

Abstract
The extracellular matrix (ECM) is dynamically involved in many aspects of cell growth and survival, and it plays an active role in cancer etiology. In comparison to healthy ECM, tumor associated ECM shows high collagen deposition and remodeling activity, which results in an increased amount of denatured collagen strands in tumor tissues. Capitalizing on this distinguishing feature, we developed tumor-localizing polymeric carriers that selectively bind to denatured collagen in the tumor ECM. We synthesized N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers with their side chains conjugated to collagen hybridizing peptides (CHPs). HPMA copolymer-CHP conjugates exhibited selective affinity to denatured collagen and localized to tumors in an orthotopic MDA-MB-231 murine breast cancer model. The conjugates had increased tumor localization compared to copolymers with scrambled peptides in the side chains, as well as increased retention compared to free CHPs. Such conjugates show promise as carriers for ECM-acting drugs and imaging agents in the management of diseases characterized by high ECM remodeling activity.
Keywords: Collagen, Extracellular matrix, HPMA, Breast cancer, Drug delivery
1. Introduction
Cancer progression is accompanied by extensive changes in the tumor extracellular matrix (ECM) [1–4]. The ECM, the complex non-cellular environment surrounding cells [5], is an active player in cell growth and survival, and it influences many types of disease etiology [1,2,5]. ECM is composed largely of cell-adhesive structural proteins, including collagen, laminin, elastin, and fibronectin, as well as glycosaminoglycans, proteoglycans, and various signaling molecules and growth factors [6–8]. It provides structural support for tissue organization and regulates signaling to cells [9,10]. In cancer, the ECM undergoes morphological and biochemical changes that distinguish it from healthy tissues [11,12], including an increased breakdown and restructuring of ECM molecules [3].
Collagen is the most abundant protein in the human body [13–15]. There are 28 types of collagen, all of which are trimers with some or all portions of the trimer folded into a triple helical super-secondary protein structure comprised of three super-coiled polyproline II helices [16], stabilized by inter-strand hydrogen bonds [17]. Collagen is remodeled as a part of normal tissue maintenance through collagenolytic proteases, specifically matrix metalloproteinases (MMP) and cathepsins [18,19]. These proteases cleave collagens at specific sites, leading to their immediate denaturation at physiological temperature and presentation of unfolded collagen strands [13]. At tumor sites, there is increased ECM remodeling activity, attributed in part to an increased expression of these degradative enzymes [13]. The proteolytic breakdown of collagen in tumors results in an increased availability of denatured (monomeric) collagen in the tumor ECM tissue [17]. Furthermore there is also increased deposition of newly synthesized collagen (desmoplasia) in tumors [20]. This offers an opportunity for selective targeting to tumor tissue and retention at the tumor site through binding to denatured collagen.
Current strategies for polymer-mediated drug delivery (such as targeting of cell surface receptors) are often limited due to restricted diffusion through the dense ECM [21,22]. An alternative approach is to intentionally target the ECM to disrupt remodeling that is essential for tumor growth in many cancers. This reduces the need for polymeric carriers to overcome a dense stroma and the interstitial fluid pressure characteristic of tumors [21], both of which limit the diffusion of therapeutics from the vasculature to the interstitial tissue and ultimately uptake by cancer cells [22,23]. Targeting collagen is particularly advantageous because it is the most abundant constituent of the ECM [21], and it is present immediately upon extravasation. Targeting and anchoring to remodeling collagen also allows for specific and sustained delivery to the ECM of drugs which therapeutically act on the matrix components to slow tumor growth and metastasis. This has potential applications in cancers where the ECM plays a particularly important role, including pancreatic, ovarian, and breast cancer [20,24,25]. Furthermore, there are many cancer drugs (e.g. monoclonal antibodies (mAbs), MMP inhibitors, etc.) [26,27] that work outside the cell, which would benefit from extracellular anchoring. Additionally, the primary means of drug resistance in cancers is through cell efflux pumps on the cell membrane [28]. Targeting therapeutics outside the cell can evade this mechanism of drug resistance. Attachment of ECM-targeting moieties to polymeric carriers provides the opportunity for multi-valency, alters biodistribution, and enables conjugation of imaging agents simultaneously with drug molecules. Therefore ECM-targeting using polymeric carriers offers several advantages for anticancer drugs. There are several publications regarding the utility of targeting ECM molecules, including collagen (intact and denatured), for localization and targeted delivery of therapeutics [4,29–31].
Collagen hybridizing peptides (CHPs) comprise a class of peptides rationally designed to mimic the native collagen structure and have been shown to specifically bind to denatured (monomeric) collagen strands through triple helical hybridization [29,32,33]. They have been shown to bind to several different types of collagens denatured by MMP activity, heat, denaturants, or mechanical damage [14,29,32,34]. CHPs have been widely explored for their utility in interrogating the properties of damaged collagens in musculo-skeletal tissues [32,34,35].
