

Abstract
Human Ferredoxin 1, also referred to as Adrenodoxin (Adx), is the sole electron carrier supporting the function of all seven mitochondrial cytochrome P450 (CYP) enzymes. Adx utilizes conserved negatively charged residues along its α helix-3 to interact with either the proximal surface of CYP enzymes or the binding surface of Adrendodoxin Reductase (AdR). However, in the oxidized state, Adx assumes a monomer-homodimer equilibrium that requires the presence of its unstructured C-terminal tail. Crystallographic structures of full-length human Adx dimers indicate that part of the binding surface necessary for its interactions with CYPs or with AdR is partially occluded by the dimer interface. In this study, protein NMR spectroscopy was used to interrogate the interactions between full-length (2–124) or truncated monomeric (2–108) human Adx and human CYP24A1 (with and without its vitamin-D substrate) as well as interactions with AdR. Here, monomeric Adx induced a similar pattern of peak broadening as that induced by addition of CYP24A1 substrate, consistent with a 1:1 Adx:CYP interaction as the functional complex. Additionally, removal of the C-terminal tail appears to enhance the interaction with AdR, despite removal of some of the AdR contacts in the tail region. This finding was also supported by an NMR competition assay. These findings suggest that the Adx dimers do not undergo meaningful interactions with either CYP or AdR, but may instead be responsible for regulating access to monomeric Adx. These conclusions are discussed in the context of a revised model of the Adx electron shuttle mechanism.
Keywords: cytochrome P450, Adrenodoxin, Ferredoxin 1, Adrenodoxin Reductase, NMR, protein-protein interactions
Graphical Abstract
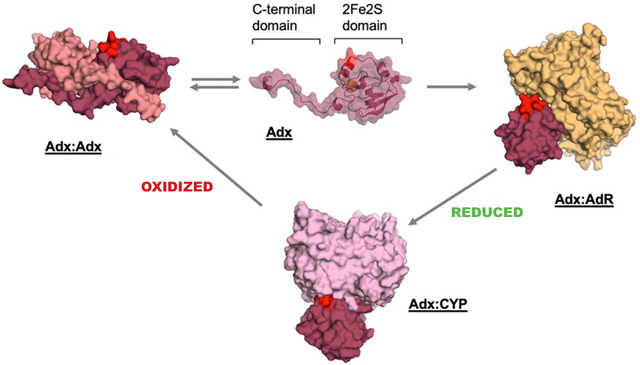
Adx-detected NMR data in this study support the formation of mutually exclusive complexes between Adx-Adx, Adx-CYP, or Adx-AdR during electron delivery. The conserved α helix 3 (highlighted in red) is necessary for CYP and AdR interactions, but is unavailable in Adx homodimers.
1. Introduction
The water-soluble mitochondrial Ferredoxin 1 protein, also known as Adrenodoxin (Adx), is the universal redox partner for all seven mammalian mitochondrial cytochrome P450 enzymes (CYPs). As such, Adx supports the function of CYPs that are responsible for the bioactivation of endogenous vitamin-D (CYP27A1 and CYP27B1), the inactivation of vitamin-D and related analogs (CYP24A1), the conversion of vitamin A1 into vitamin A2 (CYP27C1) [1], the multi-step conversion of cholesterol into pregnenolone (CYP11A1), and the biosynthesis of corticosterone and aldosterone (CYP11B1 and CYP11B2, respectively) [2, 3]. In its reduced state, Adx delivers electrons one at a time by forming a 1:1 CYP:Adx protein complex that is mediated in part by charge pairing interactions between the anionic α-helix 3 of Adx and the cationic proximal surface of the CYP [4–9]. Following Adx electron delivery to the CYP, oxidized Adx dissociates and assumes its own monomer-dimer equilibrium as driven by the unstructured C-terminal tail; removal of the last twenty residues of Adx vastly reduces its dimerization, while increasing its ability to support catalysis [10, 11]. Adx is then returned to its reduced state upon electron transfer from the flavin-containing domain of Adrenodoxin reductase (AdR) [2].
The highly conserved and negatively charged Adx α-helix 3 is known to comprise part of the binding interface for both the CYP:Adx and AdR:Adx complexes [6, 7, 12]. Therefore, a heterotrimer composed of CYP:Adx:AdR, once hypothesized to form [2, 13], is prevented due to steric hindrance and to the partially overlapping nature of the unique protein contacts. While it is now generally accepted that Adx forms mutually exclusive complexes with either the CYP or with AdR [14, 15], the extent to which homodimers of Adx interact with CYPs or with AdR is less clear. Previous studies utilizing nuclear magnetic resonance (NMR) spectroscopy reported the presence of Adx:CYP24A1 contacts that occur outside of α-helix 3, including α-helix 1, that at the time were theorized to originate from the presence of the Adx dimer [8, 16]. Interactions between CYP11B1 and the Adx dimer have also been reported as products of chemical cross-linking studies [17]. Moreover, the Adx dimer has been proposed to bind to AdR and function as a temporary electron carrier in the regeneration of reduced Adx monomers [2, 18]. Another complicating factor is that the available crystal structures of full-length Adx show two distinct arrangements of the protein. In the first structure of full-length bovine Adx (PDB 1CJE) [19], the molecules are arranged in such a way that the anionic α-helix 3 of each monomer is entirely solvent exposed and would theoretically be available to interact either with CYPs or with AdR. However, in the structure of full-length human Adx (PDB 3P1M) [20], α-helix 3 of each monomer is instead partially obstructed by the positioning of the C-terminal tail of the opposite monomer, and may therefore be unavailable for additional protein contacts.
In the enclosed study, we employ a combination of protein NMR spectroscopy, chemical crosslinking, and other biochemical and functional assays to interrogate the interactions between full-length human Adx (residues 2–124), and either the human vitamin-D inactivating enzyme CYP24A1 (hCYP24A1) or bovine AdR. We also carry out a parallel analysis of the interactions using the C-terminal truncated form of hAdx (residues 2–108) that does not form detectable dimers. The opportunity to analyze the protein-protein complex for ligand-free human CYP24A1 only recently became available upon development of a cleavable fusion construct of the enzyme with hAdx [21]. We report that the Adx:CYP complex between monomeric 15N-hAdx (2–108) and hCYP24A1 results in a similar pattern of NMR peak broadening as the corresponding complex with full-length 15N-hAdx that is formed in the presence of the substrate 1α,25-dihydroxyvitamin D (1α,25(OH)2D). This may indicate that addition of CYP substrate preferences the CYP:Adx interaction away from the hAdx dimer. The interaction of each form of hAdx with AdR was also investigated. We found that 15N-hAdx (2–108) results in increased differential peak broadening when in the presence of AdR, suggesting a complex with enhanced specificity when the monomer-dimer equilibrium of hAdx is removed. Lastly, following analysis of long-range NMR chemical shift perturbations, along with spectra from mixed-label samples of hAdx, these data support an arrangement of hAdx monomers that is consistent with the dimer interface in the crystal structure of hAdx (PDB 3P1M); notably, the dimeric arrangement likely prevents meaningful intermolecular interactions of α-helix 3 while in a dimer. Taken together, this work suggests that, even in the oxidized form, the Adx monomer, not the dimer, is the preferential binding partner for CYP:Adx and CYP:AdR complexes.
2. Results
2.1. 1α,25(OH)2D modulates the human CYP24A1 complex with human Adx
Molecular recognition between the mitochondrial electron shuttle Adx and corresponding mitochondrial CYP enzymes is known to occur via the highly conserved and negatively charged α-helix 3 of Adx and the positively charged proximal surface of the CYP [2, 16, 17, 22, 23]. However, beyond this recognition event, subtle differences do exist between particular CYP-Adx complexes, likely driven by non-conserved Adx binding sites on the CYP. In the current study, the first interaction investigated was that of a species-matched complex between human CYP24A1 and human Adx, similar to a recent study of the carbon-24 hydroxylase complex consisting of rat CYP24A1 with rat Adx[16]. To achieve this by NMR, it was first necessary to assign the backbone amide resonances of full-length human Adx. The NMR chemical shift values of a truncated human Adx (residues 4–114) had previously been reported by Pochapsky and colleagues [24]. However, the assignment of the full-length protein (124 residues) was necessary in order to examine the protein-protein complex in the presence of the C-terminal tail of Adx (see Figure S1). The Cα secondary chemical shift plot is shown in Figure S2 and indicates that the C-terminal residues don’t adopt secondary structural elements in solution.
1H-15N HSQC spectra were acquired for samples of 15N-hAdx alone and in the presence of 0.5 molar equivalents of unliganded hCYP24A1. This intermediate ratio of hCYP24A1 was selected due to its detectable change in NMR peak intensities of 15N-hAdx, while also preventing complete loss of the spectrum due to signal broadening. The spectra with added hCYP24A1 showed the expected peak broadening resulting in decreased signal intensity across the molecule (Figure 1A), as reported previously in other NMR studies of CYP-Adx interactions[16, 25]. Residues that are differentially affected by a decrease in intensity that is beyond one standard deviation of the average include regions of α-helix 3 (Figure 1A), including residues Asp-76 through Ala-81, along with regions of the following loop region comprising residues Gly-83 through Thr-85.
Figure 1. 1α,25(OH)2D modulates the human CYP24A1 complex with Adx.
