

Abstract
Objective: To compare the efficacy and safety of paroxetine controlled release (CR) (12.5 mg/day or 25 mg/day) versus placebo in premenstrual dysphoric disorder (PMDD).
Method: A double-blind, randomized, placebo-controlled trial was conducted over 3 menstrual cycles in women aged 18–45 years with confirmed DSM-IV PMDD in 47 outpatient centers across the United States and Canada from November 1999 to January 2002. The primary efficacy measure was the visual analog scale (VAS)-Mood, which is the mean of 4 core symptoms: irritability, tension, depressed mood, and affective lability.
Results: A statistically significant difference was observed in favor of paroxetine CR 25 mg versus placebo on the VAS-Mood (adjusted mean difference = −12.58 mm, 95% CI = −18.40 to −6.76; p < .001) and for paroxetine CR 12.5 mg versus placebo (adjusted mean difference = −7.51 mm, 95% CI = −13.40 to −1.62; p = .013). Paroxetine CR was generally well tolerated.
Conclusion: Paroxetine CR doses of 12.5 mg/day and 25 mg/day are effective in treating PMDD and are well tolerated.
Premenstrual dysphoric disorder (PMDD) is characterized in the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV)1 as a cluster of psychological, behavioral, and somatic symptoms that appear regularly during the 3 to 10 days prior to menstrual bleeding (luteal phase) and remit completely after the onset of menstruation (follicular phase). Essential features of PMDD include markedly depressed mood, anxiety, tension, irritability, affective lability, feeling overwhelmed or out of control, decreased concentration, decreased energy, change in appetite, change in sleep, and decreased interest in activities.1 Physical symptoms may include breast tenderness or swelling, bloating, joint/muscle pain, weight gain, and headaches. These symptoms are present in most menstrual cycles during the year and cause a significant impact on family, work, and social functioning. While an estimated 75% of women with regular menstrual cycles experience premenstrual symptoms, only 3% to 9% of women suffer from PMDD.2–6
The functional impairment associated with PMDD is comparable to that observed in subjects suffering from major depression and often leads to reduction in quality of life.7,8 Women who suffer from PMDD are high utilizers of health care resources,9,10 are twice as likely as non-PMDD counterparts to make repeat visits to their primary care doctor, and are over 4 times as likely to make repeat visits to a specialist for mental health treatment.4
The underlying pathophysiology of PMDD is thought to be associated with serotonergic dysregulation.11 This is supported by emerging data demonstrating that selective serotonin reuptake inhibitors (SSRIs) are highly effective in treating this disorder.12 Fluoxetine,13 sertraline,7 paroxetine,14,15 and citalopram16 have all demonstrated efficacy in large, double-blind, clinical trials for patients with severe premenstrual syndrome or PMDD.
PMDD is a chronic disorder with onset generally occurring in the mid-20s that continues until menopause. The continued monthly appearance and subsequent subsidence of disabling symptoms makes PMDD unique in that many patients are otherwise asymptomatic during the majority of their menstrual cycle but experience symptoms for the 3 to 10 days prior to onset of menstruation.17 Treatment should therefore be focused on reducing these vacillations and provide premenstrual symptom reduction to a level equal to the remainder of their cycle (remission).
Aims of the Study
The aims of the study were to investigate efficacy and safety of 2 fixed dosages of paroxetine controlled release (CR) formulation in the treatment of patients diagnosed with PMDD and to describe the potential for this drug to lead to remission.
METHOD
Subject Selection
This was a multicenter, randomized, double-blind, placebo-controlled, fixed-dose study to assess the efficacy and safety of paroxetine CR in women with PMDD. Eligible patients included women aged 18–45 years with regular menstrual cycles (duration between 22–35 days) and confirmed DSM-IV PMDD.1 Symptoms of the disorder were required to have been present in at least 9 out of 12 menstrual cycles over the previous year. To confirm the diagnosis of PMDD, subjects were required to prospectively rate their symptoms using daily diaries for 2 consecutive “reference” cycles prior to randomization. Subjects were considered eligible for randomization if the onset of severe premenstrual symptoms during the luteal phase was followed by symptom subsidence during the follicular phase based on the 4 core symptoms of PMDD (irritability, tension, affective lability, and depressed mood). To meet diagnostic criteria for PMDD for the purposes of this trial, patients were required to demonstrate a 200% worsening on 1 core symptom or a 100% worsening on 2 or more symptoms during the luteal phase relative to their follicular phase score. Patients were also required to have a baseline Clinical Global Impressions-Severity of Illness scale (CGI-S)18 score of ≥ 3.
