

Abstract
The gut microbiome can be thought of as a virtual organ given its immense metabolic capacity and profound effects on host physiology. Migratory birds are capable of adaptively modulating many aspects of their physiology to facilitate long-distance movements, raising the hypothesis that their microbiome may undergo a parallel remodeling process that helps to meet the energetic demands of migration.
To test this hypothesis, we investigated changes in gut microbiome composition and function over the fall migration of the Blackpoll Warbler (Setophaga striata), which exhibits one of the longest known autumnal migratory routes of any songbird and rapidly undergoes extensive physiological remodeling during migration.
Overall, our results showed that the Blackpoll Warbler microbiome differed significantly across phases of fall migration. This pattern was driven by a dramatic increase in the relative abundance of Proteobacteria, and more specifically a single 16S rRNA gene amplicon sequence variant belonging to the family Enterobacteriaceae. Further, Blackpoll Warblers exhibited a progressive reduction in microbiome diversity and within-group variance over migration, indicating convergence of microbiome composition among individuals during long-distance migration. Metagenomic analysis revealed that the gut microbiome of staging individuals was enriched in bacterial pathways involved in vitamin, amino acid, and fatty acid biosynthesis, as well as carbohydrate metabolism, and that these pathways were in turn positively associated with host body mass and subcutaneous fat deposits.
Together, these results provide evidence that the gut microbiome of migratory birds may undergo adaptive remodeling to meet the physiological and energetic demands of long-distance migration.
Keywords: 16S rRNA, birds, Blackpoll Warbler, gut microbiome, metagenomics, migration, passerine, physiology
Graphical Abstract
FAO Frank: Animal Physiological Ecology
INTRODUCTION
The microbial communities of the intestinal tract, known as the gut microbiome, can profoundly influence animal phenotypes through a myriad of effects on host physiology (e.g., development, digestion, immunity; McFall-Ngai et al., 2013). The composition of the gut microbiome is shaped by a variety of host factors (e.g., diet, environment, phylogeny; Ley et al., 2008a; Muegge et al., 2011; Rothschild et al., 2018), which can influence host phenotypes through effects on microbiome functions (Cho & Blaser, 2012; Clemente et al., 2012). However, the vast majority of research on the effect of the gut microbiome on host phenotypes has been derived from humans and model organisms under highly static conditions (Colston & Jackson, 2016; Pascoe et al., 2017). We now recognize that there is immense variation in host microbiota both within and between individuals, across habitats, and through time, and that these differences may reflect differences in host adaptive capacity and fitness in the wild (Alberdi et al., 2016; Suzuki, 2017). Therefore, studying the relationship between the gut microbiome and host phenotypes through space and time is essential to our understanding of wild vertebrate ecology and evolution (Moran et al., 2019; Trevelline, Fontaine, et al., 2019).
Animals faced with extreme energetic or physiological challenges are ideal natural models for investigating potentially adaptive microbiome-mediated host phenotypes. The gut microbiome of wild vertebrates can confer the ability for their hosts to tolerate seasonal temperature extremes (Fontaine et al., 2022), consume diets which would otherwise be toxic (Kohl et al., 2014, 2016), resist parasitic infections (Knutie et al., 2017), and balance nitrogen during fasting (Wiebler et al., 2018). Further, seasonal differences in host diet can trigger the remodeling of gut the microbiome that has been associated with host phenotypes such as intestinal epithelial structure, immune function, and energy metabolism (Carey & Assadi-Porter, 2017), though these relationships are not well understood. Many of these microbiome-mediated host phenotypes could influence vertebrate fitness in the wild, and thus adaptive microbiome functions may be favored by natural selection (Fontaine & Kohl, 2020; Groussin et al., 2017; Henry et al., 2021; Ley et al., 2008b; Lynch & Hsiao, 2019; Zilber-Rosenberg & Rosenberg, 2008).
Neotropical-Nearctic migratory birds annually complete some of the most energetically-demanding long-distance movements in the animal kingdom and can adaptively modulate aspects of their physiology to meet the extreme energetic demands of migration (McWilliams & Karasov, 2001). At the conclusion of a months-long stationary breeding period, migrants begin depositing protein (in the form of muscle and organ tissue) and fat, which provide the primary sources of metabolic energy for long-distance migration (Jenni & Jenni-Eiermann, 1998). In general, migration is initially characterized by the depletion of these energy reserves during short overnight flights followed by daytime ‘refueling’ of subcutaneous fat stores and recovery of muscle and organ tissue at intermediate “stopover” locations (McWilliams & Karasov, 2001; Schwilch et al., 2002). Many species of migratory birds repeat this depletion-recovery cycle of energy reserves at stopover sites until they reach a natural impedance (e.g., ocean, desert) where they are faced with either the extreme challenge of a multi-day, non-stop flight over thousands of kilometers without food, water, or rest, or a longer over-land journey to circumvent the impedance, where they risk higher predation risk. To overcome the energetic challenge of crossing the impedance, some migrants undergo a weeks-long form of stopover known as ‘staging’ – a period characterized by extreme hyperphagia, increased muscle and digestive system mass, and a rapid accumulation of subcutaneous fat stores in the weeks prior to long bouts of flight (Warnock, 2010). The ability to adaptively modulate phenotypes that enhance migratory performance is a unifying characteristic among migratory birds (McWilliams & Karasov, 2001; Piersma, 1998), and therefore is considered a key adaptation in the evolution of the migration (Alerstam et al., 2003).
The gut microbiome can be thought of as a virtual organ given its immense metabolic capacity and profound effects on host physiology (O’Hara & Shanahan, 2006). This raises the possibility that, like other host organs, the microbiome of migratory birds undergoes a potentially adaptive remodeling that helps to meet the energetic demands of long-distance migration. While limited to just a few descriptive studies, previous work has demonstrated that gut microbiome community composition of migratory birds differs between breeding and wintering habitats (Skeen et al., 2021; Wu et al., 2018), and is responsive to the timing and duration of migratory stopover (Lewis et al., 2016, 2017; Thie et al., 2022). Further, these studies have demonstrated that migration can lead to the enrichment of certain microbial taxa (e.g., Corynebacteria and Proteobacteria; Risely et al., 2017, 2018; Skeen et al., 2021; Turjeman et al., 2020). Importantly, the functional capacity of the microbiome is directly related to its composition (Burke et al., 2011), and thus these findings suggest that the demands of migration exert strong selective pressures leading to an adaptive enrichment of microbiome functions that help the host overcome the extreme energetic demands of long-distance migration. However, whether variation in microbiome composition over space and time is associated with the enrichment of microbial functions that could enhance host energy metabolism remains unknown.
In this study, we investigated changes in gut microbiome composition and function over the fall migration of a Neotropical-Nearctic migratory passerine, the Blackpoll Warbler (Setophaga striata). To test the hypothesis that the gut microbiome is adaptively remodeled to meet the energetic demands of migration, we characterized changes in gut microbiome composition and function from fecal samples collected at multiple sites within each distinct phase of the Blackpoll Warbler’s North American fall migratory route. We predicted that the gut microbiome of staging individuals would be enriched in microbial pathways that could enhance energy metabolism during long-distance migration, and that these pathways would be associated with metrics of energetic condition.
MATERIALS AND METHODS
Study species and sample collection
Each fall, Blackpoll Warblers complete one of the longest known migratory routes of any songbird (~20,000 km), from breeding sites in western North America to wintering habitats in northern South America (Fig. 1; DeLuca et al., 2015, 2019, 2020). Blackpoll Warbler migration from breeding sites in western Canada begins following a months-long stationary breeding phase and is initially characterized by short overnight flights to inland stopover sites where they recuperate their energy reserves (DeLuca et al., 2020; Morris et al., 2016). This process is repeated over several weeks as Blackpoll Warblers progress southeast across North America to the Atlantic Ocean, where they are faced with a non-stop transoceanic migration (up to 3,500 km over 88 hours) to wintering sites in South America (DeLuca et al., 2015, 2019, 2020; Williams et al., 1978). To successfully complete this energetically-demanding feat, Blackpoll Warblers along the Atlantic coast undergo a weeks-long process known as pre-migration staging characterized by an extensive physiological transformation that includes a rapid increase in body mass and subcutaneous fat deposits (DeLuca et al., 2020), which are important metrics of energetic condition in this species (Smetzer et al., 2017).
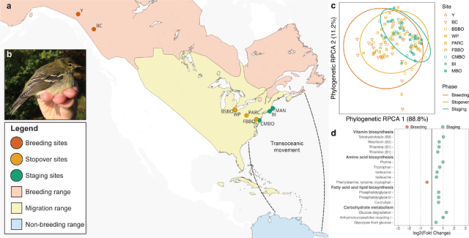
Figure 1. Sampling locations across phases of Blackpoll Warbler fall migration.
(a) Points indicate sampling locations over three phases of Blackpoll Warbler fall migration. Breeding (red points; n = 17): Yukon (Y; n = 2) and British Columbia (BC; n = 15); stopover (orange points; n = 58): Black Swamp Bird Observatory (BSBO; n = 12), Winous Point (WP; n = 8), Powdermill Avian Research Center (PARC; n = 19), and Foreman’s Branch Bird Observatory (FBBO; n = 19); Staging (green points; n = 41): Cape May Bird Observatory (CMBO; n = 15), Block Island (BI; n = 6), Manomet Bird Observatory (MBO; n = 20). (b) Staging adult Blackpoll Warbler (Setophaga striata). Blackpoll Warbler range map provided by BirdLife International and Birds of the World. Blackpoll Warbler photo provided by Manomet Bird Observatory.
