

Abstract
Nucleotidyl transfer is an archetypal enzyme reaction central to DNA replication and repair. Here we describe a variation of the nucleotidylation reaction termed “catch and release” that is used by an antibiotic modifying enzyme. The aminoglycoside nucleotidyl transferase 4’ (ANT4’) inactivates antibiotics such as kanamycin and neomycin through nucleotidylation within an active site that shares significant structural, and inferred underlying catalytic similarity, with human DNA polymerase beta. Here we follow the entire nucleotidyl transfer reaction coordinate of ANT4’ covalently inactivating neomycin using X-ray crystallography. These studies show that although the underlying reaction mechanism is conserved with polymerases, a short 2.35 A hydrogen bond is initially formed to facilitate tight binding of the aminoglycoside substrate and is subsequently disrupted by the assembly of the catalytically active ternary complex. This enables the release of products post catalysis due to a lower free energy of the product state compared to the starting substrate complex. We propose that this “catch and release” mechanism of antibiotic turnover observed in ANT4’ is a variation of nucleotidyl transfer that has been adapted for the inactivation of antibiotics.
Keywords: nucleotidyl transfer, antibiotic resistance, short hydrogen bond, aminoglycoside, DNA polymerase
Graphical Abstract
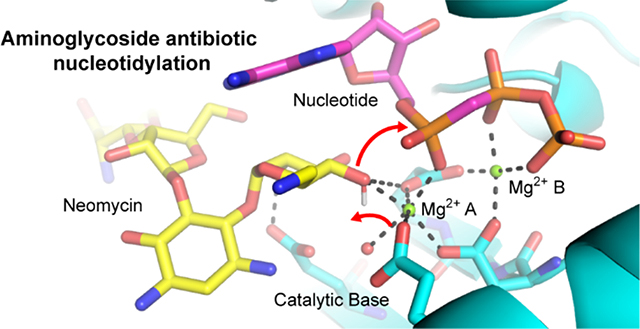
Nucleotide triphosphates (NTP) are ubiquitous enzyme substrates and cofactors in a multitude of biological processes, including DNA synthesis. NTPs are central to polymerase mediated DNA synthesis as substrates in the highly conserved nucleotidyl transfer reaction1–2. Studies of polymerase-mediated nucleotidyl transfer demonstrate how divalent metal ions and active site carboxylate groups deprotonate and activate a hydroxyl oxygen on the DNA primer terminus. Through attack of the alpha phosphate of the NTP, the activated nucleophile catalyzes the formation of a phosphodiester bond while generating a pyrophosphate leaving group3–7. A catalytic metal is involved in lowering the pKa of the DNA 3’ OH primer terminus (metal A), whereas a second nucleotide binding metal (metal B) coordinates the pyrophosphate moiety of the nucleotide (Figure 1A). The two metals are coordinated by three conserved active site carboxylate groups.
Figure 1.

Conservation of nucleotidyl transfer active sites. A. Close-up view of the Pol b ternary complex active site (PDB code 2FMS). Protein is colored in cyan, the NTP is shown with magenta carbon atoms, and the DNA primer terminus is shown with yellow carbon atoms. In Pol b, the nucleotide binding metal (Mg B) is ligated with the b and g phosphates of the nucleotide triphosphate, as well as Asp190 and Asp192. The catalytic metal (Mg A) is coordinated by all three aspartate residues, including the catalytic base (Asp256) and the activated nucleophile hydroxyl from the carbohydrate moiety. Hydrogen bonds are represented as black dashed lines. B. Close-up view of the D80Y ANT4’ ternary complex active site (PDB code 1KNY). Protein is colored in cyan, nucleotide substrate is shown with magenta carbon atoms, and kanamycin is shown with carbon atoms colored green. Conserved active site carboxylate groups of D80Y ANT4’ and Pol b are shown and labelled in the same color. The nucleophilic oxygen on neomycin is circled in red. In ANT4’ Asp50 and Glu52 are homologous to Asp190 and Asp192 in Pol b. C. The proposed nucleotidyl transfer reaction of ANT4’. The Mg2+ ions involved in the reaction coordinate are represented as green spheres. In these studies, the far left diagram was captured crystallo-graphically (ATP was replaced with non-hydrolyzable AMCPCPP) as well as the far right diagram.
