

Abstract
HIV-1 generates remarkable intra- and inter-host viral diversity during infection. In response to dynamic selective pressures of the host environment, HIV-1 will evolve distinct phenotypes – biological features that provide fitness advantages. The transmitted form of HIV-1 has been shown to require a high density of CD4 on the target cell surface (as found on CD4+ T cells) and typically uses CCR5 as a co-receptor during entry. This phenotype is referred to as R5 T cell-tropic (or R5 T-tropic); however, HIV-1 can switch to a secondary co-receptor, CXCR4, resulting in a X4 T cell-tropic phenotype. Macrophage-tropic (or M-tropic) HIV-1 can evolve to efficiently enter cells expressing low densities of CD4 on their surface (such as macrophages/microglia). So far only CCR5-using M-tropic viruses have been found. M-tropic HIV-1 is most frequently found within the central nervous system, and infection of the CNS has been associated with neurological impairment. It has been shown that interferon resistance phenotypes have a selective advantage during transmission, but the underlying mechanism of this is still unclear. During untreated infection, HIV-1 evolves under selective pressure from both the humoral/antibody response and CD8+ T cell killing. Sufficiently potent antiviral therapy will suppress viral replication, but if the antiviral drugs are not sufficiently potent to stop replication then the replicating virus will evolve drug resistance. HIV-1 phenotypes are highly relevant to treatment efforts, clinical outcomes, vaccine studies, and cure strategies. Therefore, it is critical to understand the dynamics of the host environment that drive these phenotypes and how they affect HIV-1 pathogenesis. This review will provide a comprehensive discussion of HIV-1 entry, transmission, and drug resistance phenotypes. Finally, we will assess the methods used in previous and current research to characterize these phenotypes.
Keywords: HIV-1, phenotype, T cell-tropic, Macrophage-tropic, coreceptor tropism, drug resistance, interferon resistance
1. Introduction
Viruses exist in two states: the extracellular virus particle and the intracellular replicating virus. The diversity of viral replication schemes and the myriad of ways viruses manipulate the host while replicating in the cell are fascinating topics and surprisingly rich in novel concepts given the deceptively small size of the typical viral genome. However, in this review we will focus on the virus particle, specifically the HIV-1 particle. The details of the particle are becoming well understood. HIV-1 shares the genes for the structural protein precursors (gag/Gag, env/Env) and the encoded enzymes (pro/PR, pol/RT, IN) with all retroviruses (and extending back to retrotransposons like Ty3, with the exception of env) (D’Souza and Summers, 2005). While the basic replication scheme and essential viral proteins are common among retroviruses, evolution has resulted in diversity even in these essential genes, for example retroviruses display remarkable diversity in the host cell receptor that is used to identify its target cell (Weiss and Tailor, 1995). The details of the role of each of these viral proteins in viral replication, and their locations in the assembled virion, can be found in many reviews and virology textbooks.
Our goal in this review is not to describe HIV-1 in the context of what all retroviruses do. Rather, our goal is to describe biological diversity that evolves within HIV-1 populations. HIV-1 represents what may be the most evolutionary dynamic genetic system in all of biology. This capacity for the virus to evolve existed long before we started selecting for drug resistance, which fully revealed the potential for rapid evolution. Our job is to see the diversity in viral phenotypes as a window into changing virus-host interactions. Specifically, we will review changes in viral phenotypes over the course of the infection as the relentless viral replication drives changes in the host, to which the virus responds through evolution toward a new optimum for viral replication. Fortunately, virus evolution ends once suppressive antiviral therapy is initiated. However, lots goes on if therapy is started late, and evolutionary events before the initiation of therapy are important considerations as we try to understand the nature of the latent reservoir that persists on therapy.
The virus goes through a predictable set of changes in phenotype if an infection progresses to depletion of CD4+ T cells. This predictable set of changes occurs because the same changes occur in the host, and the virus represents a large, rapidly replicating population that always finds its way to the improved version given the changed host (Coffin, 1995). These changes are most dramatic in what we will call the “entry” phenotype, changes in the use of the viral receptor (CD4) and coreceptor (CCR5 or CXCR4) as the virus changes it target cell for infection. While the virus can do this almost every time over the course of an infection, variation in the host population (i.e. us) is very different. HIV-1 entered humans about 100 years ago (Sharp and Hahn, 2011, Korber et al., 2000, Zhu et al., 1998) and less than 1% of the population has been infected (HIV/AIDS, 2021). Thus, there has not been enough time and/or selective pressure for the human population to evolve, so variation in the host that affects the outcome of infection is due to pre-existing features of human genetic variability. A few of these features become reflected in viral phenotypes and will be mentioned.
Untreated HIV-1 infection is a that leads to immunodeficiency and death. Potent, suppressive therapies have been developed that stop disease progression, and we must all be advocates for the equitable distribution of these life-saving therapies and to reduce stigma. We must also form partnerships with people at risk of HIV-1 to slow the rate of new infections. This review will largely focus on the virus but we would be remiss not to acknowledge both the suffering caused by the virus and the heroic role many people living with HIV have played in their participation as partners in research to generate the information that is discussed below. While we have used a light touch in the way we discuss this topic, our real goal is to provide an accurate view of HIV-1 that can help guide the next generation of questions as our field moves forward.
1.1. HIV-1 phenotypes
Much of the evolution of human intellectual history can be traced as misunderstanding the true nature of something due to available tools that give an incomplete view of reality. For example, in the future people will look back at us in bewilderment as we struggle to understand the nature of dark matter (even though it makes up a majority of the mass in our universe). The HIV-1 entry phenotype story has had its own jog in the road that caused the “wild type” form of the HIV-1 entry phenotype initially to be overlooked. We will briefly review the history of our evolving understanding of entry that has now largely been corrected. However, some of the downstream implications of the initially missing phenotypic form still appear in discussions and conceptualizations of pathogenesis. Thus, a major goal for us is to make these phenotypes unambiguous so that readers will be able to recognize whenever downstream concepts need to be challenged with a more appropriate framework of entry phenotype and by implication the target cell for replication. The viral env gene encodes the Env protein on the surface of the viral particle, and it is this protein that is responsible for interaction with the viral receptors. Thus, our discussion of entry phenotype is largely a discussion of the evolution of the viral Env protein.
Most of this review will focus on three entry phenotypes that HIV-1 can display and why and how they evolve. These three phenotypes and how they relate to each other are shown in Figure 1. The important point is that wild type HIV-1 is called R5 T cell-tropic, and two evolutionary variants that evolve during increasing immunodeficiency are X4 T cell-tropic and macrophage (M)-tropic. These three phenotypes reflect changing interactions with the viral primary receptor CD4 or a change in the choice of coreceptor, the coreceptor switch from using CCR5 to using CXCR4. Most of the review will focus on these three phenotypic variants that are associated with cell entry with each variant defining a different target cell. There are reports of HIV-1 and SIVs using different coreceptors but this will not be discussed as most of the biology of these viruses appears to be explained by CD4, CCR5, and CXCR4. We will discuss several settings where selective pressure changes the apparent fitness of the virus. While we may discuss the virus as being less fit, it is important to remember that fitness is maximized in the presence of the selective pressure the virus is experiencing. For example, drug resistant variants may be less fit than wild type virus in the absence of the drug but more fit in the presence of the drug (Deeks et al., 2001, Verhofstede et al., 1999).
Figure 1.
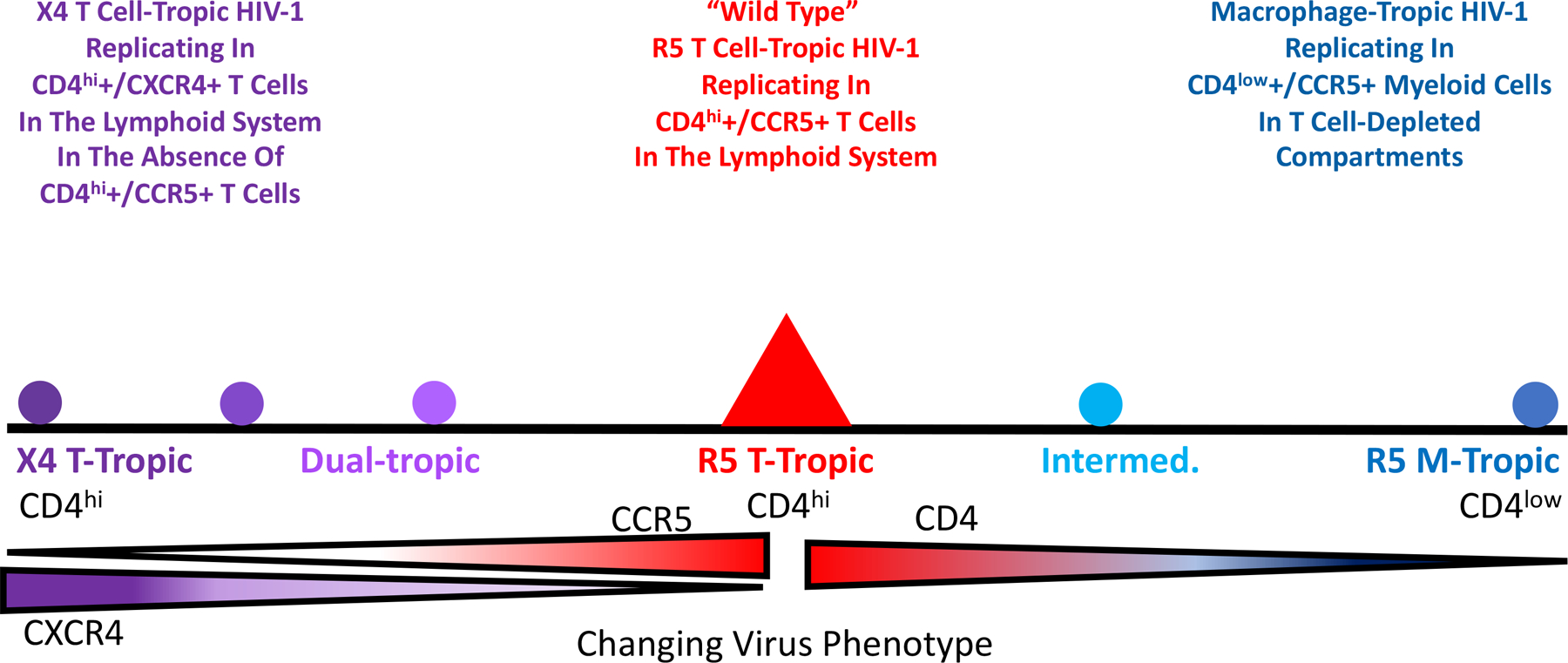
Change of HIV-1 virus entry phenotypes that are related to cell-type tropism and coreceptor tropism.
1.2. Why phenotypes are important to study
The Rosetta Stone was created around 200 BCE in Egypt to allow translations between Egyptian hieroglyphics and ancient Greek. All HIV-1 variants look the same when grown in primary CD4+ T cells, however when these variants (or their genes and gene products) are put into laboratory assays we find there are phenotypic variants. These assays become the Rosetta Stone that allows us to read the changing environment in the host that gave rise to the variant. The simple case is drug resistance where there is selection for a phenotypic variant that allows replication in the presence of the drug. This same concept can be applied to phenotypic variation that is the result of changes in the host environment. While drug resistance has the practical outcome of affecting therapy, other variants tell us about the biology of the host and the virus-host interaction. Phenotypic variation is important because it gives us insight into important features like transmission and pathogenesis. Our understanding of viral pathogenesis must explain the associated viral phenotypes; if pathogenesis and viral phenotypes don’t reinforce each other in our understanding then at a minimum our understanding is incomplete and, in some cases, potentially wrong.