Patients were considered ineligible if they met DSM-IV criteria for other Axis I disorders (except specific phobias) in the previous 6 months, were diagnosed with gynecological or other clinically significant disease, had clinically significant depressive symptomatology during the follicular phase (defined as a Montgomery-Asberg Depression Rating Scale19 score ≥ 10 at screening), were a significant risk for suicide, were taking medication for PMDD symptoms, received previous adequate treatment or participated in a clinical trial with an SSRI for PMDD, or were breastfeeding or pregnant. Use of oral or systemic contraception during the study also precluded participation.
All potential patients provided signed informed consent prior to participation. The study protocol was reviewed and approved by local regulatory authorities and local Institutional Review Boards and was conducted in accordance with the Declaration of Helsinki (1996 revision) and under the principles of good clinical practice, as laid out in the International Conference on Harmonisation document Good Clinical Practice Consolidated Guideline.
Clinical Trial Methodology
Potential study candidates were evaluated based on entry criteria at an initial screening visit (Figure 1). Eligible patients were instructed to begin rating their daily PMDD symptoms in diaries using a visual analog scale (VAS) at the start of their next menstrual cycle, for 2 consecutive reference cycles. The use of a VAS as a valid and reliable assessment for mood symptoms is well documented.20 The VAS consists of a 100-mm horizontal line with descriptors that range from “not at all” (0 mm) to “extreme” (100 mm). The following 11 symptoms were recorded: irritability, tension, affective lability, depressed mood, decreased interest, difficulty concentrating, lack of energy, change in appetite, change in sleep pattern, feeling out of control, and physical symptoms. The primary efficacy variable, the “VAS-Mood” score, is a derived variable based on the composite score of the 4 core PMDD symptoms (irritability, tension, depressed mood, and affective lability) and is considered a valid, reliable, and sensitive instrument to measure changes in premenstrual mood symptoms.21 No medication was administered during the first reference cycle, while single-blind placebo was administered and taken on a daily basis during the second reference cycle. An additional reference cycle was made available to patients who met all entry criteria but failed to achieve the predefined level of cyclical worsening of core PMDD symptoms. Patients who successfully completed 2 consecutive reference cycles and met all entry criteria were randomly assigned by computer-generated randomization code in a 1:1:1 ratio to double-blind study medication that consisted of paroxetine CR 25 mg, paroxetine CR 12.5 mg, or similar-appearing placebo. Randomized patients were instructed to take study medication orally, once daily in the morning, continuously throughout the menstrual cycle.
Figure 1.

Study Design
After randomization, study visits were scheduled to occur within the first 3 days of the onset of menses for up to 3 treatment cycles. Patients continued to document their symptoms throughout the study by recording VAS scores on a daily basis for each of the 11 items in their diaries. Other efficacy assessments conducted at the visit included the CGI-S, the CGI-Improvement scale (CGI-I),18 and a patient-rated assessment of impairment (Sheehan Disability Scale [SDS]).22 Vital signs, laboratory data, and adverse events data were also collected during the study.
Patients who completed the study were eligible to enter into a 3-month extension study. Patients who withdrew during the study or were ineligible to enter the extension study were followed for up to 28 days after their last dose of study medication (follow-up phase).
Study Objectives
The primary objective of this study was to compare the efficacy of daily treatment throughout the menstrual cycle with paroxetine CR (12.5 mg/day or 25 mg/day) versus placebo. The secondary objective of the study was to assess the safety of treatment with paroxetine CR. The primary efficacy variable was the change in the mean luteal phase VAS-Mood scores from baseline to end of treatment cycle 3. Secondary outcome measures included change from baseline to treatment cycle 3 in the sum of the 11 VAS symptoms (VAS-Total), physical symptoms, social impairment, the proportion of patients showing response defined as a ≥ 50% reduction from baseline VAS-Mood scores, the proportion of patients showing response defined as a CGI-I item score of 1 (very much improved) or 2 (much improved), and the proportion of patients in remission defined as a VAS-Mood score less than or equal to the baseline mean follicular phase score.