In collaboration with several research groups and bird banding stations across North America, Blackpoll Warbler fecal samples were collected for microbiome analysis in 2018 and 2019 at multiple locations within three distinct phases of fall migration: breeding (n = 17), migratory stopover (n = 58), and pre-migration staging (n = 41; Fig. 1; Supplemental Data S1). Importantly, previous work has shown that the microbiota of avian feces provide a reliable proxy for the bacterial communities of the lower digestive tract, but does not necessarily represent the microbiome as a whole due to spatial heterogeneity in microbial abundances along the intestine (Videvall et al., 2018; Yan et al., 2019). Fecal samples collected from breeding Blackpoll Warblers in western Canada were categorized as “breeding” (Yukon and British Columbia). Fecal samples collected at inland sites (>100 km from the Atlantic coast) were categorized as “stopover” (Ohio, Pennsylvania, and Maryland, USA), whereas those collected along the US Atlantic coast (New Jersey, Massachusetts, and Rhode Island, USA) were categorized as “staging” (DeLuca et al., 2015; Morris et al., 2016; Warnock, 2010). Stopover and staging sites were selected based on previous work showing that these locations are used by Blackpoll Warblers migrating from breeding sites in western Canada (Fig. 1; DeLuca et al., 2019). Captured birds were placed in a clean paper bag for approximately 10 minutes or until defecation, after which fecal samples were transferred to vials of Queen’s lysis buffer using sterile swabs. All fecal samples were kept frozen at −20°C in the field for less than one month before long-term laboratory storage at −80°C. Birds were aged, sexed, weighed (to nearest 0.1g), and measured for subcutaneous fat deposits (scored 0–8; Kaiser, 1993) and unflattened wing chord (to nearest 1mm). Energetic condition while controlling for structural size, as measured by wing chord, was calculated using a Scaled Mass Index (SMI) following Peig & Green (2009). The collection of Blackpoll warbler fecal samples and morphometric data were conducted under the appropriate field and/or animal research permits (Supplemental Data S2).
16S rRNA amplicon profiling
DNA was extracted from Blackpoll Warbler fecal samples using the Qiagen PowerFecal DNA Kit (Qiagen, Hilden, Germany; 12830) before amplification and sequencing by the Genome Research Core of the University of Illinois at Chicago as previously described (Trevelline, MacLeod, et al., 2019). Briefly, polymerase chain reaction (PCR) was used to amplify a portion of the bacterial 16S rRNA gene for Illumina sequencing using the Earth Microbiome Project primers 515F and 806R (Caporaso et al., 2011) targeting the V4 region of microbial small subunit ribosomal RNA gene. Amplicon libraries were sequenced using a 2×251 paired-end run on an Illumina MiSeq. Additionally, we also sequenced 2 lysis-buffer immersed swab controls and 10 ‘blank’ extractions (using only kit reagents with no samples or swabs added) to control for microbial DNA found in commercial extraction kits (Salter et al., 2014). A total of 4,582,950 raw Illumina sequencing reads (mean of 32,274 per sample [n = 142] ± 3,679 SE) were trimmed, paired, and quality filtered using default parameters in the DADA2 pipeline (Callahan et al., 2016) implemented in QIIME 2 (version 2021.11; Bolyen et al., 2019). Sequences that passed the quality filter were denoised into 1,191 unique 16S rRNA gene amplicon sequence variants (ASVs), which were identified using the SILVA reference database (release 138; Quast et al., 2013). Identified ASVs were filtered to exclude sequences of non-bacterial origin (archaea, plants, eukaryotes, and mitochondria) and sequences that could not be classified beyond the domain level. Sequences were then passed to the R package Decontam (Davis et al., 2018) for the statistical identification and removal of 23 contaminant ASVs found in both control extractions and fecal samples. Samples with fewer than 100 sequences were excluded from our analysis (n = 10), resulting in a final total of 3,389,606 reads (74% of input reads; mean of 29,221 per sample [n = 116] ± 3,416 SE) and 1,057 ASVs. All downstream analyses were conducted on unrarefied data (McMurdie & Holmes, 2014).
Shotgun metagenomics
Extracted fecal DNA from a subset of breeding (Yukon and British Columbia; n = 8) and staging Blackpoll Warblers (Block Island; n = 12) were also sequenced with shotgun metagenomics by CoreBiome, Inc. (St. Paul, MN). Samples were selected to compare metagenomics profiles between high fat (8; n = 8) or no fat (0; n = 12) individuals (Kaiser, 1993). Briefly, sequencing libraries were prepared using a procedure adapted from the Illumina Nextera Library Prep Kit (Illumina, 20018705) and sequenced on an Illumina NovaSeq using single-end 1×100 reads with the Illumina NovaSeq SP reagent kit (Illumina, 20027464). A total of 115,289,944 raw sequence reads (mean of 5,764,497 per sample (n = 20) ± 1,117,040 SE) were filtered for low quality (Q-Score < 20), trimmed of adapter sequences using cutadapt (version 2.10). To reduce the possibility of DNA contamination from hosts, reads were then mapped against the Yellow-rumped Warbler genome (Setophaga coronata coronata; assembly mywa_2.1), the closest relative of the Blackpoll Warbler with a fully sequenced genome, as well as the human genome (assembly GRCh38.p13) using bowtie2 (version 2.4.3). Reads that did not align to either genome were profiled for microbial functions using HUMAnN 3 (HMP Unified Metabolic Analysis Network; Beghini et al., 2021) and the UniRef50 database (Suzek et al., 2015).
Statistical analysis