In this work, we conjugated CHPs to N-(2-hydroxypropyl)methacrylamide (HPMA) copolymers and evaluated them for binding to denatured collagen in vitro and tumor localization in vivo. Fig. 1 depicts the rationale for the use of HPMA copolymer-CHP conjugates (HPMA-CHP) in tumor ECM targeting. HPMA copolymers are water-soluble polymers, which are generally recognized as non-toxic and non-immunogenic [36–40]. They can confer multifunctionality through synthetically adaptable side chains, in particular by incorporation of targeting moieties, imaging agents, and releasable drugs [36,40]. Because they exhibit size-dependent accumulation in the tumor site via the enhanced permeability and retention (EPR) effect [37,38,40–43], HPMA copolymers have been studied extensively for targeted drug delivery [36,37,39,44].
Fig. 1.
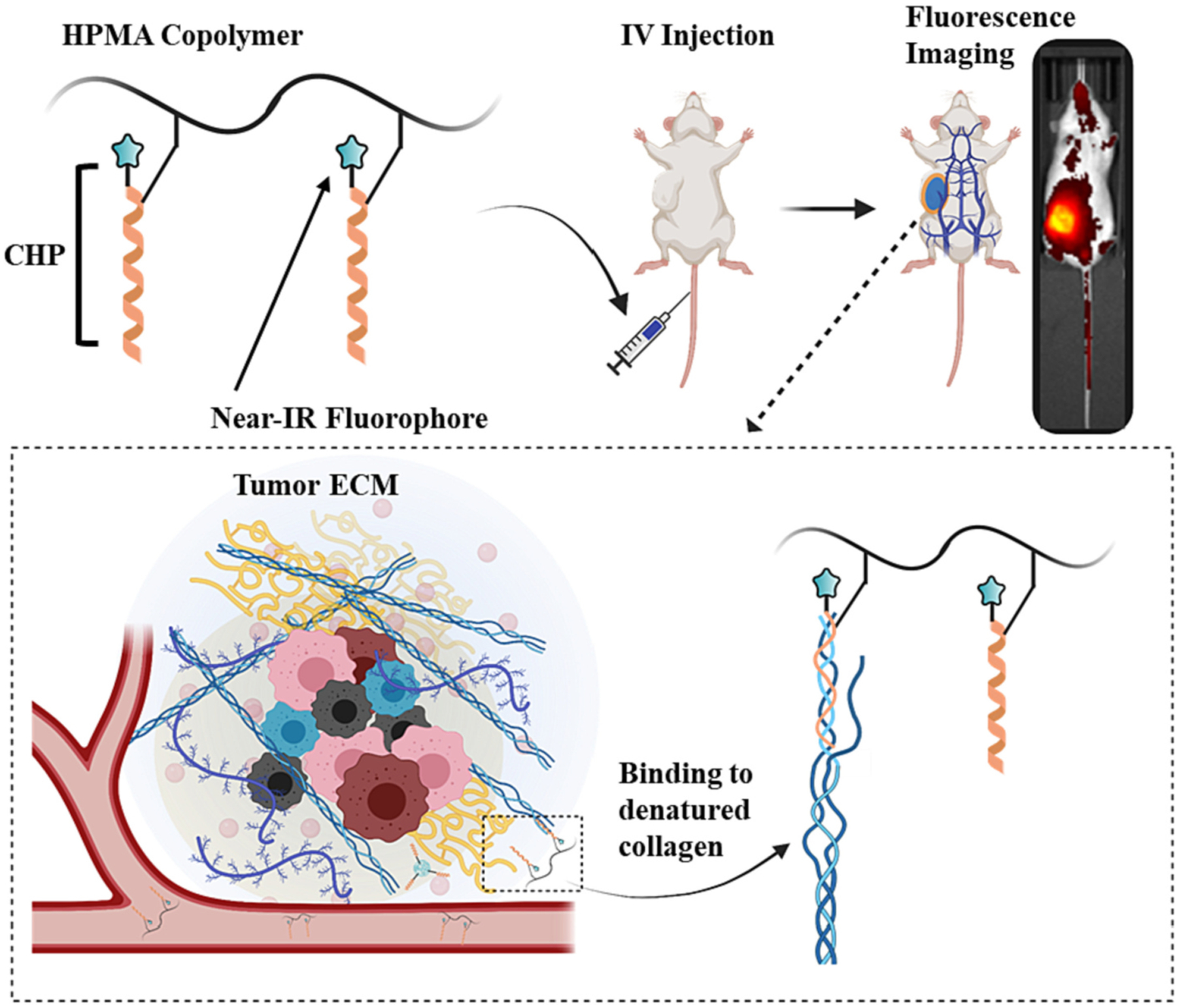
Overview of HPMA-CHP targeting denatured collagens of tumor ECM in breast tumor-bearing mice.