Statistical Methods
A sample size of 86 evaluable patients in each of the 3 study arms provided 90% power to detect a difference of 16.3 mm (common standard deviation of 33.5 mm) between either dose of paroxetine CR and placebo in the change from baseline in the primary efficacy variable VAS-Mood. Allowing for a 20% attrition rate, this resulted in a target of 108 patients randomly assigned to each treatment group, a total of 324 patients overall. To adjust for the 2 paroxetine versus placebo comparisons, a 2-sided alpha level of 0.025 was used.
A normal, linear regression model was used to analyze the primary variable, adjusting for the following pre-specified covariates regardless of their significance: treatment group, center, baseline VAS-Mood score, and age (in years) at entry to the study. Centers with few patients were grouped with centers with larger numbers of patients, the smallest center being grouped with the largest center. The same model was fitted to each continuous secondary variable with the remaining responder endpoints analyzed using logistic regression (adjusting for the same covariates, except CGI-I, for which a baseline value does not exist). All models were fitted using the SAS computer package.23
For all efficacy measures, the primary conclusions were based on the treatment cycle 3 study endpoint using the last-observation-carried-forward (LOCF) approach to handle missing data. Supportive analyses were also performed on the observed cases data at each treatment cycle. All hypothesis testing was 2-sided. For the primary endpoint alone, an adjustment for the 2 treatment comparisons was made using Hochberg's modification to the Bonferroni inequality.24
For each of the individual VAS items, the patient's mean score was calculated for each luteal phase by averaging the item score over the last 5 days of the luteal phase prior to onset of menstruation. A mean item score was derived only when scores were available for at least 4 of the 5 designated days. The patient's mean luteal phase VAS-Mood score was then calculated as the mean of the luteal phase core item scores, provided that at least 3 scores were available.
Change from baseline values were calculated as the value at a particular treatment visit minus the patient's baseline measurement. For efficacy analyses, only those patients who had at least 1 postbaseline efficacy assessment were included.
RESULTS
Patients
The study was carried out in 47 outpatient centers across the United States and Canada from November 1999 until January 2002. A total of 1974 women were screened for the study during an initial assessment, of which 1603 were screen-only patients (Figure 2): 762 patients did not meet study screening or baseline entry criteria, 368 patients were lost to follow-up, 380 withdrew for other reasons (including withdrawal of consent, pregnancy, moving, etc.), 76 were withdrawn due to protocol deviations, and 17 patients were withdrawn due to a baseline (prerandomization) adverse event. The remaining 371 patients successfully completed the reference phase and were randomly assigned to double-blind study medication. As defined in the protocol, randomized patients were to be included in the intent-to-treat (ITT) population if they received at least 1 dose of double-blind study medication treatment and had at least 1 postbaseline assessment. Twelve randomized patients did not satisfy the latter criterion and were excluded from all efficacy and safety analyses: 5 patients in the 25-mg group, 6 in the 12.5-mg group, and 1 in the placebo group. Of the remaining 359 patients, 120 were randomly assigned to 25 mg of paroxetine CR, 115 to 12.5 mg of paroxetine CR, and 124 to placebo.
Figure 2.

Study Flow
Baseline Data
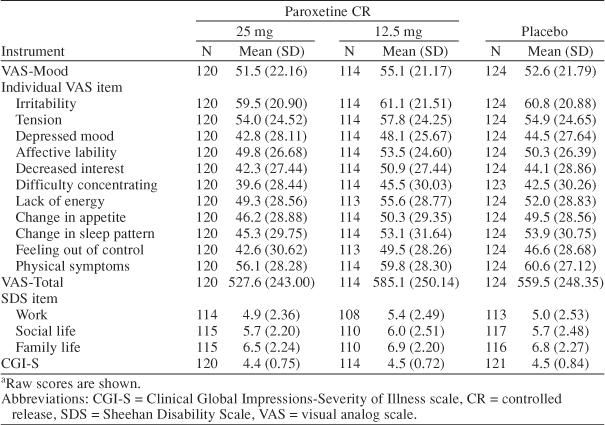
Baseline demographic characteristics and age at onset of PMDD for ITT patients were well balanced across treatment groups (Table 1). The mean age at study entry was 36 years, and the majority (94.7%) of patients were white. Additionally, there were no marked differences at baseline with respect to severity of PMDD symptoms, functional impairment as measured by the SDS items, or global assessments across treatment groups (Table 2). Global assessments of disease severity at baseline revealed the presence of moderate-to-marked illness that was accompanied by substantial impairment in areas of social functioning such as work life, social life, and family life. Mean baseline VAS-Mood scores ranged from 52 mm to 55 mm across treatment groups and were similar to baseline scores observed in previous PMDD studies.13–15,25
Table 1.
