreund J; Francl MM Organometallics 1999, 18, 3913. [Google Scholar]
- 77.Eisch JJ; Hordis CK J. Am. Chem. Soc 1971, 93, 4496. [Google Scholar]
- 78.Egger KW J. Chem. Soc., Faraday Trans. 1 1972, 68, 1017. [Google Scholar]
- 79.Eisch JJ; Burlinson NE J. Am. Chem. Soc 1976, 98, 753. [Google Scholar]
- 80.Binger P; Schäfer H Tetrahedron Lett 1975, 16, 4673. [Google Scholar]
- 81.Richey HG; Kubala B; Smith MA Tetrahedron Lett 1981, 22, 3471. [Google Scholar]
- 82.Kinoshita H; Hirai N; Miura K J. Org. Chem 2014, 79, 8171. [DOI] [PubMed] [Google Scholar]
- 83.Kinoshita H; Ishikawa T; Miura K Org. Lett 2011, 13, 6192. [DOI] [PubMed] [Google Scholar]
- 84.Kinoshita H; Yaguchi K; Tohjima T; Hirai N; Miura K Tetrahedron Lett 2016, 57, 2039. [Google Scholar]
- 85.Eisch JJ; Fichter KC J. Am. Chem. Soc 1974, 96, 6815. [Google Scholar]
- 86.Eisch JJ; Amtmann R; Foxton MW J. Organomet. Chem 1969, 16, P55. [Google Scholar]
- 87.Eisch JJ; Sexsmith SR; Fichter KC J. Organomet. Chem 1990, 382, 273. [Google Scholar]
- 88.Mole T; Surtees JR Aust. J. Chem 1964, 17, 1229. [Google Scholar]