

Abstract
The anterior segment dysgeneses are a broad group of heterogeneous disorders characterized by developmental abnormalities of the anterior segment of the eye, including primary congenital aphakia, Peters sequence, aniridia, and Axenfeld–Rieger spectrum. These conditions can have overlapping phenotypes and both genotypic and phenotypic heterogeneity. This article provides a strategy for both phenotyping and then genotyping using a targeted stepwise approach.
Keywords: Anterior segment mesenchymal dysgenesis, cornea, lens
Introduction
Anterior segment dysgenesis (ASD) encompasses a broad group of developmental disorders affecting the anterior part of the eye, particularly the cornea, iris, and lens. Understanding these conditions can be particularly complex due to variable expression, overlapping phenotypes, and both genetic and phenotypic heterogeneity. ASD relies on an accurate diagnosis and management. This paper will suggest a strategy for identifying the phenotype of these patients and how this can lead to appropriate genotyping to arrive at a final diagnosis.
Similar to other genetic conditions, a stepwise approach will achieve the most accurate phenotyping and genotyping. A comprehensive and accurate medical and family history associated with a complete systemic and ophthalmic examination is a key to establishing the phenotype, which will be the foundation of the most efficient genotyping process. By opting for this strategy, the clinician is also able to lower the risks inherent to genetic testing and minimize the false genotype rate.[1] It is essential to emphasize the role of genetic counseling during this process, before and after genetic testing.
We will focus on the most common and well-recognized disorders. Other less frequent disorders are summarized in Table 1.
Table 1.
Less common anterior segment dysgeneses
| Disorder | Inheritance | Genes* | Systemic manifestations† |
|---|---|---|---|
| Coloboma[2] | AR, AD, XLr, XLd | ALG3 SCKL TMEM67 POMT1 PITX2 PQBP1 |
Microcephaly, developmental delay, Pai syndrome, Renpenning syndrome, Skeletal dysplasia, Brain malformations |
| Cornea plana[3] | AD, AR | KERA | None reported |
| Anterior segment mesodermal dysgenesis[4] | AD | PITX3 | None reported |
| Congenital iris ectropion[5] | PAX6 | Neurofibromatosis type 1, Prader–Willi | |
| Megalocornea[6] | XL, AD, AR | CHRDL1 dup22q. 11.2 | Mucolipidosis, Neuhauser syndrome, Osteogenesis imperfecta |
| Microcornea[7,8] | AD, AR |
EPHA2
FOXE3 GJA8 CRYGD CRYGC MAB21L2 ATOH7, SLC16A12 BEST1 ARL2 MIP GJA3 MAF CRYAA CRYBB3 CRYBB2 CRYBB1, CRYBA4 NHS SOX2 |
Ehlers–Danlos syndrome, Marfan syndrome, Norrie syndrome, Turner syndrome, Waardenburg syndrome, Alport syndrome, Oculodentodigital syndrome |
| Autosomal dominant anterior stromal iris hypoplasia[9] | AD | PITX2 | None reported |
| Iris traction band | Unknown | None reported |
*Most commonly associated genes with the isolated ASD; †Syndromes listed in association although the genes for these syndromes are not typically associated with the listed ocular findings as an isolated finding. Nomenclature: AD, AR, XLd, XLr. AD=Autosomal dominant, AR=Autosomal recessive, XLd=X-linked dominant, XLr=X-linked recessive
Clinical Evaluation of Anterior Segment Dysgenesis
Clinical phenotyping of ASD requires a careful slit lamp examination, intraocular pressure, and, where indicated, imaging, to establish the clinical diagnosis. Figure 1 suggests a flow diagram to approach the identification of the phenotype. The first question the clinician should ask is whether a lens is present. This may require ultrasound biomicroscopy (UBM) or anterior segment optical coherence tomography (aOCT). If there is not, the most probable diagnosis is primary aphakia. Other clues to this diagnosis would include retinal dysplasia, a silvery gray appearance of the cornea, and a high likelihood of glaucoma. If a lens is present, the ophthalmologist may then ask a second question, if an iris is present. If there is little or no iris, perhaps only a residual stub or peripheral iris, the patient has aniridia. If a significant iris is present, the next question arises: is there posterior embryotoxon? Although this finding is seen in 8%–32% of the general population,[1,10] it is suggestive of the Axenfeld–Rieger spectrum. If there is no posterior embryotoxon, one can look for other diagnostic indicators such as iridocorneal adhesions and a posterior scallop on the cornea on UBM or aOCT suggestive of Peters anomaly. Thereafter, other findings such as pupil position, persistent fetal vasculature on the iris, or a coloboma-like iris defect can help identify alternate phenotypes.