Here we report the synthesis of the copolymer-peptide conjugates, their physicochemical properties including triple helix refolding propensity and collagen binding, and their accumulation and retention in tumors of an orthotopic murine breast cancer (MDA-MB-231) model.
2. Materials and methods
2.1. Materials
(2S,4S)-Fmoc-4-fluoro-pyrrolidine-2-carboxylic acid was purchased from Chem Impex International (Wood Dale, IL). All other Fmoc-amino acids were purchased from VWR International (Radnor, PA). Collagen Type I (rat tail) was purchased from BD Sciences (Franklin Lakes, NJ). Nunc Black 96-well Maxisorp coated plates, N,N-dimethylformamide (DMF), and N,N-diisopropylethylamine (DIPEA) were purchased from Thermo Fisher Scientific (Waltham, MA). Cyanine5.5 N-hydroxysuccinimide (NHS) ester reactive dye was purchased from Lumiprobe (Hunt Valley, MD). All other materials for this study were acquired from Sigma Aldrich (St. Louis, MO).
2.2. Synthesis of comonomers
N-(2-hydroxypropyl)methacrylamide (HPMA) and N-methacryloylglycylglycine (MA-GG-OH) comonomers were synthesized according to previously published methods [45]. MA-GG-OH was prepared for incorporation into copolymers as a carboxylic acid attachment site for CHPs. Comonomers were characterized using electrospray ionization mass spectrometry (ESI-MS) and nuclear magnetic resonance (NMR) spectroscopy.
2.3. Synthesis of HPMA copolymer precursors
HPMA copolymer precursors were synthesized using free radical copolymerization of the comonomers HPMA and MA-GG-OH. The feed ratio was 85:15 (HPMA:MA-GG-OH) molar ratio, and azobisisobutyronitrile (AIBN) was used as an initiator in a solvent mixture of DMSO: methanol (2:3), with total monomer concentration of 1 M. Specifically, 240 mg (1.70 mmol) of HPMA was combined with 60 mg (0.30 mmol) of MA-GG-OH in 2 mL of DMSO/methanol in a 5 mL glass ampule (Thomas Scientific, Swedesboro, NJ), and 12 mg of AIBN was added. The reaction mixture in an ampule was purged with nitrogen using five freeze-thaw cycles, and the glass ampule was sealed. The reaction was stirred under an inert N2 atmosphere at 50 °C in a temperature-controlled oil bath for 24 h. To terminate the reaction, the ampule was opened, and the reaction mixture was added to an 80-fold excess volume of acetone to precipitate the solid product. The precipitates were isolated through centrifugation followed by washing twice with acetone and air drying.
2.4. Synthesis of CHP
Peptide [Cy5.5-KGGG(GfO)9; f: (2S, 4S)-4-fluoroproline); O: 4-hydroxyproline] (single amino acid abbreviations are used) was synthesized by standard Fmoc-mediated solid phase peptide synthesis (SPPS) using an automated peptide synthesizer with some manual modifications. Cyanine5.5 (Cy5.5) was incorporated for in vivo fluorescence detection. Specifically, the peptide sequences K(Boc)GGG(GfO)9 were grown from TentaGel-R-RAM resin using HBTU (2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate, hexafluorophosphate benzotriazole tetramethyl uronium) coupling chemistry. Cy5.5 NHS ester was added to the N-terminus of the peptide on resin in DMF in the presence of DIPEA. Cleavage from the resin using a standard cleavage cocktail [trifluoroacetic acid (TFA):water:triisopropyl silane (TIPS) = 95:2.5:2.5 by volume] produced free peptides, with an amide at the C-terminus, and a deprotected lysine side chain. The lysine residue serves as a bifunctional linker to incorporate both Cy5.5 (via N-terminus) and to provide a primary amine for attachment to the copolymer (via side chain amine). The Gly-Gly-Gly is a flexible spacer intended to decouple peptide from polymer backbone. Peptides were purified by reverse phase high performance liquid chromatography (HPLC) [Agilent SD-1 Prepstar HPLC Pump and a Zorbax 300SB-C18 column (Agilent)]. Peptide identities were confirmed using MALDI-ToF (Bruker UltrafleXtreme) mass spectrometry (MW = 3449 g/mol). Control peptide of scrambled sequence (SCHP: Cy5.5-KGGGOfGGOfGfGfOfOGOfGOOfGGOOffG) having amino acid composition identical to targeted CHP was synthesized in a similar manner. The control scrambled peptide lacks triple helical folding propensity and is unable to bind to denatured collagen [13].