Baseline Characteristics (ITT population)
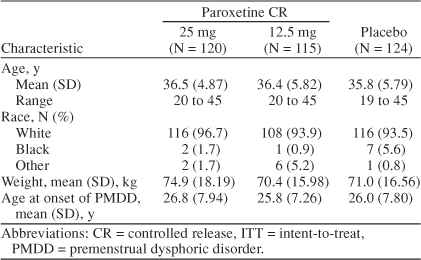
Table 2.
Baseline Scores in Efficacy Rating Scalesa
Efficacy Endpoints
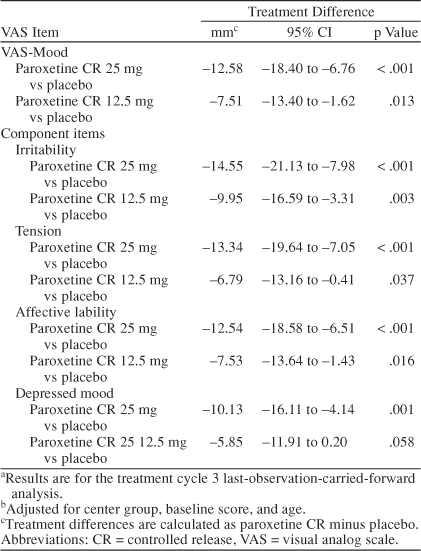
The mean scores in Table 3 show that improvements in VAS-Mood were observed within the first treatment cycle for all 3 treatment groups, and the improvements seen in the paroxetine CR groups (25 mg and 12.5 mg) were consistently superior to placebo through all treatment cycles. A summary of the adjusted mean change from baseline scores for the VAS-Mood and the component items is presented in Table 4. At study endpoint (treatment cycle 3 LOCF), a statistically significant difference was observed in favor of paroxetine CR 25 mg versus placebo on the VAS-Mood (adjusted mean difference = −12.58 mm, 95% CI = −18.40 to −6.76; p < .001). A statistically significant difference in VAS-Mood scores was also observed at endpoint in favor of paroxetine CR 12.5 mg versus placebo (adjusted mean difference = −7.51 mm, 95% CI = −13.40 to −1.62; p = .013). In a supplementary analysis of the VAS-Mood, no statistically significant interactions were found between treatment and the other main effects. As shown in Table 4, the benefit of paroxetine CR on VAS-Mood scores was observed in each of the 4 core mood symptoms of PMDD: irritability, tension, affective lability, and depressed mood.
Table 3.
VAS-Mood Scores by Treatment Cyclea
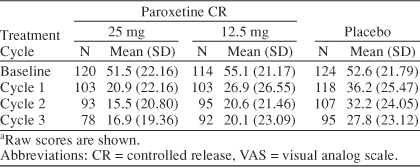
Table 4.
Summary of Adjusted Mean Change From Baseline in VAS-Mood Item Scoresa,b
Paroxetine CR also demonstrated greater mean reductions in VAS-Total scores compared with placebo at each timepoint (Figure 3). At the treatment cycle 3 LOCF endpoint, statistically significant differences in mean changes were observed in favor of paroxetine CR 25 mg versus placebo (adjusted mean difference = −123.72 mm, 95% CI = −183.21 to −64.23; p < .001) as well as for paroxetine CR 12.5 mg versus placebo (adjusted mean difference = −78.18 mm, 95% CI = −138.3 to −18.10; p = .011).
Figure 3.

Mean VAS-Total Score by Cyclea
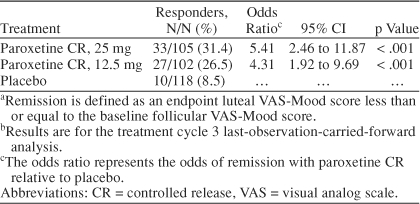
An analysis of response based on a 50% reduction from baseline luteal VAS-Mood score showed that 76% of patients randomly assigned to 25 mg of paroxetine CR and 67% of patients randomly assigned to 12.5 mg of paroxetine CR met criteria for a satisfactory treatment response at endpoint. The response rate among placebo patients was lower, at 50%. In logistic regression analysis, the estimated odds of responding on paroxetine CR treatment were between 2 to 3 times higher than responding to placebo and statistically significant for both doses of paroxetine CR when compared with placebo (Table 5). An analysis of remission defined as an endpoint luteal VAS-Mood score less than or equal to the baseline mean follicular phase score also demonstrated that paroxetine CR was superior to placebo. Remission rates for the paroxetine CR 25-mg and 12.5-mg groups were 31% and 27%, respectively, compared with a 9% rate among placebo patients. The corresponding odds of achieving remission on paroxetine CR treatment were 4 to 5 times greater than placebo (Table 6).
