

Abstract
Oncolytic virus therapy has shown activity against primary melanomas; however, its efficacy in brain metastases remains challenging, mainly because of the delivery and immunosuppressive nature of tumors in the brain. To address this challenge, we first established PTEN-deficient melanoma brain metastasis mouse models and characterized them to be more immunosuppressive compared with primary melanoma, mimicking the clinical settings. Next, we developed an allogeneic twin stem cell (TSC) system composed of two tumor-targeting stem cell (SC) populations. One SC was loaded with oncolytic herpes simplex virus (oHSV), and the other SC was CRISPR-Cas9 gene-edited to knock out nectin 1 (N1) receptor (N1KO) to acquire resistance to oHSV and release immunomodulators, such as granulocyte-macrophage colony-stimulating factor (GM-CSF). Using mouse models of brain metastatic BRAFV600E/PTEN−/− and BRAFV600E/wt/PTEN−/− mutant melanomas, we show that locoregional delivery of TSCs releasing oHSV and GM-CSF (TSC-G) activated dendritic cell– and T cell–mediated immune responses. In addition, our strategy exhibited greater therapeutic efficacy when compared with the existing oncolytic viral therapeutic approaches. Moreover, the TSCs composed of SC-oHSV and SCN1KO–releasing GM-CSF and single-chain variable fragment anti–PD-1 (TSC-G/P) had therapeutic efficacy in both syngeneic and patient-derived humanized mouse models of leptomeningeal metastasis. Our findings provide a promising allogeneic SC-based immunotherapeutic strategy against melanomas in the CNS and a road map toward clinical translation.
INTRODUCTION
Despite recent improvements in therapeutic approaches, patients with advanced melanoma still have limited survival prognosis, with brain metastases contributing to half of all melanoma-related deaths (1). Advanced-stage melanomas have a high propensity to metastasize to the brain, with 60% of patients developing brain metastases at some point (2, 3). Although multidisciplinary therapies such as radiotherapy, targeted therapy, and surgeries have been used to treat melanoma brain metastasis (MBM), the overall survival is only 4 to 6 months (4). Immunotherapy has revolutionized treatment for melanoma. Immune checkpoint inhibitors (ICIs) are one of the major advances in recent cancer therapy, especially for the treatment of metastatic melanoma (5). However, clinical studies with systemic administration of ICIs have revealed poorer intracranial responses than extracranial responses due partially to the limited penetration of ICI antibodies (Abs) into the brain and cerebrospinal fluid (CSF) (6–9). Therefore, alternative therapeutic agents and strategies are urgently needed.
Oncolytic virotherapy represents a newer form of immunotherapy, in which oncolytic viruses (OVs) are used to selectively replicate in and kill neoplastic cells (10, 11) and are among the latest therapies that have progressed to the clinic (12–16). The lytic activity of OVs promotes the release of tumor antigens and supports the development of antitumor immune responses (17). Intralesional injection of Food and Drug Administration (FDA)–approved talimogene laherparepvec (T-VEC; recombinant oncolytic herpes simplex virus, oHSV) (18) has been shown to induce antitumor immune responses for distant uninjected tumor lesions but has not improved overall patient survival of stage IV disease that has metastatic lesions (17). Systemic delivery of oHSV has been hampered by issues such as virus neutralization, sequestration, and inefficient extravasation (19). To counter these problems, we have previously shown that oHSV-loaded mesenchymal stem cells (SCs) home extensively to multiple metastatic tumor deposits in the brain, deliver oHSV locally, and have therapeutic efficacy in imageable mouse models of MBM (20). Although promising, the immunosuppressive tumor microenvironment of MBM (21), which prevents efficient antitumor immune responses, is yet to be explored. Phosphatase and tensin homolog (PTEN) loss is associated with shorter time to MBM and correlates with shorter overall survival (22). In this study, we first developed syngeneic mouse models of PTEN-deficient MBMs and profiled their unique tumor immune microenvironment to create a new platform to test the efficacy of immunotherapy.
Cytokines are potent immunomodulatory molecules and have been successfully used as adjuvants in OV therapy for cancer. Numerous preclinical and clinical studies have investigated the therapeutic efficacy of cytokine-expressing OVs, such as oHSV (18), adenovirus (23), and vaccinia virus (24) in different cancer types. ICIs can be rationally combined with oHSV therapy, because virally infected dying tumor cells release tumor antigens to the tumor microenvironment to attract innate and adaptive immune cells, offering the potential to achieve a more durable response and outcome (25–27). Similarly to cytokines, ICI genes can be incorporated into OVs to minimize toxicities associated with systemic treatment with ICIs. However, OV-mediated cytokine and ICI expression is dependent on OV infection of cancer cells, and infected cells are expected to die, making the abundance and duration of cytokine and ICI expression in the tumor microenvironment unpredictable (28, 29). SCs genetically engineered with immunomodulators have the potential of prolonging therapeutic molecule delivery at the tumor site. However, given the susceptibility of SCs to oHSV-mediated oncolysis (30), codelivery of oHSV and immunomodulators from the same SCs is not feasible. We have previously shown that both mouse and human SCs highly express nectin-1 (CD111), the most efficient entry receptor for oHSV (31). In this study, we overcame the limitation of the SC codelivery system by knocking out the nectin-1 receptor in SCs (SCN1KO) using CRISPR-Cas-9 technology and further engineered SCN1KO to release various immunomodulators, including granulocyte-macrophage colony-stimulating factor (GM-CSF) and single-chain variable fragment anti–programmed death receptor-1 (PD-1) (scFvPD-1). The major goal of this study was to assess the mechanism-based efficacy of locoregional delivery of an engineered twin SC (TSC) platform releasing oHSV and immunomodulators in the immunosuppressive syngeneic and humanized MBM mouse models.
RESULTS
PTEN deficiency is associated with MBM and immune suppression
PTEN is a phosphatase that is involved in the negative regulation of cell survival signaling through the phosphoinositide 3-kinase (PI3K)/AKT pathway (32). First, we explored the correlation between advanced melanoma and PTEN expression. Overall, TCGA data showed that 20% of melanomas express PTEN (Fig. 1A). Patients with melanoma with higher PTEN expression showed longer overall survival compared with patients with lower PTEN expression (Fig. 1B, P < 0.05). PTEN expression was significantly lost in late-stage metastatic melanoma compared with other genes, including BRAF and TP53 (Fig. 1C, P < 0.05, and fig. S1A). A high proportion of clinical MBM cases were characterized by the loss of PTEN expression in metastatic melanoma cells (Fig. 1C, P < 0.05). Another RNA dataset of patients with metastatic melanoma also demonstrated that MBM had lower gene expression of PTEN than other metastatic melanomas (Fig. 1D, P < 0.05) (33). To further explore this, we analyzed immune cell infiltration in melanoma. PTEN-low melanoma had significantly fewer CD3+ T cells (Fig. 1E, P < 0.05). Furthermore, patients with higher PI3K/AKT pathway expression had fewer CD3+ T cells, CD4+ T cells, and activated dendritic cells (DCs) and more regulatory T cells and macrophages (fig. S1B). Comparing primary melanoma with metastatic melanoma, the latter had fewer CD4+ T cells and DCs (fig. S1C). These results suggest that PTEN loss is associated with MBM and poor prognosis through immunosuppressive mechanisms. Therefore, it is critical to evaluate immunotherapy against advanced PTEN-deficient melanoma.
Fig. 1. PTEN deficiency is correlated with MBM and immune suppression.
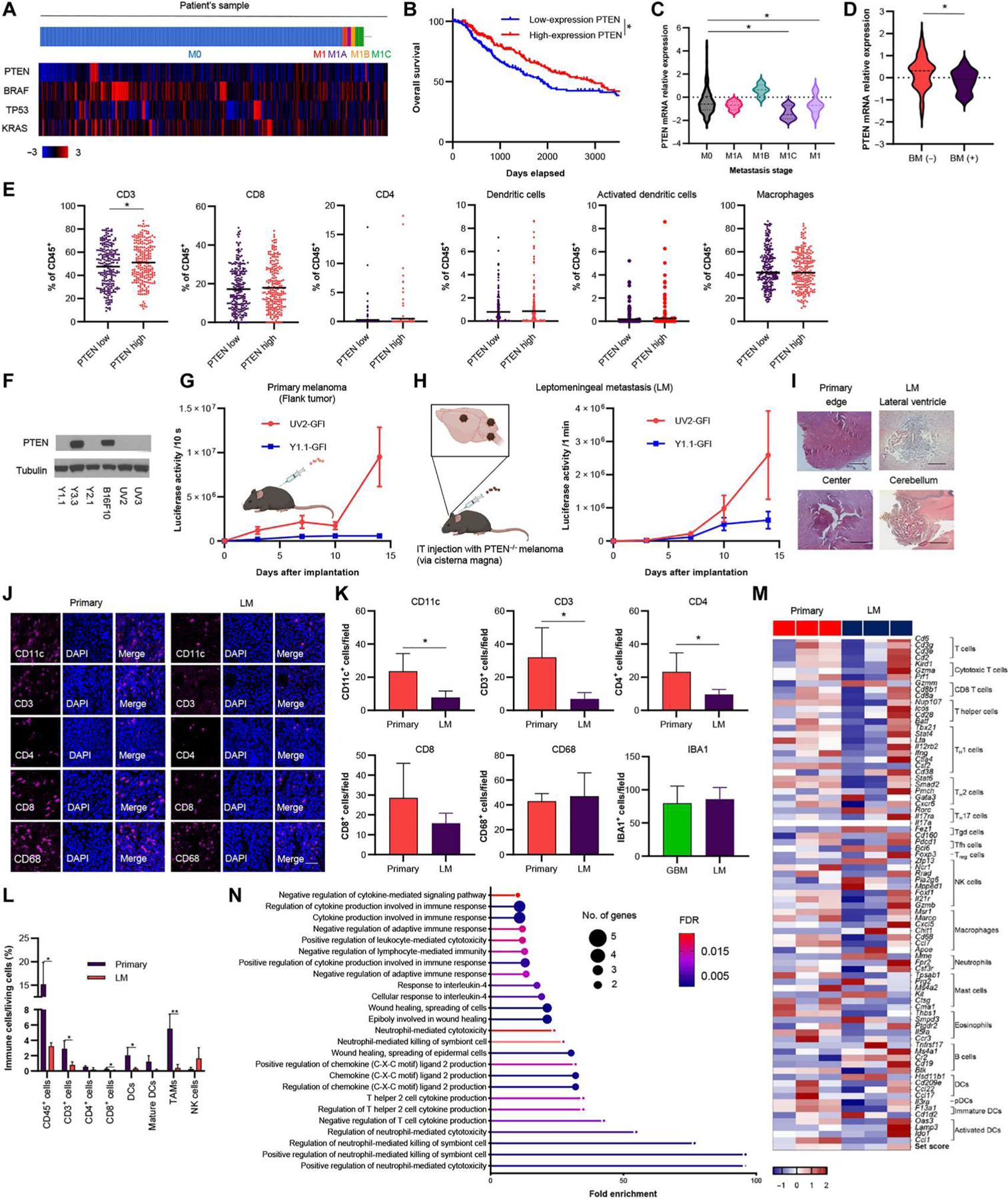
(A) Heatmap of mRNA expression of PTEN, BRAF, TP53, and KRAS in patient samples of metastatic stage from the TCGA database (n = 337). M0, no distant metastasis; M1, distant metastasis (unknown lesions); M1A, distant skin metastasis; M1B, lung metastasis; M1C, other distant metastasis including brain mRNA expression z scores relative to all samples (≧1.5) was defined as positive expression. (B) Kaplan-Meier curves of overall survival for high PTEN melanoma and low PTEN melanoma of patients from TCGA (the threshold cutoff was median). *P < 0.05. (C) Comparison of PTEN expression between metastatic stages from the TCGA database. *P < 0.05. (D) Comparison of PTEN expression in BM and other metastasis (n = 144) from the dbGaP study (phs000452.v3.p1). *P < 0.05. (E) Immune profile analysis of high–PTEN expression melanoma and low–PTEN expression melanoma from the TCGA database. *P < 0.05. (F) Western blotting of PTEN and tubulin in six murine melanoma cell lines. (G) Schematic of primary melanoma mouse models. BLI signal curve of primary UV2-GFl (n = 4)– or Y1.1-GFl (n = 3)–bearing mice. Data are presented as means + SEM. BLI, bioluminescence imaging. (H) Schematic of leptomeningeal metastasis (LM) mouse models (left). BLI signal curve of flank and IT-injected UV2-GFl (n = 3)– or Y1.1-GFl (n = 3)–bearing mice (right). Data are presented as means + SEM. (I) Representative hematoxylin and eosin (H&E) staining of primary melanoma (edge and central area) and LM mouse model (lateral ventricle and cerebellum area). Scale bar, 100 μm. (J) IF analysis of CD11c, CD3, CD4, CD8, and CD68 in UV2-GFl primary (n = 4) and LM (n = 3) mouse models. Scale bar, 6 mm. (K) Mean number of TILs expressing CD11c, CD3, CD4, CD8, CD68, and IBA1 was statistically assessed from three selected fields. IBA1 was compared with UV2-GFl LM– and glioblastoma (GBM) (CT2A-mCherry-Fluc)–bearing mice (n = 3 per group). Data are presented as means + SD. *P < 0.05. (L) Flow cytometry showing the difference of immune profiles between UV2-GFI primary (n = 4) and LM (n = 3) mouse models. Data are presented as means + SD. *P < 0.05 and **P < 0.01. (M) Heatmap of differential expression of genes associated with immune cell types in UV2-GFI flank and LM melanoma plotted as z score of normalized gene expression for each gene. (N) GO analysis for down-regulated immune-related pathway enrichment [UV2-GFI primary (n = 3) versus LM (n = 3) mouse models].