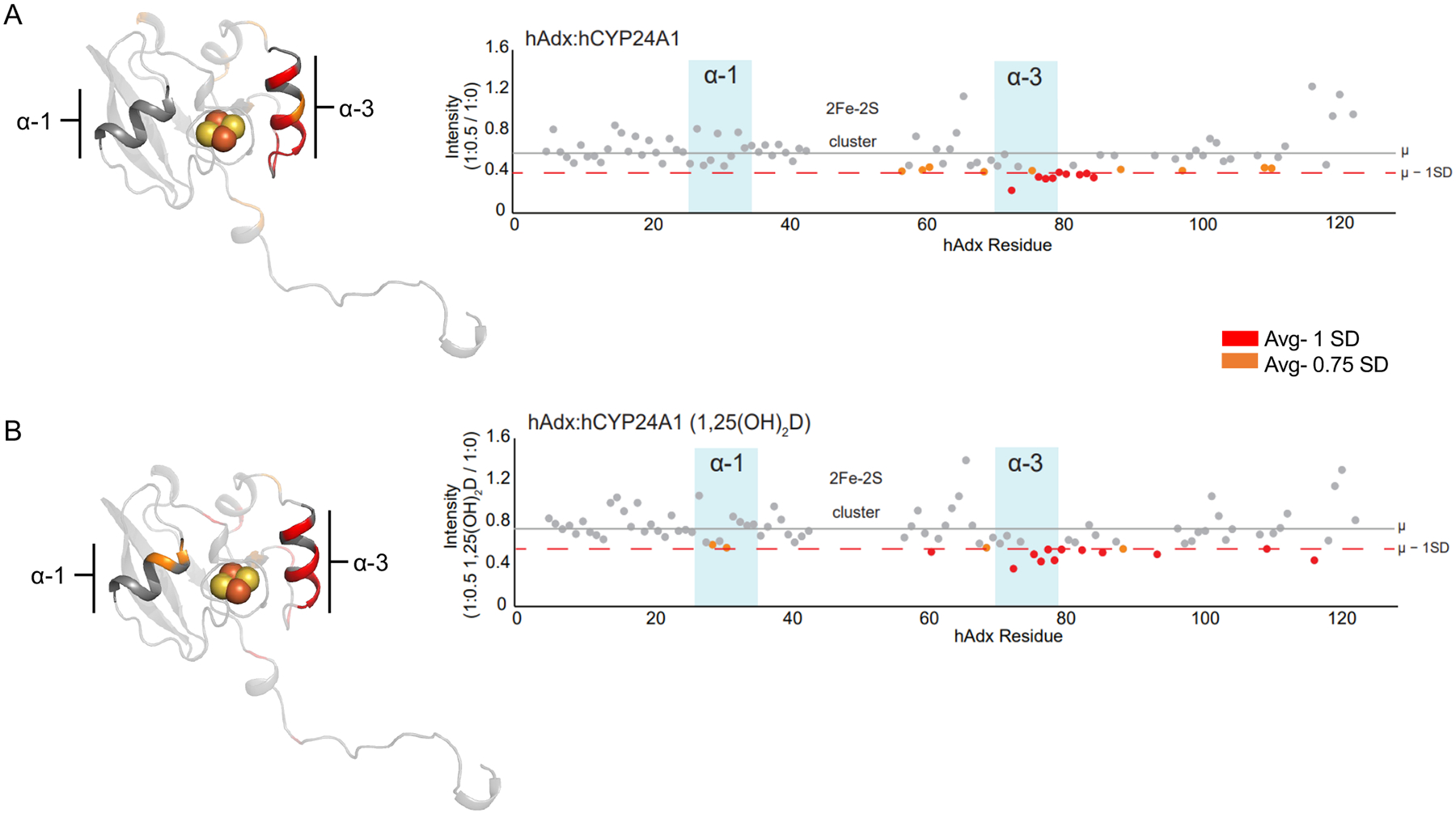
NMR peak broadening effects are mapped and quantified as ratios of remaining signal intensity following interactions between full-length 15N-hAdx (2–124) with hCYP24A1 in (A). The corresponding experiment acquired using hCYP24A1 bound to 1α,25(OH)2D is shown in (B). Intensity ratios were calculated by dividing 1H-15N HSQC peak intensities of bound Adx from unbound Adx. Most affected residues in red indicate broadening more than one standard deviation from the mean peak intensity. Residues in orange indicate more than 0.75 standard deviations from the mean. The horizontal lines mark the average per residue intensity (gray, μ) and the average minus one standard deviation (dashed orange, SD). Data are mapped onto a monomer of full-length human Adx (PDB 3P1M).
The corresponding experiment was then carried out in the presence of the hCYP24A1 substrate 1α,25(OH)2D (Figure 1B). This form of vitamin-D represents the biocactivated form of the hormone, with side-chain hydroxylation by CYP24A1 representing the primary pathway for its inactivation[26, 27]. Here, we observed what initially appeared to be a very similar pattern of peak broadening across the hAdx molecule, with most of the effect again focused on α-helix 3. However, closer examination revealed that the additional residues Leu-29 and Asp-31, which form a connected surface on α-helix 1, are also differentially affected, albeit modestly by 0.75 standard deviations from the mean. On its own, it is not possible to infer from this experiment whether these subtle differences represent a meaningful change in the complex that results from addition of substrate (although a more pronounced effect on α-helix 1 was observed in experiments described later in this study). However, in the context of previous experiments using rat CYP24A1, in which the presence and selection of CYP24A1 ligand has been shown to alter redox protein interactions[16, 28], these findings may indicate that the presence of 1,25(OH)2D induces a subtle change in the recognition of hAdx that results in additional interactions on surfaces opposite of the 2Fe-2S cluster of hAdx.
An additional observation from these spectra was that for both substrate-free and substrate-bond experiments, some residues (Glu-65 and select residues along the C-terminal tail) undergo a consistent but modest increase in peak intensity. These may signal an increase in flexibility at these sites due to the Adx:CYP interaction.
2.2. Biochemical and Functional Characterization of hAdx (2–108)
In the current study, as in previous experiments [16], it was noted that addition of CYP into samples 15N-hAdx induced peak broadening in regions of the ferredoxin beyond those at the putative CYP recognition site. For example, effects on α-helix 1 are routinely observed, along with broadening at Asp-61 of α-helix 2 (Figure 1). It has previously been theorized that these de-localized effects may arise from interactions of the Adx dimer in the NMR samples [8, 16]. To determine whether these non-putative contact sites are a result of the Adx dimer binding to hCYP24A1, NMR binding data were acquired using the truncated hAdx protein (residues 2–108).
The truncated hAdx (2–108) was first confirmed as a monomer by both native gel electrophoresis and analytical size exclusion chromatography (Figure 2 A–B). In the native gel, hAdx (2–124) resolves as three distinct bands. The larger two bands likely represent the dimer and monomer, respectively, while the smallest band likely represents a compact arrangement of the flexible C-terminal tail in the monomer. In contrast, hAdx (2–108) presents primarily as a single band that that migrates slightly less than the monomer in hAdx (2–124). It should be noted that removal of the C-terminal tail includes removal of the basic residues Arg-115 and Lys-122, which likely slows migration of the protein on a native gel by altering its net charge. Follow-on experiments using a calibrated 24 mL gel filtration column (Figure 2B) revealed that hAdx (2–124) elutes in a relatively broad peak and at a volume consistent with a molecular weight of approximately 17 kDa; slightly larger than the predicted size of 14.5 kDa of the protein with a histidine purification tag, likely representing the rapid equilibrium between monomer and dimer. In contrast, hAdx (2–108) elutes at a volume indicating a molecule at 12.9 kDa, which is truer in size to the predicted mass of 13 kDa for monomeric hAdx (2–108). The truncated form of bovine Adx (2–108) reportedly retains its ability to bind to CYPs and effectively transfers the electrons required for catalysis [12, 29]. Accordingly, hAdx (2–108) was found to not merely support, but enhance metabolism of 1α,25(OH)2D by hCYP24A1 (Figure 2C).
Figure 2. Functional activity and CYP interactions for hAdx (2–108).
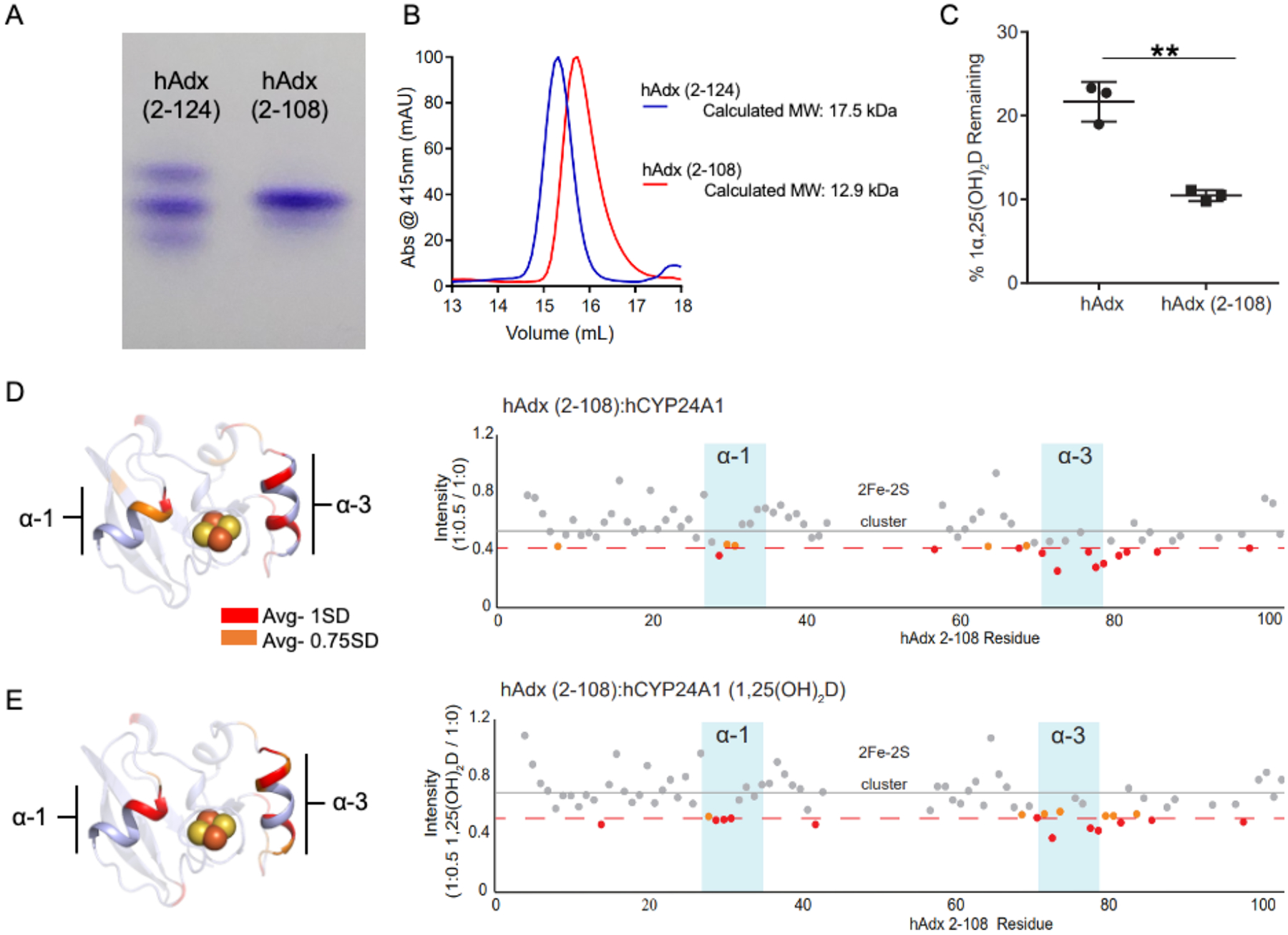
Truncation of the C-terminal tail of hAdx shifts the Adx equilibrium to a monomer, as shown by native gel electrophoresis in (A) and by analytical gel filtration chromatography in (B). hAdx (2–108) retains the ability the electrons required by hCYP24A1 (C). NMR peak broadening effects upon addition of unlabeled hCYP24A1 is shown in panel (D) for the two proteins at a half molar equivalent (1:0.5) of the CYP, and again in panel (E) for the same experiment in the presence of 1α,25(OH)2D. Residues broadened more than 1 and 0.75 standard devions from the mean are indicated in red and orange, respectively. Depletion of 1α,25(OH)2D by hCYP24A1 was analyzed using a one-way ANOVA analysis with α = 0.05, n =3, and p < 0.05.