Table 5.
Response Based on 50% Reduction From Baseline Luteal VAS-Mood Scores at Endpointa
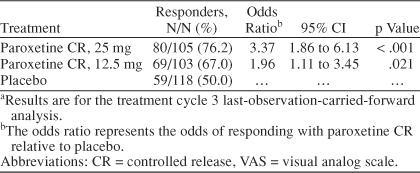
Table 6.
Remissiona Based on VAS-Mood Scoresb
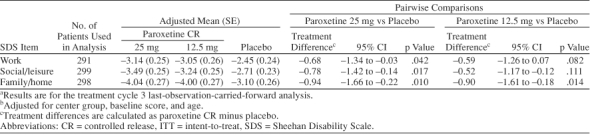
Analysis of social functioning based on changes in the SDS individual item scores (work, social, and family life) showed that paroxetine CR was superior to placebo in improving impairment associated with each of these domains. Paroxetine CR 25 mg was statistically significantly superior to placebo in each domain, while 12.5 mg showed statistical superiority over placebo in family life alone (Table 7).
Table 7.
Summary of Analysis of Adjusted Change From Baseline in SDS Individual Item Scores (ITT population)a,b
Tolerability
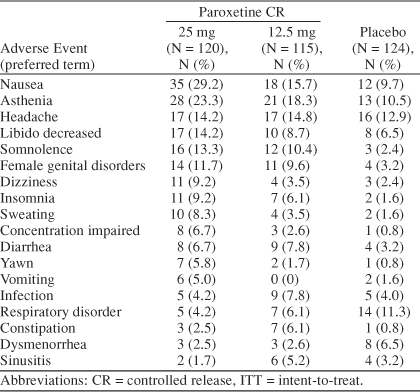
Paroxetine CR was generally well tolerated during this 3-month clinical trial. A total of 41 (11.4%) of the 359 ITT patients were withdrawn from the study due to an adverse event. These were 20 patients (17%) from the paroxetine CR 25-mg group, 12 patients (10%) from the paroxetine CR 12.5-mg group, and 9 patients (7%) from the placebo group. The most common adverse event leading to withdrawal was asthenia, which occurred in 7 patients in the 25-mg group, 3 patients in the 12.5-mg group, and 3 patients in the placebo group. The proportion of patients experiencing at least 1 treatment-emergent adverse event, defined as any adverse event that emerged during treatment or that worsened relative to the pretreatment state, was 86% in the paroxetine CR 25-mg group, 74% in the paroxetine CR 12.5-mg group, and 64% in the placebo group. The most common adverse events (≥ 5% in either paroxetine group and twice the rate of placebo) were nausea, asthenia, decreased libido, somnolence, female genital disorders (e.g., anorgasmia, difficulty reaching or achieving orgasm, delayed orgasm, and sexual dysfunction), dizziness, insomnia, sweating, concentration impaired, diarrhea, yawn, vomiting, and constipation (Table 8). Changes in mean weight from baseline to treatment cycle 3 were similar across treatment groups.
Table 8.
Treatment Phase–Emergent Adverse Events (ITT population) Most Frequently Reported (≥ 5% in any treatment group)
There were 156 patients observed during the follow-up phase of the study (25 mg, N = 52; 12.5 mg, N = 43; placebo, N = 61). The most common adverse event reported during this phase was dizziness, reported in 5 patients in the 25-mg group, 4 patients in the 12.5-mg group, and none in the placebo group.
