

Summary
Craniosynostosis is a group of disorders of premature calvarial sutural fusion, and the calvarial stem cells (CSCs) that produce fusion-driving osteoblasts in craniosynostosis are poorly understood. Here we show that both physiologic calvarial mineralization and pathologic calvarial fusion in craniosynostosis reflect the interaction of two separate stem cell lineages; a recently reported CathepsinK (CTSK) lineage CSC (CTSK+ CSC)1 and a separate Discoidin domain-containing receptor 2 (DDR2) lineage stem cell (DDR2+ CSC) identified in this study. Deletion of Twist1, a gene associated with human craniosynostosis2,3, solely in CTSK+ CSCs is sufficient to drive craniosynostosis, however the sites destined to fuse surprisingly display a depletion of CTSK+ CSCs and a corresponding expansion of DDR2+ CSCs, with DDR2+ CSC expansion being a direct maladaptive response to CTSK+ CSC depletion. DDR2+ CSCs display full stemness features, establishing the presence of two distinct stem cell lineages in the sutures, with each population contributing to physiologic calvarial mineralization. DDR2+ CSCs mediate a distinct form of endochondral ossification without the typical hematopoietic marrow formation. Implantation of DDR2+ CSCs into suture sites is sufficient to induce fusion, and this phenotype was prevented by co-transplantation of CTSK+ CSCs. Lastly, the human counterparts of DDR2+ CSCs and CTSK+ CSCs display conserved functional properties in xenograft assays. The interaction between these two stem cell populations provides a new biologic interface to modulate calvarial mineralization and suture patency.
Keywords: Stem cell, Bone, Craniosynostosis, Calvarium
Given that CTSK+ CSCs (Lin−Thy-1.2−6C3−CD200+CD105−CD51+ calvarial stem cells expressing a Ctsk-Cre driven lineage reporter) contribute to physiologic calvarial mineralization1, we hypothesized that defects in these cells may underlie calvarial disorders such as craniosynostosis. To examine this, we generated mice with a Ctsk-Cre driven conditional deletion of Twist1 (Twist1Ctsk), as TWIST1 loss of function causes one of the most common craniosynostosis disorders, Saethre-Chotzen Syndrome2,3. Indeed, deletion of Twist1 in CTSK lineage cells was sufficient to drive a suture fusion phenotype with a distribution consistent with this disorder, including fusion of the coronal (COR), lambdoid (LAM), squamosal (SQ), and occipitointerparietal (OIP) sutures (Fig. 1a, b, Extended Data Fig. 1a, b)4. While we expected expansion and activation of CTSK+ CSCs to mediate fusion, we surprisingly instead observed a marked depletion of CTSK lineage cells at the sutures destined to fuse and noted a correspondence between the degree of depletion and the completeness of fusion at each suture (Fig. 1c, Extended Data Fig. 1c, d), a finding that parallels other observations linking SSC abundance to suture patency in the Twist1+/− craniosynostosis model5. CTSK lineage cells were present at identical numbers in E16.5 Twist1Ctsk and WT control mice, but subsequently underwent postnatal apoptosis (Extended Data Fig. 1e, f). At sites displaying suture fusion and CTSK+ CSC dropout, CTSK+ CSCs and their derivatives disappeared and were replaced with another cell type not bearing the CTSK lineage reporter (Fig. 1d, Extended Data Fig. 1g–k).
Fig. 1 |. Defects in CTSK+ CSCs cause craniosynostosis and the expansion of an alternative lineage.

a, μCT of skull of WT (Twist1+/+;Ctsk-Cre;mTmG, n=6) and Twist1Ctsk (Twist1fl/fl;Ctsk-Cre;mTmG, n=6), P28. Red arrows, fusing sutures. Interfrontal (IF); coronal (COR); sagittal (SAG); lambdoid (LAM); squamosal (SQ); occipitointerparietal (OIP) sutures. b, μCT showing histology sites, red lines. LAM (i) and OIP (ii), P30. Insets, bright-field merged images. c, LAM in WT and Twist1Ctsk;mTmG mice, P14. d, FACS plots of total live CTSK+ cells (top), CTSK+ CSCs (Lin−Thy-1.2−6C3−CTSK-mGFP+CD200+CD105−CD51+) (middle), and CTSK-negative putative CSCs (CTSK− pCSCs, Lin−Thy-1.2−6C3−CTSK-mGFP−CD200+CD105−CD51+) (bottom) in microdissected sutures of WT (n=12) and Twist1Ctsk;mTmG (n=17) mice, P14. Lin indicates CD31, CD45, and Ter119. Absolute numbers and frequency of populations relative to total live cells. e, Immunostaining for COMP and Alcian Blue of LAM, P14. f, Endochondral ossification in Twist1Ctsk;mTmG mice, P7. g, FACS plots (i), total CTSK+ cells relative to total live cells (ii), (iii) LAM, iDTRCtsk;mTmG after DT or PBS (PBS, n=4; DT, n=3). h, μCT (i) and 2D cut planes (ii) iDTR (n=5) and iDTRCtsk (n=6) mice at P21 and P35 after DT. i, H&E of the sutures of iDTR and iDTRCtsk mice after DT. j, FACS plots of CTSK− pCSCs and CTSK− pCSCs relative to total live cells from iDTRCtsk mice (PBS, n=4; DT, n=4). ****P < 0.0001. Mean ± s.d., unpaired two-tailed Student’s t-test (d, g, j). All FACS experiments (d, g, j) were repeated a minimum of three times on separate cohorts. Images in a, c, e, g (iii), h, and i represent results from at least five biological replicates. b, f are representative of a minimum of three independent experiments. Scale bars, 100 μm, b, c, e, f, g, i; all μCT images, 1.0 mm.
This dropout of CTSK+ CSCs was accompanied by a major shift in the form of bone formation in the sutures. Previously, we reported that CTSK+ CSCs are specialized for the major physiologic form of calvarial bone formation, intramembranous bone formation1, where bone is deposited directly without a cartilage template. Accordingly, dropout of CTSK+ CSCs was accompanied by the pathologic induction of endochondral bone formation, a form of bone formation normally excluded from most sutures 6–8, as seen through the presence of a cartilage template formed by non-CTSK lineage cells that was subsequently remodeled by osteoclasts and chondrocyte apoptosis into mineralized bone (Fig. 1e, f, Extended Data Fig. 1l). This cartilage was fully remodeled into bone at later timepoints, thereby distinguishing this endochondral ossification process from the formation of ectopic cartilage (Fig. 1b, Extended Data Fig. 1m, n). Taken together, a second calvarial lineage not derived from CTSK+ progenitors expands to become the dominant population at fusing sutural sites displaying endochondral ossification.
Dropout of CTSK+ CSCs was sufficient to drive the expansion of this alternative non-CTSK population and suture fusion, as seen with inducible deletion of CTSK+ CSCs using diphtheria toxin (DT) administration in iDTR;Ctsk-Cre (iDTRCtsk) mice. These mice not only displayed hypomineralization of the calvarium similar to that seen after conditionally deleting the Osterix (Osx) gene required for osteoblast generation in CTSK+ cells (OsxCtsk)1, but also displayed partial suture fusion at similar sites to those fused in Twist1Ctsk mice (Fig. 1g–j, Extended Data Fig. 2a–f). Similar to findings in Twist1Ctsk mice, compensatory expansion of non-CTSK lineage sutural cells preceded fusion in iDTRCtsk mice. The iDTRCtsk model produced a porosity phenotype in the cortices of long bones similar to that reported in OsxCtsk mice, thereby supporting that this model produced the expected loss-of-function in CTSK-expressing SSCs1. Additionally, an osteopetrotic phenotype representing deletion of CTSK-expressing osteoclasts was observed in long bones, thereby providing an internal control validating the expected iDTR effect (Extended Data Fig. 2g–i). However, due to the expected turnover of mature osteoclasts, this osteoclast ablation effect was transient (Extended Data Fig. 2j). While a contribution from osteoclasts to the fusion phenotype cannot be excluded, this is unlikely to be a primary driver of the phenotype observed, as blocking osteoclast activity with bisphosphonates did not produce suture fusion (Extended Data Fig. 2k). Based on the presence of a fusion phenotype only in iDTRCtsk but not in OsxCtsk mice, we postulate that it is the absence of CTSK+ CSCs themselves and not the absence of more mature Osterix-dependent osteoblast populations in the CTSK lineage that drives the fusion phenotype.
Based on these results, we hypothesized that a second calvarial stem cell exists within the pool of non-CTSK lineage sutural cells. To identify this putative second non-CTSK calvarial stem cell, candidate markers of this population were identified by transcriptional analysis, immunostaining, and flow cytometry. Among these candidates, the transmembrane collagen receptor DDR2 was prioritized based on reports that Ddr2 is expressed in the sutural cells and reports that Ddr2 mutant mice display calvarial mineralization defects9–11. Additionally, several other candidate markers were evaluated and found to display inadequate selectivity in labeling the non-CTSK cell fraction or were found to mark populations without robust bone formation capacity (Extended Data Fig. 3a–g). DDR2 marked a sutural population not overlapping with CTSK lineage cells and was present at sites of sutural endochondral ossification (Fig. 2a, b, Extended Data Fig. 3h). Consistent with the expansion of non-CTSK lineage cells observed above, both Twist1Ctsk and iDTRCtsk mice, as well as the Twist1+/− craniosynostosis model, all displayed marked expansion of a DDR2+ population within the pool of sutural non-CTSK lineage cells. This population of sutural DDR2+ cells included a subpopulation bearing the same surface immunophenotype as CTSK+ CSCs, Lin−Thy-1.2−6C3−CD200+CD105−CD51+ cells that we hypothesized to represent a new calvarial stem cell type expressing DDR2 (DDR2+ CSCs) (Fig. 2c, d, Extended Data Fig. 3i–l).
Fig. 2 |. Identification of a sutural DDR2+ CSC.
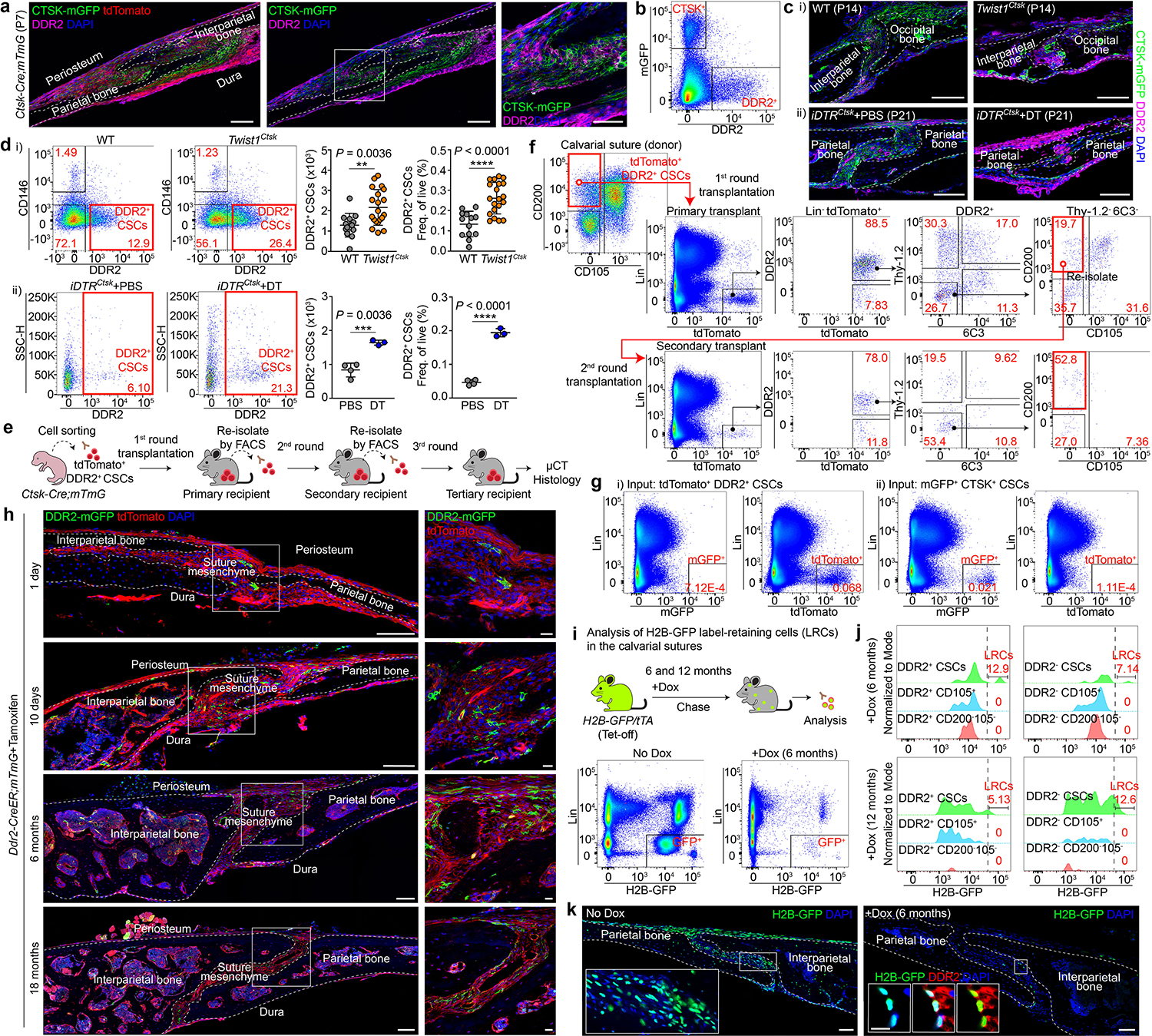
a, DDR2+ cells, LAM, Ctsk-Cre;mTmG mice, P7. b, FACS, P7. Pregated on Lin−Thy-1.2−6C3−. c, DDR2, WT and Twist1Ctsk;mTmG mice, P14 (i), iDTRCtsk mice, P21 (ii). d, DDR2+ CSCs by FACS (Lin−6C3−Thy-1.2−CTSK-mGFP−CD200+CD105−CD51+DDR2+) in the sutures of WT (n=13), Twist1Ctsk (n=20), P7 to P10 (i), and iDTRCtsk mice, PBS (n=4) or DT (n=3) at P21 (ii). e, In vivo intramuscular serial transplantation. f, FACS of DDR2+ CSC-derived cells after serial transplantation. Black arrows, parent/daughter gates. Red boxes, DDR2+ CSCs. Representative of 4 independent experiments. g, FACS showing no interconversion between tdTomato+ DDR2+ CSCs and mGFP+ CTSK+ CSCs isolated from Ctsk-Cre;mTmG mice after the first round of transplantation. h, Lineage tracing of sutural DDR2+ cells using Ddr2-CreER;mTmG mice over an 18 month chase. i, FACS plots showing total H2B-GFP label-retaining cells after 6 months of chase post-doxycycline (Dox). j, GFP signal intensity histogram of Lin−Thy-1.2−6C3−DDR2+ or DDR2− populations after 6 (n=9) or 12 months (n=8) of chase. Label-retaining cells (LRCs) were determined as gated. Green, CD200+CD105− (CSC); Blue, CD200variableCD105+; Red, CD200−CD105−. k, LAM, H2B-GFP mice after 6 months of chase with DDR2 immunostaining. In all graphs, each of dot represents an individual mouse. ****P < 0.0001. Mean ± s.d., unpaired two-tailed Student’s t-test (d). Images in a, c, h, k are representative of at least three independent experiments. Scale bars, 100 μm, a, c, h (left), k; 50 μm, a (far-right); 20 μm, c, h (right), k (inset).