Figure 1.
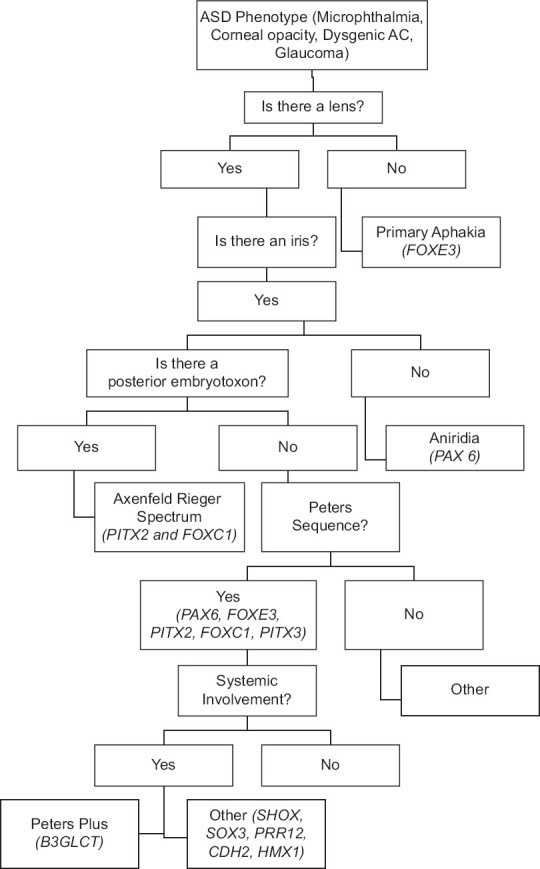
Diagnostic algorithm for phenotyping and genotyping anterior segment dysgeneses. Most frequent genes indicated
Congenital primary aphakia
Congenital primary aphakia is a severe panocular dysgenesis characterized primarily by aborted lens development during the 4th week of gestation due to failure of lens placode induction from the surface ectoderm.[11] As the lens has a critical role in the induction and stromal differentiation of the cornea,[10,12,13] this results in severe dysgenesis of the anterior segment and microphthalmia.[14] Often these patients present with a silvery corneal appearance probably secondary to thin corneal stroma and disorganized collagen lamellae.[15] As the anterior and posterior structures visualization usually is poor, aOCT or UBM can identify the absence of a lens. Some patients present with buphthalmos due to glaucoma, which can occur in over 20%.[16] Patients may also present with features of Peters anomaly and/or retinal dysplasia.
Congenital primary aphakia is due to pathologic variants in FOXE3. This gene encodes a transcription factor with a DNA-binding domain, the forkhead domain. In a 2022 series, 82% of patients showed pathogenic heterozygous variants in this gene.[12] There have been reports of autosomal dominant and recessive inheritance. Usually, nonsense variants follow an autosomal recessive pattern, with more severe ocular and systemic manifestations. Autosomal dominant variants usually result in a C-terminal extension of the protein, with a milder and isolated ocular phenotype. Reported extraocular manifestations to include Arnold–Chiari malformation, global developmental delay, polycystic ovary syndrome, ventricular septal defect, and renal pelvic dilation. Variants in this gene have also been associated with thoracic aortic aneurysms.[17]
Aniridia
Aniridia is an autosomal dominant disorder, almost always secondary to heterozygous variants or deletions in PAX6, with high penetrance, variable expression, and phenotypic heterogeneity.[12,18] PAX6 is the master control gene for eye morphogenesis, so pathogenic variants result in a broad spectrum of eye manifestations.[19] The principal clinical manifestation is the underdevelopment of the iris, but the disorder is also associated with cataracts, keratopathy, glaucoma, nystagmus, and optic nerve and foveal hypoplasia.[20] Aniridia secondary to large deletions may involve both PAX6 and the Wilms tumor gene (WT1), resulting in WAGR syndrome: Wilms tumor, aniridia, genitourinary abnormalities, and the relative difference in development. Genetic testing gives critical information about cancer risk: either through chromosomal microarray that identifies a deletion involving WT1 or sequencing of PAX6 identifying intragenic pathogenic variants. In cases where genetic testing is unavailable, abdominal ultrasound screening protocols should be used.[21] Deletion of the region downstream of PAX6 and WT1, involving the genes ELP4 and/or DCDC1, which affect the expression of PAX6, may also result in aniridia. The presence of aniridia plus posterior embryotoxon should suggest the rare aniridic form of the Axenfeld–Rieger spectrum due to deletions or duplications involving 6p25.[22] There have been reports of a condition characterized by blepharophimosis and ASD, including near-total absence of the iris, microphthalmia, and macular dysgenesis/hypoplasia due to missense variants in gene MAB21L1.[23]
Axenfeld–Rieger spectrum
This spectrum is characterized by the variable combination of posterior embryotoxon, iridocorneal adhesions, iris hypoplasia, and corectopia/polycoria. Systemic features include anomalies of the teeth, redundant periumbilical skin, facial dysmorphism, hearing impairment, and heart abnormalities.[24] It is autosomal dominant with high penetrance and variable expression.[25] Pathogenic variants in PITX2, a fundamental transcription factor in eye development, are more often associated with systemic manifestations. Congenital heart defects and hearing loss are more common with pathogenic variants in FOXC1. These two genes may be disease causing by deletion or duplication as well. They are responsible for approximately 70% of cases.[13,24] Less frequent genes in which pathogenic variants have been associated with Axenfeld–Rieger spectrum phenotypes include RIEG2,[26] GJA1,[27] COL4A1,[28] FGD1, PIK3R1,[29] and FOXC2, CYB1P1, LAMB2.[30]