2.5. Conjugation of peptide to HPMA copolymer precursors
CHPs were conjugated to copolymer precursors using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) coupling chemistry. For the targeted peptide, the peptide and copolymer precursor were separately dissolved in 10% DIPEA in DMF and heated to 55 °C. EDC (12 μmol) was added to the copolymer solution (1.2 mM, 1 mL) and stirred for 5 min. The peptide solution (6 mM, 1 mL) was then added, and the reaction was stirred for 24 h at 55 °C. The reaction mixture was added to excess ether to precipitate the product, which was isolated through centrifugation and decanting. The solid precipitates were dissolved in water and dialyzed at 50 °C for 2 d to dissociate and remove any unreacted CHP which may have bound to pendant CHPs of the HPMA-CHP. The dialyzed solution was lyophilized to yield the final copolymer-peptide conjugate. A schematic of the HPMA-CHP synthesis, including peptide structure, is shown in Fig. 2.
Fig. 2.
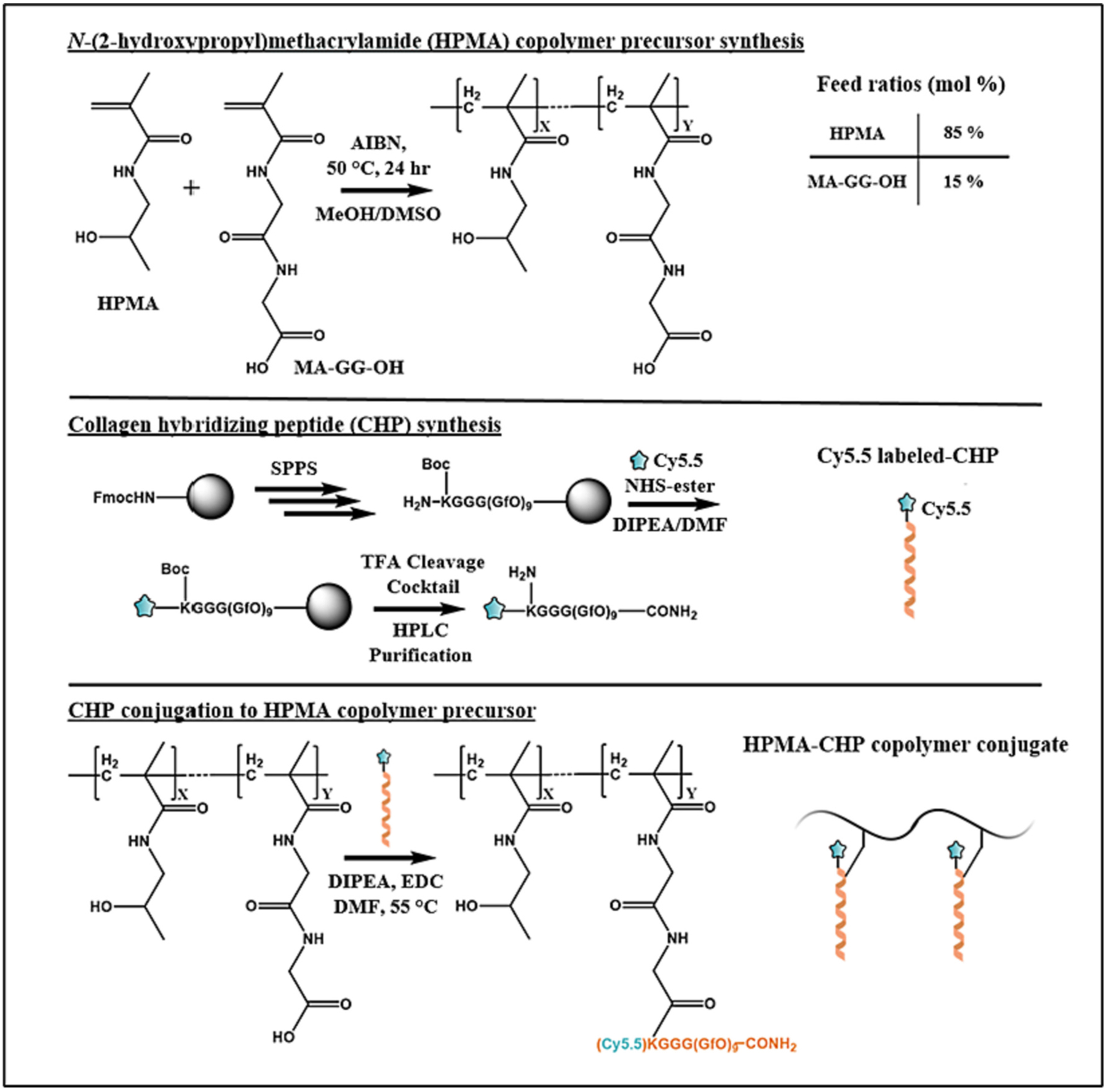
Synthetic scheme of HPMA-CHP conjugates.
