

Abstract
Synthetic approaches to novel 4,8-dimethyl-4’-halomethyl-4’,5’-dihydropsoralens as synthetic precursors to 4,8-dimethyl-4’-(N-pyridiniummethyl)-4’,5’-dihydropsoralens are described. The compounds are potential therapeutic agents for improved psoralen ultraviolet radiation therapy with reduced mutagenicity.
In previous papers, we reported new methods for the synthesis of 4,8-dimethyl-5’-halomethyl-4’,5’-dihydropsoralens and their amine derivatives and also psoralen analogs.1–4 In this paper, we report synthetic procedures for the generation of novel 4,8-dimethyl-4’-halomethyl-4’,5’-dihydropsoralens and their amine derivatives designed to selectively target the psoralen receptor on the cell membrane of epithelial cells. Use of water-soluble salts promoted increased cell surface effects with the minimization of the mutagenic and carcinogenic effects.5
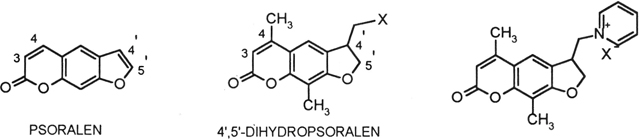
Though naturally occurring psoralens in combination with ultraviolet light have been utilized for centuries in the treatment of skin conditions such as psoriasis, vitiligo and eczema, synthetic psoralens have been developed to enhance activity and minimize side effects.6 The accepted mode of action of psoralen ultraviolet radiation therapy has been the intercalation of DNA, followed by photoactivated crosslinking of the 3,4-double bond and the 4’,5’-double bond of the psoralen with pyrimidine base pairs in the DNA forming mono- and biscyclobutane dimers.5 Previous papers from our group have focused on an alternative mode of action, a psoralen specific receptor (a 22,000 Dalton protein) which has been identified on the cytoplasmic membrane of epidermal target cell.4, 8, 9 When a psoralen molecule binds to the receptor and is activated by ultraviolet light, the binding of the epidermal growth factor is prohibited.4, 8, 9 A limitation to psoralen ultraviolet A therapy has been the concern that DNA crosslink repair is highly error prone and may result in post treatment cancer induction.10 The synthesis of psoralen molecules that would be unable to photo-crosslink, though still maintaining efficacy, has been the goal of this research. Heindel has shown that unsaturation of the furan ring, or even the presence of the furan ring, is not required for photoactivity of psoralens.3,4 Synthesis of psoralens with a saturated furan ring would eliminate the possibility of crosslink formation and could reduce the undesired side effects of psoralen ultraviolet A therapy. Attachment to the psoralen ring system of a hydrophilic moiety, such as a pyridinium iodide, bromide or chloride substituent, would increase cell surface effects. To maintain the desired high water solubility combined with the inability to form covalent diadducts, the 4,8-dimethyl-4’-halomethyl-4’,5’-dihydropsoralens served as promising synthetic targets on the pathway to 4’-pyridiniummethyl-4’,5’-dihydropsoralen salts.11
RESULTS AND DISCUSSION
Diazonium Tetrafluoroborate Route
The research published by Beckwith on o-allyloxybenzenediazonium tetrafluoroborates as intermediates in formation of dihydrobenzofurans was the precedent for the synthesis of 4,8-dimethyl-4’-halomethyl-4’,5’-dihydropsoralens using the allyloxycoumarindiazonium tetrafluoroborate salts.12, 13 In the cyclization of aryldiazonium salts, the nucleophile transferred is derived from an added reagent such as sodium iodide, copper(II) chloride, copper(II) bromide, or copper(I) cyanide, allowing the generation of different products from a single diazo precursor. High yields resulted from the efficiency of the radical generation and cyclization. In psoralen generation from the allyloxycoumarindiazonium tetrafluoroborate (4), sodium iodide in acetone promoted the closure of the furan ring of the psoralen. The reaction product recovered indicated that the rate of cyclization of the radical was faster than the iodination of the uncyclized aryl radicals (Scheme 1). In psoralen synthesis, the generation of five membered rings by the exo trig mode occurred exclusively, with no formation of six membered rings by the endo mode. The synthetic route began with the nitration of 4,8-dimethyl-7-hydroxycoumarin with nitric acid in sulfuric acid to give 4,8-dimethyl-7-hydroxy-6-nitrocoumarin (1) in 81% yield (Scheme 1).14 Treatment with allyl bromide and potassium carbonate in dimethyl sulfoxide gave an 83% recovery of 7-allyloxy-4,8-dimethyl-6-nitrocoumarin (2). The reduction of 2 was accomplished with tin(II) chloride, tin, and concentrated hydrochloric acid in ethanol giving 7-allyloxy-6-amino-4,8-dimethylcoumarin (3) in 83% yield.15 The reduction was attempted unsuccessfully with palladium on carbon in cyclohexene and also with ammonium hydroxide / sodium bisulfite in isopropanol. 6-(7-Allyloxy-4,8-dimethylcoumarin)-diazonium tetrafluoroborate (4), a highly stable diazonium tetrafluoroborate salt from sodium nitrite, formed rapidly in tetrafluoroboric acid in 82% yield in a method developed by Roe on iodoallyloxybenzenes.16
Scheme 1.

