

Abstract
Proteins with potential roles in meiotic recombination often have essential or important functions during the mitotic cell cycle. In addition, proteins may have different functions at different times during meiosis. In such cases, it can be challenging to precisely determine protein function during meiosis using null or hypomorphic mutants. One example is the Sgs1–Top3–Rmi1 helicase–decatenase complex, which is required for normal vegetative growth and genome stability. In such cases, conditional loss-of-function mutants can be useful. In this chapter, we describe the construction of two types of conditional mutants, meiotic depletion alleles and auxin-induced degradation alleles, that allow protein depletion specifically during budding yeast meiosis, and illustrate their use with Sgs1. We also describe a modified method for the isolation of meiotic recombination intermediates that combines previous psoralen cross-linking and cetyltrimethylammonium bromide isolation methods.
1. INTRODUCTION
In the budding yeast Saccharomyces cerevisiae, meiotic recombination intermediate metabolism and the crossover/noncrossover pathway choice are regulated by an evolutionarily conserved complex that contains the BLM helicase homolog, Sgs1, and the Top3–Rmi1 topoisomerase/decatenase (STR complex; De Muyt et al., 2012; Jessop & Lichten, 2008; Jessop, Rockmill, Roeder, & Lichten, 2006; Kaur, De Muyt, & Lichten, 2015; Oh et al., 2007; Oh, Lao, Taylor, Smith, & Hunter, 2008; Rockmill, Fung, Branda, & Roeder, 2003; Tang, Wu, Zhang, & Hunter, 2015). This complex (and its homologs in other species) plays important roles in homologous recombination (HR) and genome stability during the mitotic cell cycle (Larsen & Hickson, 2013). Null mutants lacking any member of the STR complex display growth and genome stability defects that preclude their use in analyses of meiotic recombination (Gangloff, McDonald, Bendixen, Arthur, & Rothstein, 1994; Jessop et al., 2006; Mullen, Kaliraman, Ibrahim, & Brill, 2001). To address this problem, conditional expression and depletion approaches have been developed and applied to the study of STR meiotic function. These allow functional STR to be present during premeiotic growth, while allowing subsequent events of meiosis to occur in its absence. In this chapter, we describe the application of two different approaches to conditional STR protein expression: promoter replacement, so that a gene of interest is expressed only during mitotic growth; and auxin-induced degradation, which allows the time-controlled conditional degradation of a target protein during meiosis. The use of these approaches for conditional depletion of Sgs1 is described as an example, but these approaches should be applicable to the analysis of meiotic function of many proteins that are normally present in both mitotic and meiotic cells.
2. MEIOSIS-SPECIFIC PROTEIN DEPLETION
Many cellular proteins perform important functions during both meiosis and the mitotic cell cycle. Because of this, the null mutants and heat-sensitive conditional mutants usually used to study mitotic functions are often impractical for the study of their meiotic functions. For example, the poor growth and genome instability shown by null mutants lacking Sgs1, Top3, or Rmi1, particularly in diploids in the widely used SK1 background, are so severe that the mating-type heterozygosity necessary for meiosis cannot be maintained during the growth of bulk cultures (Jessop et al., 2006; Kaur et al., 2015). Moreover, because many yeast strains, including SK1, sporulate poorly above 34°C, and higher temperatures affect meiotic recombination (Börner, Kleckner, & Hunter, 2004; Chan, Borts, & Hoffmann, 2009; Honigberg & Esposito, 1994), the use of heat-sensitive mutants is often impractical. For these reasons, several strategies have been adopted to allow conditional depletion of proteins during meiosis.
2.1. Strategies for Meiotic Protein Depletion
2.1.1. Meiotic Depletion Alleles
The term “meiotic depletion allele” is generally used to refer to constructs, where the promoter of a gene is replaced by one that is expressed only during the mitotic cell cycle. In practice, promoters from MCD1 (also called SCC1; Clyne et al., 2003) and from CLB2 (Lee & Amon, 2003) have been used. Both genes are poorly expressed during meiosis (Chu et al., 1998; Primig et al., 2000), but are expressed at different times during the mitotic cell cycle. MCD1 is expressed during S-phase, while CLB2 is first expressed in late S-phase and continues to be expressed during G2 (Cho et al., 1998; Grandin & Reed, 1993). Both promoters are available in plasmids containing the relevant 5ʹ upstream region (−840 to −1 for MCD1; −1000 to −1 for CLB2) and an upstream drug-resistance cassette (Goldstein & McCusker, 1999; Wach, 1996) to allow selection of transformants (see Sections 2.2 and 2.3, for construct and transformation details). We and others have used such alleles successfully to deplete Sgs1, Top3, and Rmi1 (Jessop et al., 2006; Kaur et al., 2015; Oh et al., 2008; Tang et al., 2015; see Fig. 1, for an example), the repair protein Mms4 (Jessop & Lichten, 2008; Oh et al., 2008), and polo kinase Cdc5 (Clyne et al., 2003; Lee & Amon, 2003). Others have used meiotic depletion alleles to deplete many other proteins; a partial list includes the replicative helicase loader Cdc6 (Blitzblau & Hochwagen, 2013), the kinetochore proteins Iml3, Dsn1, and Mtw1 (Vincenten et al., 2015), SUMO-specific proteases Ulp1 and Ulp2 (Leung et al., 2015), the spindle pole body protein Mps3 (Li, Shao, Jin, & Yu, 2015), topoisomerase II (Zhang et al., 2014), the Smc5/6 complex proteins Smc5 and Nse4 (Copsey et al., 2013), the ATR kinase Mec1 (Gray, Allison, Garcia, Goldman, & Neale, 2013), the PP2a subunit Cdc55 (Bizzari & Marston, 2011), the cohesin loading and maintenance factors Scc2, Scc3, and Pds5 (Jin, Guacci, & Yu, 2009; Lin, Jin, Liu, Hampton, & Yu, 2011; Lin, Wang, Jin, & Yu, 2011), the condensin component Brn1 (Brito, Yu, & Amon, 2010), the APC/C adaptor Cdc20 (Lee & Amon, 2003), the Aurora B kinase Ipl1 (Yu & Koshland, 2007), and the centromeric cohesin protector Sgo1 (Katis et al., 2010).
Fig. 1.
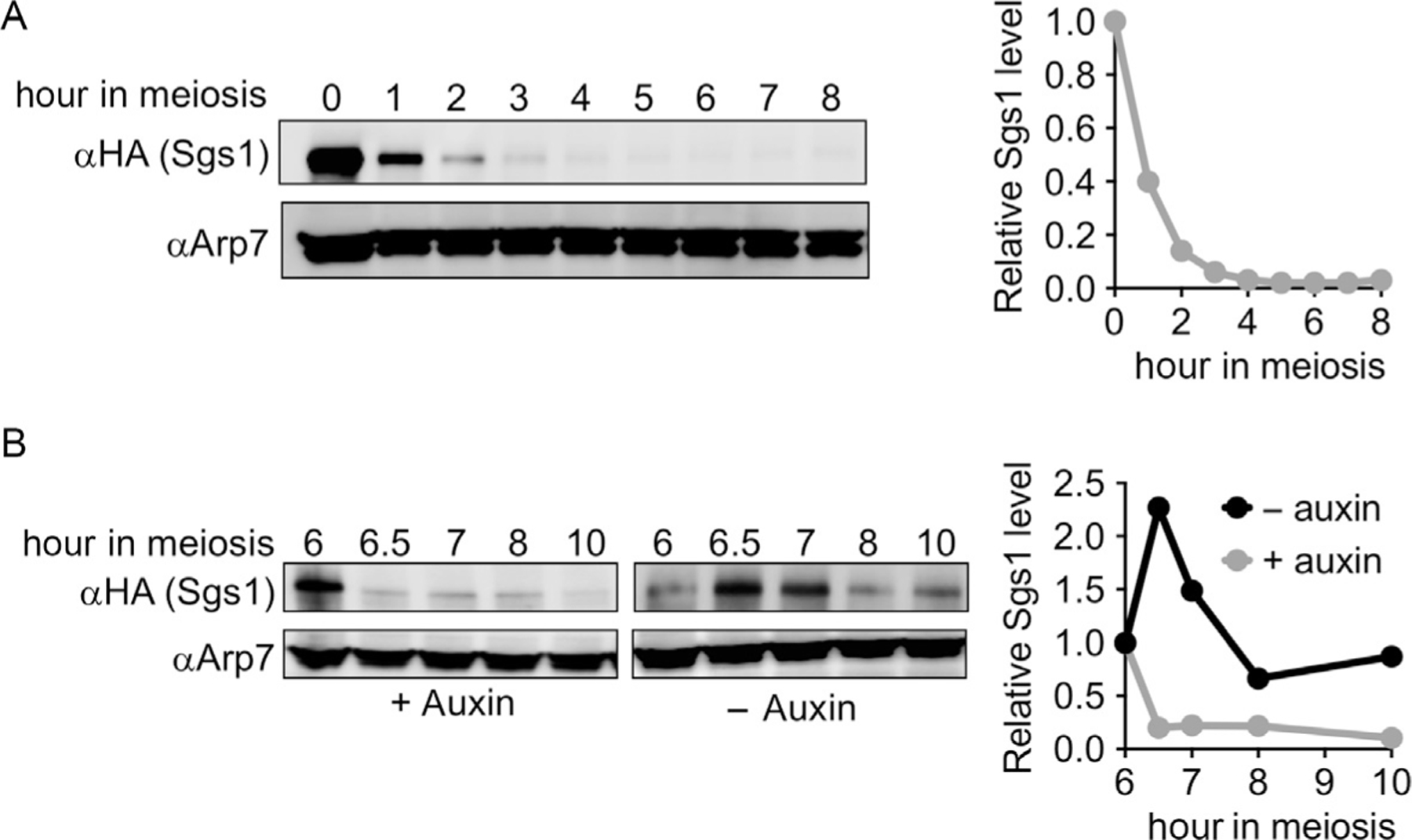
Western blots illustrating meiotic depletion of Sgs1. (A) Meiotic depletion, using a strain where an HA epitope-tagged Sgs1 protein is expressed from the CLB2 promoter (kanMX-pCLB2-3HA-SGS1, see Fig. 2A). Arp7 is used as a loading control. Arp7-normalized levels of Sgs1, relative to the premeiotic culture (0h), are plotted. (B) Auxin-induced depletion, using a strain where Sgs1 protein contains C-terminal HA epitope tags and the IAA17 auxin recognition domain (SGS1-3HA-IAA17-hygMX, see Fig. 2C), and the osTIR1 F-box protein is expressed from the TEF1 promoter (pTEF1-osTIR1). At 6h in meiosis, the culture was split, auxin (3-indoleacetic acid) was added to one half, and vehicle (DMSO) was added to the other. Arp7 is used as a loading control. Sgs1 levels, relative to the time of auxin addition (6h), are plotted.