1H-15N HSQC spectra of 15N-hAdx (2–108) were acquired in the presence and absence of unliganded hCYP24A1 (Figure 2D). Notably, the distributed peak broadening effects that have been observed previously in experiments using hAdx (2–124) are also present when the complex is formed with the truncated protein. This confirms that some of the effects on these regions are not due to the presence of the hAdx dimer, but are instead a result of the hAdx monomer binding to hCYP24A1. Of most interest was the introduction of a differential effect between Leu-29 and Asp-31 of α-helix 1, which is similar to the effects observed by introducing 1α,25(OH)2D into samples of hCYP24A1 with hAdx (2–124) (comparison of Figures 1B and 2D). Next, corresponding spectra were acquired using hCYP24A1 bound to 1α,25(OH)2D. In this case, the effect on α-helix 1 becomes more distinctive and the effect on α-helix 3 remains (Figure 2E). These data indicate that the non-putative (non α-helix 3) contact sites observed in NMR binding spectra actually arise from interactions with the hAdx monomer, not the dimer as was previously suspected, and may instead arise from secondary or steering interactions. Moreover, elimination of the hAdx dimer by using hAdx (2–108) forms a CYP-Adx complex with a peak broadening pattern that resembles the substrate-bound complex with hAdx (2–124) (compare to Figure 1B).
2.3. The AdR complex with hAdx (2–108) induces increased NMR peak broadening
Next, the complex between full-length and truncated hAdx with Adrenodoxin reductase was investigated. The 1:1 Adx-AdR complex is necessary for the reduction of Adx and plays a central role in the shuttle model of Adx electron delivery [2, 6, 22, 23]. This complex is well characterized by structural and mutational studies that include a crystallographic structure of Adx in complex with AdR, in addition to a solution model of hAdx (2–108) with AdR that was based on paramagnetic NMR-derived constraints [30, 31]. Through these studies, it is understood that the AdR binding surface on Adx encompasses α-helix 3, and therefore overlaps with the binding surface used for molecular recognition of CYPs. In this study, it was surmised that the analysis of the interaction between full length and hAdx (2–108) with AdR would provide an informative comparison with the effects seen with hCYP24A1; specifically, to determine whether distributed contacts (outside of α-helix 3) also occur during formation of the Adx-AdR complex, or whether they are unique to molecular recognition in CYP-Adx complexes.
1H-15N HSQC spectra of 15N-hAdx (2–124) and 15N-hAdx (2–108) were acquired in the presence of 0.5 and 1 molar equivalents of AdR. Here again, there was a decrease in peak intensities (Figure 3A and 3C), similar to that observed upon addition of hCYP24A1, and with only modest chemical shift perturbations of approximately 0.15 ppm in the 15N dimension for the most affected residues. However, peak broadening was also generally more differentially affected than with hCYP24A1. In Figure 3C, peak broadening effects at the 1:0.5 ratio are mapped onto hAdx. For additional context, the same residues are also highlighted on the crystallized complex between bovine Adx with AdR (PDB: 1E6E) [30]. Effects were noted at residues along α-helix 3 including Thr-71, Asp-72, and Asp-79. These residues are in general agreement with the orientation of Adx in the crystallographic complex. However, it was also noted that additional peak broadening does occur in regions that do not appear to interact with AdR in the crystal structure. These included the loop region following α-helix 1, encompassing residues Leu-37 to Ile-40, and Asp-31 of α-helix 1.
Figure 3. Role of the C-terminal tail of Adx in the interaction with AdR.
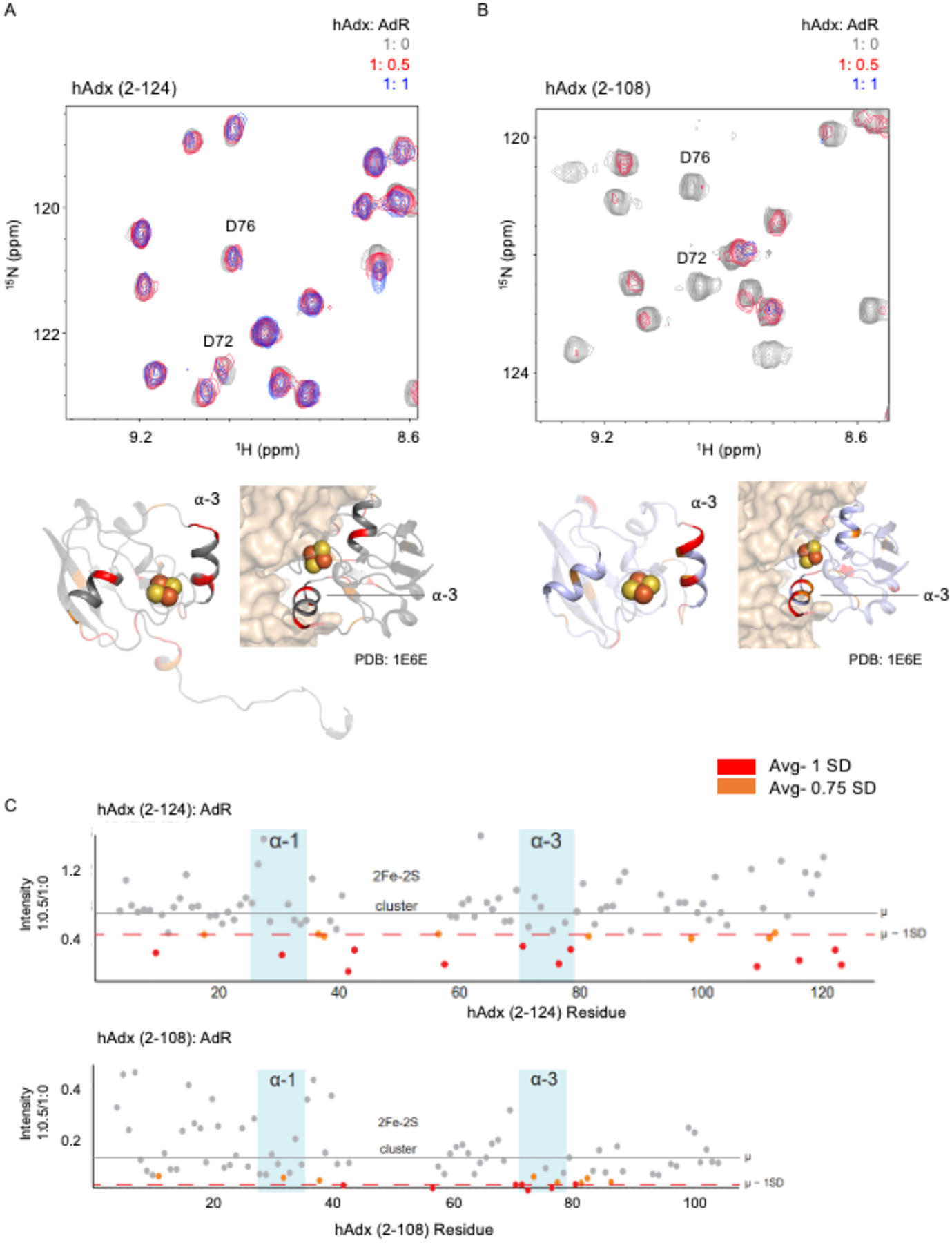
Representative overlaid 1H-15N HSQC spectra of 15N-hAdx (2–124) and 15N-hAdx (2–108) titrated with AdR are shown in panels A and B. Differentially affected residues are mapped onto a monomer from the structure human Adx (PDB 3P1M), with the C-terminal tail removed in (B), alongside a mapping of residues on the crystal structure of the bovine AdR:Adx complex (PDB 1E6E). Quantification of individual residue peak broadening upon addition of one half equivalents of AdR is shown in panel (C).