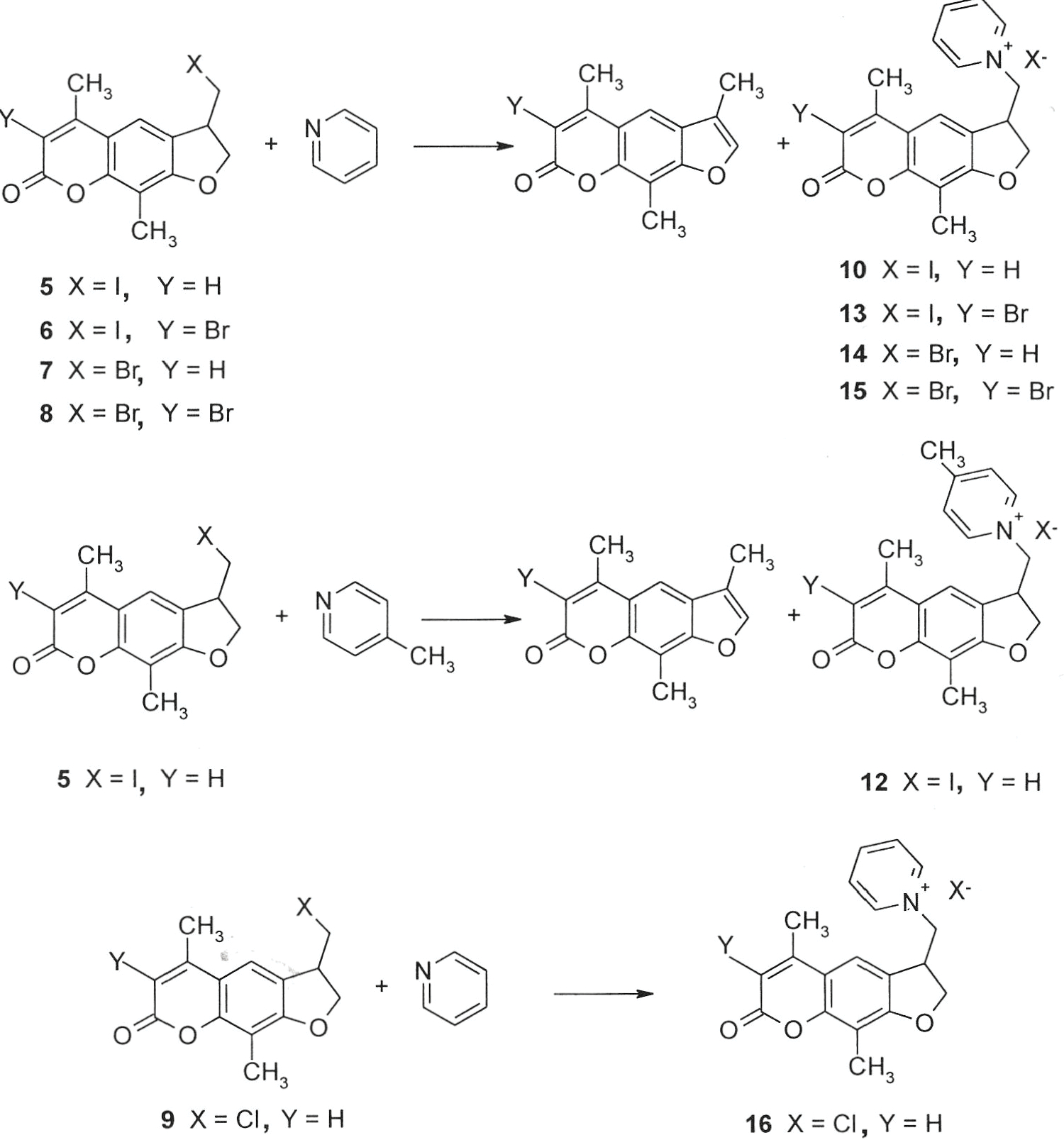
Compound (4) cyclized readily with sodium iodide in acetone to generate 4,8-dimethyl-4’-iodomethyl-4’,5’-dihydropsoralen (5) in 95% crude yield or with copper(II) bromide in DMSO to generate 4’-bromomethyl- 4,8-dimethyl-4’,5’-dihydropsoralen (7) in 56% yield with a procedure developed by Beckwith for formation of functionalized dihydrobenzofurans by radical cyclization. The 4,8-dimethyl-4’-halomethyl-4’,5’-dihydropsoralens showed C3 substitution when treated with NBS in chloroform or methylene chloride.11 4,8-Dimethyl-4’-iodomethyl-4’,5’-dihydropsoralen (5) gave 3-bromo-4,8-dimethyl-4’-iodomethyl-4’,5’-dihydropsoralen (6) in 92% yield and 4’-bromomethyl-4,8-dimethyl-4’,5’-dihydropsoralen (7) gave 3-bromo-4’-bromomethyl-4,8-dimethyl-4’,5’-dihydropsoralen (8) in 76 % yield when treated with NBS. Use of acetone as the solvent in the copper(II) bromide cyclization gave greatly reduced yields. Copper(II) chloride cyclized compound (4) in DMSO to generate 4’-chloromethyl-4,8-dimethyl-4’,5’-dihydropsoralen (9) in 79% yield. The 4,8-dimethyl-4’-cyanomethyl-4’,5’-dihydropsoralen (18) was generated in 42% yield with copper(I) cyanide in DMSO and pyridine.