Several considerations are important to the success of this promoter-replacement strategy. First, it relies on endogenous processes to deplete the protein of interest before it can function during meiosis, and one cannot assume that the protein will be absent during meiosis simply because the replacement promoter is not active. Thus, the protein of interest must either have a short half-life or be specifically degraded during the mitotic division prior to initiation of meiosis; long-lived proteins that remain at high levels during meiosis in the absence of further transcription are not amenable to this approach. It is important to monitor levels of the protein of interest during meiosis to confirm effective depletion, either using antibodies against the endogenous protein or with antibodies against an antigenic tag that is included in the promoter-replacement construct. In the case of the pCLB2-SGS1 allele described here, the target protein is still present at significant levels during the first 2h of meiosis, when DNA replication occurs, but is substantially depleted at later times, when meiotic recombination occurs (Fig. 1A).
Second, it is important to ensure that the promoter replacement does not have collateral consequences on gene function during the mitotic cell cycle that might carry over into meiosis. This is best determined empirically, by examining promoter-replacement strains to see if they display mutant phenotypes, during vegetative growth, expected for full or partial loss-of-function alleles. In practice, this has not been a concern for pCLB2-SGS1 promoter-replacement constructs (as well as for pCLB2-TOP3 and pCLB2-RMI1 constructs), even though the time of peak expression from the CLB2 promoter (late S- and G2-phases) comes after the time (S-phase) when Sgs1–Top3–Rmi1 complex activity is important. However, it is conceivable that use of a CLB2 promoter might create a mutant phenotype, especially for a gene whose protein product is degraded during mitosis but is required earlier in the cell cycle. In such a case, the MCD1 promoter may be more suitable.
Finally, it is important to recognize that deletion of a gene’s promoter and/or its replacement with the CLB2 or MCD1 promoter may impact expression of the gene immediately upstream of the target gene, particularly when the two genes are divergently transcribed. While this has proven not to be an issue in the constructs we have used, it remains a formal concern. One possible way to address this concern would be to insert the mitosis-specific promoter immediately upstream of the gene of interest, without deleting any upstream sequences, and to rely on transcription terminators present in the drug-resistance cassette, used to select transformants, to block read-through transcription.
2.1.2. Conditional Depletion Using Auxin-Inducible Degrons
While meiotic depletion alleles generally confer loss of function for a target protein throughout meiosis, some analyses (for example, execution point analysis) require wild-type function during early stages of meiosis, followed by target protein depletion at later stages. Two different conditional depletion strategies have been used in studies of yeast meiosis. The first approach, called anchor-away (Haruki, Nishikawa, & Laemmli, 2008), removes a protein from the nucleus by tethering it to a cytoplasmic protein via a rapamycin-dependent dimerization module. This approach has been used to conditionally relocate several nuclear proteins (Zip1, Rad54, Psy2) during yeast meiosis, resulting in loss-of-function phenotypes (Subramanian et al., 2016). We have not used this method, which requires additional mutations (tor1–1 to tolerate rapamycin, fpr1 to remove an endogenous rapamycin-binding protein) to function, and which requires cytological assays to confirm effective nuclear depletion.
A second approach uses a plant-derived auxin-inducible degron to conditionally target a protein of interest for ubiquitin-mediated proteolysis (Nishimura, Fukagawa, Takisawa, Kakimoto, & Kanemaki, 2009; Nishimura & Kanemaki, 2014). This system has been used in a variety of organisms and has been modified specifically for use in budding yeast. It contains two components: a degron, based on the IAA17 transcription repressor, that is fused to the protein of interest, and an SCF E3 ligase component, TIR1, usually from rice (osTIR1). Auxins (usually indole acetic acid, IAA) promote the interaction of the IAA17-based tag with TIR1, leading to the rapid polyubiquitylation of the target protein and subsequent degradation by the proteasome, often in a matter of minutes in mitotically growing cells. Unfortunately, constructs and conditions that support rapid degradation of IAA17-tagged proteins in mitotic cells work poorly, if at all, in meiotic yeast cells. Efficient degradation appears to require high levels of osTIR1, and the original expression vectors developed by Nishimura and Kanemaki do not meet this need during meiosis. Two approaches have been used to address this problem. Tang et al. (2015) have constructed strains where osTIR1 expression is driven by the CUP1 promoter, with osTIR1 expression being induced by adding CuSO4 to cultures about 1h before the addition of auxin. As an alternative, we have constructed strains where osTIR1 is constitutively expressed at high levels in both mitotic and meiotic cells, using either the TEF1 or the ZEO1 promoters; these strains can be used for auxin-inducible degradation of target proteins during both meiosis and the mitotic cell cycle, without the need for other factors (Fig. 1B). Construction of strains with IAA17-protein fusions, pTEF1-osTIR1 or pZEO1-osTIR1, and use of these strains for meiotic depletion are described later.
2.2. Construction of Strains for Meiotic Protein Depletion
In the following sections, construction of meiotic depletion and of auxin-inducible degron yeast strains is described, using SGS1 as an example.
2.2.1. CLB2 Promoter Replacement
The strategy used for promoter replacement is illustrated in Fig. 2 and starts with a plasmid (pMJ787, originally Amon plasmid 455, a gift from Angelika Amon) that contains a kanMX module for selecting transformants with G418, nt −1000 to +6 from the CLB2 locus, and sequences encoding three tandem influenza hemagglutinin epitopes (3XHA). The kanMX-pCLB2-3xHA module is inserted at a locus by PCR amplification with primers corresponding to the edges of the module with an additional 40nt of sequence from the target locus (see Section 2.3.3). While this homology is in theory sufficient to direct integration in the yeast genome (Manivasakam, Weber, McElver, & Schiestl, 1995), we have found that the yield of transformants can be substantially increased by including two additional DNA fragments that contain 150–200nt of sequence flanking the site of insertion. The cotransforming DNA fragments contain sufficient homology to frequently recombine with each other (Kunes, Ma, Overbye, Fox, & Botstein, 1987), creating a kanMX-pCLB2-3xHA fragment with 150–200nt of flanking homology that is able to integrate in the genome with greater efficiency (Fig. 2A). If the N-terminal 3xHA tag is found to interfere with protein function, the same plasmid can be used for amplification with an alternative set of primers to create a promoter replacement without the N-terminal HA tag (Fig. 2B).
Fig. 2.