Differences in metrics of microbiome alpha diversity across phases of migration (Chao1 ASV richness and Shannon diversity) were assessed using mixed effects models with random effects for age, sex, and collection site (where applicable) with Tukey HSD correction in JMP Pro version 17.0 (SAS Institute Inc., Cary, NC). Phylogenetic Robust Aitchison PCA (Martino et al., 2019; hereafter, phylogenetic RPCA) was used to test for differences in microbiome community composition across phases of migration with permutational multivariate analysis of variance (PERMANOVA) and permutational analysis of dispersion (PERMDISP) in QIIME 2. Pairwise geographic distances between collection sites were calculated using the R package swaRm (version 0.6) before testing for significant association with pairwise mean phylogenetic RPCA distances using linear models in JMP. ASV count tables were converted into relative abundances to identify differentially abundant bacterial taxa across migration phases and metrics of avian energetic condition (SMI and fat scores). Differentially-abundant taxa were identified using a mixed-model approach in MaAsLin2 (Microbiome Multivariable Associations with Linear Models; Mallick et al., 2021; analysis_method = “CPLM” [Compound Poisson Linear Model], transform = “AST” [Arcsine Transformation]) with age, sex, and collection site as random effects and corrected for multiple comparisons using False Discovery Rate (FDR). Significant associations between migration phase, SMI, and fat score with HUMAnN 3.0 gene pathway abundances were identified using MaAsLin2 with negative binomial model (minimum prevalence = 0.5) and FDR corrections on cumulative sum scaled data. Heatmaps of HUMAnN 3 bacterial pathways that were significantly associated with migration phase, SMI, and fat score were generated in the R package pheatmap (version 1.0.12) using log10(pathway abundance +1) and clustered using Ward’s minimum variance as implemented in the hclust() function in R. For all statistical analyses, P/Q-values ≤ 0.05 were defined as ‘significant’.
RESULTS
Gut microbiome community composition and structure
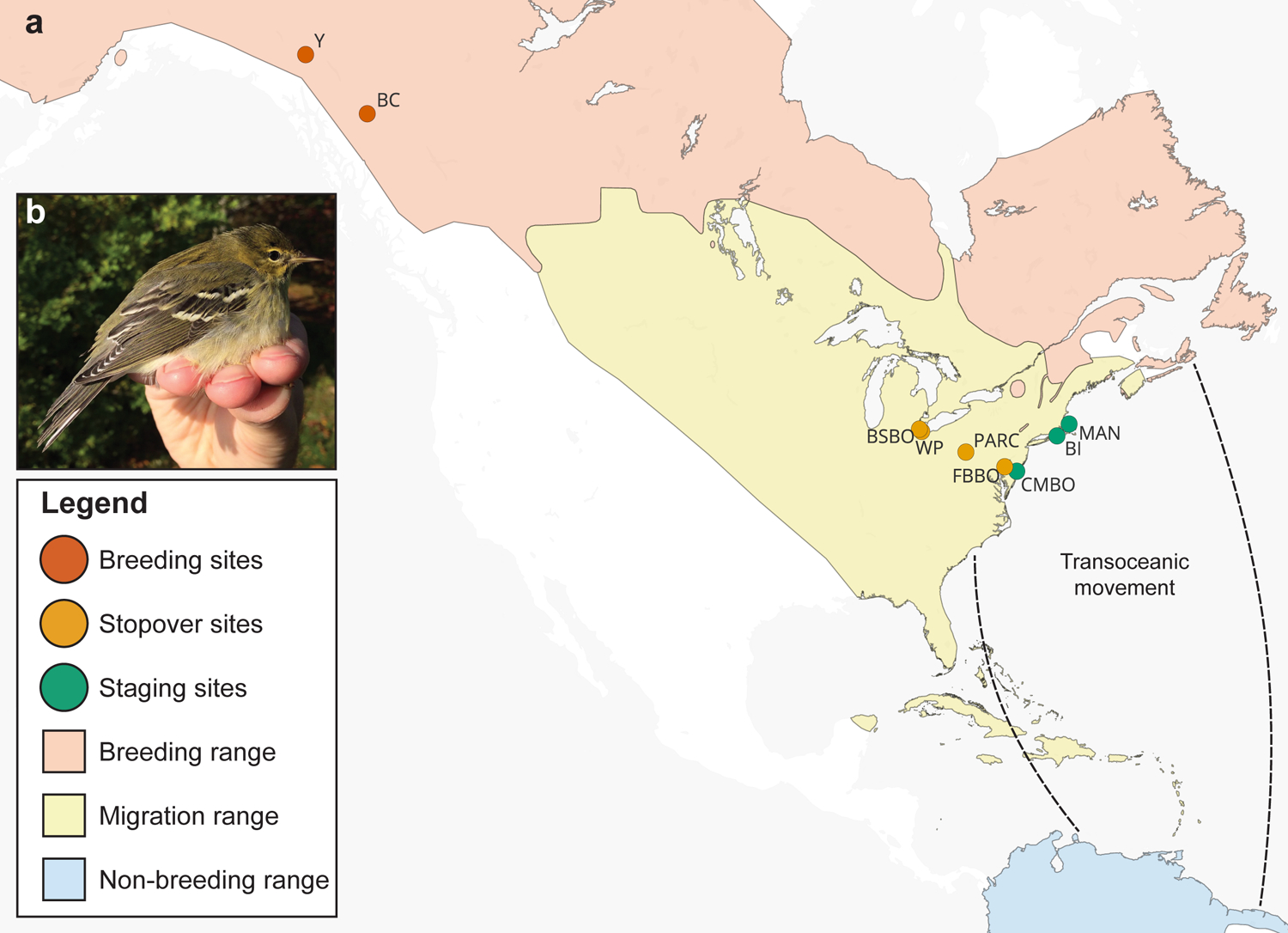
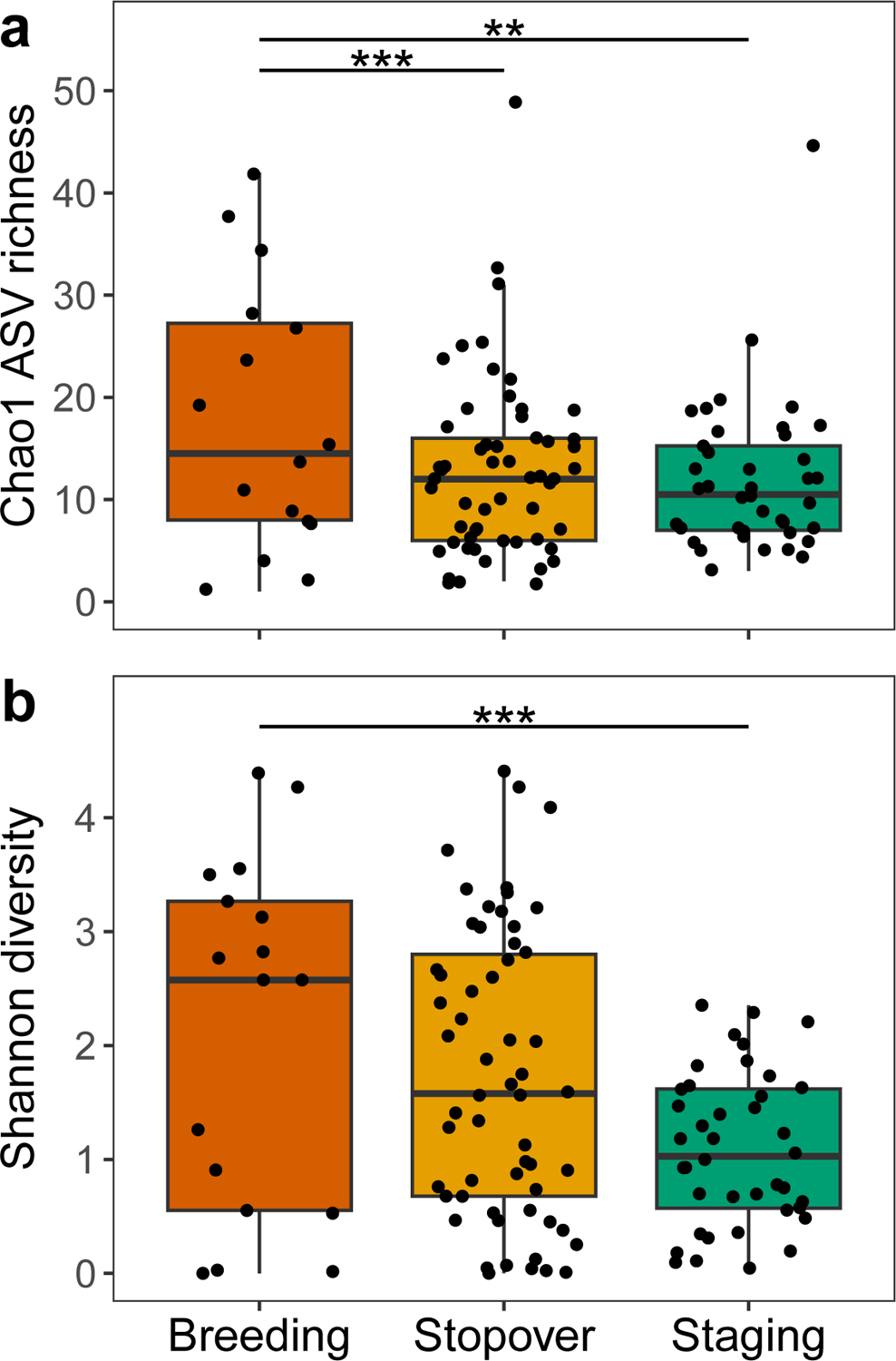
We first investigated how Blackpoll Warbler gut microbiota differed across three phases of fall migration. For metrics of microbiome alpha diversity, there was a significant reduction in Chao1 ASV richness (F = 10.25, P < 0.0001; Fig. 2a) and Shannon diversity (F = 9.67, P = 0.0001; Fig. 2b) across phases of migration. There were no significant differences in alpha diversity across sampling sites within each phase of migration (Supplemental Fig S1). We also observed significant differences in microbiome community composition (PERMANOVA pseudo-F = 16.88, P = 0.001; Fig. 3a) and a reduction of within-group microbiota dissimilarities (PERMDISP pseudo-F = 5.89, P = 0.004; Fig. 3b) across phases of migration, indicating an overall reduction of inter-individual microbiome variation as Blackpoll Warblers migrate from breeding to staging habitats. Importantly, there was no significant association between the community dissimilarities between microbiomes and the geographic distances between the collection sites from which the microbiomes were sampled (Supplemental Fig. S2; Supplemental Data S3).
Figure 2. The Blackpoll Warbler gut microbiome exhibited reduced alpha diversity over phases of fall migration.
(a) Chao1 ASV richness (F = 10.25, P < 0.0001) and (b) Shannon diversity (F = 9.67, P = 0.0001) differed significantly across phases of migration: Breeding (n = 17), stopover (n = 58), and staging (n = 41). *** denotes P ≤ 0.001 and * denotes P ≤ 0.05 after Tukey HSD correction.
Figure 3. Convergence of the Blackpoll Warbler gut microbiome community composition over phases of fall migration.
(a) Phylogenetic RPCA distances differed significantly across phases of migration (PERMANOVA pseudo-F = 16.88, P = 0.001). Ellipses represent 95% confidence level of group centroids. Breeding (red triangles; n = 17): Yukon (Y; n = 2) and British Columbia (BC; n = 15); Stopover (orange squares; n = 58): Black Swamp Bird Observatory (BSBO; n = 12), Winous Point (WP; n = 8), Powdermill Avian Research Center (PARC; n = 19), and Foreman’s Branch Bird Observatory (FBBO; n = 19); Staging (green circles; n = 41): Cape May Bird Observatory (CMBO; n = 15), Block Island (BI; n = 6), Manomet Bird Observatory (MBO; n = 20). (b) Within-group phylogenetic RPCA distances differed significantly across phases of migration (PERMDISP pseudo-F = 5.89, P = 0.004). ** denotes P ≤ 0.01 after FDR correction.
Across all stages of migration, the Blackpoll Warbler microbiome was dominated by taxa in the phylum Proteobacteria (96%), followed by Firmicutes (3%) and 18 others with total relative abundances less than 1% (Fig. 4a; Supplemental Data S4). For phyla with relative abundances greater than 1%, Proteobacteria progressively increased as Blackpoll Warblers migrated from breeding to coastal staging sites (Fig. 5a), while Firmicutes progressively decreased (Fig. 5b). At the family level, the gut microbiome was dominated by three families of Proteobacteria: Enterobacteriaceae (55%), Yersiniaceae (20%), and Morganellaceae (10%; Fig. 4b). The most dominant bacterial family Enterobacteriaceae (phylum Proteobacteria) increased as Blackpoll Warblers migrated to staging sites (Fig. 5c and 5d). This increase in the relative abundance of Enterobacteriaceae was primarily driven by a single ASV that could not be identified beyond the family level (Fig. 5c); however, Enterobacteriaceae remained differentially abundant even after excluding this undescribed ASV from the analysis (Fig. 5d). There was no significant association between metrics of host energetic condition (SMI and fat score) with the relative abundance of bacterial phyla, families, or ASVs.
Figure 4. Remodeling of Blackpoll Warbler gut microbiome composition over phases of fall migration.
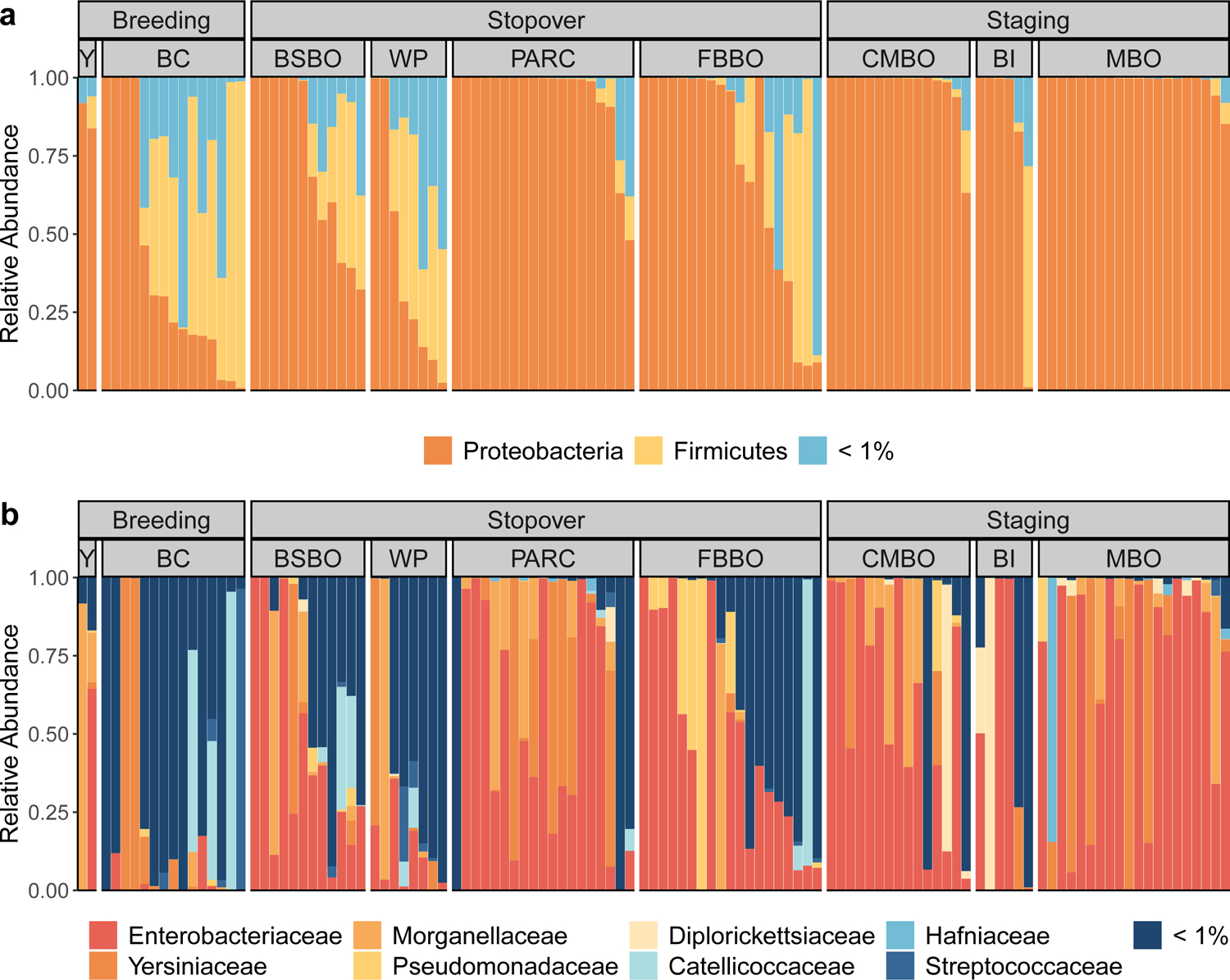
(a) Relative abundances of bacterial phyla and (b) families across phases of migration. Letter codes denote sampling locations within phases of migration. Breeding (n = 17): Yukon (Y; n = 2) and British Columbia (BC; n = 15); Stopover (n = 58): Black Swamp Bird Observatory (BSBO; n = 12), Winous Point (WP; n = 8), Powdermill Avian Research Center (PARC; n = 19), and Foreman’s Branch Bird Observatory (FBBO; n = 19); Staging (n = 41): Cape May Bird Observatory (CMBO; n = 15), Block Island (BI; n = 6), Manomet Bird Observatory (MBO; n = 20).