Development and characterization of primary and metastatic mouse tumor models of PTEN-deficient melanoma
We examined the expression of PTEN in different murine melanoma cell lines that were previously generated (34, 35). Western blot analysis revealed that several murine cell lines, YUMM1.1 (Y1.1), YUMM2.1 (Y2.1), D4MUV2 (UV2), and D4MUV3 (UV3) cells, did not express PTEN (Fig. 1F). In previous studies, Y1.1 and Y2.1 cells have been used as models of BRAFV600E/wt/PTEN−/− melanomas, whereas UV2 and UV3 cells represent BRAFV600E/PTEN−/− melanomas (34, 35, 36). On the other hand, YUMM3.3 (Y3.3) and B16F10 (B16) cells simulate BRAFV600E/wt/PTEN+/+ and BRAFwt/PTEN+/+ melanomas, respectively (34, 36). We selected Y1.1 and UV2 to generate mouse PTEN-deficient melanoma cell lines expressing a bimodal green fluorescent protein (GFP)–firefly luciferase (Fluc) fusion protein (fig. S2) and used these cells to develop C57BL/6 mouse models of both primary tumors and leptomeningeal metastasis (LM) tumors (Fig. 1, G to I). UV2-GFP-Fluc (UV2-GFI) cells grew efficiently as an LM mouse model, whereas Y1.1-GFP-Fluc (Y1.1-GFI) cells often grew as extracranial tumors after intrathecal injection of tumor cells. To assess the composition of immune cells in the tumor microenvironment of primary melanoma and LM in these UV2-GFI–bearing mouse tumor models, we performed immunofluorescence (IF) analysis for DCs (CD11c), T cells (CD3, CD4, and CD8), and macrophages (CD68 and IBA1). IF analysis showed that the LM mouse model had fewer CD11c-, CD3-, CD4-, and CD8-positive cells than the primary tumor mouse model (Fig. 1, J and K). There was no difference in CD68+ cells between primary and LM mouse models. Comparison of IBA1+ microglial concentrations in the LM mouse model and PTEN-deficient glioblastoma (CT2A) mouse model found no significant difference (Fig. 1K, P = 0.7326). Furthermore, flow cytometry (FCM) analysis revealed that the LM mouse model had significantly fewer DCs, CD4+, and CD8+ T cells than the primary melanoma mouse model (Fig. 1L; DCs, P < 0.05; CD4+ T cells, P < 0.05; CD8+ T cells, P < 0.05). Moreover, transcriptome analysis also showed that two of three UV2-GFI–derived LM samples had down-regulation of these immune cells compared with primary melanoma (Fig. 1M). Furthermore, several immunological pathways, including cytokine-related pathways, were down-regulated in LM samples (Fig. 1N). These results suggested that LM represents more immunosuppressive tumors compared with primary melanoma in the mouse models tested.
SC-loaded oHSVs have therapeutic effects in vitro and in vivo
We explored the efficacy of recombinant oHSVs using G47Δ, a third-generation oHSV type 1, in melanoma cells. To characterize the oncolytic activity of oHSV, we studied its cytopathic effects in human melanoma cell lines, Mewo and M12 (fig. S3A), as well as the previously used murine melanoma (Y1.1, Y2.1, UV2, UV3, and B16) cells in vitro (Fig. 2A). Cell viability assay revealed that Y1.1, Y2.1, UV2, and UV3 cells were more sensitive to oHSV, whereas B16 and Y3.3 cells were more resistant (Fig. 2A). On the other hand, temozolomide, one of the standard chemotherapies for melanoma, did not induce cell death in any of the cell lines (fig. S3B). Therefore, we chose Y1.1, Y2.1, and UV2 cells as oHSV-sensitive, PTEN-deficient melanoma cells in this study. The clinically approved oHSV, T-VEC, expresses GM-CSF, so we also created oHSV that encodes mouse GM-CSF cDNA (oHSV-GM-CSF). oHSV-GM-CSF also induced cell death in Y1.1, Y2.1, and UV2 cells (fig. S3C). Next, we tested the effect of oHSV on the PI3K/AKT pathway in melanoma cells. Western blot analysis showed that oHSV increased p-AKT, p-mTOR, and PI3K in Y1.1, Y2.1, and UV2 cells (fig. S3D). We confirmed that oHSV-mediated tumor cell death was associated with induction of apoptosis, whereas it was not associated with necroptosis, as indicated by up-regulation of cleaved poly(adenosine diphosphate–ribose) polymerase (PARP)/PARP, but no change in pRIP3 expression (fig. S3E). Next, we determined whether oHSV could induce immunogenic cell death (ICD) through the release of damage-associated molecular patterns, such as adenosine triphosphate (ATP) and high-mobility group box 1 protein (HMGB1). oHSV significantly induced ATP release from Y1.1, Y2.1, and UV2 cells 24 and 48 hours after oHSV infection (Fig. 2B; Y1.1, P < 0.05; Y2.1, P < 0.0001; UV2, P < 0.0001). Further, Western blot analysis also showed that oHSV increased HMGB1 from Y1.1 and UV2 cells 24 hours after oHSV infection (fig. S3E). To explore the induction of ICD with oHSV in vivo, oHSV-infected UV2-GFI cells were used as a vaccine in mice receiving UV2-GFI cells (Fig. 2C). Compared with phosphate-buffered saline (PBS)–treated mice, tumor growth was significantly suppressed in mice vaccinated with oHSV-infected cells (Fig. 2D, P < 0.01). These results show that oHSV induced immunogenic oncolytic cell death in our melanoma models.
Fig. 2. Stem cells are efficient carriers of oHSV.
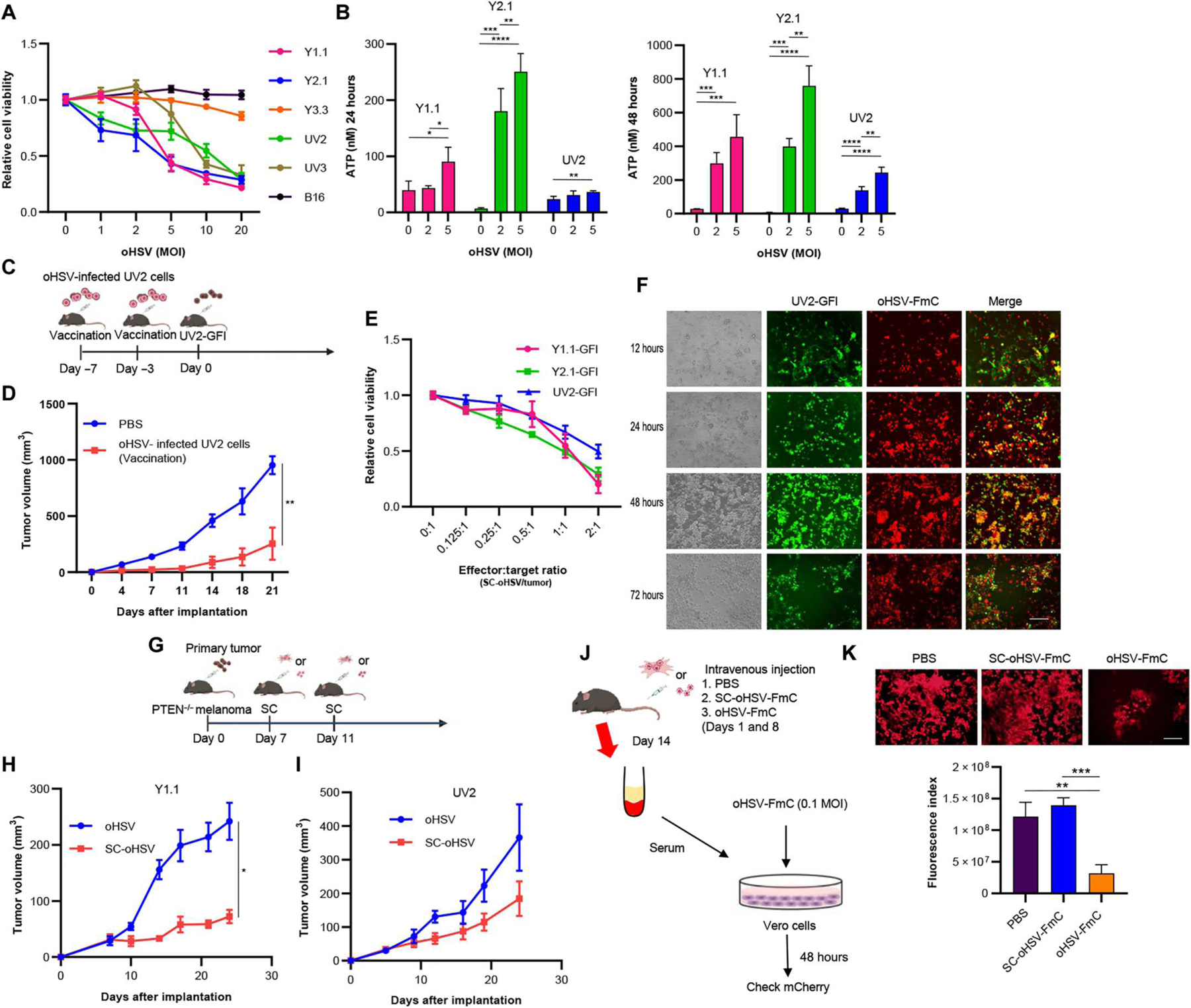
(A) Cell viability assay of six melanoma cells was assessed 3 days after oHSV treatment at the indicated doses (MOI) (n = 5 per group, technical replicates). Data are presented as means ± SD. (B) Extracellular ATP secreted from Y1.1, Y2.1, and UV2 cells was measured using a luminescence assay 24 and 48 hours after oHSV treatment (0, 2, and 5 MOI, n = 4 per group). Data are presented as means + SD. *P < 0.05. **P < 0.01, ***P < 0.001, ****P < 0.0001. (C) In the vaccination study, UV2-GFI cells treated with oHSV (5 MOI) for 2 days were administered subcutaneously into the flanks of C57/BL6 mice on days −7 and −3 for vaccination, and UV2-GFI cells (1 × 106 cells) were inoculated subcutaneously on day 0. (D) Tumor volume was monitored and compared between PBS (n = 5) and vaccinated mice (n = 5). Data are presented as means ± SEM. **P < 0.01. (E) PTEN mutant melanoma cells were investigated by cell viability assay 3 days after SC-oHSV treatment in vitro (n = 5 per group, technical replicates). Data are presented as means ± SD. (F) Coculture showing killing of melanoma cells (green) infected with oHSV-FmC (red) released from SCs for 12, 24, 48, and 72 hours, respectively. Scale bar, 100 μm. (G) Experimental design. Y1.1-GFI or UV2-GFl subcutaneous melanoma tumors were treated with oHSV (4 × 105 PFU per mouse) or SC-oHSV (4 × 105 cells per mouse) intratumorally 7 and 11 days after tumor inoculation. Tumor volumes were measured every 3 to 4 days after implantation. (H) Plots showing subcutaneous Y1.1-GFI tumor growth in mice treated with oHSV (n = 8) or SC-oHSV (n = 8). Data are presented as means ± SEM. *P < 0.05. (I) Plots showing subcutaneous UV2-GFI tumor growth in mice treated with oHSV (n = 5) or SC-oHSV (n = 5). Data are presented as means ± SEM. (J) Stealth effect (reduction of antiviral Ab) of SC-oHSV providing protection from the immune system in vivo. Experimental design: PBS, SC-oHSV-FmC, or oHSV-FmC was systemically injected in C57BL/6 mice twice every week (days 1 and 8), and the blood was collected from each mouse at day 14. Vero cells were infected with oHSV-FmC for 2 days with the serum collected from the mice. (K) mCherry spots observed on fluorescence microscopy and the intensity of mCherry spots measured by ImageJ (n = 3 per group). Data are represented as means + SD. Scale bar, 100 μm. **P < 0.01 and ***P < 0.001.
We explored the use of adipose-derived mesenchymal SCs as delivery carriers for oHSV (fig. S2F). SCs were incubated with oHSV at different multiplicities of infection [MOIs: plaque-forming unit (PFU) per cell], and their survival was measured in vitro. SCs infected at 1, 2, 5, 10, and 20 MOI survived for 24 hours, followed by a rapid decrease in viable populations by 48 hours after infection (fig. S2G). SCs infected at 1 and 2 MOI retained ~50% of the original population at 48 hours, whereas SCs infected at MOIs of 5 or higher retained 25% or fewer of the original population at the same time point. Furthermore, fluorescence microscopy showed that almost all SCs infected at 2 and 5 MOI expressed reporter mCherry 24 hours after infection (fig. S3H). Infection with 5 MOI led to a higher infection rate but a larger decline in SC viability at the 24-hour time point, posing a challenge for their use in in vivo treatment. When cocultured with murine melanoma cells, SC-oHSV at 2 and 5 MOI comparably decreased the viability of Y1.1-GFl cells (fig. S3I). Therefore, oHSV at 2 MOI was chosen for treatment studies. SC-oHSV (2 MOI) effectivity killed Y1.1, Y2.1, and UV2-GFl in a dose-dependent manner (Fig. 2E). Imaging of coculture with melanoma cells and SC-oHSV-FmC showed the spread of oHSV infection and subsequent killing of melanoma cells (Fig. 2F).
In vivo, in the primary tumor mouse models of Y1.1-GFI or UV2-GFl, intratumoral administration of SC-oHSV led to a significant decrease in Y1.1-GFI tumor growth compared with oHSV alone (Fig. 2, G, H, and I, H; P < 0.05). In addition, in vitro multicytokine and chemokine assays showed that SC-oHSV induced the release of several chemokines, such as C-C motif chemokine ligand 5, C-X-C motif chemokine ligand 10/interferon gamma–induced protein 10 (IP-10), interferon gamma, and tumor necrosis factor–α (TNF-α), which are known to play important roles in recruiting tumor-infiltrating lymphocytes (TILs) (fig. S3I) (37, 38). One of the clinical limitations of oHSV is that it cannot be given systemically because of neutralizing Abs–mediated immune elimination of the viruses (39). We, thus, examined the production of anti-oHSV Abs after intravenous injections of SC-oHSV-FmC or oHSV-FmC in C57BL/6 mice. Vero cells were infected with oHSV-FmC in vitro for 2 days in the presence of serum collected from the mice (Fig. 2J). The sera collected from the mice treated systemically twice with SC-oHSV-FmC did not impede oHSV infection, whereas the sera collected from the mice treated twice with oHSV-FmC significantly reduced infection (Fig. 2K, P < 0.01), indicating that SC loading of oHSV reduced the production of anti-oHSV Abs in immune-competent mice. These results suggested that SC-oHSV has the potential to induce ICD through the active release of immunogenic molecules and chemokines, leading to activation of the host immune response against tumors while minimizing the clearance of the oHSV by the host’s immune system. Together, these data show that SCs are suitable oHSV carriers for virotherapy.