Antibiotic resistance enzymes, such as aminoglycoside modifying enzymes (AGMEs) use the nucleotidyl transfer reaction to inactivate antibiotics8. The covalently modified antibiotic is no longer able to form the requisite interactions with its molecular target, the bacterial ribosome, thus rendering the antibiotic ineffective9. One such AGME is the aminoglycoside nucleotidyl transferase 4’ (ANT4’) from Staphylococcus aureus. ANT4’ is among the more promiscuous AGMEs10–11 which inactivates aminoglycosides from both of the two main structural classes of aminoglycoside antibiotics (Figure S1) through transfer of a nucleoside monophosphate group from ATP to the 4’-hydroxyl group of the antibiotic12–14. A two metal ion nucleotidyl transfer reaction mechanism similar to polymerases has been proposed for ANT4’, as the structural homology between the ANT4’ catalytic site and the human base-excision repair DNA polymerase, polymerase beta (Pol b), was identified when the X-ray crystal structure was first solved15–16 (Figure 1B). More recently, this mechanism has been suggested to be used by several related aminoglycoside nucleotidyl transfer enzymes17–20, despite lacking a complete structural understanding of the reaction coordinate. Demonstrating the similarity underlying nucleotidyl transfer reaction among ANT4’ and Pol b, the active site carboxylate groups and substrates are also arranged in a similar manner within these enzymes (Figure 1). In ANT4’ Glu76 coincides with the Pol b catalytic base (Asp256), and kinetic studies have demonstrated the importance of this residue in ANT4’21. However, unlike polymerases, ANT4’ forms a homodimeric quaternary structure. Although there are ANT enzymes that are monomeric17–18, the ANT4’ homodimer leads to the placement of an additional carboxylate group into the active site (Glu145) from an adjacent monomer. Glu145 also functions as an alternate catalytic base in the proposed ANT4’ reaction mechanism14· 21 (Figure IB).
A detailed understanding of the various catalytic mechanisms utilized by modifying enzymes is required to decipher the basis of antibiotic inactivation, which can aid in the design of new aminoglycosides. Here, we determined the crystal structure of ANT4’ in the binary (neomycin), ternary substrate (neomycin and AMPCPP), and product complex (AMP-neomycin and pyrophosphate), thus permitting a complete picture of the reaction coordinate of this enzyme. Based on these structures and structures of an active site mutant, as well as isothermal calorimetry (ITC) binding studies, we demonstrate how ANT4’ uses a variation of the classical nucleotidyl transfer mechanism, which we call “catch and release”. Through modulation of a strong, short hydrogen bond (SHB), which is either a short-ionic hydrogen bond (SIHB) or a low-barrier hydrogen bond (LBHB)22, ANT4’ tightly binds the aminoglycoside antibiotic in the absence of a fully-assembled active site. Formation of the complete active complex results in disruption of the SHB and a lower free energy of the product state.
The processivity of polymerases is evolutionarily encoded in the need to bind to a DNA template and perform multiple rounds of nucleotidyl transfer without release, yet we propose this variation in the reaction is a mechanism to aid in product release and antibiotic turnover. These new insights into this reaction archetype not only lead to a better understanding of nucleotidyl transfer, but also shed light on the strategies that can be used to evade the action of antibiotics by the pathologically relevant AGME family23.
Results and Discussion
Crystallographic snapshots of wild-type ANT4’ catalysis
A total of 6 structures of a thermally stable T130K mutant of ANT4’ were determined with resolutions ranging between 1.65 A and 2.50 A (Tables S1 and S2). A binary neomycin complex was also determined in the wild-type ANT4’ enzyme to confirm aminoglycoside binding is not perturbed by this mutation (Figure 2A and Table S1). Polder omit maps (Figure S2) as well as LigPlot+24 interaction analysis (Figures S3 and S4) for all ligands are shown in the Supplement.
Figure 2.

Comparison of ANT4’ mutant structures. A. Superposition of wild-type ANT4’ (magenta carbon atoms) bound to neomycin (green carbon atoms) with T130K ANT4’ (cyan carbon atoms) bound to neomycin (yellow carbon atoms). Hydrogen bonds to the T130K-neomycin complex are shown as black dashed lines. B. Superposition of D80Y ANT4’ and the T130K ANT4’ ternary complexes. The D80Y ANT4’ mutant is shown with kanamycin (green carbon atoms) and AMPNPP (magenta carbon atoms). The T130K ANT4’ mutant is shown with neomycin (yellow carbon atoms) and AMPCPP (magenta carbon atoms). The 4’ hydroxyl, the site of adenylation, is circled in black, and the alpha phosphate is circled in red.
The T130K mutation is on the surface of ANT4’ and more than 21 A away from the enzyme active site. Earlier studies have shown that the thermodynamics of ligand binding and solvent effects25–26 as well as protein dynamics27 of wild-type ANT4’ and the T130K variant are very similar. Another thermostable mutant of ANT4’ (D80Y), was used in the previously determined structure of ANT4’ in the presence of kanamycin16. Neomycin was used in our studies with the T130K mutant because D80Y has significantly different thermodynamics of enzyme-ligand, protein-solvent interactions and protein dynamics relative to wild-type and T130K variants of ANT4’25–26.