2. Entry phenotypes
2.1. Discovery of entry phenotypes
If we had quickly come to understand the three HIV-1 entry phenotypes there would not be much of a history to tell. However, our understanding of entry phenotypes is a story that spans 20+ years. The original isolation of HIV-1 was done using cultures of peripheral blood nuclear cells (PBMCs)(Barré-Sinoussi et al., 1983), and it is fair to say that all isolates of HIV-1 will grow in PBMCs. An important discovery was that HIV-1 uses CD4 as its key component of the receptor (Dalgleish et al., 1984). However, at that time there was no understanding (or precedent) that HIV-1 might use two receptors. To make growing HIV-1 in the lab more convenient, there was an effort to find continuous cell lines that would support viral replication. It soon became possible to grow some strains of HIV-1 in CD4+ T cell lines (derived from T cell cancers). However, only some viral isolates would grow in these cell lines, but when the virus did grow the effect was visually distinctive (Asjö et al., 1986). As shown in Figure 2, the right combination of virus and cell line would lead to infected cells with the viral Env protein on the surface of the cell fusing with nearby uninfected cells to cause multinucleated cells, or syncytia. Isolates that could grow in these cells were labeled syncytium-inducing (SI) while those that did not induce syncytia (because they did not grow in the cells) were called non-syncytium-inducing (NSI) (Zhu et al., 1993). Further work showed that SI viruses were associated with advancing immunodeficiency (St Clair et al., 1993, Shankarappa et al., 1999, Tersmette et al., 1989). Given the growth in T cell lines, these SI viruses were also called T cell-tropic (or more accurately T cell line-tropic). We now understand that these T cell lines only expressed CXCR4 and not CCR5, but this was not yet part of the HIV-1 story.
Figure 2.
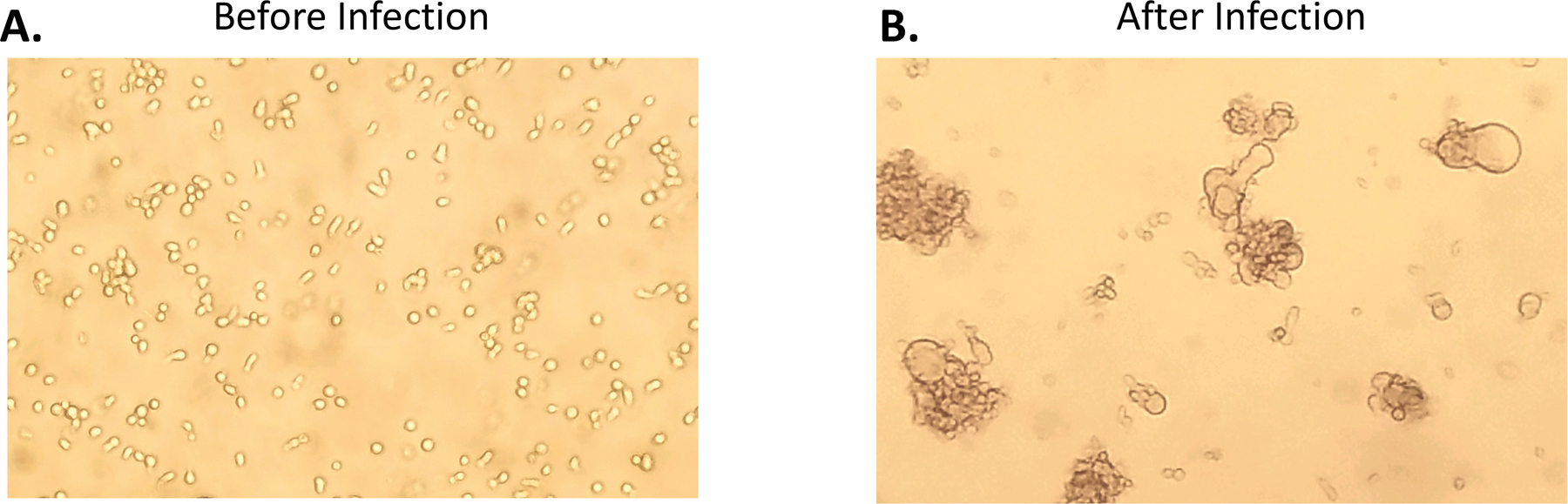
CEM174 cell line before after HIV-1 infection in vitro, indicating the formation of syncytium
Early on a virus isolate was generated after passage on macrophages securing the idea that HIV-1 could infect macrophage as well as CD4+ T cells (Gartner et al., 1986). This led to a test of NSI viruses, which could not grow in T cells lines, to see if as a group they could infect macrophages, which they could although with widely varying efficiency. Thus the NSI viruses as a group became macrophage-tropic (Zhu et al., 1993, van’t Wout et al., 1994).
The inability of all HIV-1 isolates to infect any cell that expressed CD4 indicated something was missing in our understanding of HIV-1 infectivity. This void in our understanding was filled with the discovery that HIV-1 required a coreceptor, with CXCR4 being discovered first (Feng et al., 1996) followed quickly by the discovery that CCR5 functioned as the coreceptor for most isolates (Alkhatib et al., 1996, Deng et al., 1996, Dragic et al., 1996). This quickly led to the insight that most T cell lines express CXCR4 but not CCR5. Adding the co-receptor specificity to the isolate name gave rise to X4 T cell-tropic (or X4 T cell line-tropic) isolates. That left the viruses using CCR5 as the NSI viruses and now the R5 macrophage-tropic viruses. It is important to note that this gives rise to two HIV-1 entry phenotypes, not the three we presented at the very beginning.
In the absence of antiviral treatment, which was the case early in the epidemic, most people progressed to “AIDS-defining illnesses”. One of these conditions was HIV-associated dementia (HAD) (Navia et al., 1986b, Navia et al., 1986a, Kaul et al., 2001, Ghafouri et al., 2006). This led to an interest in a “neuro-tropic” form of HIV-1 that could account for the dramatic clinical state of HAD. Viral genomes recovered from brain tissue at autopsy from some people who had suffered neurocognitive impairment prior to death had a distinctive property – they could infect cells with a low density of CD4 on the cell surface (such as macrophage) more efficiently than the viruses found in the blood (Martín-García et al., 2006, Dunfee et al., 2006, Duenas-Decamp et al., 2009, Schnell et al., 2011, Joseph et al., 2014, Joseph et al., 2015). Both types of viruses used CCR5 but, given the right assay, there were clearly two distinct phenotypic groups of viruses, the ones that could efficiently infect macrophages (appropriately called M-tropic) and the ones that only poorly infected macrophages and for which there was no name. While many people working with virus from the blood continued to think of the non-X4 viruses as R5 M-tropic virus, those working with virus from the brain came to understand that the virus in the blood was not M-tropic relative to the virus had evolved in the CNS environment of a subset of people.
It took the development of two tools to clearly place this unnamed form of HIV-1 as the wild type form of the virus, i.e. virus using CCR5 and requiring a high density of CD4 for efficient entry (as found on CD4+ T cells). The first tool was the development of methods to monitor the efficiency of infection as a function of CD4 density. As noted above, all “NSI” viruses can infect macrophages at some level but it is hard to account for differences in efficiency between different virus isolates. The ability to manipulate the level of CD4 on the cell surface either through transient transfection (Gorry et al., 2002, Thomas et al., 2007) or by creating cell lines with different levels of CD4 (Kabat et al., 1994, Platt et al., 1998, Peters et al., 2004) became an important tool for defining two entry phenotypes based on the cell surface density of CD4. The second tool was the application of end-point dilution PCR, also called SGS or SGA, to amplify individual viral genomes or genes to get genes without needing to culture virus, i.e. to get genes as they existed in vivo. When this was applied to the viral env gene (Salazar-Gonzalez et al., 2008), it became possible to isolate both types of phenotypic variants from the same person demonstrating that one was the evolutionary variant of the other (Schnell et al., 2011). In retrospect we can point to such isolates being created first by molecular cloning (Koyanagi et al., 1987, Li et al., 1991, Westervelt et al., 1991), but their true place in HIV-1 biology had to await the ability to use the more convenient PCR to get a large enough database to create generalizations. Today the most convenient entry assay is to use a cell line called Affinofile cells (Johnston et al., 2009, Joseph et al., 2014), which will be described below. The effect of this work was to define the third phenotypic variant which we have called R5 T cell-tropic and which in reality is the wild type form of HIV-1. The three phenotypic variants and their use of receptor and co-receptor are listed in Table 1. The corollary of the three phenotypes is that if you can’t demonstrate the presence of a virus with the M-tropic phenotype then it is unlikely that infection of macrophage (or monocytes or microglia) is a significant feature of the viral biology you are trying to understand. It is this concept that has been slow to spread through all of our beliefs of viral pathogenesis and latency. In Figure 3, we demonstrate a structure of the Env trimer binding to CD4 (bottom pink) and CCR5 (right pink). (Zhang et al., 2018) The CD4 binding site is shown in blue, and the partial V3 loop is shown in purple, with rest of the Env in green.
Table 1.
HIV-1 entry phenotypes by cell-type tropism and coreceptor tropism
| Target Cell type/CD4 tropism | |||
|---|---|---|---|
| CD4+ T Cell - High CD4 Density | Macrophage/Microglia - Low CD4 Density | ||
| coreceptor tropism | CCR5 | R5 T Cell-Tropic | R5 Macrophage-Tropic |
| CXCR4 | X4 T Cell-Tropic | none | |
Figure 3.
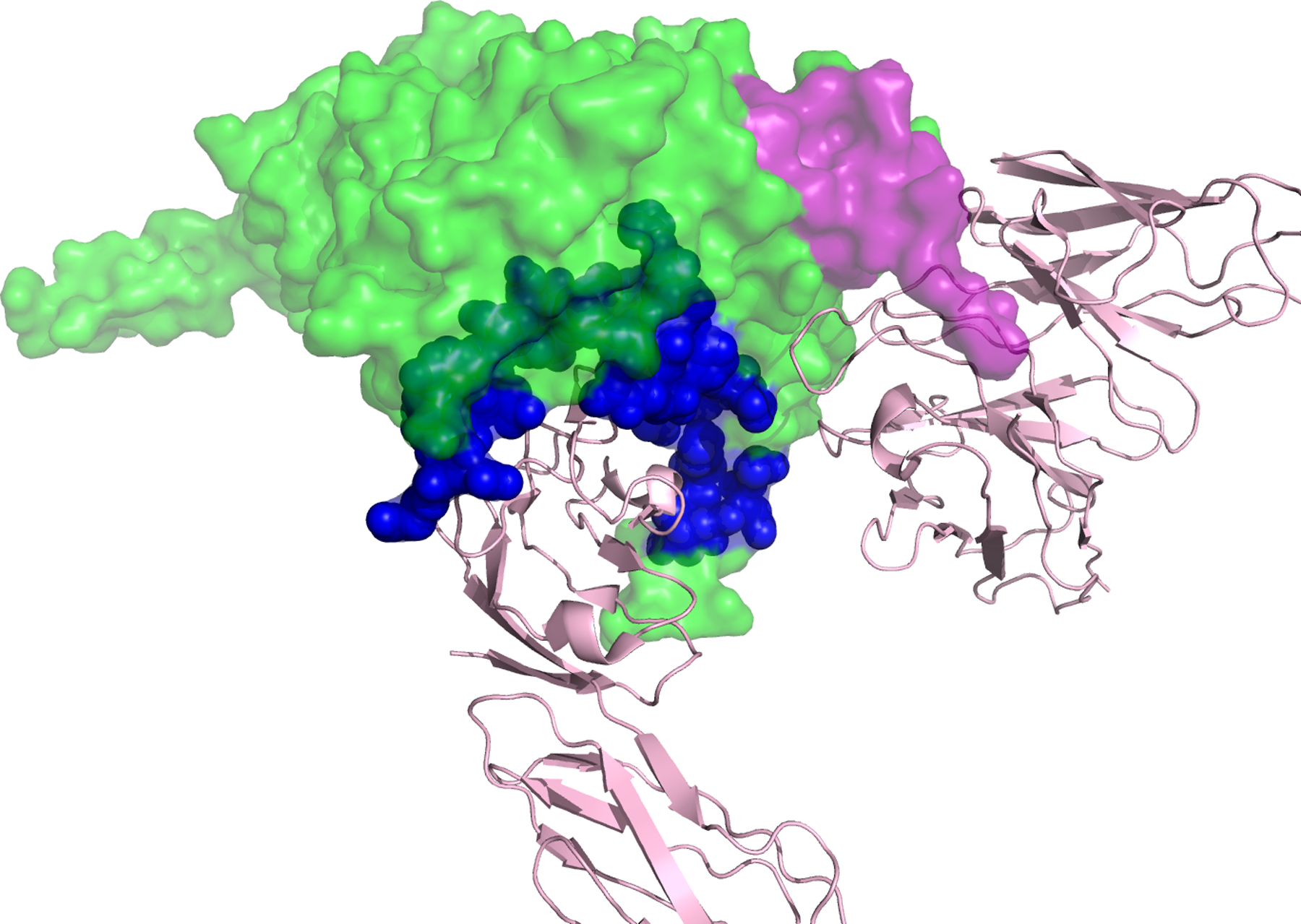
A structure of HIV-1 Env trimer binding to CD4 and CCR5. CD4 is shown in pink at the bottom. CCR5 is shown in pink to the right. CD4 binding site on Env is shown in blue, and the partial V3 loop is shown in purple. Of note, in this structure, only half of the V3 loop is solved.