When reacting the 4’-halomethyldihydropsoralens (5, 6, 7 or 8) with pyridine, recovery of the desired product was 10–52% (Scheme 2). Dehydrohalogenation gave the major products, 4,8,4’-trimethylpsoralen or 3-bromo-4,8,4’-trimethylpsoralen. The 4’-iodomethyldihydropsoralen (5) with pyridine gave 12–13% yield of the pyridinium iodide salt (10) and with 4-methylpyridine gave 18 % of the methylpyridinium iodide salt (12). The 3-bromo-4’-iodomethyldihydropsoralen (6) gave 11% yield of the pyridinium iodide salt (13). When forming the pyridinium iodide salt (10), a 1H-NMR study confirmed the existence of a transient exocyclic methylene intermediate leading to the generation of 4,8,4’-trimethylpsoralen as the major product (between 60–80%).11 In refluxing morpholine, the 4,8,4’-trimethylpsoralen dehydrohalogenation product was recovered in greater than 85% yield from 7. Stronger bases and morehindered amines had a greater tendency to promote dehydrohalogenation through the exocyclic methylene psoralen transient intermediate. The 4’-bromomethyl-4,8-dimethyl-4’,5’-dihydropsoralens showed a reduced tendency to dehydrohalogenate in the presence of pyridine with 4’-bromomethyl-4,8-dimethyl-4’,5’-dihydropsoralen (7) yielding 43 % of the pyridinium bromide salt (14) and 3-bromo-4,8-dimethyl-4’-bromomethyl-4’,5’-dihydropsoralen (8) yielding 52 % of the pyridinium bromide salt (15). The 4’-chloromethyl-4,8-dimethyl-4’,5’-dihydropsoralen (9) did not dehydrohalogenate but evidenced a reduced tendency for substitution with pyridine, giving a yield of 56% after extended reaction times.
Scheme 2.
PHARMACOLOGIC TESTING AND RESULTS
Psoralens (10, 12, 13, 15) were evaluated in cell culture for their ability to inhibit the growth of PAM 212 malignant murine keratinocytes. Photoactivated psoralens are established inhibitors of keratinocyte growth.17 Compounds (10) (IC50 1.8 uM) and (12) (IC50 2.6 uM) were the most active. Compound (14), the bromide counter ion version of 10, and compound (16), the chloride counter ion version of 10, dissociate in vivo to the same organic ligand as 10 and hence would possess the same pharmacology. Compounds (13) and (15) showed no activity up to their solubility maxima of approximately 100 uM. Mutagenicity/ carcinogenicity is a major problem for psoralens which cross the cell membrane and bind to nuclear DNA.18 These pyridinium psoralens, however, are water-soluble, quaternary salts which express their activity in an extra-cellular mechanism and as such hold promise as non-mutagenic phototherapeutics.
CONCLUSION
The 4’-iodomethyl-4’,5’-dihydropsoralen formed in high yield through a diazonium tetrafluoroborate intermediate. It was highly prone to dehydrohalogenation in the amine substitution reaction. The 4’-bromomethyl-4’,5’-dihydropsoralens were substituted more readily with greater yield of the desired quaternary compound and showed a tendency for dehydrohalogenation (Scheme 2). The 4’-chloromethyl-4’,5’-dihydropsoralens were not prone to dehydrohalogenation but were slow to react with pyridine. This route involved the first generation of 4’-halomethyl-4’,5 ‘-dihydropsoralens as photo-activated agonists for the psoralen receptor of epidermal cells. IC50 measurements of these 4’-pyridiniummethyl-4,8-dimethylpsoralens have shown impressive activity in the photoactivated keratinocyte assay.
EXPERIMENTAL GENERAL
The 1H and 13C NMR spectra were recorded on a Bruker AC 250 operated at 250.13 MHz and 62.89 MHz in the FT mode. Chemical shifts for hydrogen and carbon resonances were reported in ppm (δ) relative to tetramethylsilane. Structural determination also included COSY, DEPT and HETCOR experiments. Thin-layer chromatographies were performed with fluorescent silica gel plates. Silica gel (230–400 mesh) was used for flash chromatography separations. High performance liquid chromatography analyses were run on a Hewlett Packard HP 1050 Series model. Gas chromatography/mass spectrometry was run on a Varian gas chromatograph 3300 model in series with a Finnegan ITS 40™ Magnum Ion Trap Mass Spectrometer. Melting points were determined on a Mettler FP 81 MBC cell with a Mettler FP 80 central processor. Elemental analyses were determined by Oneida Research Services, Whitesboro, New York. Chemicals and solvents were obtained from commercial sources and used without further purification unless otherwise stated. Starting materials were purchased from Aldrich Chemical, unless otherwise specified.
SYNTHESIS OF PRECURSORS FOR THE DIAZONIUM TETRAFLUOROBORATE RING CLOSURE
4.8-Dimethyl-7-hydroxy-6-nitrocoumarin (1).