The ANT4’ neomycin binding site in the wild-type crystal structure is identical to the T130K mutant complex and occurs within a pocket lined with negatively charged hydrogen bonding groups that form salt-bridges with the positively charged aminoglycoside (Figure 2A). The prime ring of neomycin is placed directly into the active site composed of three carboxylate groups in a manner identical to the canonical DNA polymerase active site, yet it differs from the orientation of the kanamycin 4’ hydroxyl in the active site of the D80Y mutant of ANT4’16 (Figure 2B). In the T130K mutant the position of the 4’ hydroxyl of neomycin, the site of adenylation, overlaps with the position of the 3’ OH of the DNA primer terminus of Pol p. This places the neomycin 4’ nucleophile in a catalytically competent position (3.1 Â between atoms) to attack the alpha phosphate of the nucleotide. This is in contrast to the orientation of kanamycin in the D80Y ANT4’ mutant (Figure 2B), where the 4’ hydroxyl is positioned 5.3 A away from the alpha phosphate site of attack28. The neomycin ANT4’ complex was crystallized in the presence of high concentrations of Mg2+ (100–200 mM). A Mg ion (termed Mg -A) is present and coordinated by three water molecules, Glu52, Glu145 and the 4’ OH of neomycin (Figure 3A). One of the active site carboxylate groups (Glu52) forms a short 2.35 A hydrogen bond with the 3’ hydroxyl of neomycin (shown as red dotted lines in Figure 3A). The distance between heavy atoms suggests this rather short hydrogen bond is either a SIHB or a LBHB, where matched pKa values of heavy atoms lead to resonance stabilization and an additional ~5–10 kcal more energy versus a canonical hydrogen bond22.
Figure 3.

Crystallographic snapshots of the T130K ANT4’ reaction coordinate. A. The T130K ANT4’ neomycin complex active site. The SHB formed between the Glu52 and the neomycin 3’ OH is represented as a red dashed line. B. The ternary (neomycin-AMPCPP) ANT4’ active site. C. The product state (adenylated neomycin) ANT4’ active site. T130K ANT4’ is shown with cyan ribbon representation with yellow carbon atoms for neomycin and magenta carbon atoms for AMPCPP or AMP. Select hydrogen bonds in the active site are represented as black dashed lines. The complete hydrogen bonding interactions are diagramed in Figure S3.
Upon formation of a non-hydrolyzable ternary complex (neomycin/AMPCPP) the catalytic Mg2+ (Mg A) and nucleotide binding Mg2+ (Mg B) coordinate to the 4’ OH and the b and g phosphates respectively (Figure 3B). Mg A is coordinated by Glu145, Asp50, Glu52 and the 4’ OH of the neomycin, which is likely in a deprotonated form as the distance from Mg A is 2.1 A. Glu52 also coordinates Mg B, thus this residue forms a bidentate hydrogen bond with both metal ions when the active site is fully assembled for catalysis. The engagement of the two metal ions by Glu52 causes the SHB to the 3’ OH to be lost, with the electronegativity of the carboxylate group drawn to the two positively charged metal ions, rather than forming interactions with the aminoglycoside. The adenine ring of the nucleotide stacks directly above the prime ring of neomycin where it forms van der Waals interactions that likely partially compensate for the loss of the SHB upon engagement of the two Mg ions by Glu52.
In a manner similar to the time-resolved study of nucleotidyl transfer in polymerases3, 5, the adenylated neomycin ANT4’ product as well as the pyrophosphate leaving group were crystallo-graphically captured by soaking ANT4’ ternary crystals (ATP and neomycin) in magnesium, which were initially inhibited from turning over substrate due to presence of Ca2+ (Figure 3C). In addition to the two products (AMP-neomycin and pyrophosphate) the catalytic metal and the nucleotide binding metal were still present in the active site. Rather than forming a SHB with neomycin, Glu52 completely engages the catalytic Mg ion. Only a single hydrogen bond (Glu76) remains formed with the adenylated prime ring of neomycin. The product-mediated reorganization of the hydrogen bonding network, including loss of the SHB, likely leads to a significantly weaker interaction energy with the product state. Indeed, multiple rounds of catalysis occurred within the confines of the crystal, which is evident by an additional adenylated neomycin located adjacent to the active site (Figure S5).
Crystallographic snapshots of ANT4’ catalysis lacking the short hydrogen bond
To test the proposed role of the SHB in ANT4’ catalysis the crystal structures of a T130K/E52D double mutant were determined in a binary neomycin form, a ternary (neomycin and AMPCPP) form and a product state (AMP-neomycin and pyrophosphate). Polder omit maps as well as LigPlot+ interaction analysis for all T130K/E52D structures are shown in Figures S2 and S4, respectively24. The lack of the delta carbon in aspartic acid results in lengthening of the SHB in the T130K/E52D binary complex (Figure 4A), where instead a longer and weaker 3.2 A canonical hydrogen bond is found. Upon formation of the non-hydrolyzable ternary complex, the geometry of the active site is identical to the T130K variant (Figure 4B). We note that both the T130K and the T130K/E52D mutants are able to perform catalysis in the crystal, and the organization of the active site and products are similar (Figure 4C) with the exception of Asp52 forming a bifurcated hydrogen bond to the both metal ions.
Figure 4.