Serial transplantation, analysis of differentiation hierarchy, lineage tracing, and label retention studies, in addition to in vitro colony formation, clonal multipotency, and serial mesensphere formation assays were all used to identify that putative DDR2+ CSCs are indeed stem cells. DDR2+ CSCs also displayed in vitro features of stemness. Single cell-derived clones of DDR2+ CSCs were capable of in vitro tri-lineage differentiation into osteoblasts, adipocytes, and, separately, chondrocytes. DDR2+ CSCs also displayed in vitro colony forming (CFU-F) activity and osteoblast colony forming activity (CFU-Ob) similar to that of CTSK+ CSCs. Lastly, DDR2+ CSCs and CTSK+ CSCs displayed serial mesensphere formation capacity, a correlate of stemness in skeletal cells1,12 (Extended Data Fig. 4a–g). In vivo transplantation showed that DDR2+ CSCs, but not other DDR2+ populations, were able to both self-renew and generate all of the other DDR2+ populations present in the native suture after the first round of transplantation. When re-isolated after the first round of transplantation and subjected to the second round of transplantation, DDR2+ CSCs were again capable of similar self-renewal and differentiation (Fig. 2e, f, Extended Data Fig. 4h, i), demonstrating serial transplantation and self-renewal capacity similar to that of other skeletal stem cell (SSC) populations identified in long bones1,13. Finally, DDR2+ CSCs self-renewing after the first two rounds of transplantation were yet again capable of bone formation after re-isolation and the third round of transplantation. Both DDR2+ CSCs and CTSK+ CSCs formed mineralized bone organoids after each round of transplantation (Extended Data Fig. 4j). Transplantation also clarified that DDR2 is present on the entire lineage of cells derived from DDR2+ CSCs. No evidence of interconversion between DDR2+ CSCs or CTSK+ CSCs was observed, as DDR2+ CSCs did not upregulate expression of a CTSK reporter, and, likewise, CTSK+ CSC-derived cell types remained DDR2-negative (Fig. 2g, Extended Data Fig. 4k, l).
In vivo pulse-chase lineage tracing also supported the stemness of DDR2+ CSCs, producing durable labeling that disseminated to downstream populations as opposed to the transient labeling seen in non-stem populations 14. Ddr2-CreER;mTmG mice were used to target pulse-chase labeling to DDR2 lineage calvarial cells (Fig. 2h, Extended Data Fig. 5a–j). Ddr2-CreER labeled both suture resident cells and bone-adjacent osteoblasts (Fig. 2h, Extended Data Fig. 5c–f), showing >90% concordance with DDR2 flow cytometry (Extended Data Fig. 5g). Consistent with DDR2 lineage cells including a stem population, a single early tamoxifen pulse provided durable labeling that was retained in the suture at least 18 months later, with the labeled cells including calvarial osteoblasts (Extended Data Fig. 5f). Moreover, the labeled DDR2 lineage cells persisting after a 12 month chase specifically included DDR2+ CSCs (Extended Data Fig. 5h–j). Taken together, DDR2+ CSCs are long-lived cells with a continual contribution to the calvarial osteoblast pool.
Label retention in the H2B-GFP system has been used across many organs as a sensitive method to define slow-cycling stem cells15–18. Consistent with the result of transplantation-based stemness assays, H2B-GFP retention was observed in DDR2+ CSCs but not other DDR2+ populations at 6 and 12 months (Fig. 2i–k, Extended Data Fig. 5k–q). In line with the model that the suture contains two separate stem cells, DDR2+ CSCs and CTSK+ CSCs, additional, separate label-retaining cells were observed in the fraction of DDR2− cells bearing surface markers consistent with CTSK+ CSCs (Lin−CD200+CD105−Thy-1.2−6C3− cells). As it was not possible to use GFP-based genetic reporters of the CTSK lineage together with the H2B-GFP system, we sought alternative markers of the CTSK+ CSC lineage to confirm the additional presence of label-retaining CTSK+ CSCs, identifying that Galectin-1 immunostaining displayed high concordance with Ctsk-Cre based lineage reporters (Extended Data Fig. 5p). Using Galectin-1, the label retention in DDR2− CSCs was confirmed to localize to CTSK lineage Galectin-1+ cells and therefore represent labeling of CTSK+ CSCs (Extended Data Fig. 5q). Thus, there are separate pools of calvarial label-retaining cells corresponding to both DDR2+ CSCs and CTSK+ CSCs, consistent with the presence of two stem cells in the calvarium.
Next, we evaluated whether DDR2+ CSCs are capable of differentiating into bone-forming osteoblasts and whether they mediate endochondral ossification accounting for the inappropriate sutural endochondral ossification seen in Twist1Ctsk mice. Indeed, DDR2+ CSCs isolated by FACS and transplanted to the kidney capsule of secondary hosts formed robust bone organoids (Fig. 3a, b, Extended Data Fig. 6a, b). This bone was derived from graft DDR2+ CSCs, as osteocytes and osteoblasts in the organoid bore the tdTomato marker of graft DDR2+ CSCs (Fig. 3c). Additionally, no interconversion between DDR2+ CSCs and CTSK+ CSCs could be observed as there was no induction of Ctsk-Cre driven mGFP expression in the DDR2+ CSCs over time. Likewise, CTSK+ CSC-derived cells displayed no induction of DDR2 expression (Fig. 3c, Extended Data Fig. 6c, d).
Fig. 3 |. DDR2+ CSCs are endochondral-specialized.

a, b, Bone organoids after renal capsule transplantation, 4 weeks. c, Cryo-sectioned bone organoids derived from DDR2+ CSCs (red) from Ctsk-Cre;mTmG mice. No interconversion of DDR2+ CSCs (red) into CTSK+ CSCs (green) is seen. Inset, additional DDR2 immunostaining. d, Timecourse of the bone organoids. von Kossa staining (black) for mineralized bone and Safranin O (red) for cartilage in adjacent serial sections. e, Timecourse immunofluorescence for markers of endochondral ossification in DDR2+ CSC-derived bone organoids 2, 4, and 8 weeks post-transplantation. f, H&E and immunostaining for markers of hematopoietic cells in organoids from femur SSCs, DDR2+ CSCs, or CTSK+ CSCs. g, FACS plots indicating recruitment of hematopoietic populations to femoral SSC-derived (n=4) or DDR2+ CSC-derived (n=3) bone organoids. Lin, CD3, Gr-1, CD11b, B220, and Ter119. Total Lin−cKit+ cells (LK); Total Lin−Sca1+cKit+ cells (LSK); Long-term hematopoietic stem cells (LT-HSC, Lin−Sca1+cKit+CD150+CD48−); Short-term HSC (ST-HSC, Lin−Sca1+cKit+CD150−CD48−); Multipotent progenitors (MPP, Lin−Sca1+cKit+CD150−CD48+). h, Principal component analysis (PCA) of the CTSK+ CSC and DDR2+ CSC transcriptome profiling after isolation from P7 suture (n=5). i, Heatmap of gene expression. The color scale bar shows Z-score values on normalized read counts across the samples. ****P < 0.0001, P values are shown. Mean ± s.d., unpaired two-tailed Student’s t-test. Images in b-f are representative of a minimum of three (d, e) and five (b, c, and f) independent experiments. Scale bars, 1.0 mm, b; 100 μm, c, e, f; 200 μm, d.
Whereas CTSK+ CSCs mediated the expected intramembranous ossification after transplantation1, DDR2+ CSCs underwent endochondral ossification consistent with being mediators of the inappropriate sutural endochondral ossification seen preceding suture fusion in Twist1Ctsk mice (Fig. 3d, e, Extended Data Fig. 6e–i). However, while endochondral ossification is typically intrinsically coupled with the recruitment of hematopoietic marrow elements1,19, DDR2+ CSCs mediate a distinct form of endochondral ossification without recruitment of hematopoietic cells, consistent with their inability to recruit long-term hematopoietic stem cells (Fig. 3f, g, Extended Data Fig. 6j). While the etiology of this difference in hematopoietic support capacity is likely multifactorial, DDR2+ CSC-derived organoids did not include cells expressing the hematopoietic niche factor CXCL12 that were present in organoids derived from long bone SSCs20 (Extended Data Fig. 6k). Likewise, DDR2 lineage cells displayed reduced expression of other mediators of hematopoietic support relative to femoral SSCs, including Cxcl12, Angpt1 and Kitlg (Extended Data Fig. 6l). Taken together, this indicates that a third fundamental form of bone formation, endochondral ossification without hematopoiesis, exists alongside traditional intramembranous and endochondral ossification, and DDR2+ CSCs are specialized for this third form of bone formation.
DDR2+ CSCs and CTSK+ CSCs display broadly distinct transcriptional profiles, reinforcing that DDR2+ CSCs and CTSK+ CSCs are distinct cell types (Fig. 3h). Consistent with the chondrogenic capacity of DDR2+ CSCs, these cells expressed higher levels of classically chondrocyte-associated transcripts such as Sox9, Col2a1, and Acan, though these transcripts were also present at lower levels in CTSK+ CSCs. Both CTSK+ CSCs and DDR2+ CSCs expressed several transcripts associated with SSCs at other skeletal sites, including Myc, Runx2, Klf4, and Nes1,12,21,22. Interestingly, many of these shared genes were also present in both periosteal and endosteal stem cells1 in addition to growth plate resident SSCs23 in long bones, indicating convergence on a set of SSC-defining genes shared among multiple SSC types at different skeletal sites. GLI1 was expressed in both CTSK+ CSCs and DDR2+ CSCs, suggesting that this schema resolves the pool of Gli1 lineage calvarial progenitors into multiple discrete stem cell types24. Similarly, DDR2+ CSCs and CTSK+ CSCs also co-expressed other genes previously used to define calvarial progenitor populations, including Axin225 and Prrx126, indicating that the DDR2+ CSC versus CTSK+ CSC definitions cut across several established schemas for calvarial progenitors. DDR2+ CSCs and CTSK+ CSCs also both co-expressed several genes involved in embryonic suturogenesis including Msx1/227, Acta2, Lgr5, and Lrig128 (Fig. 3i, Extended Data Fig. 7a–c). Lastly, a number of genes associated with human craniosynostosis are expressed at higher levels in DDR2+ CSCs, including Efnb1, Ihh, Msx2, Fgfr2, and Fgfr3, raising interest in the role of DDR2+ CSCs in the associated disorders 29–31 (Extended Data Fig. 7d). These DDR2+ CSC and CTSK+ CSC expression signatures mapped onto distinct populations in several scRNA-Seq datasets of the murine calvarium, providing additional support for the co-existence of DDR2+ CSC and CTSK+ CSC lineages in the calvarial sutures (Extended Data Fig. 7e, f).
In addition to a role in craniosynostosis, the DDR2+ CSC lineage has an important contribution to calvarial mineralization distinct from that of the CTSK+ CSC lineage. Inducible postnatal deletion of DDR2+ CSCs in Ddr2-CreER;DTA mice resulted in enlargement of the anterior fontanelle due to hypomineralization of the frontal and parietal bones, and severe hypomineralization of the interparietal bone (Fig. 4a). Similarly, deletion of Osx specifically in DDR2+ CSCs (Osxfl/fl;Ddr2-CreER) resulted in calvarial hypomineralization similar to that seen in Ddr2-CreER;DTA mice (Fig. 4b). Interestingly, DDR2 itself appears to be functional on DDR2+ CSCs, as mice with the spontaneously occurring smallie mutation causing a loss of DDR2 expression (Ddr2slie mice) display calvarial hypomineralization10 especially evident in the parietal and frontal bones widened sutures. Ddr2slie mice also displayed an absence or reduction in the cartilage structures in the lateral portions of the interparietal and occipital bones, implicating the DDR2+ CSC lineage in the formation of the cartilaginous elements of the calvarium (Extended Data Fig. 8a–c).
Fig. 4 |. DDR2+ CSCs contribute to calvarial mineralization and suture fusion.

a, b, μCT, DTA, DTADdr2−ER (a) and Osxfl/fl, Osx Ddr2−ER (b) mice, P14, 7 days after TAM (DTA, n=5; DTADdr2−ER, n=6; Osxfl/fl, n=7; Osx Ddr2−ER, n=6). c, Orthotopic transplantation of FACS-purified CTSK+ CSCs and DDR2+ CSCs into 4-week-old suture-ablated mice. d, μCT, 8 weeks post-transplantation. Black arrowhead, primary implantation site. Red arrows, fusing sutures. e, 2D cut plane, skull 16 weeks post-transplantation of indicated populations. Asterisks, fused sutures. f, μCT, 16 weeks after implantation of DDR2+ CSCs. tdTomato for DDR2+ CSC engraftment and fusion at the implantation site (i), but a contralateral control (ii). Red lines, sites of histologic sampling. g, CTSK+ CSCs and iDTR;CTSK+ CSCs were isolated from P7–10 Ctsk-Cre;mTmG or iDTRCtsk;mTmG mice, respectively. All recipients were administered DT 8 weeks post-transplantation. h, μCT 16 weeks post-transplantation (Control, n=6; CTSK+, n=6; DDR2+, n=5; CTSK+ and DDR2+, n=5; iDTR;CTSK+ and DDR2+, n=4). i, H&E and fluorescence imaging, primary implantation site 16 weeks post-transplantation and DT administration. Asterisks, the region of fusion. j, The percentage of fused LAM (i) or OIP (ii) suture length relative to total LAM or OIP suture length on the ipsilateral side. n=3 independent experiments. ****P < 0.0001, P values are shown. Mean ± s.d., one-way ANOVA with Tukey’s posttest (j). e, f, h, and i are representative of a minimum of three independent experiments. Scale bars, 100 μm, f (i,ii), i; all μCT images, 1.0 mm.
Using orthotopic transplantation model5,32 where the native lambdoid suture is surgically ablated and replaced by a defined set of cells implanted into the ablation site (Fig. 4c, Extended Data Fig. 8d), implantation of DDR2+ CSCs was sufficient to promote fusion in a manner that is restrained by co-implantation with CTSK+ CSCs (Fig. 4d, e, Extended Data Fig. 8e, f). DDR2+ CSCs, but not CTSK+ CSCs, were sufficient to mediate fusion not only at the implantation site (the LAM suture), but also in an extended region of the SAG, OIP, and SQ sutures adjacent to the implantation site, despite both populations being capable of robust engraftment (Fig. 4f, Extended Data Fig. 8g–i). This fusion in adjacent regions reflected the migration of graft DDR2+ CSCs to these sites, in addition to the retention of graft cells at the primary implantation site. Consistent with the finding that depletion of CTSK+ CSCs was sufficient to induce suture fusion, co-implantation of CTSK+ CSCs with DDR2+ CSCs prevented the fusion-promoting activity of DDR2+ CSCs, while not impacting their ability to engraft. This fusion-suppressing ability of CTSK+ CSCs reflects continuous, direct regulation and not induced death or differentiation of DDR2+ CSCs as co-implantation of CTSK+ CSCs and DDR2+ CSCs followed by post-transplantation ablation of the implanted CTSK+ CSCs using a diphtheria toxin (DT) system was able to restore the fusion activity of DDR2+ CSCs (Fig. 4g–j, Extended Data Fig. 8j, k).