Interestingly, a comparison with NMR spectra of the complex with hAdx (2–108) shows differences in both the location of the disproportionately affected residues as well as the overall extent of peak broadening (Figure 3C). First, there is an overall more focused effect on residues in or near α-helix 3. For example, residues Glu-73 and Glu-74 are now differentially affected in addition to Asp-72 and Asp-79. This is accompanied by a decreased effect in many of the non-putative sites. These differences may arise from adjustments at the protein-protein interface in response to removal of secondary contacts between the C-terminal tail and AdR. Moreover, these spectra in the presence of AdR undergo more significant loss of signal. For comparison, for 15N-hAdx (2–108), the 1:1 spectrum with AdR is broadened nearly beyond detection. Taken together, these data suggest that the interaction with AdR results in distributed secondary effects, similar to that observed with hCYP24A1, but more so when the hAdx dimer is present. In the presence of the monomer, the complex formed appears to be increasingly specific to the Adx binding surface as represented by the co-crystal structure with AdR.
2.4. The solution dimer interface of hAdx partially occludes the CYP recognition site
NMR spectra of the complexes between hAdx (2–108): hCYP24A1 and the hAdx (2–108): AdR both result in relatively more focused patterns of peak broadening on the hAdx molecule, suggesting an increase in the specificity of the complex when hAdx is present as a monomer. These findings are consistent with the crystallographic arrangement of hAdx (2–124) (PDB: 3P1M) [20], in that the respective CYP and AdR binding surfaces on hAdx both appear to be partially obstructed by the dimer interface. Since this arrangement of Adx molecules in the dimer is distinct from that reported in a different structure (PDB: 1CJE) [19], we wanted to confirm that this crystallographic arrangement of hAdx is also present in solution.
First, in a variation of the previous NMR binding experiments, the 1H-15N HSQC spectrum of full-length 15N-hAdx was acquired alone and then in the presence of a 10-fold excess of either unlabeled hAdx (2–124) or unlabeled hAdx (2–108). The rationale for this mixed label approach was that by increasing the concentration of total hAdx, it would drive the 15N labeled protein towards the dimeric state, as predicted by Le Chatelier’s Principle, and therefore reflect changes in the NMR spectra. The concentration of combined hAdx in these samples is also higher than the concentration at which the proportion of hAdx dimers are reported to stabilize in solution ([10]. While this experiment did not result in chemical shift perturbations, possibly due to the rapid exchange equilibrium between the monomeric and dimeric states, it did result in a focused decreased in net peak intensities for some of the C-terminal tail residues of the protein, but only when in the presence of excess full-length hAdx (2–124). The full intensity ratio plots are shown in Figure S3. The most affected region is summarized in Figure 4A. The excess addition of unlabeled hAdx (2–124) (blue icons) results in minimal peak broadening for most of the labeled protein, with the exception of regions of the unstructured C-terminal tail. Various C-terminal residues, including A114, I118-G121, and T122 are broadened to approximately one half of the original peak intensity. By contrast, for spectra recorded in the presence of excess hAdx (2–108) (Figure 4A, red icons), the same C-terminal residues are no longer differentially affected. This experiment confirms the role of the C-terminal tail in forming part of the dimer interface. Moreover, the distribution of affected residues closely matches the crystallographic arrangement of the interlocking C-terminal tail in contact with the core domain of the opposite monomer as predicted in PDB 3P1M. By contrast, in the alternative crystallographic arrangement of full-length bovine Adx (PDB 1CJE), the electron density beyond residue 110 is missing, thus suggesting that the residues with differential peak broadening in the current study don’t participate in that dimer interface.
Figure 4. NMR Verification of the hAdx solution dimer interface.
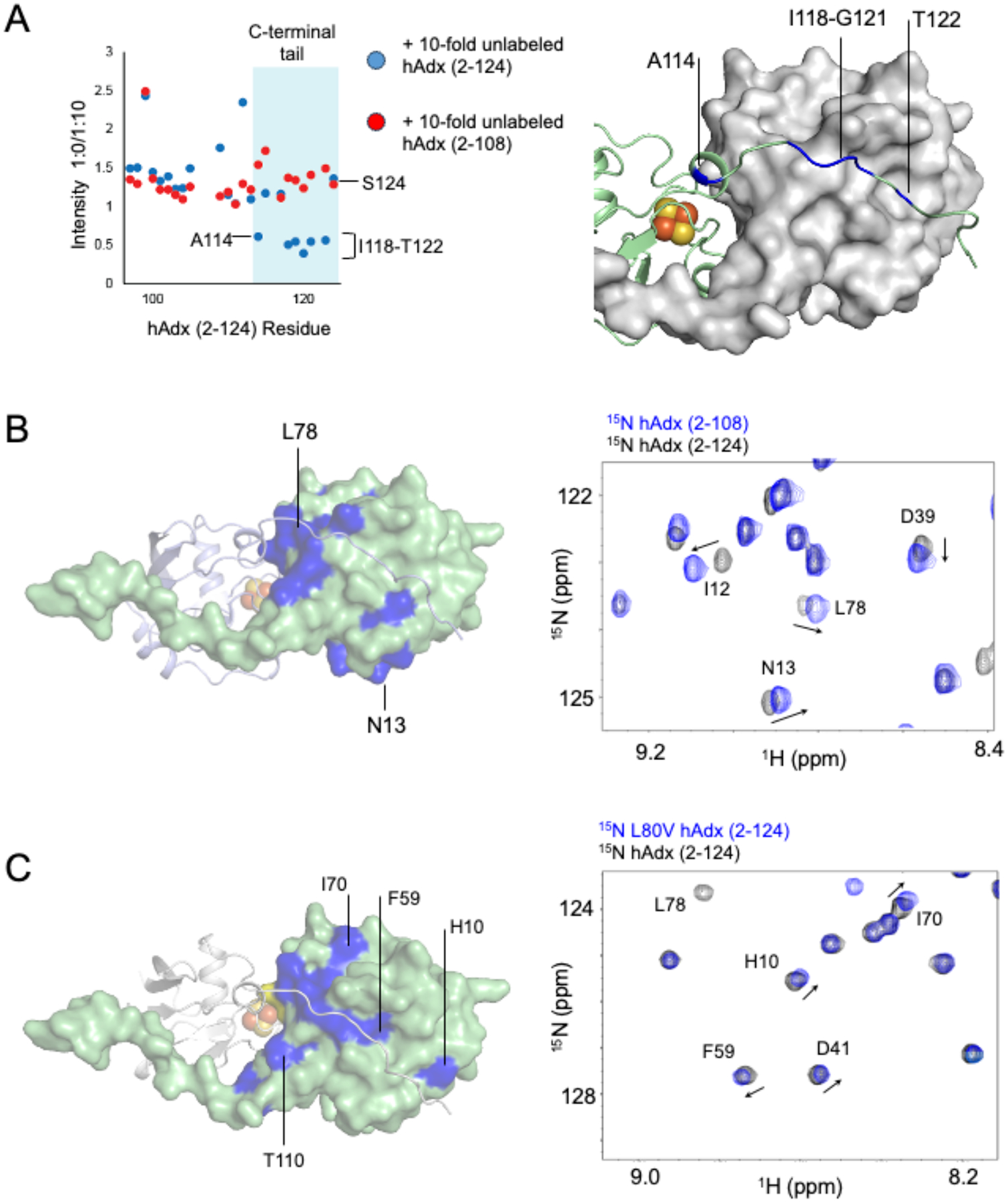
(A) 1H-15N HSQC peak intensity ratios of 15N-hAdx (2–124) in the presence of either a 10-fold excess of unlabeled hAdx (2–124) or hAdx (2–108). An overlay of the 1H-15N HSQC spectra of full-length and truncated hAdx is shown in (B) and an overlay of the full-length wild-type hAdx and hAdx containing the L80V mutation is shown in (C). All affected residues are mapped onto the structure of an hAdx dimer (PDB 3P1M).
Next, the 1H-15N HSQC spectrum of 15N-hAdx (2–108) was overlaid with the spectrum of full-length 15N-hAdx (Figure 4B, full spectra in Figure S4) as a way to map changes in the environment near the dimer interface. Upon removal of the C-terminal 16 residues, a large number of chemical shift perturbations occur in the core domain of hAdx and at residues that are not restricted to the site of the truncation. These include, among other residues, a connected surface formed by Asp-79 and Gly-83 through Asp-86, and an additional surface formed by Ile-12 and Asn-13. This region includes Leu-84, which according to the crystallographic arrangement is predicted to participate in a hydrophobic contact with Ile-118 of the opposite monomer. These regions are mapped onto one molecule of hAdx (2–124) in the crystallographic dimer in Figure 4B. Notably, much of the surface pattern aligns closely with the arrangement of the interlocking C-terminal tail from the opposite monomer.
Last, a 1H-15N HSQC spectrum was acquired for a point mutation that was predicted to perturb the dimer interface according to the interlocking tail arrangement of the dimer. Even conservative mutations at Leu-80 hAdx are known to affect function in multiple CYPs [17, 21, 28], presumably by disrupting nonpolar contacts with the proximal surface of the enzyme. The spectrum of an L80V mutation, overlaid with that of wild-type protein, resulted in long-range chemical shift perturbations of the core hAdx domain that, interestingly, resemble the pattern that results from removing the C-terminal tail (Figure 4B and 4D, full spectra are shown in Figure S5) and that trace the arrangement of the C-terminal tail on the crystallographic dimer. To be sure, during purification L80V hAdx was found to elute from a size exclusion chromatographic column at a similar volume as wild-type hAdx, indicating that the mutation does not disrupt dimerization of hAdx. Therefore, the long-range NMR perturbations like those observed at Phe-59 and His-10 are interpreted to represent a subtle adjustment to the dimer interface. Together, these findings provide supporting evidence that the arrangement of hAdx in a solution dimer aligns closely with the arrangement present in the PDB 3P1M crystal structure.