4.8-Dimethyl-7-hydroxycoumarin (7.50 g, 39.4 mmol) was dissolved in 75 mL of concentrated sulfuric acid at rt and chilled to −20°C before the addition of chilled nitrating mixture (3 mL of concentrated nitric acid added to 9 mL of concentrated sulfuric acid). Stirring was continued for 3 h at −20°C with the mixture warming before pouring onto ice. Bright yellow crystals were filtered, washed with water and dried to recover 7.50 g (81% yield). The product was recrystallized from ethanol to give yellow green crystals: mp 229.5–231.5°C; 1H-NMR (DMSO-d6): δ 2.29 (s, 3H), 2.41 (s, 3H), 6.37 (s, 1 H), 8.19 (s, 1H), 11.30 (s, 1H); 13C-NMR (DMSO-d6): δ 8.8, 18.0, 112.4, 112.9, 115.1, 119.9, 132.8, 152.9, 154.2, 155.1, 159.0; MS: (EI) m/z (relative intensity) 236 (M+, 23), 235 (M+, base) 207 (84), 77 (30). Anal. Calcd for C11H9NO5: C, 56.18; H, 3.86; N, 5.96. Found: C, 55.94; H, 3.79; N, 5.96.
7-Allyloxy-4,8-dimethyl-6-nitrocoumarin (2).
Compound (1) (5.00 g, 21.2 mmol), dried K2CO3 (12.8 g, 93.0 mmol) and 100 mL of dimethyl sulfoxide were heated to reflux before the addition of allyl bromide (12.9 g, 106 mmol). Refluxing was continued overnight. Solids were removed by filtration and solvent evaporated to isolate the product. The crude product was placed under vacuum to remove residual dimethyl sulfoxide. The yield was 4.85 g (83% yield). Recrystallization from isopropanol yielded bright yellow crystals: mp 158.3–158.8°C; 1H-NMR (DMSO-d6): 6 2.31 (s, 3H), 2.41 (s, 3H), 4.54 (d, J = 5.6 Hz, 2H), 5.25 (d, J = 8.5 Hz, 1H), 5.38 (d, J = 18 Hz, 1H), 5.98–6.10 (m, 1H), 6.48 (s, 1H), 8.20 (s, 1H); 13C-NMR (DMSO-d6): δ 9.4, 18.1,75.8, 114.4, 115.8, 119.1, 119.5, 122.0, 132.7, 140.6, 151.6, 152.7, 154.2, 158.9. Anal. Calcd for C14H13NO5 × 0.17 H2O: C, 60.40; H, 4.83; N, 5.03. Found: C, 60.40; H, 4.74; N, 5.02.
7-Allyloxy-6-amino-4,8-dimethylcoumarin (3).
Finely ground 2 (3.00 g, 10.9 mmol), tin (3.24 g, 27.3 mmol), tin(II) chloride (3.04 g, 16.0 mmol), 9 mL of concentrated hydrochloric acid and 200 mL of ethanol were stirred overnight, during which all tin dissolved. Most of the ethanol was removed in vacuo. As the solution was allowed to cool, a gel formed which was filtered to recover a small amount of pearlized crystals (the hydrochloride salt). Water was added and a drop of isoamyl alcohol was added to prevent frothing as solid sodium bicarbonate was added with stirring until the solution was basic. Ether was added to the filtrate and a precipitate formed that was filtered and rinsed with ether to remove tin (II) chloride. The solids were dried and extracted with hot ethanol on the funnel of a filtration flask. Evaporation of the ethanol gave a crude product that was recrystallized from ethanol to provide 2.20 g (82% yield) of bright mustard yellow crystals: mp 145.6–147.4° C; 1H-NMR (DMSO-d6): δ 2.28 (s, 3H), 2.49 (s, 3H), 4.54 (d, J = 5.6 Hz, 2H), 5.28 (d, J = 8.6 Hz, 1 H), 5.48 (d, J = 18.0 Hz, 1H), 5.86 (br s, 2H), 5.98–6.32 (m, 1H), 6.39 (s, 1H), 7.58 (s, 1H); 13C-NMR (DMSO-d6): δ 9.4, 18.1, 74.6, 113.6, 115.1, 116.0, 118.7, 120.1, 125.5, 133.5, 147.0, 151.5, 152.5, 159.6. Anal. Calcd for C14H15NO3: C, 68.56; H, 6.93; N, 5.66. Found: C, 68.34; H, 6.98; N, 5.52.
6-(7-Allyloxy-4,8-dimethylcoumarin)diazonium tetrafluoroborate (4)
Compound (3) (1.00 g, 4.08 mmol) and 3.36 mL of tetrafluoroboric acid (24% aqueous) were chilled in an ice / acetone bath before the dropwise addition of a 40% aqueous sodium nitrite solution (0.714 g, 4.13 mmol) in 1 mL of water. A precipitate formed instantly with much frothing. After 15 min, the solids were collected by filtration, rinsed with a minimal amount of 5% cold tetrafluoroboric acid, ice cold methanol, and ether. The solids were used without further drying. The weight recovered was 0.685 g (82% crude yield). Recrystallization of solids from acetone / ether afforded tan crystals: mp 139–141° C; 1H-NMR (CD3OD): δ 2.50 (br s, 6H), 5.02 (d, J = 12.8 Hz, 2H), 5.48 (d, J = 10.6 Hz, 1H), 5.61 (d, J = 16.4 Hz, 1H), 6.14–6.35 (m, 1H), 6.57 (s, 1H), 8.91 (s, 1H). Anal. Calcd for C14H13N2O3BF4: C, 48.87; H, 3.81; N, 8.14. Found: C, 48.68; H, 3.89; N, 7.94.