Isolated posterior embryotoxon, usually without iridocorneal adhesions, can be an isolated finding in between 8% and 32% of the general population.[10] One systemic association is Alagille syndrome secondary to JAG1 variants.[31]
Peters anomaly/sequence
Peters anomaly/sequence is characterized by posterior corneal defects with secondary opacity associated with iridocorneal and possible keratolenticular adhesions with or without cataracts. This condition is the result of a defective lens formation from surface ectoderm, followed by secondary disruption of neural crest migration during embryogenesis.[32] The majority of cases are sporadic, but autosomal recessive and dominant inheritance has been described.[33] This spectrum is characterized by broad genotypic heterogeneity. The genes more frequently involved in this condition are PAX6, FOXE3, PITX2, FOXC1, and PITX3. However, there have been reported variants in COL4A1, CYB1P1, FLNA, HCCS, NDP, SLC4A11, and TFAP2.[34]
Peters-Plus syndrome refers to several disorders. The designation most accurately describes autosomal recessive Peters sequence in the setting of skeletal dysplasia, developmental delay, and other malformations due to biallelic disruption of function of the B3GLCT on 13q.[35] Peters sequence can also be seen with a variety of other malformations, in particular cardiac, due to disruption of a variety of genes (e.g., SHOX, SOX3, PRR12, CDH2, and HMX1), some of which have yet to be discovered. Genetic testing should be driven by the specific systemic phenotype.
Iris coloboma is often isolated but may be part of a larger picture, including ocular coloboma with microphthalmia, optic nerve involvement, and other ocular developmental disorders such as cataract, persistent fetal vasculature, and uncommonly glaucoma, with or without systemic manifestations.[36] The genetic testing strategy is informed by the complete phenotype. Cornea plana is characterized by flattening of the corneal surface, associated with variable degrees of hyperopia, peripheral corneal opacity with or without glaucoma.[37] Anterior segment mesodermal dysgenesis is characterized by variable degrees of corneal opacity, posterior embryotoxon, and corneal-irido-lenticular adhesions. It has been associated with the development of glaucoma, making routine screening examinations mandatory.[38] Congenital iris ectropion is an isolated, often unilateral, disorder associated with angle dysgenesis with secondary development of glaucoma.[5] The main diagnostic criteria for megalocornea are a corneal diameter >12.5 mm in the presence of normal pachymetry and axial length. This can be associated with radial iris transillumination. It is not associated with glaucoma, but the differential diagnosis of primary congenital glaucoma should be considered. Microcornea features a corneal diameter <10 mm (<9 mm in newborns) with/without other ocular and/or systemic malformations.[7] Genotyping is again targeted toward the complete phenotype. Autosomal dominant anterior stromal iris hypoplasia is characterized by a poorly developed iris stroma that allows for the baring of the iris sphincter. There is a high incidence of glaucoma.[9]
Genotyping