Figure 5. Taxonomic shifts in the Blackpoll Warbler gut microbiota over phases of fall migration.
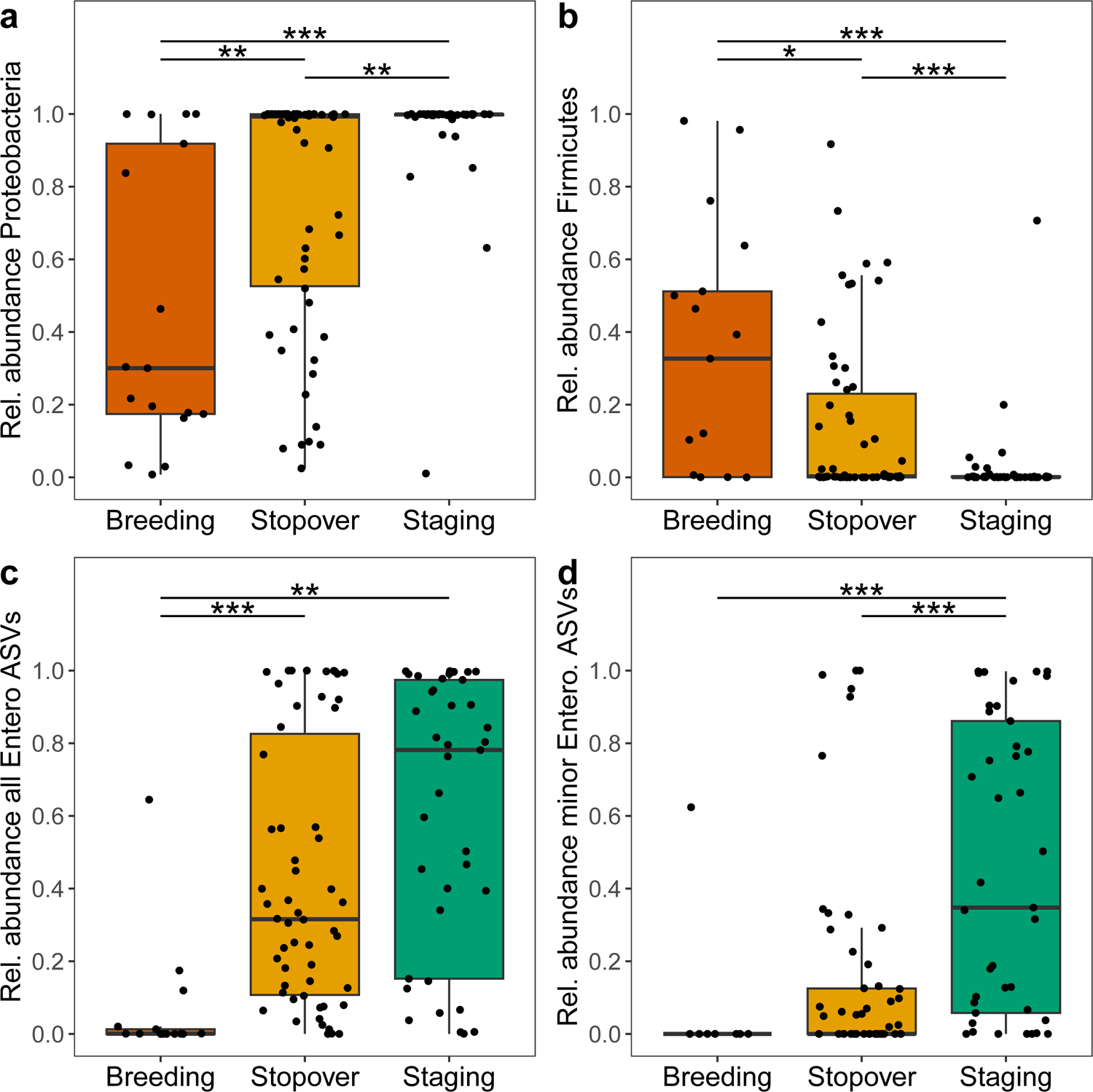
The relative abundance of the bacterial phyla (a) Proteobacteria increased while (b) Firmicutes decreased significantly over phases of migration. (c) The relative abundance of the bacterial family Enterobacteriaceae increased significantly over phases of migration. (d) The relative abundance of the bacterial family Enterobacteriaceae increased significantly after the removal of a dominant undescribed ASV. Breeding (n = 17), stopover (n = 58), and staging (n = 41). *** denotes P ≤ 0.001, ** denotes P ≤ 0.01, and * denotes. P ≤ 0.05 after FDR correction.
Gut microbiome function
Next, we used metagenomics to compare functional profiles of the Blackpoll Warbler gut microbiome between a subset of breeding (n=8) and staging (n=12) individuals. Our approach revealed a significant enrichment of 34 MetaCyc pathways between breeding and staging individuals (Fig. 6; Supplemental Data S5; Supplemental Fig. S3). The gut microbiome of staging individuals was enriched in a diversity of bacterial pathways including those involved in vitamin, amino acid, and fatty acid biosynthesis, as well as nucleoside/nucleotide cycling. Staging Blackpoll Warblers were also enriched in carbohydrate degradation and homolactic fermentation of carbohydrates to the short-chain fatty acid lactate. In contrast, breeding individuals were primarily enriched in bacterial metabolic pathways involved in nucleotide degradation and polyprenoid biosynthesis.
Figure 6. The gut microbiome of staging Blackpoll Warblers is enriched in pathways related to energy metabolism.
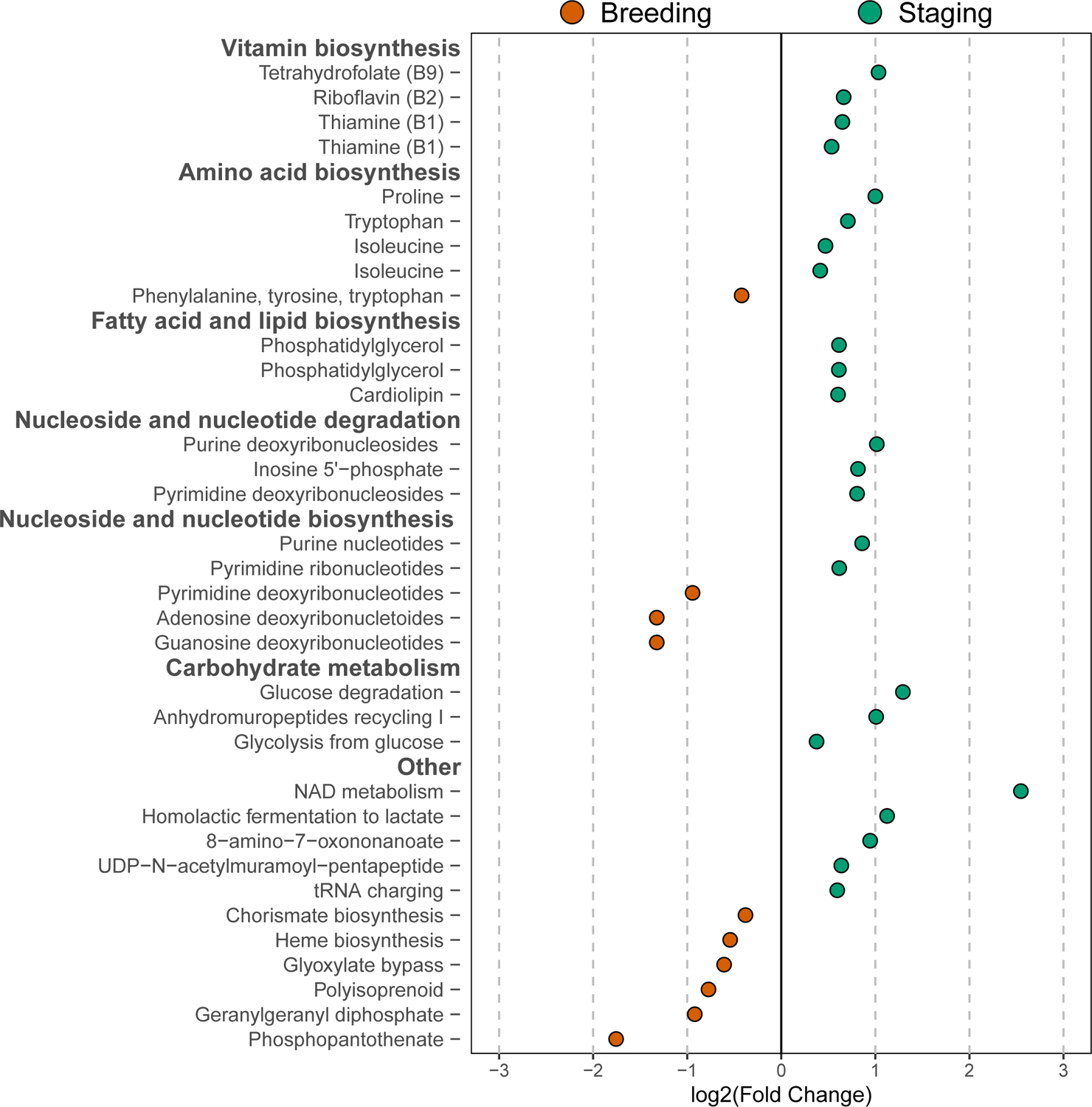
Log2 fold change plot illustrating 34 differentially enriched bacterial metabolic pathways between breeding (n = 8; red points) and staging (n = 12; green points) Blackpoll Warblers. Red points represent pathways enriched in breeding individuals, while green points represent those enriched in staging individuals. All pathways were significant at the threshold of P ≤ 0.05 after FDR correction.