SYNTHESIS OF 4,8-DIMETHYL-4’-HALOMETHYL-4’,5’-DIHYDROPSORALENS
4,8-Dimethyl-4’-iodomethyl-4’,5’-dihydropsoralen (5).
Sodium iodide (0.680 g, 4.50 mmol) and a small crystal of I2 were added to 15 mL of acetone. Separately, 4 (0.775 g, 2.23 mmol) was dissolved in 5 mL of acetone and added dropwise to the sodium iodide / I2 solution. After 15 min, the solvent was removed in vacuo. The solids were taken up in chloroform, washed with water and aqueous sodium bisulfite and dried over magnesium sulfate. Evaporation of the solvent recovered pink tinged crystals that weighed 0.750 g (95% crude yield). Recrystallization from ethanol yielded white crystals: mp 179–180° C; 1H-NMR (CDCl3): δ 2.16 (s, 3H), 2.42 (s, 3H), 3.19 (t, J = 9.5 Hz, 1H), 3.35 (dd, J1 = 10 Hz, J2 = 5 Hz, 1 H), 3.78–3.92 (m, 1H), 4.38 (dd, J1 = 10 Hz, J2 = 5 Hz, 1H), 4.65 (t, J = 9.1 Hz, 1H), 6.10 (s, 1H), 7.20 (s, 1H); 13C-NMR (DMSO-d6): δ 8.7, 11.8, 19.5, 44.7, 79.7, 108.1, 111.6, 114.8, 119.7, 126.9, 154.6, 154.8, 161.5, 163.1; MS: (EI) m/z (relative intensity) 357 (M+, 22), 356 (M+, 55) 230 (14), 229 (base), 187 (26). Anal. Calcd for C14H13O3I: C, 47.21; H, 3.68. Found: C, 47.27; H, 3.64.
3-Bromo-4,8-dimethyl-4’-iodomethyl-4’,5’-dihydropsoralen (6).
NBS (68 mg, 0.38 mmol) and compound (5) (135 mg, 0.379 mmol) in 15 mL of methylene chloride were stirred overnight. The solvent was evaporated and the resulting solids were taken up in chloroform, washed with saturated aqueous sodium bisulfite, twice with 1 mL portions of water, and then dried over magnesium sulfate before the solvent was evaporated. After recrystallization from ethanol, the weight recovered was 165 mg (92% yield) tan crystals: mp 205.7–205.9° C; 1H-NMR (CDCl3): δ 2.29 (s, 3H), 2.51 (s, 3H), 3.19 (t, J = 9.5 Hz, 1H), 3.39 (dd, J1 = 10 Hz, J2 = 5 Hz, 1 H), 3.75–3.87 (m, 1H), 4.40 (dd, J1 = 10 Hz, J2 = 5 Hz, 1H), 4.67 (t, J = 9.1 Hz, 1H), 7.16 (s, 1H); MS: (EI) m/z (relative intensity) 437 (M+, 23), 436 (M+, 86) 435 (base), 309 (15), 308 (49), 307 (19). Anal. Calcd for C14H12O3BrI: C, 38.65; H, 2.78. Found: C, 38.66; H, 2.61.
4’-Bromomethyl-4,8-dimethyl-4’,5’-dihydropsoralen (7).
Copper(II) bromide (260 mg, 1.16 mmol) in 5 mL of dimethyl sulfoxide was added dropwise to compound (4) (400 mg, 1.16 mmol) in 4 mL of dimethyl sulfoxide at rt. Immediate effervescence was evidenced and stirring was continued overnight. The solvent was removed in vacuo and the product was recovered as a red oil. The oil was dissolved in chloroform, washed with two 1 mL portions of water, 1 mL of saturated aqueous sodium bisulfite and dried over magnesium sulfate. The solvent was evaporated and the crude yield was 0.200 g (56% yield). The mixture was purified by silica gel chromatography with elution by 5% methanol / 95% chloroform. Tan crystals had mp 134–135.5°C; 1H-NMR (CDCl3): δ 2.18 (s, 3H), 2.31 (s, 3H), 3.38 (t, J = 6 Hz, 1H), 3.54 (t, J = 9.7 Hz, 1H), 3.78–3.92 (m, 1 H), 4.44–4.56 (m, 1H), 4.68 (t, J = 9 Hz, 1H), 6.03 (s, 1H), 7.15 (s, 1H); 13C-NMR (CDCl3): δ 8.8, 19.5, 35.1, 44.6, 77.4, 111.7, 114.3, 117.7, 124.3, 125.1, 153.1, 154.1, 161.7, 162.2; MS: (EI) m/z (relative intensity) 311 (M+, 20), 310 (M+, 70) 309 (23), 308 (70), 215 (base), 187 (48). Anal. Calcd for C14H13O3Br: C, 54.39; H, 4.24. Found: C, 54.55; H, 4.29.
3-Bromo-4’-bromomethyl-4,8-dimethyl −4’,5’-dihydropsoralen (8).