2.2. Assay to detect the entry phenotypes
2.2.1. Assay For Entry Dependency On CD4 Density
We have used target cell names to identify different entry phenotypes, i.e. T cell-tropic and M-tropic. We would argue that from the point of view of viral evolution this is appropriate; in each case this is the cell type the virus is best adapted to. It is also true that while M-tropic viruses are adapted to entry using a low density of CD4, nothing keeps the virus from infecting cells with a high density of CD4, i.e. CD4+ T cells. Should M-tropic viruses be called dual-tropic for macrophages and T cells? We would argue no, it is the evolutionary step the virus has undergone to use a low density of CD4 that defines its most salient feature. Conversely, early confusion about macrophage tropism centered on the fact that all CCR5-using viruses could infect culture-differentiated monocyte-derived macrophages (MDM) with at least some detectable level of infection. When we compared paired T-tropic and M-tropic viruses from the person we found that on average there was a 25-fold difference in the efficiency of infection of MDMs between M- and T-tropic viruses (Arrildt et al., 2015). Again, it is this evolutionary step that defines the M-tropic variant. However, using MDMs is not the most convenient infection system where quantification is important for comparisons of infection efficiency. Since the basis for the M-tropic phenotype is the efficiency of CD4 use, the alternative to MDMs is to have a system where other variables are controlled and only the density of CD4 is manipulated to be able to assess infection efficiency at different CD4 levels. An initial approach used transfection to express CD4 at different levels. An alternative approach was to create stable cell lines that expressed different levels of CD4. We have found a cell system that has regulatable levels of surface CD4 to be a robust system to measure the macrophage tropism phenotype.
Ben-Hur Lee and his colleagues created a cell line that has both regulatable CD4 and CCR5, using different inducible transcription expression systems for each protein (Pugach et al., 2009, Johnston et al., 2009). The cell line, called Affinofile cells (for Affinity Profiling) has been especially useful for its inducible CD4 levels. At the highest induced level, the surface density of CD4 is slightly lower than that found on CD4+ T cells (“high”), while the uninduced basil level is slightly lower than that found on MDMs (“low”) (Lee et al., 1999, Joseph et al., 2014). It is possible to look at a dose-response of infectivity as a function of CD4 density, however a simpler assay is to look at the ratio of infectivity at high CD4 density to that at low CD4 density. A typical T-tropic virus will have a residual infectivity at low CD4 density of 1–2% of that at the high CD4 density. In contrast, an M-tropic virus will have residual infectivity at low CD4 density ranging from 10–40% that of the infectivity at high CD4 density (Joseph et al., 2014). It is relatively rare to find viruses with infectivities between 2–10% (Sturdevant et al., 2015), a phenotype we call intermediate M tropism, and these viruses deserve more study as to whether they are intermediates in the evolution of M tropism or have some other biological meaning. However, classifying viruses as T- and M-tropic using this system provides a robust phenotypic readout for most viruses. In this description we have referred to “viruses” when in reality this assay is most conveniently done using env gene expression vectors of viral env genes generated by end-point dilution PCR from in vivo material in a pseudotype strategy of a reporter virus genome (Joseph et al., 2014).
There are two other points to raise about the evolution of M-tropic virus. The Env protein on the surface of these viruses is especially sensitive to inactivation/neutralization with soluble CD4, a property that must ultimately be accounted for in understanding the mechanistic basis of M tropism (Arrildt et al., 2015). The other point is that while there is strong evidence that the viral Env protein has new properties that allow the efficient infection of macrophages, we do not know very much about how other viral proteins (or cis-acting genomic regions) may evolve to accommodate the change in target cell from T cells to macrophages.
2.2.2. Assay for Co-Receptor Tropism
There are three ways viruses are assessed to determine their coreceptor use: i) cells that express CD4 and either CCR5 or CXCR4, ii) infection of cells that express CD4 and both CCR5 and CXCR4 and test the sensitivity of infection with inhibitors maraviroc and/or AMD-3100, and iii) genotyping.
The coreceptor tropism can be determined using phenotypic or genotypic approaches. The phenotypic assays often involve PCR amplification of the env gene, molecular cloning and in vitro culture of pseudotyped virus. The MT-2 assay was the initial method to distinguish “SI” (X4) and “NSI” (R5) phenotypes. Patient-derived cells or viral isolates are co-cultured with the MT-2 cells, which express only CXCR4. The presence of X4 cells were determined by the forming of syncytia (Kootstra and Schuitemaker, 2005). This assay lacks a CCR5-only cell control, thus it cannot distinguish true negative results (R5 virus) from other factors preventing the infection of MT-2 cells. Several other recombinant or pseudovirus phenotypic assays were later developed to replace the MT-2 assays. In these assays, partial or whole env genome is amplified by PCR from viral RNA or DNA, followed by molecular cloning to make recombinant or pseudotyped viruses. These viruses are tested in CD4+ cells lines expressing either CCR5 or CXCR4 to determine the coreceptor phenotypes. Several of these assays have been used in clinical practice, such as the Trofile assay (Whitcomb et al., 2007). A more sophisticated phenotypic assay is to use SGA/SGS instead of bulk PCR to amplify HIV-1 env gene, followed by molecular cloning of each single genome isolated. Pseudotyped viruses can be tested in the presence of CCR5 inhibitor maraviroc (Fätkenheuer et al., 2005) and/or CXCR4 inhibitor AMD3100 (Donzella et al., 1998) to determine the coreceptor tropism (Zhou et al., 2016). Viruses that can be suppressed by CXCR4 inhibitor are considered as X4 virus. Viruses that can be suppressed in the presence of CCR5 inhibitor are considered as R5 virus. Viruses that can only be suppressed when both CCR5 and CXCR4 inhibitors are present in the cell culture are considered as dual-tropic. However, because SGA/SGS and molecular cloning are labor-intensive, this approach is reserved for research only (Butler et al., 2009).
In the same way drug resistance to HIV-1 therapeutics is monitored by sequence analysis, X4/R5 designation can be predicted through genotyping assays. The binding of gp120 and CD4 displaces the variable loop V1V2 and exposes the V3 loop, creating the co-receptor binding site. Thus, the V3 loop is the major determinant for the coreceptor tropism. However, in contrast with HIV-1 drug resistance variants, the genetic determination of HIV-1 coreceptor tropism cannot be simply explained by a few point mutations of the V3 sequence. Therefore, several bioinformatics approaches were developed to predict coreceptor tropisms based on the V3 sequences. The 11/25 charge rule is the simplest algorithm based on the amino acid change to basic amino acids at positions 11 and/or 25 of V3 loop. However, this assay only had a moderate correlation with the results from phenotypic assays (Low et al., 2007), with an Arg residue at position 25 being the least predictive. The Position-Specific Scoring Matrix (PSSM) assesses entire V3 sequences and forecasts X4 variants by evaluating the scoring of each amino acid residue within the V3 loop (Garrido et al., 2008). The Geno2Pheno algorithm (Lengauer et al., 2007) also analyzes the entire V3 sequence and provides a quantitative score (false positive rate, FPR) as the probability of predicting a R5 variant as a X4 variant by mistake. The FPR value ranges from 0% (has to be X4) to 100% (must be R5). The Geno2Pheno algorithm has gained increasing popularity among clinicians and researches in recent years, and it has been assessed in several clinical trials (Swenson et al., 2011a, Swenson et al., 2011b, Sanchez et al., 2011, McGovern et al., 2012, Kagan et al., 2014). This algorithm requires a preset FPR cut-off for calling X4 variants, which is usually determined by data from the CCR5 inhibitor Maraviroc clinical trials. However, the actual coreceptor tropism of variants with different FPR values has not been widely examined using phenotypic assays. Our data has shown that FPR values below 2% were reliably X4 while those above FPR 10% were reliably R5. However, for env genes with FPR scores between 2% and 10%, about half were X4 and half were R5 by phenotypic assessment (Zhou et al., 2016). This study had a relatively small sample size so the assessment of 50% is a rough estimate but it does show the significant limitation of picking a single FPR value to bin R5 and X4 viruses.
There is a curious feature of X4 T-tropic viruses, most of them can use either CCR5 or CXCR4 for entry; for this reason this group of viruses are often called dual-tropic. However, when we tested sensitivity to a CCR5 inhibitor for this group of viruses we found that they became increasingly sensitive (Zhou et al., 2016). We interpreted this result to mean that as viruses evolve to use CXCR4, they are becoming less efficient at interacting with CCR5 (and thus more sensitive to competition to binding CCR5 by maraviroc). X4 variants with the residual ability to use CCR5 just means that there has not yet been a mutation that enhances interaction with CXCR4 at the expense of any interaction with CCR5. From this perspective viruses that start using CXCR4 are doing so because of a lack of selective pressure to continue to use CCR5. For this reason, we regard the capacity to use CXCR4 as an indicator for an X4 virus, signifying its progression towards CXCR4 utilization with or without fixation of mutations that preclude CCR5 utilization. We believe that X4 viruses will all require a high density of CD4 for efficient entry, marking CD4+/CXCR4+ T cells as their target. However, the importance of CD4 density for X4 viruses needs to be examined with a larger sample size of isolates to be able to generalize this point.
2.3. Biology of viruses with different entry tropism
In our view, viruses with different entry phenotypic properties appear because of a changing environment in the host generating new selective pressures. Understanding what the phenotypic properties represent in terms of the changing selective pressures thus allows one to use the phenotypic properties of the virus to gauge the state of the host environment. For this discussion we will focus on M-tropic versus R5 T-tropic virus, then R5 T-tropic versus X4 T-tropic virus.
2.3.1. When and where are different HIV-1 phenotypes observed?
Transmission properties of M-tropic HIV-1 versus T-tropic HIV-1.
To be transmitted the virus has to appear in genital secretions (most HIV-1 transmission events occur through sex acts), and it must be able to establish and sustain a new infection in the recipient, initially at a mucosal site.
As noted above, the field of HIV-1 virology struggled with the concept of M-tropic versus R5 T-tropic virus as two different evolutionary states. As these two variants have become better understood and studied it has become possible to distinguish between them in different settings. Two studies carried out surveys of transmitted/founder (T/F) viruses and failed to find any T/F M-tropic viruses, in one case using infection of macrophages to characterize subtype B T/F viruses (Keele et al., 2008) and in the other case using Affinofile cells to characterize subtype C T/F viruses (Abrahams et al., 2009). In all cases, the T/F viruses were poor at infecting macrophages or poor at infecting cells that have a low density of CD4. This leads to the conclusion that M-tropic virus is rarely (if ever) transmitted.
Why aren’t M-tropic viruses transmitted? As noted below, M-tropic viruses are rarely found outside of the CNS and thus are unlikely to be present in genital secretions which would preclude them from being transmitted. While “unlikely” is an appropriate term it does not mean never; we observed a highly compartmentalized virus in semen of one man (i.e. a different genetic lineage compared to the virus in this person’s blood) and it turned out that this was an M-tropic virus (Bednar et al., 2015). While the M/T tropism of virus in semen and vaginal secretions has not been surveyed in the same way as T/F viruses, it seems likely this one case is the exception that proves the rule, i.e. M-tropic viruses are unlikely to be transmitted because they, in general, do not evolve in the genital tract.