Establishment of oHSV-resistant SCs secreting immunomodulators
The cell surface adhesion molecule nectin-1 (CD111) is known as the most efficient entry receptor for oHSV (31), and we have previously shown that both mouse and human SCs highly express this protein (20). To allow prolonged secretion of immunomodulators in the presence of oHSV and to enhance the therapeutic efficacy of SC-oHSV for PTEN-deficient melanoma, we created oHSV-resistant SCs by CRISPR-Cas9–mediated knockout of nectin-1 (SCN1KO) (Fig. 3A). FCM and Western blot analyses confirmed the knockout of nectin-1 in SCs (Fig. 3B), and cell viability assay after treatment with oHSV confirmed the resistance of SCN1KO to oHSV as compared with unmodified SCs (Fig. 3C). SCN1KO were further engineered to secrete interleukin-12 (IL-12), IL-15, GM-CSF, or 4–1BB ligand (Fig. 3D). To determine which immunomodulator synergically suppressed PTEN-deficient melanoma when combined with SC-oHSV, a mixture composed of Y1.1-GFl cells, SC-oHSV, and SCN1KO immunomodulator was inoculated into the flanks of C57BL/6 mice, and tumor growth was monitored for 7 days. In this screening test, a combination therapy of SCN1KO-GM-CSF (SCN1KO-G) and SC-oHSV [TSCs releasing oHSV and GM-CSF (TSC-G)] showed the most potent therapeutic efficacy compared with the other combination therapies and SC-oHSV-GM-CSF on its own (Fig. 3E). Therefore, we focused on investigating TSC-G therapy for PTEN-deficient melanoma. SCN1KO-G retained the oHSV-resistant phenotype of SCN1KO (fig. S4A). FCM analysis demonstrated the presence of cell surface expression of the GM-CSF receptor in a murine macrophage cell line, RAW264.7 cells, but not in Y1.1-GFl, Y2.1-GFl, and UV2-GFl cells (fig. S4B). Furthermore, conditioned medium (CM) derived from SCN1KO-G induced higher growth of RAW264.7 compared with CM from control SCs, SC-Rluc-mCherry (RmC), whereas no significant difference in cell growth was observed for Y1.1-GFl, Y2.1-GFl, and UV2-GFl cells in vitro (Fig. 3F and fig. S4C). Intratumoral administration of SCN1KO-G did not significantly change the growth of Y1.1-GFl flank tumors in vivo (fig. S4D, P = 0.7945).
Fig. 3. oHSV-resistant stem cells secreting GM-CSF target DCs and macrophages.
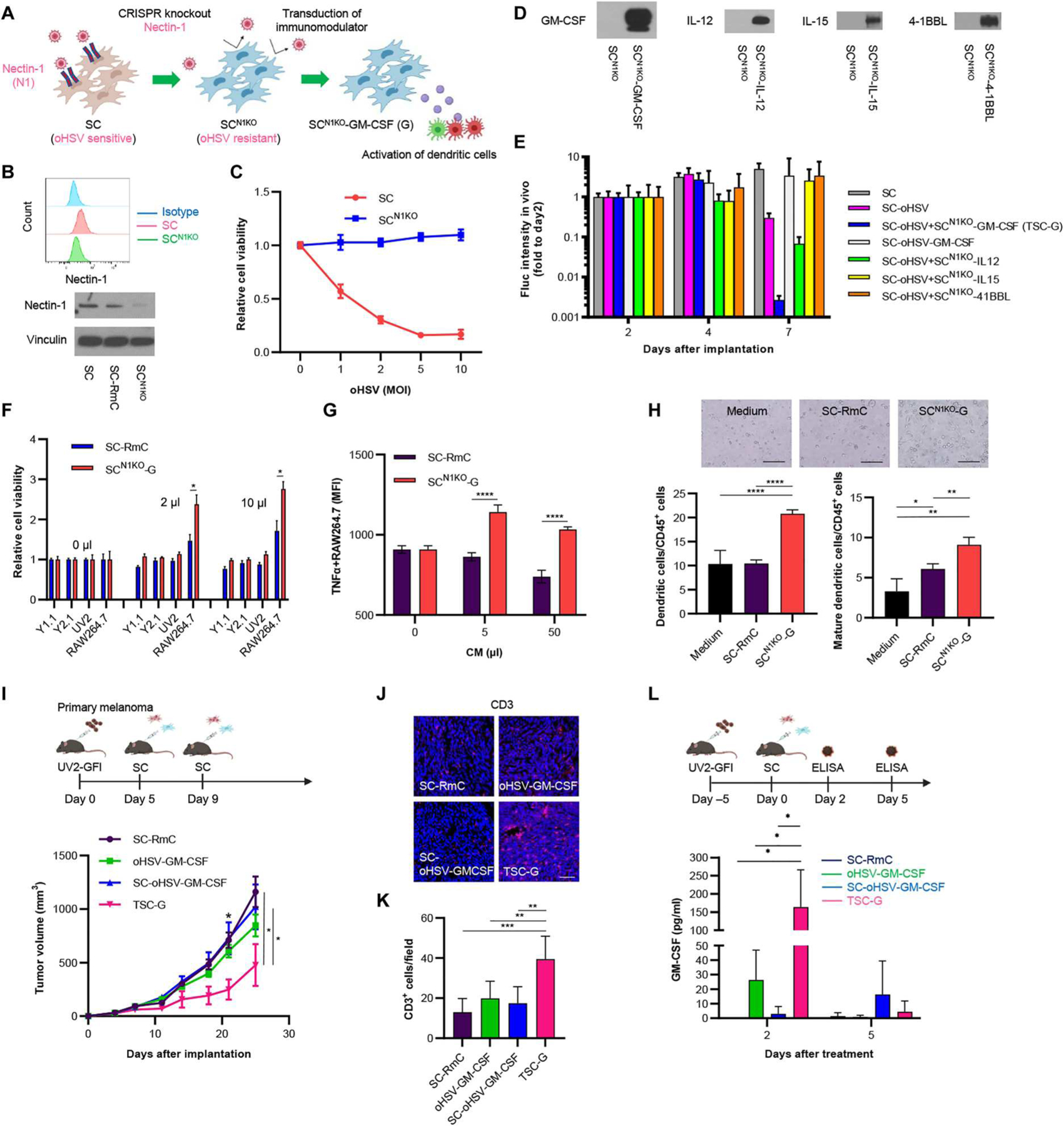
(A) Scheme showing the creation of SCN1KO-expressing immunomodulators. To establish oHSV-resistant SC-GM-CSF, nectin-1 on SCs was deleted by CRISPR-Cas9 gene editing, and SCN1KO was subsequently transduced with GM-CSF. (B) Flow cytometry (FCM, top) and Western blotting (bottom) showing expression of nectin-1 in SCs. (C) Cell viability assays showing SCN1KO resistance to oHSV compared with SCs. Data are presented as means ± SD (n = 5 per group). (D) Expression of immunomodulators in SCN1KO by Western blotting). (E) In vivo screening showing the antitumor activity of SCN1KO-secreting immunomodulators combined with SCs loaded with oHSV in Y1.1-GFI–bearing primary melanoma mouse tumors. Data are presented as means + SEM. (n = 5 per group). (F) Cell viability assays showing the influence of SCN1KO-G and SC-Rluc-mCherry (RmC) on murine macrophages (RAW264.7) and melanoma (Y1.1-GFl, Y2.1-GFl, and UV2-GFl). (n = 5 per group, technical replicates). Data are presented as means + SD. *P < 0.05. (G) Plot showing the effect of SCN1KO-G conditioned medium (CM) on TNF-α–positive RAW264.7 cells by FCM after incubation for 4 days. Data are presented as means + SD (n = 3 per group). ****P < 0.0001. (H) Plot showing the effect of SCN1KO-G CM on differentiation to dendritic cells (DCs) and mature DCs from murine bone marrow cells (CD45+ cells) by FCM after incubation for 4 days (n = 3 or 4 per group). Data are presented as means + SD. Scale bar, 100 μm. *P < 0.05. **P < 0.01, and ****P < 0.0001. (I) Experimental design (top). In brief, in the UV2-GFl subcutaneous melanoma mouse model, the tumor was treated with SC-oHSV and SCN1KO-G intratumorally 5 and 9 days after tumor cell inoculation. Then, tumor volumes were measured every 3 to 4 days after implantation. Plots showing subcutaneous UV2-GFl tumor growth in mice (bottom) treated with control SC-RmC (n = 6), oHSV-GM-CSF (n = 6), SC-oHSV-GM-CSF (n = 7), or SC-oHSV and SCN1KO-G (n = 7). Data are presented as means ± SEM. *P < 0.05. (J) Representative images of immunofluorescence (IF) analysis for CD3+ TILs in tumor tissues harvested 30 days after UV2-GFl tumor cell inoculation. Scale bar, 6 μm. (K) Mean number of TILs expressing CD3 was statistically assessed from three selected fields as represented in (J) (n = 5 per group). Data are presented as means + SD. **P < 0.01 and ***P < 0.001. (L) In vivo concentrations of GM-CSF after treatment with SC-RmC (n = 3), oHSV-GM-CSF (n = 3 or 4), SC-oHSV-GM-CSF (n = 3), or SC-oHSV and SCN1KO-G (n = 4). The tumors were collected on days 2 and 5 after treatment. Data are presented as means + SD. *P < 0.05.
Next, we performed functional assays using RAW264.7 and bone marrow cells to examine the activity of SCN1KO-G–secreted GM-CSF on the differentiation and activation of macrophages and DCs. FCM showed that CM of SCN1KO-GM-CSF significantly induced more TNF-α–producing RAW264.7 cells than control medium or CM of SC-RmC (Fig. 3G and fig. S4E; P < 0.0001). To explore the ability of the CM to differentiate mouse bone marrow cells to M1 (CD11b+F4/80+CD86+) or M2 (CD11b+F4/80+CD206+) macrophages, we incubated mouse bone marrow cells with SCN1KO-G CM for 3 days ex vivo. FCM analysis of macrophage populations showed that SCN1KO-G CM significantly induced differentiation of macrophages to M1 and activated M1 (CD80+) and M2 macrophages (fig. S4, F and G; M1, P < 0.001; activated M1, P < 0.001; M2, P < 0.01). We also found that mouse bone marrow cells’ exposure to SCN1KO-G CM for 4 days significantly increased populations of DCs (CD45+CD11b+CD11c+, P < 0.0001) and mature DCs [CD45+CD11b+CD11c+ major histocompatibility complex (MHC) II I-A/I-E+, P < 0.01] (Fig. 3H and fig. S4H). These results confirmed the biological functions of SCN1KO-G–secreted GM-CSF.
When cocultured with Y1.1-GFI cells, SCN1KO-G and oHSV-GM-CSF–infected SC-RmC induced similar concentrations of GM-CSF in supernatant at 24 hours (fig. S4I). However, at 48 hours, GM-CSF concentrations decreased with oHSV-GM-CSF compared with SCN1KO-G, reflecting cell death of oHSV-GM-CSF–infected SCs. Intratumoral injection of TSC-G therapy significantly suppressed tumor growth compared with SC-RmC, oHSV-GM-CSF, or SC-oHSV-GM-CSF in the UV2-GFl primary tumor mouse model (Fig. 3I, P < 0.05), which was consistent with the Y1.1-GFl mouse model (Fig. 3E). IF images showed that TSC-G therapy led to the recruitment of more CD3-positive TILs compared to controls 25 days after therapy (Fig. 3, J and K). TSC-G therapy released more GM-CSF than oHSV-GM-CSF or SC-oHSV-GM-CSF 2 days after intratumoral injection, whereas the secretion of GM-CSF by TSC-G or oHSV-GM-CSF on its own was reduced 5 days after intratumoral injection (Fig. 3L, P < 0.05). Thus, the efficacy of TSC-G therapy in PTEN-deficient melanoma mouse models was superior to oHSV-GM-CSF or SC-oHSV-GM-CSF and was associated with rapid, robust secretion of GM-CSF from SCN1KO-G.
Abscopal effects of TSC-G in a bilateral subcutaneous tumor model
The abscopal effect is an interesting phenomenon in which tumor shrinkage at metastatic or distant sites is achieved after local therapy of the primary tumor. We next assessed the antitumor effects, including abscopal effects, of TSC-G therapy in two different bilateral subcutaneous melanoma mouse models, Y1.1-GFl and UV2-GFl (Fig. 4A). In the Y1.1-GFl tumor mouse model, TSC-G therapy showed significantly increased therapeutic effects compared with SC-RmC, oHSV-GM-CSF, or SC-oHSV alone in directly treated tumors (Fig. 4B and fig. S5A; P < 0.001). Furthermore, the suppression of tumor growth by TSC-G therapy was better than SC-RmC at the nontreatment site (P < 0.05), whereas oHSV-GM-CSF or SC-oHSV alone did not show efficacy. Similarly, TSC-G therapy decreased tumor growth in injected tumors and mediated abscopal effects in the UV2-GFl tumor mouse model (Fig. 4C and fig. S5B; treatment site: P < 0.05; nontreatment site: P < 0.05). When these mice with Y1.1-GFl tumors were rechallenged with Y1.1-GFI cells in the brain after each treatment, all the mice receiving TSC-G therapy (n = 4) remained tumor free (Fig. 4D). To understand the mechanism underlying the therapeutic efficacy, we performed cytotoxic T lymphocyte (CTL) assays using splenocytes after treatment. The splenocytes (effector) collected from the spleen of treated mice showed significant cytotoxicity against Y1.1-GFl cells (target) as compared with control SC-RmC treatment, whereas no cytotoxicity was observed against murine lung TC-1-GFl cells ex vivo (Fig. 4E and fig. S4C; P < 0.05). These results suggested that the TSC-G therapy induced tumor-specific and systemic immune responses in the treated mice.
Fig. 4. TSC-G therapy generates systemic immunity against bilateral flank PTEN-deficient melanoma in vivo.