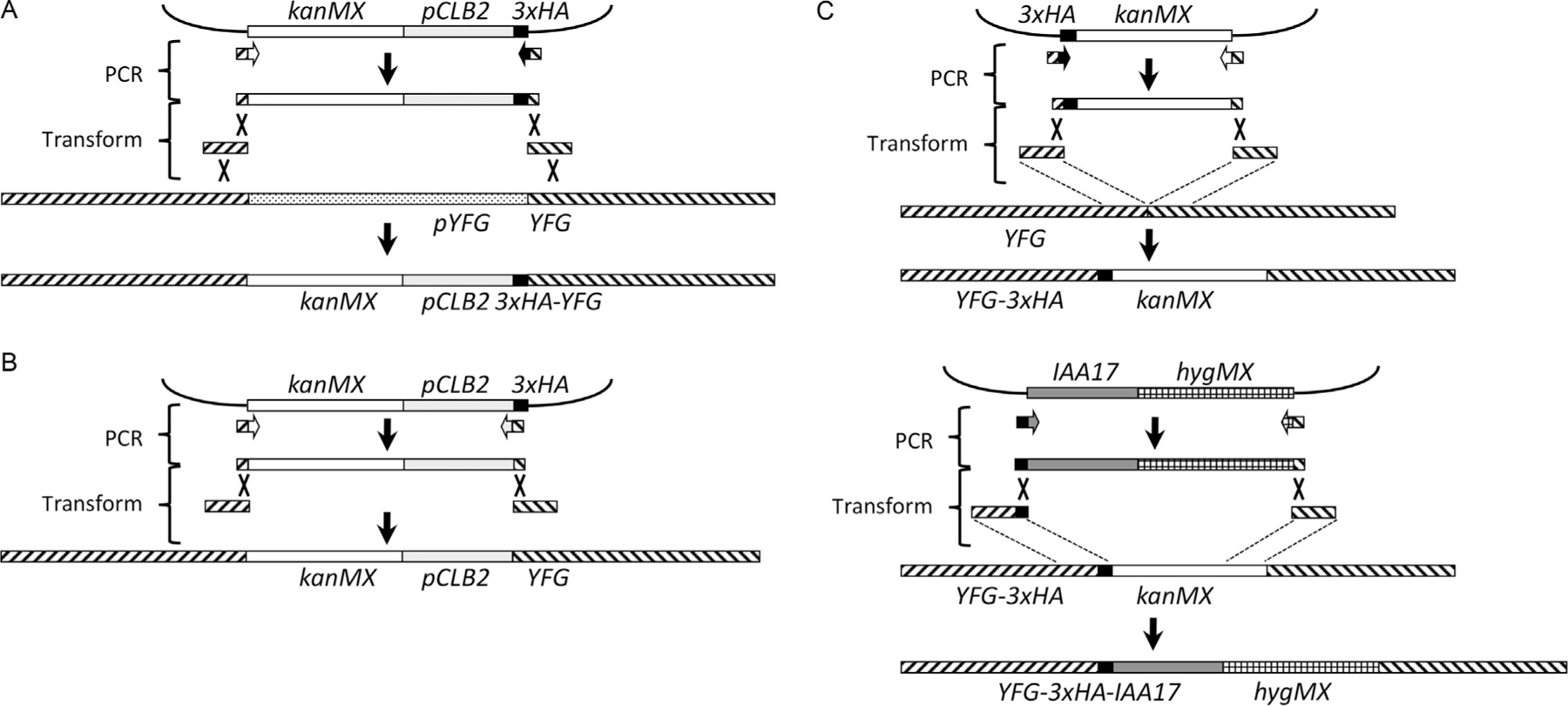
Three-fragment transformation strategy for promoter replacement and IAA17 tagging. (A) Replacement of a target gene (YFG) promoter with the CLB2 promoter and addition of an N-terminal HA tag (see text for details). A kanMX-pCLB2-3xHA fragment is amplified from plasmid DNA, using hybrid primers with 3ʹ homology to fragment boundaries and 5ʹ homology to the boundaries of the desired insertion site. This fragment is mixed with two fragments (150nt or greater) that contain sequences at the boundaries of the insertion site, and transformed into yeast. The additional homology provided by the two fragments increases the recovery of integrants, which replace the endogenous promoter with kanMX and the CLB2 promoter, and also add a 3xHA epitope tag at the N-terminus of the protein. (B) If desired, the 3xHA tag can be omitted by amplifying with a hybrid primer that contains 3ʹ homology to the right-hand CLB2 promoter boundary, and 5ʹ homology to the right-hand boundary of the insertion site. (C) Strategy for creating a C-terminal hybrid 3xHA-IAA17 tag. First, a 3xHA epitope tag is inserted at the C-terminus of the gene linked to kanMX, using three-fragment transformation. The same strategy is used to replace the kanMX moiety with a hygMX-linked IAA17 insert.
2.2.2. IAA17 Tagging
The strategy used for tagging with the IAA17 auxin-response element has been described (Nishimura & Kanemaki, 2014); in our work, we have generally used C-terminal IAA17 tags with a 3xHA epitope tag between the gene’s coding sequences and the IAA17 tag. This has the advantage of allowing protein expression from its native promoter, and of monitoring protein levels on Western blots (see Section 3.3). Construction is done in two steps (Fig. 2C). First, the protein is C-terminally tagged using pFA6a-3HA-kanMX6 (Longtine et al., 1998), using the same flanking homology fragment strategy as described earlier (Section 2.2.1). Second, the IAA17 tag is added to the C-terminal 3xHA tag, again using the flanking homology fragment strategy, using a derivative of pMK43 (Nishimura et al., 2009) in which the kanMX cassette is replaced by either a hygMX (pMJ894) or a natMX (pMJ893) drug-resistance cassette (Goldstein & McCusker, 1999).
2.2.3. osTIR1 Expression
The second element of the auxin-inducible expression system, the rice-derived TIR1 F-box protein, was originally obtained as a yeast codon-optimized version whose expression is driven by the GAL1 promoter (pYK6; Kubota, Nishimura, Kanemaki, & Donaldson, 2013). It appears that this protein must be expressed at high levels in order to function optimally, and the GAL1 promoter is not compatible with such high levels of expression during meiosis. Using genome-wide meiotic expression data (Primig et al., 2000), we identified two genes, TEF1 and ZEO1, that displayed uniformly high levels of expression both during the mitotic cell cycle and during meiosis; plasmids containing these promoters were constructed (Fig. 3) and integrated at the URA3 locus.
Fig. 3.
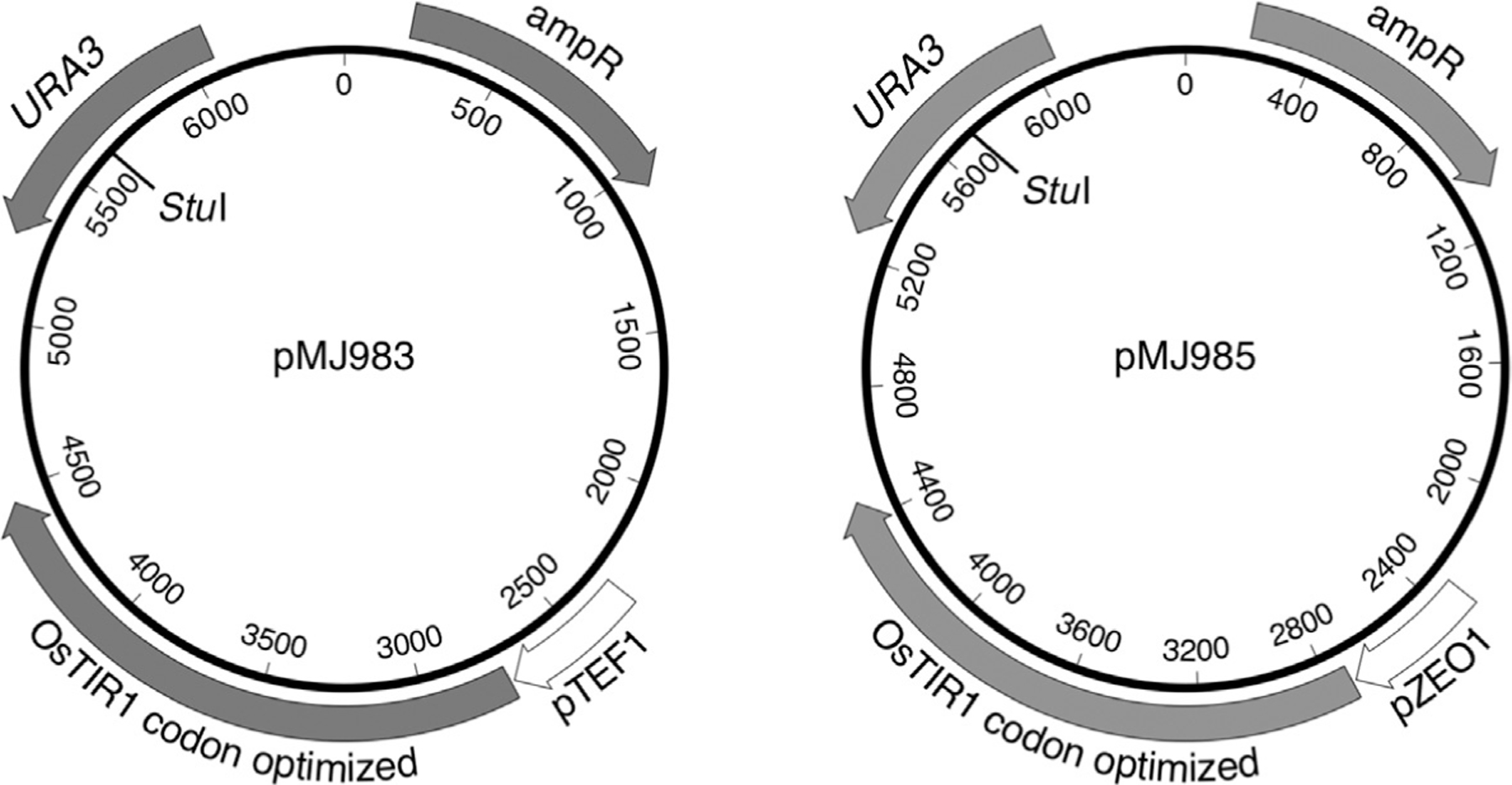
Plasmids with osTIR1 expressed from high-level, constitutive promoters. pMJ983 contains TEF1 promoter sequences from −409 to −1nt; pMJ985 contains ZEO1 promoter sequences from −405 to −1nt. The unique StuI site, used to direct integration to the URA3 locus, is indicated.