Crystallographic snapshots of the T130K/E52D ANT4’ reaction coordinate. A. Superposition of the T130K/E52D ANT4’ neomycin bound active site with the T130K ANT4’ neomycin complex. The 3.2 Â hydrogen bond to the neomycin 3’ OH is represented as a red dashed line. B. Superposition of the T130K/E52D ANT4’ ternary complex (neomycin and AMPCPP) active site with the T130K ANT4’ ternary complex. C. Superposition of the T130K/E52D ANT4’ product state (adenylated neomycin) active site with T130K ANT4’ product complex. The T130K/E52D ANT4’ is shown with magenta ribbon representation with yellow carbon atoms for neomycin and magenta carbon atoms for AMPCPP or AMP. T130K ANT4’ is shown with all gray carbon atoms. Select hydrogen bonds in the active site are represented as black dashed lines. The complete hydrogen bonding interactions are diagramed in Figure S4.
Thermodynamic role of the short hydrogen bond in ANT4’
To support our structural interpretation of ANT4’ catalysis, substrate dissociation constants (Kd) were determined using isothermal calorimetry (ITC) for the T130K and T130K/E52D ANT4’ mutants (Table 1, Figure S6). Consistent with replacement of the SHB with a canonical longer hydrogen bond, the T130K/E52D mutant binds to neomycin 2.5-fold weaker than the T130K enzyme (Table 1, Kd values in the presence of 4.0 mM Mg2+ for T130K and T130K/E52D). However, ANT4’ crystallized in the presence of high amounts of divalent metal ions (100–200mM), which is far higher than the typical free cellular Mg2+ concentration of <1.0 mM. ITC titrations of the apo protein with Mg2+ failed to produce measurable heats of injection, which suggests the Mg2+ in the neomycin complex is due to crystallization conditions. The neomycin Kd values were therefore also measured in the absence of magnesium (Table 1). The strength of neomycin binding is modulated in response to the magnesium in the binding site as the Kd for neomycin binding to T130K ANT4’ in the absence of Mg2+ is five times smaller, indicating tighter binding than when titrated in the presence of 4 mM magnesium (Table 1). Moreover, in the absence of Mg2+ the T130K/E52D mutant binds to neomycin 4.3 times weaker than the T130K enzyme, equivalent to a loss 0.9 kcal/mol for substitution of the SHB for a canonical hydrogen bond (Table 1, Kd values in the absence of magnesium for T130K and T130K/E52D). In direct support of the loss of the hydrogen bond between residue 52 and neomycin upon formation of the ternary state, the Kd values for neomycin binding in the presence of AMPCPP are identical in the T130K and T130K/E52D mutants (Table 1). Interestingly, the T130K Kd for binding to this state in the absence of Mg2+ is only two-fold weaker than the formation of the binary aminoglycoside complex. It is likely that the loss of the SHB from the binary neomycin complex is partially compensated by the stacking interaction between the adenine ring and the prime ring of neomycin in the ternary non-catalytic complex.
Table 1.
ITC binding parameters of ANT4’
| Enzyme | Ligand | N | Kd (μM) |
|---|---|---|---|
| T130K | neomycin | 0.9 ± 0.01 | 0.34 ± 0.03 |
| T130K (No Mg2+) | neomycin | 1.0 ± 0.01 | 0.07 ± 0.01 |
| T130K/E52D | neomycin | 0.8 ± 0.01 | 0.87 ± 0.08 |
| T130K/E52D (No Mg2+) | neomycin | 1.0 ± 0.02 | 0.30 ± 0.10 |
| T130K/AMPCPP | neomycin | 0.7 ± 0.004 | 0.13 ± 0.02 |
| T130K/E52D/AMPCPP | neomycin | 0.7 ± 0.001 | 0.12 ± 0.03 |
Conclusions
The variation of the nucleotidyl transfer reaction found in ANT4’ demonstrates how this ubiquitous reaction archetype can be tuned to function in biological processes beyond nucleic acid synthesis29. The SHB found in ANT4’ may be the result of adapting an ancient DNA polymerase to perform chemistry on what in comparison would be a unique class of substrates. Indeed, DNA polymerases perform multiple rounds of nucleotidyl transfer on DNA substrates that are at high concentrations in the nucleus, which also consist of thousands of atoms that form numerous interactions with the protein, yet for example, the Kd for the Pol b binary oligo-nucleotide complex is around 0.5–2.0 pM30. This represents a ~10-fold weaker interaction when compared to the ANT4’ neomycin Kd, although there are well over 10-fold fewer atoms found in neomycin versus a double stranded DNA oligo-nucleotide.