Further investigation revealed IGF1 as a candidate CTSK+ CSC-derived factor regulating the expansion and fusion-promoting activity of DDR2+ CSCs. Within these CSC populations, Igf1 was largely expressed on CTSK+ CSCs, and its cognate receptor Igf1r was largely expressed on DDR2+ CSCs (Extended Data Fig. 9a). In addition to the number of Igf1-expressing CTSK+ CSCs being decreased in Twist1Ctsk mice, the per-cell expression of Igf1 expression was downregulated in the residual Twist1Ctsk CTSK+ CSCs remaining (Extended Data Fig. 9b). Injection of recombinant IGF1 over the calvarium was able to partially prevent the fusion phenotype in Twist1Ctsk mice, thus repleting IGF1 was sufficient to ameliorate the Twist1Ctsk craniosynostosis phenotype (Fig. 5a, b and Extended Data Fig. 9c). Moreover, CTSK+ CSC-derived IGF1 was necessary to prevent suture fusion as Igf1fl/fl;Ctsk-Cre (Igf1Ctsk) mice displayed spontaneous suture fusion and expansion of DDR2+ CSCs, mirroring observations in Twist1Ctsk mice (Fig. 5c–f, Extended Data Fig. 9d, e). Thus, IGF1 is, in this context, a CTSK+ CSC-derived regulator of DDR2+ CSCs, though other such regulators may also exist.
Fig. 5 |. Identification of IGF1 as a CTSK+ CSC-derived factor regulating DDR2+ CSCs.
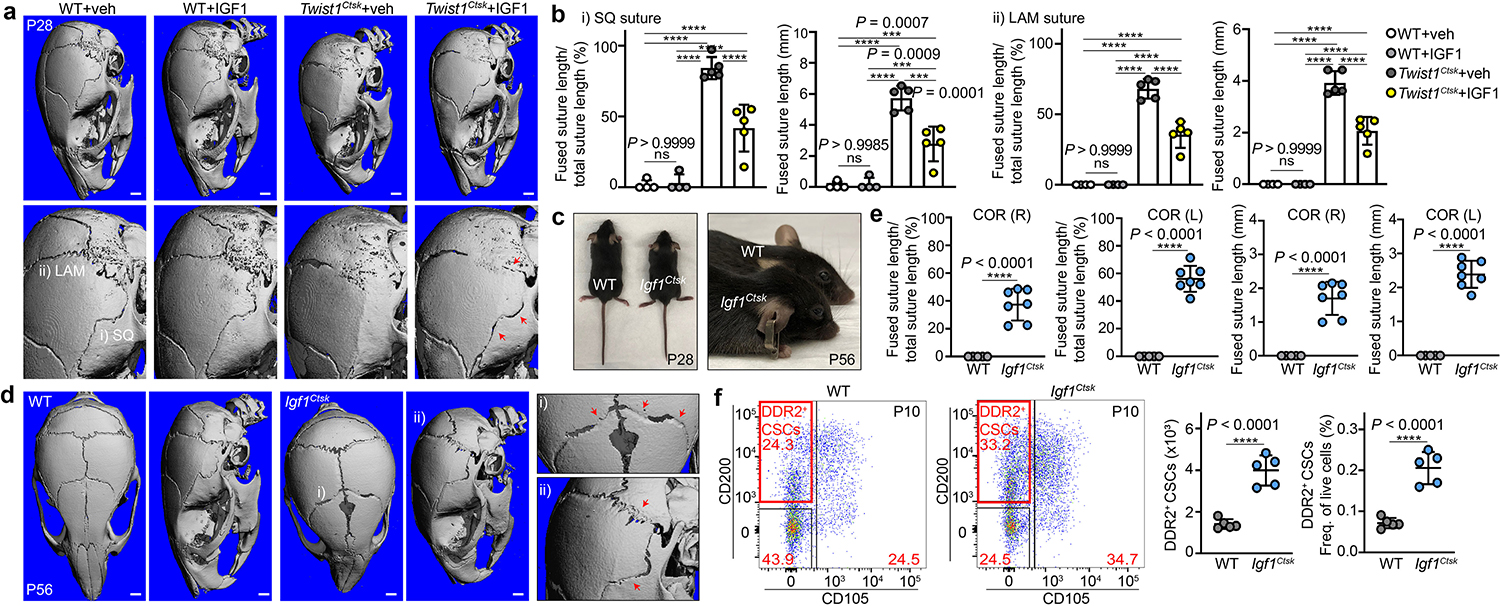
a, μCT, Twist1Ctsk mice at P28 after IGF1 treatment, showing rescue of fusion. Experimental design in Extended Data Fig. 9c. Arrows, sites of suture patency (WT+veh, n=4; WT+IGF1, n=4; Twist1Ctsk+veh, n=5; Twist1Ctsk+IGF1, n=5). b, Relative percentage of fused suture length (WT+veh, n=4; WT+IGF1, n=4; Twist1Ctsk+veh, n=5; Twist1Ctsk+IGF1, n=5). SQ (i) and LAM (ii) (****P < 0.0001). c, Gross appearance of Igf1fl/fl;Ctsk-Cre (Igf1Ctsk) mice, P28 and P56. d, μCT 3D scans of the skull of WT and Igf1Ctsk mice at P56 (WT, n=5; Igf1Ctsk, n=7). Red arrows, fusing sutures. e, The relative percentage of fused coronal suture length in WT (n=5) and Igf1Ctsk (n=7) mice at P56. f, FACS plots and quantification of DDR2+ CSCs in the sutures of WT and Igf1Ctsk mice at P10 (n=5), showing expansion of sutural DDR2+ CSCs. Each of dot represents an individual mouse. ****P < 0.0001. Mean ± s.d., unpaired two-tailed Student’s t-test (e, f), one-way ANOVA with Tukey’s posttest (b). FACS plots represent results from five biological replicates. Images in a, d represent results from at least four biological replicates. Scale bars in all μCT images denote 1.0 mm.
Human counterparts of DDR2+ CSCs were present in the sutures of patients with craniosynostosis, establishing the clinical relevance of this SSC type. Consistent with prior literature8,33 and observations in Twist1Ctsk mice, calvarial specimens from patients with sporadic craniosynostosis contained abundant DDR2+ cells and displayed inappropriate endochondral ossification with cartilage undergoing active remodeling into bone (Fig. 6a–c, Extended Data Fig. 10a–c). Human sutural tissue also included both DDR2+ and DDR2− cells bearing a surface immunophenotype nearly identical to that of murine DDR2+ CSCs and CTSK+ CSCs (Lin−Thy-1−CD200+CD105− cells that are DDR2+ or DDR2−, hereafter hDDR2+ CSCs and hDDR2− CSCs) (Extended Data Fig. 10d). Given the inability to utilize CTSK-based lineage reporters in humans, hDDR2− CSCs were here utilized as a proxy for murine CTSK+ CSCs. In support of this, hDDR2+ CSCs and hDDR2− CSCs displayed conserved gene expression features similar to that of their respective murine counterparts, including expression of IGF1R on hDDR2+ CSCs (Fig. 6d, Extended Data Fig. 10e, f). Interestingly, this hCSC definition also largely overlapped with another set of markers used to define stem cells in human fetal long bones (PDPN+CD146−CD164+CD73+)34 (Extended Data Fig. 10d). Thus, hDDR2+ and hDDR2− CSCs show a convergent immunophenotype that fulfills criteria from multiple schema for identifying SSCs.
Fig. 6 |. Identification of human DDR2+ CSCs.

a, H&E (n=14) and Alcian Blue (n=7), b, immunostaining (n=7), c, d, immunofluorescence, human SAG craniosynostosis specimens. e, In vivo serial transplantation of hDDR2− CSCs (left)and hDDR2+ CSCs (right), FACS plots showing the self-renewal and differentiation. FACS from initial donor (top), primary transplants (middle), and secondary transplants (bottom). Black and red arrows indicate parent/daughter gates and transplanted populations, respectively. μCT, bone organoids derived from the tertiary transplants. f, Flow cytometry histograms of dye dilution proliferation analysis. g, CFU-F, hDDR2− CSCs, hDDR2+ CSCs, and other human calvarial non-stem populations (n≥3 independent experiments). h, Bright-field, initially seeded, primary, secondary, and tertiary mesenspheres (left). Quantification of cell numbers per sphere (right, n≥9). i, Gross image of xenograft bone organoid in the renal capsule of NSG-EGFP host (left). μCT timecourse, hDDR2− CSC or hDDR2+ CSC-derived bone organoids (right). j, Endochondral ossification in xenograft bone organoids from hDDR2+ CSCs by von Kossa and Safranin O. k, Immunostaining, on xenograft bone organoids derived from hDDR2+ CSCs, (human nuclear antigen, HNA) implanted into the renal capsule NSG-EGFP hosts. (i) Confocal image showing the graft stained as indicated. (ii) High magnification of i. (iii) COL2A1+ chondrogenic cells (left) and MMP13+ apoptotic cells (right) in xenograft organoids derived from hDDR2+ CSCs. ****P < 0.0001. Mean ± s.d., one-way (g), two-way (h) ANOVA with Tukey’s posttest. FACS plots (e, f) and images (a-d, g-k) are representatives of a minimum of three independent experiments. Scale bars, 200 μm, a (left); 25 μm, a (middle and right), b; 100 μm, c, d, j, k; 20 μm, d (inset); 500 μm, g; 50 μm, h; all μCT images, 1.0 mm.
Both hDDR2+ CSCs and hDDR2− CSCs displayed robust functional evidence of stemness, as both were able to respectively generate all of the skeletal human DDR2+ and DDR2− populations present in the initial donor suture tissue in addition to undergoing proliferative self-renewal over the course of 3 rounds of serial transplantation (Fig. 6e, f, Extended Data Fig. 11a–f). Both hDDR2− and hDDR2+ CSCs were also capable of serially forming bone organoids in xenograft systems over the course of 3 successive rounds of transplantation and re-isolation (Fig. 6e). Similarly, both hDDR2+ CSCs and hDDR2− CSCs displayed in vitro features of stemness including CFU-F activity, clonal multipotency, and the capacity to serially form mesenspheres (Fig. 6g, h, Extended Data Fig. 11g–i). Consistent with observations that murine DDR2+ CSCs are specialized for endochondral ossification, hDDR2+ CSC-derived organoids included both graft-derived osteoblasts and chondrocytes and characteristically included the transient presence of an initial cartilage template that underwent vascular invasion and remodeling into mineralized bone (Fig. 6i–k, Extended Data Fig. 11j, k).
In summary, we here find that the calvarial sutures contain two distinct stem cell types. One is a CTSK+ CSC that is specialized for intramembranous bone formation. A second type identified here is a DDR2+ CSC that characteristically mediates endochondral ossification without marrow formation. These definitions provide a starting point for further iterative refinement of stem cell identity in the calvarium. This also provides support for an emerging consensus that skeletal physiology broadly reflects a diverse set of distinct progenitor cell types35–37. Interactions between these two cells are critical for the pathogenesis of craniosynostosis and suture patency, as dropout of CTSK+ CSCs triggers a maladaptive expansion and activation of DDR2+ CSCs, which in turn results in inappropriate endochondral ossification in the sutures and, ultimately, fusion. This model also potentially explains widespread clinical and animal model findings that calvarial hypomineralization can accompany craniosynostosis38–40. Counterparts of both of these cell types are also present in human calvarial sutures. Given their role in craniosynostosis, agents that inhibit DDR2+ CSCs or otherwise block their compensatory expansion offer an attractive strategy for adjunct medical therapy of craniosynostosis. Moreover, this provides an example of how multiple skeletal stem cell lineages can co-exist and interact at a single site, with disruptions in these stem cell to stem cell interactions serving as a new mechanism for skeletal disease.
Methods
Animals.
Cathepsin K-Cre (Ctsk-Cre) mice were a gift from S. Kato (University of Tokyo) and T. Nakamura (Tokyo Dental College)41, Floxed Twist1 (Twist1fl/fl, Stock 016842-UNC) mice were obtained from the Mutant Mouse Resource & Research Centers (MMRRC)42, Floxed Osterix (Osxfl/fl) mice were a gift from B. de Crombrugghe (University of Texas M.S. Anderson Cancer Center)43, Ddr2slie mice on the C57BL/6J background were a gift from R. T. Franceschi10. mTmG (Stock 007676), ROSA26iDTR (Stock 007900), ROSA-DTA (Stock 009669), MIP-GFP (Stock 006864), NOD scid gamma (NSG)-EGFP mice (Stock 021937), pTRE-H2BGFP (Stock 005104), and Floxed Igf1 (Igf1fl/fl, Stock 016831) mice were purchased from the Jackson Laboratories. ROSA:LNL:tTA mice (JAX, Stock 011008) were a gift from L. Puglielli (University of Wisconsin-Madison)44. Ddr2-CreER (MerCreMer) mice were generated by R. T. Cowling and B. H. Greenberg with assistance from the Transgenic Core and Embryonic Stem Cell shared resource at UCSD45. Briefly, a MerCreMer cassette was inserted in-frame into exon 2 of Ddr2 using homologous recombination following a strategy similar to what was previously described46. Ddr2-CreER mice crossed with mTmG mice for lineage tracing studies. Recipient heterozygous MIP-GFP mice were generated by breeding homozygous MIP-GFP males with wild type C57BL/6J females. All mice were maintained on a C57BL/6J background throughout the study. All animals were maintained in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were handled according to protocols approved by the Weill Cornell Medical College institutional animal care and use committee (IACUC). All animal experiments followed all relevant guidelines and regulations.
Chemical reagents administration.
iDTR;Ctsk-Cre;mTmG pups were given 50 ng of diphtheria toxin (DT) (Sigma, cat. D0564) intraperitoneally as indicated in Extended Data Fig. 2a. PBS was used as a vehicle. For functional inhibition of osteoclasts, C57BL/6J mice were given alendronate sodium trihydrate (1 mg/kg; Sigma, cat. A4978) intraperitoneally as illustrated in Extended Data Fig. 2k. For lineage tracing analysis, Ddr2-CreER activity was induced at postnatal day 2 (P2). Tamoxifen (TAM) (100 mg/kg; Sigma, cat. T5648) was prepared in corn oil and subcutaneously injected into animals for 2 consecutive days as described in Extended Data Fig. 5a. To investigate the physiologic contribution of DDR2+ CSCs, ROSA-DTA, and Osxfl/fl mice were crossed with Ddr2-CreER;mTmG mice and TAM was delivered subcutaneously into P7 pups. Recombinant IGF1 (1 mg/kg; R&D Systems, cat. 291-G1) was subcutaneously administered over the calvarium of Twist1Ctsk mice at P3, as described in Extended Data Fig. 9c. PBS was used as a vehicle.
Histone H2B-GFP labeling retention system.
pTRE-H2BGFP mice were previously described47. To label slow-cycling cells under the control of a tetracycline response element, pTRE-H2BGFP mice were crossed with ROSA:LNL:tTA (tet-off) mice. 8-week-old mice were fed with a doxycycline rodent diet (200 mg/kg; Bio-Serv, cat. S3888) for 6 and 12 month chase periods. The littermates without doxycycline feeding were used as a control. Additional controls of mice that were continuously exposed to doxycycline, including during the embryonic period by providing doxycycline to pregnant female mice, were used as a negative control of H2B-GFP expression.
Human specimens.
This study was carried out under the institutional review board (IRB) protocol (no. 1402014802), with all patients providing informed consent through the Center for Neurogenetics at Weill Cornell Medical Center. Human calvarial tissue used for scientific analyses was obtained from 37 cases of craniosynostosis surgical repair procedures. Participants had an age range of less than 1 year old and were 70% male. Detailed annotation of the clinical specimens including age, sex, and the primary diagnosis is provided in Supplementary Table 1. All human tissue experiments followed all relevant guidelines and regulations. Written consent was provided by the parents or guardians of all study participants.