Compound (7) (450 mg, 1.45 mmol) was dissolved in 20 mL of chloroform before the addition of NBS (0.258 g, 1.449 mmol) at rt. The reaction mixture was stirred overnight and washed twice with 1 mL portions of saturated aqueous sodium bisulfite followed by washing twice with 1 mL portions of water and drying with magnesium sulfate. Elution of product by 5 % methanol / 95% chloroform on silica gel, followed by recrystallization from methanol, recovered 0.430 g (76% yield) tan crystals: mp 148–150°C; 1H-NMR (CDCl3): δ 2.17 (s, 3H), 2.48 (s, 3H), 3.50 (d, J = 9 Hz, 1H), 3.58–3.70 (m, 1H), 3.89–4.05 (m, 1 H), 4.53–4.64 (m, 1H), 4.79 (t, J = 9 Hz, 1H), 7.38 (s, 1H); MS: (EI) m/z (relative intensity) 391 (M+, 35), 390 (M+, 40), 389 (M+, base) 388 (38), 1 15 (10). Anal. Calcd for C14H12O3Br2: C, 43.33; H, 3.12. Found: C, 43.56; H, 3.26.
4’-ChloromethyI-4,8-dimethyl-4’,5’-dihydropsoralen (9).
A solution of compound (4) (500 mg, 1.45 mmol) in 8 mL of dimethyl sulfoxide was added to a solution of anhydrous copper(II) chloride (193 mg, 1.45 mmol) in 3 mL dimethyl sulfoxide. Immediate evolution of gas was evidenced and the reaction was stirred at rt overnight. The solvent was removed in vacuo and the resultant red solid was taken up in methylene chloride, washed with two 1 mL portions of water and dried with magnesium sulfate. Removal of solvent gave 340 mg of tan crystals (89 % crude yield). Purification on a silica gel column with 20% ethyl acetate / 80% hexane eluent gave 299 mg white crystals (79% yield) which were then recrystallized from ethanol: mp 149.5– 150.7°C; 1H-NMR (CDCl3): δ 2.31 (s, 3H), 2.55 (s, 3H), 3.66 (t, J = 9 Hz, 1H), 3.79–3.81 (m, 1H), 3.90–4.10 (m, 1 H), 4.63–4.65 (m, 1H), 4.90–4.95 (m, J = 9 Hz, 1H), 6.14 (s, 1H), 7.34 (s, 1H); 13C-NMR (CDCl3): δ 8.8, 19.5,44.8,46.77, 76.2, 109.0, 111.7, 114.2, 1 17.8, 123.7, 153.1, 154.4, 161.8, 162.2; MS: (EI) m/z (relative intensity) 266 (M+, 11), 264 (M+, 33), 215 (base), 187 (36). Anal. Calcd for C14H13O3Cl: C, 63.52; H, 4.95. Found: C, 63.30; H, 4.96.
SYNTHESIS OF 4,8-DIMETHYL-4’-PYRIDINIUMMETHYL-4’,5’-DIHYDROPSORALEN HALIDE SALTS
4.8-Dimethyl-4,-pyridiniummethyl-4’,5’-dihydropsoralen Iodide Salt (10).
Two mL of pyridine was added to 5 (150 mg, 0.421 mmol), and the mixture refluxed for 2 h. Solids formed upon cooling and pyridine was evaporated in vacuo. Residual pyridine was removed by placing the flask on a vacuum pump overnight. Solids were refluxed in chloroform for 1 h to solubilize 4,8,4’-trimethylpsoralen, which formed as the major product. Undissolved quaternary compound was recovered by filtration and washed with 0.5 mL of acetone and 0.5 mL of ether to remove the slight red coloration. The 4,8-dimethyl-4’-pyridiniummethyl-4’,5’-dihydropsoralen iodide salt was recrystallized from ethanol slowly and 22 mg (12% yield) bright tan crystals were recovered: mp 290–292° C; 1H-NMR (CD3OD): δ 2.21 (s, 3H), 2.33 (s, 3H), 4.20–4.28 (m, 1H), 4.73 (d, J = 6.1 Hz, 2H), 4.87–4.97 (m, 2H), 6.16 (s, 1H), 7.15 (s, 1H), 8.13 (t, J = 6.1 Hz, 2H), 8.63 (t, J = 8.5 Hz, 1H), 8.93 (d, J = 6.1 Hz, 2H). Anal. Calcd for C19H18NO3I: C, 52.43; H, 4.17; N, 3.22. Found: C, 52.26; H, 4.02, N, 3.21.
The major product, 4,8,4’-trimethylpsoralen (11), was recovered from the chloroform layer by removal of solvent in vacuo and recrystallization from ethanol: mp 187–188° C. 1H-NMR (CDCl3): δ 2.19 (s, 3H), 2.43 (s, 3H), 2.46 (s, 3H), 6.16 (s, 1H), 7.44 (s, 1H), 7.51 (s, 1H).
4.8-Dimethyl-4’-(N-4”-methylpyridiniummethyl)-4’,5’-dihydropsoralen Iodide Salt (12).
Compound (5) (138 mg, 0.389 mmol) was dissolved in 2 mL of 4-methylpyridine and the mixture was reacted for 2 h at 105° C. The mixture was then allowed to cool down to rt and the pyridine was removed in vacuo. The crude product was recrystallized from chloroform to yield the pyridinium salt as a pale yellow solid (31 mg, 18 % yield): 1H-NMR (CD3OD): δ: 2.20 (s, 3H), 2.35 (d, J = 0.9 Hz, 3H), 2.70 (s, 3H), 4.20–4.28 (m, 1H), 4.68–4.73 (m, 2H), 4.82–4.92 (m, 2H), 6.16 (d, J = 0.8 Hz, 1H), 7.22 (s, 1H), 7.95 (d, J = 6.3 Hz, 2H), 8.75 (d, J = 6.5 Hz, 2H). Anal. Calcd for C20H20NO3I x 0.25 H2O: C, 52.99; H, 4.56; N, 3.09. Found: C, 52.88; H, 4.50; N, 3.04.