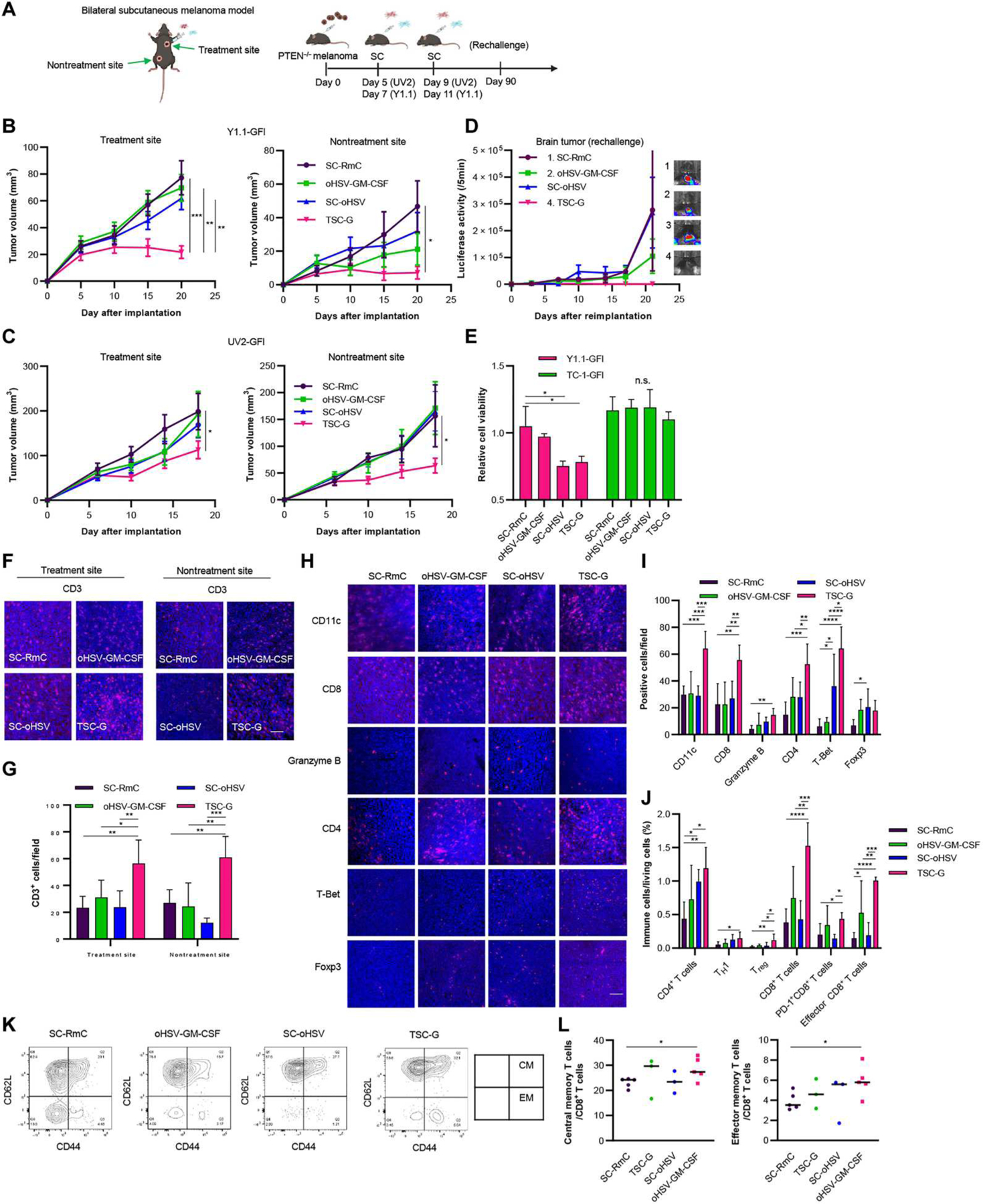
(A) Experimental design. In brief, in the bilateral Y1.1-GFl and UV2-GFl subcutaneous melanoma mouse model, one tumor was treated with SC-oHSV and SCN1KO-GM-CSF (TSC-G) intratumorally twice starting 5 days after inoculation, and the other tumor was left untreated. Tumor volumes were measured every 3 to 5 days after implantation. (B) Plots showing subcutaneous Y1.1-GFl tumor growth in mice treated with control RmC (n = 7), oHSV-GM-CSF (n = 7), SC-oHSV (n = 8), or TSC-G (n = 8). (Left) Subcutaneous tumor growth in treated tumors. (Right) Subcutaneous tumor growth in untreated tumors. Data are represented as means ± SEM. *P < 0.05, **P < 0.01, and ***P < 0.001. (C) Plots showing subcutaneous UV2-GFl tumor growth in mice treated with control SC-RmC (n = 7), oHSV-GM-CSF (n = 7), SC-oHSV (n = 8), or TSC-G (n = 8). (Left) Subcutaneous tumor growth in treated tumors. (Right) Subcutaneous tumor growth in untreated tumors. Data are represented as means ± SEM. *P < 0.05. (D) Y1.1-GFl–bearing mice after treatment (n = 4 per group) were rechallenged with Y1.1-GFI cells on day 90 into the brain. Plot showing brain metastatic tumor growth measured by in vivo Fluc bioluminescence (representative images shown right). Data are represented as means ± SEM. (E) Splenocytes from mice on day 90 after treatment (n = 3 per group) were incubated at 37°C with either Y1.1-GFl melanoma cells or TC-1-GFl lung at effector cell:target cell ratios (8:1). Data are represented as means + SD. *P < 0.05. n.s., not significant. (F) Representative images of IF analysis of CD3 in UV2-GFl tumors 14 days after the indicated treatment (n = 4 or 5 per group). Scale bar, 6 μm. (G) Quantified mean number of TILs expressing CD3 was statistically assessed from three selected fields as represented in (F). Data are represented as means + SD. *P < 0.05, **P < 0.01, and ***P < 0.001. (H) Representative images of IF analysis of CD11c, CD8, granzyme B, CD4, T-Bet, and Foxp3 in UV2-GFl tumors 14 days after indicated treatment (n = 4 or 5 per group). Scale bar, 6 μm. (I) Quantified mean number of CD11c, CD8, granzyme B, CD4, T-Bet, and Foxp3. Data are represented as means + SD. *P < 0.05. **P < 0.01, ***P < 0.001, and ****P < 0.0001. (J) Flow cytometric analysis of immune cells collected from UV2-GFl–bearing subcutaneous tumor 7 days after treatment. Data are represented as means + SD (n = 4 to 6 per group). *P < 0.05. **P < 0.01, ***P < 0.001, and ****P < 0.0001. (K) Representative FCM plots of central memory (CM) CD8+ T cells and effector memory (EM) CD8+ T cells on splenocytes after treatment. (L) FCM analysis of CM and EM CD8+ T cells on splenocytes after treatment (n = 3 or 5 per group). Data are represented as means + SD. *P < 0.05.
We next profiled tumor-infiltrating immune cells after treatment in the UV2-GFl mouse model. IF analysis demonstrated an increase in CD3-positive cells at the treatment site and the nontreatment site in the TSC-G group (Fig. 4, F and G, P < 0.01). TSC-G therapy increased granzyme B and T-Bet–positive cells as well as CD4-, CD8-, and CD11c-positive cells at the treatment site, suggesting the activation of CTLs and type 1 T helper (TH1) cells (Fig. 4, H and I). FCM analysis also revealed that TSC-G increased TH1 cells (CD45+ CD3+CD4+T-Bet+), CTLs (CD45+CD3+CD8+), and effector memory CD8 T cells (CD45+CD3+CD8+CD62L-CD44+) (Fig. 4J). Further, we characterized memory T cells in the spleen 30 days after treatment (fig. S5D). The TSC-G therapy significantly induced effector memory CD8 T cells (CD45+CD3+CD8+CD62L−CD44+) and central memory CD8 T cells (CD45+CD3+CD8+CD62L+CD44+) when compared with SC-RmC treatment (Fig. 4, K and L, P < 0.05). Immunotherapies such as ICIs have been associated with severe adverse effects in several major organs, such as the lung and pancreas (40). No body weight loss was noted, and no detectable toxicity was observed in major organs after TSC-G therapy (fig. S5, E and F). These results indicated that our TSC-G therapy drives efficacy through the activation of systemic antitumor T cell immunity.
TSC-G/P therapy has therapeutic efficacy in immunosuppressive LM
LM is one of the severe disease types in brain metastasis (BM) (41), and patients with LM have a very poor prognosis due to lack of therapeutic options (42). To address the immunosuppressive nature of LM that we created with UV2-GFl cells, we further transduced SCN1KO-G cells with an scFv against PD-1 (Fig. 5A). FCM showed programmed cell death-1 ligand 1 (PD-L1) expression on PTEN-deficient melanoma cell lines (Y1.1, Y2.1, and UV2) (fig. S6A). Western blot assay confirmed that SCN1KO-GM-CSF/scFvPD-1 (SCN1KO-G/P) cells expressed both scFvPD-1 and GM-CSF (Fig. 5B). FCM showed that scFvPD-1 blocked anti–PD-1 Ab binding to PD-1 on splenocytes, confirming the function of scFvPD-1 (fig. S6B). In vivo, we confirmed that intrathecally (IT; delivered through cisterna magna) injected SCN1KO-G/P cells survived in the CSF space for 7 days and were cleared out afterward (Fig. 5C). The utility of SCs as carriers for oHSV in LM is unknown. To determine the influence of loading oHSV in SCs on the persistence of oHSV in vivo, SC-oHSV-FmC or oHSV-FmC was IT-injected 5 days after inoculation in the UV2-GFP-Rluc–bearing LM mouse model. IT injection of SC-oHSV-FmC resulted in longer oHSV persistence than IT injection with oHSV-FmC, peaking around day 12 in mice bearing LM tumors (Fig. 5D). To explore the efficacy of SC delivery of oHSV, GM-CSF, and scFvPD-1 for LM, we tested IT injection of TSC-G/P (TSCs releasing oHSV, GM-CSF, and scFvPD-1) in the mouse model of LM (Fig. 5E). A significant suppression of tumor growth was observed when tumors were treated with TSC-G/P or TSC-G compared with control SC-RmC (Fig. 5F, P < 0.05), which also translated into a significantly prolonged overall survival (Fig. 5G, P < 0.001). On the other hand, treatment with SC-oHSV with and without SCN1KO-scFvPD-1 did not reduce tumor growth indicated by luciferase activity or prolong overall survival compared with SC-RmC alone in this LM mouse model (fig. S6, C and D). The therapeutic efficacy of TSC-G/P or TSC-G was diminished in nonobese diabetic severe combined immunodeficient (NOD/SCID) mice (Fig. 5H and fig. S6E), suggesting the critical role of the immune system in the efficacy of TSC-G/P or TSC-G therapies. To further investigate the mechanism underlying these therapies, FCM immune profiling was performed 7 days after treatment (Fig. 5I and fig. S6F). Both TSC-G/P and TSC-G therapies significantly increased CD45+ cells compared with control SC-RmC (Fig. 5I; TSC-G/P, P < 0.01; TSC-G, P < 0.05). In addition, TSC-G/P therapy increased DCs (CD45+ CD11b+CD11c+; P < 0.05), mature DCs (CD45+CD11b+CD11c+ MHC II I-A/I-E+; P < 0.05), CD3+ T cells (P < 0.01), CD4+ T cells (P < 0.05), and CD8+ (P < 0.05) T cells compared with SC-RmC alone (Fig. 5I). We also performed RNA sequencing (RNA-seq) analysis on tumors to study the immune profile and understand the mechanism of action of GM-CSF. The TSC-G/P or TSC-G therapies up-regulated expression of immune cell–associated genes, including T cells (cytotoxic, helper, TH1, and TH2), macrophages, and DCs (Fig. 5J). The TSC-G/P–treated group had higher ICD-related gene expression than the control group, suggesting TSC-G/P–induced ICD (Fig. 5K). Further, Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed that treatment with TSC-G/P or TSC-G activated necroptosis signaling, apoptosis signaling, and cytokine-cytokine receptor interaction signaling (table S1). Gene Ontology (GO) analysis also indicated activation of the immune system after IT injection with TSC-G/P or TSC-G (fig. S6G). GO and KEGG analysis also revealed that these therapies down-regulated Janus kinase–signal transducer and activator of transcription (JAK-STAT) and PI3K-AKT pathways (fig. S6, H and I). Further, the IT injection with TSC-G/P also suppressed tumor growth and improved overall survival in Y2.1-GFl–bearing LM mouse model (fig. S6, J and K). Last, we also confirmed that TSC-G and TSC-G/P treatment did not induce any toxicity measured by unremarkable histology in major organs and the central nervous system (CNS) and lack of body weight loss (fig. S7, A to C). These results revealed that IT injection with TSC-G/P or TSC-G induced anti-LM activity through the activation of the antitumor immune system, including DCs and T cells, and down-regulated JAK-STAT and PI3K-Akt pathways in immunosuppressive PTEN-deficient LM mouse model.
Fig. 5. SCs secreting dual immunomodulators with SC-oHSV (TSC) have therapeutic efficacy in immunosuppressive LM mouse models.