The SHB of ANT4’ plays a unique role in this variant of nucleotidyl transfer where trace amount of antibiotics needs to bind tightly pre-catalysis and only a single round of catalysis needs to occur per binding event. The ANT4’ SHB modulates the free energy of the enzyme substrate complexes during the reaction coordinate, a phenomenom which has also recently been demonstrated to be a factor in determining substrate specificity and catalytic efficiency in an acetyl transferase AGME31–32. In ANT4’ the SHB is present in the pre-catalytic binary drug complex, where tight, specific recognition of the bactericidal antibiotics would be most important. Indeed, the neomycin minimum inhibitory concentration against susceptible Staphylococcus aureus is approximately 10-fold weaker than the ANT4’ neomycin Kd33. Upon formation of the catalytically relevant ternary complex which includes the nucleotide triphosphate and cations, the SHB to the aminoglycoside is lost, due to the engagement of the carboxylate group with both of the canonical nucleotidyl transfer Mg ions (Figure 2). The Mg ions in ANT4’ play a unique role in the reaction, where the catalytic magnesium (Mg A), not only functions in activating the nucleotidyl transfer nucleophile, but also in modulating the free energy of the reaction coordinate through disruption of the SHB formed with the neomycin (Figure 4). Concomitant with the loss of this interaction is the formation of the ternary state where the Kd is two-fold weaker although no chemistry has taken place. Once nucleotidyl transfer occurs only one of the three hydrogen bonds remains between ANT4’ and the modified prime ring of the aminoglycoside, which serves to further reduce the energy of the modified drug product complex from that of the starting state that contains the SHB. Thus, the modulation of this SHB serves as a thermodynamic signal that also mediates the “catch and release” of the antibiotic substrates.
ANT4’ is a promiscuous AGME that inactivates a broad spectrum of aminoglycosides11, including tobramycin, which lacks the 3’ hydroxyl moiety involved in forming the observed SHB. Interestingly, tobramycin was shown to be one of the best turned over substrates of ANT4’. So how ubiquitous is the proposed “catch and release” mechanism of ANT4’? Our present studies, in combination with earlier work on ANT4’, suggest that the active site does not restrict substrates to bind in an identical location16 (Figure 2), even though these aminoglycosides (kanamycin and neomycin) share a common core (Figure S1). It is therefore possible that the drugs that lack the SHB-forming 3’ hydroxyl could be repositioned within the active site to facilitate the SHB-mediated “catch and release”. Future structural studies will aim to address these outstanding questions in ANT4’ antibiotic inactivation, which will ultimately contribute important knowledge to the design of new aminoglycoside medications.
Methods
Protein expression and purification
All the ANT4’ variants used in this study (WT, T130K and T130K/E52D) were cloned in a pGEX-4T-3 vector with a TEV (Tobacco Etch Virus) protease cleavage site at the C- terminus of the GST affinity tag. The proteins were expressed in BL21 RIL E. coli cells and grown in Terrific Broth media at 37 °C and transferred to 21 °C after induction with 1mM IPTG. The cells were grown for 16 h, harvested, resuspended in lysis buffer (1* PBS buffer, 1 mM DTT and 1% Triton) and lysed by sonication. The cell lysate was clarified with centrifugation and the supernatant was then incubated with Glutathione Sepharose 4B resin for 2–3 hours before being loaded into a column. The column was washed with wash buffer (1* PBS buffer, 1 mM DTT) before TEV protease was added. The protein-loaded GST resin was incubated with TEV protease for 16 h at 4 °C. The cleaved protein was collected in the flow through and treated with the HiTrap Ni2+ chelating HP column to remove the TEV protease. The flow through was collected and dialyzed in 50 mM Tris pH 8.0, 100 mM NaCl and 5mM EDTA to remove imidazole and any nucleotide contaminants. The dialyzed protein was further purified with a Superdex 75 10/300 column (GE healthcare Life Sciences) equilibrated in 50 mM Tris pH 8.0 and 100 mM NaCl. The purity of the enzymes was evaluated by SDS PAGE prior to being flash-frozen in liquid nitrogen and stored at −80 °C.
Crystallization and X-ray data collection
All the ANT4’ crystals were grown by the hanging drop vapor diffusion method at 12 °C, using droplets containing 1:1 ratio of protein solution and mother liquor. WT, T130K and T130K/E52D enzymes were used at a concentration of 15 mg/ml in 50 mM Tris pH 8.0 and 100 mM NaCl buffer for crystallization. The crystals were cryo-protected using respective mother liquor with 30% glycerol prior to mounting and flash freezing in liquid nitrogen. Binary complex crystals were obtained by incubating the enzyme with 2–3 mM neomycin for 2 h at room temperature and mixed with an equal amount of precipitant (16–17% PEG 8K, 0.1 M Tris pH 7–8, 0.1–0.2 M MgCk). For the preparation of the non-hydrolyzable ternary complex crystals of ANT4’-Neo- AMPCPP, the enzyme (15 mg/ml in 50 mM Tris pH 8.0 and 100 mM NaCl buffer) was incubated with 2 mM neomycin and 4 mM AMPCPP for 2 h at room temperature and mixed with the precipitant solution (12% PEG 8K, 0.1 M Tris pH 8.0, 0.1–0.2 M MgCl2). For the ternary complex crystals of ANT4’-adenylated Neo-PP, the enzyme was dialyzed in 50 mM Tris pH 8.0 and 100 mM NaCl buffer containing 10 mM CaCl2 before setting up crystallization trays. The Cardialyzed enzyme was then crystallized in 9–10% PEG 5K MME, 0.1 M Tris pH 7.5–8.0, 0.1–0.2 M CaCl2. The crystals were transferred to a cryo-solution containing 9–10% PEG 5K MME, 0.1 M Tris pH 7.5–8.0, 10 mM MgCl2 and 30% glycerol for 45min. This resulted in the formation of a ternary product complex with pyrophosphate. The reaction was stopped by freezing the crystals in liquid nitrogen prior to data collection. All the binary and ternary complex crystals were collected at 100K at different beamlines (23ID-D, 23ID-B, 21ID-F) at the Advanced Photon source (APS) using a Pilatus 3–6 M detector, Eiger 16M and Rayonix MX300 detectors respectively. Diffraction data were processed with XDS34 or HKL-300035 or MOSFLM36 and scaled with SCALA or AIMLESS from the CCP4 suite37. Space groups, cell parameters, and data collection statistics are given in Tables S1 and S2.