The infection of macrophages in mucosal tissue at the site of transmission in the recipient is a topic of some interest. There is much we don’t know about initial infections that ultimately lead to a transmission event. Most of the time the infection is initiated by a single variant as viewed by the initial virus in the blood (Derdeyn et al., 2004, Keele et al., 2008, Abrahams et al., 2009). Are multiple cells infected at the site of transmission but the virus in one cell “wins out” to become the dominate transmitted virus? Are macrophages fortuitously infected by R5 T-tropic viruses that are otherwise inefficient at infecting cells at a low CD4 density? Is the initial infection of a CD4+ T cell specifically or alternatively a macrophage required to establish the subsequent systemic infection? These are questions that continue to be debated and studied. Finally, even if an M-tropic virus were transmitted it is possible that the selective pressures that cause R5 T-tropic virus to predominate in the systemic infection of lymphoid tissue would cause the M-tropic virus to revert to a virus that requires a high density of CD4 for efficient infection, i.e. to target CD4+ T cells.
Transmission properties of X4 T-tropic HIV-1 versus R5 T-tropic HIV-1.
While there is no evidence for transmission of M-tropic HIV-1, there are numerous reports of transmission of X4 T-tropic viruses. However, while there is significant interest in knowing the frequency with which X4 T-tropic viruses are transmitted in the population, there is an important methodological limitation in how we define X4 viruses and this impacts the quality of the transmission estimates that have been made.
Several studies have estimated the frequency of transmission of X4 viruses by examining T/F virus sequences. In one study (Keele et al., 2008) the phenotypic assessment was on a large number of env genes from T/F viruses which gave a value of 2% of T/F viruses being X4; a Geno2Pheno assessment of the sequences from these T/F viruses showed these phenotypically X4 T/F viruses all had a Geno2Pheno FPR less than 2% (Zhou et al., 2016). In another study (Chalmet et al., 2012), a total of 539 recently diagnosed HIV-1 positive individuals were genotyped for their coreceptor tropism. Using two FPR cut-off at 5.75% and 10%, a total of 12% and 19% participants were considered having X4 virus, respectively. We have to point out that the study population in this report were recently diagnosed individuals, many of whom could already have been at a relatively late stage of infection. Moreover, the use of these higher FPR values likely overestimated the true prevalence of X4 viruses in these samples. While there are other studies similar to these, the main point is that X4 viruses can be transmitted although the available data do not reveal whether they are transmitted at the frequency with which they appear in genital secretions or if they are transmitted with lower efficiency than R5 T cell-tropic viruses.
Presence of M-tropic HIV-1 versus T-tropic HIV-1 during asymptomatic infection.
Little is known about how or when M-tropic viruses start to evolve. M-tropic viruses have been detected most often in the CNS, including when they are being shed into the CSF. We examined people within the first two years of infection and found that a small fraction (10%) had compartmentalized virus in the CSF that had the ability to use a low density of CD4 marginally better than the virus in the blood (Sturdevant et al., 2015). We have referred to this type of virus as having an intermediate M-tropic phenotype. However, this study did not include longitudinal data to see if these intermediate viruses were direct precursors to M-tropic viruses.
It is a reasonable assumption that the evolution of M-tropic viruses requires four features of the host: i) reduced immune surveillance, such as in the CNS; ii) immunodeficiency that further compromises immune surveillance; iii) an absence of CD4+ T cells as the favored target of replication (again a feature of the CNS); and iv) the presence of myeloid cells that express at least a low level of CD4 on the surface. The CNS has reduced immune surveillance, plenty of myeloid cells (microglia and perivascular macrophages) and, in the absence of pleocytosis (i.e. an influx of inflammatory cells from the periphery to the CNS) only modest numbers of CD4+ T cells to support viral replication. It is likely that replication in terminally differentiated myeloid cells is slower/more difficult than in activated CD4+ T cells such that even a modest level of immunocompetence is able to suppress replication and thus preclude evolution of M-tropic virus. However, as a person approaches immunodeficiency, virus replication could take hold in myeloid cells and the evolutionary process would begin to increase entry efficiency with low CD4 density. There is a need for a large cross-sectional analysis of CSF from viremic people over the course of their asymptomatic infection to assess how often and when M-tropic virus evolves. As a corollary, it seems likely that detection of M-tropic virus in the periphery will be infrequent and thus anecdotal and probably linked to advanced states of immunodeficiency.
Presence of X4-tropic HIV-1 versus R5 T-tropic HIV-1 during asymptomatic infection.
Most CD4+ T cells have CXCR4 while only a subset has CCR5 (Bleul et al., 1997, Lee et al., 1999). Thus it seems odd that the virus, which is capable of using CXCR4 as a coreceptor, spends most of its time targeting CCR5 as the coreceptor. However, CCR5 has been reported to be a marker of activated T cells (Alkhatib et al., 1996) making it the better choice of a coreceptor if the goal is to replicate in a metabolically active cell. In this view, it is the loss of CD4+ CCR5+ T cells with advancing immunodeficiency that allows selection for replication in CD4+ T cells using CXCR4 as the coreceptor. As noted above, early work with HIV-1 isolates fortuitously used transformed T cell lines that expressed CD4 and CXCR4 but not CCR5. The X4 viruses that grew in these cells would induce syncytia and were called syncytia-inducing viruses (SI). The presence of these variants in people with decreasing CD4+ T cells (Shankarappa et al., 1999) links the appearance of X4 variants being associated with increasing immunodeficiency.
As noted above, X4 viruses can be observed as the transmitted virus. It is not clear if there is accelerated disease progression in people who are initially infected with an X4 virus or even if the virus reverts to an R5 virus as would normally be seen during the early phases of an infection. Another feature of the appearance of X4 variants is that they appear as distinct lineages that can co-exist with lineages of R5 viruses (Zhou et al., 2016). It is possible that the X4 viruses are coming from local environments (for instance, individual lymph nodes) where the CCR5+ CD4+ T cells were lost to a greater degree than other lymphoid tissues that are still producing R5 T-tropic virus.
There is evidence that the frequency of X4 viruses varies in different subtype lineages. It may be that the appearance of X4 viruses, compared to the subtype B lineage, is less frequent in the subtype C lineage and more frequent in the subtype D lineage (Parrish et al., 2012, Ping et al., 1999). This would be counter-intuitive if the appearance of X4 viruses were solely a function of advancing immunodeficiency of the host (and loss of CCR5+ CD4+ T cells in the host) since we would assume the lineages are mostly similar to each other in terms of their potential to induce pathogenesis. If these differences in the appearance of X4 lineages truly exist then it may be a difference in the virus that is a determinant. In looking for X4 viruses in subtype C variants we noted that they often include small deletions in the Env protein V3 loop, the portion of the Env protein that interacts with the coreceptor (Cilliers et al., 2003). It may be that the V3 loop conformation, which can interact with CXCR4, is more difficult to adopt in the subtype C virus Env protein, requiring a deletion in V3. Conversely, the subtype D Env protein V3 loop may be in a conformation (or may shift between conformations) that fortuitously allows interaction with CXCR4 while normally using CCR5 (Huang et al., 2007). In this case it would be that much easier to evolve and X4 virus for subtype D lineage viruses.
2.3.2. How do these entry phenotypes correlate with disease progression?
It is one thing to note that these variants can evolve to infect new cell types during the course of advancing immunodeficiency, however it is something else to ask the question of whether these variants contribute to disease progression. We will try to address this question below.
Pathogenesis associated with M-tropic HIV-1 versus T-tropic HIV-1.
There is a clear case to be made for the evolution of M-tropic virus and CNS pathogenesis. Often the virus in the CSF is present at 1–10% of the viral load in the blood and the two populations are well mixed (Peterson et al., 2014, Sturdevant et al., 2015). We consider this to be a default state of virus in the CNS, i.e. the release of virus from infected T cells that have wandered into the CNS. There is no evidence that this equilibrated viral population is undergoing any replication in the CNS, and thus is likely to be of little consequence to inducing pathogenesis in the CNS, as the virus is not adapted to enter myeloid cells that become the target in the CNS (Sturdevant et al., 2015). The one exception to this model is where there is pleocytosis (Spudich et al., 2005). When this happens the viral load in the CSF can increase but again this is likely due to the presence of infected T cells migrating into the CNS, although there is the possibility of amplification of virus in the population of CD4+ T cells that entered the CNS as part of an inflammatory response. While pleocytosis is elevated in its frequency in untreated people with HIV-1, it is not the normal state making the impact of R5 T cell-tropic virus on the CNS likely transitory.
In contrast to R5 T cell-tropic virus and the influx of inflammatory cells into the CNS, the M-tropic virus evolves to replicate within the CNS compartment. Little is known about how this process starts. We have observed the presence of compartmentalized viruses with an intermediate phenotype in CSF in a subset of people during early infection (Sturdevant et al., 2015). However, we do not know if these people are uniquely able to support an evolving M-tropic virus infection in the brain or if these early compartmentalized viruses disappear and true M-tropic virus lineages only appear late.
The most common place to find M-tropic virus is in the CNS late in disease. The end-point diseases in the CNS are viral encephalitis and HIV-associated dementia (HAD). When we examined the virus in the CSF of people diagnosed with HAD we found that about one-half of them had T-tropic virus and the other half had M-tropic virus (Schnell et al., 2011). It is unclear if the T-tropic virus was due to pleocytosis and was hiding an M-tropic virus infection deeper in the brain, which could make a link between M-tropic virus in the CNS and apparent disease. It is also possible that there are pleocytosis/inflammatory paths to CNS pathology that are distinct from M-tropic virus infection of brain macrophage/microglia as a pathogenic process. It is also not known what level of infection is needed to induce a CNS disease state. It is possible that the initial evolution of M-tropic virus in the CNS by itself does not induce a disease state but that this happens when the CNS infection reaches some type of a threshold level.
Pathogenesis associated with X4 T-tropic HIV-1 versus R5 T-tropic HIV-1.
Does disease progression accelerate because X4 viruses evolve, or does the appearance of X4 viruses simply represent reaching a certain stage of advanced immunodeficiency? This question has persisted since the earliest association of X4 viruses with increasing immunodeficiency. Presumably the selective pressure for the appearance of X4 viruses is the disappearance of CCR5+ CD4+ T cells. CXCR4 is much more widely distributed among CD4+ T cells than is CCR5. This implies that the virus has an advantage when replicating in CCR5+ CD4+ T cells, otherwise it would just be an X4 virus all the time. Thus we can infer that replication in CXCR4+ CD4+ T cells is suboptimal and selected against when there are CCR5+ CD4+ T cells available. To be provocative, this could mean that infection of non-CCR5+ T cells is less pathogenic with slower viral replication properties. This interpretation would favor a model where X4 viruses are a marker for immunodeficiency and accelerated disease progression, not causal. About 1% of northern Europeans carry a deletion in both alleles of the CCR5 gene making them resistant to infection with an R5 virus (Galvani and Novembre, 2005). However, such people can be infected with an X4 virus. When we studied one such person who was infected early in the epidemic, we noted that disease progression had been typical for an HIV-1 infection (Michael et al., 1998). It will be interesting to see if people infected with an X4 virus at transmission maintain that phenotype or revert to an R5 virus as the phenotype of virus that replicates best when CCR5+ CD4+ T cells are available. In a study of two individuals, one with CCR5wt/wt and the other with CCR5∆32/∆32, were infected with two X4 founder viruses. After approximately 4 years of infection, the HIV-1 population in the CCR5wt/wt individual reverted from 100% X4 to approximately 60% R5, but in the CCR5∆32/∆32 individual, the viral population remained as X4 virus over the disease course (Le et al., 2015).
3. Phenotypes Associated With Transmitted/Founder Viruses
3.1. Usually a single viral particle will establish an infection.
HIV-1 is primarily transmitted through sexual contact. This process is largely influenced by the physical barriers present during sexual transmission, which significantly limit the total number of viruses and genetic diversity of the viral population transmitted from the donor to the recipient (Derdeyn et al., 2004, Keele et al., 2008, Abrahams et al., 2009). Understanding the virological determinants that influence the success of HIV-1 transmission is important to understand for developing successful prevention strategies.
It is not possible to study the exact virus present during a transmission event that leads to a systemic infection. The alternative approach is to look at the earliest detectable virus (usually in the blood) and infer what its properties were at the time of transmission. In a study of 102 participants infected with subtype B HIV-1, sequence diversity in the viral env increased as a function of time from transmission (Keele’s paper). Extrapolating back to the time of transmission indicated that in most cases a single virus established the infection. Phenotyping experiments using pseudotypes of these Env sequences revealed that most of these viruses were R5 T-tropic, but a few R5/X4 dual-tropic viruses were identified as well. The assessment of R5 T-tropic was based on the fact that the viruses infected macrophages very poorly.