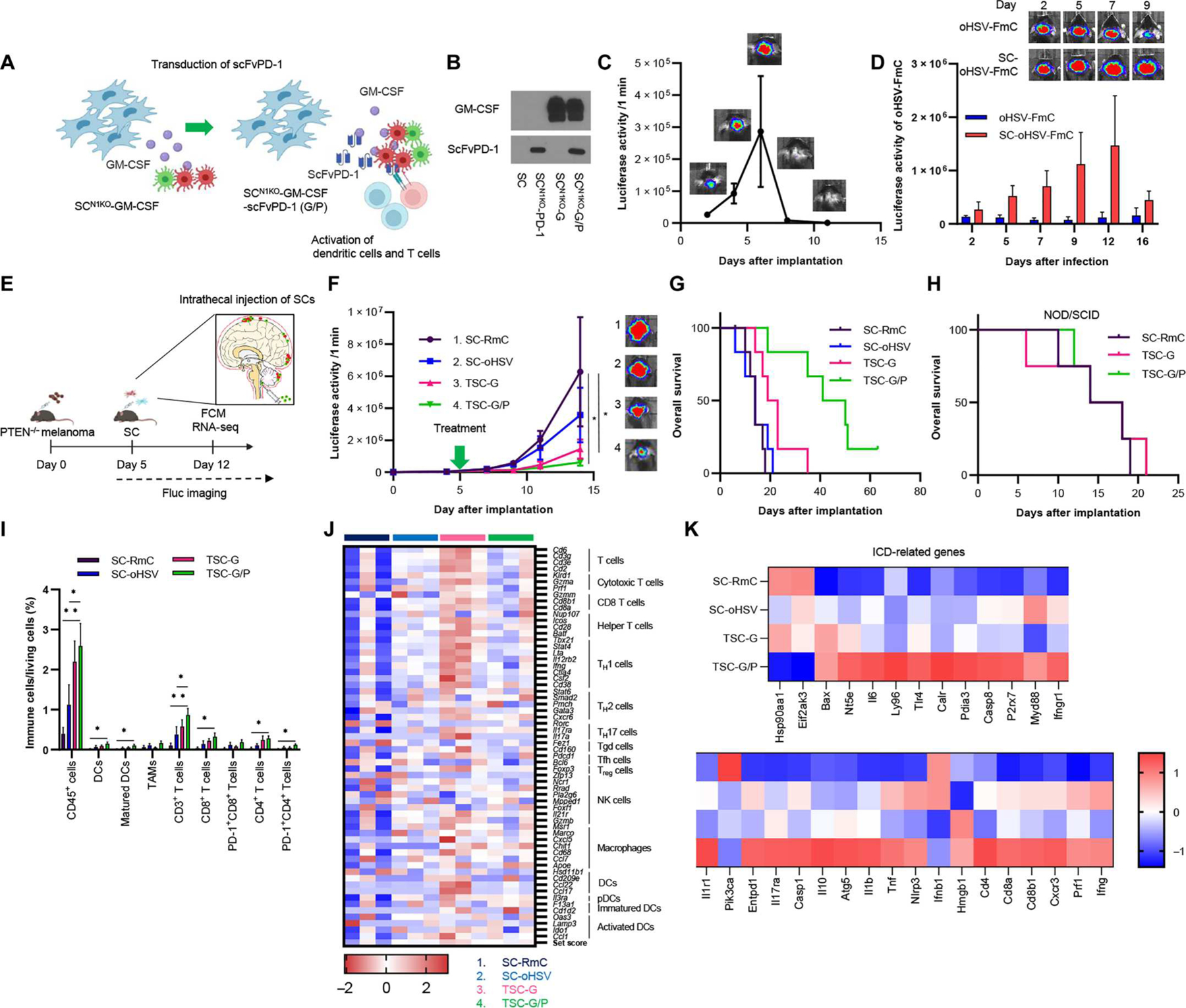
(A) Scheme showing creation of SCN1KO-GM-CSF/scFvPD-1 (G/P). SCN1KO-GM-CSF were transduced with scFvPD-1 for codelivery of GM-CSF and scFvPD-1 to immunosuppressive LM tumors. (B) Expression of GM-CSF and scFvPD-1 in supernatant from SCN1KO-G/P by Western blotting. (C) BLI signal and photographs of IT-injected SCN1KO-G/P-Fluc–bearing mice (n = 3). Data are represented as means ± SEM. (D) A representative bioluminescence plot (top) showing changes in Fluc activity as a measure of virally infected cells after IT injection of oHSV-Fluc (1 × 105 PFU) and SC-oHSV-Fluc (2 × 105 cells) (n = 4 per group) in a UV2-GFP-Rluc–bearing LM mouse model was quantified (bottom). Data are represented as means + SEM. (E) Experimental design. In brief, in the UV2-GFl LM mouse model, SCs were IT administrated one time 5 days after implantation of tumors. Tumor volumes were measured every 2 to 3 days by BLI. FCM and RNA sequencing (RNA-seq) analysis were performed day 12 after treatment. (F) Fluc signal curves (left) and representative BLI images (right) of mice bearing UV2-GFl tumors treated with SC-RmC (n = 6), SC-oHSV (n = 6), TSC-G, or TSC-G/P (n = 7). Data are represented as means ± SEM. *P < 0.05. (G) Kaplan-Meier curves of overall survival of mice. (H) Kaplan-Meier curves of overall survival of UV2-GFI–bearing NOD/SCID mice after treatment with SC-RmC (n = 4), TSC-G (n = 4), or TSC-G/P (n = 4). P < 0.001. (I) Flow cytometric analysis of TILs collected from UV2-GFl–bearing LM tumor 7 days after treatment. Data are represented as means + SD (n = 4 to 7 per group). *P < 0.05 and **P < 0.01. (J) Heatmap of differential expression of genes associated with immune cell types after treatment in LM mouse tumors by RNA-seq. (n = 3 per group). (K) Heatmap of differential expression of genes associated with ICD after treatment in an LM mouse model.
Human allogeneic stem cells releasing oHSV, GM-CSF, and scFvPD-1 have therapeutic efficacy in patient-derived PTEN-deficient MBM
To explore the human immune system in preclinical studies of OV therapy, humanized mouse models have been tested (43), with most studies using flank tumor humanized mouse models (44, 45). Therefore, we first implanted patient-derived PTEN-deficient brain metastatic melanoma M12 cells expressing GFP-Fluc (M12-GFl) intracranially in NOD-SCID mice and confirmed that M12-GFl cells grew well in the brain (fig. S8A). Next, to establish the M12-GFl–bearing humanized mouse model, we implanted M12-GFl cells intracranially in bone marrow–liver–thymic (BLT) humanized mice (Fig. 6A) and confirmed that M12-GFl cells grew well in the brain of this mouse model, too (Fig. 6B, left). IF of tumor-infiltrating immune cells indicated the presence of human immune cells such as CD11c- and CD3-positive cells (Fig. 6C). Immune profiling of melanoma brain tumors and splenocytes, mandibular and cervical lymph nodes, and bone marrow cells showed the presence of human DCs and T cells (CD4+ and CD8+ cells) (Fig. 6D, and fig. S8, B and C).
Fig. 6. Allogeneic SCs releasing human GM-CSF/scFvPD-1-TK and SC-oHSV (hTSC-G/P-TK) have therapeutic efficacy in humanized patient-derived PTEN-deficient MBM mice.
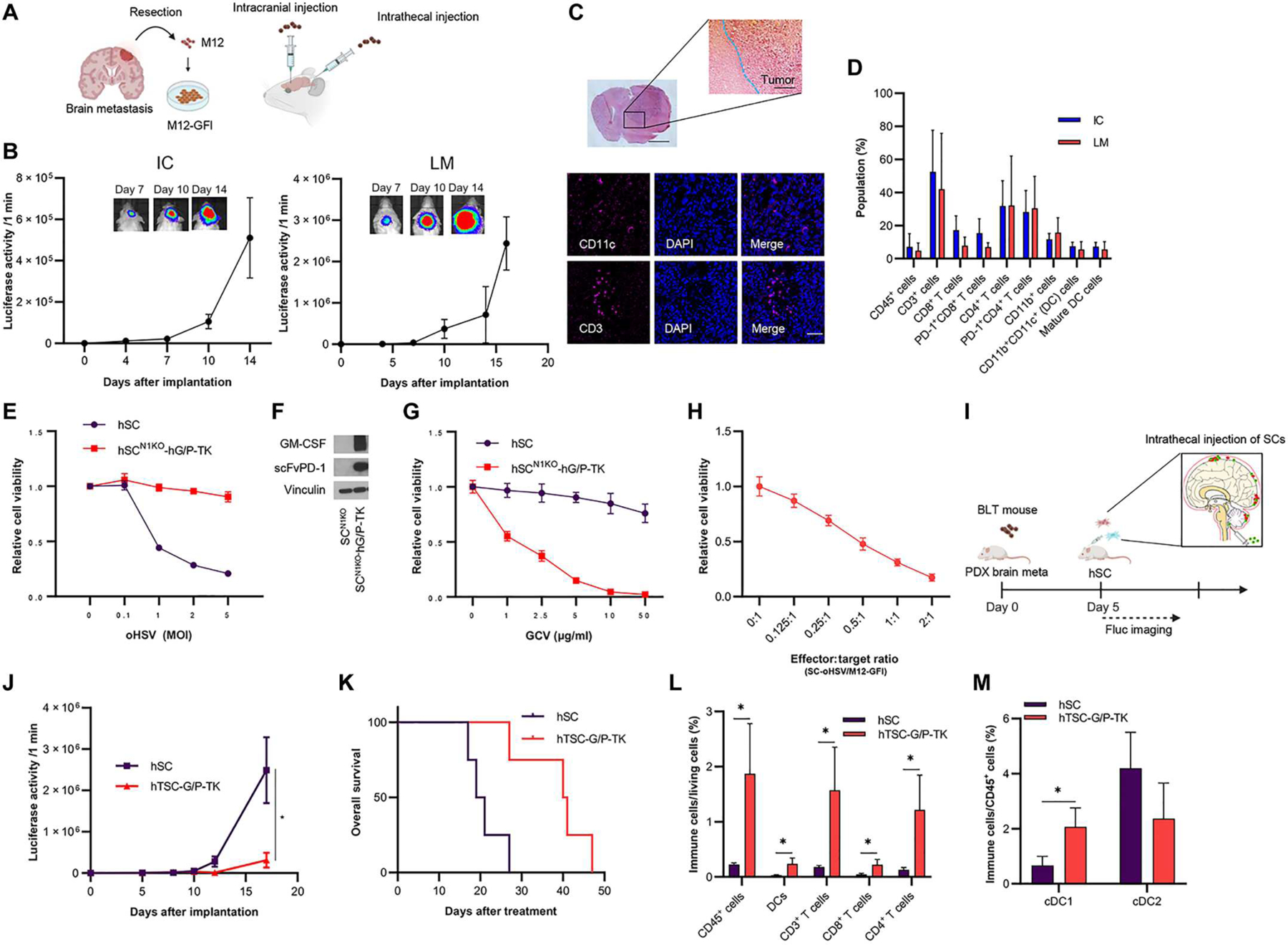
(A) Scheme showing intracranial injection (IC) and intrathecal injection as LM with patient-derived MBM (M12-GFl) cells. M12 cells were isolated from a patient with melanoma metastasized to the brain, expanded in culture, and engineered to introduce an in vivo imaging marker, GFP-Luciferase (GFI). (B) BLI signal curve of intracranially (let) or intrathecally (right) injected M12-GFl–bearing BLT humanized mice (n = 3 or 4 per each group). Data are represented as means ± SEM. (C) Representative H&E staining of the brain M12-GFl mouse model (top). IF analysis of CD11c and CD3 in the brain M12-GFl tumor BLT humanized model (bottom). Scale bar, 6 μm. (D) Flow cytometric (FCM) analysis of immune profiling of the brain M12-GFI mouse tumor model (IC) and the LM mouse tumor model (LM) (n = 4 per group). Data are represented as means + SD. (E) Cell viability assays showing hSCN1KO-hG/P-TK resistance to oHSV compared with human SCs (hSC) in vitro (n = 5 per group). Data are represented as means ± SD. (F) Expression of human GM-CSF and scFvPD-1 in supernatant from hSCN1KO-G/P-TK by Western blotting. (G) Cell viability assay of SCN1KO-hG/P-TK in the absence or presence of ganciclovir (GCV) for 2 days (n = 5 per group, technical replicates). Data are represented as means ± SD. (H) M12-GFI cells were investigated by cell viability assay 3 days after hSC-oHSV treatment in vitro (n = 5 per group). Data are represented as means ± SD. (I) Experimental design. In brief, in the M12-GFl LM mouse model, SCs were intrathecally administrated one time 5 days after implantation of M12-GFl cells. Tumor volumes were measured every 2 to 4 days by BLI. (J) Fluc signal curves and representative BLI images of BLT humanized mice bearing M12-GFl tumors treated with hSCs (n = 4) or hTSC-G/P-TK (n = 4). Data are represented as means ± SEM. *P < 0.05. (K) Kaplan-Meier curves of overall survival of mice. *P < 0.05. (L) FCM analysis of immune cells collected from M12-GFl LM tumor 10 days after treatment (n = 3 per group). *P < 0.05. (M) FCM analysis of conventional (c) DC1 and cDC2 collected from M12-GFl LM tumor 10 days after treatment (n = 3 per group). *P < 0.05.
In parallel, we also created an M12-GFl LM mouse model using BLT humanized mice (Fig. 6, A and B, right), in which the immune profiling analysis showed human DCs and T cells in LM tumors as well as in splenocytes, mandibular and cervical lymph nodes, and bone marrow cells (Fig. 6D, and fig. S8, B and C). Thus, we successfully established BM and LM melanoma humanized mouse models that allowed us to investigate the interaction between the human immune system and human melanoma cells during immunotherapy.
To advance the clinical translation of our murine studies, we also created human allogeneic mesenchymal stem cells (hSCs) by CRISPR-Cas9 (hSCN1KO) (fig. S8D). The hSCN1KO were resistant to oHSV compared with hSCs (fig. S8E). Next, we created hSCN1KO secreting human GM-CSF and scFvPD-1 (hSCN1KO-hG/P) and confirmed that these cells were also resistant to oHSV when compared with hSCs (Fig. 6E). Western blot assay showed that hSCN1KO-hG/P cells expressed both scFvPD-1 and GM-CSF (Fig. 6F). We also incorporated an HSV thymidine kinase (HSV-TK) gene into hSCN1KO-hG/P as a safety switch and confirmed that ganciclovir killed hSCsN1KO-hG/P-TK in a dose-dependent manner in vitro (Fig. 6G). On the other hand, hSC-oHSV (2 MOI) was able to kill cocultured M12-GFl cells in vitro (Fig. 6H). Then, we tested IT injection of hSC-oHSV and hSCN1KO-hG/P-TK (hTSC-G/P-TK) to test the efficacy of SC delivery of oHSV, GM-CSF, and scFvPD-1 for LM (Fig. 6I). IT injection of hTSC-G/P-TK significantly suppressed tumor growth and resulted in longer overall survival (Fig. 6, J and K, P < 0.05). Last, immune profiling analysis of the tumors showed that hTSC-G/P-TK therapy significantly increased CD45+ cells, T cells (CD4+CD3+, and CD8+CD3+ cells), and DCs (CD11c+ cells) compared with control hSCs (Fig. 6L, and fig. S8, F to H; P < 0.05). hTSC-G/P-TK therapy significantly induced an increased ratio of conventional DC1 (cDC1) to total CD45+ cells (Fig. 6M, P < 0.05), indicating that hGM-CSF and oHSV successfully activated human DCs. In addition, we confirmed that the hTSC-G/P-TK therapy did not cause body weight loss (fig. S8I). These results showed that IT injection of hTSC-G/P-TK is a safe and effective approach that activated DCs and T cells in a humanized patient-derived melanoma LM mouse model (fig. S9).
DISCUSSION
In this study, we created and characterized immunosuppressive syngeneic LM mouse models and explored SC-mediated delivery of oHSV in combination with immunomodulators to treat primary and metastatic melanomas. Using CRISPR-Cas-9 technology, we created oHSV-resistant nectin-1 receptor knockout SCs (SCN1KO) and showed that SCN1KO can be efficiently used to codeliver immunomodulators with SC-oHSV. We showed that SCN1KO-G augmented the therapeutic efficacy of SC-oHSV in orthotopic mouse models of primary and brain metastatic PTEN-deficient melanomas in vivo through activation of the immune system. Furthermore, SCN1KO releasing both GM-SCF and scFvPD-1 effectively boosted SC-oHSV immunotherapy for immunosuppressive PTEN-deficient BM with both human and mouse immune systems.
PTEN-deficient melanoma is associated with developing BM and poor overall survival in patients with melanoma (46). Activation of the PI3K pathway through loss of PTEN results in resistance to ICIs (47). We examined the correlation between PTEN expression and BM using TCGA and clinical samples. In accordance with previous reports (22, 48, 49), our analysis showed that patients with BM had lower PTEN expression than patients without BM, suggesting further investigation of PTEN status as a potential biomarker that predicts the development of BM. Moreover, we found that activation of the PI3K/AKT pathway in patients with low PTEN expression was correlated with an immunosuppressive phenotype in melanoma. Some clinical studies showed PTEN-deficient melanoma to be resistant to immunotherapy (47). Therefore, developing strategies for PTEN-deficient MBM is crucial.