2.6. Characterization of HPMA-CHP
Size exclusion chromatography (SEC) (Akta Fast Protein Liquid Chromatography, Superose 6 column, 10 × 300 mm, phosphate buffered saline (PBS), pH 7.4, eluent) connected to a refractive index detector (OptiLab Rex, Wyatt Technologies Corporation, Santa Barbara, Ca) was used to determine the molecular weight and polydispersity of copolymer precursors. The peptide content of the conjugates was calculated using UV-VIS spectrophotometry.
2.7. Circular dichroism
The circular dichroism (CD) melting temperature of HPMA-CHP was measured on a JASCO J-1500 CD spectrophotometer, using quartz cells with a path length of 1 cm. All samples were dissolved in deionized (DI) water at a concentration of 4 mg/mL. Thermal melting point curves were generated by monitoring the CD ellipticity at 225 nm from 4 °C to 90 °C at a heating rate of 0.5 °C/min. The melting temperature (Tm) was determined as the minimum of the first derivative of the melting curve. Additionally, a kinetic study was done to measure the refolding rate of HPMA-CHP at 4 °C and at 37 °C. For refolding kinetics, polymer solutions (same concentration as above) in CD cuvette were incubated in 90 °C water for 10 min. The cuvette was then immediately placed in the CD spectrophotometer set at 37 °C or 4 °C. The ellipticity at 225 nm was monitored over 3 h. A smoothing curve was used prior to the analysis of CD data.
2.8. Collagen binding
Wells of 96-well Nunc black Maxisorp plates (Thermo Fisher Scientific, Waltham, MA) were coated with either denatured or intact type I collagen. For intact collagen, 50 μL type I collagen (rat tail, 4 mg/mL, 0.02 N acetic acid) was added directly to the wells. For denatured collagen, the collagen solution was heated at 70 °C for 15 min prior to adding to wells. The wells were allowed to dry completely under a fan. The wells were then washed with PBS to neutralize, followed by DI water, and then allowed to dry completely.
To each collagen-coated well, 75 μL of HPMA-CHP or HPMA-SCHP in PBS (1 mg/mL) was added. Prior to addition to the wells, solutions were heated to 70 °C for 10 min, then cooled in ice for 30 s. The well plates were covered and placed at 4 °C for 1 h. Excess solution was pipetted out, and wells were washed with 200 μL of PBS, followed by 200 μL of DI water. Fluorescence intensity of the wells was measured on a SpectraMax M-2 Microplate Reader (Molecular Devices, LLC, San Jose, Ca) at excitation of 630 nm and emission of 710 nm. All samples were measured in triplicate.
For studying retention after washing, 200 μL of PBS was added to each well prepared as above, and the plate was placed in an incubator at 37 °C and shaken for 5 min. The solution was pipetted out from the wells, and the wells were washed with DI water and allowed to dry completely. The fluorescence was read as above, and the percentage of the fluorescence was calculated compared to the initial fluorescence. The procedure was repeated for a total of five times. All samples were tested in triplicate.
2.9. Animal experiment
All experiments were approved by and performed in accordance with the University of Utah’s Institutional Animal Care and Use Committee (IACUC) guidelines (IACUC #21–10,007), with the assistance of the Preclinical Research Resource (PRR) at the Huntsman Cancer Institute (HCI). In order to generate an orthotopic breast cancer murine model, non-obese diabetic/scid (NOD/SCID) mice were implanted with MDAMB-231 cells (0.5 × 106 cells/tumor) suspended in Matrigel in the mammary fat pad of the mice. Tumors were allowed to grow for 7 wk. until they were between 150 and 300 mm3, measured by calipers. Mice were fed a non-alfalfa-containing diet to avoid interference during fluorescence imaging.
Mice were treated with hair remover (Nair) to reduce auto-fluorescence and divided into three experimental groups, with tumor sizes spread similarly in the three groups. Mice were tail-vein injected with 7.5 nmol (in terms of CHP content and Cy5.5 content, which were equivalent) of HPMA-CHP, HPMA-SCHP, or CHP in 175 uL of PBS solution. Fluorescence of the injected compound from the animal was imaged using an In Vivo Imaging System (IVIS) Spectrum (Caliper Life-Sciences) using wavelengths of 675 nm for excitation and 720 nm for emission, at t = 0 (before injection), 0.2 (immediately after injection), 6, 12, 24, 48, 72, and 168 h. Regions of interest (ROIs) of identical area encompassing the tumors were selected, and total radiant efficiency was quantified for analysis. Animals were euthanized after 168 h, using CO2 inhalation and cervical dislocation, and organs (heart, lung, liver, kidneys, spleen, and stomach/intestines) and tumors were harvested and imaged for biodistribution. ROIs, identical across each type of organ, were selected, and total radiant efficiency was quantified for analysis.