3.2. Features of the particle and viral genome.
Two minor phenotypes have been linked to T/F viruses. First, T/F viruses appear to be modestly under-glycosylated in the viral Env protein (Abrahams et al., 2009, Derdeyn et al., 2004). Typically this protein has around 30 N-linked glycosylation sites. The average among a population of T/F viruses is slightly below this, although the significance of this observation is not known. Another feature of the T/F virus that has been reported is that the virus is closer to the consensus sequence of the viral clade than is the diversity of the sequences seen in the donor viral population (Carlson et al., 2014). This observation implies there is a selection for fitness at some point in the transmission process, although whether this occurs in the donor in the genital tract or in the recipient during the first few rounds of replication is not known.
3.3. Interferon Resistance.
T/F viruses have the intriguing property of being interferon resistant relative to the virus in later stages of the infection. This observation was initially made using virus isolates (Fenton-May et al., 2013) and then extended to molecular clones of viral genomes made from longitudinal samples (Iyer et al., 2017). A longitudinal study in 26 HIV-infected individuals characterized IFN resistance over the course of infection (Gondim et al., 2021), and showed that, during untreated infection, IFN resistance is highly dynamic, with the highest levels of resistance occurring during early/acute infection and also late-stage disease progression associated with lower CD4 T cell counts. Following suppression of viral replication via. antiretroviral therapy, a treatment interruption study showed that viruses that successfully rebounded displayed equivalently heightened resistance to type-I IFNs compared to viruses present during early acute infection. Together, these data demonstrate HIV-1 variants with heightened resistance to type I IFNs and increased fitness may have a selective advantage during transmission events, may be more frequently transmitted, or more efficiently replicate during periods where the host antiviral capacity is highest.
One point that is not yet clear is whether the observed IFN resistance phenotype is a specific adaptation, or an indirect effect of the result of increased replicative fitness (Fenton-May et al., 2013, Deymier et al., 2015). More recent data from in vitro IFN resistance selection experiments have demonstrated that it is possible for T/F-like viruses (similar to those isolated from donor genital tracts) to evolve IFN-alpha 2 resistance independently of increased replicative capacity (Iyer et al., 2017). This observation was only seen with selection experiments against IFN-alpha 2, not IFN-β. Thus the nature of IFN-resistance remains a complex question but an important one in understanding the phenotype of the viruses that most readily establish a new infection. Of note, all these studies were based on R5-T tropic virus. The viruses in the brain may be under significantly different selective pressure adapting to replication in myeloid cells and under reduced humoral and T cell killing surveillance, although features of innate immunity may be prominent.
3.4. Phenotypic Variation Of T/F Viruses To A Broadly Neutralizing Antibody: The AMP Trials
Reducing transmission of HIV-1 is an important field of study predicated on blocking the T/F variant from establishing an infection. Antivirals have been shown to be effective in pre-exposure prophylaxis (Fonner et al., 2016, Riddell et al., 2018, McCormack et al., 2016, Van Damme et al., 2012). However, these multi-drug therapies are frequently taken as a daily regimen. Immune-based prevention using broadly neutralizing antibodies (bnAbs) present a potential longer-acting alternative to a daily oral regimens of antivirals. Previous work has demonstrated that people with HIV-1 can develop broadly neutralizing antibodies to the viral Env protein, although their own virus is resistant through escape via evolution. These broadly neutralizing antibodies cluster in their binding to a limited number of sites on the viral Env protein. Recently, an IgG1 bnAb targeting the CD4 binding site, VRC01, was tested as a way of preventing transmission when given intravenously in the Antibody Mediated Prevention trials (AMP) (Corey et al., 2021). The primary endpoint of the trial showed no difference in accrual of infections in the placebo arm or in either of the arms with two different doses of VRC01. However, a more nuanced analysis of the data revealed several important lessons. The first was that there was significant heterogeneity in the population of transmitted viruses across people who got infected with respect to sensitivity to VRC01. The second was that although heterogeneity had been anticipated, the levels of VRC01 needed to provide broad protection were significantly underestimated; the assay measuring viral neutralization with VRC01 made the antibody appear more potent than what it was capable of being in vivo. Third, when the shift in apparent sensitivity was accounted for then it was possible to observe that VRC01 did block the transmission of the most sensitive variants in the treated arms relative to the control arms, with a reduced frequency of incidence with a sensitive virus from 30% to 9%. Viral load was also lower at first detection in VRC01 treatment groups compared to placebo. Although VRC01 did not lower overall incidence of infection compared to control groups in the AMP trials, it neutralized viruses that were appropriately sensitive to it in vivo and significantly influenced the population of T/F viruses that could establish infection. Overall, the AMP trial demonstrated that antibody-based prevention can effectively neutralize T/F viruses, but also highlighted the inherent difficulties of assessing the amount of antibody needed to neutralize virus in vivo and accounting for HIV-1 diversity.
4. HIV-1 Phenotypes Related to Treatment/Resistance-Associated Mutations
4.1. The development of HIV-1 drug resistance phenotypes
After the discovery of HIV-1, several small-molecule nucleotide/nucleoside analogs were developed in the 1980s to treatment the infection. Zidovudine (AZT) was the first approved antiviral therapy for HIV-1 infection. AZT showed efficacy in the suppression of viral replication and restoration of CD4+ T cell counts. However, resistant phenotypes against AZT quickly emerged in patients with prolonged therapy (Rooke et al., 1989). Soon, resistant phenotypes against other antiretroviral drugs were discovered in in vitro cell culture and in viruses present in people taking the drugs. Treating patients with only one antiviral drug often led to treatment failure with drug resistant viral phenotypes. The strategy of using multiple antiviral drugs acting on different viral targets, i.e., the highly active antiviral therapy (HAART) was introduced in mid-1990s and it drastically changed the HIV-1 treatment strategies (Gulick et al., 1997). Successful HAART completely suppresses the viral replication, eliminates the chance for the virus to develop drug resistant phenotypes, restores the CD4+ T cell numbers and function, prevents opportunistic infections, and greatly prolongs the life expectancy of people living with HIV-1 (Collaboration, 2003, Lu et al., 2018, Collaboration, 2008).
Combination therapy significantly diminishes the likelihood of drug resistance evolving. First, a HAART regimen is typically composed of drugs that target different proteins involved in the HIV-1 replication cycle. Thus, multiple mechanisms must simultaneously fail for resistance to emerge against all the drugs in the treatment regimen. Furthermore, combination therapy has the capacity to completely suppress viral replication. When viral replication is halted, the generation of new variants is also effectively prevented. However, drug resistant variants can still occur with combination therapy due to insufficient level of drug exposure, often from poor adherence to HAART (Bangsberg et al., 2004). HIV-1 displays extensive genetic diversity due to several factors including the error-prone viral reverse transcriptase, a short viral replication cycle, selective pressure from the host immune system, and viral recombination when two or more viruses co-infect the same cell (Holmes et al., 1992, Perelson et al., 1996, Preston et al., 1988, Burke, 1997, Jung et al., 2002). HIV-1 shows a dynamic change in the genetic diversity within individual hosts as infection progresses, allowing for the selection of variants exhibiting resistant phenotypes against HIV-1 treatment. Insufficient levels of antivirals can select variants that confers an advantage in the presence of the drug, and these variants are more likely to persist and become dominant if the selective pressure from the drug continues. Eventually, the intra-host viral population becomes increasingly resistant to the therapy, resulting in treatment failure of the therapy (Clavel and Hance, 2004). In addition, if the individuals carrying these variants transmit the virus to others, the recipient can become infected with a founder variant that already carries the resistance mutation(s) (Supervie et al., 2010, Castro et al., 2013).
Recent studies have showed certain polymorphisms on HIV-1 Env protein may reduce the viral susceptibility to certain antivirals, including dolutegravir (DTG), without any resistance mutations on the pol gene after selection in vitro (Van Duyne et al., 2019, Hikichi et al., 2021). Alternative mechanisms of drug resistance beyond the target genes are still being studied. The prevalence of these mutations in people receiving ART, and their contribution to resistance and treatment failure needs to be carefully assessed.
4.2. HIV-1 resistance testing
HIV-1 resistance testing was initially conducted using phenotyping assays. Phenotyping assays require in vitro isolation of virus from the patient samples, molecular cloning of the HIV-1 genomes, and culturing pseudotyped virus in the presence of different antiviral drugs to test their replicative abilities (Bronze et al., 2012). However, this procedure is usually time and labor consuming. The genetic determinants of resistance were later discovered for the resistant variants against different antiviral drugs, and prediction algorithms have been developed to predict drug resistant phenotypes given the viral sequence using tools such as the Stanford HIV-1 Drug Resistance Database (Rhee et al., 2003). Maraviroc represents a special case as it only inhibits R5 viruses and is ineffective against X4 viruses. There are several genotype-to-phenotype prediction algorithms for X4 coreceptor tropism (discussed above) but none of them have 100% accuracy. Thus, clinical testing of maraviroc resistance (i.e., coreceptor tropism) sometimes needs to confirmed by phenotyping assay (Vandekerckhove et al., 2011). However, identifying resistance phenotypes is still relevant in the development of new antiretroviral drugs, as the genetic determinants are unknown. Researchers often combine the selection of resistant variants and viral genomic sequencing to identify the genetic determinants and potential resistance pathways to certain antivirals (Spielvogel et al., 2023).
4.3. Fitness cost of the resistance variants
Patients who failed antiviral therapy often have drug resistant variants in the viral population. When HAART is discontinued in the face of virus replication and drug resistance, it has been observed that wild-type virus can replace the variants that were resistant to the previous HAART regimen, evidence that the drug resistant variants are less fit than wild type in the absence of therapy. It was seen after 16 weeks when a protease inhibitor (PI)-based regimen was discontinued and variants that were resistant to the PI were replaced by wild-type variants that were sensitive to the previous antivirals (Deeks et al., 2001). In another study, wild-type virus replaced resistant variants against reserve transcriptase (RT) inhibitors and PI 2–8 weeks after the treatment interruption (Verhofstede et al., 1999). These findings suggest that variants with drug resistance mutations are associated with decreased viral replication fitness when the selective pressure from the treatment is removed. Viral fitness is the ability to survive and replicate in a given environment (Clementi and Lazzarin, 2010). Multiple studies have shown that the majority of the resistance mutations against HIV-1 antivirals are associated with reduced viral fitness, while some the presence of a subset of mutations allow the virus to maintain high fitness (Clavel et al., 2000, Menzo et al., 2003, Nijhuis et al., 2001). It was also observed that secondary/compensatory mutations could occur to variants with certain resistance mutations, particularly against PIs, increasing the viral fitness. Moreover, the variants with resistance and compensatory mutations could persist without the selective pressure from the previous PIs (Boeri et al., 2003). These data suggest that HIV-1 often develops resistance mutations in vitro and in vivo against antivirals at the cost of viral fitness. The decrease in viral fitness caused by specific drug resistance mutations could be leveraged in the development of HAART strategies.
Variants with K65R mutation on RT exhibit resistance to several nucleoside/nucleotide RT inhibitors (NRTIs) including tenofovir (TFV) and abacavir (Squires et al., 2003, Gulick et al., 2004, Gallant et al., 2004). However, variants with K65R have reduced viral fitness, and more importantly, increased susceptibility to AZT (Parikh et al., 2006). Thus, AZT could become a treatment option for those who develop K65R mutation. Another example is the RT mutation M184V. M184V is associated with the use of lamivudine (3TC) and emtricitabine (FTC). M184V reduces the viral susceptibility to 3TC/FTC by more than 200 fold (Boucher et al., 1993, Tisdale et al., 1993). However, M184V is associated with reduced viral fitness both in vitro and in vivo, and it increases the susceptibility to AZT and TFV by 2 fold. In a randomized study, participants with M184V mutation received 3TC monotherapy or complete treatment interruption (TI) for 48 weeks. The recovery of viral fitness was observed in the TI group after week 24 but not in the 3TC group. The de-selection of M184V mutation was observed starting from week 12, only in the TI group. Moreover, despite the continuing replication of HIV-1, participants in the 3TC group had better clinical and immunology outcomes than those in the TI group (Castagna et al., 2006, Gianotti et al., 2008). This result strongly suggested that HIV-1 variants with low viral fitness were also less pathogenic.