2.3. Insertion of Constructs by Transformation
In the following section, transformation by electroporation, using the flanking homology fragment strategy (see Fig. 2), is described. We have found this approach to be suitable for strain SK1 derivatives; in other strains, other approaches may be used (Gietz & Woods, 2001).
2.3.1. Equipment
High-capacity refrigerated tabletop or floor centrifuge with adaptors for 50-mL conical-bottom tubes
Vortex mixer
Electroporator capable of 1.5kV, 200 Ω, 25μF pulse (BioRad Micro-Pulser or equivalent)
Electroporation cuvettes, 0.2cm gap (BioRad 1652082 or equivalent). Keep at 4°C.
Shaking incubator or water bath, set at 30°C.
2.3.2. Reagents
YEPAD broth: 1% Bacto™ yeast extract, 2% Bacto™ peptone, 2% D-glucose, 0.004% adenine, pH 5.5 (adjusted with 1 N HCl). Prepare as a 1.1 × stock without glucose, and add glucose from a sterile 20% stock before use.
YEPAD+ 1 M sorbitol broth; make as above but include sorbitol at 1 M.
1 M sorbitol, filter-sterilize and store at 4°C
Sonicated carrier DNA. We usually use either salmon or herring sperm DNA, dissolved in 10mM TRIS, 1mM EDTA, pH 7.5 (TE), sonicated until no longer viscous, phenol-extracted and ethanol precipitated, and resuspended in TE at 5mg/mL. Chill to 4°C before use.
Selective media, made with 1 M sorbitol.
DNA fragments, made either by PCR or by digestion of plasmids, ideally at a concentration of 1–2μg/μL (see Note 1). Chill to 4°C before use.
2.3.3. Procedure
Inoculate a single colony of the desired strain into 5mL YEPAD; grow overnight with aeration at 30°C.
Wash three times with cold 1 M sorbitol by centrifuging 2min at 3220 × g at 4°C, draining thoroughly.
After the last centrifugation, decant as much supernatant as possible, and resuspend the pellet by vortexing. The final yield should be about 30μL of cells per 1mL of original overnight culture.
Transfer 40μL of cell suspension to a cold microfuge tube. At this point, cells can be frozen at −80°C.
If frozen cells are used, thaw cell suspension on ice.
Add 12.5μg sonicated carrier DNA and 1–2μg of each transforming DNA fragment.
Add the cell+DNA mix to a precooled electroporation cuvette (see Note 1).
Pulse-shock in electroporator at 1.5kV, 200 Ω, 25μF.
Immediately add 0.4mL ice-cold 1 M sorbitol and mix.
If nutritional selection is used, cells can be immediately plated on the relevant dropout plates made with 1 M sorbitol (see Note 2).
If a drug-resistance selection is used, add the transformation mix to 4.5mL of YEPAD+ 1 M sorbitol and aerate at 30°C for 4–5h before plating (see Note 2).
Incubate plates at 30°C for 2–3 days. Integrative transformants will be visible as larger colonies in a background of smaller microcolonies that contain abortive transformants.
3. SYNCHRONOUS MEIOSIS AND DNA, PROTEIN, AND CYTOLOGICAL ANALYSIS
A basic requirement for the real-time analysis of protein function during meiosis is the ability to generate populations of cells that undergo meiosis synchronously. Much of the work done in this regard has used strains of the SK1 genetic background (Alani, Cao, & Kleckner, 1987; Kane & Roth, 1974), where sporulation can be induced in liquid cultures with relative synchrony. The methods described later are the ones currently used by our lab to sporulate SK1, for isolation of DNA from meiotic cultures in a way that preserves recombination intermediates, for Western blots to monitor protein depletion, and for nuclear staining to monitor meiotic divisions.
3.1. Synchronous Sporulation
Most past and current studies induce SK1 strains (Kane & Roth, 1974) to sporulate by pregrowth in rich medium with acetate as the carbon source, to the point of transition between exponential growth and stationary phase. Cells are then shifted into starvation medium with acetate as the sole carbon source and are aerated vigorously to induce sporulation and meiosis. There is considerable variation between groups in terms of the composition of both presporulation and sporulation medium (Börner & Cha, 2015; Lichten, 2014; Oh et al., 2009). The protocol below uses buffered presporulation medium to limit the alkalinization of cultures that occurs during growth in non-fermentable carbon sources. High pH prevents nutrient uptake (McCusker & Haber, 1977) and can result in premature entry into meiosis.
3.1.1. Equipment
Rotating tissue culture drum (Bellco 7736–10115 or equivalent) in a 30°C incubator
Glass culture tubes, 20 × 150mm
2-L Erlenmeyer flasks (autoclaved)
250-mL centrifuge bottle (autoclaved)
High-capacity tabletop or floor centrifuge with adaptors for 250-mL bottles
2.8-L triple-baffled Fernbach flask (Bellco 2551–02800 or equivalent)
2-L triple-baffled Erlenmeyer flasks (Bellco 2542–02000 or equivalent)
Shaking incubator or shaking water bath capable of sustained operation at 375rpm (see Note 3)
Spectrophotometer with at least 1cm distance between the cuvette and detector (see Note 4) and 1.5mL semimicro disposable cuvettes, light path 10mm
3.1.2. Reagents (See Note 5)
YEPAD broth and plates: Broth—see Section 2.3.2. Plates—YEPAD plates are made the same way, but Bacto™ agar is added to 2% before autoclaving.
Presporulation broth (SPS): 1% Bacto™ peptone, 0.5% Bacto™ yeast extract, 1% potassium acetate, 0.17% Bacto™ yeast nitrogen base without amino acids, 1% ammonium sulfate, 0.5% potassium hydrogen phthalate, pH 5.5 (adjusted with 10 N KOH).
Sporulation medium (KAc): 1% potassium acetate, 0.001% polypropylene glycol 2000 (Sigma 202339, from a 1% stock suspension), supplemented as required by the specific auxotrophies of the strain being used. Supplements are added at 1/5 the concentration used to support vegetative growth in minimal media. Concentrations of common supplements used in our lab: histidine, 0.0004%; lysine, 0.0006%; arginine, 0.0004%; leucine, 0.0012%; uracil, 0.0002%. Add polypropylene glycol and supplements after autoclaving (see Note 5).
3-Indoleacetic acid (Auxin, Sigma I3750), 500mM in DMSO. Make fresh the day of use.
3.1.3. Procedure (See Note 6)
(–4 days) Streak diploid strains to be used on YEPAD agar plates. Incubate 48h at 30°C
(–2 days) Inoculate a single colony into 5mL YEPAD broth culture in a 20 × 150mm glass culture tube and incubate 24h at 30°C on a roller drum.
(–1 day) Inoculate four SPS cultures (240mL of SPS in a 2-L Erlenmeyer flask) with a range of dilutions of the overnight culture. We generally inoculate at 1/500, 1/1000, 1/1333, and 1/2000 dilutions. Incubate with vigorous shaking (300rpm) for ~18h at 30°C. Also, aliquot 400mL supplemented KAc into a 2.8-L triple-baffled Fernbach flask and incubate at 30°C. Reserve 240mL unsupplemented KAc for washing and incubate at 30°C.
About 18h after initiating SPS cultures, measure culture OD600. Grow cultures until one reaches an OD600 of 1.4 (~2 × 107 cells/mL).
Transfer culture to a 250-mL centrifuge bottle and centrifuge 3min, 3220 × g. Decant the supernatant, drain briefly, and resuspend in 240mL prewarmed KAc without supplements. Remove 35mL sample for DNA extraction, 3mL sample for protein extraction, and 1mL sample for DAPI staining.
Centrifuge remaining culture as above and resuspend in a portion of the 400mL supplemented KAc from the 2.8-L Fernbach flask, return to the flask, and incubate at 30°C with very vigorous shaking (375rpm).
Take samples at time points as needed for DNA, protein, and DAPI staining, as in step 5.
For auxin-induced protein depletion: at desired time, split culture in half, into two 2-L baffled Erlenmeyer flasks, adding to one 500mM auxin to a final concentration of 2mM and an identical volume of DMSO to the other. Continue additions every hour to maintain auxin concentrations.
Alternatively, if greater sample volumes are required, start two SPS cultures for each dilution (step 3). On the day of the sporulation, pool the two cultures whose OD600 is closest to 1.4, split into two centrifuge bottles, and process as above (steps 5–6) using two 2.8-L Fernbach flasks. At the desired time, inoculate one flask with auxin and the other with DMSO as in step 8.