2.10. Statistical analysis
Significance was determined using unpaired t-test between groups, using GraphPad Prism Software. A significance level of α = 0.05 was used for all statistical tests.
3. Results and discussion
3.1. Synthesis and characterization of HPMA-CHPs
HPMA copolymer precursors were synthesized by free radical copolymerization of HPMA and MA-GG-OH using AIBN initiator with a mol percent feed ratio of 85:15 (HPMA:MA-GG-OH). Weight average molecular weight, number average molecular weight, and polydispersity as determined by SEC were 85 kDa, 47 kDa, and 1.8, respectively. Fluorescently labeled collagen hybridizing peptide [targeted CHP: Cy5.5-KGGG(GfO)9; f: (2S, 4S)-4-fluoroproline); O: 4-hydroxyproline] and scrambled peptide (SCHP: Cy5.5-KGGGOfGGOfGfGfOfOGOfGOOfGGOOffG) were synthesized on solid support, and purified peptides were conjugated to the copolymer precursor using 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC)-mediated coupling chemistry. After purification of HPMA-CHP conjugates, peptide contents were measured using UV–Vis spectrophotometry. On average, HPMA-CHP had 1.8 peptides per polymer chain, and HPMA-SCHP had 3.7 peptides per polymer chain.
Under identical reaction conditions, fewer targeted peptides were conjugated to polymers compared to scrambled peptides. Increasing the targeted peptide concentration during the conjugation reaction did not result in conjugation of additional peptides per polymer. Despite this difference, all experiments showed significantly more collagen binding for the targeted conjugates compared to scrambled conjugates. The large peptide size (~3.5 kDa) compared to smaller peptides often used in other HPMA conjugate systems [38,41] may have resulted in low conjugation yield. We note that the number of peptides per polymer chain determined by UV–Vis reflects an average value, not an absolute value.
3.2. CHP’s triple helical folding behavior allows HPMA-CHP to bind to denatured collagen strands at 37 °C
The CD melting temperatures of HPMA-CHP and HPMA-SCHP conjugates in DI water (4 mg/mL) were determined by measuring the ellipticity at 225 nm from 4 °C to 90 °C at a heating rate of 0.5 °C/min (Fig. 3a). HPMA-CHP exhibited a sigmoidal transition between 30 °C and 60 °C with a midpoint melting Tm at 48 °C which corresponded to the minimum of the first derivative of the melting curve. HPMA-SCHP did not exhibit a sigmoidal melting curve. The rate of refolding of HPMA-CHP was measured by CD (Fig. 3b). At 4 °C, refolding was fast with refolding half-time of 7.5 min. At 37 °C, HPMA-CHP rapidly refolded only up to 68% folded (ellipticity of 4 °C sample after 3 h was used as 100% folded), and then remained close to this value for the remaining 3 h.