Isolation of suture cells.
For murine samples, microdissected calvarial sutures were subjected to both mechanical and enzymatic digestion. The harvested tissues were minced using razor blades and digested for up to 20 minutes with digestion cocktail buffer containing Collagenase P (1 mg/ml; Roche, cat. 11213857001) and Dispase II (2 mg/ml; Roche, cat. 04942078001) in medium containing 2% serum at 37 °C with agitation. Medium containing 2% serum was added to the digest and the tubes were centrifuged to pellet cells. The supernatant was carefully removed, and the pellet was resuspended in DNase I (2 units/ml; Roche, cat. 04716728001) solution and briefly incubated for 5 min at 37 °C. Media was added to the tube and the digested tissue was re-suspended thoroughly by pipetting and then filtered through a 70-micron nylon mesh. Tubes were centrifuged and the resulting cell pellet was subjected to FACS. For human calvarium specimens, tissues were subjected to mechanical and enzymatic digestion with Collagenase A (1 mg/ml; Roche, cat. 10103586001) and Dispase II (2 mg/ml) in a medium containing 2% serum for 30 min at 37 °C under agitation, and the cells were then isolated following the same procedure as described above.
Fluorescence-activated cell sorting (FACS).
Prepared single-cell suspensions were washed twice with ice-cold FACS buffer (2% serum + 1 mM EDTA in PBS) and incubated with Fc blocking buffer (1:100 dilution; BD bioscience, cat. 553142 for mouse and cat. 564765 for human) for 15 min at 4 °C. Primary antibody dilutions were prepared in Brilliant Stain Buffer (BD bioscience, cat. 563794). Cells were incubated in the dark for 40 min at 4 °C with primary antibody solution, washed 2 times with FACS buffer, and incubated with secondary antibody solution for 20 min, if needed. Detailed information of all antibodies for FACS of murine and human samples is stated in the Reporting Summary. Validation for cell sorting and the specificity of selected antibodies was initially performed using HEK-293 cells (ATCC, cat. CRL-1573). Cells were then washed several times and re-suspended in FACS buffer with DAPI (BD bioscience, cat. 564907). FACS was performed using a Becton Dickinson Aria II equipped with 5 lasers (BD bioscience). Beads (Invitrogen, cat. 01-3333-42) were used to set initial compensation. Fluorescence minus one (FMO) controls were used for additional compensation and to assess background levels for each stain. Gates were drawn as determined by internal FMO controls to separate positive and negative populations for each cell surface marker. Typically, 2.5 million events were recorded for each FACS analysis, and the data were analyzed using FlowJo (v10.6.1). To better depict the FACS gating strategy, arrows have been added to the plot to illustrate parent/daughter gates. Additionally, Extended Data Figures have been provided to show the full details of the FACS profiles and gating strategies used (Extended Data Fig. 1g, 3i, 4h, i, l, 5n, 10d, 11b, c, e).
Primary cell culture and clonal differentiation assays.
The clonal multipotency of sorted cell populations was evaluated under normoxic conditions. Mouse primary cells were grown in a complete MesenCult medium (Stem Cell Technologies, cat. 05501) with stimulatory supplements (Stem Cell Technologies, cat. 05502, 05500). After initial cell plating, cells were left undisturbed and allowed to grow at 37 °C under humidified conditions for a week. Half of the medium was replaced every 3 days. Cells were passaged once they were 60–70% confluent using a cell dissociation reagent (Gibco, cat. A1110501). Cells sorted from human calvarial tissue were cultured under conditions similar to those described above using a commercial medium preparation (Stem Cell Technologies, cat. 05401) with stimulatory supplements (Stem Cell Technologies, cat. 05402). The differentiation potential of 10 colonies derived from single FACS-isolated cell populations was examined. In brief, single sorted cells were plated at a density of 100 cells in 6-well culture plates and allowed to form individual colonies. Initial dispersion of the plated cells as single cells was confirmed by light microscopy. Each of the selected colonies was extracted using a cloning cylinder. The extracted cells were regrown for 3 days in 12-well plates and then allowed to differentiate under both osteogenic and adipogenic conditions as described below. For osteogenic differentiation and Alizarin Red S staining, sorted cells were expanded and then allowed to differentiate using an osteogenic differentiation medium (Gibco, cat. A1007201) for 21 days. The medium was changed every other day. At the end of this period, cells were washed with cold PBS and fixed with 4% PFA for 30 min on ice. Cells were washed with distilled water and stained with Alizarin Red S for 2 min. Cells were then washed thoroughly with water and air-dried before microscopic visualization. Adipogenic differentiation studies were conducted with sorted cells using methods similar to those described above. In brief, cells were allowed to differentiate in an adipogenic differentiation medium (Gibco, cat. A1007001). Fresh medium was added every other day for a total of 14–20 days. The medium was removed, and cells were washed with PBS and fixed with 4% PFA for 30 min at room temperature. Cells were rinsed again with PBS and stained for 30 min with Oil Red O working solution (3:2 dilution with water). After washing 5 times with PBS, cells were then observed under a light microscope. For chondrogenic differentiation and Alcian Blue staining, cells were grown as pellet culture. 1 × 104 cells in chondrogenic differentiation medium (Gibco, cat. A1007101) were centrifuged at 1500 rpm for 5 min in 15 ml conical tubes. Cultures were re-fed every 3 days and allowed to differentiate for a minimum of 14 days. At the endpoint of this study, the medium was removed, and pellets were harvested and washed carefully with PBS and fixed with 4% PFA for 30 min, then embedded in the OCT compound. After cryo-sectioning, slides were stained for 30 min with 1% Alcian Blue solution, washed three times with 0.1 N HCl, and then with PBS.
Mesensphere assays.
Mesensphere assays were performed as described previously1. Briefly, single cells isolated by FACS from mouse or human calvarial sutures were plated at a density of 100 cells per cm2 and allowed to grow in ultra-low adherence culture dishes (Stem Cell Technologies, cat. 27145). Plates were incubated at 37 °C with 5% CO2 and left undisturbed for a week. Half of the medium was replaced every 5 days. 10 days later, mesenspheres formation was observed under the microscopy and dissociated into single cells using Accutase solution (Gibco, cat. A11105–01), then was subsequently re-plated to generate secondary and tertiary mesenspheres.
Colony-forming unit-fibroblast (CFU-F) and osteoblast (CFU-Ob) culture and differentiation.
Freshly FACS-isolated murine cells were seeded at a clonal density of 2×103 cells per well in 6-well culture plates with MEM α (Gibco, cat. A1049001) containing 10% serum, and triplicate cultures were established. Half of the medium was replaced every 3 days. CFU-F was measured after 10 days of culture by Crystal violet staining and CFU-Ob was measured at 28 days of culture by Alizarin Red S staining after light fixation with 4% PFA. The number of colonies comprising at least 50 cells was counted in each well on day 10 (CFU-F) and day 28 (CFU-Ob). For human cells, cells were seeded at a clonal density of 1×102 cells per well in 6-well culture plates and then subjected following the same procedure as described above.
μCT scans and analysis.
μCT analysis was conducted on a Scanco Medical μCT 35 system at the Citigroup Biomedical Imaging Core as previously described48. Briefly, a Scanco Medical μCT 35 system with an isotropic voxel size of 7 μm and 20 μm was used to image the distal femur/tibia and skull, respectively. Scans were conducted in PBS and used an X-ray tube potential of 55 kVp, an X-ray intensity of 0.145 mA, and an integration time of 600 ms. μCT analysis was performed by an investigator blinded to the genotypes of the animals.
Renal capsule transplantation for bone organoids.
Freshly FACS-purified mouse tdTomato-expressing DDR2+ CSCs and mGFP-expressing CTSK+ CSCs from Ctsk-Cre;mTmG mice were transplanted underneath the renal capsules of 8 to 10-week-old MIP-GFP mice. To avoid the potential immunogenicity of GFP itself, all transplantation studies were conducted in MIP-GFP hosts that have baseline immunologic tolerance to GFP variants. For human DDR2+ CSCs and DDR2− CSCs, cells freshly isolated by FACS were transplanted under the renal capsule of 8 to 12-week-old immunodeficient NSG-EGFP mice. 1 to 2×104 murine or human cells were implanted underneath the renal capsule. The number of cells implanted for serial transplantation studies is included in the corresponding figure legends. Renal capsule transplantation was performed as previously described1. Briefly, mice were anesthetized and shaved on the left flank and abdomen before sterilization at the surgical site. The kidney was externalized through a 1 cm incision and a 2 mm pocket was made in the renal capsule. A matrigel plug (Corning, cat. 356231) containing the cells of interest was implanted underneath the capsule and the hole was sealed using a cauterizer before replacing the kidney back into the body cavity. Approximately more than 75 donor mice were sacrificed to harvest the desired number of CSCs for every organoid-based transplantation experiment. Animals were euthanized by CO2 after the indicated experimental duration. After sacrifice, kidneys were lightly fixed with 4% PFA for 6 hours and mineralized bone formation was detected by μCT scans and fluorescent stereomicroscopes. Samples were subjected to infiltration, embedding, and sectioning as described below. Human xenografts derived from hDDR2+ CSCs were detected by both staining with human-specific nuclear antigen and observing the absence of a host-expressed GFP transgene.
Intramuscular transplantation for determination of stemness and differentiation hierarchy.
In validation studies, we found that the intramuscular site was more suitable than the kidney capsule for serial transplantation studies, as it was easier to physically separate graft tissue from surrounding host parenchyma during re-isolation after each round of transplantation (data not shown). Intramuscular transplantation was performed in 6 to 8-week-old MIP-GFP or immunodeficient NSG-EGFP mice. A 1 mm longitudinal incision was made on the right hindlimb and the right anterolateral femur was exposed. The vastus lateralis muscle was dissected and a 2–3 mm muscle pouch was surgically created. A surgifoam absorbable gelatin sponge (Ethicon, cat. 1972) containing sorted cell populations of interest was placed into the muscle pouch. For human DDR2+ CSCs and DDR2− CSCs, cells isolated by FACS were labeled with DiD lipophilic cell membrane labeling dye-AF647 (Invitrogen, cat. V22887) prior to transplantation. The overlying fascia was closed by using 4–0 polyglactin 910 absorbable sutures (Ethicon, cat. J386), and wound clips were used to close the skin incision. For serial transplantation studies, animals were euthanized by CO2 narcosis 7–10 days post-surgery, and the muscle housing the grafts was dissociated for re-isolation of graft cells by FACS for the second round of transplantation. After digestion of the primary grafts, the resulting single-cell suspension was subsequently processed for sorting as described above. Cell populations of interest self-renewing after the first round of transplantation were again re-isolated and transplanted into new hosts. For mouse studies, 2 to 3×104 DDR2+ CSCs were initially transplanted into primary recipients. 1 to 3×103 DDR2+ CSCs were re-isolated from the primary transplants and then re-transplanted. Less than approximately 1×103 DDR2+ CSCs were re-isolated from the secondary transplants. Grafts were explanted and analyzed 7 days after transplantation. For human studies, 2 to 5×105 hDDR2− CSCs and hDDR2+ CSCs were initially transplanted into the immunodeficient NSG-EGFP primary recipients. 2 to 5×104 hDDR2− CSCs and hDDR2+ CSCs were re-isolated from the primary transplants and then transplanted. 1 to 3×103 hDDR2− CSCs and hDDR2+ CSCs were re-isolated from the secondary transplants and then transplanted into the kidneys of tertiary recipients. Grafts were explanted and analyzed 10 days (for FACS) and 21 days (for bone organoid formation) after transplantation.
Suture ablation model.
4-week-old MIP-GFP mice were anesthetized and had the skin overlying the skull shaved before sterilization of the surgical site. A scalpel was used to carefully remove the periosteum of the skull prior to removing the lambdoid sutures and surrounding bones on the right side of the skull using a micro hand drill with 0.2 mm drill bits (Stoelting, cat. 58610). Cell populations of interest (2×104 cells) were isolated by FACS from calvarial sutures of Ctks-Cre;mTmG or iDTR;Ctsk-Cre;mTmG mice and were mixed with 2 μl of a gel mixture (GelMA:Matrigel:Collagen I, 6:3:1) (Cellink, cat. IK3051020303, Corning, cat. 356231, Gibco, cat. A1048301, respectively) prior to implantation at the defect site. The gel mixture, with or without cells, was cross-linked by exposure to UV light for a few seconds after transplantation and the skin incision was closed using an absorbable suture. Animals were euthanized by CO2 and the specimens were collected after 8 and 16 weeks.
Sample preparation for cryo-sectioning and imaging.
Freshly extracted mouse samples were fixed with 4% PFA for 4–6 hours at 4 °C. Samples were washed with PBS and decalcified with daily changes of 0.5 M EDTA for 1–5 days depending on the age of the samples. Samples were incubated with infiltration medium (20% sucrose + 2% polyvinylpyrrolidone in PBS) with rocking until they sank to the bottom of the tube. Embedding was performed with a customized embedding medium (OCT + 15% sucrose in PBS) and samples were preserved at −80 °C. Sections, 10–20 μm in thickness, were cut using a Leica cryostat.
Immunohistochemistry.
Immunohistochemical analysis was conducted on Zeiss instruments and systems at the Optical Microscopy Core using the previously described49. In brief, frozen samples were thawed at room temperature and rehydrated with PBS, permeabilized with 0.3% Triton X-100 in PBS for 10 min, and blocked for 30 min with 5% donkey serum in PBS. Dilutions of primary antibodies (1:50–1:100) were freshly prepared in 0.3% Triton X-100 in PBS. Samples were incubated overnight with primary antibodies at 4 °C and, then washed three times with PBS. Secondary antibodies (1:1000 dilution) were added to samples for 1 hour, followed by washing three times with PBS. Samples were finally mounted with ProLong Gold antifade reagent with or without DAPI (Invitrogen, cat. P36931). Detailed information of all antibodies used for the immunohistochemistry of murine and human samples is stated in the Reporting Summary. Imaging was performed with a Zeiss LSM 880 laser scanning confocal microscope. All data were processed using Zeiss ZEN 2.3 SP1 software. In Figure 6a, b, Extended Data Fig. 10a, formalin-fixed paraffin-embedded (FFPE) human calvarium specimens were used for histology and stained with the antibodies as indicated in images.
Bulk RNA Sequencing.
Total mRNA was freshly extracted from FACS-isolated CSC populations from P7 Ctsk-Cre mice (n=5) using RNeasy Plus Micro Kit (Qiagen, cat. 74034). Total RNA integrity was checked using a 2100 Bioanalyzer (Agilent Technologies). cDNA synthesis and amplification were performed by SMART-Seq v4 ultra-low input RNA kit (Takara Bio, USA) starting with 1 ng of total RNA from each sample. 150 pg of qualified full-length double-strand cDNA was used and processed for Illumina library construction with the Nextera XT DNA Library Preparation Kits (Illumina). Then the normalized cDNA libraries were pooled and sequenced on an Illumina NovaSeq6000 sequencer with pair-end 50 cycles. Bulk RNA sequencing was performed on a total of 10 FACS-isolated CSC populations (5 CTSK+ and 5 DDR2+) from mice, and a counts matrix was obtained. DESeqDataSet was generated using this unnormalized counts matrix. Differential expression analysis was performed using the ‘DESeq’ function and results tables were generated using the ‘results’ function to compare the DDR2+ vs. CTSK+ groups50. Log fold change shrinkage was performed using the ‘lfcShrink’ function with the ‘apeglm’ method51 for visualization and ranking of genes. Results were exported as .csv files (one with all the genes, and one with the subset of genes with adjusted P value < 0.05). 2-D PCA plots of the samples were generated by the first two principal components using the ‘plotPCA’ function. Normalized read counts were used for generating heatmap plots. The data was visualized by heatmap with hierarchical clustering in R v3.6.2 with the pheatmap package v1.0.12. GO analysis was performed using DAVID Bioinformatics Resource tools v6.852,53.