BM still has high mortality, although advances in chemotherapy, targeted therapies, and immunotherapies have improved survival (50). Further, melanoma LM has the poorest prognosis in BM despite the use of BRAF inhibitors or ICIs (42). The lack of understanding of the tumor microenvironment of LM has limited the development of therapies. However, there are no reports of the establishment of syngeneic LM mouse models. In a previous study, RNA-seq analysis of CSF from patients with LM identified the activation of the PI3K/AKT pathway (51). Here, we created and characterized LM and primary melanoma models using PTEN-deficient melanoma cells and reported immune profiles in mouse melanoma LM models. Our immune profiling of UV2-based models revealed major differences in the tumor microenvironment because the LM model was immunosuppressive compared with the flank model, suggesting that the immunosuppressive LM model might be resistant to immunotherapy, as was shown in patients (47). This LM mouse model recapitulates the LM disease in clinical settings, offers a platform to test existing and new therapeutic agents, and serves as a bridge for rapid translation to the clinic.
T-VEC, an oHSV expressing GM-CSF, was approved by the U.S. FDA for the local treatment of unresectable melanoma and nodule lesions. GM-CSF is recognized as an inflammatory cytokine, which modulates DC differentiation and macrophage activation (52). Although T-VEC mediated oncolytic cell death and enhanced antitumor immune responses (53), it failed to show an improvement in the overall survival of patients with brain, liver, and lung metastasis (18). To overcome the limitation of T-VEC, we created oHSV-resistant SCs by knocking out nectin-1 receptor using CRISPR-Cas9 and engineered them to secrete immunomodulators to further enhance the activation of antitumor immunity. Our findings revealed that SCN1KO-G boosted oHSV efficacy by releasing higher concentrations of GM-CSF immediately after implantation compared with oHSV-GM-CSF or SC-oHSV-GM-CSF. Early robust expression of GM-CSF from SCN1KO-G compared with oHSV-GM-CSF or SC-oHSV-GM-CSF might underlie the enhanced T cell activation and therapeutic efficacy of SCN1KO-G–facilitated SC-oHSV therapy against primary melanoma and skin metastasis.
Recently, a phase 2 clinical trial of pembrolizumab in patients with LM (NCT02886585) (54) revealed safety and limited neurological toxicity. Pembrolizumab increased the abundance of CD8+ T cells in the CSF compared with pretreatment in a small fraction of patients (55). In addition, a phase 2 study of ipilimumab and nivolumab (NCT02939300) revealed an acceptable safety profile and promising efficacy in patients with LM (56). However, clinical benefits, such as longer survival, were limited to patients with LM derived from breast cancer. We previously established LM mouse models using patient-derived breast cancer cells and showed that SC delivery of targeted therapeutics is a promising approach for LM (57). In this study, we engineered SCN1KO secreting GM-CSF and scFvPD-1 to treat immunosuppressive PTEN-deficient melanoma LM mouse models and investigated the immune profile and therapeutic efficacy after IT injection of TSC-G/P. Our findings indicate that the IT injection of TSC-G/P prolonged overall survival through the activation of cytokine-cytokine receptor interaction signaling and expression of immune cell–associated genes, including T cells (cytotoxic, helper, TH1, and TH2), macrophages, and DCs. To the best of our knowledge, our findings are the first to develop IT injection of SC-based OVs and immune profile post-treatment LM tumors. Moreover, our data suggested a role for immunomodulation by TSC-G/P or TSC-G in contributing to the modulation of oncologic pathways and anti-melanoma activities such as JAK-STAT and PI3K-AKT. Further research is needed to understand the mechanism of down-regulation of these pathways.
The presence of an adaptive immune system in syngeneic mice provides an excellent platform for studies of treatments modulating antitumor immune responses. However, the lack of a wide range of tumor models carrying clinically relevant oncogenic molecular alterations limits a thorough investigation of therapeutics in such models. Furthermore, the discrepancy in immune system activation and responses in human and animals might preclude preclinical studies from accurately predicting clinical trial results. To overcome this gap, humanized mouse models represent a next-generation preclinical oncology platform that enables us to study how human immune cells respond to human tumors in vivo (58, 59). Surgical transplantation of human fetal liver and thymus tissue fragments into immunodeficient mice generates a BLT humanized mouse model that portrays a more robust and closer representation of the human immune system and response. Recent studies used humanized xenografted mice to demonstrate the infiltration of human T, B, and natural killer cells within the tumor after the treatment with oncolytic vaccinia virus (44). Most previous reports on humanized mouse model used flank tumors to study immunotherapy for cancers (45, 60). In this study, we first confirmed that patient-derived melanoma cells could grow in the brain of a BLT model. Tumor infiltration of DCs and T cells was observed in our melanoma brain tumor humanized model. Furthermore, we successfully established a patient-derived PTEN-deficient melanoma LM BLT mouse model and performed immune profiling analysis to show human DCs and T cells in LM tumors and in the spleen, mandibular and cervical lymph nodes, and bone marrow. This is the first report to date that describes an approach to establish BM and LM BLT mouse models. To translate our findings from syngeneic mice to clinical settings, we created oHSV-resistant human SCs and transduced them with human GM-CSF and scFvPD-1. In addition, SCN1KO-human GM-CSF/scFvPD-1 was armed with HSV-TK, which has shown promise as a safety switch in patients with cell-based therapies (61). Our findings reveal that hTSC-G/P therapy increased overall survival in our patient-derived PTEN-deficient melanoma LM BLT mouse model, suggesting that IT injection of two populations of allogeneic SCs has a therapeutic benefit in the context of a human immune system.
Allogeneic mesenchymal SCs can be easily obtained and banked and, following the good manufacturing practice (GMP) standards (62), offer “off-the-shelf” cellular therapy options for tumors. Our previous study with allogeneic human GMP grade SCs was performed to support an Investigational New Drug application, presented to the FDA to start a first-in-human (FIH) study (63). This FIH study of engineered SCs will assess safety and tolerability in patients with primary and recurrent glioblastoma after surgical resection. More recently, SC delivery of OVs was confirmed to be feasible and safe in clinical trials in patients with brain tumors (NCT03072134) (64). Furthermore, there are several ongoing phase 1/2 clinical trials testing OV-loaded SCs for different cancer types (NCT02068794, NCT01844661, and NCT03896568). To improve the therapeutic efficacy for distant metastasis, our findings reveal that SCs are rationale carriers of oHSV because they protect oHSV from the humoral immune system. IT injection for patients with LM is feasible because the IT technique is common and clinically used for pain control after surgery (65). This is supported by the previous studies in which IT-delivered chemotherapy was tested in patients with LM and CNS lymphoma (66, 67). Our results support that allogeneic SC–based OV immunotherapy is widely applicable to varying forms of clinical metastases, including LM.
Our study has some limitations. Although we show that TSC-G/P therapy induced antitumor immunity through activation of DCs and T cells in both mouse and human PTEN–deficient LM mouse models, the molecular mechanism behind this activity has not been fully elucidated. We speculate that treatment-induced down-regulation of the JAK-STAT and PI3K-AKT pathways could play a role in stimulating tumor-specific T cell immune responses, but this will need to be validated. The current work focused on PTEN-deficient melanomas, and characterizing the therapeutic potential of TSC-G/P therapy for MBM tumors that are driven by other oncogenes such as KRAS and BRAF will be necessary. In addition, the therapeutic efficacy of the TSC-G/P platform should be explored for other metastatic lesions and other cancer types to broaden its the therapeutic applications. However, we anticipate that optimization of immunomodulators and delivery routes will be required for different disease conditions.
In summary, we have established human and murine immunocompetent leptomeningeal preclinical platforms to test therapeutic agents and serve as a bridge for rapid translation of new therapies. Using these platforms, our study reveals that locoregional delivery of oHSV and immunomodulators from two SC populations successfully induces activation of DCs and CD4+ and CD8+ cells in the human and the mouse immune system. These results support translation of our TSC-G/P strategy to a phase 1 clinical trial for patients with LM to confirm the safety of IT injection of these therapeutic SCs and to ultimately help improve overall survival and quality of life. Toward this end, we are performing preclinical toxicological assessments testing TSCs created under GMP conditions.
MATERIALS AND METHODS
Study design
The purpose of this study was to develop a translational therapeutic strategy that can simultaneously induce a direct killing of tumor cells and induce antitumor immune responses to combat the immunosuppressive microenvironment. First, we developed and characterized preclinical immune-component LM mouse models, mimicking the clinical settings. Next, we hypothesized that genetically engineered SCs can be used as cellular vehicles for treating local and disseminated cancers. All experiments performed in this study have been replicated to demonstrate biological reproducibility and to ensure adequate statistical methods for comparisons. All animals were randomly assigned to each group with comparable tumor sizes. The study was not blinded, and no statistical methods were used to predetermine the sample size. The methods for in vivo experiments and statistical tests are described below in Materials and Methods and in the figure legends.
Cell lines and cell cultures
YUMM1.1 (Y1.1), YUMM2.1 (Y2.1), YUMM3.3 (Y3.3), D3UV2 (UV2), D3UV3 (UV3), and B16F10 (B16), murine melanoma cells and M12 patient–derived MBM lines (provided by J. Sarkaria, Mayo Clinic, Rochester) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin. Mouse adipose tissue–derived mesenchymal SCs were cultured in low-glucose DMEM supplemented with 15% (v/v) FBS, 1% (v/v) L-glutamine, 1% (v/v) non-essential amino acid solution, and 1% (vol/vol) penicillin-streptomycin. Human adipose tissue–derived mesenchymal SCs were grown in DMEM/F-12 supplemented with 10% (v/v) FBS, 1% (v/v) L-glutamine, 1% (v/v) penicillin-streptomycin, and recombinant human fibroblast growth factor (40 ng/ml; R & D Systems). Neither cell line was cultured for more than 2 months after resuscitation. Cell authentication was not performed by the authors.
Lentiviral transduction and engineering of stable cell lines
Lentiviral packaging (LV-mouse GM-CSF-GFP, LV-scFvPD-1-GFP, and LV-human GM-CSF/scFvPD-1-mCherry) was performed by transfection of 293T cells, and cells were transduced with lentiviral vectors in medium containing protamine sulfate (2 μg/ml). For bioluminescence imaging (BLI), cells were transduced with LV-Pico2-GFP-Fluc or LV-GFP-Fluc. They are selected by fluorescence-activated cell sorting using a BD FACS Aria Fusion cell sorter or by puromycin selection in culture. GFP expression was visualized by fluorescence microscopy.
CRISPR knockout of nectin-1
To establish human and mouse nectin-1 knockout lines using CRISPR-Cas9, SCs were transduced with lentiviral Cas9 expression vectors coding for constitutively expressed Cas9 protein and lentiviral single-guide RNA (sgRNA) expression vector pLKO.DEST.hygro containing the sgRNA target sequences described above for nectin-1. For targeting of mouse nectin-1, CCTTCATCGACGGCACCATCCGCCTCTCCGGTC or CTGGAGGACGAGGGCATGTACATCTGTGA sgRNA was used. For human nectin-1 knockout, top and bottom strands of sgRNA oligos were aligned as previously described (68), followed by cloning strategy into a lentiCRISPRv2GFP plasmid (Addgene, plasmid 82416) using the restriction enzyme Bsm BI. All destination sgRNA expression vectors were sequenced to confirm correct U6-sgRNA inserts before proceeding with third-generation lentiviral packaging. For targeting of human nectin-1, TGGCTTCATCGGCACAGACG sgRNA was used. Both mouse and human SCN1KO were selected on hygromycin (50 to 200 μg/ml) and by GFP+ cell sorting using a BD FACS Aria Fusion cell sort in culture, respectively.
Oncolytic HSV
The G47Δ bacterial artificial chromosome (BAC) contains the genome of G47Δ (γ34.5-, ICP6-, and ICP47-). G47Δ-mouse GM-CSF (oHSV-GM-CSF) is also a BAC-based recombinant oHSV vector with the genomic backbone of G47Δ (γ34.5-, ICP6-, and ICP47-). Briefly, the respective shuttle plasmid carrying mouse GM-CSF was integrated into G47ΔBAC using Cre-mediated recombination in DH10B Escherichia coli and proper recombination confirmed by restriction analysis of BAC clones. Next, the resulting BAC and an Flpe-expressing plasmid were cotransfected into Vero cells to remove the BAC-derived sequences and the enhanced green fluorescent encoding sequences and allow virus to be produced. Recombinant virus oHSV-GM-CSF was plaque-purified and expanded. oHSV-GM-CSF express E. coli lacZ driven by endogenous ICP6 promoter and GM-CSF driven by the HSV immediate early 4/5 promoter. GM-CSF secretion from oHSV-GM-CSF–infected Vero cells was confirmed by enzyme-linked immunosorbent assay. MOI and PFUs were used as virus units in vitro and in vivo, respectively. Titers of infectious oHSV were determined by plaque assay on Vero cells (American Type Culture Collection).
In vivo mouse experiments
All in vivo experiments were approved by the Subcommittee on Research Animal Care at Brigham and Women’s Hospital. Mice that died or were euthanized for ethical reasons before defined experimental end points were excluded. Animals were randomly allocated to cages and experimental groups.
Bilateral primary melanoma model
Y1.1-GFl cells (2 × 106 cells per mouse) or UV2-GFl (1 × 106 cells per mouse) were subcutaneously implanted into the bilateral flanks of 6- to 8-week-old female C57BL/6 mice (Charles River Laboratories). Treatment was initiated 7 days after implantation. One side was treated with SC-RmC (4 × 105 cells), SC-oHSV (2 MOI, 6 hours) + SC-RmC (2 × 105 cells), oHSV-GM-CSF (2 × 105 PFU) + SC-RmC (4 × 105 cells), and combination therapy with SC-oHSV (2 × 105 cells) and SCN1KO-G (2 × 105 cells) intratumorally on days 7 and 11. The perpendicular diameter of each tumor was then measured every 3 to 5 days, and tumor volume was calculated using the following formula: tumor volume (mm3) = a × b2 × 0.5, where a represents the longest diameter, b represents the shortest diameter, and 0.5 is a constant used to calculate the volume of an ellipsoid. In the Y1.1-GFl melanoma mouse model, these mice were further rechallenged intracranially Y1.1-GFl cells (2 × 105 cells per mouse) on day 40, and the tumor growth was monitored by In Vivo Imaging System (IVIS).