DISCUSSION
This study demonstrated that paroxetine CR, at doses of 25 mg/day and 12.5 mg/day, is effective in treating the core mood symptoms of PMDD as measured by the primary measure of efficacy, the VAS-Mood. This primary outcome parameter is comprised of measures of irritability, tension, depressed mood, and affective lability. A statistically significant improvement was demonstrated in all of these symptoms individually, as well as on the composite VAS-Mood scale for both doses of paroxetine CR compared with placebo. Results also showed that both doses of paroxetine CR were significantly superior to placebo on promoting changes on the VAS-Total, the sum of all 11 symptoms that compose the DSM-IV criteria for PMDD (irritability, tension, affective lability, depressed mood, decreased interest, difficulty concentrating, lack of energy, change in appetite, change in sleep pattern, feeling out of control, and physical symptoms). Similar to previous findings with fluoxetine13 and sertraline,7 the improvement in PMDD symptoms was observed within the first treatment cycle.
Additional corroborative evidence supporting the efficacy of 25 mg and 12.5 mg of paroxetine CR was provided by a responder analysis based on clinically relevant reductions in PMDD symptomatology. Up to 76% of patients treated with paroxetine CR 25 mg and 67% of patients treated with paroxetine CR 12.5 mg were considered responders, defined as a 50% reduction from baseline VAS-Mood scores. These response rates are comparable to the response rates seen with other SSRIs in PMDD such as fluoxetine (52%)13 and sertraline (62%)7.
In this study, patients receiving paroxetine CR were up to 5 times more likely to achieve remission compared with patients receiving placebo. Given that only 9% of placebo patients realized this level of improvement, it could be suggested that in addition to being a clinically meaningful endpoint, remission, as defined here, serves as a valuable discriminator between drug and placebo.
While these results suggest additional improvement for the 25-mg paroxetine CR group relative to the 12.5-mg paroxetine CR group for efficacy, the safety data suggest increased tolerability for the 12.5-mg paroxetine CR group, with only 10% of patients dropping out due to adverse events (17% for paroxetine CR 25 mg and 7% for placebo). This is in contrast to previous reports, which indicated that higher doses of fluoxetine (60 mg/day) were of no additional benefit in treating PMDD compared with a lower dose (20 mg/day) and caused increased adverse events.13
The strict entry criteria for this study assured that the appropriate population of PMDD subjects was studied. The mean follicular phase VAS-Mood score at baseline for all 3 treatment groups was between 6.0 and 6.5, while the corresponding mean luteal phase score was between 51.5 and 55.1, representing a substantial change between follicular and luteal phases, consistent with the diagnosis of PMDD. The number of subjects randomly assigned in relation to the number of patients screened (371/1974, 19%) demonstrates the stringent requirements for a diagnosis of PMDD and the complexity of prospective assessments of the cyclical nature of such symptoms.
Consistent with previous studies, this study population of subjects with PMDD demonstrated a substantial level of functional impairment at baseline. Baseline SDS scores were among the highest documented levels of impairment associated with a mental disorder.26 This was particularly apparent for the family life domain, which measures impairment in the patient's ability to relate to family members and conduct routine daily activities such as managing the home, shopping, and cleaning. Paroxetine CR was associated with improvement in the patient's family, social, and work life.
Although data from this study, conducted in both the United States and Canada, are comparable to a recent study of controlled-release paroxetine conducted solely in the United States,15 interpretation of these data is limited by several factors. One constraint is the homogeneous population studied in this trial (i.e., 95% white). The population in this trial also met criteria for moderate-to-severe PMDD without psychiatric comorbidity. Future studies should include ethnic minorities, as well as a broader spectrum of symptom severity and psychiatric co-morbidity, to increase the generalizability of the benefit observed in patients in this study.
In conclusion, paroxetine CR at doses of both 12.5 mg/day and 25 mg/day is effective in treating symptoms of PMDD and in improving social functioning. Response to treatment can be expected within the first treatment cycle, and most patients (up to 76% in this sample) demonstrated a significant improvement at endpoint in the key symptoms of irritability, tension, affective lability, and depressed mood.
Drug names: citalopram (Celexa and others), fluoxetine (Sarafem and others), paroxetine (Paxil and others), sertraline (Zoloft).
Financial disclosure: Dr. Pearlstein has received grant/research support from Ortho-McNeil, Berlex, Warner Chilcott, and GlaxoSmithKline; has received honoraria from Warner Chilcott and GlaxoSmithKline; and has participated in speakers/advisory boards for Berlex, GlaxoSmithKline, and Warner Chilcott. Mr. Bellew is an employee and major stock shareholder of GlaxoSmithKline. Dr. Endicott is a consultant for and has received honoraria from Pfizer, Eli Lilly, Berlex, GlaxoSmithKline, AstraZeneca, Merck, Otsuka, Wyeth, Sanofi-Synthelabo, and Janssen; has received grant/research support from Pfizer; and has received other financial or material support from Eli Lilly. Dr. Steiner has been a consultant for, received grant/research support and honoraria from, and has participated in speakers/advisory boards for Eli Lilly, Pfizer, GlaxoSmithKline, Lundbeck, Novartis, Wyeth, Barr Laboratories, Proctor & Gamble, Ortho-McNeil, Warner Chilcott, AstraZeneca, and Serenix.