3.2. DNA Preparation by Combined CTAB Extraction and Psoralen Cross-Linking
Methods for the preparation of DNA for the analysis of meiotic recombination products and intermediates have been described elsewhere (Ahuja & Börner, 2011; Allers & Lichten, 2000; Oh et al., 2009; Schwacha & Kleckner, 1994). Holliday junction (HJ)-containing intermediates are vulnerable to loss by branch migration, and three main strategies have been used to stabilize HJ-containing intermediates: psoralen cross-linking, which forms interstrand cross-links that limit branch migration; preparation in the presence of multivalent cations (including CTAB and hexamminecobalt(III) chloride, CoHex), which limits branch migration by stabilizing the stacked-X HJ conformation; and preparation in agarose plugs, which stabilizes HJs by limiting arm movement (Oh et al., 2009). Psoralen cross-linking has the advantage of allowing harsher downstream extraction procedures to be used, but has the disadvantage of requiring cross-link reversal if single-strand analyses are desired; in addition, cross-links at a restriction enzyme site block digestion, resulting in a low but unavoidable level of partial digestion products. Conversely, CTAB extraction is considerably more demanding technically, but yields DNA that requires no further processing for downstream analyses. We recently found that a combined CTAB extraction and psoralen cross-linking approach confers modest improvements in recombination intermediate yields (J. S. A. and M. L., unpublished), and describe it here. Methods for downstream analysis, including 1D- and 2D-gel electrophoresis, blotting, and probing, have been described elsewhere (Ahuja & Börner, 2011; Oh et al., 2009).
3.2.1. Equipment
High-capacity tabletop or floor centrifuge with adaptors for 50-mL conical-bottom tubes and 5-mL round-bottom tubes
Variable-speed microfuge
Long-wavelength UV (366nm) transilluminator, with output calibrated using a long-wave UV meter (UVP Blak-Ray, model J-221 or equivalent)
Qubit 3.0 minifluorometer, Nanodrop 2000 microvolume spectrophotometer, or equivalent
50-mL conical-bottom polypropylene tubes (Falcon 352070 or equivalent)
5-mL round-bottom polypropylene tubes (Falcon 352063 or equivalent)
1.5-mL microfuge tubes
35-mm Petri dish (Falcon 353001 or equivalent)
Fine-tip transfer pipettes (Samco 23220S)
3.2.2. Reagents
1mg/mL psoralen (Trioxsalen, Sigma-Aldrich T6137) in 100% ethanol (see Note 7)
50mM Tris–HCl, 50mM EDTA, pH 8
Storage solution: 1 M sorbitol, 50mM potassium phosphate, 10mM EDTA, 20% glycerol, pH 7.5. Filter-sterilize and store at 4°C
Spheroplasting solution: 1 M sorbitol, 50mM potassium phosphate, 10mM EDTA, 5mM hexamine cobalt chloride, pH 7.5. Filter-sterilize and store at RT
Zymolyase 100T
β-Mercaptoethanol
-
CTAB extraction solution: 3% CTAB, 100mM Tris, 25mM EDTA, 2 M NaCl, 2% polyvinylpyrrolidone (avg. MW 40,000, PVP40), 20mM CoHex. Preparation method is critical, as the final solution is too viscous to filter-sterilize, and components can precipitate if not added in the correct order
For 100mL:
Weigh out 11.69g NaCl; add 10mL 1 M Tris–HCl pH 7.5 and 5mL 0.5 M EDTA pH 8 and adjust to 40mL total with water. Add 10mL 10% PVP40 to dissolve the NaCl.
Filter-sterilize in the following order, adding each component after the previous has completely passed through the filter:
10mL 200mM CoHex
10mL 10% PVP40
50mL NaCl/Tris/EDTA/PVP40 from above
30mL 10% CTAB
Store at 37°C. If a precipitate forms, discard.
DNase-free RNase, 10mg/mL
Proteinase K, 20mg/mL in 10mM Tris, 20mM CaCl2, 50% glycerol, pH 7.5. Store aliquots at −20°C
Chloroform:isoamyl alcohol, 24:1
CTAB dilution solution: 1% CTAB, 50mM Tris, 1mM EDTA, 4mM CoHex, pH 7.5. Filter-sterilize and store at RT
0.4 M NaCl, 10mM Tris, 1mM EDTA, 1mM CoHex, pH 7.5. Filter-sterilize and store at 4°C
1.42 M NaCl, 10mM Tris, 1mM EDTA, 1mM CoHex, pH 7.5. Filter-sterilize and store at 4°C
95% ethanol
70% ethanol, 0.3mM CoHex
70% ethanol, 3mM MgCl2
10mM Tris, 10mM MgCl2, 200mM NaCl, pH 7.5. Filter-sterilize and store at 4°C
10mM Tris, 2mM MgCl2, 50μM spermidine, pH 7.5. Filter-sterilize and store at 4°C
3.2.3. Procedure
Place a 35-mL aliquot of sporulation or presporulation culture in a 50-mL conical tube and centrifuge 2min at 3220 × g. Drain well, resuspend in 1.5mL psoralen solution, made fresh the same day by adding 1mL of 1mg/mL psoralen stock to 9mL of 50mM Tris, 50mM EDTA. Vortex the psoralen working solution before each use to maintain suspension.
Place cells in a 35mm Petri dish on long-wave UV transilluminator. Cross-link 10min, 0.6mW/cm2, agitating plates every 2min to resuspend settled cells (see Note 8).
Transfer cells to 1.5-mL microfuge tubes, centrifuge 2min, 1200 × g.
Remove supernatant and resuspend in storage solution. Recentrifuge as above, remove supernatant, freeze pellet on dry ice, and store at −80°C.
Thaw frozen pellet on ice. While pellets are thawing, prepare zymolyase solution: 0.5mg/mL zymolyase 100T, 1% β-mercaptoethanol in spheroplasting solution. Keep on ice until use.
Resuspend cell pellet in 0.5mL zymolyase solution and transfer to a 5-mL round-bottom tube.
Incubate at room temperature for 2.5–3.5min, monitoring degree of spheroplasting by microscope (see Note 9).
When 80% of cells are spheroplasted (see Note 9), centrifuge 2min at 3200 × g at room temperature. Thoroughly remove supernatant. Gently resuspend cell pellet in 0.5mL CTAB extraction solution by flicking or by pipetting up and down. Add 12.5μL proteinase K solution, mix gently. Add 1μL RNase solution, mix gently.
Incubate at 37°C for 15 \min, mixing gently every 5min.
Transfer to a 1.5-mL microfuge tube that contains 0.3mL chloroform:isoamyl alcohol. Vortex vigorously for 10s, shake, and vortex again for 5s. Leave at room temperature for 2min and vortex again for 5s.
Centrifuge at top speed in microfuge for 3min. Promptly remove upper aqueous layer, avoiding white interface layer, and transfer to a 5-mL round-bottom tube.
Layer 1.5mL CTAB dilution solution on top, mix by inverting five times and then let sit at room temperature for 10min. Invert again 20 times, until a precipitate forms. If a precipitate does not form, add another 0.5mL CTAB dilution solution and repeat mixing and 10min rest. The precipitate should sink to the bottom of the tube.
Using a fine-tip transfer pipette, carefully remove the supernatant. If the precipitate is loose and not all supernatant can be removed, pulse-spin very briefly in the microfuge to make the precipitate settle to the bottom of the tube. Immediately add 2mL ice-cold 0.4 M NaCl, 10mM Tris, 1mM EDTA, 1mM CoHex. Mix briefly until the pellet floats. Carefully remove the supernatant, avoiding drying the pellet. Repeat this wash.
Promptly add 0.5mL ice-cold 1.42 M NaCl, 10mM Tris, 1mM EDTA, 1mM CoHex, and shake tube gently until the pellet becomes translucent.
Transfer to a prechilled 1.5-mL microfuge tube and add 1mL 95% ethanol. Invert gently until a precipitate forms. Allow precipitate to settle to the bottom of the tube. Pulse-spin if necessary. Remove supernatant with a fine-tip transfer pipette, add 1mL 70% ethanol, 0.3mM CoHex. Vortex briefly until pellet floats, then let settle. Pulse-spin if necessary. Remove supernatant.
If white CTAB particles are visible, repeat steps 14 and 15.
Completely remove ethanol and allow to air-dry briefly. The pellet should still be damp; overdrying will make resuspending difficult.
Add 0.1mL ice-cold 10mM Tris, 10mM MgCl2, 200mM NaCl, soak pellet on ice for at least 30min. Add 0.2mL 95% ethanol, invert gently until precipitate forms. Allow precipitate to settle, pulse-spinning if necessary. Remove supernatant, wash pellet twice with 0.2mL 70% ethanol, 3mM MgCl2 as in step 15.
Remove ethanol completely and air-dry briefly as in step 17.
Resuspend pellet in 50–100μL 10mM Tris, 2mM MgCl2, 50μM spermidine (see Note 10).