2.5. AdR preferentially binds monomeric hAdx
NMR spectra of full-length 15N-hAdx (2–124) and truncated 15N-hAdx (2–108) titrated with AdR indicate that the interaction between AdR and hAdx that is missing the C-terminal tail may be stronger than the interaction with full-length hAdx (Figure 5). While increased binding affinity is consistent with the observed increase in peak broadening for 15N-hAdx (2–108) (Figure 3B), the interpretation of these data is not straight-forward since these comparisons are not quantitative.
Figure 5. Competitive NMR binding and EDC cross-linking between AdR and hAdx.
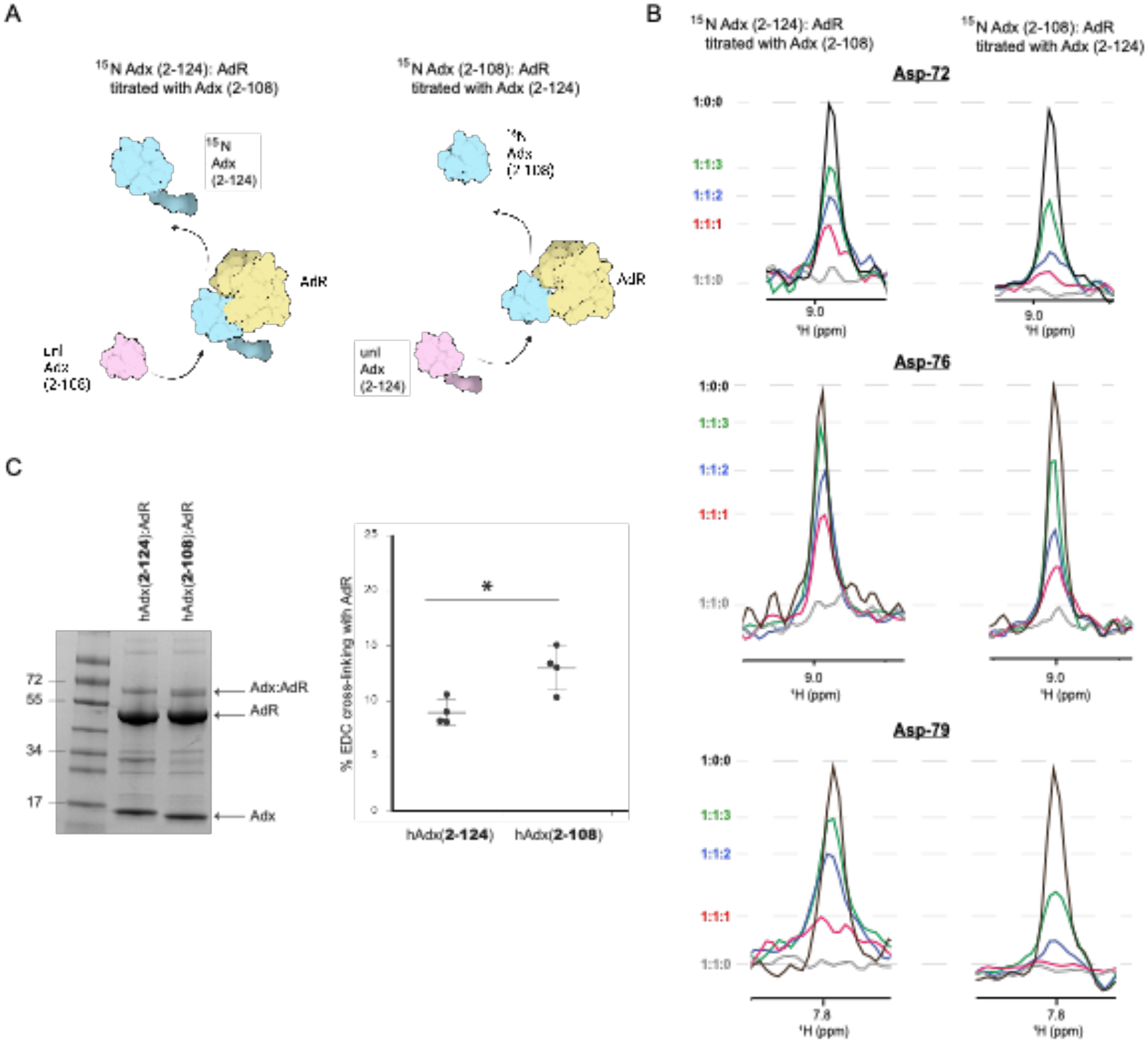
(A) Diagram showing competitive displacement of labeled 15N-hAdx(2–108) or 15N-hAdx(2–124), (B) normalized 1D traces from 1H-15N HSQC spectra showing increased relative displacement of full-length hAdx (2–124) (left panel) and relative displacement of truncated hAdx (2–108) (right panel). A comparative cross-linking experiment with AdR using the zero-length linker EDC is shown in panel (C). The diagram in panel (A) was created using BioRender. Quantification of cross-linking efficiency was measured by visualization of cross-linked bands (ImageJ) and analyzed for significance using a one-way ANOVA analysis with α = 0.05, n =4, and p < 0.05.
To confirm what appears to be enhanced binding to AdR upon truncation of the C-terminal tail, a competition experiment was designed in which recovery of the NMR signal from 1H-15N HSQC spectra was used as an indicator of displaced labeled protein (Figure 5A). Briefly, complexes were formed between AdR and either full-length 15N-hAdx (2–124) or truncated 15N-hAdx (2–108), thus leading to broadening of the NMR spectra. Subsequently, additional samples were prepared containing incrementally higher concentrations of the unlabeled counterpart of hAdx, such that the unlabeled protein out-competes the isotopically labeled protein, resulting in a recovery of the NMR peak intensities as the labeled protein is displaced from its interaction with AdR. Here, it was observed that addition of unlabeled 15N-hAdx (2–108) leads to a greater recovery of the free 15N-hAdx (2–124) signal, as highlighted in Figure 5B by one-dimensional traces for three of the α-helix 3 residues Asp-72, Asp-76, and Asp-79. By comparison, addition of corresponding concentrations of unlabeled hAdx (2–124) results in a smaller recovery of displaced truncated 15N-hAdx (2–108). These data indicate that the complex between AdR with C-terminal truncated hAdx (2–108) persists longer in the presence of competing full-length hAdx (2–124), consistent with what appears to be a greater affinity for the AdR:hAdx (2–108) complex.
Next, a zero-length chemical cross-linker (EDC) was used to measure accumulation of cross-linked product resulting from either form of hAdx with AdR (Figure 5C). Following a 2-hour incubation, both forms of hAdx formed cross-linked products with AdR that are consistent in size with a 1:1 complex. However, quantification of the complex gel band from multiple independent reactions confirms that hAdx (2–108) results in a modest but statistically significant increase in overall cross-linking efficiency. This result in agreement with the NMR competition assay in Figure 5A and 5B, indicating an apparent higher affinity for the truncated hAdx by AdR.
3. Discussion
3.1. Role of Adx dimers in CYP function
While human Adx, also known as Ferredoxin 1, has recently been implicated in the biogenesis of iron-sulfur clusters [32, 33], it is best characterized as the sole electron carrier in support of all seven mitochondrial cytochrome P450 enzymes. The protein-protein interaction between mitochondrial CYPs and Adx is a requirement for proper CYP function. As one example, mutations on the Adx binding surface of the vitamin-D activating enzyme CYP27B1 correlate with the onset of vitamin-D dependent Rickets Type-I [34]. Upon being reduced, Adx delivers electrons individually twice during each round of CYP catalysis. To be sure, there are several aspects of the role of Adx as an electron shuttle that are now well established. It is known, for example, that conserved anionic residues along α-helix 3 of Adx recognize cationic residues located on the proximal surface of the CYP [35], with the variability that has been documented between individual CYP-Adx complexes, like those described for the closely related CYP11B1 and CYP11B2 enzymes [36], likely arising from the lower degree of conservation on the CYP proximal surface.
However, beyond this basic paradigm, the details that govern the controlled reduction of Adx within the inner mitochondrial space are not well known. Adx interacts with a dedicated reductase in the form of AdR, whereby it becomes reduced and is then able to shuttle electrons to all seven mitochondrial CYPs [2]. However, while in the oxidized state immediately following release from the CYP, Adx is also free to assume a monomer-homodimer equilibrium that is absent or negligible when the protein is in its reduced state [10]. The amount of dimeric Adx at equilibrium is high enough that dimers are present in both crystal structures of full-length human and full-length bovine Adx [19, 20]. Moreover, Adx resolved by native gel electrophoresis in this study, and at lower protein concentrations than those required for NMR or crystallography, also shows a significant higher molecular weight band consistent with that of a dimer (Figure 2A). However, whether the interaction of dimeric Adx undergoes meaningful interactions with either CYP or with AdR is unknown. Chemical cross-linking studies using CYP11B1 indicate a complex that is consistent with 2:1 Adx:CYP arrangement [17], yet similar studies using CYP24A1 reflect only the 1:1 interaction [28]. Earlier models of the electron shuttle mechanism predict that AdR interacts with dimers of Adx that then participate in a step-wise reduction of each attached Adx molecule [18] [2]. However, these models relied on an alternate arrangement Adx dimers (1CJE) and the structure that is most consistent with our NMR data (3P1M) was not yet available.
3.2. Substrate primes CYP24A1 for the interaction with monomeric Adx
In the current study, we utilized the 1H-15N HSQC spectrum of human Adx as a reporter of its protein-protein interactions in solution with either human CYP24A1 or with AdR. Since removal of the unstructured C-terminal residues is known to prevent homodimer formation while preserving electron delivery [10, 11, 37] (Figure 2), the enclosed study includes Adx both with the intact C-terminal tail (hence Adx in a monomer-homodimer equilibrium) and Adx with the C-terminal truncation (monomeric Adx).