Paroxetine CR Study Investigators: Phyllis Marx, M.D.; Jay Cooper, M.D.; Donald England, M.D.; Linda Frison, M.D.; Walter Hood, M.D.; Matthew Menza, M.D.; Peter Holland, M.D.; Gary Horowitz, M.D.; Rakesh Jain, M.D.; James Kyserm, M.D.; Robert Nordland, M.D.; Stephanie Seremetis, M.D.; David Franck, M.D.; Ward Smith, M.D.; Diana Dell, M.D.; Anna Stout, Ph.D.; Lawrence Adler, M.D.; Raul Jimenez, M.D.; Barbara Levy, M.D.; Paras Harashawat, M.D.; Abbey Strauss, M.D.; Anil Patel, M.D.; Steven Targum, M.D.; Max Maizels, M.D.; Irwin Kerber, M.D.; Marc Hertzman, M.D.; Lance Holeman, M.D.; Ari Albala, M.D.; William Fuller, M.D.; Neil Kaye, M.D.; Clifford Goldman, M.D.; Sneh Kapila, M.D.; Janice Miller, M.D.; Richard Bergeron, M.D.; Louise Laberge, M.D.; Pratap Chokka, M.D.; Norman Costigan, M.D.; Dan Dattani; Marie-Jose Filteau; Kevin Kjernisted, M.D.; Paul Lesperance, M.D.; Autar Munshi, M.D.; Margaret Oakander, M.D.; Jeannette Janzen, M.D.; Paul Whitsitt, M.D.; Louis van Zyl; Benjamin Lasko; Joanne MacDonald, M.D.; Anthony Levitt, M.D.; Geeta Achyuthan, M.D.; Celine Bouchard, M.D.; and Manon Therien, M.D.
Footnotes
Funding for this study (protocol number, 29060/689) was provided by GlaxoSmithKline, King of Prussia, Pa.
Presented in part at the 41st annual meeting of the American College of Neuropsychopharmacology (ACNP), December 8–12, 2002, San Juan, Puerto Rico.
Financial disclosure appears at the end of the article.
Study investigators are listed at the end of the article.
REFERENCES
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. Washington, DC: American Psychiatric Association. 1994 [Google Scholar]
- Johnson SR, McChesney C, Bean JA.. Epidemiology of premenstrual symptoms in a non-clinical sample, 1: prevalence, natural history and help-seeking behavior. J Reprod Med. 1988;33:340–346. [PubMed] [Google Scholar]
- Rivera-Tovar AD, Frank E.. Late luteal phase dysphoric disorder in young women. Am J Psychiatry. 1990;147:1634–1636. doi: 10.1176/ajp.147.12.1634. [DOI] [PubMed] [Google Scholar]
- Wittchen HU, Becker E, and Lieb R. et al. Prevalence, incidence and stability of premenstrual dysphoric disorder in the community. Psychol Med. 2002 32:119–132. [DOI] [PubMed] [Google Scholar]
- Sternfeld B, Swindle R, and Chawla A. et al. Severity of premenstrual symptoms in a health maintenance organization population. Obstet Gynecol. 2002 99:1014–1024. [DOI] [PubMed] [Google Scholar]
- Cohen LS, Soares CN, and Otto MW. et al. Prevalence and predictors of premenstrual dysphoric disorder (PMDD) in older premenopausal women: The Harvard Study of Moods and Cycles. J Affect Disord. 2002 70:125–132. [DOI] [PubMed] [Google Scholar]
- Yonkers KA, Halbreich U, and Freeman E. et al. Symptomatic improvement of premenstrual dysphoric disorder with sertraline treatment: a randomized controlled trial. JAMA. 1997 278:983–988. [PubMed] [Google Scholar]
- Pearlstein TB, Halbreich U, and Batzar ED. et al. Psychosocial functioning in women with premenstrual dysphoric disorder before and after treatment with sertraline or placebo. J Clin Psychiatry. 2000 61:101–109. [DOI] [PubMed] [Google Scholar]
- Robinson RL, Swindle RW.. Premenstrual symptom severity: impact on social functioning and treatment-seeking behaviors. J Womens HealtV Gend Based Med. 2000;9:757–768. doi: 10.1089/15246090050147736. [DOI] [PubMed] [Google Scholar]