Lastly, we investigated whether bacterial pathways were associated with host energetic condition (SMI) and subcutaneous fat deposits. SMI and fat score were significantly associated with a total of 36 unique bacterial metabolic pathways (Fig. 7). SMI was positively associated with the enrichment of 16 pathways, primarily those involved in bacterial carbohydrate metabolism, as well as vitamin and amino acid biosynthesis (Fig. 7a). In contrast, fat scores were associated with the enrichment of 15 pathways, 3 of which were also positively associated with SMI: proline biosynthesis, sugar biosynthesis, and glycolysis (Fig. 7b).
Figure 7. The enrichment of bacterial pathways is associated with metrics of Blackpoll Warbler energetic condition.
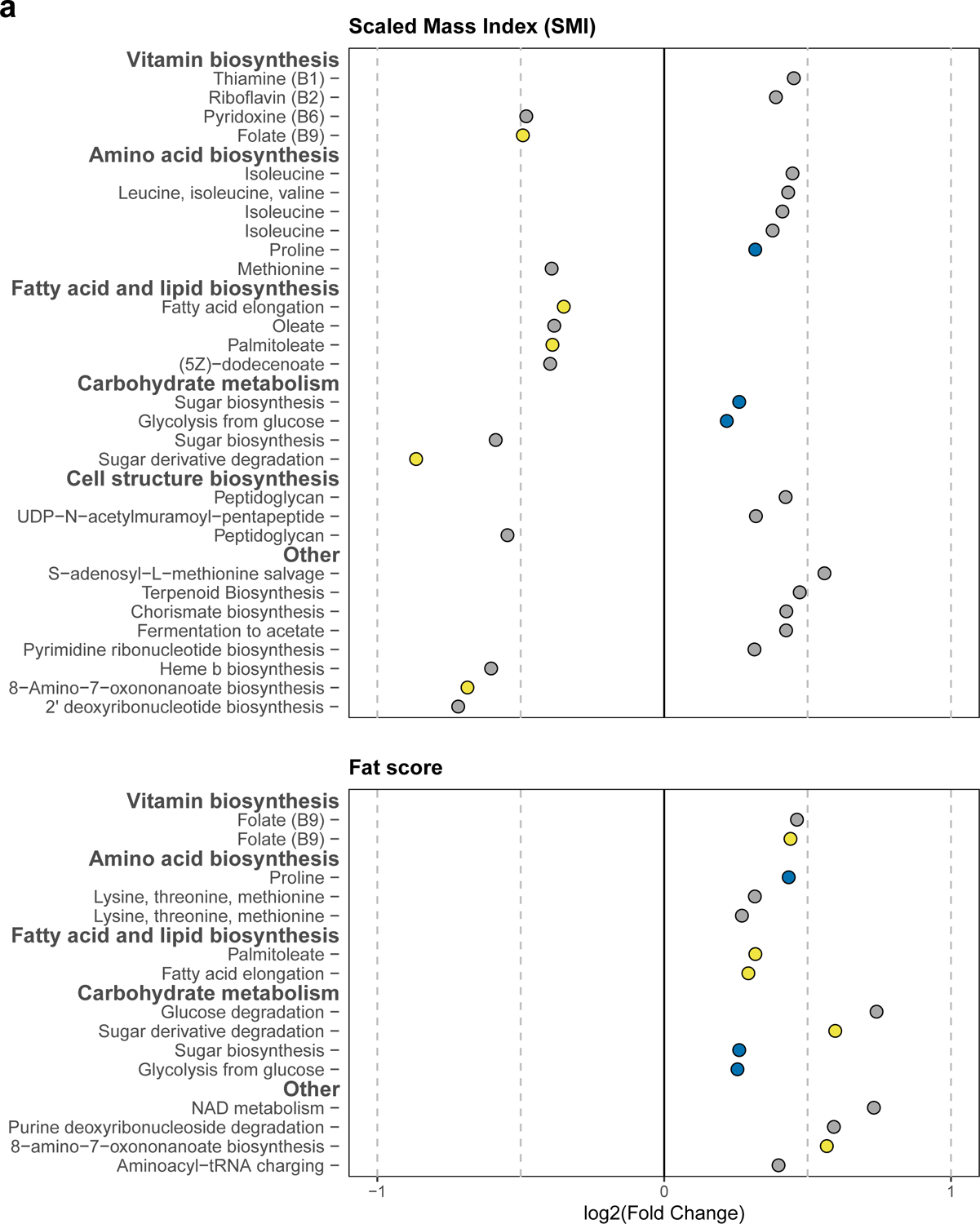
Log2 fold change plot illustrating 36 unique bacterial metabolic pathways significantly associated with (a) Scaled Mass Index (SMI) and (b) subcutaneous fat scores. Yellow points denote bacterial pathways that were significantly associated with both SMI and fat score. Blue points denote pathways that had a significant positive association with both SMI and fat score. Gray points denote pathways significantly associated with either SMI or fat score. All pathways were significant at the threshold of P ≤ 0.05 after FDR correction.
DISCUSSION
In this study, we demonstrated that the Blackpoll Warbler gut microbiome undergoes a dramatic and potentially adaptive remodeling during long-distance migration. Specifically, we showed that the gut microbiota exhibited a reduction in microbiome diversity and inter-individual microbiome dissimilarity, as well as significant differences in composition, across three distinct phases of migration. Notably, these patterns were driven by a significant increase in the relative abundance of Proteobacteria, and more specifically a single ASV belonging to the family Enterobacteriaceae. Metagenomic analysis revealed that the gut microbiome of staging Blackpoll Warblers was enriched in several bacterial pathways with potential relevance for the metabolic needs of migrating birds. Indeed, we found significant associations between several of these bacterial pathways and important indicators of energetic condition and migratory performance. Together, these results support the hypothesis that the gut microbiota of migratory birds is adaptively remodeled to meet the physiological and energetic demands of long-distance migration.
Remodeling of microbiome community composition
We observed robust and significant changes in the Blackpoll Warbler gut microbiota across phases of migration. While the dominance of Proteobacteria in the Blackpoll Warbler gut microbiome is generally consistent with previous descriptions of wild passerine gut microbiota (Bodawatta et al., 2022; Grond et al., 2018), there were dramatic differences in microbiome community composition over phases of migration. Consistent with previous studies showing that migrating birds exhibit reduced gut microbial diversity (Risely et al., 2018; Skeen et al., 2021), Blackpoll Warbler gut microbiota exhibited a progressive reduction in diversity from breeding sites in western Canada to staging sites along the US Atlantic coast (Fig. 2). Moreover, microbiome community composition using phylogenetic RPCA was distinct across migration phases, and was characterized by a reduction in inter-individual microbiome dissimilarity across phases of migration, particularly in the staging phase (Fig. 3). These findings suggest that the challenges imposed by long-distance migration result in a convergence of microbiome composition mediated by natural selection favoring specific microbial lineages. Supporting this conclusion, differences in gut microbial community composition were driven by dramatic shifts in the relative abundance of bacterial phyla belonging to Proteobacteria and Firmicutes among individuals at stopover and staging sites (Fig. 4 and Fig. 5). The increase in the relative abundance of Proteobacteria across phases of migration was primarily driven by a single ASV that belonged to the family Enterobacteriaceae (Fig. 5d), but whose species-level identification and function are unknown. The consistency across individuals with which this ASV came to dominate the microbiota in staging birds is consistent with the possibility that it may harbor functions important for host migration performance. Notably, an increased relative abundance of Proteobacteria among migrating individuals was also observed in Kirtland’s Warbler (Setophaga kirtlandii; Skeen et al., 2021), a species closely related to the Blackpoll Warbler, suggesting that similar host factors (e.g., diet) during migration may result in convergent shifts in gut microbiota (Muegge et al., 2011). In contrast, our results differ from previous studies in Calidris shorebirds showing that migratory individuals were enriched in the bacterial family Corynebacteriaceae (phylum Actinobacteria; Risely et al., 2017, 2018), as the relative abundance of this family in Blackpoll Warblers did not differ across migration phases. Cumulatively, these results suggest that microbiome remodeling may be a widespread phenomenon among migratory birds, but that the enrichment of potentially beneficial microbial lineages may be specific to the phylogeny and life history traits of the host (e.g., diet, phenology).
Potentially adaptive microbiome functions
Compared to breeding individuals, the gut microbiome of staging individuals was enriched in bacterial pathways involved in the biosynthesis of amino acids, which have been identified as important biomolecules for migrating birds (Jenni & Jenni-Eiermann, 1998; Klaassen, 1996). For example, staging migratory birds accumulate fat-free mass in the form of muscle and certain organ tissues (Lindström & Piersma, 1993; Piersma, 1990), which can be catabolized (along with fat and glucose) as a source of energy during migration (Elowe et al., 2023; Jenni & Jenni-Eiermann, 1998). Migrating birds primarily catabolize protein from pectoral muscle, digestive organs, and the gastrointestinal tract (Battley et al., 2000; Bauchinger & Biebach, 2001; Biebach, 1998; Jenni & Jenni-Eiermann, 1998; Schwilch et al., 2002), leading to corresponding reductions in organ mass and function that must be recovered during stopover (Karasov & Pinshow, 2000; Muñoz-Garcia et al., 2012). The availability of plasma amino acids is the limiting factor in protein synthesis in birds (Scanes & Dridi, 2022) and previous work has shown that microbial amino acid synthesis can contribute to the host plasma amino acid pool (Metges, 2000). Alternatively, birds can also synthesize glucose from plasma amino acids via gluconeogenesis, providing an additional source of energy in fasting birds (Scanes & Dridi, 2022). In this study, the gut microbiome of staging individuals was enriched in the biosynthesis of several essential amino acids identified as critical for tissue growth in birds (Scanes & Dridi, 2022), and these pathways were also significantly associated with host body mass and subcutaneous fat deposits. These results suggest the possibility that the microbiome of staging Blackpoll Warblers provides a supplementary source of amino acids that could be used to rebuild catabolized digestive organs, which must be fully functional to digest and assimilate dietary nutrients during fall migration (McWilliams & Karasov, 2001). This hypothesis is supported by the positive association between several amino acid biosynthesis pathways and host body mass (Fig. 7).