Interestingly, the pattern of peak broadening mapped onto the surface of truncated hAdx (2–108) resembles the pattern observed with full-length hAdx, but only when the CYP24A1 substrate 1α,25(OH)2D is present. This comparison is summarized in Figure 6. Both complexes result in the expected differential peak broadening along the primary CYP binding site composed of α-helix 3. However, peak broadening also occurs for residues on α-helix 1 and is present both when the C-terminal tail is removed as well as when the substrate is present. Addition of 1α,25(OH)2D to the interaction with 15N-hAdx (2–108) only increases this trend. While initially surprising, this finding is consistent with what we understand of Adx function; specifically, since the productive complex between the Adx (albeit in a reduced state) and the CYP is that of a stoichiometric 1:1 interaction, it is reasonable to conclude that the presence of substrate primes CYP24A1 for the interaction with monomeric human Adx. The impact of ligand binding on the CYP24A1-Adx interaction has previously been documented for the rat isoform (a carbon-24 vitamin-D hydroxylase) [16]. In this context, it is possible that the presence of Adx:Adx dimers in the full-length sample competes with the CYP:Adx interaction to the extent that secondary binding effects along α-helix 1 are not observable until substrate is added.
Figure 6. Summary of NMR peak broadening effects mapped onto hAdx.
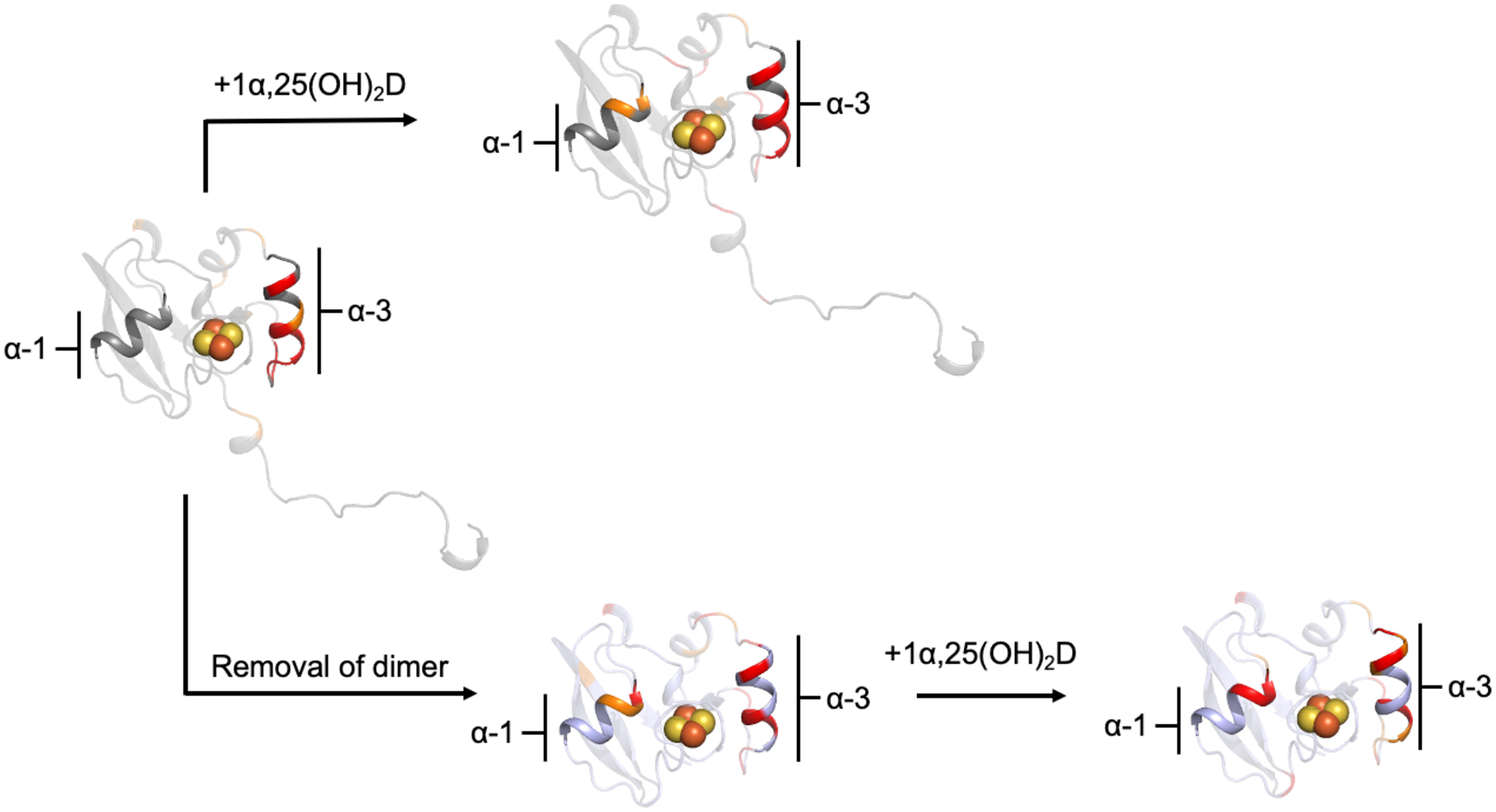
Mapped ieffects are derived from the ratios of remaining intensities from the preceding experiments. Addtion of the hCYP24A1 substrate, 1α,25(OH)2D has a similar effect along α-helix 1 as removing the C-terminal tail. All effects were mapped onto a monomer of hAdx from PDB 3P1M.
3.3. The Adx dimer equilibrium may regulate access of 1:1 Adx-AdR interactions
Another key finding from this study is that removal of the C-terminal tail of hAdx increases the apparent affinity toward AdR, as indicated by the extent of NMR peak broadening (Figure 3) and supported by both an NMR-based competition assay (Figure 5B), and by comparative chemical cross-linking (Figure 5C).
Interestingly, this finding is counter to what is expected based on an examination of the protein contacts in the crystal structure of bovine AdR with bovine Adx (PDB 1E6E) [12], in which some C-terminal tail residues, including Arg-115 and Glu-116 (Gln-116 in human Adx), may participate in hydrogen bonding interactions with AdR. A role for the C-terminal tail in interactions with AdR is also supported by differential peak broadening along the tail residues Ser-117, Thr-124, and Ser-125 observed in this study (Figure 3C). Nonetheless, removal of these residues along with the rest of the tail appears to enhance the interaction, rather than disrupt it. Our interpretation of these findings is that the increased affinity for truncated hAdx that is implied by our results may actually be due to prevention of the hAdx dimer, which likely competes with a 1:1 AdR, hAdx complex. We theorize that when the dimer is removed via truncation of the tail, the concentration of monomeric hAdx that is available for AdR has effectively increased, thereby promoting more binding, even though the net concentration of hAdx is unaltered. Based on the migration of the full-length protein on a native gel (Figure 2), the monomer is likely the predominant species of hAdx at equilibrium. However, the percentage of dimer present appears to be sufficient to significantly sequester monomeric hAdx and thereby prevent all of hAdx from interacting with AdR or hCYP24A1. The net effect is that fewer interactions occur with AdR when the dimer is present and these average out into less peak broadening overall, as shown in Figure 3C.
3.4. Mutually exclusive complexes of Adx inform a revised model of electron shuttling
In order for human Adx dimerization to compete with either the Adx:CYP24A1 or the Adx:AdR complex, as our data suggest, we surmise that the dimer itself must not form meaningful or specific interactions with either binding partner. In the course of this study, we attempted to engineer a pure dimer of hAdx via judicious incorporation of disulfide bond linkages along the putative dimer interface (data not shown). However, these efforts resulted in non-specific disulfide bond formation leading to soluble aggregates. Therefore, we do not have a dimer-only version of the protein. However, an analysis of the dimer interface from the crystal structure of full-length human Adx (PDB 3P1M) points toward a structural basis for why 2:1 dimer interactions with either of the other proteins are unlikely to occur. This structure was deposited in 2010 without a corresponding publication, and therefore has not been adequately discussed. In that dimeric arrangement, which is supported in solution by the NMR data (Figure 4), α-helix 3 (and particularly Asp-79, which is required to bind both CYPs and AdR), is positioned toward the dimer interface and is therefore partially occluded (as summarized in Figure 7). Such an arrangement is also consistent with structural changes that are known to occur, particularly along α -helix 3, upon reduction of Adx [18]; α -helix 3 adopts a more disordered helical struture in the reduced state and this may make formation of an Adx dimer unfavorable. Therefore, the monomer-dimer equilibrium in the oxidized state may regulate access of potential binding sites for either Adx:CYP or Adx:AdR interactions. In such a model, the Adx dimer would not contribute meaningful interactions in either step. Despite this, the mutually exclusive nature of all three Adx complexes, as shown in Figure 7, hints toward a functional role for Adx dimers, potentially by sequestering monomeric Adx. One possibility is that dimerization provides a reservoir of non-reduced Adx that is available for AdR in the mitochondrial matrix, and that the nature of the dimer equilibrium may regulate access to monomers of Adx when required by changes in cellular metabolism without having to increase expression of the protein; maintenance of the equilibrium would provide a steady supply of monomeric Adx.
Figure 7. Summary model of protein-protein interactions of Adx.