3.3. Protein Isolation and Western Blotting
Monitoring target protein levels during meiosis is important to determine the extent of depletion or induction. Western blots are used, probing either with antibodies specific to the protein of interest or using epitope-tagged constructs and probing with antiepitope antibodies. For Sgs1, Top3, and Rmi1, we use N- or C-terminal 3xHA tags, detected with anti-HA mouse monoclonal antibody (clone 12CA5, Roche). Polyclonal antisera are used to detect Arp7 (sc-8961, Santa Cruz Biotechnology) as a loading control. Blots are either probed first with the protein of interest, stripped, and reprobed with anti-Arp7, or separate gels are run with identical loadings of the same sample and are separately probed. To allow gel-to-gel comparisons, a common sample of the same or related strain expressing the tagged protein (usually a mitotic culture) should be loaded on every gel.
Proteolysis is a major concern in isolating nuclear proteins from meiotic cells, due to the tight vacuole-nuclear association that forms during meiosis (Pan et al., 2000). The protocol below, adapted from Foiani, Marini, Gamba, Lucchini, and Plevani (1994), uses trichloroacetic acid (TCA) pre-treatment and lysis in the presence of TCA to minimize proteolysis.
3.3.1. Equipment
High-capacity tabletop or floor centrifuge with adaptors for 5-mL round-bottom tubes
Variable-speed microfuge
5-mL round-bottom polypropylene tubes (Falcon 352063 or equivalent)
1.5-mL screw-top microfuge tubes
Acid-washed glass beads, 425–600μm (Sigma G8772)
Benchtop votexer
Mini-Beadbeater (Biospec) or equivalent
Block-type incubator or water bath, set to 95°C
Vertical minigel box and transfer apparatus (XCel Mini-Cell and XCell II™ Blot Module, Thermo Fisher Scientific)
Gel cassettes (Life Technologies NC2010)
Electrophoresis and transfer apparatus (XCell II Blot Module, Invitrogen)
Orbital platform shaker
Invitrolon™ PVDF/Filter paper sandwich, 0.4μm pore, 8.3 × 7.3cm (Thermo Fisher Scientific, LC2005)
CCD camera or scanning fluorimeter
3.3.2. Reagents
10% glycerol
20% and 5% TCA
1.5 M Tris–HCl, pH 8.8
Sample buffer: 0.1 M Tris–HCl, 0.2% SDS, 5% β-mercaptoethanol, 10% glycerol, 0.01% bromophenol blue
Protogel 29:1 acrylamide:bisacrylamide mix (National Diagnostics)
Tetramethylethylenediamine (TEMED)
2 × Stacking buffer: 0.125M Tris–HCl, 0.2% SDS, pH 6.8
2 × Resolving buffer: 0.75M Tris–HCl, 0.2% SDS, pH 8.8
10% ammonium persulfate
Precast 8%–16% Tris–Glycine Mini Gels, 12 well (Thermo Fisher Scientific, XP08162BOX)
Running buffer: 0.192 M glycine, 25mM Tris–HCl, 0.1%SDS, pH 8.3
Transfer buffer: 0.096 M glycine, 12.5mM Tris–HCl, 0.1% SDS, 20% methanol, pH 8.3. Chill to 4°C before use.
Phosphate-buffered saline (PBS): 137mM NaCl, 2.7mM KCl, 10mM Na2HPO4, 1.8mM KH2PO4, pH 7.4.
Biotin-free milk powder (I-Block™, Thermo Fisher Scientific, T2015)
Tween 20
Anti-HA mouse monoclonal antibody (clone 12CA5, Roche; 11583816001, used at 1:10,000 dilution)
Anti-Arp7 goat polyclonal antibody (Santa Cruz Biotechnology, sc-8961, used at 1:1000 dilution)
Alkaline phosphatase-conjugated goat-anti-mouse (Sigma A3562, used at 1:5000 dilution)
Alkaline phosphatase-conjugated rabbit-anti-goat (Sigma A4187, used at 1:5000 dilution)
Blocking buffer: PBS+ 2mg/mL biotin-free milk powder (I-block), 0.05% Tween 20. Add milk powder slowly to PBS until completely dissolved and then add Tween 20.
Alkaline solution: 50mM Tris–HCl, 100mM NaCl, 1mM MgCl2, pH 9.5
CDP-Star Chemiluminescent Substrate, 0.25mM (Thermo Fisher Scientific T2147)
Restore PLUS Western Blot Stripping Buffer (Thermo Fisher Scientific 46430)
3.3.3. Procedure
3.3.3.1. Sample Preparation
Collect 3mL samples from a sporulating culture, centrifuge 2min at 3220 × g, and drain. Resuspend in 1mL 10% glycerol, transfer to 1.5mL screw-top microfuge, and centrifuge in microfuge 2min at top speed, drain, freeze on dry ice, and store at −80°C.
Thaw the pellet on ice, add 100μL of 20% TCA, and vortex to resuspend.
Add glass beads such that there will be no free liquid on the top of the beads. Disrupt cells by shaking continuously on a Bead beater (preferred) or vortex at highest speed for 2min.
Add 400μL of 5% TCA to the broken cells+beads, vortex, and transfer the cloudy supernatant to a fresh 1.5-mL screw cap tube.
Add 200μL of 5% TCA to beads, vortex, recover the supernatant, and add it to the previous supernatant.
Centrifuge the combined supernatants 5min at top speed in the microfuge. Discard the clear supernatant, removing as much as is possible.
Add 80μL of sample buffer to the pellet and vortex vigorously. Much of the pellet is composed of cellular debris that will not go back into solution. If the sample turns yellow, add 5–20μL Tris 1.5 M pH 8.8, until the blue color is restored.
Heat the sample at 95°C for 5min. Centrifuge 3min at top speed in the microfuge. Transfer supernatant containing the SDS-solubilized proteins to a new 1.5mL screw cap tube.
Typically, 7.5μL of extract is loaded per lane.
3.3.3.2. Gel Preparation and Electrophoresis
We use 6% gels to detect Sgs1, 10% gels to detect all other proteins. Alternatively, precast 8%–16% gradient gels can be used, but resolution will be somewhat compromised.
For a 10% 10mL gel, mix 5mL 2 resolving buffer, 3.4mL Protogel mix, 1.4mL water, 150μL 10% ammonium persulfate, 15μL TEMED. For a 6% gel, 2mL Protogel and 2.8mL water are used. Pour acrylamide mix up to the last line on the cassette and immediately overlay with 100μL of water. Allow to polymerize at least 30min.
Prepare 5% stacking gel: mix 2.5mL stacking buffer, 0.83mL Protogel mix, 1.6mL water, 75μL 10% ammonium persulfate, 7.5μL TEMED. Pour water off resolving gel and add stacking gel. Insert comb and allow 30min to polymerize.
Remove comb and sealing tape and place gel in gel box. Add running buffer to cover wells, approximately 500mL.
Heat samples to 95°C, 3min. Load 5–7.5μL sample per well.
Run gels at 180V, room temperature, 1–1.5h, until tracking dye reaches the bottom of the gel.
3.3.3.3. Western Blotting and Detection
Disassemble gel cassette; if stacking gels are used, remove the stacking layer. Place gel in 10mL transfer buffer on orbital shaker, 10min at 50rpm.
Soak PVDF membrane in 10mL 100% methanol for 1min. Wash in 20mL water on orbital shaker, 2min at 50rpm, and then place in 10mL transfer buffer.
Soak filter paper from the membrane/filter kit and scotch pads from the transfer apparatus in transfer buffer in a glass beaker (or equivalent), using enough transfer buffer to completely submerse them.
Place two or three soaked scotch pads on the cathode side of the transfer apparatus, followed by the presoaked filter paper. Pour some buffer on the filter paper and place the gel over it. Pour some buffer on the gel, and place the PVDF membrane on it, followed by presoaked filter paper. Remove all air bubbles from the assembly and place two or three soaked scotch pads on top. Close the apparatus with the anode and place it in the gel tank. Pour transfer buffer inside the transfer apparatus until it covers the transfer sandwich. The outer chamber can be filled with either buffer or water.
Transfer at 15V, 16h at 4°C (for Sgs1) or at 25V, 1.5h at room temperature (for other proteins).
Disassemble transfer apparatus, place the membrane in 20mL of I-block blocking buffer and incubate for at least 1h at room temperature or overnight at 4°C on an orbital shaker.
Discard blocking buffer and add 20mL of blocking buffer with the appropriate dilution of primary antibody. Incubate at least 3h at room temperature or overnight at 4°C on an orbital shaker (recommended for Sgs1).
Discard antibody solution, add 20mL blocking buffer, and wash 5min at room temperature on an orbital shaker. Repeat for five additional washes.
Add 20mL of blocking buffer with the appropriate dilution of secondary antibody and incubate 1h at room temperature on an orbital shaker.
Discard secondary antibody solution and wash as in step 8.
Place the membrane in 10mL alkaline buffer for 2min. Remove the membrane, drain excess solution and place into 3mL of CDP-star, making sure that membrane is complexly coated. Remove membrane, drain excess substrate, and wrap in plastic wrap or a developing folder.
Scan using a Fuji LAS3000 CCD camera or equivalent.
To strip blots, place membrane in 20mL blocking buffer, wash 10min at room temperature on an orbital shaker. Washed blots can be stored at 4°C in blocking buffer before stripping.