7. Funding
This work was supported by National Institute of Allergy and Infectious Diseases at the National Institutes of Health (R01-AI40970 to R. S.). These authors received infrastructure support from the UNC CFAR (P30-AI050410) and the UNC Lineberger Comprehensive Cancer Center (P30-CA016086).
Footnotes
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Abrahams MR, Anderson JA, Giorgi EE, Seoighe C, Mlisana K, Ping LH, Athreya GS, Treurnicht FK, Keele BF, Wood N, Salazar-Gonzalez JF, Bhattacharya T, Chu H, Hoffman I, Galvin S, Mapanje C, Kazembe P, Thebus R, Fiscus S, Hide W, Cohen MS, Karim SA, Haynes BF, Shaw GM, Hahn BH, Korber BT, Swanstrom R & Williamson C 2009. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J Virol, 83, 3556–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM & Berger EA 1996. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science, 272, 1955–1958. [DOI] [PubMed] [Google Scholar]
- Arrildt KT, Labranche CC, Joseph SB, Dukhovlinova EN, Graham WD, Ping LH, Schnell G, Sturdevant CB, Kincer LP, Mallewa M, Heyderman RS, Rie AV, Cohen MS, Spudich S, Price RW, Montefiori DC & Swanstrom R 2015. Phenotypic Correlates of HIV-1 Macrophage Tropism. J Virol, 89, 11294–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asjö B, Morfeldt-Månson L, Albert J, Biberfeld G, Karlsson A, Lidman K & Fenyö EM 1986. Replicative capacity of human immunodeficiency virus from patients with varying severity of HIV infection. Lancet, 2, 660–662. [PubMed] [Google Scholar]
- Bangsberg DR, Moss AR & Deeks SG 2004. Paradoxes of adherence and drug resistance to HIV antiretroviral therapy. Journal of Antimicrobial Chemotherapy, 53, 696–699. [DOI] [PubMed] [Google Scholar]
- Barré-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, Dauguet C, Axler-Blin C, Vézinet-Brun F, Rouzioux C, Rozenbaum W & Montagnier L 1983. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science, 220, 868–871. [DOI] [PubMed] [Google Scholar]
- Bednar MM, Hauser BM, Ping LH, Dukhovlinova E, Zhou S, Arrildt KT, Hoffman IF, Eron JJ, Cohen MS & Swanstrom R 2015. R5 Macrophage-Tropic HIV-1 in the Male Genital Tract. J Virol, 89, 10688–10692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleul CC, Wu L, Hoxie JA, Springer TA & Mackay CR 1997. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc Natl Acad Sci U S A, 94, 1925–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeri E, Gianotti N, Canducci F, Hasson H, Giudici B, Castagna A, Lazzarin A & Clementi M 2003. Evolutionary characteristics of HIV type 1 variants resistant to protease inhibitors in the absence of drug-selective pressure. AIDS Res Hum Retroviruses, 19, 1151–1153. [DOI] [PubMed] [Google Scholar]
- Boucher CA, Cammack N, Schipper P, Schuurman R, Rouse P, Wainberg MA & Cameron JM 1993. High-level resistance to (−) enantiomeric 2’-deoxy-3’-thiacytidine in vitro is due to one amino acid substitution in the catalytic site of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother, 37, 2231–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronze M, Steegen K, Wallis CL, De Wolf H, Papathanasopoulos MA, Van Houtte M, Stevens WS, De Wit TR & Stuyver LJ 2012. HIV-1 phenotypic reverse transcriptase inhibitor drug resistance test interpretation is not dependent on the subtype of the virus backbone. PLoS One, 7, e34708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke DS 1997. Recombination in HIV: an important viral evolutionary strategy. Emerg Infect Dis, 3, 253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler DM, Pacold ME, Jordan PS, Richman DD & Smith DM 2009. The efficiency of single genome amplification and sequencing is improved by quantitation and use of a bioinformatics tool. J Virol Methods, 162, 280–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson JM, Schaefer M, Monaco DC, Batorsky R, Claiborne DT, Prince J, Deymier MJ, Ende ZS, Klatt NR, Deziel CE, Lin TH, Peng J, Seese AM, Shapiro R, Frater J, Ndung’u T, Tang J, Goepfert P, Gilmour J, Price MA, Kilembe W, Heckerman D, Goulder PJ, Allen TM, Allen S & Hunter E 2014. HIV transmission. Selection bias at the heterosexual HIV-1 transmission bottleneck. Science, 345, 1254031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagna A, Danise A, Menzo S, Galli L, Gianotti N, Carini E, Boeri E, Galli A, Cernuschi M, Hasson H, Clementi M & Lazzarin A 2006. Lamivudine monotherapy in HIV-1-infected patients harbouring a lamivudine-resistant virus: a randomized pilot study (E-184V study). AIDS, 20, 795–803. [DOI] [PubMed] [Google Scholar]
- Castro H, Pillay D, Cane P, Asboe D, Cambiano V, Phillips A, Dunn DT, Resistance FTUCGOHD, Aitken C, Asboe D, Webster D, Cane P, Castro H, Chadwick D, Churchill D, Clark D, Collins S, Delpech V, Geretti AM, Goldberg D, Hale A, Hué S, Kaye S, Kellam P, Lazarus L, Leigh-Brown A, Mackie N, Orkin C, Rice P, Pillay D, Smit E, Templeton K, Tilston P, Tong W, Williams I, Zhang H, Zuckerman M, Greatorex J, Wildfire A, O’shea S, Mullen J, Mbisa T, Cox A, Tandy R, Hale T, Fawcett T, Hopkins M, Ashton L, Garcia-Diaz A, Shepherd J, Schmid ML, Payne B, Chadwick D, Hay P, Rice P, Paynter M, Clark D, Bibby D, Kaye S, Kirk S, Maclean A, Aitken C & Gunson R 2013. Persistence of HIV-1 Transmitted Drug Resistance Mutations. The Journal of Infectious Diseases, 208, 1459–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmet K, Dauwe K, Foquet L, Baatz F, Seguin-Devaux C, Van Der Gucht B, Vogelaers D, Vandekerckhove L, Plum J & Verhofstede C 2012. Presence of CXCR4-using HIV-1 in patients with recently diagnosed infection: correlates and evidence for transmission. J Infect Dis, 205, 174–184. [DOI] [PubMed] [Google Scholar]
- Cilliers T, Nhlapo J, Coetzer M, Orlovic D, Ketas T, Olson WC, Moore JP, Trkola A & Morris L 2003. The CCR5 and CXCR4 coreceptors are both used by human immunodeficiency virus type 1 primary isolates from subtype C. J Virol, 77, 4449–4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel F & Hance AJ 2004. HIV drug resistance. N Engl J Med, 350, 1023–1035. [DOI] [PubMed] [Google Scholar]
- Clavel F, Race E & Mammano F 2000. HIV drug resistance and viral fitness. Adv Pharmacol, 49, 41–66. [DOI] [PubMed] [Google Scholar]
- Clementi M & Lazzarin A 2010. Human immunodeficiency virus type 1 fitness and tropism: concept, quantification, and clinical relevance. Clin Microbiol Infect, 16, 1532–1538. [DOI] [PubMed] [Google Scholar]
- Coffin JM 1995. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science, 267, 483–489. [DOI] [PubMed] [Google Scholar]
- Collaboration ATC 2008. Life expectancy of individuals on combination antiretroviral therapy in high-income countries: a collaborative analysis of 14 cohort studies. Lancet, 372, 293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaboration C 2003. Determinants of survival following HIV-1 seroconversion after the introduction of HAART. Lancet, 362, 1267–1274. [DOI] [PubMed] [Google Scholar]
- Corey L, Gilbert PB, Juraska M, Montefiori DC, Morris L, Karuna ST, Edupuganti S, Mgodi NM, Decamp AC, Rudnicki E, Huang Y, Gonzales P, Cabello R, Orrell C, Lama JR, Laher F, Lazarus EM, Sanchez J, Frank I, Hinojosa J, Sobieszczyk ME, Marshall KE, Mukwekwerere PG, Makhema J, Baden LR, Mullins JI, Williamson C, Hural J, Mcelrath MJ, Bentley C, Takuva S, Gomez Lorenzo MM, Burns DN, Espy N, Randhawa AK, Kochar N, Piwowar-Manning E, Donnell DJ, Sista N, Andrew P, Kublin JG, Gray G, Ledgerwood JE, Mascola JR & Cohen MS 2021. Two Randomized Trials of Neutralizing Antibodies to Prevent HIV-1 Acquisition. N Engl J Med, 384, 1003–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’souza V & Summers MF 2005. How retroviruses select their genomes. Nature Reviews Microbiology, 3, 643–655. [DOI] [PubMed] [Google Scholar]
- Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF & Weiss RA 1984. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature, 312, 763–767. [DOI] [PubMed] [Google Scholar]
- Deeks SG, Wrin T, Liegler T, Hoh R, Hayden M, Barbour JD, Hellmann NS, Petropoulos CJ, Mccune JM, Hellerstein MK & Grant RM 2001. Virologic and immunologic consequences of discontinuing combination antiretroviral-drug therapy in HIV-infected patients with detectable viremia. N Engl J Med, 344, 472–480. [DOI] [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR & Landau NR 1996. Identification of a major co-receptor for primary isolates of HIV-1. Nature, 381, 661–666. [DOI] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Bibollet-Ruche F, Mokili JL, Muldoon M, Denham SA, Heil ML, Kasolo F, Musonda R, Hahn BH, Shaw GM, Korber BT, Allen S & Hunter E 2004. Envelope-constrained neutralization-sensitive HIV-1 after heterosexual transmission. Science, 303, 2019–2022. [DOI] [PubMed] [Google Scholar]
- Deymier MJ, Ende Z, Fenton-May AE, Dilernia DA, Kilembe W, Allen SA, Borrow P & Hunter E 2015. Heterosexual Transmission of Subtype C HIV-1 Selects Consensus-Like Variants without Increased Replicative Capacity or Interferon-α Resistance. PLoS Pathog, 11, e1005154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donzella GA, Schols D, Lin SW, Esté JA, Nagashima KA, Maddon PJ, Allaway GP, Sakmar TP, Henson G & Declercq E 1998. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nature Medicine, 4, 72–77. [DOI] [PubMed] [Google Scholar]
- Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP & Paxton WA 1996. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature, 381, 667–673. [DOI] [PubMed] [Google Scholar]
- Duenas-Decamp MJ, Peters PJ, Burton D & Clapham PR 2009. Determinants flanking the CD4 binding loop modulate macrophage tropism of human immunodeficiency virus type 1 R5 envelopes. J Virol, 83, 2575–2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunfee R, Thomas ER, Gorry PR, Wang J, Ancuta P & Gabuzda D 2006. Mechanisms of HIV-1 neurotropism. Curr HIV Res, 4, 267–278. [DOI] [PubMed] [Google Scholar]
- Fätkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, Saag MS, Goebel FD, Rockstroh JK & Dezube BJ 2005. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nature Medicine, 11, 1170–1172. [DOI] [PubMed] [Google Scholar]
- Feng Y, Broder CC, Kennedy PE & Berger EA 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science, 272, 872–877. [DOI] [PubMed] [Google Scholar]
- Fenton-May AE, Dibben O, Emmerich T, Ding H, Pfafferott K, Aasa-Chapman MM, Pellegrino P, Williams I, Cohen MS, Gao F, Shaw GM, Hahn BH, Ochsenbauer C, Kappes JC & Borrow P 2013. Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology, 10, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonner VA, Dalglish SL, Kennedy CE, Baggaley R, O’reilly KR, Koechlin FM, Rodolph M, Hodges-Mameletzis I & Grant RM 2016. Effectiveness and safety of oral HIV preexposure prophylaxis for all populations. AIDS, 30, 1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallant JE, Staszewski S, Pozniak AL, Dejesus E, Suleiman JM, Miller MD, Coakley DF, Lu B, Toole JJ & Cheng AK 2004. Efficacy and safety of tenofovir DF vs stavudine in combination therapy in antiretroviral-naive patients: a 3-year randomized trial. JAMA, 292, 191–201. [DOI] [PubMed] [Google Scholar]