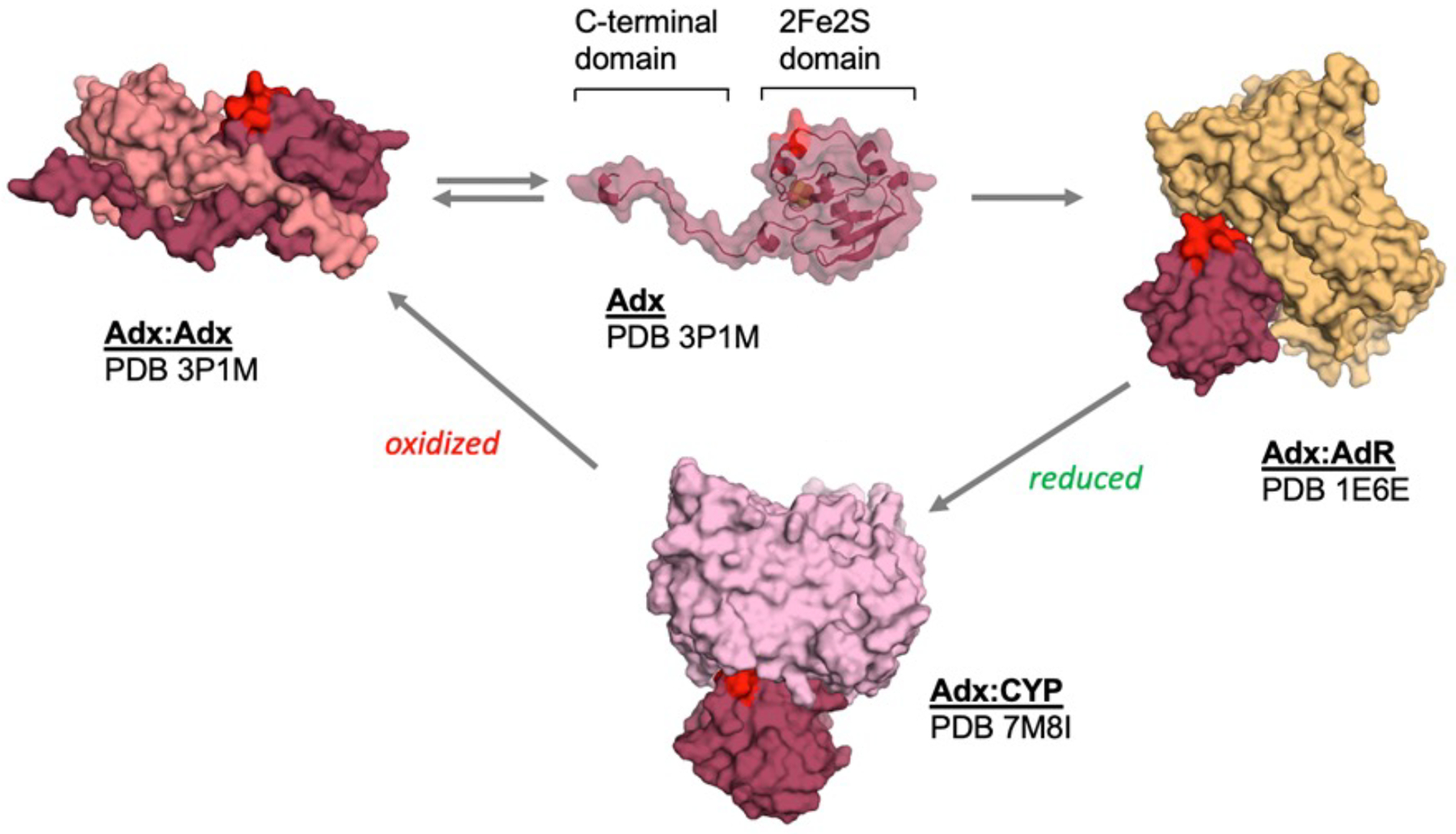
The involvement of α -helix 3 (which is highlighted in red) as part of the interface in the hAdx dimer, the Adx:AdR electron transfer complex, and the functional Adx:CYP complex, suggests a series of mutually exclusive interactions as part of its electron shuttle mechanism. The complexes shown are that of the structure with bovine AdR (PDB 1E6E) and the structure with CYP11B2 (PDB 7M8I).
4. Conclusions
This work characterizes inter-molecular interactions of human Adx in solution that are critical for electron delivery in support of the function of all seven mitochondrial CYPs. Included in these findings is evidence that Adx undergoes mutually exclusive interactions with either hCYP24A1, AdR, or with another monomer of Adx, while homodimers of Adx do not appear to play a direct role in functional interactions. These findings also inform a revised model of mitochondrial electron delivery that accounts for dimer formation in Adx. This study also provides evidence that supports the dimeric arrangement of human Adx as reported in the crystal structure (3P1M). Importantly, such an arrangement, in which regions of the CYP and AdR binding surfaces are not accessible, explains the ability of Adx dimerization to disrupt formation of the functionally relevant complexes, as indicated in this study.
5. Experimental procedures
5.1. Materials
CYP24A1 substrate, 1α,25(OH)2D, was procured from MedChem Express. Isotope labeling reagents 15N NH4Cl and 13C-D-glucose were from Cambridge Isotopes. Cloning of human Adx and point mutations were generated at Genscript.
5.2. Protein production
Production of hCYP24A1 was carried out as recently described [21] using a fused hAdx_hCYP24A1 construct with a tobacco etch virus (TEV) protease cut site located in the linker and a 6 X polyhistidine tag on the N-terminus of the hAdx. Briefly, the protein was expressed in E. coli JM109 cells and extracted from the membrane using (3-((3-cholamidopropyl) dimethylammonio)-1-propanesulfonate) (CHAPS) detergent. The extracted fusion protein was purified on a Ni-NTA column, followed by separation of the hAdx domain by overnight incubation with TEV protease and subsequent isolation of hCYP24A1 by size exclusion chromatography. The hCYP24A1 was diluted to between 4–10 μM and stored at in −80 °C until use.
Production of full-length hAdx (non-fusion) and AdR was completed as previously described[21]. The mutations to full-length hAdx and truncation of the C-terminal tail were generated at GenScript and the mutant proteins were expressed and purified using the same protocols as wild-type proteins.
Production of 15N labeled hAdx (see full amino acid numbering in Supplementary Figure S6) was carried out using the published protocol [21]. The overnight cultures of pET-15 plasmids containing either full-length or truncated hAdx were transfected into BL21(DE3) E. coli and grown overnight in Luria Broth media. Expressions were carried out in 1 liter cultures of minimal media containing 1 g/L of 15N-NH4Cl. Cultures expression double labeled 13C, 15N samples of hAdx for backbone NMR assignment were also supplemented with 2 g/L of 13C-D glucose (Cambridge Isotopes). Cultures were grown at 37 °C with shaking at 220 RPM until OD600= 1.0, then induced with 1 mM Isopropyl β-D-1-thiogalactopyranoside. Expressions were continued at 26 °C for 48 hours. Cells were harvested and protein was purified as previously described for unlabeled protein.
5.3. NMR backbone assignments of 13C,15N-hAdx (2–124)
The 13C,15N-hAdx was exchanged into NMR buffer consisting of 50 mM potassium phosphate (a mixture of monobasic KH2PO4 and dibasic K2HPO4), 50 mM NaCl, pH 7.4, 5% D2O. A sample at a protein concentration of 1 mM was used to acquire a three-dimensional HNCACB [38] data set at 25 °C on an a Bruker Avance III 800 MHz spectrometer with a TCI Cryo Probe. Acquisition parameters consisted of 16 scans, 48 increments in the 15N dimension, and 128 increments in the 13C dimension. Data sets were processed using nmrPipe and analyzed on NMRViewJ [39, 40]. Backbone assignments for hAdx (2–124) were aided significantly by comparison with assignments previously reported for human Adx (4–114) [24]. Secondary chemical shift values from Cα were used to determine secondary structural elements of the unstructured C-terminal tail [41, 42].
5.4. NMR 2D data acquisition and processing
All samples of 15N-hAdx for 2D 1H-15N HSQC acquisition were prepared in the low salt NMR buffer described above. Labeled protein concentrations were kept at 50 μM and the concentrations of hCYP24A1 and AdR were adjusted accordingly. For samples containing hCYP24A1, care was taken to keep the CHAPS concentration below the micellar concentration and thus prevent transferring detergent micelles into NMR samples. The enzyme was first transferred into a high salt, detergent free buffer consisting of 500 mM potassium phosphate, 300 mM NaCl, and 20% glycerol at pH 7.4 and passed through a 50 kDa cutoff concentrator (Millipore). Subsequently, 15N-hAdx was added and the mixture transferred into a 10 kDa cutoff concentrator prior to transfer into the low salt NMR buffer. For samples containing substrate, 1α,25(OH)2D (MedChemExpress) was added at super-saturating concentrations during the final buffer exchange. Data acquisition was carried out on a Bruker Avance III 800 MHz spectrometer at 25 °C, using 32 scans and 128 increments as described previously [28]. To measure the effects of binding, 1H-15N HSQC peak intensities were extracted using NMRViewJ [40] and used to calculate the ratios of remaining intensities using the formula of Intensitybound / Intensityfree. The average per residue intensity loss was calculated and used to determine a standard deviation from the mean. Residues reduced in intensity by either 0.75 or 1 standard deviation from the mean were mapped onto the crystal structure of hAdx (3P1M) [20].
5.5. Native gel electrophoresis
hAdx (2–124) and hAdx (2–108) were resolved on a native gel in a buffer consisting of 25 mM TrisHCl, 192 mM glycine, pH 8.3. Protein samples were prepared at a 1:1 volume with BioRad native PAGE sample buffer and at a concentration of 5 μM. The gel was stained with coomassie blue for visualization.
5.6. Analytical gel filtration chromatography
Previously purified hAdx (2–124) and hAdx (2–108) were resolved on a calibrated and pre-equilibrated 24 ml BioRad Enrich 650 gel filtration column. The running buffer consisted of 50 mM potassium phosphate, 5% glycerol, pH 7.4. The column was run at a flow rate of 0.5 ml/min with detection at 280 nm and 415 nm. The column itself was calibrated using a low molecular weight calibration kit from Cytiva.