Structure determination and refinement
The structures were solved by molecular replacement with PHASER38 using PDB:1KNY as the template. Model building and refinement were carried out with COOT39 and Phe- nix40. Model analysis was done with MolProbity41 and figures were prepared with Pymol. Atomic coordinates and the structure factors for all complexes have been deposited in the Protein Data Bank (PDB) (Tables S1 and S2). Crystallographic refinement statistics are listed in Tables S1 and S2.
Binding studies
All ITC experiments were performed at 25°C using the Microcal VP-ITC (Microcal Inc, USA) with a cell volume of 2 ml. All solutions were degassed prior to the experiments. 13–40 ^M of ANT4’ enzymes (T130K and T130K/E52D) were dialyzed in 50 mM MOPS pH 8.0, 100 mM NaCl and with or without 4 mM MgCl2 buffer and titrated against 0.3 mM of neomycin. For the set of experiments with AMPCPP, both the enzyme and neomycin were premixed with 0.5 mM AMPCPP and degassed prior to the titration. The titration data were fitted to a single set of identical sites model using MicroCal ORIGIN software supplied with the instrument. Base-line correction, peak integration and binding parameters (number of binding sites (N), binding constant (Ka), change in binding enthalpy (AH) and change in binding entropy (AS)) were obtained using the ORIGIN software.
Accession Codes
Structures and reflection data of ANT4’ complexes reported here have been deposited to the Protein Data Bank under the accession codes: 6UN8, 6NMK, 6NML, 6NMM, 6PO4, 6PO6, and 6PO8.
Supplementary Material
Table S1. Data collection statistics of wild type and T130K ANT4’.
Table S2. Data collection statistics of T130K/E52D ANT4’.
Figure S1. Two classes of aminoglycoside antibiotics.
Figure S2. Polder omit maps of ANT4’ ligands.
Figure S3. LigPlot+ interaction analysis of T130K ANT4’ ligands.
Figure S4. LigPlot+ interaction analysis of T130K/E52D ANT4’ ligands.
Figure S5. Additional adenylated neomycin in ANT4’ structure.
Figure S6. ITC based dissociation constants (Kd) for T130K and T130K/E52D ANT4’.
ACKNOWLEDGMENTS
The Oak Ridge National Laboratory Center for Structural Molecular Biology (FWP ERKP291) is supported by the Office of Biological and Environmental Research of the US Department of Energy. This research was supported in part by an appointment to the HERE Program at Oak Ridge National Laboratory. The authors would like to thank Edward Wright for help with ITC data collection and Prashasti Kumar for helpful discussions. The GM/CA beamline at the APS was used for some crystallographic data collection. The GM/CA beamlines at APS has been funded in whole or in part with Federal funds from the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Funding Sources
This work was supported by a grant from the National Science Foundation (MCB-1662080).