From the remaining organic layer, 70 mg (78 %) 4,8,4’-trimethylpsoralen (11) was purified by flash silica gel column chromatography (chloroform).
3-Bromo-4,8-dimethyl-4’-pyridiniummethyl-4’,5’-dihydropsoralen Iodide Salt (13).
Compound (6) (130 mg, 0.308 mmol) was added to 1 mL of pyridine and refluxed for 2 h. Solids formed upon cooling, and pyridine was evaporated in vacuo. Residual pyridine was removed by placing the flask on a vacuum pump overnight. Solids were refluxed in chloroform for 1 h to solubilize 3-bromo-4,8,4’-trimethylpsoralen which formed as the major product (65 mg recovered). Undissolved quaternary compound was recovered by filtration and washed with acetone and ether to remove the slight red coloration. The 4,8-dimethyl-4’-pyridiniummethyl-4’,5’-dihydropsoralen iodide salt was recrystallized from ethanol slowly, and 18 mg (11% yield) of tan crystals were recovered: mp > 270° C; 1H-NMR (CD3OD): δ 2.28 (s, 3H), 2.52 (s, 3H), 4.22–4.30 (m, 1H), 4.73 (d, J = 6.1 Hz, 2H), 4.87–4.97 (m, 2H), 7.22 (s, 1H), 8.13 (t, J = 6.1 Hz, 2H), 8.66 (t, J = 8.5 Hz, 1H), 8.91 (d, J = 6.7 Hz, 2H). Anal. Calcd for C19H17NO3BrI: C, 44.39; H, 3.33, N, 2.72. Found: C, 44.39; H, 3.24, N, 2.68.
4.8-Dimethyl-4’-pyridiniummethyl-4’,5’-dihydropsoralen Bromide Salt (14).
Compound (7) (180 mg, 0.580 mmol) was added to 2 mL of pyridine and heated at 110° C for 2 h. The residual pyridine was removed in vacuo and remaining solids were refluxed in 20 mL of chloroform. Undissolved solids were collected by filtration and recrystallized from ethanol. The recovered yield was 99 mg (43 % yield), tan crystals: mp 283.2–283.8° C; 1H-NMR (DMSO-d6): δ 2.13 (s, 3H), 2.45 (s, 3H), 4.16–4.36 (m, 1H), 4.64–4.70 (m, 2H), 4.86–4.92 (m, 2H), 6.06 (s, 1H), 7.28 (s, 1H), 8.15 (t, J = 7 Hz, 2H), 8.61 (dd, J1 = 8 Hz, J2 = 8 Hz 1H), 8.95 (d, J = 6.2 Hz, 2H). Anal. Calcd for C19H18NO3Br x 0.36 H2O: C, 57.81; H, 4.77; N, 3.55. Found: C, 57.81; H, 4.61; N, 3.46.
The chloroform layer was dried to yield 4,8,4’-trimethylpsoralen (11) as the major product.
3-Bromo-4,8-dimethyl-4’-pyridiniummethyl-4’,5’-dihydropsoralen Bromide Salt (15).
Compound (8) (125 mg, 0.332 mmol) was added to 2 mL of pyridine and heated at 110° C for 2 h with stirring. The residual pyridine was removed in vacuo and remaining solids were refluxed in 20 mL of chloroform. Undissolved solids were collected by filtration and recrystallized from ethanol. The recovered yield was 79 mg (52 % yield): mp > 280° C (decomp); 1H-NMR (CD3OD): δ 2.21 (s, 3H), 2.33 (s, 3H), 4.20–4.28 (m, 1H), 4.73 (d, 2H), 4.87–4.97 (m, 2H), 7.15 (s, 1H), 8.13 (t, J = 6.1 Hz, 2H), 8.63 (t, J = 8.5 Hz, 1H), 8.93 (d, J = 6.1 Hz, 2H). Anal. Calcd for Cl9H17NO3Br2 × 0.2 H2O: C, 48.46; H, 3.72; N, 2.98. Found: C, 48.46; H, 3.67; N, 2.94.
The chloroform layer was dried to yield 3-bromo-4,8,4’-trimethylpsoralen (16) as the other major product. Recrystallization from methanol yielded tan crystals: mp 280–281° C; 1H-NMR (CDCl3): δ 2.30 (s, 3H), 2.60 (s, 3H), 2.73 (s, 3H), 7.48 (s, 1H), 7.59 (s, 1H). Anal. Calcd for C14H11O3Br: C, 54.75; H, 3.61. Found: C, 54.77; H, 3.39.
4.8-Dimethyl-4’-pyridiniummethyl-4’,5’-dihydropsoralen Chloride Salt (17).