Analysis of single-cell RNA Sequencing (scRNA-Seq).
Single-cell RNA sequencing data using public datasets published54,55 was analyzed by Seurat v4.3.0 for the standard cell clustering and visualization. Briefly, the clustering in the Ayturk et al. report55 was used as reported in that manuscript for the analysis here without modification. For the Farmer et al. report54, clustering analysis was repeated due to the distinct experimental focus of the present report. FindNeighbors() and FindClusters() functions were used for this clustering. In both cases, filtering based on mitochondrial and ribosomal RNA content was not modified from the analysis performed in the initial studies reporting this data. The AddModuleScore() function was performed to annotate all cells for both CTSK+ CSC and DDR2+ CSC-associated gene expression signatures. The gene list comprising the CTSK+ CSC and DDR2+ CSC expression signatures was generated by taking the top 50 differentially expressed genes in each of these cell types in the bulk RNA sequencing data in Fig. 3i. This gene list was further trimmed by evaluating the expression of all genes in the Ayturk et al. and Farmer et al. scRNA-Seq datasets, genes displaying negligible expression were dropped from the gene signature as noncontributory. Components of the gene expression signatures used for this score are individually displayed in Extended Data Fig. 7e (iii), f (iii), and the full list of genes comprising these scores is provided in Supplementary Table 2. Expression of selected individual components of these gene expression signatures, focusing on genes with biologic relevance to craniosynostosis, is displayed in violin plots in Extended Data Fig. 7e (iii), f (iii). For visualization of the clusters and the projection of the CTSK+ CSC and DDR2+ CSC gene expression signature score on these cells, UMAP was used for the Farmer et al. and tSNE was used for the Ayturk et al. reports to mirror the data visualization methods used in the initial reports and thereby facilitate comparison to the analysis originally reported.
Statistical analyses.
All data are shown as mean ± s.d. as indicated. For comparisons between two groups, unpaired, two-tailed Student’s t-tests were used. For comparisons of three or more groups, one-way or two-way ANOVA was used if normality tests passed, followed by Tukey’s multiple comparison test for all pairs of groups, unless otherwise specified. GraphPad PRISM v8.1.2 was used for statistical analysis. P < 0.05 was considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Extended Data
Extended Data Fig. 1 |. Analysis of skeletal CTSK lineage cells in the calvarial sutures of wildtype (WT) and Twist1Ctsk mice.

a, μCT images of the skulls of WT and Twist1Ctsk mice (P0 WT, n=6 Twist1Ctsk, n=8; P7 WT, n=8 Twist1Ctsk, n=5; P14 WT, n=6 Twist1Ctsk, n=6; P21 WT, n=6 Twist1Ctsk, n=5; P56 WT, n=9 Twist1Ctsk n=6). Twist1Ctsk mice display 100% penetrance of fusion at the squamous (SQ), occipitointerparietal (OIP), and lambdoid (LAM) sutures from 14 days of age and onwards. Partial penetrance of coronal (COR) suture fusion (16–40%) is observed. b, H&E images of the OIP suture in WT and Twist1Ctsk mice at P21. The black arrow indicates the fused suture. c, Fluorescence images of the major sutures including the Interfrontal (IF), coronal (COR), sagittal (SAG), and lambdoid (LAM) sutures in WT (Twist1+/+;Ctsk-Cre;mTmG) and Twist1Ctsk (Twist1fl/fl;Ctsk-Cre;mTmG) mice at P21. d, FACS plots (left) and absolute and relative quantification (right) of the amount of skeletal CTSK lineage cells (CD31−CD45−Ter119−CTSK-mGFP+) from the major sutures of WT (n=4) and Twist1Ctsk (n=4) mice at P14. Lin indicates CD31, CD45, and Ter119 for all FACS analyses. e, FACS plots and absolute cell number of the skeletal CTSK lineage cells from the calvarial sutures of WT (n=4) and Twist1Ctsk (n=4) mice at E16.5. f, FACS plots (left) and absolute and relative quantification (right) of apoptotic cells within the skeletal CTSK lineage cells from the sutures of WT (n=3) and Twist1Ctsk (n=3) mice at P2. g, Gating strategy for FACS analysis of CTSK+ CSC and CTSK− CSC populations in the sutures of WT (Ctsk-Cre;mTmG) mice. h, Twist1 gene expression levels in FACS-isolated CTSK+ CSCs and CTSK lineage cells of WT (n=3) and Twist1Ctsk (n=3) mice as determined by RT-PCR. QuantStudio 6 Flex RT-PCR Software v1.3 was used for mRNA analysis. i, FACS analysis of the frequency of cell populations in the calvarial suture. i) CTSK+ CSCs and CTSK− putative CSCs (CTSK− pCSCs) relative to total skeletal lineage cells in the sutures of WT (n=12) and Twist1Ctsk;mTmG (n=17) mice. ii) Absolute and relative quantification of the amount of CTSK+ PP1 (Lin−Thy-1.2−6C3−CTSK+CD200−CD105−) and PP2 (Lin−Thy-1.2−6C3−CTSK+CD105+) populations in the sutures of WT (n=7) and Twist1Ctsk;mTmG (n=9) mice. j, Images of immunostaining for Ki-67 of the LAM sutures in WT (n=3) and Twist1Ctsk;mTmG (n=3) mice at P10. Quantification of the number of tdTomato and Ki-67 double-positive cells per high power field in the LAM and OIP sutures (bottom). k, Confocal fluorescence imaging for Osteocalcin (OCN) staining of the COR (i) and LAM sutures (ii) in WT and Twist1Ctsk;mTmG mice. Two regions in the LAM sutures from each group (n=4) were measured for quantification. l, Confocal immunofluorescence imaging for hallmarks of active endochondral ossification including staining for osteoclasts (TRAP), chondrocytes (COL2A1, COMP), molecular mediators of endochondral ossification (MMP13), markers of hypertrophic chondrocyte apoptosis (Cleaved Caspase-3 (CC3)), and vascular invasion (CD31) in consecutive slices of the OIP sutures of Twist1Ctsk;mTmG mice at P7. The insets show high magnification of the region demonstrating that sutural cartilage templates derived from non-CTSK lineage cells in Twist1Ctsk mice. m, n, Timecourse Safranin O staining (red) and representative low-power (i) and high-power (ii) images of the OIP (m) and LAM (n) sutures in WT (n=3) and Twist1Ctsk (n=3) mice. In the OIP and LAM sutures, the penetrance of this sutural endochondral ossification phenotype is 100% in Twist1Ctsk;mTmG mice at P10. Far-right, enlarged view of the box in each panel. Each of dots shown in the graph represents an individual mouse. ****P < 0.0001, P values are shown. Mean ± s.d., unpaired, two-tailed Student’s t-test (d, e, h-k), two-way ANOVA with Sidak’s corrections for multiple comparisons test (f). All images and FACS plots are representative of at least three independent experiments. Scale bars are denoted in images.
Extended Data Fig. 2 |. In vivo depletion of CTSK+ cells using an inducible DTR (iDTR) system.

a, Schematic representation of the experimental design and gross appearance of ROSA26iDTR and iDTRCtsk mice before and after DT administration. The red arrow in image indicates an abnormal dome-shaped skull. b, Immunostaining for Cleaved Caspased-3 (CC3) in the sutures of iDTRCtsk;mTmG mice with PBS or DT. n=3 independent experiments. Green, CTSK+ (mGFP) cells; Red, non-CTSK lineage (tdTomato) cells; Magenta, CC3. Far-right, enlarged view of the box. c, FACS analysis of the absolute amount of CTSK+ cells and CTSK− pCSCs in the sutures of iDTRCtsk mice with PBS (n=4) or DT (n=4) administration (i). Normalized absolute quantification of the indicated cell types by analyzing the same number of cells passing the FSC/SSC, doublet, and live gates in all specimens (ii). d, μCT scan 3D images of the skull of ROSA26iDTR and iDTRCtsk mice at P11, P21, and P35 after DT administration. Red arrows indicate suture defects including abnormal fusion and hypomineralization (ROSA26iDTR, n=5; iDTRCtsk, n=5). e, μCT scan 2D images of the skull with an enlarged view of the region outlined in blue of ROSA26iDTR and iDTRCtsk mice at P21 and P35. f, Confocal immunofluorescence images of Ki-67 and quantification of the number of tdTomato and Ki-67 double-positive cells per high-power field of the OIP and LAM sutures in iDTRCtsk;mTmG mice at P14 after PBS (n=3) or DT (n=3) administration. Dots in graphs represent an individual mouse. g-i, Representative μCT images of the cortical (g), trabecular bone (h), and tibias (i) in ROSA26iDTR and iDTRCtsk mice after DT administration. Arrows show cortical defects including uneven periosteal surfaces and a double cortex. Quantification of bone volume/total volume (BV/TV), trabecular number (Tb.N), trabecular thickness (Tb.Th), and trabecular separation (Tb.Sp) of trabecular bones in ROSA26iDTR (n=4) and iDTRCtsk (n=4) mice at P21 after DT administration (h). j, Representative confocal fluorescent images of CTSK-expressing osteoclasts in iDTRCtsk;mTmG mice at P35 after PBS or DT administration. Red arrows indicate CTSK+ osteoclasts in the interparietal bones. Quantification of the number of osteoclasts per high-power field (HPF) of the sutures iDTRCtsk;mTmG mice after PBS or DT administration. Two or three regions from each group (n=3) were measured for quantification. k, Experimental scheme for functional inhibition of osteoclast activity using alendronate (1 mg/kg) and μCT scan images of the long bones and skulls of C57BL/6J mice at P42 after alendronate treatment. (Vehicle (PBS), n=3; Alendronate, n=5). ****P < 0.0001, P values are shown. Mean ± s.d., unpaired, two-tailed Student’s t-test (c, f, h, j). All images and FACS plots are representative of at least three independent experiments. Scale bars are denoted in images.
Extended Data Fig. 3 |. Characterization of DDR2+ CSCs.
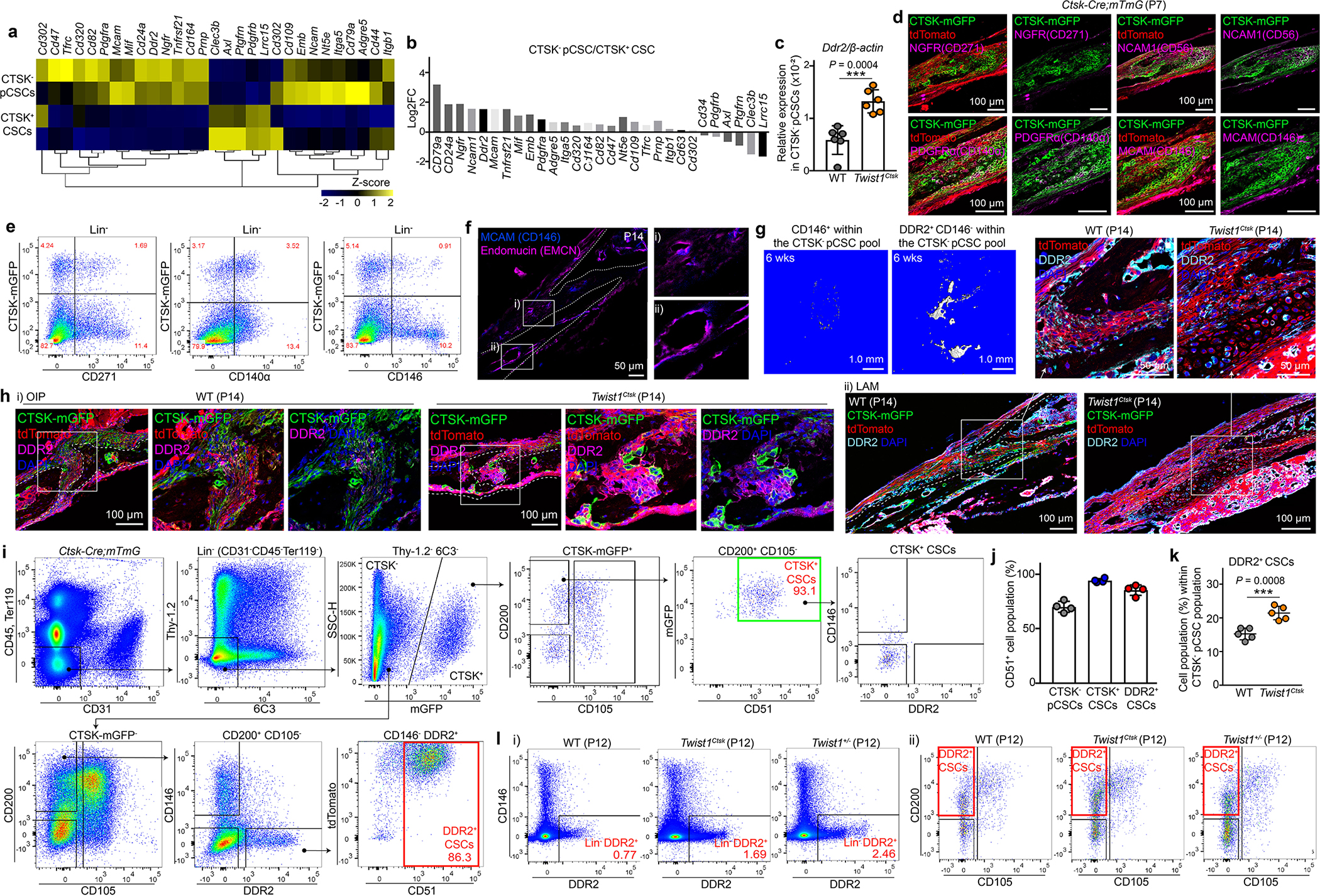
a, b, Candidate cell surface markers from gene expression analysis of FACS-purified CTSK+ CSC and CTSK− pCSC populations determined by RNA-Seq. c, Ddr2 mRNA expression in FACS-isolated CTSK− pCSCs of WT (n=6) and Twist1Ctsk (n=6) mice analyzed by RT-PCR. d, Immunostaining for candidate cell surface markers including NGFR (CD271), NCAM1 (CD56), PDGFRα (CD140α), and MCAM (CD146) in the calvarial suture of Ctsk-Cre;mTmG mice at P7. Experiments were repeated 3 independent times. e, Flow cytometry for CD271, CD140α, and CD146 versus CTSK lineage cells in the calvarial sutures. f, Immunofluorescence staining for CD146 and Endomucin (EMCN) in the calvarial sutures. g, μCT 3D images of the bone formed by CD146+ (left) and DDR2+CD146− (right) populations isolated from the pool of CTSK− pCSCs 6 weeks after transplantation. Images are representative of 3 independent experiments. h, Immunostaining for DDR2 in the OIP (i) and LAM (ii) sutures of WT and Twist1Ctsk;mTmG mice at P14. i, Flow cytometry gating strategy for CTSK-negative DDR2+ CSCs in the calvarial sutures of P7 Ctsk-Cre;mTmG mice. Black arrows indicate parent/daughter gates. j, Flow cytometry analysis of the percentage of CD51+ cells within CTSK− pCSC, CTSK+ CSC, and DDR2+ CSC populations (n=4). k, Quantification of the percentage of DDR2+ CSCs within the pool of the CTSK− putative stem cell fraction in the sutures of WT (n=5) and Twist1Ctsk;mTmG (n=5) mice as determined by flow cytometry. l, FACS plots showing skeletal DDR2+ cells (i) and DDR2+ CSCs (ii) in the sutures of WT, Twist1Ctsk, and Twist1+/− mice at P12. ****P < 0.0001, P values are shown. Mean ± s.d., unpaired, two-tailed Student’s t-test (c, k). Images in d, f, h are representative of a minimum of three biological replicates, and FACS plots are representative of three independent experiments. Scale bars are denoted in images.