5.7. EDC chemical cross-linking
Purified hAdx (2–124) and hAdx (2–108) (10 μM) were individually mixed with AdR (10 μM) in a low salt buffer consisting of 10 mM potassium phosphate, pH 7.4. The crosslinking agent 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC) was added at a final concentration of 2 mM and the the final mixture (50 μl volume) was incubated for two hours at 25 °C with gentle agitation. Each reaction was run individually in quadruplicate and resolved by denaturing SDS-PAGE. Coomassie stained gels were imaged on a a ChemiDoc Touch imaging system (Bio-Rad) and the protein bands quantified by ImageJ. Crosslinking efficiency was calculated as a ratio of the areas of the crosslinked product gel band divided by the sum of all band values using the following formula: (Area of Adx-AdR cross-linked product) divided by the sum areas of all bands (free Adx + free Adx-Adx dimer + free AdR + the Adx-AdR complex). Final ratios were analyzed for significance using a one-way ANOVA using an α =0.5.
5.8. CYP24A1 Functional Assays
Reconstituted hCYP24A1 functional assays were carried out as previously described with a few modifications [21]. Briefly, assays contained 1.5 μM hCYP24A1, 1.5 μM AdR and 1.0 μM hAdx. The reactions were run in 100 mM potassium phosphate with 5 μM 1α,25(OH)2D. All reactions were pre-incubated at 37 °C and started by the addition of 1 mM NADPH. The reactions were quenched after 60 seconds by addition of acetonitrile. The zero minute controls were quenched with acetonitrile prior to the addition of NADPH. 25(OH)D (2 μM) was added as an internal standard. Samples were resolved on a Poroshell C18 column (Agilent) using an Agilent Infinity 1260 ii HPLC. The mobile phase consisted of 70% acetonitrile and 30% water. Substrate detection was by UV-Vis monitoring at 264 nm. 1α,25(OH)2D depletion was calculated as a percentage of the substrate remaining compared to the zero minute reactions. All samples were run in triplicate and a One-way ANOVA test (α=0.5) was used to determine significance.
Supplementary Material
Acknowledgements
Constructs for expression of human Adx were originally gifts from the laboratory of Dr. Emily Scott at the University of Michigan. We would also like to acknowledge Dr. Dave Kiemle, director at the SUNY-ESF High Field NMR facility, for facilitating acquisition of HSQC spectra.
Source of funding:
This work was supported by NIH grant R35GM133375 (DFE).
Uncommon abbreviations:
- HSQC
Heteronuclear Single Quantum Coherence
- Adx
Adrenodoxin
- AdR
Adrenodoxin Reductase
Footnotes
AI Disclosure Statement
AI-assisted technologies were not used in the course of writing or editing this manuscript.
Data availability statement
Raw and processed NMR spectra are available on request.
References
- [1].Enright JM, Toomey MB, Sato S.-y., Temple SE, Allen JR, Fujiwara R, Kramlinger VM, Nagy LD, Johnson KM, Xiao Y, Current Biology, vol. 25, 2015, pp. 3048–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ewen KM, Kleser M, Bernhardt R, Biochim Biophys Acta, vol. 1814, 2011, pp. 111–125. [DOI] [PubMed] [Google Scholar]
- [3].Omura T, Chem Biol Interact, vol. 163, 2006, pp. 86–93. [DOI] [PubMed] [Google Scholar]
- [4].Usanov SA, Graham SE, Lepesheva GI, Azeva TN, Strushkevich NV, Gilep AA, Estabrook RW, Peterson JA, Biochemistry, vol. 41, 2002, pp. 8310–8320. [DOI] [PubMed] [Google Scholar]
- [5].Strushkevich NV, Azeva TN, Lepesheva GI, Usanov SA, Biochemistry (Mosc), vol. 70, 2005, pp. 664–671. [DOI] [PubMed] [Google Scholar]
- [6].Strushkevich N, MacKenzie F, Cherkesova T, Grabovec I, Usanov S, Park HW, Proc Natl Acad Sci U S A, vol. 108, 2011, pp. 10139–10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Brixius-Anderko S, Scott EE, J Biol Chem, vol. 296, 2021, pp. 100794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Estrada DF, J Biol Chem, vol. 293, 2018, pp. 4167–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lambeth JD, Kriengsiri S, J Biol Chem, vol. 260, 1985, pp. 8810–8816. [PubMed] [Google Scholar]
- [10].Behlke J, Ristau O, Müller E-C, Hannemann F, Bernhardt R, Biophysical chemistry, vol. 125, 2007, pp. 159–165. [DOI] [PubMed] [Google Scholar]
- [11].Schiffler B, Kiefer M, Wilken A, Hannemann F, Adolph HW, Bernhardt R, Journal of Biological Chemistry, vol. 276, 2001, pp. 36225–36232. [DOI] [PubMed] [Google Scholar]
- [12].Muller JJ, Lapko A, Bourenkov G, Ruckpaul K, Heinemann U, J Biol Chem, vol. 276, 2001, pp. 2786–2789. [DOI] [PubMed] [Google Scholar]
- [13].Kido T, Kimura T, Journal of Biological Chemistry, vol. 254, 1979, pp. 11806–11815. [PubMed] [Google Scholar]
- [14].Muller EC, Lapko A, Otto A, Muller JJ, Ruckpaul K, Heinemann U, Eur J Biochem, vol. 268, 2001, pp. 1837–1843. [PubMed] [Google Scholar]
- [15].Lambeth JD, Geren LM, Millett F, J Biol Chem, vol. 259, 1984, pp. 10025–10029. [PubMed] [Google Scholar]
- [16].Kumar A, Estrada DF, Drug Metabolism and Disposition, vol. 47, 2019, pp. 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Peng H-M, Auchus RJ, Biochemistry, vol. 56, 2017, pp. 2282–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Beilke D, Weiss R, Löhr F, Pristovšek P, Hannemann F, Bernhardt R, Rüterjans H, Biochemistry, vol. 41, 2002, pp. 7969–7978. [DOI] [PubMed] [Google Scholar]
- [19].Pikuleva IA, Tesh K, Waterman MR, Kim Y, Archives of biochemistry and biophysics, vol. 373, 2000, pp. 44–55. [DOI] [PubMed] [Google Scholar]
- [20].A.J. Chaikuad C; Krojer T; Yue WW; Phillips C; Bray JE; Pike ACW; Muniz JRC; Vollmar M; Weigelt J; Arrowsmith CH; Edwards AM; Bountra C; Kavanagh K; Oppermann U, To be published.
- [21].Jay N, Duffy SR, Estrada DF, Biochemistry, vol. 61, 2022, pp. 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Child SA, Reddish MJ, Glass SM, Goldfarb MH, Barckhausen IR, Guengerich FP, Archives of Biochemistry and Biophysics, 2020, pp. 108596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Glass SM, Webb SN, Guengerich FP, Archives of Biochemistry and Biophysics, 2021, pp. 109076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kostic M, Pochapsky SS, Obenauer J, Mo H, Pagani GM, Pejchal R, Pochapsky TC, Biochemistry, vol. 41, 2002, pp. 5978–5989. [DOI] [PubMed] [Google Scholar]
- [25].Hollingsworth SA, Batabyal D, Nguyen BD, Poulos TL, Proc Natl Acad Sci U S A, vol. 113, 2016, pp. 8723–8728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pedersen JI, Shobaki HH, Holmberg I, Bergseth S, Bjorkhem I, J Biol Chem, vol. 258, 1983, pp. 742–746. [PubMed] [Google Scholar]
- [27].Jones G, Prosser DE, Kaufmann M, Journal of lipid research, vol. 55, 2014, pp. 13–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Kumar A, Wilderman PR, Tu C, Shen S, Qu J, Estrada DF, Biochemistry, vol. 59, 2020, pp. 1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Schiffler B, Zöllner A, Bernhardt R, European Biophysics Journal, vol. 40, 2011, pp. 1275–1282. [DOI] [PubMed] [Google Scholar]
- [30].Muller JJ, Lapko A, Bourenkov G, Ruckpaul K, Heinemann U, Journal of Biological Chemistry, vol. 276, 2001, pp. 2786–2789. [DOI] [PubMed] [Google Scholar]
- [31].Chu J-W, Kimura T, Journal of Biological Chemistry, vol. 248, 1973, pp. 2089–2094. [PubMed] [Google Scholar]
- [32].Schulz V, Freibert SA, Boss L, Muhlenhoff U, Stehling O, Lill R, FEBS Lett, vol. 597, 2023, pp. 102–121. [DOI] [PubMed] [Google Scholar]
- [33].Shi Y, Ghosh M, Kovtunovych G, Crooks DR, Rouault TA, Biochim Biophys Acta, vol. 1823, 2012, pp. 484–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zalewski A, Ma NS, Legeza B, Renthal N, Flück CE, Pandey AV, The Journal of Clinical Endocrinology & Metabolism, vol. 101, 2016, pp. 3409–3418. [DOI] [PubMed] [Google Scholar]
- [35].Muller A, Muller JJ, Muller YA, Uhlmann H, Bernhardt R, Heinemann U, Structure, vol. 6, 1998, pp. 269–280. [DOI] [PubMed] [Google Scholar]
- [36].Loomis CL, Brixius-Anderko S, Scott EE, J Inorg Biochem, vol. 235, 2022, pp. 111934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Uhlmann H, Kraft R, Bernhardt R, J Biol Chem, vol. 269, 1994, pp. 22557–22564. [PubMed] [Google Scholar]
- [38].Grzesiek SB, A Journal of Magnetic Resonance, vol. 99, 1992, pp. 201–207. [Google Scholar]
- [39].Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A, Journal of biomolecular NMR, vol. 6, 1995, pp. 277–293. [DOI] [PubMed] [Google Scholar]
- [40].Johnson BA, Protein NMR Techniques, Springer, 2004, pp. 313–352. [Google Scholar]
- [41].Mielke SP, Krishnan V, Progress in nuclear magnetic resonance spectroscopy, vol. 54, 2009, pp. 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wishart DS, Sykes BD, Journal of biomolecular NMR, vol. 4, 1994, pp. 171–180. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed NMR spectra are available on request.