ABBREVIATIONS
- AGMEs
aminoglycoside modifying enzymes
- ANT4’
aminoglycoside nucleotidyl transferase 4’
- LBHB
low-barrier hydrogen bond
- SHB
short hydrogen bond
- SIHN
short-ionic hydrogen bond
REFERENCES
- 1.Yang W, Portraits of a Y-family DNA polymerase. FEBS Lett 2005, 579 (4), 868–72. [DOI] [PubMed] [Google Scholar]
- 2.Yamtich J; Sweasy JB, DNA polymerase family X: function, structure, and cellular roles. Biochim Biophys Acta 2010, 1804 (5), 1136–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakamura T; Zhao Y; Yamagata Y; Hua YJ; Yang W, Watching DNA polymerase eta make a phosphodiester bond. Nature 2012, 487 (7406), 196–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jamsen JA; Beard WA; Pedersen LC; Shock DD; Moon AF; Krahn JM; Bebenek K; Kunkel TA; Wilson SH, Time-lapse crystallography snapshots of a double-strand break repair polymerase in action. Nat Commun 2017, 8 (1), 253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freudenthal BD; Beard WA; Shock DD; Wilson SH, Observing a DNA polymerase choose right from wrong. Cell 2013, 154 (1), 157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freudenthal BD; Beard WA; Wilson SH, New structural snapshots provide molecular insights into the mechanism of high fidelity DNA synthesis. DNA Repair (Amst) 2015, 32, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Whitaker AM; Smith MR; Schaich MA; Freudenthal BD, Capturing a mammalian DNA polymerase extending from an oxidized nucleotide. Nucleic Acids Res 2017, 45 (11), 6934–6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wright GD, Aminoglycoside-modifying enzymes. Curr Opin Microbiol 1999, 2 (5), 499–503. [DOI] [PubMed] [Google Scholar]
- 9.Taber HW; Mueller JP; Miller PF; Arrow AS, Bacterial uptake of aminoglycoside antibiotics. Microbiol Rev 1987, 51 (4), 439–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumar P; Selvaraj B; Serpersu EH; Cuneo MJ, Encoding of Promiscuity in an Aminoglycoside Acetyltransferase. J Med Chem 2018, 61 (22), 10218–10227. [DOI] [PubMed] [Google Scholar]
- 11.Porter VR; Green KD; Zolova OE; Houghton JL; Garneau-Tsodikova S, Dissecting the cosubstrate structure requirements of the Staphylococcus aureus aminoglycoside resistance enzyme ANT(4’). Biochem Biophys Res Commun 2010, 403 (1), 85–90. [DOI] [PubMed] [Google Scholar]
- 12.Le Goffic F; Martel A; Capmau ML; Baca B; Goebel P; Chardon H; Soussy CJ; Duval J; Bouanchaud DH, New plasmid-mediated nucleotidylation of aminoglycoside antibiotics in Staphlococcus aureus. Antimicrob Agents Chemother 1976, 10 (2), 258–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Goffic F; Baca B; Soussy CJ; Dublanchet A; Duval J, [ANT(4’)I: a new aminoglycoside nucleotidyl transferase found in “staphylococcus aureus” (author’s transl)]. Ann Microbiol (Paris) 1976, 127 (3), 391–9. [PubMed] [Google Scholar]
- 14.Chen-Goodspeed M; Vanhooke JL; Holden HM; Raushel FM, Kinetic mechanism of kanamycin nucleotidyl transferase from Staphylococcus aureus. Bioorg Chem 1999, 27 (5), 395–408. [Google Scholar]
- 15.Sakon J; Liao HH; Kanikula AM; Benning MM; Rayment I; Holden HM, Molecular structure of kanamycin nucleotidyl transferase determined to 3.0-A resolution. Biochemistry 1993, 32 (45), 11977–84. [DOI] [PubMed] [Google Scholar]
- 16.Pedersen LC; Benning MM; Holden HM, Structural investigation of the antibiotic and ATP-binding sites in kanamycin nucleotidyl transferase. Biochemistry 1995, 34 (41), 13305–11. [DOI] [PubMed] [Google Scholar]
- 17.Cox G; Stogios PJ; Savchenko A; Wright GD, Structural and molecular basis for resistance to aminoglycoside antibiotics by the adenylyltransferase ANT(2”)-Ia. MBio 2015, 6 (1), e02180–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y; Nasvall J; Wu S; Andersson DI; Selmer M, Structure of AadA from Salmonella enterica: a monomeric aminoglycoside (3”)(9) adenyltransferase. Acta Crystallogr D Biol Crystallogr 2015, 71 (Pt 11), 2267–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stogios PJ; Evdokimova E; Morar M; Koteva K; Wright GD; Courvalin P; Savchenko A, Structural and functional plasticity of antibiotic resistance nucleotidylyltransferases revealed by molecular characterization of lincosamide nucleotidylyltransferases lnu(A) and lnu(D). J Mol Biol 2015, 427 (12), 2229–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morar M; Bhullar K; Hughes DW; Junop M; Wright GD, Structure and mechanism of the lincosamide antibiotic adenylyltransferase LinB. Structure 2009, 17 (12), 1649–1659. [DOI] [PubMed] [Google Scholar]
- 21.Matesanz R; Diaz JF; Corzana F; Santana AG; Bastida A; Asensio JL, Multiple keys for a single lock: the unusual structural plasticity of the nucleotidyltransferase (4’)/kanamycin complex. Chemistry-A European Journal 2012, 18 (10), 2875–2889. [DOI] [PubMed] [Google Scholar]