- Hylan TR, Sundell K, Judge R.. The impact of premenstrual symptomatology on functioning and treatment-seeking behavior: experience from the United States, United Kingdom, and France. J Womens Health Gender Based Med. 1999;8:1043–1052. doi: 10.1089/jwh.1.1999.8.1043. [DOI] [PubMed] [Google Scholar]
- Steiner M, Pearlstein T. Premenstrual dysphoria and the serotonin system: pathophysiology and treatment. J Clin Psychiatry. 2000 61suppl 12. 17–21. [PubMed] [Google Scholar]
- Pearlstein T.. Selective serotonin reuptake inhibitors for premenstrual dysphoric disorder: the emerging gold standard? Drugs. 2002;62:1869–1885. doi: 10.2165/00003495-200262130-00004. [DOI] [PubMed] [Google Scholar]
- Steiner M, Steinberg S, and Stewart D. et al. Fluoxetine in the treatment of premenstrual dysphoria. Canadian Fluoxetine/Premenstrual Dysphoria Collaborative Study Group. N Engl J Med. 1995 332:1529–1534. [DOI] [PubMed] [Google Scholar]
- Eriksson E, Hedberg MA, and Andersch B. et al. The serotonin inhibitor paroxetine is superior to the noradrenaline reuptake inhibitor maprotiline in the treatment of premenstrual syndrome. Neuropsychopharmacology. 1995 12:167–176. [DOI] [PubMed] [Google Scholar]
- Cohen LS, Soares CN, and Yonkers KA. et al. Paroxetine controlled release for premenstrual dysphoric disorder: a double-blind, placebo-controlled trial. Psychosom Med. 2004 66:707–713. [DOI] [PubMed] [Google Scholar]
- Wikander I, Sundblad C, and Andersch B. et al. Citalopram in premenstrual dysphoria: is intermittent treatment during luteal phases more effective than continuous medication throughout the menstrual cycle? J Clin Psychopharmacol. 1998 18:390–398. [DOI] [PubMed] [Google Scholar]
- Endicott J, Amsterdam J, and Eriksson E. et al. Is premenstrual dysphoric disorder a distinct clinical entity? J Womens Health Gend Based Med. 1999 8:663–679. [DOI] [PubMed] [Google Scholar]
- Guy W. ed. ECDEU Assessment Manual for Psychopharmacology, Revised. US Dept Health, Education, and Welfare publication (ADM) 76-338. Rockville, Md: National Institute of Mental Health. 1976 [Google Scholar]
- Montgomery SA, Asberg MC.. A new depression rating scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382–389. doi: 10.1192/bjp.134.4.382. [DOI] [PubMed] [Google Scholar]
- McCormack HM, Horne DJ, Sheather S.. Clinical applications of visual analogue scales: a critical review. Psychol Med. 1988;18:1007–1019. doi: 10.1017/s0033291700009934. [DOI] [PubMed] [Google Scholar]
- Steiner M, Streiner DL, and Steinberg S. et al. The measurement of premenstrual mood symptoms. J Affect Disord. 1999 53:269–273. [DOI] [PubMed] [Google Scholar]
- Sheehan DV, Harnett-Sheehan K, and Raj BA. The measurement of disability. Int Clin Psychopharmacol. 1996 11suppl 3. 89–95. [DOI] [PubMed] [Google Scholar]
- SAS Version 8.1. SAS Institute Inc: Cary, NC. 1999. [Google Scholar]
- Hochberg Y.. A sharper Bonferroni procedure for multiple tests of significance. Biometrica. 1988;75:800–802. [Google Scholar]
- Sundblad C, Wikander I, and Andersch B. et al. A naturalistic study of paroxetine in premenstrual syndrome: efficacy and side-effects during ten cycles of treatment. Eur Neuropsychopharmacol. 1997 7:201–206. [DOI] [PubMed] [Google Scholar]
- Olfson M, Fireman B, and Weissman MM. et al. Mental disorders and disability among patients in a primary care group practice. Am J Psychiatry. 1997 154:1734–1740. [DOI] [PubMed] [Google Scholar]