The gut microbiome of staging individuals was also enriched in the biosynthesis of several B vitamins, most notably three separate pathways producing thiamine (Fig. 6). Thiamine (vitamin B1) is known to promote immune performance in vertebrates (Kumar & Axelrod, 1978), and thiamine deficiencies have been linked to disease susceptibility in wild birds (Balk et al., 2009). The energetic challenges of long-distance migration reduce several metrics of immune performance in birds (Owen & Moore, 2006), raising the possibility that the enrichment of thiamine biosynthesis pathways among staging Blackpoll Warblers is an adaptive property of their microbiome that helps to maintain immune function under periods of intense metabolic demand. Interestingly, we also observed a significant negative association between some microbial vitamin biosynthesis pathways and host body mass, suggesting that these pathways could have beneficial functions for Blackpoll Warblers during periods of lower body weight (e.g., during breeding and raising offspring).
Lastly, the gut microbiome of staging Blackpoll Warblers was enriched in several bacterial pathways involved in cell membrane phospholipid biosynthesis. Phospholipids are readily digested and absorbed in the avian intestinal tract before being transported to the liver, where they are metabolized into triglycerides – the primary form of stored fat and the primary fuel source for long-distance migration in birds (Price, 2010). While it is tempting to speculate that microbial synthesized phospholipids could help facilitate fat storage in staging Blackpoll Warblers, experimental evidence has demonstrated that dietary phospholipid content has no effect on body mass of migratory birds (Cerasale & Guglielmo, 2006). Regardless, the overall enrichment of microbial pathways with plausible contributions to migratory physiology among staging individuals supports the hypothesis that the remodeling of Blackpoll Warbler gut microbiome confers adaptive advantages to hosts facing the energetic challenges of migration.
Potential mechanisms
The remodeling of the gut microbiome among migrating Blackpoll Warblers could be triggered by seasonal frugivory during migratory stopover. Many species of migratory birds (including Blackpoll Warblers) shift from a protein-rich insectivorous diet during breeding to a carbohydrate-rich frugivorous diet during migration (Bairlein & Gwinner, 1994). The gut microbiome responds rapidly to changes in carbohydrate consumption (David et al., 2014; Smits et al., 2017), and recent work in bats provides evidence that fruit consumption can lead to the functional enrichment of carbohydrate degradation pathways (Ingala et al., 2021). In our study, we observed a robust remodeling of the Blackpoll Warbler microbiome over fall migration that was characterized by a significant increase in the relative abundance of an unidentified ASV in the family Enterobacteriaceae (Fig. 4 and Fig. 5). Interestingly, this family of bacteria has been previously associated with frugivorous bird species (García-Amado et al., 2018), supporting the idea that seasonal diet shifts may trigger a remodeling of the Blackpoll Warbler microbiome. Further, we showed that staging Blackpoll Warblers were enriched in bacterial metabolic pathways involved in sugar degradation, homolactic fermentation, and glycolysis, indicating that the gut microbiome of staging individuals may be specifically adapted to a fruit-based diet rich in carbohydrates. Notably, the gut microbiome of staging individuals was enriched in the fermentation of fructose to the short-chain fatty acid lactate, which is readily absorbed in the avian intestine and essential to hepatic gluconeogenesis (Scanes & Dridi, 2022). The association of host energetic condition with bacterial carbohydrate metabolism (sugar biosynthesis and glycolysis) suggests that these pathways may facilitate energy storage during migration (Fig. 7). While seasonal frugivory is thought to provide an important source of carbohydrates for migratory birds, these fruits are comparatively low in protein (known as the “frugivory paradox”; Bairlein, 2002), which is necessary to rebuild catabolized digestive organs during stopover (Muñoz-Garcia et al., 2012). As noted above, the gut microbiome of staging Blackpoll Warblers was enriched in several amino acids synthesis pathways, raising the possibility that gut microbiota help migrants overcome the nutritional constraints of their diet during stopover and staging. Overall, these results suggest that the widespread consumption of carbohydrate-rich fruit among migratory birds could trigger a remodeling of the microbiome that represents an additional adaptive response to the energetic demands of migration, but further study of how diet and microbiota covary (e.g., Schmiedová et al., 2022) over different Blackpoll Warbler migration routes is necessary.
Digestive systems are energetically-expensive to maintain, and previous work has established that phenotypic flexibility is an important mechanism by which migratory birds adaptively reduce certain digestive functions to help meet the energetic demands of long-distance flight (McWilliams & Karasov, 2001). Our results suggest that this paradigm can be extended to the gut microbiome, which we demonstrate exhibits remarkable flexibility in composition leading to several potentially adaptive functions during Blackpoll Warbler migration. For instance, the enrichment of genes involved in bacterial protein and lipid metabolism over migration is consistent with the hypothesis that the gut microbiome acts as a phenotypically flexible trait that reduces the energetic burden of maintaining these metabolic processes. The proximate mechanisms driving the enrichment of these pathways could be initiated by changes in host diet or exposure to environmental microbiota (Bodawatta et al., 2022; Grond et al., 2018) over migration. In this study, Blackpoll Warblers underwent a reduction in microbiome diversity over fall migration, providing additional support for our hypothesis that the gut microbiome is responding to the energetic demands of migration rather than environmental factors. However, repeated sampling of individuals over migration (e.g., Skeen et al., 2021) and experimental techniques such as fecal microbiome transplants into gnotobiotic animal models (e.g., Sommer et al., 2016; Trevelline & Kohl, 2022) are necessary to definitively establish the functional effects of particular microbial taxa on host digestive physiology and migration performance.
Conclusions
Overall, our study demonstrates that the gut microbiome of a long-distance migratory bird undergoes a compositional and functional remodeling over fall migration. Whereas previous work on migrants has focused on compositional differences across just a few sites, we coordinated with researchers and bird banding stations across North America to show that gut microbiota of a staging migrant was enriched in bacterial taxa capable of synthesizing biomolecules important for migrating birds (Jenni & Jenni-Eiermann, 1998; Klaassen, 1996). Moreover, we observed several significant associations between microbial pathways and metrics of host migratory condition, highlighting the importance of using metagenomic techniques in combination with 16S rRNA microbial inventories. These findings build upon numerous descriptive studies showing immense variation in gut microbiota among migratory birds (Bodawatta et al., 2022; Grond et al., 2018) and contribute to a growing body of evidence that host-microbe interactions shape the ecology and evolution of wild birds. Given that the energetic state of migratory birds is a critical determinant of migration performance, our results suggest that the gut microbiome of migratory birds may be an important yet unrecognized factor influencing the survival, fitness, and conservation of migratory birds.
Supplementary Material
ACKNOWLEDGMENTS
We thank Lila Tauzer and Hilary Cooke for the collection of fecal samples for metagenomics at breeding sites in Western Canada, as well as Clara Cooper-Mullin and Scott McWilliams for fecal samples collected at staging sites on Block Island. We also thank Trevor-Lloyd Evans of Manomet Bird Observatory, Annie Lindsay of Powdermill Avian Research Center, Mark Shieldcastle and staff of Black Swamp Bird Observatory, Liz Ames and staff of Winous Point Marsh Conservancy, Maren Gimpel of Foreman’s Branch Bird Observatory, David LaPuma and staff at New Jersey Audubon’s Cape May Bird Observatory for the collection of blackpoll fecal samples at stopover and staging sites. Blackpoll warbler range map provided by BirdLife International and Birds of the World. The authors have no conflicts of interest to declare.
FUNDING
This work was funded by a grant from the British Ecological Society (SR21\100252), a Rose Fellowship from the Cornell Lab of Ornithology to BKT, funds to CMT from Ohio Agricultural Research and Development Center, and grant R35GM138284 from the National Institutes of Health National Institute of General Medical Sciences to AHM.
Footnotes
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
DATA AVAILABILITY
Sequencing data have been deposited in the National Center for Biotechnology Information Sequence Read Archive Database and are publicly available at BioProject PRJNA971188.