Extended Data Fig. 4 |. Determination of the stemness and differentiation hierarchy of DDR2+ CSCs.

a, Fluorescence images of a timecourse of single cell-derived clonal expansion in FACS-purified CTSK+ CSCs and DDR2+ CSCs isolated from Ctsk-Cre;mTmG mice (i) and no induction of Ctsk-Cre driven mGFP was observed in DDR2+ CSCs (ii). b, Alcian Blue (i), Alizarin Red S (ii), and Oil Red O (iii) staining for in vitro clonal multipotency. 10 single FACS-isolated CTSK+ CSCs or DDR2+ CSCs were expanded and able to undergo both osteoblast and adipocyte differentiation. All cells were derived from the expansion of a single FACS-isolated CTSK+ CSC or DDR2+ CSC. Bar graphs show the clonal efficiency demonstrating that all colonies were capable of both osteoblasts and adipocytes differentiation from the same clone of cells. 10 colonies were examined in triplicate experiments. c, Images showing the fibroblast colony-forming units (CFU-F) derived from FACS-isolated CTSK+ CSCs or DDR2+ CSCs. The number of CFU-F colonies per well was quantified at day 10 after seeding (n=5 single FACS-isolated CTSK+ CSCs or DDR2+ CSCs). d, In vitro limiting dilution analysis and CFU-F formation. Dilutions of 500, 200, 100, 50, 20, and 10 CTSK+ CSC or DDR2+ CSCs were seeded onto 6-well culture plates. e, Clonal expansion and differentiation into osteoblasts in FACS-isolated CTSK+ CSCs and DDR2+ CSCs. Colonies were stained for Alizarin Red S at day 28. The number of CFU-Ob colonies per well was quantified (n=6 single FACS-isolated CTSK+ CSCs or DDR2+ CSCs). f, Bright-field and fluorescent images of initial seeded, primary, secondary, and tertiary mesenspheres derived from CTSK+ CSCs (GFP, green), DDR2+ CSCs (RFP, red), and non-stem fraction of calvarial cells (Thy-1.2+ here) in CTSK+ or DDR2 lineage isolated from Ctsk-Cre;mTmG mice. g, Quantification of individual cell numbers per sphere (top) and the percentage of cells able to form spheres (bottom) for CTSK+ CSCs, DDR2+ CSCs, CTSK+Thy-1.2+, and DDR2+Thy-1.2+ cells. n=3 independent experiments. h, Experimental strategy for in vivo stemness and differentiation hierarchy study and gating strategy showing serially transplanted cell populations including DDR2+ CSCs (i), DDR2+Thy-1.2−6C3−CD200variableCD105+ (ii), DDR2+Thy-1.2−6C3−CD200−CD105− (iii), and DDR2+Thy-1.2+6C3− (iv) of the calvarial suture of the primary donor (Ctsk-Cre;mTmG mice). Freshly FACS-isolated 2 to 3×104 cells were transplanted into the muscle of primary recipients. i, FACS analysis of serially transplanted cells derived from ii), iii), and iv) populations after the first and second rounds of intramuscular transplantation. 1 to 3×103 cells after the first round of transplantation were re-isolated and re-implanted into the muscle of secondary recipients. FACS plots marked by black boxes show differentiation of the indicated populations after the first round of transplantation and plots marked by red boxes show these populations in secondary recipients after the second round of transplantation. Black and red arrows indicate parent/daughter gates and transplanted populations, respectively. Experiments underwent 5 independent repeats. j, μCT images of bone formation from transplanted CTSK+ CSCs and DDR2+ CSCs under the renal capsule of recipients after each round of serial transplantation. The number of transplanted cells is indicated in images. k, FACS plots of tdTomato-expressing DDR2+ CSC and mGFP-expressing CTSK+ CSC populations isolated from Ctsk-Cre;mTmG mice after the first round of transplantation. Collagen sponges without cells were used as a negative control. l, FACS plots demonstrating that CTSK+ CSCs did not induce expression of DDR2 over the course of serial transplantation and that CTSK+ CSCs were capable of self-renewal and generating the entire lineage of CTSK lineage cells present in initial donors after the first round of transplantation. ****P < 0.0001, P values are shown. Mean ± s.d., unpaired, two-tailed Student’s t-test (c, d), two-way ANOVA with Tukey’s multiple comparisons test (g). Images in a-f, j are representative of a minimum of three biological replicates, and FACS plots are representative of at least three independent experiments. Scale bars are denoted in images.
Extended Data Fig. 5 |. Long-term lineage tracing and label-retention studies in the calvarial suture.

a, Experimental design for lineage tracing of tamoxifen (TAM)-induced Ddr2-CreER;mTmG mice pulsed at P2. b, Confocal fluorescence imaging of the calvarial suture in Ddr2-CreER;mTmG mice before TAM induction. c, Short-term chase of calvarial DDR2 lineage cells in the area of lambdoid sutures in Ddr2-CreER;mTmG mice. Low-power (top) and high-power images (bottom) of the suture mesenchyme, calvarial bone, and marrow showing DDR2 lineage sutural resident cells and bone-adjacent osteoblasts 10 days after an initial pulse. Green, DDR2 lineage (mGFP) cells; Red, non-DDR2 lineage (tdTomato) cells; Blue, DAPI. d, e, Long-term chase for 6 (d) or 18 months (e) to visualize calvarial DDR2 lineage cells in Ddr2-CreER;mTmG mice. f, Timecourse fluorescence imaging of osteoblasts (i) and CD200-expressing cells (ii) derived from DDR2 lineage cells (green) in the lambdoid sutures of Ddr2-CreER;mTmG mice. g, Flow cytometry plot showing the concordance between DDR2 antibody staining and Ddr2-CreER pulse-chase studies in calvarial-lineage cells. In addition to DDR2 expression on skeletal cells, expression of DDR2 in subsets of Lineage+ leukocytes is noted. h, FACS analysis of genetic lineage tracing of DDR2 lineage cells in the calvarial sutures of Ddr2-CreER;mTmG mice. Red boxes indicate calvarial DDR2+ fractions (upper panel) and DDR2+ CSCs (Lin−DDR2-mGFP+ Thy1.2−6C3−CD200+CD105−, lower panel). i, Percentage of DDR2 ineage cell types including DDR2+ CSCs, DDR2+CD200varialbeCD105+, and DDR2+CD200−CD105− cells within the parent gate (Lin−DDR2+Thy-1.2−6C3− cells) 1 day (n=5), 10 days (n=6), 1 month (n=6), 3 months (n=5), and 12 months (n=7) after TAM induction in the calvarial sutures as determined by FACS. j, FACS quantification of genetic lineage tracing for DDR2 lineage cells in the calvarial sutures of Ddr2-CreER;mTmG mice 1 day (n=5), 10 days (n=6), 1 month (n=6), 3 months (n=5), and 12 months (n=7) after TAM administration. The relative and absolute amount of calvarial DDR2 lineage cells from the pool of total lineage-negative cells over a chase period in i and ii. The amount of specific DDR2 lineage cell types including DDR2+ CSCs, DDR2+CD200varialbeCD105+, DDR2+CD200−CD105−, DDR2+Thy-1.2+, and DDR2+6C3+ cells over the chase period is provided in iii. k, Flow cytometry plot showing H2B-GFP baseline expression in the calvarial sutures in the absence of doxycycline (dox). l, Plots showing H2B-GFP+ cells at baseline without dox exposure (i), H2B-GFPhigh label-retaining calvarial sutural cells exposed to dox over 6 months (ii). Plots for additional control mice that were continuously exposed to dox demonstrating a negligible level of H2B-GFP expression and no expression of DDR2 (iii). Black arrows indicate parent/daughter gates. m, FACS quantification of the absolute and relative amount of skeletal H2B-GFPhigh labeling in the calvarial sutures after dox exposure (H2B/tTA, n=7). Controls of mice that were continuously exposed to dox (tTA, n=2; H2B, n=2; H2B/tTA, n=5), including during the embryonic period by providing dox to pregnant female mice, continuing dox exposure while pups were breastfeeding and then subsequently continuing direct dox exposure after weaning. Freq, frequency. n, o, Gating strategy for H2B-GFPhigh label-retaining cells within DDR2+ CSC and DDR2− CSC populations and offset histogram overlays displaying GFP fluorescence in Lin−Thy-1.2−6C3−DDR2+ or DDR2− populations including CD200+CD105− (CSC), CD200variableCD105+, and CD200−CD105− after 6 (i) or 12 month (ii) chase periods. Black arrows indicate parent/daughter gates. p, Representative images (left) and quantification (right) of immunostaining for Galectin-1 (Gal-1) overlapping with CTSK+ (mGFP) cells in Ctsk-Cre;mTmG mouse calvarium at P14 (n=5). Green, CTSK+ (mGFP) cells; Magenta, Galectin-1; Blue, DAPI. A relative amount of Galectin-1+ cells overlapping with CTSK+ (mGFP) or non-CTSK lineage (tdTomato) cells was quantified per high power field. q, Representative immunostaining images for Galectin-1 in the calvarial sutures of H2B-GFP mice after 12 months of dox exposure. Co-localization is shown by white arrows. Green, H2B-GFP; Red, Galectin-1; Blue, DAPI. Each of dots shown in the graph represents an individual mouse. ****P < 0.0001, P values are shown. Mean ± s.d., two-tailed Student’s t-test (p), one-way ANOVA with Tukey’s multiple comparisons test (j, m). Images in b-f, p, q and FACS plots are representative of at least three independent experiments. Scale bars are denoted in images.
Extended Data Fig. 6 |. Functional characterization of DDR2+ CSCs.
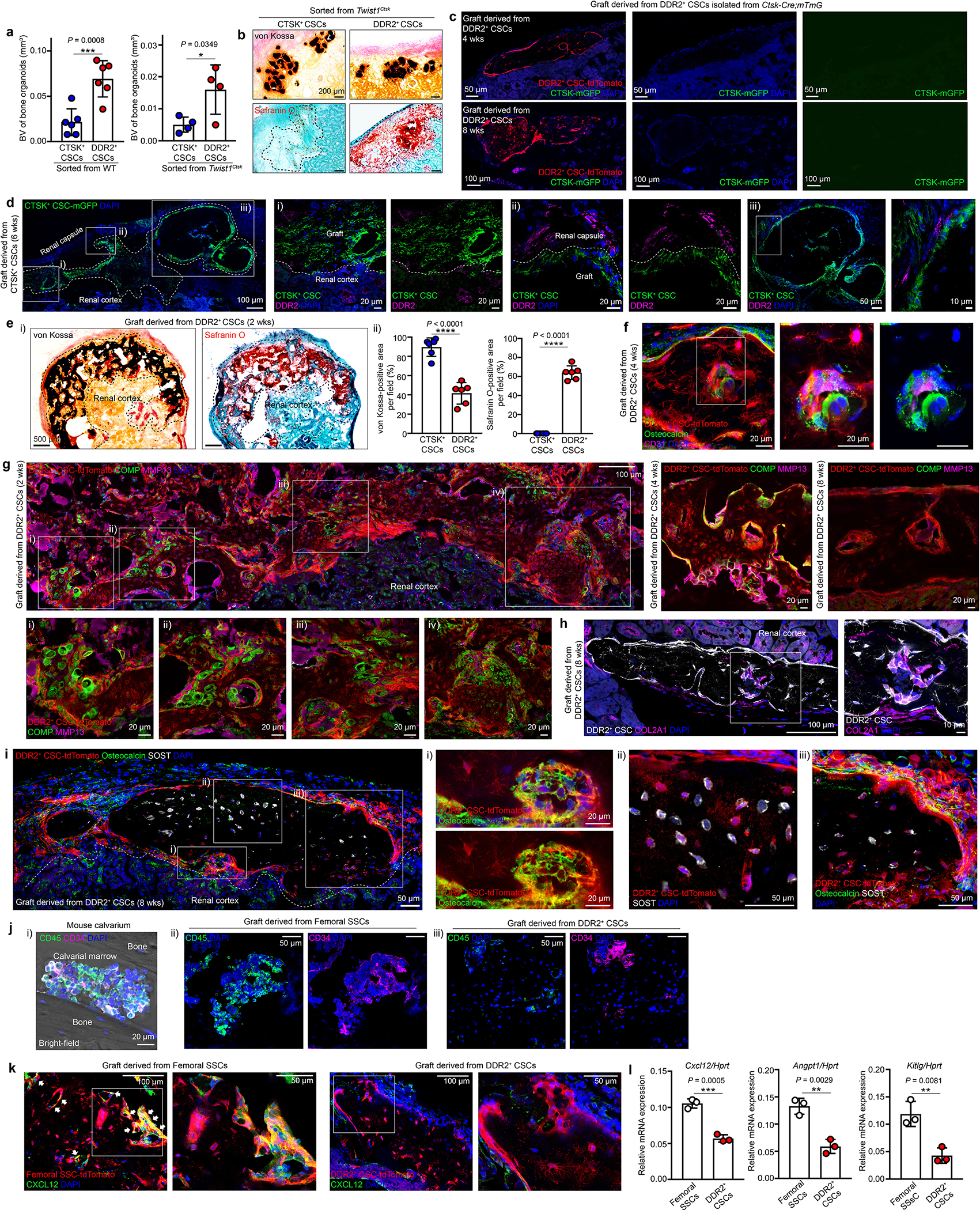
a, Quantification of bone volume (BV) when equal numbers of CTSK+ CSCs and DDR2+ CSCs were individually isolated from WT (left, n=6) or Twist1Ctsk (right, n=6) mice 4 weeks after transplantation. Each of dots shown in the graph represents an individual graft. b, Representative images of von Kossa staining (top, black) for mineralized bone and Safranin O staining (bottom, red) for cartilage in adjacent serial sections from renal capsule bone organoids derived from CTSK+ CSCs and DDR2+ CSCs isolated from Twist1Ctsk mice 4 weeks after transplantation. c, Fluorescence images of grafts derived from DDR2+ CSCs (red) isolated from Ctsk-Cre;mTmG mice by FACS 4 or 8 weeks after transplantation into the renal capsule of secondary recipients. Green, CTSK+ mGFP; Red, Primary graft (DDR2+ CSCs); Blue, DAPI. d, Representative immunostaining images for DDR2 (magenta) in the graft 6 weeks after transplantation of CTSK+ CSCs (green) into the renal capsule. e, Images of von Kossa and Safranin O staining in the adjacent serial sections of the same renal capsule bone organoid grafts derived from CTSK+ CSC or DDR2+ CSCs 2 weeks after transplantation (i). Quantification of the area of von Kossa and Safranin O staining is provided in ii. f-i, Endochondral ossification with active cartilage remodeling is seen in the grafts derived from DDR2+ CSCs (red) via immunostaining for Osteocalcin (green) and CD31 (magenta) 4 weeks after transplantation (f), COMP (green) and MMP13 (magenta) 2, 4, and 8 weeks after implantation (g), DDR2+ CSCs and their derivatives (white) and COL2A1 (magenta) (h), Osteocalcin (green) and SOST (white) at 8 weeks post-transplantation (i) into the renal capsules of primary recipients. Higher magnification images are provided with the corresponding Roman numerals. j, k, Endochondral ossification without recruitment of hematopoietic elements in DDR2+ CSC-derived bone organoids: Immunostaining for hematopoietic cells with CD45 (green) and CD34 (magenta) in mouse calvarium at P21 (i), grafts derived from freshly sorted femoral skeletal stem cells (SSCs) (ii), or DDR2+ CSCs (iii) (j). Immunostaining for CXCL12 (green) in the grafts derived from femoral skeletal stem cells (SSCs, red) (left) or DDR2+ CSCs (red) (right) (k) 4 weeks after the renal capsule transplantation. Co-localization is shown by white arrows. l, RT-PCR analysis of hematopoietic niche factors including Cxcl12, Angpt1, and Kitlg in FACS-isolated femoral SSCs (n=3) and DDR2+ CSCs (n=3). Each of dots shown in the graph represents the cells of interest isolated from an individual mouse. ****P < 0.0001, P values are shown. Mean ± s.d., unpaired, two-tailed Student’s t-test (a, e (ii), l). All images shown in this figure are representative of a minimum of three independent experiments. Scale bars are included in microscopy images.