Once a complete phenotype has been established, genetic testing can proceed, preferably using a strategy with a narrow gene hypothesis to minimize the false genotype rate.[1] This process should only be pursued with proper pretest genetic counseling. The goals of genetic counseling are to provide complete information about testing options, recurrence risk, risk to other family members, cost, expectations for result accuracy and timing, incidental results, and possible variants of uncertain significance. Genetic counseling aims to be nondirective.[39] Genetic counselors have expertise in facilitating appropriate test selection, incorporating the phenotype information from the ophthalmologist, and considering other factors, including patient preference and motivations, certification and licensing standards met by performing laboratories, and insurance coverage and payment options. They can also identify research opportunities or other supportive resources for patients and families.[40]
Genetic counseling is also essential after the results are received. One of the fundamental steps in this whole process is the interpretation of results. The results should be interpreted following the American College of Genomics and the Association of Molecular Pathology guidelines who define a five-tier framework of classification of gene variants as: pathogenic, likely pathogenic, uncertain significance, likely benign, and benign.[41] This analysis should be comprehensive, looking at the function of the gene involved, if the variants identified are reported in medical literature, population frequency of the variant, evolutionary conservation of the affected gene location, and other characteristics. While testing laboratories generally provide interpretation according to these guidelines, it is important for the clinical team to review this carefully and analyze the match between the genotype and phenotype observed in their patient to ensure consistency. Testing of other family members may be necessary to understand the results of the proband. This process requires a team with expertise in interpreting genomic variant results and disclosing them to patients and families in a meaningful way. It is the latter which requires proper genetic counseling to effectively communicate, and interpret the impact of, the genetic test results to the patient and their family.
Conclusion
The anterior segment dysgeneses are a complex and sometimes the overlapping collection of developmental disorders which may be isolated or occur in the setting of associated systemic disease. A comprehensive clinical evaluation is mandatory to achieve a robust phenotype which can then lead to appropriate genetic testing. The process should be supported by genetic counseling.
Data availability statement
Data Sharing not applicable to this article as no datasets were generated or analyzed during the current study.
Financial support and sponsorship
This work was partly funded by the Adeline Lutz-Steven S.T. Ching, M.D. Distinguished Professorship in Ophthalmology (AVL) and an unrestricted grant from Research to Prevent Blindness to the Department of Ophthalmology at the University of Rochester.
Conflicts of interest
The authors declare that there are no conflicts of interests of this paper.
References
- 1.Stone EM, Andorf JL, Whitmore SS, DeLuca AP, Giacalone JC, Streb LM, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017;124:1314–31. doi: 10.1016/j.ophtha.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lingam G, Sen AC, Lingam V, Bhende M, Padhi TR, Xinyi S. Ocular coloboma-a comprehensive review for the clinician. Eye (Lond) 2021;35:2086–109. doi: 10.1038/s41433-021-01501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khan AO, Aldahmesh M, Meyer B. Recessive cornea Plana in the Kingdom of Saudi Arabia. Ophthalmology. 2006;113:1773–8. doi: 10.1016/j.ophtha.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 4.Summers KM, Withers SJ, Gole GA, Piras S, Taylor PJ. Anterior segment mesenchymal dysgenesis in a large Australian family is associated with the recurrent 17 bp duplication in PITX3. Mol Vis. 2008;14:2010–5. [PMC free article] [PubMed] [Google Scholar]
- 5.Laaks D, Freeman N. Congenital iris ectropion uveae presenting with glaucoma in infancy. J AAPOS. 2013;17:214–6. doi: 10.1016/j.jaapos.2012.11.010. [DOI] [PubMed] [Google Scholar]