Fig. 3.
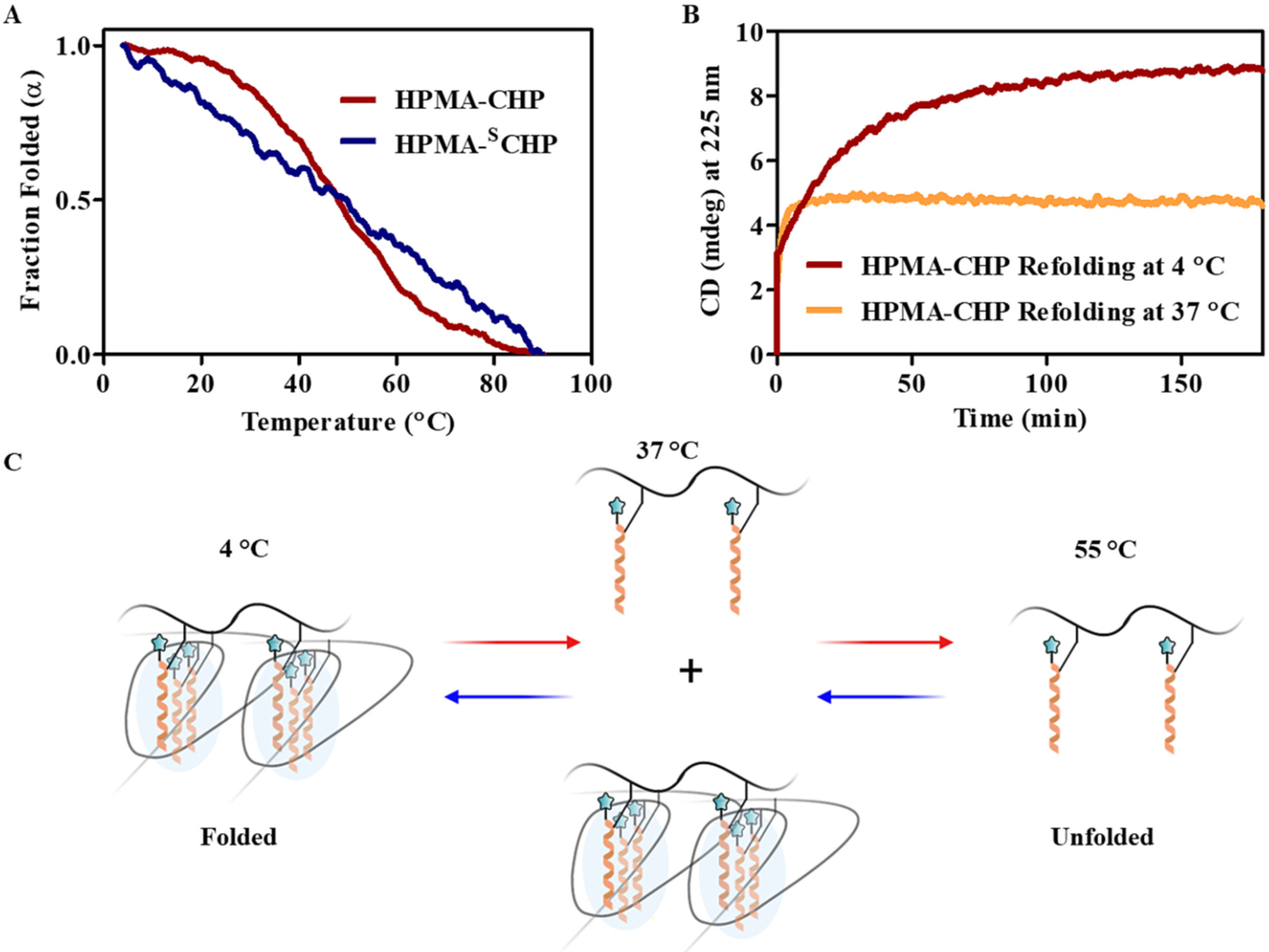
HPMA-CHP exhibits a broad melting curve associated with CHP self-trimerization with Tm around 48 °C. (A) CD melting curves of HPMA-CHP and HPMA-SCHP, (B) CD refolding curves of HPMA-CHP at 4 °C and 37 °C, and (C) schematic illustration of folding behavior of HPMA-CHP at different temperatures: CHPs are unfolded at 55 °C, partially folded at 37 °C, and fully folded at 4 °C.
CHPs are known to self-trimerize to form homotrimers [13], which precludes their binding to collagen strands. Tendency for such homo-trimerization is significantly enhanced when more than two CHPs are bound to a template molecule or to the same polymer backbone, due to neighboring group effect [46,47]. Initially, we prepared HPMA copolymer-peptide conjugates comprised of conventional CHP with GlyProHyp repeats which have high triple helical propensity. These conjugates exhibited fast refolding and high Tm of 62 °C, indicative of homotrimerization of CHPs templated to the HPMA backbone. In fact, when tested, these compounds showed low affinity to denatured collagen due to fast self-homotrimerization in both plate binding assay and in vivo mouse experiments (data not shown). These results suggested that, to make polymer-conjugated CHPs with active collagen binding, CHPs with low tendency for self-trimerization were needed such as those with GfO repeats reported previously [32]. The CD refolding data indicated that the melted HPMA-CHP (which has the GfO repeats) only fold partially even when incubated at 37 °C for >1 h. This suggests that at body temperature, a portion of CHPs displayed on HPMA backbone are not trimerized and are able to bind to denatured collagen strands present in tumor ECMs (Fig. 3c).
3.3. HPMA-CHP binds to denatured collagen strands by triple helical folding
Binding assay using collagen coated plates (Fig. 4) demonstrated that HPMA-CHP showed more binding to thermally denatured type I collagen compared to intact type I collagen. HPMA-SCHP showed negligible binding to both collagens with no difference between the two.
Fig. 4.