- Galvani AP & Novembre J 2005. The evolutionary history of the CCR5-Δ32 HIV-resistance mutation. Microbes and Infection, 7, 302–309. [DOI] [PubMed] [Google Scholar]
- Garrido C, Roulet V, Chueca N, Poveda E, Aguilera A, Skrabal K, Zahonero N, Carlos S, García F, Faudon JL, Soriano V & De Mendoza C 2008. Evaluation of eight different bioinformatics tools to predict viral tropism in different human immunodeficiency virus type 1 subtypes. J Clin Microbiol, 46, 887–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC & Popovic M 1986. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science, 233, 215–259. [DOI] [PubMed] [Google Scholar]
- Ghafouri M, Amini S, Khalili K & Sawaya BE 2006. HIV-1 associated dementia: symptoms and causes. Retrovirology, 3, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianotti N, Tiberi S, Menzo S, Danise A, Boeri E, Galli L, Clementi M, Lazzarin A & Castagna A 2008. HIV-1 replication capacity and genotype changes in patients undergoing treatment interruption or lamivudine monotherapy. J Med Virol, 80, 201–208. [DOI] [PubMed] [Google Scholar]
- Gondim MVP, Sherrill-Mix S, Bibollet-Ruche F, Russell RM, Trimboli S, Smith AG, Li Y, Liu W, Avitto AN, Devoto JC, Connell J, Fenton-May AE, Pellegrino P, Williams I, Papasavvas E, Lorenzi JCC, Salantes DB, Mampe F, Monroy MA, Cohen YZ, Heath S, Saag MS, Montaner LJ, Collman RG, Siliciano JM, Siliciano RF, Plenderleith LJ, Sharp PM, Caskey M, Nussenzweig MC, Shaw GM, Borrow P, Bar KJ & Hahn BH 2021. Heightened resistance to host type 1 interferons characterizes HIV-1 at transmission and after antiretroviral therapy interruption. Sci Transl Med, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorry PR, Taylor J, Holm GH, Mehle A, Morgan T, Cayabyab M, Farzan M, Wang H, Bell JE, Kunstman K, Moore JP, Wolinsky SM & Gabuzda D 2002. Increased CCR5 affinity and reduced CCR5/CD4 dependence of a neurovirulent primary human immunodeficiency virus type 1 isolate. J Virol, 76, 6277–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, Mcmahon D, Richman DD, Valentine FT, Jonas L & Meibohm A 1997. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. New England Journal of Medicine, 337, 734–739. [DOI] [PubMed] [Google Scholar]
- Gulick RM, Ribaudo HJ, Shikuma CM, Lustgarten S, Squires KE, Meyer WA 3rd, Acosta EP, Schackman BR, Pilcher CD, Murphy RL, Maher WE, Witt MD, Reichman RC, Snyder S, Klingman KL & Kuritzkes DR 2004. Triple-nucleoside regimens versus efavirenz-containing regimens for the initial treatment of HIV-1 infection. N Engl J Med, 350, 1850–1861. [DOI] [PubMed] [Google Scholar]
- Hikichi Y, Van Duyne R, Pham P, Groebner JL, Wiegand A, Mellors JW, Kearney MF & Freed EO 2021. Mechanistic Analysis of the Broad Antiretroviral Resistance Conferred by HIV-1 Envelope Glycoprotein Mutations. mBio, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiv/Aids, J. U. N. P. O. 2021. Global HIV & AIDS statistics—Fact sheet UNAIDS: Geneva, Switzerland. [Google Scholar]
- Holmes EC, Zhang LQ, Simmonds P, Ludlam CA & Brown AJ 1992. Convergent and divergent sequence evolution in the surface envelope glycoprotein of human immunodeficiency virus type 1 within a single infected patient. Proc Natl Acad Sci U S A, 89, 4835–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Eshleman SH, Toma J, Fransen S, Stawiski E, Paxinos EE, Whitcomb JM, Young AM, Donnell D & Mmiro F 2007. Coreceptor tropism in human immunodeficiency virus type 1 subtype D: high prevalence of CXCR4 tropism and heterogeneous composition of viral populations. J Virol, 81, 7885–7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SS, Bibollet-Ruche F, Sherrill-Mix S, Learn GH, Plenderleith L, Smith AG, Barbian HJ, Russell RM, Gondim MV, Bahari CY, Shaw CM, Li Y, Decker T, Haynes BF, Shaw GM, Sharp PM, Borrow P & Hahn BH 2017. Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proc Natl Acad Sci U S A, 114, E590–e599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston SH, Lobritz MA, Nguyen S, Lassen K, Delair S, Posta F, Bryson YJ, Arts EJ, Chou T & Lee B 2009. A quantitative affinity-profiling system that reveals distinct CD4/CCR5 usage patterns among human immunodeficiency virus type 1 and simian immunodeficiency virus strains. J Virol, 83, 11016–11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SB, Arrildt KT, Sturdevant CB & Swanstrom R 2015. HIV-1 target cells in the CNS. J Neurovirol, 21, 276–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SB, Arrildt KT, Swanstrom AE, Schnell G, Lee B, Hoxie JA & Swanstrom R 2014. Quantification of entry phenotypes of macrophage-tropic HIV-1 across a wide range of CD4 densities. J Virol, 88, 1858–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung A, Maier R, Vartanian JP, Bocharov G, Jung V, Fischer U, Meese E, Wain-Hobson S & Meyerhans A 2002. Recombination: Multiply infected spleen cells in HIV patients. Nature, 418, 144. [DOI] [PubMed] [Google Scholar]
- Kabat D, Kozak SL, Wehrly K & Chesebro B 1994. Differences in CD4 dependence for infectivity of laboratory-adapted and primary patient isolates of human immunodeficiency virus type 1. J Virol, 68, 2570–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan RM, Johnson EP, Siaw MF, Van Baelen B, Ogden R, Platt JL, Pesano RL & Lefebvre E 2014. Comparison of genotypic and phenotypic HIV type 1 tropism assay: results from the screening samples of Cenicriviroc Study 202, a randomized phase II trial in treatment-naive subjects. AIDS Res Hum Retroviruses, 30, 151–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Garden GA & Lipton SA 2001. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature, 410, 988–994. [DOI] [PubMed] [Google Scholar]
- Keele BF, Giorgi EE, Salazar-Gonzalez JF, Decker JM, Pham KT, Salazar MG, Sun C, Grayson T, Wang S, Li H, Wei X, Jiang C, Kirchherr JL, Gao F, Anderson JA, Ping LH, Swanstrom R, Tomaras GD, Blattner WA, Goepfert PA, Kilby JM, Saag MS, Delwart EL, Busch MP, Cohen MS, Montefiori DC, Haynes BF, Gaschen B, Athreya GS, Lee HY, Wood N, Seoighe C, Perelson AS, Bhattacharya T, Korber BT, Hahn BH & Shaw GM 2008. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc Natl Acad Sci U S A, 105, 7552–7557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kootstra NA & Schuitemaker H 2005. Determination of cell tropism of HIV-1. Methods Mol Biol, 304, 317–325. [DOI] [PubMed] [Google Scholar]
- Korber B, Muldoon M, Theiler J, Gao F, Gupta R, Lapedes A, Hahn BH, Wolinsky S & Bhattacharya T 2000. Timing the ancestor of the HIV-1 pandemic strains. Science, 288, 1789–1796. [DOI] [PubMed] [Google Scholar]
- Koyanagi Y, Miles S, Mitsuyasu RT, Merrill JE, Vinters HV & Chen IS 1987. Dual infection of the central nervous system by AIDS viruses with distinct cellular tropisms. Science, 236, 819–822. [DOI] [PubMed] [Google Scholar]
- Le AQ, Taylor J, Dong W, Mccloskey R, Woods C, Danroth R, Hayashi K, Milloy MJ, Poon AFY & Brumme ZL 2015. Differential evolution of a CXCR4-using HIV-1 strain in CCR5wt/wt and CCR5∆32/∆32 hosts revealed by longitudinal deep sequencing and phylogenetic reconstruction. Scientific Reports, 5, 17607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B, Sharron M, Montaner LJ, Weissman D & Doms RW 1999. Quantification of CD4, CCR5, and CXCR4 levels on lymphocyte subsets, dendritic cells, and differentially conditioned monocyte-derived macrophages. Proc Natl Acad Sci U S A, 96, 5215–5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengauer T, Sander O, Sierra S, Thielen A & Kaiser R 2007. Bioinformatics prediction of HIV coreceptor usage. Nat Biotechnol, 25, 1407–1410. [DOI] [PubMed] [Google Scholar]
- Li Y, Kappes JC, Conway JA, Price RW, Shaw G & Hahn B 1991. Molecular characterization of human immunodeficiency virus type 1 cloned directly from uncultured human brain tissue: identification of replication-competent and-defective viral genomes. J Virol, 65, 3973–3985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low AJ, Dong W, Chan D, Sing T, Swanstrom R, Jensen M, Pillai S, Good B & Harrigan PR 2007. Current V3 genotyping algorithms are inadequate for predicting X4 co-receptor usage in clinical isolates. AIDS, 21, F17–24. [DOI] [PubMed] [Google Scholar]
- Lu D-Y, Wu H-Y, Yarla NS, Xu B, Ding J & Lu T-R 2018. HAART in HIV/AIDS treatments: future trends. Infectious Disorders-Drug Targets (Formerly Current Drug Targets-Infectious Disorders), 18, 15–22. [DOI] [PubMed] [Google Scholar]
- Martín-García J, Cao W, Varela-Rohena A, Plassmeyer ML & González-Scarano F 2006. HIV-1 tropism for the central nervous system: Brain-derived envelope glycoproteins with lower CD4 dependence and reduced sensitivity to a fusion inhibitor. Virology, 346, 169–179. [DOI] [PubMed] [Google Scholar]
- Mccormack S, Dunn DT, Desai M, Dolling DI, Gafos M, Gilson R, Sullivan AK, Clarke A, Reeves I, Schembri G, Mackie N, Bowman C, Lacey CJ, Apea V, Brady M, Fox J, Taylor S, Antonucci S, Khoo SH, Rooney J, Nardone A, Fisher M, Mcowan A, Phillips AN, Johnson AM, Gazzard B & Gill ON 2016. Pre-exposure prophylaxis to prevent the acquisition of HIV-1 infection (PROUD): effectiveness results from the pilot phase of a pragmatic open-label randomised trial. Lancet, 387, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcgovern RA, Thielen A, Portsmouth S, Mo T, Dong W, Woods CK, Zhong X, Brumme CJ, Chapman D, Lewis M, James I, Heera J, Valdez H & Harrigan PR 2012. Population-based sequencing of the V3-loop can predict the virological response to maraviroc in treatment-naive patients of the MERIT trial. J Acquir Immune Defic Syndr, 61, 279–86. [DOI] [PubMed] [Google Scholar]
- Menzo S, Monachetti A, Balotta C, Corvasce S, Rusconi S, Paolucci S, Baldanti F, Bagnarelli P & Clementi M 2003. Processivity and drug-dependence of HIV-1 protease: determinants of viral fitness in variants resistant to protease inhibitors. AIDS, 17, 663–671. [DOI] [PubMed] [Google Scholar]
- Michael NL, Nelson JA, Kewalramani VN, Chang G, O’brien SJ, Mascola JR, Volsky B, Louder M, White GC 2nd, Littman DR, Swanstrom R & O’brien TR 1998. Exclusive and persistent use of the entry coreceptor CXCR4 by human immunodeficiency virus type 1 from a subject homozygous for CCR5 delta32. J Virol, 72, 6040–6047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navia BA, Cho ES, Petito CK & Price RW 1986a. The AIDS dementia complex: II. Neuropathology. Ann Neurol, 19, 525–535. [DOI] [PubMed] [Google Scholar]
- Navia BA, Jordan BD & Price RW 1986b. The AIDS dementia complex: I. Clinical features. Ann Neurol, 19, 517–524. [DOI] [PubMed] [Google Scholar]
- Nijhuis M, Deeks S & Boucher C 2001. Implications of antiretroviral resistance on viral fitness. Current Opinion in Infectious Diseases, 14, 23–28. [DOI] [PubMed] [Google Scholar]