REFERENCES
- Alberdi A, Aizpurua O, Bohmann K, Zepeda-Mendoza ML, & Gilbert MTP (2016). Do vertebrate gut metagenomes confer rapid ecological adaptation? Trends in Ecology & Evolution, 31(9), Article 9. 10.1016/j.tree.2016.06.008 [DOI] [PubMed] [Google Scholar]
- Alerstam T, Hedenström A, & Åkesson S (2003). Long-distance migration: Evolution and determinants. Oikos, 103(2), 247–260. 10.1034/j.1600-0706.2003.12559.x [DOI] [Google Scholar]
- Bairlein F (2002). How to get fat: Nutritional mechanisms of seasonal fat accumulation in migratory songbirds. Naturwissenschaften, 89(1), 1–10. 10.1007/s00114-001-0279-6 [DOI] [PubMed] [Google Scholar]
- Bairlein F, & Gwinner E (1994). Nutritional mechanisms and temporal control of migratory energy accumulation in birds. Annual Review of Nutrition, 14, 187–215. 10.1146/annurev.nu.14.070194.001155 [DOI] [PubMed] [Google Scholar]
- Balk L, Hägerroth P-Å, Åkerman G, Hanson M, Tjärnlund U, Hansson T, Hallgrimsson GT, Zebühr Y, Broman D, Mörner T, & Sundberg H (2009). Wild birds of declining European species are dying from a thiamine deficiency syndrome. Proceedings of the National Academy of Sciences, 106(29), 12001–12006. 10.1073/pnas.0902903106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battley PF, Piersma T, Dietz MW, Tang S, Dekinga A, & Hulsman K (2000). Empirical evidence for differential organ reductions during trans-oceanic bird flight. Proceedings of the Royal Society B: Biological Sciences, 267(1439), 191–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauchinger U, & Biebach H (2001). Differential catabolism of muscle protein in Garden Warblers (Sylvia borin): Flight and leg muscle act as a protein source during long-distance migration. Journal of Comparative Physiology B: Biochemical, Systemic, and Environmental Physiology, 171(4), 293–301. 10.1007/s003600100176 [DOI] [PubMed] [Google Scholar]
- Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, Mailyan A, Manghi P, Scholz M, Thomas AM, Valles-Colomer M, Weingart G, Zhang Y, Zolfo M, Huttenhower C, Franzosa EA, & Segata N (2021). Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife, 10, e65088. 10.7554/eLife.65088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biebach H (1998). Phenotypic organ flexibility in Garden Warblers (Sylvia borin) during long-distance migration. Journal of Avian Biology, 29(4), 529–535. 10.2307/3677172 [DOI] [Google Scholar]
- Bodawatta KH, Hird SM, Grond K, Poulsen M, & Jønsson KA (2022). Avian gut microbiomes taking flight. Trends in Microbiology, 30(3), 268–280. 10.1016/j.tim.2021.07.003 [DOI] [PubMed] [Google Scholar]
- Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, … Caporaso JG (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nature Biotechnology, 37(8), Article 8. 10.1038/s41587-019-0209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke C, Steinberg P, Rusch D, Kjelleberg S, & Thomas T (2011). Bacterial community assembly based on functional genes rather than species. Proceedings of the National Academy of Sciences, 108(34), 14288–14293. 10.1073/pnas.1101591108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, & Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nature Methods, 13(7), Article 7. 10.1038/nmeth.3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, & Knight R (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proceedings of the National Academy of Sciences, 108(Supplement_1), 4516–4522. 10.1073/pnas.1000080107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carey HV, & Assadi-Porter FM (2017). The hibernator microbiome: Host-bacterial interactions in an extreme nutritional symbiosis. Annual Review of Nutrition, 37, 477–500. [DOI] [PubMed] [Google Scholar]
- Cerasale DJ, & Guglielmo CG (2006). Plasma metabolite profiles: Effects of dietary phospholipids in a migratory passerine (Zonotrichia leucophrys gambelii). Physiological and Biochemical Zoology, 79(4), 754–762. 10.1086/505510 [DOI] [PubMed] [Google Scholar]
- Cho I, & Blaser MJ (2012). The human microbiome: At the interface of health and disease. Nature Reviews Genetics, 13(4), 260–270. 10.1038/nrg3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente JC, Ursell LK, Parfrey LW, & Knight R (2012). The Impact of the Gut Microbiota on Human Health: An Integrative View. Cell, 148(6), 1258–1270. 10.1016/j.cell.2012.01.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colston TJ, & Jackson CR (2016). Microbiome evolution along divergent branches of the vertebrate tree of life: What is known and unknown. Molecular Ecology, 25(16), 3776–3800. 10.1111/mec.13730 [DOI] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, Biddinger SB, Dutton RJ, & Turnbaugh PJ (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature, 505(7484), Article 7484. 10.1038/nature12820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis NM, Proctor DM, Holmes SP, Relman DA, & Callahan BJ (2018). Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome, 6(1), 226. 10.1186/s40168-018-0605-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca WV, Holberton R, Hunt PD, & Eliason BC (2020). Blackpoll Warbler (Setophaga striata). In Birds of the World. Cornell Lab of Ornithology. [Google Scholar]
- DeLuca WV, Woodworth BK, Mackenzie SA, Newman AEM, Cooke HA, Phillips LM, Freeman NE, Sutton AO, Tauzer L, McIntyre C, Stenhouse IJ, Weidensaul S, Taylor PD, & Norris DR (2019). A boreal songbird’s 20,000 km migration across North America and the Atlantic Ocean. Ecology, 100(5), 1–4. [DOI] [PubMed] [Google Scholar]
- DeLuca WV, Woodworth BK, Rimmer CC, Marra PP, Taylor PD, McFarland KP, Mackenzie SA, & Norris DR (2015). Transoceanic migration by a 12 g songbird. Biology Letters, 11(4), 20141045. 10.1098/rsbl.2014.1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elowe CR, Groom DJE, Slezacek J, & Gerson AR (2023). Long-duration wind tunnel flights reveal exponential declines in protein catabolism over time in short- and long-distance migratory warblers. Proceedings of the National Academy of Sciences, 120(17), e2216016120. 10.1073/pnas.2216016120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine SS, & Kohl KD (2020). Optimal integration between host physiology and functions of the gut microbiome. Philosophical Transactions of the Royal Society B: Biological Sciences, 375(1808), Article 1808. 10.1098/rstb.2019.0594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine SS, Mineo PM, & Kohl KD (2022). Experimental manipulation of microbiota reduces host thermal tolerance and fitness under heat stress in a vertebrate ectotherm. Nature Ecology & Evolution, 6(4), 405–417. 10.1038/s41559-022-01686-2 [DOI] [PubMed] [Google Scholar]
- García-Amado MA, Shin H, Sanz V, Lentino M, Martínez LM, Contreras M, Michelangeli F, & Domínguez-Bello MG (2018). Comparison of gizzard and intestinal microbiota of wild neotropical birds. PLOS ONE, 13(3), e0194857. 10.1371/journal.pone.0194857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grond K, Sandercock BK, Jumpponen A, & Zeglin LH (2018). The avian gut microbiota: Community, physiology and function in wild birds. Journal of Avian Biology, 49(11), e01788. 10.1111/jav.01788 [DOI] [Google Scholar]
- Groussin M, Mazel F, Sanders JG, Smillie CS, Lavergne S, Thuiller W, & Alm EJ (2017). Unraveling the processes shaping mammalian gut microbiomes over evolutionary time. Nature Communications, 8(1), 14319. 10.1038/ncomms14319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry LP, Bruijning M, Forsberg SKG, & Ayroles JF (2021). The microbiome extends host evolutionary potential. Nature Communications, 12(1), Article 1. 10.1038/s41467-021-25315-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingala MR, Simmons NB, Dunbar M, Wultsch C, Krampis K, & Perkins SL (2021). You are more than what you eat: Potentially adaptive enrichment of microbiome functions across bat dietary niches. Animal Microbiome, 3(1), 82. 10.1186/s42523-021-00139-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenni L, & Jenni-Eiermann S (1998). Fuel supply and metabolic constraints in migrating birds. Journal of Avian Biology, 29(4), Article 4. [Google Scholar]
- Kaiser A (1993). A New Multi-Category Classification of Subcutaneous Fat Deposits of Songbirds (Una Nueva Clasificación, con Multi-categorías, para los Depósitos de Grasa en Aves Canoras). Journal of Field Ornithology, 64(2), 246–255. [Google Scholar]
- Karasov WH, & Pinshow B (2000). Test for physiological limitation to nutrient assimilation in a long‐distance passerine migrant at a springtime stopover site. Physiological and Biochemical Zoology, 73(3), 335–343. 10.1086/316746 [DOI] [PubMed] [Google Scholar]
- Klaassen M (1996). Metabolic constraints on long-distance migration in birds. Journal of Experimental Biology, 199(1), Article 1. [DOI] [PubMed] [Google Scholar]
- Knutie SA, Wilkinson CL, Kohl KD, & Rohr JR (2017). Early-life disruption of amphibian microbiota decreases later-life resistance to parasites. Nature Communications, 8(1), 86. 10.1038/s41467-017-00119-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl KD, Connelly JW, Dearing MD, & Forbey JS (2016). Microbial detoxification in the gut of a specialist avian herbivore, the Greater Sage-Grouse. FEMS Microbiology Letters, 363(14), Article 14. 10.1093/femsle/fnw144 [DOI] [PubMed] [Google Scholar]
- Kohl KD, Weiss RB, Cox J, Dale C, & Dearing MD (2014). Gut microbes of mammalian herbivores facilitate intake of plant toxins. Ecology Letters, 17(10), 1238–1246. 10.1111/ele.12329 [DOI] [PubMed] [Google Scholar]
- Kumar M, & Axelrod AE (1978). Cellular antibody synthesis in thiamin, riboflavin, biotin and folic acid-deficient rats. Proceedings of the Society for Experimental Biology and Medicine, 157(3), 421–423. 10.3181/00379727-157-40068 [DOI] [PubMed] [Google Scholar]
- Lewis WB, Moore FR, & Wang S (2016). Characterization of the gut microbiota of migratory passerines during stopover along the northern coast of the Gulf of Mexico. Journal of Avian Biology, 47(5), Article 5. 10.1111/jav.00954 [DOI] [Google Scholar]
- Lewis WB, Moore FR, & Wang S (2017). Changes in gut microbiota of migratory passerines during stopover after crossing an ecological barrier. The Auk, 134(1), Article 1. 10.1642/AUK-16-120.1 [DOI] [Google Scholar]
- Ley RE, Hamady M, Lozupone C, Turnbaugh P, Ramey RR, Bircher JS, Schlegel Michael. L., Tucker TA, Schrenzel MD, Knight R, & Gordon JI (2008a). Evolution of mammals and their gut microbes. Science, 320(5883), Article 5883. 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Hamady M, Lozupone C, Turnbaugh P, Ramey RR, Bircher JS, Schlegel Michael. L., Tucker TA, Schrenzel MD, Knight R, & Gordon JI (2008b). Evolution of mammals and their gut microbes. Science (New York, N.Y.), 320(5883), 1647–1651. 10.1126/science.1155725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström Å, & Piersma T (1993). Mass changes in migrating birds: The evidence for fat and protein storage re-examined. Ibis, 135(1), 70–78. 10.1111/j.1474-919X.1993.tb02811.x [DOI] [Google Scholar]
- Lynch JB, & Hsiao EY (2019). Microbiomes as sources of emergent host phenotypes. Science, 365(6460), 1405–1409. 10.1126/science.aay0240 [DOI] [PubMed] [Google Scholar]
- Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, Tickle TL, Weingart G, Ren B, Schwager EH, Chatterjee S, Thompson KN, Wilkinson JE, Subramanian A, Lu Y, Waldron L, Paulson JN, Franzosa EA, Bravo HC, & Huttenhower C (2021). Multivariable association discovery in population-scale meta-omics studies. PLOS Computational Biology, 17(11), Article 11. 10.1371/journal.pcbi.1009442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino C, Morton JT, Marotz CA, Thompson LR, Tripathi A, Knight R, & Zengler K (2019). A Novel Sparse Compositional Technique Reveals Microbial Perturbations. mSystems, 4(1), e00016–19. 10.1128/mSystems.00016-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFall-Ngai M, Hadfield MG, Bosch TCG, Carey HV, Domazet-Lošo T, Douglas AE, Dubilier N, Eberl G, Fukami T, Gilbert SF, Hentschel U, King N, Kjelleberg S, Knoll AH, Kremer N, Mazmanian SK, Metcalf JL, Nealson K, Pierce NE, … Wernegreen JJ (2013). Animals in a bacterial world, a new imperative for the life sciences. Proceedings of the National Academy of Sciences, 110(9), Article 9. 10.1073/pnas.1218525110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, & Holmes S (2014). Waste not, want not: Why rarefying microbiome data is inadmissible. PLoS Computational Biology, 10(4), Article 4. 10.1371/journal.pcbi.1003531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams SR, & Karasov WH (2001). Phenotypic flexibility in digestive system structure and function in migratory birds and its ecological significance. Comparative Biochemistry and Physiology Part A: Molecular & Integrative Physiology, 128(3), 577–591. 10.1016/S1095-6433(00)00336-6 [DOI] [PubMed] [Google Scholar]
- Metges CC (2000). Contribution of microbial amino acids to amino acid homeostasis of the host. The Journal of Nutrition, 130(7), 1857S–1864S. 10.1093/jn/130.7.1857S [DOI] [PubMed] [Google Scholar]
- Moran NA, Ochman H, & Hammer TJ (2019). Evolutionary and Ecological Consequences of Gut Microbial Communities. Annual Review of Ecology, Evolution, and Systematics, 50, 451–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris SR, Covino KM, Jacobs JD, & Taylor PD (2016). Fall migratory patterns of the Blackpoll Warbler at a continental scale. The Auk, 133(1), 41–51. 10.1642/AUK-15-133.1 [DOI] [Google Scholar]
- Muegge BD, Kuczynski J, Knights D, Clemente JC, González A, Fontana L, Henrissat B, Knight R, & Gordon JI (2011). Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science, 332(6032), 970–974. 10.1126/science.1198719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Garcia A, Aamidor SE, McCue MD, McWilliams SR, & Pinshow B (2012). Allocation of endogenous and dietary protein in the reconstitution of the gastrointestinal tract in migratory blackcaps at stopover sites. Journal of Experimental Biology, 215(7), 1069–1075. 10.1242/jeb.062547 [DOI] [PubMed] [Google Scholar]
- O’Hara AM, & Shanahan F (2006). The gut flora as a forgotten organ. EMBO Reports, 7(7), 688–693. 10.1038/sj.embor.7400731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen JC, & Moore FR (2006). Seasonal differences in immunological condition of three species of thrushes. The Condor, 108(2), 389–398. 10.1093/condor/108.2.389 [DOI] [Google Scholar]
- Pascoe EL, Hauffe HC, Marchesi JR, & Perkins SE (2017). Network analysis of gut microbiota literature: An overview of the research landscape in non-human animal studies. The ISME Journal, 11(12), 2644–2651. 10.1038/ismej.2017.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peig J, & Green AJ (2009). New perspectives for estimating body condition from mass/length data: The scaled mass index as an alternative method. Oikos, 118(12), 1883–1891. 10.1111/j.1600-0706.2009.17643.x [DOI] [Google Scholar]
- Piersma T (1990). Pre‐migratory “fattening” usually involves more than the deposition of fat alone. Ringing & Migration, 11(2), 113–115. 10.1080/03078698.1990.9673972 [DOI] [Google Scholar]
- Piersma T (1998). Phenotypic flexibility during migration: Optimization of organ size contingent on the risks and rewards of fueling and flight? Journal of Avian Biology, 29(4), Article 4. [Google Scholar]
- Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, & Glöckner FO (2013). The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Research, 41(Database issue), D590–D596. 10.1093/nar/gks1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risely A, Waite D, Ujvari B, Klaassen M, & Hoye B (2017). Gut microbiota of a long‐distance migrant demonstrates resistance against environmental microbe incursions. Molecular Ecology, 26(20), 5842–5854. 10.1111/mec.14326 [DOI] [PubMed] [Google Scholar]
- Risely A, Waite DW, Ujvari B, Hoye BJ, & Klaassen M (2018). Active migration is associated with specific and consistent changes to gut microbiota in Calidris shorebirds. Journal of Animal Ecology, 87(2), Article 2. 10.1111/1365-2656.12784 [DOI] [PubMed] [Google Scholar]
- Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N, Shilo S, Lador D, Vila AV, Zmora N, Pevsner-Fischer M, Israeli D, Kosower N, Malka G, Wolf BC, … Segal E (2018). Environment dominates over host genetics in shaping human gut microbiota. Nature, 555(7695), 210–215. 10.1038/nature25973 [DOI] [PubMed] [Google Scholar]
- Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, Turner P, Parkhill J, Loman NJ, & Walker AW (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biology, 12(1), 87. 10.1186/s12915-014-0087-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanes CG, & Dridi S (2022). Protein metabolism. In Sturkie’s Avian physiology (6th ed., pp. 455–467). Academic Press. [Google Scholar]
- Schmiedová L, Tomášek O, Pinkasová H, Albrecht T, & Kreisinger J (2022). Variation in diet composition and its relation to gut microbiota in a passerine bird. Scientific Reports, 12(1), Article 1. 10.1038/s41598-022-07672-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwilch R, Grattarola A, Spina F, & Jenni L (2002). Protein loss during long-distance migratory flight in passerine birds: Adaptation and constraint. Journal of Experimental Biology, 205(5), Article 5. 10.1242/jeb.205.5.687 [DOI] [PubMed] [Google Scholar]
- Skeen HR, Cooper NW, Hackett SJ, Bates JM, & Marra PP (2021). Repeated sampling of individuals reveals impact of tropical and temperate habitats on microbiota of a migratory bird. Molecular Ecology, 30(22), Article 22. 10.1111/mec.16170 [DOI] [PubMed] [Google Scholar]
- Smetzer JR, King DI, & Taylor PD (2017). Fall migratory departure decisions and routes of blackpoll warblers Setophaga striata and red-eyed vireos Vireo olivaceus at a coastal barrier in the Gulf of Maine. Journal of Avian Biology, 48(11), 1451–1461. 10.1111/jav.01450 [DOI] [Google Scholar]
- Smits SA, Leach J, Sonnenburg ED, Gonzalez CG, Lichtman JS, Reid G, Knight R, Manjurano A, Changalucha J, Elias JE, Dominguez-Bello MG, & Sonnenburg JL (2017). Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science, 357(6353), 802–806. 10.1126/science.aan4834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer F, Ståhlman M, Ilkayeva O, Arnemo JM, Kindberg J, Josefsson J, Newgard CB, Fröbert O, & Bäckhed F (2016). The gut microbiota modulates energy metabolism in the hibernating Brown bear Ursus arctos. Cell Reports, 14(7), Article 7. 10.1016/j.celrep.2016.01.026 [DOI] [PubMed] [Google Scholar]
- Suzek BE, Wang Y, Huang H, McGarvey PB, Wu CH, & the UniProt Consortium. (2015). UniRef clusters: A comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics, 31(6), 926–932. 10.1093/bioinformatics/btu739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki TA (2017). Links between natural variation in the microbiome and host fitness in wild mammals. Integrative and Comparative Biology, 57(4), Article 4. 10.1093/icb/icx104 [DOI] [PubMed] [Google Scholar]
- Thie N, Corl A, Turjeman S, Efrat R, Kamath PL, Getz WM, Bowie RCK, & Nathan R (2022). Linking migration and microbiota at a major stopover site in a long-distance avian migrant. Movement Ecology, 10(1), 46. 10.1186/s40462-022-00347-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelline BK, Fontaine SS, Hartup BK, & Kohl KD (2019). Conservation biology needs a microbial renaissance: A call for the consideration of host-associated microbiota in wildlife management practices. Proceedings of the Royal Society B: Biological Sciences, 286(1895), 20182448. 10.1098/rspb.2018.2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelline BK, & Kohl KD (2022). The gut microbiome influences host diet selection behavior. Proceedings of the National Academy of Sciences, 119(17), e2117537119. 10.1073/pnas.2117537119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelline BK, MacLeod KJ, Langkilde T, & Kohl KD (2019). Gestation alters the gut microbiota of an oviparous lizard. FEMS Microbiology Ecology, 95(7), fiz086. 10.1093/femsec/fiz086 [DOI] [PubMed] [Google Scholar]
- Turjeman S, Corl A, Wolfenden A, Tsalyuk M, Lublin A, Choi O, Kamath PL, Getz WM, Bowie RCK, & Nathan R (2020). Migration, pathogens and the avian microbiome: A comparative study in sympatric migrants and residents. Molecular Ecology, 29(23), 4706–4720. 10.1111/mec.15660 [DOI] [PubMed] [Google Scholar]
- Videvall E, Strandh M, Engelbrecht A, Cloete S, & Cornwallis CK (2018). Measuring the gut microbiome in birds: Comparison of faecal and cloacal sampling. Molecular Ecology Resources, 18(3), 424–434. 10.1111/1755-0998.12744 [DOI] [PubMed] [Google Scholar]
- Warnock N (2010). Stopping vs. staging: The difference between a hop and a jump. Journal of Avian Biology, 41(6), 621–626. 10.1111/j.1600-048X.2010.05155.x [DOI] [Google Scholar]
- Wiebler JM, Kohl KD, Lee RE, & Costanzo JP (2018). Urea hydrolysis by gut bacteria in a hibernating frog: Evidence for urea-nitrogen recycling in Amphibia. Proceedings of the Royal Society B: Biological Sciences, 285(1878), 20180241. 10.1098/rspb.2018.0241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TC, Williams JM, Ireland LC, & Teal JM (1978). Estimated flight time for transatlantic autumnal migrants. American Birds, 32(3), 275–280. [Google Scholar]
- Wu Y, Yang Y, Cao L, Yin H, Xu M, Wang Z, Liu Y, Wang X, & Deng Y (2018). Habitat environments impacted the gut microbiome of long-distance migratory swan geese but central species conserved. Scientific Reports, 8(1), Article 1. 10.1038/s41598-018-31731-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Sun C, Zheng J, Wen C, Ji C, Zhang D, Chen Y, Hou Z, & Yang N (2019). Efficacy of Fecal Sampling as a Gut Proxy in the Study of Chicken Gut Microbiota. Frontiers in Microbiology, 10. https://www.frontiersin.org/articles/10.3389/fmicb.2019.02126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilber-Rosenberg I, & Rosenberg E (2008). Role of microorganisms in the evolution of animals and plants: The hologenome theory of evolution. FEMS Microbiology Reviews, 32(5), 723–735. 10.1111/j.1574-6976.2008.00123.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data have been deposited in the National Center for Biotechnology Information Sequence Read Archive Database and are publicly available at BioProject PRJNA971188.