Remove blocking buffer, add 15mL stripping buffer, and incubate at room temperature 15min. Remove blot from stripping buffer and wash with 20mL PBS, 1min at room temperature at least three times. Wash the blot once with 20mL blocking buffer, 1min at room temperature. Remove the blot and block in 20mL blocking buffer for at least 1h. To reprobe, repeat steps 7–12.
3.4. Monitoring Nuclear Divisions by DAPI Staining
3.4.1. Equipment
Variable-speed microfuge
1.5-mL microfuge tubes
Prewashed glass slides
Cover slips, 18 × 18mm or 24 × 50mm
Epifluorescence microscope with blocks and filters for DAPI detection, with 60 × or 100 × objective
3.4.2. Reagents
95% ethanol
1μg/mL 4ʹ,6-diamidino-2-phenylindole dihydrochloride (DAPI). Store at 4°C in the dark
3.4.3. Procedure
Place a 1mL culture sample in a microfuge tube and centrifuge 2min at top speed.
Discard supernatant and resuspend cells in 50μL water. Add 1mL 95% ethanol (see Note 12).
Add 1μL of 1μg/mL DAPI. Incubate at room temperature, 10min, protected from light.
Centrifuge 2min at top speed; discard supernatant and resuspend in 1mL water. Centrifuge 1min at 7000rpm; discard supernatant and repeat 1mL water wash.
Discard supernatant, resuspend in 50μL water. Store protected from light at 4°C.
Place 5μL of cells on glass slide, cover with cover slip, and visualize using microscope (see Note 13).
ACKNOWLEDGMENTS
All plasmids, plasmid sequences, and sequences of primers mentioned in this chapter are available from the authors upon request. We thank Angelika Amon and Masato Kanemaki for plasmids, and Anura Shodhan and Matan Cohen for comments. This work was supported by the Intramural Research Program of the NIH through the Center for Cancer Research at the National Cancer Institute.
ABBREVIATIONS
- 3XHA
triple influenza hemagglutinin epitopes
- CoHex
hexamminecobalt(III) chloride
- CTAB
cetyltrimethylammonium bromide
- DAPI
4ʹ,6-diamidino-2-phenylindole dihydrochloride
- DMSO
dimethyl sulfoxide
- HJ
Holliday junction
- HR
homologous recombination
- KAc
potassium acetate sporulation medium
- PBS
phosphate-buffered saline
- SPS
presporulation medium
- STR
Sgs1–Top3–Rmi1
- TCA
trichloroacetic acid
- TE
10mM Tris, 1mM EDTA, pH 7.5
- TEMED
tetramethylethylenediamine
- YEPAD
yeast extract, peptone, adenine, dextrose growth medium
Footnotes
To prevent arcing, all DNA should be in either TE or nuclease-free water. In order to minimize sorbitol dilution, DNA should be as concentrated as possible. Briefly wiping electroporation cuvettes to remove condensation will also minimize arcing.
Transformant yields can be reduced if cells are plated at too high a concentration. We recommend spreading two selective plates with 100μL of the transformation mix, and as many selective plates with 20μL as convenient.
Use of a shaking incubator with cooling capacity (New Brunswick Innova 43 or equivalent) is recommended; incubators with limited ventilation or cooling can overheat.
Because light scattering, not absorbance, is measured here, a spectrophotometer with some distance between the cuvette and the detector should be used. We use a Pharmacia Ultrospec 2000, with ~7cm between the cuvette and the detector.
All presporulation and sporulation media should be made fresh, preferably on the day before sporulation.
To minimize experiment-to-experiment variation in timing of events during sporulation, adherence to a fixed schedule, starting when diploids are struck on YEPAD plates, is highly recommended. For example, one such routine would streak diploids at 3:00pm, incubate plates for exactly 48h, inoculate YEPAD overnights at 3:00pm, aerate exactly 24h, and inoculate SPS presporulation cultures at 3:00pm.
Psoralen is toxic; use appropriate precautions when handling.
Exposure time should be adjusted so that total exposure is 6mWmin/cm2 (0.36J/cm2).
Earlier time points take less time to spheroplast than later time points, so the spheroplasting state should be monitored frequently. Cells are sufficiently spheroplated when 80%–90% of cells are phase dark; spheroplasting can also be checked by mixing cells with 20% SDS and examining their lysis under the microscope.
DNA pellets can be difficult to resuspend. We usually incubate pellets with buffer overnight at 4°C, followed by gentle mixing by gently pipetting the resuspension mix up and down ~20 times.
If nonfat milk powder is added too rapidly, it will form clumps and never dissolve. If blocking solution is urgently needed, the PBS solution can be warmed by microwaving (~1min) beforehand.
Stain ethanol-fixed cells with DAPI and wash as soon as possible (i.e., the same day), as prolonged incubation in ethanol leads to nucleus distortion and mislocalization. Formaldehyde fixation followed by a brief ethanol permeabilization may be useful if it is impossible to stain cells on the same day.
To maximize the fraction of cells with nuclei in the same focal plane, use a small volume (~5μL) of cell suspension and a large (20 × 40mm) cover slip.
REFERENCES
- Ahuja JS, & Börner GV (2011). Analysis of meiotic recombination intermediates by two-dimensional gel electrophoresis. Methods in Molecular Biology (Clifton, NJ: ), 745, 99–116 [Chapter 7]. 10.1007/978-1-61779-129-1_7. [DOI] [PubMed] [Google Scholar]
- Alani E, Cao L, & Kleckner N (1987). A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics, 116(4), 541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allers T, & Lichten M (2000). A method for preparing genomic DNA that restrains branch migration of Holliday junctions. Nucleic Acids Research, 28(2), e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizzari F, & Marston AL (2011). Cdc55 coordinates spindle assembly and chromosome disjunction during meiosis. The Journal of Cell Biology, 193(7), 1213–1228. 10.1083/jcb.201103076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blitzblau HG, & Hochwagen A (2013). ATR/Mec1 prevents lethal meiotic recombination initiation on partially replicated chromosomes in budding yeast. eLife, 2, e00844. 10.7554/eLife.00844.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börner GV, & Cha RS (2015). Induction and analysis of synchronous meiotic yeast cultures. Cold Spring Harbor Protocols, 2015(10), 908–913. 10.1101/pdb.prot085035. [DOI] [PubMed] [Google Scholar]
- Börner GV, Kleckner N, & Hunter N (2004). Crossover/noncrossover differentiation, synaptonemal complex formation, and regulatory surveillance at the leptotene/zygotene transition of meiosis. Cell, 117(1), 29–45. [DOI] [PubMed] [Google Scholar]
- Brito IL, Yu H-G, & Amon A (2010). Condensins promote coorientation of sister chromatids during meiosis I in budding yeast. Genetics, 185(1), 55–64. 10.1534/genetics.110.115139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AC, Borts RH, & Hoffmann E (2009). Temperature-dependent modulation of chromosome segregation in msh4 mutants of budding yeast. PLoS One, 4(10), e7284. 10.1371/journal.pone.0007284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, et al. (1998). A genome-wide transcriptional analysis of the mitotic cell cycle. Molecular Cell, 2(1), 65–73. [DOI] [PubMed] [Google Scholar]
- Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown PO, et al. (1998). The transcriptional program of sporulation in budding yeast. Science (New York, NY: ), 282(5389), 699–705. [DOI] [PubMed] [Google Scholar]
- Clyne RK, Katis VL, Jessop L, Benjamin KR, Herskowitz I, Lichten M, et al. (2003). Polo-like kinase Cdc5 promotes chiasmata formation and cosegregation of sister centromeres at meiosis I. Nature Cell Biology, 5(5), 480–485. 10.1038/ncb977. [DOI] [PubMed] [Google Scholar]
- Copsey A, Tang S, Jordan PW, Blitzblau HG, Newcombe S, Chan AC-H, et al. (2013). Smc5/6 coordinates formation and resolution of joint molecules with chromosome morphology to ensure meiotic divisions. PLoS Genetics, 9(12), e1004071 10.1371/journal.pgen.1004071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Muyt A, Jessop L, Kolar E, Sourirajan A, Chen J, Dayani Y, et al. (2012). BLM helicase ortholog Sgs1 is a central regulator of meiotic recombination intermediate metabolism. Molecular Cell, 46(1), 43–53. 10.1016/j.molcel.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foiani M, Marini F, Gamba D, Lucchini G, & Plevani P (1994). The B subunit of the DNA polymerase alpha-primase complex in Saccharomyces cerevisiae executes an essential function at the initial stage of DNA replication. Molecular and Cellular Biology, 14(2), 923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff S, McDonald JP, Bendixen C, Arthur L, & Rothstein R (1994). The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: A potential eukaryotic reverse gyrase. Molecular and Cellular Biology, 14(12), 8391–8398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, & Woods RA (2001). Genetic transformation of yeast. BioTechniques, 30, 816–831. [DOI] [PubMed] [Google Scholar]
- Goldstein AL, & McCusker JH (1999). Three new dominant drug resistance cassettes for gene disruption in Saccharomyces cerevisiae. Yeast (Chichester, England: ), 15(14), 1541–1553. . [DOI] [PubMed] [Google Scholar]