- Parikh UM, Bacheler L, Koontz D & Mellors JW 2006. The K65R mutation in human immunodeficiency virus type 1 reverse transcriptase exhibits bidirectional phenotypic antagonism with thymidine analog mutations. J Virol, 80, 4971–4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish NF, Wilen CB, Banks LB, Iyer SS, Pfaff JM, Salazar-Gonzalez JF, Salazar MG, Decker JM, Parrish EH, Berg A, Hopper J, Hora B, Kumar A, Mahlokozera T, Yuan S, Coleman C, Vermeulen M, Ding H, Ochsenbauer C, Tilton JC, Permar SR, Kappes JC, Betts MR, Busch MP, Gao F, Montefiori D, Haynes BF, Shaw GM, Hahn BH & Doms RW 2012. Transmitted/founder and chronic subtype C HIV-1 use CD4 and CCR5 receptors with equal efficiency and are not inhibited by blocking the integrin α4β7. PLoS Pathog, 8, e1002686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelson AS, Neumann AU, Markowitz M, Leonard JM & Ho DD 1996. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science, 271, 1582–6. [DOI] [PubMed] [Google Scholar]
- Peters PJ, Bhattacharya J, Hibbitts S, Dittmar MT, Simmons G, Bell J, Simmonds P & Clapham PR 2004. Biological analysis of human immunodeficiency virus type 1 R5 envelopes amplified from brain and lymph node tissues of AIDS patients with neuropathology reveals two distinct tropism phenotypes and identifies envelopes in the brain that confer an enhanced tropism and fusigenicity for macrophages. J Virol, 78, 6915–6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson J, Gisslen M, Zetterberg H, Fuchs D, Shacklett BL, Hagberg L, Yiannoutsos CT, Spudich SS & Price RW 2014. Cerebrospinal fluid (CSF) neuronal biomarkers across the spectrum of HIV infection: hierarchy of injury and detection. PLoS One, 9, e116081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping LH, Nelson JA, Hoffman IF, Schock J, Lamers SL, Goodman M, Vernazza P, Kazembe P, Maida M, Zimba D, Goodenow MM, Eron JJ Jr., Fiscus SA, Cohen MS & Swanstrom R 1999. Characterization of V3 sequence heterogeneity in subtype C human immunodeficiency virus type 1 isolates from Malawi: underrepresentation of X4 variants. J Virol, 73, 6271–6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B & Kabat D 1998. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J Virol, 72, 2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston BD, Poiesz BJ & Loeb LA 1988. Fidelity of HIV-1 reverse transcriptase. Science, 242, 1168–71. [DOI] [PubMed] [Google Scholar]
- Pugach P, Ray N, Klasse PJ, Ketas TJ, Michael E, Doms RW, Lee B & Moore JP 2009. Inefficient entry of vicriviroc-resistant HIV-1 via the inhibitor-CCR5 complex at low cell surface CCR5 densities. Virology, 387, 296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SY, Gonzales MJ, Kantor R, Betts BJ, Ravela J & Shafer RW 2003. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res, 31, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddell J, Amico KR & Mayer KH 2018. HIV preexposure prophylaxis: a review. JAMA, 319, 1261–1268. [DOI] [PubMed] [Google Scholar]
- Rooke R, Tremblay M, Soudeyns H, Destephano L, Yao XJ, Fanning M, Montaner JS, O’shaughnessy M, Gelmon K, Tsoukas C & et al. 1989. Isolation of drug-resistant variants of HIV-1 from patients on long-term zidovudine therapy. Canadian Zidovudine Multi-Centre Study Group. AIDS, 3, 411–415. [DOI] [PubMed] [Google Scholar]
- Salazar-Gonzalez JF, Bailes E, Pham KT, Salazar MG, Guffey MB, Keele BF, Derdeyn CA, Farmer P, Hunter E & Allen S 2008. Deciphering human immunodeficiency virus type 1 transmission and early envelope diversification by single-genome amplification and sequencing. J Virol, 82, 3952–3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez V, Masia M, Robledano C, Padilla S, Lumbreras B, Poveda E, De Mendoza C, Soriano V & Gutierrez F 2011. A highly sensitive and specific model for predicting HIV-1 tropism in treatment-experienced patients combining interpretation of V3 loop sequences and clinical parameters. J Acquir Immune Defic Syndr, 56, 51–8. [DOI] [PubMed] [Google Scholar]
- Schnell G, Joseph S, Spudich S, Price RW & Swanstrom R 2011. HIV-1 replication in the central nervous system occurs in two distinct cell types. PLoS Pathog, 7, e1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, Huang XL & Mullins JI 1999. Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J Virol, 73, 10489–10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PM & Hahn BH 2011. Origins of HIV and the AIDS pandemic. Cold Spring Harb Perspect Med, 1, a006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielvogel E, Lee SK, Zhou S, Lockbaum GJ, Henes M, Sondgeroth A, Kosovrasti K, Nalivaika EA, Ali A, Yilmaz NK, Schiffer CA & Swanstrom R 2023. Selection of HIV-1 for resistance to fifth-generation protease inhibitors reveals two independent pathways to high-level resistance. Elife, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spudich SS, Nilsson AC, Lollo ND, Liegler TJ, Petropoulos CJ, Deeks SG, Paxinos EE & Price RW 2005. Cerebrospinal fluid HIV infection and pleocytosis: relation to systemic infection and antiretroviral treatment. BMC Infectious Diseases, 5, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squires K, Pozniak AL, Pierone G Jr., Steinhart CR, Berger D, Bellos NC, Becker SL, Wulfsohn M, Miller MD, Toole JJ, Coakley DF & Cheng A 2003. Tenofovir disoproxil fumarate in nucleoside-resistant HIV-1 infection: a randomized trial. Ann Intern Med, 139, 313–20. [DOI] [PubMed] [Google Scholar]
- St Clair MH, Hartigan PM, Andrews JC, Vavro CL, Simberkoff MS & Hamilton JD 1993. Zidovudine resistance, syncytium-inducing phenotype, and HIV disease progression in a case-control study. The VA Cooperative Study Group. Journal of Acquired Immune Deficiency Syndromes, 6, 891–897. [PubMed] [Google Scholar]
- Sturdevant CB, Joseph SB, Schnell G, Price RW, Swanstrom R & Spudich S 2015. Compartmentalized replication of R5 T cell-tropic HIV-1 in the central nervous system early in the course of infection. PLoS Pathog, 11, e1004720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supervie V, García-Lerma JG, Heneine W & Blower S 2010. HIV, transmitted drug resistance, and the paradox of preexposure prophylaxis. Proc Natl Acad Sci U S A, 107, 12381–12386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson LC, Mo T, Dong WW, Zhong X, Woods CK, Jensen MA, Thielen A, Chapman D, Lewis M, James I, Heera J, Valdez H & Harrigan PR 2011a. Deep sequencing to infer HIV-1 co-receptor usage: application to three clinical trials of maraviroc in treatment-experienced patients. J Infect Dis, 203, 237–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swenson LC, Mo T, Dong WW, Zhong X, Woods CK, Thielen A, Jensen MA, Knapp DJ, Chapman D, Portsmouth S, Lewis M, James I, Heera J, Valdez H & Harrigan PR 2011b. Deep V3 sequencing for HIV type 1 tropism in treatment-naive patients: a reanalysis of the MERIT trial of maraviroc. Clin Infect Dis, 53, 732–42. [DOI] [PubMed] [Google Scholar]
- Tersmette M, Gruters RA, De Wolf F, De Goede RE, Lange JM, Schellekens PT, Goudsmit J, Huisman HG & Miedema F 1989. Evidence for a role of virulent human immunodeficiency virus (HIV) variants in the pathogenesis of acquired immunodeficiency syndrome: studies on sequential HIV isolates. J Virol, 63, 2118–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas ER, Dunfee RL, Stanton J, Bogdan D, Taylor J, Kunstman K, Bell JE, Wolinsky SM & Gabuzda D 2007. Macrophage entry mediated by HIV Envs from brain and lymphoid tissues is determined by the capacity to use low CD4 levels and overall efficiency of fusion. Virology, 360, 105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale M, Kemp SD, Parry NR & Larder BA 1993. Rapid in vitro selection of human immunodeficiency virus type 1 resistant to 3’-thiacytidine inhibitors due to a mutation in the YMDD region of reverse transcriptase. Proc Natl Acad Sci U S A, 90, 5653–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Damme L, Corneli A, Ahmed K, Agot K, Lombaard J, Kapiga S, Malahleha M, Owino F, Manongi R & Onyango J 2012. Preexposure prophylaxis for HIV infection among African women. New England Journal of Medicine, 367, 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Duyne R, Kuo LS, Pham P, Fujii K & Freed EO 2019. Mutations in the HIV-1 envelope glycoprotein can broadly rescue blocks at multiple steps in the virus replication cycle. Proc Natl Acad Sci U S A, 116, 9040–9049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van’t Wout AB, Kootstra NA, Mulder-Kampinga GA, Albrecht-Van Lent N, Scherpbier HJ, Veenstra J, Boer K, Coutinho RA, Miedema F & Schuitemaker H 1994. Macrophage-tropic variants initiate human immunodeficiency virus type 1 infection after sexual, parenteral, and vertical transmission. J Clin Invest, 94, 2060–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandekerckhove LP, Wensing AM, Kaiser R, Brun-Vézinet F, Clotet B, De Luca A, Dressler S, Garcia F, Geretti AM, Klimkait T, Korn K, Masquelier B, Perno CF, Schapiro JM, Soriano V, Sönnerborg A, Vandamme AM, Verhofstede C, Walter H, Zazzi M & Boucher CA 2011. European guidelines on the clinical management of HIV-1 tropism testing. Lancet Infect Dis, 11, 394–407. [DOI] [PubMed] [Google Scholar]
- Verhofstede C, Van Wanzeele F, Van Der Gucht B, De Cabooter N & Plum J 1999. Interruption of reverse transcriptase inhibitors or a switch from reverse transcriptase to protease inhibitors resulted in a fast reappearance of virus strains with a reverse transcriptase inhibitor-sensitive genotype. AIDS, 13, 2541–2546. [DOI] [PubMed] [Google Scholar]
- Weiss RA & Tailor CS 1995. Retrovirus receptors. Cell, 82, 531–533. [DOI] [PubMed] [Google Scholar]
- Westervelt P, Gendelman HE & Ratner L 1991. Identification of a determinant within the human immunodeficiency virus 1 surface envelope glycoprotein critical for productive infection of primary monocytes. Proc Natl Acad Sci U S A, 88, 3097–3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcomb JM, Huang W, Fransen S, Limoli K, Toma J, Wrin T, Chappey C, Kiss LD, Paxinos EE & Petropoulos CJ 2007. Development and characterization of a novel single-cycle recombinant-virus assay to determine human immunodeficiency virus type 1 coreceptor tropism. Antimicrob Agents Chemother, 51, 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Gorman J, Geng H, Liu Q, Lin Y, Tsybovsky Y, Go EP, Dey B, Andine T, Kwon A, Patel M, Gururani D, Uddin F, Guzzo C, Cimbro R, Miao H, Mckee K, Chuang GY, Martin L, Sironi F, Malnati MS, Desaire H, Berger EA, Mascola JR, Dolan MA, Kwong PD & Lusso P 2018. Interdomain Stabilization Impairs CD4 Binding and Improves Immunogenicity of the HIV-1 Envelope Trimer. Cell Host Microbe, 23, 832–844.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Bednar MM, Sturdevant CB, Hauser BM & Swanstrom R 2016. Deep Sequencing of the HIV-1 env Gene Reveals Discrete X4 Lineages and Linkage Disequilibrium between X4 and R5 Viruses in the V1/V2 and V3 Variable Regions. J Virol, 90, 7142–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T, Korber BT, Nahmias AJ, Hooper E, Sharp PM & Ho DD 1998. An African HIV-1 sequence from 1959 and implications for the origin of the epidemic. Nature, 391, 594–597. [DOI] [PubMed] [Google Scholar]
- Zhu T, Mo H, Wang N, Nam DS, Cao Y, Koup RA & Ho DD 1993. Genotypic and phenotypic characterization of HIV-1 patients with primary infection. Science, 261, 1179–1181. [DOI] [PubMed] [Google Scholar]