Compound (9) (76 mg, 0.285 mmol) was added to 2 mL of pyridine and heated at 115° C for 3 days with stirring. The residual pyridine was removed in vacuo and remaining solids were refluxed in 20 mL of chloroform. Undissolved solids were collected by filtration and recrystallized from ethanol. The recovered yield was 55 mg (56 % yield): mp > 280° C (decomp); 1H-NMR (CD3OD): δ 2.31 (s, 3H), 2.43 (s, 3H), 4.33 (t, J = 6.4 Hz, 1H), 4.82 (d, J = 5.7 Hz, 2H), 5.00–5.04 (m, 2H), 6.26 (s, 1H), 7.23 (s, 1H), 8.22 (t, J = 7.0 Hz, 2H), 8.76 (t, J = 7.8 Hz, 1H), 9.00 (d, J = 5.8 Hz, 2H). Anal. Calcd for C19H18NO3Cl x 0.33 H2O: C, 65.28; H, 5.30; N, 4.00. Found: C, 65.28; H, 5.11; N, 4.10.
4,8-Dimethyl-4’-cyanomethyl-4’,5’-dihydropsoralen (18).
A solution of compound (4) (100 mg, 0.298 mmol) in 1.4 mL of dimethyl sulfoxide was added to copper(I) cyanide (26 mg, 0.29 mmol) in 1.4 mL of pyridine which had been stirred for 10 min. An immediate evolution of gas was accompanied by the darkening of the solution. After 20 min the solvents were removed in vacuo. The organic portion was dissolved in 4 mL of chloroform and washed with two 1 mL portions of water. The chloroform was removed in vacuo and the resulting orange solid was purified by silica gel column chromatography, with elution by 5% methanol / 95% chloroform. The crystals, recrystallized from ethanol, weighed 32 mg (42% yield): mp 156–157 °C; 1H-NMR (CDCl3): δ 2.23 (s, 3H), 2.42 (s, 3H), 2.73 (t, J = 6 Hz, 2H), 3.78–3.97 (m, 1H), 4.40–4.54 (m, 1 H), 4.74–4.88 (m, 1H), 6.16 (s, 1H), 7.44 (s, 1H); MS: (EI) m/z (relative intensity) 257 (M+, 16), 256 (M+, Base) 255 (19), 215 (26), 181 (48), 181 (17). Anal. Calcd for C15H13NO3 × 0.33 H2O: C, 68.99; H, 5.26. Found: C, 68.99; H, 5.29.
ACKNOWLEDGEMENT
This study was supported by NIH grants ES03647, ES06897 and ES05022.
REFERENCES AND NOTES
- 1.Jabin I, Heindel ND, Rapp RD, and Laskin JD, J. Heterocycl. Chem, 2000, 37, 31. [Google Scholar]
- 2.Whittemore MS, Heindel ND, Jabin I, Guillon CD, McNeel TE, Rapp RD, and Laskin JD, J. Heterocycl. Chem. 2001, 38, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jetter MM, Heindel ND, and Laskin JD, J. Heterocycl. Chem, 1990, 27, 995. [Google Scholar]
- 4.Heindel ND, Van Dongen JMAM, Sachais BS, Phillips JH, Gallo MA, and Laskin JD, J. Pharm. Sci, 1991, 80, 686. [DOI] [PubMed] [Google Scholar]
- 5.Laskin JD, Mermelstein FH, Heindel ND, and Ron Y, J. Leukocyte Biol, 1993, 54, 138. [DOI] [PubMed] [Google Scholar]
- 6.Isaacs ST, Sheen CJ, Hearst JE, and Rapport H, Biochem, 1977, 16, 1058. [DOI] [PubMed] [Google Scholar]
- 7.Photochemotherapeutic Aspects of Psoralens, NIH Publications No. 84–2692, U. S. Government Printing Offices, Scientific Editors: Pathak MA and Dunnick JK, December, 1984, 66. [Google Scholar]
- 8.Laskin JD, Lee E, Yurkow EJ, Laskin DL, and Gallo MA, Proc. Natl. Acad. Sci. USA, 1985, 82, 6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Laskin JD, Lee E, Yurkow EJ, Laskin DL, and Gallo MA, Proc. Natl. Acad. Sci. USA, 1986, 83, 8211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stern R, Nichols KT, and Vakeva L, N. Engl. J. Med. 1997, 336, 1041. [DOI] [PubMed] [Google Scholar]
- 11.Whittemore MS, “Synthesis, Structure-Activity Relationships And Photochemical Studies of Novel Coumarins, Dihydropsoralens and Dihydroangelicins as Photo-Activated Agonists of the Psoralen Receptor”, Ph.D. Dissertation, Department of Chemistry, Lehigh University, Bethlehem. PA. U. S. A. 1998. [Google Scholar]
- 12.Meijs G and Beckwith A, J. Am. Chem. Soc, 1986, 108, 5890. [DOI] [PubMed] [Google Scholar]
- 13.Beckwith A and Meijs G, J. Org. Chem, 1987, 52, 1922. [Google Scholar]
- 14.Moffett RB, J. Med. Pharm. Chem. 1962, 5, 335. [DOI] [PubMed] [Google Scholar]
- 15.Adams R and Mecorney J, J. Am. Chem. Soc, 1923, 44, 1781. [Google Scholar]
- 16.Roe A, in Organic Reactions, Vol. 5, ed. By Adams R, John Wiley and Sons, New York, 1949, pp. 204–206. [Google Scholar]
- 17.Sousa C, Melo TS, Maziere JC, and Santus R, Photochem. Photobiol., 1998, 67, 561. [PubMed] [Google Scholar]
- 18.Stern RS, Photodermatol. Photoimmunol. Photomed, 1999, 15, 37. [DOI] [PubMed] [Google Scholar]