Extended Data Fig. 7 |. Transcriptional analysis of CTSK+ CSCs and DDR2+ CSCs isolated from mouse calvarial sutures.
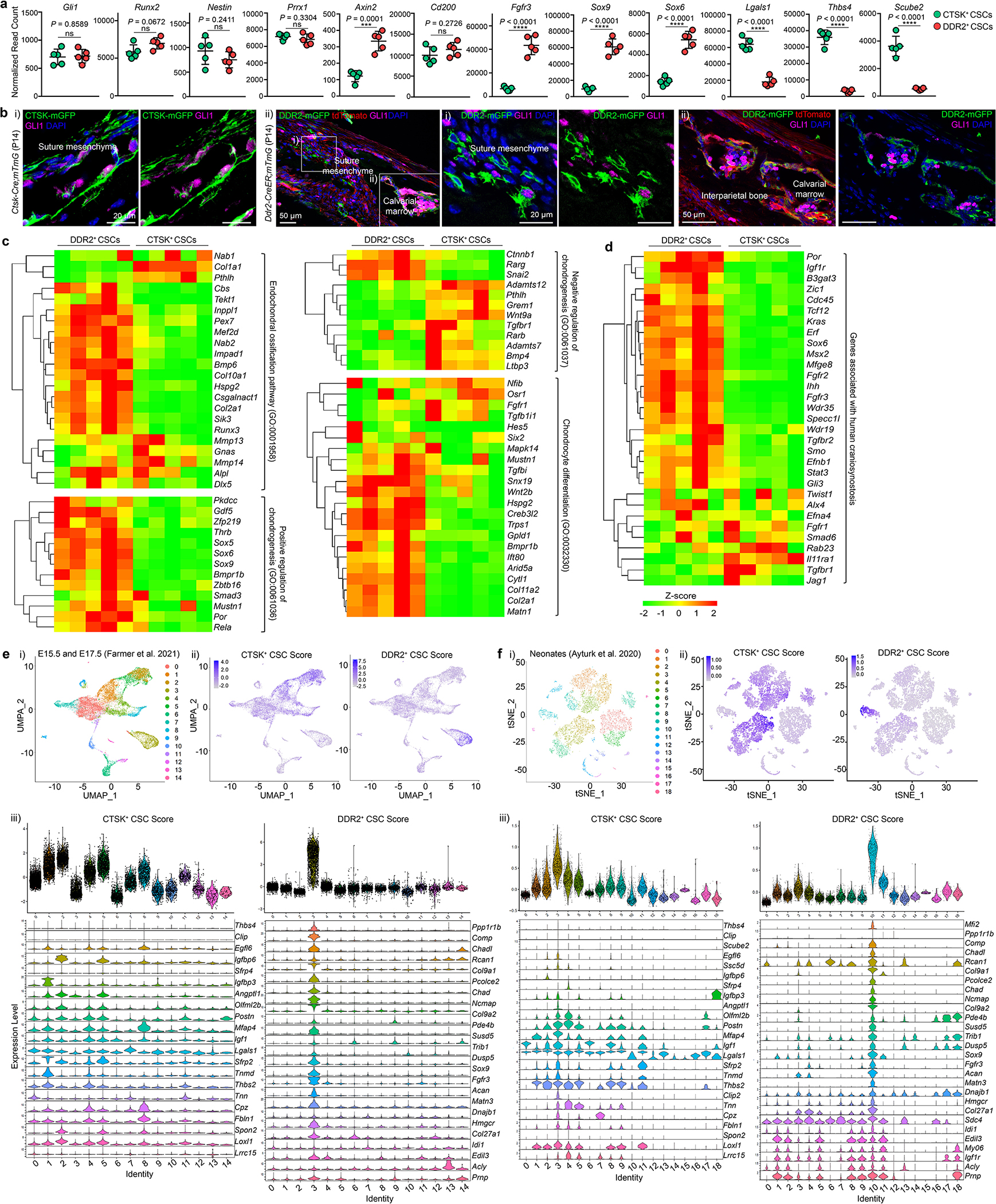
a, Bar graphs showing normalized read counts of Gli1, Runx2, Nestin, Prrx1, Axin2, Cd200, Fgfr3, Sox9, Sox6, Lgals1, Thbs4, and Scube2 as determined by RNA-Seq analysis of FACS-isolated CTSK+ CSCs (n=5) and DDR2+ CSCs (n=5) in the calvarial suture at P7. ****P < 0.0001, P values are shown. Mean ± s.d., unpaired, two-tailed Student’s t-test. b, Immunostaining for GLI1 in the calvarial sutures of Ctsk-Cre;mTmG (i) and Ddr2-CreER;mTmG (ii) mice at P14. Green, CTSK+ (mGFP) (i); DDR2+ (mGFP) (ii); Magenta, GLI1; Blue, DAPI. Images represent the results of 3 independent experiments. Scale bars are included in microscopy images. c, Hierarchical clustering of expression heatmaps showing differentially expressed genes associated with bone development, including endochondral ossification (GO:0001958), positive or negative regulation of cartilage development (GO:0061036; GO:0061037, respectively), regulation of chondrocyte differentiation (GO:0032330) by Gene Ontology Biological Process analysis from RNA-Seq in FACS-purified CTSK+ CSCs and DDR2+ CSCs at P7. d, Heatmap generated from murine bulk RNA-Seq analysis of sorted CTSK+ CSCs and DDR2+ CSCs showing differentially expressed genes associated with human craniosynostosis. e, f, scRNA-Seq analysis using public datasets published by Farmer et al. 202154 (e) and Ayturk et al. 202055 (f). i) UMAP (e) and tSNE (f) plots of calvaria single cells. ii) Pseudocoloring of the corresponding UMAP (e) or tSNE (f) plots for the CTSK+ CSC and DDR2+ CSC combined gene expression score used to identify cells expressing a CTSK+ CSC-like or a DDR2+ CSC-like transcriptional profile. iii) Violin plots showing the per cluster CTSK+ CSC and DDR2+ CSC score, including normalized expression of selected genes comprising the CTSK+ CSC score (left) and the DDR2+ CSC score (right).
Extended Data Fig. 8 |. Analysis of Ddr2slie mice and orthotopic transplantation of calvarial stem cells.
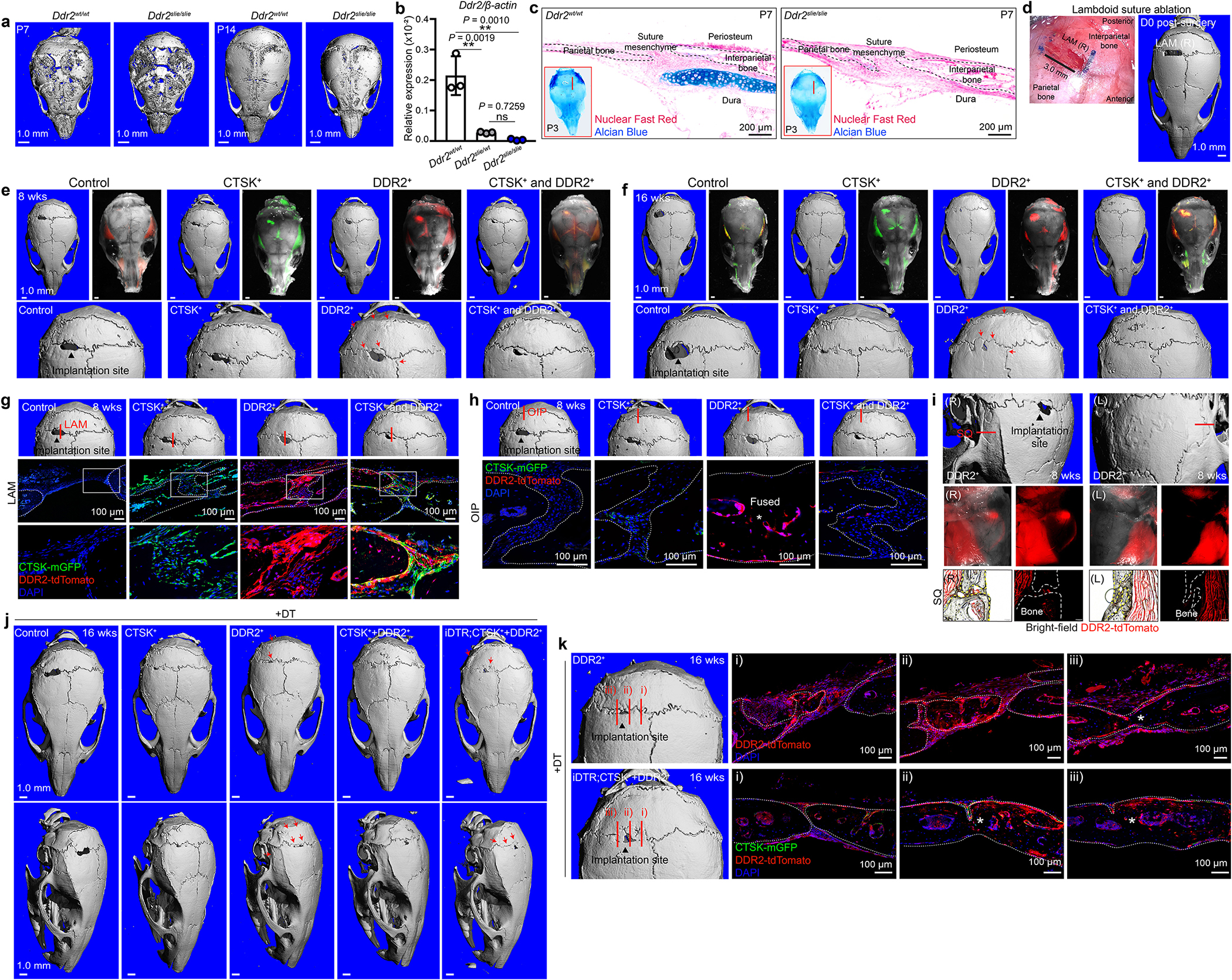
a, μCT scan 3D images of the skull of Ddr2wt/wt (WT) and Ddr2slie/slie (Ddr2slie) mice at P7 and P14. (P7 WT, n=7 Ddr2slie, n=8; P14 WT, n=5 Ddr2slie, n=6). b, RT-PCR analysis of Ddr2 expression in Ddr2wt/wt (n=3), Ddr2slie/wt (n=3), and Ddr2slie/slie (n=3) mice. Each of dots shown in the graph represents an individual mouse. P values are shown. Mean ± s.d., one-way ANOVA with Tukey’s multiple comparisons test. c, Alcian Blue staining of the lambdoid sutures in WT and Ddr2slie mice at P7. Nuclear Fast Red was used as a counterstain. Red boxes images: whole-mount P3 skull preparations with Alcian Blue staining of WT (n=8) and Ddr2slie (n=9) mice. Red lines in images indicate the sectioning area and direction. d, Surgical field and μCT scan 3D image after ablation of the right lambdoid (LAM) suture in 4-week-old MIP-GFP mice. e, f, μCT scans and stereomicroscopic fluorescence images of mouse skulls at 8 (e) or 16 weeks (f) post-transplantation. mGFP-expressing CTSK+ CSCs and tdTomato-expressing DDR2+ CSCs isolated from the calvarial sutures of P7 to P10 Ctsk-Cre;mTmG mice. Each indicated population was implanted into the suture ablation area after carrier encapsulation. The control group was implanted with only the carrier gel mixture without cells. Red arrows in μCT images indicate the fusing and fused sutures. g-i, Confocal fluorescence imaging of graft cells, showing the presence of CTSK+ CSC (green) and DDR2+ CSC (red) graft cells both at the initial site of implantation (LAM, g) and migration of graft cells from the initial site of implantation to adjacent sutures (OIP, h; SQ, i) at 8 weeks post-transplantation. Each red line in μCT images indicates the sectioning area and direction. Asterisk in the image denotes site of suture fusion. j, 3D reconstruction of μCT scans of the skull 16 weeks after orthotopic transplantation with subsequent DT administration. (Control, n=6; CTSK+, n=6; DDR2+, n=5; CTSK+ and DDR2+, n=5; iDTR;CTSK+ and DDR2+, n=4). Red arrows indicate the fusing and fused sutures. k, Fluorescent images of the sutures in mice receiving DDR2+ CSC or co-transplanted iDTR;CTSK+ CSC and DDR2+ CSC grafts 16 weeks after orthotopic transplantation with subsequent DT administration. tdTomato and GFP visualization showing engrafted cells at sites of newly formed cranial bone. Each red line in μCT images indicates the sectioning area and direction. Green, CTSK+ (mGFP); Red, DDR2+ (tdTomato); Blue, DAPI. Images are representative of at least five (a, c) biological replicates and four (e-k) independent experiments. Scale bars are denoted in images.
Extended Data Fig. 9 |. IGF1 secreted by CTSK+ CSCs suppresses fusion activity.

a, Heatmap showing differential expression of candidate signaling mediators (i) and graphs displaying normalized read counts of Igf1 and Igf1r in CTSK+ CSCs and DDR2+ CSCs determined by RNA-Seq at P7 mice (n=5). b, RT-PCR analysis of Igf1 gene expression in CTSK+ CSCs and CTSK lineage cells (Thy-1.2+/CD90+ here) from WT (n=3) and Twist1Ctsk (n=3) mice at P10. c, Schematic of the experimental design for subcutaneous injection of recombinant IGF1 over the calvarium of Twist1Ctsk mice. d, e, Characterization of Igf1fl/fl;Ctsk-Cre (Igf1Ctsk) mice: μCT scan 3D images (d) of the skull of WT and Igf1Ctsk mice at P28. Red arrows indicate fusing and fused sutures. μCT scans of 2D images (e, left) and 2D cut planes (e, right), The asterisk in e indicates fusing sutures. Each of dots shown in the graph represents an individual mouse. ****P < 0.0001, P values are shown. Mean ± s.d., unpaired, two-tailed Student’s t-test (a (ii)), one-way ANOVA with Tukey’s multiple comparisons test (b). Scale bars are denoted in images.
Extended Data Fig. 10 |. Endochondral ossification in human craniosynostosis and characterization of human DDR2+ CSCs.