- 6.Ong AP, Zhang J, Vincent AL, McGhee CN. Megalocornea, anterior megalophthalmos, keratoglobus and associated anterior segment disorders: A review. Clin Exp Ophthalmol. 2021;49:477–97. doi: 10.1111/ceo.13958. [DOI] [PubMed] [Google Scholar]
- 7.Lin ZB, Li J, Ye L, Sun HS, Yu AY, Chen SH, et al. Novel SOX2 mutation in autosomal dominant cataract-microcornea syndrome. BMC Ophthalmol. 2022;22:70. doi: 10.1186/s12886-022-02291-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lambert SR, Lyons CJ, Taylor D, Hoyt CS. Taylor &Hoyt's Pediatric Ophthalmology and Strabismus. 5th ed. Amsterdam: Elsevier; 2016. p. e1044. [Google Scholar]
- 9.Alward WL, Semina EV, Kalenak JW, Héon E, Sheth BP, Stone EM, et al. Autosomal dominant iris hypoplasia is caused by a mutation in the Rieger syndrome (RIEG/PITX2) gene. Am J Ophthalmol. 1998;125:98–100. doi: 10.1016/s0002-9394(99)80242-6. [DOI] [PubMed] [Google Scholar]
- 10.Rennie CA, Chowdhury S, Khan J, Rajan F, Jordan K, Lamb RJ, et al. The prevalence and associated features of posterior embryotoxon in the general ophthalmic clinic. Eye (Lond) 2005;19:396–9. doi: 10.1038/sj.eye.6701508. [DOI] [PubMed] [Google Scholar]
- 11.Kaushik S, Snehi S, Kaur S, Kaur A, Choudhary S, Thattaruthody F, et al. Primary aphakia: Clinical recognition is the key to diagnosis. J AAPOS. 2022;26:298.e1–5. doi: 10.1016/j.jaapos.2022.07.012. [DOI] [PubMed] [Google Scholar]
- 12.Ernst J, Medsinge A, Scanga HL, Hiasat J, Moore W, Ali A, et al. Congenital primary aphakia. J AAPOS. 2022;26:4.e1–5. doi: 10.1016/j.jaapos.2021.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Beebe DC, Coats JM. The lens organizes the anterior segment: Specification of neural crest cell differentiation in the avian eye. Dev Biol. 2000;220:424–31. doi: 10.1006/dbio.2000.9638. [DOI] [PubMed] [Google Scholar]
- 14.Manschot WA. Primary Congenital Aphakia. Arch Ophthal. 1963;69:571–7. [Google Scholar]
- 15.Chaurasia S, Jakati S, Ramappa M, Mishra DK, Edward DP. Anterior segment alterations in congenital primary aphakia-a clinicopathologic report of five cases. Indian J Ophthalmol. 2020;68:1564–8. doi: 10.4103/ijo.IJO_2078_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Badakere SV, Aulakh S, Achanta DS, Chary R, Senthil S, Chaurasia S, et al. Secondary developmental glaucoma in eyes with congenital aphakia. Indian J Ophthalmol. 2022;70:834–6. doi: 10.4103/ijo.IJO_1782_21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuang SQ, Medina-Martinez O, Guo DC, Gong L, Regalado ES, Reynolds CL, et al. FOXE3 mutations predispose to thoracic aortic aneurysms and dissections. J Clin Invest. 2016;126:948–61. doi: 10.1172/JCI83778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lim HT, Kim DH, Kim H. PAX6 aniridia syndrome: Clinics, genetics, and therapeutics. Curr Opin Ophthalmol. 2017;28:436–47. doi: 10.1097/ICU.0000000000000405. [DOI] [PubMed] [Google Scholar]
- 19.Gehring WJ, Ikeo K. Pax 6: Mastering eye morphogenesis and eye evolution. Trends Genet. 1999;15:371–7. doi: 10.1016/s0168-9525(99)01776-x. [DOI] [PubMed] [Google Scholar]
- 20.Nascimento ES, Shen LQ, Chiou CA, Shanbhag SS, Paschalis EI, Pasquale LR, et al. Glaucoma management in patients with aniridia and Boston type 1 keratoprosthesis. Am J Ophthalmol. 2019;207:258–67. doi: 10.1016/j.ajo.2019.06.018. [DOI] [PubMed] [Google Scholar]
- 21.Spreafico F, Fernandez CV, Brok J, Nakata K, Vujanic G, Geller JI, et al. Wilms tumour. Nat Rev Dis Primers. 2021;7:75. doi: 10.1038/s41572-021-00308-8. [DOI] [PubMed] [Google Scholar]
- 22.Sadagopan KA, Liu GT, Capasso JE, Wuthisiri W, Keep RB, Levin AV. Anirdia-like phenotype caused by 6p25 dosage aberrations. Am J Med Genet A. 2015;167 A:524–8. doi: 10.1002/ajmg.a.36890. [DOI] [PubMed] [Google Scholar]