- Grandin N, & Reed SI (1993). Differential function and expression of Saccharomyces cerevisiae B-type cyclins in mitosis and meiosis. Molecular and Cellular Biology, 13(4), 2113–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray S, Allison RM, Garcia V, Goldman ASH, & Neale MJ (2013). Positive regulation of meiotic DNA double-strand break formation by activation of the DNA damage checkpoint kinase Mec1(ATR). Open Biology, 3(7), 130019. 10.1098/rsob.130019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, & Laemmli UK (2008). The anchor-away technique: Rapid, conditional establishment of yeast mutant phenotypes. Molecular Cell, 31(6), 925–932. 10.1016/j.molcel.2008.07.020. [DOI] [PubMed] [Google Scholar]
- Honigberg SM, & Esposito RE (1994). Reversal of cell determination in yeast meiosis: Postcommitment arrest allows return to mitotic growth. Proceedings of the National Academy of Sciences of the United States of America, 91(14), 6559–6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessop L, & Lichten M (2008). Mus81/Mms4 endonuclease and Sgs1 helicase collaborate to ensure proper recombination intermediate metabolism during meiosis. Molecular Cell, 31(3), 313–323. 10.1016/j.molcel.2008.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessop L, Rockmill B, Roeder GS, & Lichten M (2006). Meiotic chromosome synapsis-promoting proteins antagonize the anti-crossover activity of sgs1. PLoS Genetics, 2(9), e155 10.1371/journal.pgen.0020155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Guacci V, & Yu H-G (2009). Pds5 is required for homologue pairing and inhibits synapsis of sister chromatids during yeast meiosis. The Journal of Cell Biology, 186(5), 713–725. 10.1083/jcb.200810107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane SM, & Roth R (1974). Carbohydrate metabolism during ascospore development in yeast. Journal of Bacteriology, 118(1), 8–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katis VL, Lipp JJ, Imre R, Bogdanova A, Okaz E, Habermann B, et al. (2010). Rec8 phosphorylation by casein kinase 1 and Cdc7-Dbf4 kinase regulates cohesin cleavage by separase during meiosis. Developmental Cell, 18(3), 397–409. 10.1016/j.devcel.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur H, De Muyt A, & Lichten M (2015). Top3-Rmi1 DNA single-strand decatenase is integral to the formation and resolution of meiotic recombination intermediates. Molecular Cell, 57(4), 583–594. 10.1016/j.molcel.2015.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, Nishimura K, Kanemaki MT, & Donaldson AD (2013). The Elg1 replication factor C-like complex functions in PCNA unloading during DNA replication. Molecular Cell, 50(2), 273–280. [DOI] [PubMed] [Google Scholar]
- Kunes S, Ma H, Overbye K, Fox MS, & Botstein D (1987). Fine structure recombinational analysis of cloned genes using yeast transformation. Genetics, 115(1), 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen NB, & Hickson ID (2013). RecQ helicases: Conserved guardians of genomic integrity. In Spies M (Ed.), DNA helicases and DNA motor proteins: Vol. 973 (pp. 161–184). New York, NY: Springer, New York. 10.1007/978-1-4614-5037-5_8. [DOI] [PubMed] [Google Scholar]
- Lee BH, & Amon A (2003). Role of polo-like kinase CDC5 in programming meiosis I chromosome segregation. Science (New York, NY: ), 300(5618), 482–486. 10.1126/science.1081846. [DOI] [PubMed] [Google Scholar]
- Leung W-K, Humphryes N, Afshar N, Argunhan B, Terentyev Y, Tsubouchi T, et al. (2015). The synaptonemal complex is assembled by a polySUMOylation-driven feedback mechanism in yeast. The Journal of Cell Biology, 211(4), 785–793. 10.1083/jcb.201506103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Shao Y, Jin H, & Yu H-G (2015). Ndj1, a telomere-associated protein, regulates centrosome separation in budding yeast meiosis. The Journal of Cell Biology, 209, 247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichten M (2014). Tetrad, random spore, and molecular analysis of meiotic segregation and recombination. Methods in Molecular Biology (Clifton, NJ: ), 1205, 13–28. 10.1007/978-1-4939-1363-3_2. [DOI] [PubMed] [Google Scholar]
- Lin W, Jin H, Liu X, Hampton K, & Yu H-G (2011). Scc2 regulates gene expression by recruiting cohesin to the chromosome as a transcriptional activator during yeast meiosis. Molecular Biology of the Cell, 22(12), 1985–1996. 10.1091/mbc.E10-06-0545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W, Wang M, Jin H, & Yu H-G (2011). Cohesin plays a dual role in gene regulation and sister-chromatid cohesion during meiosis in Saccharomyces cerevisiae. Genetics, 187(4), 1041–1051. 10.1534/genetics.110.122358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A, Demarini DJ, Shah NG, Wach A, Brachat A, et al. (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast (Chichester, England: ), 14(10), 953–961. . [DOI] [PubMed] [Google Scholar]
- Manivasakam P, Weber SC, McElver J, & Schiestl RH (1995). Micro-homology mediated PCR targeting in Saccharomyces cerevisiae. Nucleic Acids Research, 23(14), 2799–2800. 10.1093/nar/23.14.2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker JH, & Haber JE (1977). Efficient sporulation of yeast in media buffered near pH 6. Journal of Bacteriology, 132(1), 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen JR, Kaliraman V, Ibrahim SS, & Brill SJ (2001). Requirement for three novel protein complexes in the absence of the Sgs1 DNA helicase in Saccharomyces cerevisiae. Genetics, 157(1), 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, & Kanemaki M (2009). An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nature Methods, 6(12), 917–922. 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- Nishimura K, & Kanemaki MT (2014). Rapid depletion of budding yeast proteins via the fusion of an auxin-inducible degron (AID). Current Protocols in Cell Biology, 64, 20.9.1–16. 10.1002/0471143030.cb2009s64. [DOI] [PubMed] [Google Scholar]
- Oh SD, Jessop L, Lao JP, Allers T, Lichten M, & Hunter N (2009). Stabilization and electrophoretic analysis of meiotic recombination intermediates in Saccharomyces cerevisiae. Methods in Molecular Biology (Clifton, NJ: ), 557, 209–234. 10.1007/978-1-59745-527-5_14. [DOI] [PubMed] [Google Scholar]
- Oh SD, Lao JP, Hwang PY, Taylor AF, Smith GR, & Hunter N (2007). BLM ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules. Cell, 130(2), 259–272. 10.1016/j.cell.2007.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SD, Lao JP, Taylor AF, Smith GR, & Hunter N (2008). RecQ helicase, Sgs1, and XPF family endonuclease, Mus81-Mms4, resolve aberrant joint molecules during meiotic recombination. Molecular Cell, 31(3), 324–336. 10.1016/j.molcel.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Roberts P, Chen Y, Kvam E, Shulga N, Huang K, et al. (2000). Nucleus-vacuole junctions in Saccharomyces cerevisiae are formed through the direct interaction of Vac8p with Nvj1p. Molecular Biology of the Cell, 11(7), 2445–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primig M, Williams RM, Winzeler EA, Tevzadze GG, Conway AR, Hwang SY, et al. (2000). The core meiotic transcriptome in budding yeasts. Nature Genetics, 26(4), 415–423. 10.1038/82539. [DOI] [PubMed] [Google Scholar]
- Rockmill B, Fung JC, Branda SS, & Roeder GS (2003). The Sgs1 helicase regulates chromosome synapsis and meiotic crossing over. Current Biology: CB, 13(22), 1954–1962. [DOI] [PubMed] [Google Scholar]
- Schwacha A, & Kleckner N (1994). Identification of joint molecules that form frequently between homologs but rarely between sister chromatids during yeast meiosis. Cell, 76(1), 51–63. [DOI] [PubMed] [Google Scholar]
- Subramanian VV, Macqueen AJ, Vader G, Shinohara M, Sanchez A, Borde V, et al. (2016). Chromosome synapsis alleviates Mek1-dependent suppression of meiotic DNA repair. PLoS Biology, 14(2), e1002369 10.1371/journal.pbio.1002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Wu MK, Zhang R, & Hunter N (2015). Pervasive and essential roles of the Top3-Rmi1 decatenase orchestrate recombination and facilitate chromosome segregation in meiosis. Molecular Cell, 57(4), 607–621. 10.1016/j.molcel.2015.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenten N, Kuhl L-M, Lam I, Oke A, Kerr AR, Hochwagen A, et al. (2015). The kinetochore prevents centromere-proximal crossover recombination during meiosis. eLife, 4, 923. 10.7554/eLife.10850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wach A (1996). PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast (Chichester, England: ), 12(3), 259–265. . [DOI] [PubMed] [Google Scholar]
- Yu H-G, & Koshland D (2007). The aurora kinase Ipl1 maintains the centromeric localization of PP2A to protect cohesin during meiosis. The Journal of Cell Biology, 176(7), 911–918. 10.1083/jcb.200609153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Wang S, Yin S, Hong S, Kim KP, & Kleckner N (2014). Topoisomerase II mediates meiotic crossover interference. Nature, 511(7511), 551–556. 10.1038/nature13442. [DOI] [PMC free article] [PubMed] [Google Scholar]