- 22.Cleland WW; Frey PA; Gerlt JA, The low barrier hydrogen bond in enzymatic catalysis. J Biol Chem 1998, 273 (40), 25529–32. [DOI] [PubMed] [Google Scholar]
- 23.Shimizu K; Kumada T; Hsieh WC; Chung HY; Chong Y; Hare RS; Miller GH; Sabatelli FJ; Howard J, Comparison of aminoglycoside resistance patterns in Japan, Formosa, and Korea, Chile, and the United States. Antimicrob Agents Chemother 1985, 28 (2), 282–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laskowski RA; Swindells MB, LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 2011, 51 (10), 2778–86. [DOI] [PubMed] [Google Scholar]
- 25.Kocaman S; Serpersu EH, The Thermodynamics of Ligand Binding to the Aminoglycoside O-Nucleotidyltransferase(4’) and Variants Yields Clues about Thermophilic Properties. Biochemistry 2019, 58 (12), 1579–1586. [DOI] [PubMed] [Google Scholar]
- 26.Jing X; Serpersu EH, Solvent reorganization plays a temperature-dependent role in antibiotic selection by a thermostable aminoglycoside nucleotidyltransferase-4’. Biochemistry 2014, 53 (34), 5544–50. [DOI] [PubMed] [Google Scholar]
- 27.Jing X; Evangelista Falcon W; Baudry J; Serpersu EH, Thermophilic Enzyme or Mesophilic Enzyme with Enhanced Thermostability: Can We Draw a Line? J Phys Chem B 2017, 121 (29), 7086–7094. [DOI] [PubMed] [Google Scholar]
- 28.Marti S; Bastida A; Swiderek K, Theoretical Studies on Mechanism of Inactivation of Kanamycin A by 4’-O- Nucleotidyltransferase. Frontiers in chemistry 2018, 6, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Von Hippel PH; Fairfield FR; Dolejsi MK, On the processivity of polymerases. Ann N Y Acad Sci 1994, 726, 118–31. [DOI] [PubMed] [Google Scholar]
- 30.Huang J; Alnajjar KS; Mahmoud MM; Eckenroth B; Doublie S; Sweasy JB, The nature of the DNA substrate influences pre-catalytic conformational changes of DNA polymerase beta. J Biol Chem 2018, 293 (39), 15084–15094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kumar P; Serpersu EH; Cuneo MJ, A low-barrier hydrogen bond mediates antibiotic resistance in a noncanonical catalytic triad. Sci Adv 2018, 4 (4), eaas8667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar P; Agarwal PK; Waddell MB; Mittag T; Serpersu EH; Cuneo MJ, Low-Barrier and Canonical Hydrogen Bonds Modulate Activity and Specificity of a Catalytic Triad. Angew Chem Int Ed Engl 2019, 58 (45), 16260–16266. [DOI] [PubMed] [Google Scholar]
- 33.Sabath LD; Garner C; Wilcox C; Finland M, Susceptibility of Staphylococcus aureus and Staphylococcus epidermidis to 65 antibiotics. Antimicrob Agents Chemother 1976, 9 (6), 962–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kabsch W, XDS. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 2), 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Minor W; Cymborowski M; Otwinowski Z; Chruszcz M, HKL-3000: the integration of data reduction and structure solution- -from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 2006, 62 (Pt 8), 859–66. [DOI] [PubMed] [Google Scholar]
- 36.Battye TG; Kontogiannis L; Johnson O; Powell HR; Leslie AG, iMOSFLM: a new graphical interface for diffraction- image processing with MOSFLM. Acta Crystallogr D Biol Crystallogr 2011, 67 (Pt 4), 271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Potterton L; Agirre J; Ballard C; Cowtan K; Dodson E; Evans PR; Jenkins HT; Keegan R; Krissinel E; Stevenson K; Lebedev A; McNicholas SJ; Nicholls RA; Noble M; Pannu NS; Roth C; Sheldrick G; Skubak P; Turkenburg J; Uski V; von Delft F; Waterman D; Wilson K; Winn M; Wojdyr M, CCP4i2: the new graphical user interface to the CCP4 program suite. Acta Crystallogr D Struct Biol 2018, 74 (Pt 2), 68–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bunkoczi G; Echols N; McCoy AJ; Oeffner RD; Adams PD; Read RJ, Phaser.MRage: automated molecular replacement. Acta Crystallogr D Biol Crystallogr 2013, 69 (Pt 11), 2276–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P; Lohkamp B; Scott WG; Cowtan K, Features and development of Coot. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Afonine PV; Grosse-Kunstleve RW; Echols N; Headd JJ; Moriarty NW; Mustyakimov M; Terwilliger TC; Urzhumtsev A; Zwart PH; Adams PD, Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr 2012, 68 (Pt 4), 352–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen VB; Arendall WB 3rd; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010, 66 (Pt 1), 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Data collection statistics of wild type and T130K ANT4’.
Table S2. Data collection statistics of T130K/E52D ANT4’.
Figure S1. Two classes of aminoglycoside antibiotics.
Figure S2. Polder omit maps of ANT4’ ligands.
Figure S3. LigPlot+ interaction analysis of T130K ANT4’ ligands.
Figure S4. LigPlot+ interaction analysis of T130K/E52D ANT4’ ligands.
Figure S5. Additional adenylated neomycin in ANT4’ structure.
Figure S6. ITC based dissociation constants (Kd) for T130K and T130K/E52D ANT4’.