- 23.Wang P, Wu P, Wang J, Zeng Y, Jiang Y, Wang Y, et al. Missense mutations in MAB21L1: Causation of novel autosomal dominant ocular BAMD syndrome. Invest Ophthalmol Vis Sci. 2023;64:19. doi: 10.1167/iovs.64.3.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Reis LM, Maheshwari M, Capasso J, Atilla H, Dudakova L, Thompson S, et al. Axenfeld-Rieger syndrome: More than meets the eye. J Med Genet. 2023;60:368–79. doi: 10.1136/jmg-2022-108646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alward WL. Axenfeld-Rieger syndrome in the age of molecular genetics. Am J Ophthalmol. 2000;130:107–15. doi: 10.1016/s0002-9394(00)00525-0. [DOI] [PubMed] [Google Scholar]
- 26.Borges AS, Susanna R., Jr, Carani JC, Betinjane AJ, Alward WL, Stone EM, et al. Genetic analysis of PITX2 and FOXC1 in Rieger syndrome patients from Brazil. J Glaucoma. 2002;11:51–6. doi: 10.1097/00061198-200202000-00010. [DOI] [PubMed] [Google Scholar]
- 27.Cella W, de Vasconcellos JP, de Melo MB, Kneipp B, Costa FF, Longui CA, et al. Structural assessment of PITX2, FOXC1, CYP1B1, and GJA1 genes in patients with Axenfeld-Rieger syndrome with developmental glaucoma. Invest Ophthalmol Vis Sci. 2006;47:1803–9. doi: 10.1167/iovs.05-0979. [DOI] [PubMed] [Google Scholar]
- 28.Sibon I, Coupry I, Menegon P, Bouchet JP, Gorry P, Burgelin I, et al. COL4A1 mutation in Axenfeld-Rieger anomaly with leukoencephalopathy and stroke. Ann Neurol. 2007;62:177–84. doi: 10.1002/ana.21191. [DOI] [PubMed] [Google Scholar]
- 29.Solheim MH, Clermont AC, Winnay JN, Hallstensen E, Molven A, Njølstad PR, et al. Iris malformation and anterior segment dysgenesis in mice and humans with a mutation in PI 3-kinase. Invest Ophthalmol Vis Sci. 2017;58:3100–6. doi: 10.1167/iovs.16-21347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reis LM, Semina EV. Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol. 2011;22:314–24. doi: 10.1097/ICU.0b013e328349412b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohut TJ, Gilbert MA, Loomes KM. Alagille syndrome: A focused review on clinical features, genetics, and treatment. Semin Liver Dis. 2021;41:525–37. doi: 10.1055/s-0041-1730951. [DOI] [PubMed] [Google Scholar]
- 32.Elbaz U, Ali A, Strungaru H, Mireskandari K. Phenotypic spectrum of peters anomaly: Implications for management. Cornea. 2022;41:192–200. doi: 10.1097/ICO.0000000000002768. [DOI] [PubMed] [Google Scholar]
- 33.Bhandari R, Ferri S, Whittaker B, Liu M, Lazzaro DR. Peters anomaly: Review of the literature. Cornea. 2011;30:939–44. doi: 10.1097/ICO.0b013e31820156a9. [DOI] [PubMed] [Google Scholar]
- 34.Weh E, Reis LM, Happ HC, Levin AV, Wheeler PG, David KL, et al. Whole exome sequence analysis of Peters anomaly. Hum Genet. 2014;133:1497–511. doi: 10.1007/s00439-014-1481-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lesnik Oberstein SA, Kriek M, White SJ, Kalf ME, Szuhai K, den Dunnen JT, et al. Peters plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am J Hum Genet. 2006;79:562–6. doi: 10.1086/507567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Daich Varela M, Huryn LA, Hufnagel RB, Zein WM, Blain D, Brooks BP. Ocular and systemic findings in adults with uveal coloboma. Ophthalmology. 2020;127:1772–4. doi: 10.1016/j.ophtha.2020.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sahin BO, Seymenoglu G, Baser EF. Cornea Plana associated with open-angle glaucoma: A case report. Int Ophthalmol. 2011;31:505–8. doi: 10.1007/s10792-011-9490-4. [DOI] [PubMed] [Google Scholar]
- 38.Falkenstein RJ, Henkind P. Mesodermal dysgenesis and hyaline membranes. Am J Ophthalmol. 1973;76:462–7. doi: 10.1016/0002-9394(73)90731-9. [DOI] [PubMed] [Google Scholar]
- 39.Jamal L, Schupmann W, Berkman BE. An ethical framework for genetic counseling in the genomic era. J Genet Couns. 2020;29:718–27. doi: 10.1002/jgc4.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Capasso JE. The cost of genetic testing for ocular disease: Who pays? Curr Opin Ophthalmol. 2014;25:394–9. doi: 10.1097/ICU.0000000000000085. [DOI] [PubMed] [Google Scholar]
- 41.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data Sharing not applicable to this article as no datasets were generated or analyzed during the current study.