a, Histological analysis of active cartilage remodeling occurring at sites of active suture fusion using H&E (top) and Alcian Blue (bottom) staining of FFPE calvarial specimens from patients with craniosynostosis. The specimen displayed represents a portion of the lambdoid suture. Higher magnification images are provided with the corresponding Roman numerals indicated in lower magnification images. b, c, Representative images demonstrating active endochondral ossification in patients with craniosynostosis: human calvarial specimens were immunostained for DDR2 (green) and Osteocalcin (red) (b) and DDR2 (green), COL2A1 (red), and Osteopontin (OPN, magenta) (c). d, FACS plots for human calvarium tissues. Lin indicates CD31, CD45, and CD235a staining. Red boxes indicate hDDR2− CSCs and hDDR2+ CSCs. Orange boxes indicate markers defining human SSC populations (PDPN+CD146−CD164+CD73+), reported in human fetal long bones34. Black arrows demonstrate parent/daughter gates. Experiments underwent a minimum of 5 independent repeats. e, Heatmaps showing gene expression features of human DDR2− or DDR2+ CSCs displaying conservation of gene expression signatures observed in their murine counterpart cell types (murine CTSK+ CSCs or DDR2+ CSCs, respectively), including expression of IGF1 on hDDR2− CSCs and IGF1R on hDDR2+ CSCs as analyzed by RT-PCR. hDDR2+ CSCs and hDDR2− CSCs were freshly isolated by FACS from five individual patients with craniosynostosis. The color scale (from low expression in blue to high expression in red) depicts the log2 fold change of gene expression. The sequence of the primers is provided in Supplementary Table 3. f, Representative images of immunostaining for DDR2 (green) and IGF1R (red) in human calvarial tissues of three individual patients with craniosynostosis. Higher magnification images are provided. Results are representative of a minimum of five (a-d) and three (f) independent experiments. Scale bars are denoted in images.
Extended Data Fig. 11 |. Determination of self-renewal, differentiation hierarchy, and functional characterization of hDDR2+ CSCs.

a, Schematic representation of the intramuscular transplantation model for human sutural cells. b, FACS plots showing the differentiation of the indicated input populations including hDDR2− CSCs (i), hDDR2+ CSCs (ii), hDDR2+ CD200− (iii), and matrigel only (iv) in primary recipients immunodeficient NSG-EGFP mice 10 days after intramuscular transplantation. 1 to 3×104 hDDR2− CSCs and hDDR2+ CSCs were initially transplanted into immunodeficient NSG-EGFP primary recipients. 1 to 5×103 hDDR2+ CSCs were re-isolated from the primary transplants. Matrigel without cells was used as a control. Black arrows demonstrate parent/daughter gates. Experiments underwent 3 independent repeats. c, Flow cytometry plots showing gating strategy for human DDR2+ and DDR2− CSCs with human-specific cell surface markers including human beta-2 microglobulin (β2M) and pan-human leukocyte antigen complex (HLA-ABC) in human calvarium. d, A schematic representation of in vivo serial transplantation studies of hDDR2− CSCs and hDDR2+ CSCs. To test the proliferative self-renewal and differentiation capacity of hDDR2− CSCs and hDDR2+ CSCs, freshly FACS-isolated cells were labeled with a lipophilic cell membrane dye prior to the intramuscular transplantation. Gross images of the primary grafts (arrowheads) explanted 10 days after transplantation. e, FACS plots showing that transplanted hDDR2− CSCs (top plots) and hDDR2+ CSCs (bottom plots) underwent multiple rounds of proliferation during the assays without interconversion in the primary grafts (i) or secondary grafts (ii). f, Immunostaining for the proliferation marker Ki-67 (green) in hDDR2− CSCs and hDDR2+ CSCs (red, stained by cytoskeleton protein, α-Tubulin) isolated after the second round of transplantation. g, In vitro limiting dilution analysis and CFU-F formation. Dilutions of 500, 200, 100, 50, 20, and 10 hDDR2− CSC or hDDR2+ CSCs were seeded onto 6-well culture plates. Colonies were stained with Crystal violet and analyzed 10 days after seeding. h, Images of Alizarin Red S (upper, red) and Alcian Blue (lower, blue) staining for in vitro clonal multipotency. All cells were derived from the expansion of a single FACS-isolated hDDR2− CSC or hDDR2+ CSC. i, Quantification of the percentage of cells able to form mesenspheres from hDDR2− CSCs, hDDR2+ CSCs, hDDR2− CD200−, and hDDR2+ CD200− cells. n=3 independent experiments. j, Quantification of the area of von Kossa and Safranin O staining in the renal capsule after transplantation of hDDR2− CSCs and hDDR2+ CSCs at 2 weeks post-transplantation. n=3 independent experiments. k, Cellular and molecular features of endochondral ossification of hDDR2+ CSCs: immunostaining for skeletal cell type markers and key markers of endochondral bone development including Osteocalcin (i) and Endomucin (EMCN) (ii) in human xenografts derived from FACS-isolated hDDR2+ CSCs. Images in f-h represent five individual experiments and k represent a minimum of three independent experiments. Plots in b, c, e are representative of results from three independent experiments. ****P < 0.0001, P values are shown. Mean ± s.d., unpaired, two-tailed Student’s t-test (j), two-way ANOVA with Tukey’s multiple comparisons test (i). Scale bars are included in microscopy images.
Supplementary Material
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2021R1A6A3A14038667 to S.B.); Arthritis Grant Program from the Arthritis National Research Foundation (ANRF) (1065843 to S.B.); National Institutes of Health (NIH) under awards DP5OD021351 and R01AR075585 (to M.B.G.); 1K99 DE031819-01 by NIDCR (to S.D.); 2T32AR071302-07 from NIAMS (to A.R.Y); T32-AR078751 from NIAMS (to K.W.M); R01DE029465 (to R.T.F.); JumpStart Research Career Development Award from Weill Cornell Medicine (to S.B. and S.D.); Study Abroad Scholarships from the Mogam Science Scholarship Foundation (to S.B.); Career Award for Medical Scientists from the Burroughs Welcome Foundation (to M.B.G.); the Pershing Square Sohn Cancer Research Alliance and the Maximizing Innovation in Neuroscience Discovery (MIND) Prize (to M.B.G.); ASBMR Career-Life Balance Initiative award (to S.D.); Tri-Institutional Breakout Prize for Junior Investigators (to S.D.). This content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health. We thank Kyong-Tai Kim at POSTECH for his support and mentorship, Ren Xu, Na Li, Yuzhe Niu, Jessica Ling Zheng, Emile-Victor Kuyl, Wei Zhang, Seungjoon Noh, Bing He, Leticia Dizon, Taotao Zhang, Sushmita Mukherjee, Leona Cohen-Gould for their technical support, Rodolfo Ricart Arbona, Anna Rodriguez, George Salvador for animal care, Michelle M. Buontempo, Ashley Rowley O’Connor for clinical specimen management, and the Center for Translational Pathology, Flow Cytometry Core, Genomics Resources Core, Optical Microscopy Core, the Citigroup Biomedical Imaging Core, and the Research Animal Resource Center at Weill Cornell Medicine for their support.
Footnotes
Competing interest declaration The authors declare no competing interests.
Reporting summary. Further detailed information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
RNA-Seq data has been deposited at the Gene Expression Omnibus (GEO) under accession number GSE232652. Source data are provided with this article.
Main references
- 1.Debnath S et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 562, 133–139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Howard TD et al. Mutations in TWIST, a basic helix–loop–helix transcription factor, in Saethre-Chotzen syndrome. Nat. Genet. 15, 36–41 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Ghouzzi V El et al. Mutations of the TWIST gene in the Saethre-Chotzene syndrome. Nat. Genet. 15, 42–46 (1997). [DOI] [PubMed] [Google Scholar]
- 4.Carver EA, Oram KF & Gridley T Craniosynostosis inTwist heterozygous mice: A model for Saethre-Chotzen syndrome. Anat. Rec. 268, 90–92 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Menon S et al. Skeletal stem and progenitor cells maintain cranial suture patency and prevent craniosynostosis. Nat. Commun. 12, 4640 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Behr B, Longaker MT & Quarto N Craniosynostosis of coronal suture in Twist1 +/−mice occurs through endochondral ossification recapitulating the physiological closure of posterior frontal suture. Front. Physiol. (2011) doi: 10.3389/fphys.2011.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Henry Goodnough L et al. Twist1 mediates repression of chondrogenesis by β-catenin to promote cranial bone progenitor specification. Dev. (2012) doi: 10.1242/dev.081679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He F & Soriano P Dysregulated PDGFRα signaling alters coronal suture morphogenesis and leads to craniosynostosis through endochondral ossification. Development 144, 4026–4036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Binrayes A, Ge C, Mohamed FF & Franceschi RT Role of Discoidin Domain Receptor 2 in Craniofacial Bone Regeneration. J. Dent. Res. 002203452110074 (2021) doi: 10.1177/00220345211007447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge C et al. Discoidin Receptor 2 Controls Bone Formation and Marrow Adipogenesis. J. Bone Miner. Res. 31, 2193–2203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mohamed F, Ge C, Binrayes A & Franceschi RT Discoidin domain receptor 2 functions in skeletal progenitor cells and chondrocytes to control cartilage growth and postnatal bone formation. in ASBMR 2019 Annual Meeting (J. Bone Miner. Res. 32, 2019). [Google Scholar]
- 12.Méndez-Ferrer S et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466, 829–34 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan CKF et al. Identification and specification of the mouse skeletal stem cell. Cell 160, 285–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ono N, Ono W, Nagasawa T & Kronenberg HM A subset of chondrogenic cells provides early mesenchymal progenitors in growing bones. Nat. Cell Biol. 16, 1157–1167 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanda T, Sullivan KF & Wahl GM Histone-GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Curr. Biol. 8, 377–85 (1998). [DOI] [PubMed] [Google Scholar]
- 16.Foudi A et al. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol. 27, 84–90 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blanpain C, Lowry WE, Geoghegan A, Polak L & Fuchs E Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 118, 635–48 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Wilson A et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–29 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Chan CKF et al. Endochondral ossification is required for haematopoietic stem-cell niche formation. Nature 457, 490–4 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Greenbaum A et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y et al. Murine embryonic stem cell differentiation is promoted by SOCS-3 and inhibited by the zinc finger transcription factor Klf4. Blood 105, 635–637 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Cartwright P et al. LIF/STAT3 controls ES cell self-renewal and pluripotency by a Myc-dependent mechanism. Development 132, 885–896 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Hallett SA et al. Chondrocytes in the resting zone of the growth plate are maintained in a Wnt-inhibitory environment. Elife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao H et al. The suture provides a niche for mesenchymal stem cells of craniofacial bones. Nat. Cell Biol. 17, 386–96 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maruyama T, Jeong J, Sheu T-J & Hsu W Stem cells of the suture mesenchyme in craniofacial bone development, repair and regeneration. Nat. Commun. 7, 10526 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilk K et al. Postnatal Calvarial Skeletal Stem Cells Expressing PRX1 Reside Exclusively in the Calvarial Sutures and Are Required for Bone Regeneration. Stem Cell Reports 8, 933–946 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alappat S, Zhang ZY & Chen YP Msx homeobox gene family and craniofacial development. Cell Res. 13, 429–42 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Holmes G et al. Integrated Transcriptome and Network Analysis Reveals Spatiotemporal Dynamics of Calvarial Suturogenesis. Cell Rep. 32, 107871 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klopocki E et al. Copy-Number Variations Involving the IHH Locus Are Associated with Syndactyly and Craniosynostosis. Am. J. Hum. Genet. 88, 70–75 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Twigg SRF & Wilkie AOM A Genetic-Pathophysiological Framework for Craniosynostosis. Am. J. Hum. Genet. 97, 359–77 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkie AOM, Johnson D & Wall SA Clinical genetics of craniosynostosis. Curr. Opin. Pediatr. 29, 622–628 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu M et al. Cranial Suture Regeneration Mitigates Skull and Neurocognitive Defects in Craniosynostosis. Cell 184, 243–256.e18 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Goodnough LH et al. Twist1 mediates repression of chondrogenesis by β-catenin to promote cranial bone progenitor specification. Development 139, 4428–38 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chan CKF et al. Identification of the Human Skeletal Stem Cell. Cell 175, 43–56.e21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ambrosi TH et al. Distinct skeletal stem cell types orchestrate long bone skeletogenesis. Elife 10, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shu HS et al. Tracing the skeletal progenitor transition during postnatal bone formation. Cell Stem Cell 28, 2122–2136.e3 (2021). [DOI] [PubMed] [Google Scholar]
- 37.Sivaraj KK et al. Regional specialization and fate specification of bone stromal cells in skeletal development. Cell Rep. 36, 109352 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kreiborg S & Cohen MM Characteristics of the infant Apert skull and its subsequent development. J. Craniofac. Genet. Dev. Biol. 10, 399–410 (1990). [PubMed] [Google Scholar]
- 39.Connerney J et al. Twist1 dimer selection regulates cranial suture patterning and fusion. Dev. Dyn. (2006) doi: 10.1002/dvdy.20717. [DOI] [PubMed] [Google Scholar]
- 40.Ko JM, Yang J-A, Jeong S-Y & Yoon SH Two Children with Saethre-Chotzen Syndrome Confirmed by the TWIST1 Gene Analysis. J. Genet. Med. 8, 130–134 (2011). [Google Scholar]
Additional references
- 41.Nakamura T et al. Estrogen Prevents Bone Loss via Estrogen Receptor α and Induction of Fas Ligand in Osteoclasts. Cell 130, 811–823 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Chen Y-T, Akinwunmi PO, Deng JM, Tam OH & Behringer RR Generation of aTwist1 conditional null allele in the mouse. genesis 45, 588–592 (2007). [DOI] [PubMed] [Google Scholar]
- 43.Nakashima K et al. The Novel Zinc Finger-Containing Transcription Factor Osterix Is Required for Osteoblast Differentiation and Bone Formation. Cell 108, 17–29 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Peng Y et al. Increased transport of acetyl-CoA into the endoplasmic reticulum causes a progeria-like phenotype. Aging Cell 17, e12820 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mohamed FF et al. The collagen receptor, discoidin domain receptor 2, functions in Gli1-positive skeletal progenitors and chondrocytes to control bone development. Bone Res. 10, 11 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cowling RT et al. Discoidin domain receptor 2 germline gene deletion leads to altered heart structure and function in the mouse. Am. J. Physiol. Heart Circ. Physiol. 307, H773–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tumbar T et al. Defining the epithelial stem cell niche in skin. Science 303, 359–63 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun J et al. SLITRK5 is a negative regulator of hedgehog signaling in osteoblasts. Nat. Commun. 12, 4611 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bok S et al. MEKK2 mediates aberrant ERK activation in neurofibromatosis type I. Nat. Commun. 11, 5704 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu A, Ibrahim JG & Love MI Heavy-tailed prior distributions for sequence count data: removing the noise and preserving large differences. Bioinformatics 35, 2084–2092 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang DW, Sherman BT & Lempicki RA Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009). [DOI] [PubMed] [Google Scholar]
- 53.Huang DW, Sherman BT & Lempicki RA Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Farmer DT et al. The developing mouse coronal suture at single-cell resolution. Nat. Commun. 12, 4797 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ayturk UM et al. Single-Cell RNA Sequencing of Calvarial and Long-Bone Endocortical Cells. J. Bone Miner. Res. 35, 1981–1991 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data has been deposited at the Gene Expression Omnibus (GEO) under accession number GSE232652. Source data are provided with this article.