LM model
To inject tumor cells intrathecally, female C57BL/6 or NOD/SCID mice (6 to 10 weeks of age) were immobilized on a surgical platform after anesthesia with ketamine-xylazine. Midline skin incision was made behind the neck, and occipital muscles were dissected. The dura mater between skull and atlas vertebra was exposed. Under the observation of cerebellum and brainstem through the dura mater, a catheter connected to a microsyringe (Hamilton) was inserted into the cisterna magna. UV2-GFl (5 × 104 cells per mouse) in 4 μl was injected slowly through the catheter. The hole in the dura mater was closed with a small muscle piece immediately after removing the catheter. On day 5, SC-RmC (4 × 105 cells per mouse), SC-oHSV (2MOI, 6 hours per mouse) + SC-RmC (2 × 105 cells per mouse), combination therapy with SC-oHSV (2 × 105 cells per mouse) + SCN1KO-G (2 × 105 cells per mouse), and combination therapy with SC-oHSV (2 × 105 cells per mouse) + SCN1KO-G/P (2 × 105 cells per mouse) were injected in a similar manner through the same opening from the previous injection. Then, the tumor growth was monitored by IVIS.
Patient-derived melanoma humanized BLT mouse model
BLT mice were generated as previously described (69). Briefly, NOD/SCID mice or NOD/SCID/c/mice at 6 to 8 weeks of age were conditioned with sublethal (2 Gy) whole-body irradiation. They were anesthetized the same day, and fragments of human fetal thymus and liver were implanted under the recipient kidney capsules bilaterally. Then, CD34+ cells were injected intravenously. After 8 weeks, the human immune cell engraftment was monitored by FCM by determining the percentages of human CD45+ cells in peripheral blood. Then, BLT mice with over 25% of human CD45/human and mouse CD45 ratio (mean, 60%) were used in this study. To establish a patient-derived melanoma humanized BLT mouse model, M12-GFl cells were intracranially implanted into the brain. Next, M12-GFl cells (5 × 104 cells per mouse) were intrathecally implanted as the patient-derived melanoma LM model. To explore the therapeutic efficacy of IT injection of hSCs, hSCs (4 × 105 cells per mouse) or hSC-oHSV (2 × 105 cells per mouse) and hSCN1KO-hG/P-TK (2 × 105 cells per mouse) were intrathecally injected on day 5. Then, the tumor growth was monitored by IVIS. Further, brain tumors, splenocytes, mandibular and cervical lymph nodes, and bone marrow cells were collected to ensure the immune profiling by FCM.
Bone marrow–derived DCs
Femurs and tibias were collected from C57BL/6 mice (6 to 10 weeks of age). Bone marrows were harvested by flashing DMEM (DMEM containing 10% FBS and 1% penicillin-streptomycin) using a 23G needle. Bone marrow cells were centrifuged, resuspended in the DMEM, and seeded in a six-well plate. They were incubated with SC-RmC or SCN1KO-G condition medium for 4 days. Then, the population of DCs (CD45+CD11b+CD11c+ cells) and mature DCs (CD45+CD11b+CD11c+MHC II+ cells) was determined by FCM.
CTL assay
Mice were sacrificed about 60 days after initiation of treatment (n = 4). Splenocytes, collected by homogenization of the spleen, were treated by red blood cell (RBC) lysis buffer (BioLegend). Then, splenocytes were coincubated with Y1.1-GFl, and TC-1-GFl cells (target) were incubated with splenocytes for 24 hours at 37°C at different effector:target ratios (0:1, 1:1, 2:1, 4:1, and 8:1). Cell viability assay was performed by measuring the in vitro Fluc bioluminescence.
Immune profile experiments
C57BL/6 mice or BLT mice were intrathecally implanted with UV2-GFl (5 × 104 cells per mouse) or M12-GFl (5 × 104 cells per mouse), respectively, and treated with intrathecal injection of SC-based therapy on day 5. On day 12 or 15, mice were euthanized, and tumors were collected. Tumor tissues or spleens were harvested from mice and mashed through a 100-μm strainer. For splenocytes, RBCs were lysed using mouse RBC lysis buffer (Boston BioProducts, IBB-198). Live/dead cell discrimination was performed using the Zombie UV Fixable Viability Kit (BioLegend). Cells were incubated with FcR blocking reagent (Miltenyi Biotec) or Human TruStain FcX (Fc receptor blocking solution) (BioLegend), followed by cell surface staining with fluorochrome-conjugated anti-mouse Abs or anti-human Abs. Stained cells were fixed with 4% paraformaldehyde before running them on BD Fortessa. FCS files were analyzed on FlowJo (version 10.3.1).
Statistical analysis
Statistical analysis was performed using JMP software (SAS Institute, Cary, NC, USA). Student’s t test was used to assess the significance of differences in most continuous variables. The log-rank test was used for Kaplan-Meier survival analyses. Differences were considered significant at *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Supplementary Material
Acknowledgments:
We thank M. Schroeder and J. Sarkaria (Myco clinic) for providing the patient-derived M12 tumor line. We thank D. Bhere for helpful suggestions with the generation of human SC. All illustrations have been created for this manuscript using BioRender with a license to use the illustrations (K.S.). We thank the NeuroTechnology Studio at Brigham and Women’s Hospital for providing the InCell analyzer and Mesoscope instrument access and consultation on data acquisition and data analysis.
Funding:
This study was supported by DOD-CA180698 (to K.S.), NIH 5P01CA163222-08 (to D.F.), and Overseas Research Fellowships from Uehara Memorial Foundation (to N.K.).
Footnotes
Competing interests: K.S. owns equity in and is a member of the Board of Directors of AMASA Therapeutics, a company developing stem cell–based therapies for cancer. D.F. has a financial interest in Soltego. K.S.’s and D.F.’s interests were reviewed by Brigham and Women’s Hospital and Mass General Brigham in accordance with their conflict-of-interest policies. The other authors declare that they have no competing interests. The patent application entitled “Gene editing and engineering stem cell for drug delivery” (PCT/US22/49523) has been filed.
Other Supplementary Material for this manuscript includes the following:
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. All the cell lines generated in this study are available under a material transfer agreement from the corresponding author, K.S.
REFERENCES AND NOTES
- 1.Chamberlain MC, Brain metastases: A medical neuro-oncology perspective. Expert Rev. Neurother 10, 563–573 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Zakrzewski J, Geraghty LN, Rose AE, Christos PJ, Mazumdar M, Polsky D, Shapiro R, Berman R, Darvishian F, Hernando E, Pavlick A, Osman I, Clinical variables and primary tumor characteristics predictive of the development of melanoma brain metastases and post-brain metastases survival. Cancer 117, 1711–1720 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bedikian AY, Wei C, Detry M, Kim KB, Papadopoulos NE, Hwu WJ, Homsi J, Davies M, McIntyre S, Hwu P, Predictive factors for the development of brain metastasis in advanced unresectable metastatic melanoma. Am. J. Clin. Oncol 34, 603–610 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Coit DG, Thompson JA, Albertini MR, Barker C, Carson WE, Contreras C, Daniels GA, DiMaio D, Fields RC, Fleming MD, Freeman M, Galan A, Gastman B, Guild V, Johnson D, Joseph RW, Lange JR, Nath S, Olszanski AJ, Ott P, Gupta AP, Ross MI, Salama AK, Skitzki J, Sosman J, Swetter SM, Tanabe KK, Wuthrick E, McMillian NR, Engh AM, Cutaneous melanoma, version 2.2019, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Cancer Netw 17, 367–402 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, Drake CG, Camacho LH, Kauh J, Odunsi K, Pitot HC, Hamid O, Bhatia S, Martins R, Eaton K, Chen S, Salay TM, Alaparthy S, Grosso JF, Korman AJ, Parker SM, Agrawal S, Goldberg SM, Pardoll DM, Gupta A, Wigginton JM, Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med 366, 2455–2465 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gampa G, Vaidhyanathan S, Sarkaria JN, Elmquist WF, Drug delivery to melanoma brain metastases: Can current challenges lead to new opportunities? Pharmacol. Res 123, 10–25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rochet NM, Dronca RS, Kottschade LA, Chavan RN, Gorman B, Gilbertson JR, Markovic SN, Melanoma brain metastases and vemurafenib: Need for further investigation. Mayo Clin. Proc 87, 976–981 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pluim D, Ros W, van Bussel MTJ, Brandsma D, Beijnen JH, Schellens JHM, Enzyme linked immunosorbent assay for the quantification of nivolumab and pembrolizumab in human serum and cerebrospinal fluid. J. Pharm. Biomed. Anal 164, 128–134 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Kim R, Keam B, Kim S, Kim M, Kim SH, Kim JW, Kim YJ, Kim TM, Jeon YK, Kim DW, Chung DH, Lee JS, Heo DS, Differences in tumor microenvironments between primary lung tumors and brain metastases in lung cancer patients: Therapeutic implications for immune checkpoint inhibitors. BMC Cancer 19, 19 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aghi M, Martuza RL, Oncolytic viral therapies—The clinical experience. Oncogene 24, 7802–7816 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Selznick LA, Shamji MF, Fecci P, Gromeier M, Friedman AH, Sampson J, Molecular strategies for the treatment of malignant glioma—Genes, viruses, and vaccines. Neurosurg. Rev 31, 141–155 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buijs PR, Verhagen JH, van Eijck CH, van den Hoogen BG, Oncolytic viruses: From bench to bedside with a focus on safety. Hum. Vaccin. Immunother 11, 1573–1584 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu TC, Galanis E, Kirn D, Clinical trial results with oncolytic virotherapy: A century of promise, a decade of progress. Nat. Clin. Pract. Oncol 4, 101–117 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, Nabors LB, Markiewicz M, Lakeman AD, Palmer CA, Parker JN, Whitley RJ, Gillespie GY, Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol. Ther 17, 199–207 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Markert JM, Razdan SN, Kuo HC, Cantor A, Knoll A, Karrasch M, Nabors LB, Markiewicz M, Agee BS, Coleman JM, Lakeman AD, Palmer CA, Parker JN, Whitley RJ, Weichselbaum RR, Fiveash JB, Gillespie GY, A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther 22, 1048–1055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pecora AL, Rizvi N, Cohen GI, Meropol NJ, Sterman D, Marshall JL, Goldberg S, Gross P, O’Neil JD, Groene WS, Roberts MS, Rabin H, Bamat MK, Lorence RM, Phase I trial of intravenous administration of PV701, an oncolytic virus, in patients with advanced solid cancers. J. Clin. Oncol 20, 2251–2266 (2002). [DOI] [PubMed] [Google Scholar]
- 17.Harrington KJ, Puzanov I, Hecht JR, Hodi FS, Szabo Z, Murugappan S, Kaufman HL, Clinical development of talimogene laherparepvec (T-VEC): A modified herpes simplex virus type-1-derived oncolytic immunotherapy. Expert Rev. Anticancer Ther 15, 1389–1403 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Andtbacka RH, Kaufman HL, Collichio F, Amatruda T, Senzer N, Chesney J, Delman KA, Spitler LE, Puzanov I, Agarwala SS, Milhem M, Cranmer L, Curti B, Lewis K, Ross M, Guthrie T, Linette GP, Daniels GA, Harrington K, Middleton MR, Miller WH Jr., Zager JS, Ye Y, Yao B, Li A, Doleman S, VanderWalde A, Gansert J, Coffin RS, Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 33, 2780–2788 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Russell SJ, Peng KW, Bell JC, Oncolytic virotherapy. Nat. Biotechnol 30, 658–670 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du W, Seah I, Bougazzoul O, Choi G, Meeth K, Bosenberg MW, Wakimoto H, Fisher D, Shah K, Stem cell-released oncolytic herpes simplex virus has therapeutic efficacy in brain metastatic melanomas. Proc. Natl. Acad. Sci. U.S.A 114, E6157–E6165 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischer GM, Jalali A, Kircher DA, Lee WC, McQuade JL, Haydu LE, Joon AY, Reuben A, de Macedo MP, Carapeto FCL, Yang C, Srivastava A, Ambati CR, Sreekumar A, Hudgens CW, Knighton B, Deng W, Ferguson SD, Tawbi HA, Glitza IC, Gershenwald JE, Vashisht Gopal YN, Hwu P, Huse JT, Wargo JA, Futreal PA, Putluri N, Lazar AJ, DeBerardinis RJ, Marszalek JR, Zhang J, Holmen SL, Tetzlaff MT, Davies MA, Molecular profiling reveals unique immune and metabolic features of melanoma brain metastases. Cancer Discov 9, 628–645 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bucheit AD, Chen G, Siroy A, Tetzlaff M, Broaddus R, Milton D, Fox P, Bassett R, Hwu P, Gershenwald JE, Lazar AJ, Davies MA, Complete loss of PTEN protein expression correlates with shorter time to brain metastasis and survival in stage IIIB/C melanoma patients with BRAFV600 mutations. Clin. Cancer Res 20, 5527–5536 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koski A, Kangasniemi L, Escutenaire S, Pesonen S, Cerullo V, Diaconu I, Nokisalmi P, Raki M, Rajecki M, Guse K, Ranki T, Oksanen M, Holm SL, Haavisto E, Karioja-Kallio A, Laasonen L, Partanen K, Ugolini M, Helminen A, Karli E, Hannuksela P, Pesonen S, Joensuu T, Kanerva A, Hemminki A, Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol. Ther 18, 1874–1884 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Parviainen S, Ahonen M, Diaconu I, Kipar A, Siurala M, Vaha-Koskela M, Kanerva A, Cerullo V, Hemminki A, GMCSF-armed vaccinia virus induces an antitumor immune response. Int. J. Cancer 136, 1065–1072 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Lawler SE, Speranza MC, Cho CF, Chiocca EA, Oncolytic viruses in cancer treatment: A review. JAMA Oncol 3, 841–849 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, Kirkwood JM, Gajewski TF, Chen L, Gorski KS, Anderson AA, Diede SJ, Lassman ME, Gansert J, Hodi FS, Long GV, Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 170, 1109–1119.e10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wheeler SS, Boyce WM, Reisen WK, Field methods and sample collection techniques for the surveillance of west nile virus in avian hosts. Methods Mol. Biol 1435, 207–220 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Hu JC, Coffin RS, Davis CJ, Graham NJ, Groves N, Guest PJ, Harrington KJ, James ND, Love CA, McNeish I, Medley LC, Michael A, Nutting CM, Pandha HS, Shorrock CA, Simpson J, Steiner J, Steven NM, Wright D, Coombes RC, A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res 12, 6737–6747 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Malhotra S, Kim T, Zager J, Bennett J, Ebright M, D’Angelica M, Fong Y, Use of an oncolytic virus secreting GM-CSF as combined oncolytic and immunotherapy for treatment of colorectal and hepatic adenocarcinomas. Surgery 141, 520–529 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duebgen M, Martinez-Quintanilla J, Tamura K, Hingtgen S, Redjal N, Wakimoto H, Shah K, Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J. Natl. Cancer Inst 106, dju090 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Krummenacher C, Baribaud F, Ponce de Leon M, Baribaud I, Whitbeck JC, Xu R, Cohen GH, Eisenberg RJ, Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology 322, 286–299 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Hoxhaj G, Manning BD, The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 20, 74–88 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu D, Schilling B, Liu D, Sucker A, Livingstone E, Jerby-Arnon L, Zimmer L, Gutzmer R, Satzger I, Loquai C, Grabbe S, Vokes N, Margolis CA, Conway J, He MX, Elmarakeby H, Dietlein F, Miao D, Tracy A, Gogas H, Goldinger SM, Utikal J, Blank CU, Rauschenberg R, von Bubnoff D, Krackhardt A, Weide B, Haferkamp S, Kiecker F, Izar B, Garraway L, Regev A, Flaherty K, Paschen A, Van Allen EM, Schadendorf D, Integrative molecular and clinical modeling of clinical outcomes to PD1 blockade in patients with metastatic melanoma. Nat. Med 25, 1916–1927 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meeth K, Wang JX, Micevic G, Damsky W, Bosenberg MW, The YUMM lines: A series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res 29, 590–597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, Mullins DW, Brinckerhoff CE, Multiple murine BRaf(V600E) melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res 27, 495–501 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Homet Moreno B, Zaretsky JM, Garcia-Diaz A, Tsoi J, Parisi G, Robert L, Meeth K, Ndoye A, Bosenberg M, Weeraratna AT, Graeber TG, Comin-Anduix B, Hu-Lieskovan S, Ribas A, Response to programmed cell death-1 blockade in a murine melanoma syngeneic model requires costimulation, CD4, and CD8 T cells. Cancer Immunol. Res 4, 845–857 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Comerford I, McColl SR, Mini-review series: Focus on chemokines. Immunol. Cell Biol 89, 183–184 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Zlotnik A, Yoshie O, Chemokines: A new classification system and their role in immunity. Immunity 12, 121–127 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Totsch SK, Schlappi C, Kang KD, Ishizuka AS, Lynn GM, Fox B, Beierle EA, Whitley RJ, Markert JM, Gillespie GY, Bernstock JD, Friedman GK, Oncolytic herpes simplex virus immunotherapy for brain tumors: Current pitfalls and emerging strategies to overcome therapeutic resistance. Oncogene 38, 6159–6171 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Naidoo J, Page DB, Li BT, Connell LC, Schindler K, Lacouture ME, Postow MA, Wolchok JD, Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Ann. Oncol 26, 2375–2391 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taillibert S, Chamberlain MC, Leptomeningeal metastasis. Handb. Clin. Neurol 149, 169–204 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Smalley KS, Fedorenko IV, Kenchappa RS, Sahebjam S, Forsyth PA, Managing leptomeningeal melanoma metastases in the era of immune and targeted therapy. Int. J. Cancer 139, 1195–1201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morton JJ, Bird G, Refaeli Y, Jimeno A, Humanized mouse xenograft models: Narrowing the tumor-microenvironment gap. Cancer Res 76, 6153–6158 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsoneva D, Minev B, Frentzen A, Zhang Q, Wege AK, Szalay AA, Humanized mice with subcutaneous human solid tumors for immune response analysis of vaccinia virus-mediated oncolysis. Mol. Ther. Oncol 5, 41–61 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakao S, Arai Y, Tasaki M, Yamashita M, Murakami R, Kawase T, Amino N, Nakatake M, Kurosaki H, Mori M, Takeuchi M, Nakamura T, Intratumoral expression of IL-7 and IL-12 using an oncolytic virus increases systemic sensitivity to immune checkpoint blockade. Sci. Transl. Med 12, eaax7992 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Cabrita R, Mitra S, Sanna A, Ekedahl H, Lövgren K, Olsson H, Ingvar C, Isaksson K, Lauss M, Carneiro A, Jönsson G, The role of PTEN loss in immune escape, melanoma prognosis and therapy response. Cancers 12, 742 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, Xu C, McKenzie JA, Zhang C, Liang X, Williams LJ, Deng W, Chen G, Mbofung R, Lazar AJ, Torres-Cabala CA, Cooper ZA, Chen PL, Tieu TN, Spranger S, Yu X, Bernatchez C, Forget MA, Haymaker C, Amaria R, McQuade JL, Glitza IC, Cascone T, Li HS, Kwong LN, Heffernan TP, Hu J, Bassett RL Jr., Bosenberg MW, Woodman SE, Overwijk WW, Lizee G, Roszik J, Gajewski TF, Wargo JA, Gershenwald JE, Radvanyi L, Davies MA, Hwu P, Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discov 6, 202–216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wikman H, Lamszus K, Detels N, Uslar L, Wrage M, Benner C, Hohensee I, Ylstra B, Eylmann K, Zapatka M, Sauter G, Kemming D, Glatzel M, Müller V, Westphal M, Pantel K, Relevance of PTEN loss in brain metastasis formation in breast cancer patients. Breast Cancer Res 14, R49 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang L, Zhang S, Yao J, Lowery FJ, Zhang Q, Huang WC, Li P, Li M, Wang X, Zhang C, Wang H, Ellis K, Cheerathodi M, McCarty JH, Palmieri D, Saunus J, Lakhani S, Huang S, Sahin AA, Aldape KD, Steeg PS, Yu D, Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 527, 100–104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taillibert S, Le Rhun É, Epidemiology of brain metastases. Cancer Radiother 19, 3–9 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Smalley I, Law V, Wyatt C, Evernden B, Fang B, Koomen JM, Welsh EA, Macaulay RJB, Forsyth PA, Smalley KSM, Proteomic analysis of CSF from patients with leptomeningeal melanoma metastases identifies signatures associated with disease progression and therapeutic resistance. Clin. Cancer Res 26, 2163–2175 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becher B, Tugues S, Greter M, GM-CSF: From growth factor to central mediator of tissue inflammation. Immunity 45, 963–973 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Liu BL, Robinson M, Han ZQ, Branston RH, English C, Reay P, McGrath Y, Thomas SK, Thornton M, Bullock P, Love CA, Coffin RS, ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther 10, 292–303 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Brastianos PK, Lee EQ, Cohen JV, Tolaney SM, Lin NU, Wang N, Chukwueke U, White MD, Nayyar N, Kim A, Alvarez-Breckenridge C, Krop I, Mahar MK, Bertalan MS, Shaw B, Mora JL, Goss N, Subramanian M, Nayak L, Dietrich J, Forst DA, Nahed BV, Batchelor TT, Shih HA, Gerstner ER, Moy B, Lawrence D, Giobbie-Hurder A, Carter SL, Oh K, Cahill DP, Sullivan RJ, Single-arm, open-label phase 2 trial of pembrolizumab in patients with leptomeningeal carcinomatosis. Nat. Med 26, 1280–1284 (2020). [DOI] [PubMed] [Google Scholar]
- 55.Prakadan SM, Alvarez-Breckenridge CA, Markson SC, Kim AE, Klein RH, Nayyar N, Navia AW, Kuter BM, Kolb KE, Bihun I, Mora JL, Bertalan MS, Shaw B, White M, Kaplan A, Stocking JH, Wadsworth MH II, Lee EQ, Chukwueke U, Wang N, Subramanian M, Rotem D, Cahill DP, Adalsteinsson VA, Miller JW, Sullivan RJ, Carter SL, Brastianos PK, Shalek AK, Genomic and transcriptomic correlates of immunotherapy response within the tumor microenvironment of leptomeningeal metastases. Nat. Commun 12, 5955 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brastianos PK, Strickland MR, Lee EQ, Wang N, Cohen JV, Chukwueke U, Forst DA, Eichler A, Overmoyer B, Lin NU, Chen WY, Bardia A, Juric D, Dagogo-Jack I, White MD, Dietrich J, Nayyar N, Kim AE, Alvarez-Breckenridge C, Mahar M, Mora JL, Nahed BV, Jones PS, Shih HA, Gerstner ER, Giobbie-Hurder A, Carter SL, Oh K, Cahill DP, Sullivan RJ, Phase II study of ipilimumab and nivolumab in leptomeningeal carcinomatosis. Nat. Commun 12, 5954 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kitamura Y, Kanaya N, Moleirinho S, Du W, Reinshagen C, Attia N, Bronisz A, Revai Lechtich E, Sasaki H, Mora JL, Brastianos PK, Falcone JL, Hofer AM, Franco A, Shah K, Anti-EGFR VHH-armed death receptor ligand-engineered allogeneic stem cells have therapeutic efficacy in diverse brain metastatic breast cancers. Sci. Adv 7, eabe8671 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wege AK, Ernst W, Eckl J, Frankenberger B, Vollmann-Zwerenz A, Mannel DN, Ortmann O, Kroemer A, Brockhoff G, Humanized tumor mice—A new model to study and manipulate the immune response in advanced cancer therapy. Int. J. Cancer 129, 2194–2206 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Tian H, Lyu Y, Yang YG, Hu Z, Humanized rodent models for cancer research. Front. Oncol 10, 1696 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang M, Yao LC, Cheng M, Cai D, Martinek J, Pan CX, Shi W, Ma AH, De Vere White RW, Airhart S, Liu ET, Banchereau J, Brehm MA, Greiner DL, Shultz LD, Palucka K, Keck JG, Humanized mice in studying efficacy and mechanisms of PD-1-targeted cancer immunotherapy. FASEB J 32, 1537–1549 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tiberghien P, Ferrand C, Lioure B, Milpied N, Angonin R, Deconinck E, Certoux JM, Robinet E, Saas P, Petracca B, Juttner C, Reynolds CW, Longo DL, Hervé P, Cahn JY, Administration of herpes simplex-thymidine kinase-expressing donor T cells with a T-cell-depleted allogeneic marrow graft. Blood 97, 63–72 (2001). [DOI] [PubMed] [Google Scholar]
- 62.Lechanteur C, Briquet A, Bettonville V, Baudoux E, Beguin Y, MSC manufacturing for academic clinical trials: From a clinical-grade to a full GMP-compliant process. Cell 10, 1320 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bhere D, Choi SH, van de Donk P, Hope D, Gortzak K, Kunnummal A, Khalsa J, Revai Lechtich E, Reinshagen C, Leon V, Nissar N, Bi WL, Feng C, Li H, Zhang YS, Liang SH, Vasdev N, Essayed W, Quevedo PV, Golby A, Banouni N, Palagina A, Abdi R, Fury B, Smirnakis S, Lowe A, Reeve B, Hiller A, Chiocca EA, Prestwich G, Wakimoto H, Bauer G, Shah K, Target receptor identification and subsequent treatment of resected brain tumors with encapsulated and engineered allogeneic stem cells. Nat. Commun 13, 2810 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fares J, Ahmed AU, Ulasov IV, Sonabend AM, Miska J, Lee-Chang C, Balyasnikova IV, Chandler JP, Portnow J, Tate MC, Kumthekar P, Lukas RV, Grimm SA, Adams AK, Hébert CD, Strong TV, Amidei C, Arrieta VA, Zannikou M, Horbinski C, Zhang H, Burdett KB, Curiel DT, Sachdev S, Aboody KS, Stupp R, Lesniak MS, Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: A first-in-human, phase 1, dose-escalation trial. Lancet Oncol 22, 1103–1114 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nair A, Implications of intrathecal chemotherapy for anaesthesiologists: A brief review. Scientifica 2016, 3759845 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang X, Chen F, Yang SB, Wang YL, Qian ZH, Li Y, Lin H, Li P, Peng YC, Wang XM, Li WB, Intrathecal methotrexate in combination with systemic chemotherapy in glioblastoma patients with leptomeningeal dissemination: A retrospective analysis. World J. Clin. Cases 10, 5595–5605 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rusconi C, Cheah CY, Eyre TA, Tucker D, Klener P, Giné E, Crucitti L, Muzi C, Iadecola S, Infante G, Bernard S, Auer RL, Pagani C, Duglosz-Danecka M, Mocikova H, van Meerten T, Cencini E, Marin-Niebla A, Williams ME, Angelillo P, Nicoli P, Arcari A, Morello L, Mannina D, Vitagliano O, Sartori R, Chiappella A, Sciarra R, Stefani PM, Dreyling M, Seymour JF, Visco C, Ibrutinib improves survival compared with chemotherapy in mantle cell lymphoma with central nervous system relapse. Blood 140, 1907–1916 (2022). [DOI] [PubMed] [Google Scholar]
- 68.La Ferlita A, Alaimo S, Di Bella S, Martorana E, Laliotis GI, Bertoni F, Cascione L, Tsichlis PN, Ferro A, Bosotti R, Pulvirenti A, RNAdetector: a free user-friendly stand-alone and cloud-based system for RNA-Seq data analysis. BMC Bioinformatics 22, 298 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brainard DM, Seung E, Frahm N, Cariappa A, Bailey CC, Hart WK, Shin HS, Brooks SF, Knight HL, Eichbaum Q, Yang YG, Sykes M, Walker BD, Freeman GJ, Pillai S, Westmoreland SV, Brander C, Luster AD, Tager AM, Induction of robust cellular and humoral virus-specific adaptive immune responses in human immunodeficiency virus-infected humanized BLT mice. J. Virol 83, 7305–7321 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Yang THO, Porta-Pardo E, Gao GF, Plaisier CL, Eddy JA, Ziv E, Culhane AC, Paull EO, Sivakumar IKA, Gentles AJ, Malhotra R, Farshidfar F, Colaprico A, Parker JS, Mose LE, Vo NS, Liu J, Liu Y, Rader J, Dhankani V, Reynolds SM, Bowlby R, Califano A, Cherniack AD, Anastassiou D, Bedognetti D, Mokrab Y, Newman AM, Rao A, Chen K, Krasnitz A, Hu H, Malta TM, Noushmehr H, Pedamallu CS, Bullman S, Ojesina AI, Lamb A, Zhou W, Shen H, Choueiri TK, Weinstein JN, Guinney J, Saltz J, Holt RA, Rabkin CS, Lazar AJ, Serody JS, Demicco EG, Disis ML, Vincent BG, Shmulevich I, The immune landscape of cancer. Immunity 48, 812–830.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL, Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol 37, 907–915 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ge SX, Jung D, Yao R, ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. All the cell lines generated in this study are available under a material transfer agreement from the corresponding author, K.S.