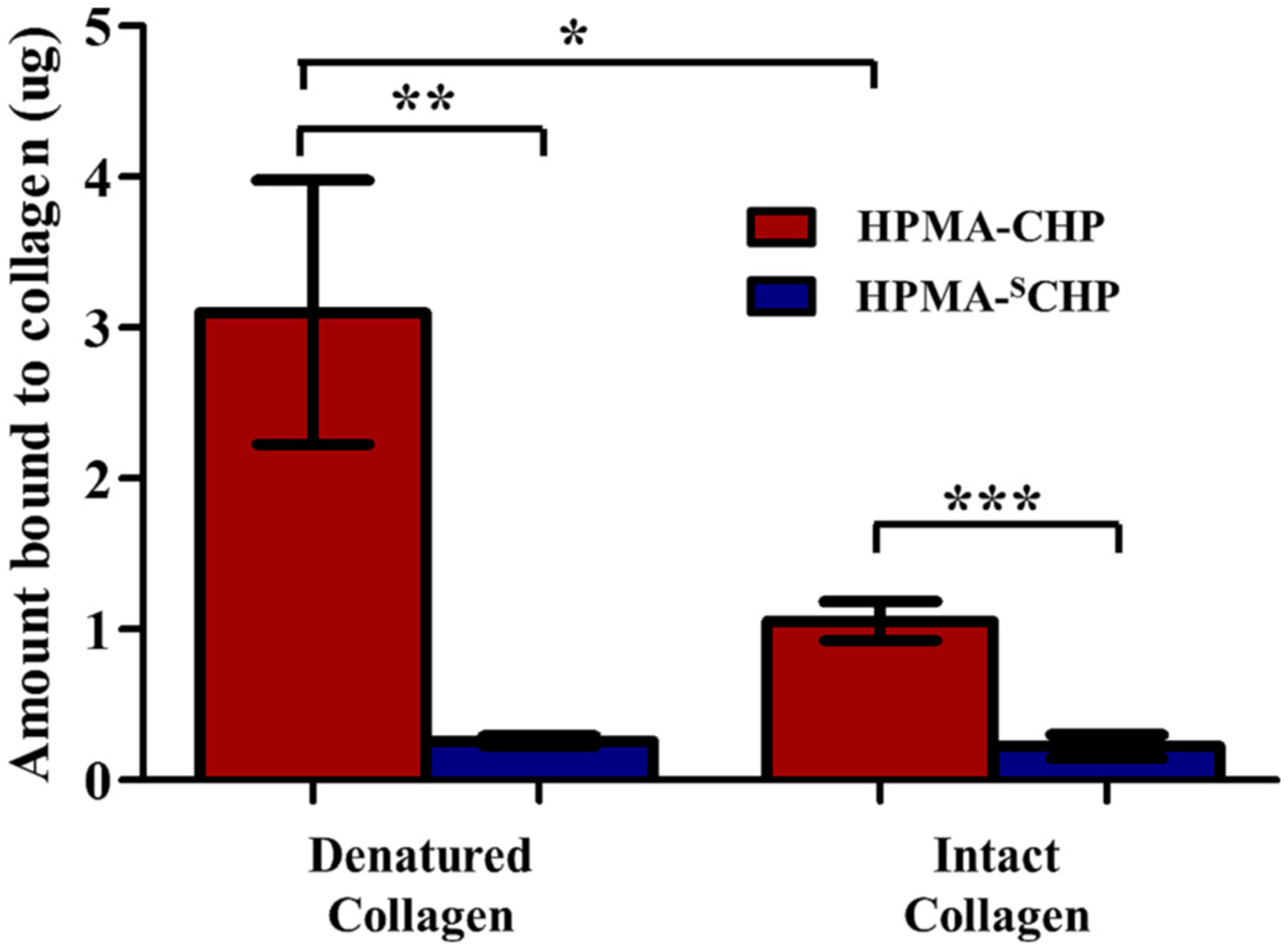
HPMA-CHP conjugates bind selectively to denatured collagen over intact collagen, and significantly more than HPMA-SCHP conjugates. HPMA-SCHP showed no difference in binding between intact and denatured collagen. (Mean ± SD, N = 3, *p < 0.05, **p < 0.01, ***p < 0.001).
HPMA-CHP binding to denatured collagen validates that tethering of CHPs to HPMA copolymer side chains did not restrict their binding to collagen strands. There was low but measurable binding to intact collagen (likely due to a small amount of denatured collagen in the rat tail collagen solution); however, the binding was three-fold higher for denatured collagen, demonstrating the selectivity for denatured collagen as previously reported [13]. The lack of collagen binding for the HPMA-SCHP confirms that the binding was due to CHP’s triple helical hybridization to collagen strands and not a result of polymer entrapment in a gelatin matrix or non-specific polymer adhesion to denatured collagen.
3.4. HPMA-CHP and CHP bound to denatured collagen exhibited similar retention against washing
HPMA-CHP and CHP which were bound to gelatin-coated wells were subjected to washing with blank PBS to test and compare their affinity to denatured collagen. After five washes, HPMA-CHP and CHP showed no difference in retention to denatured collagen (Fig. 5).
Fig. 5.
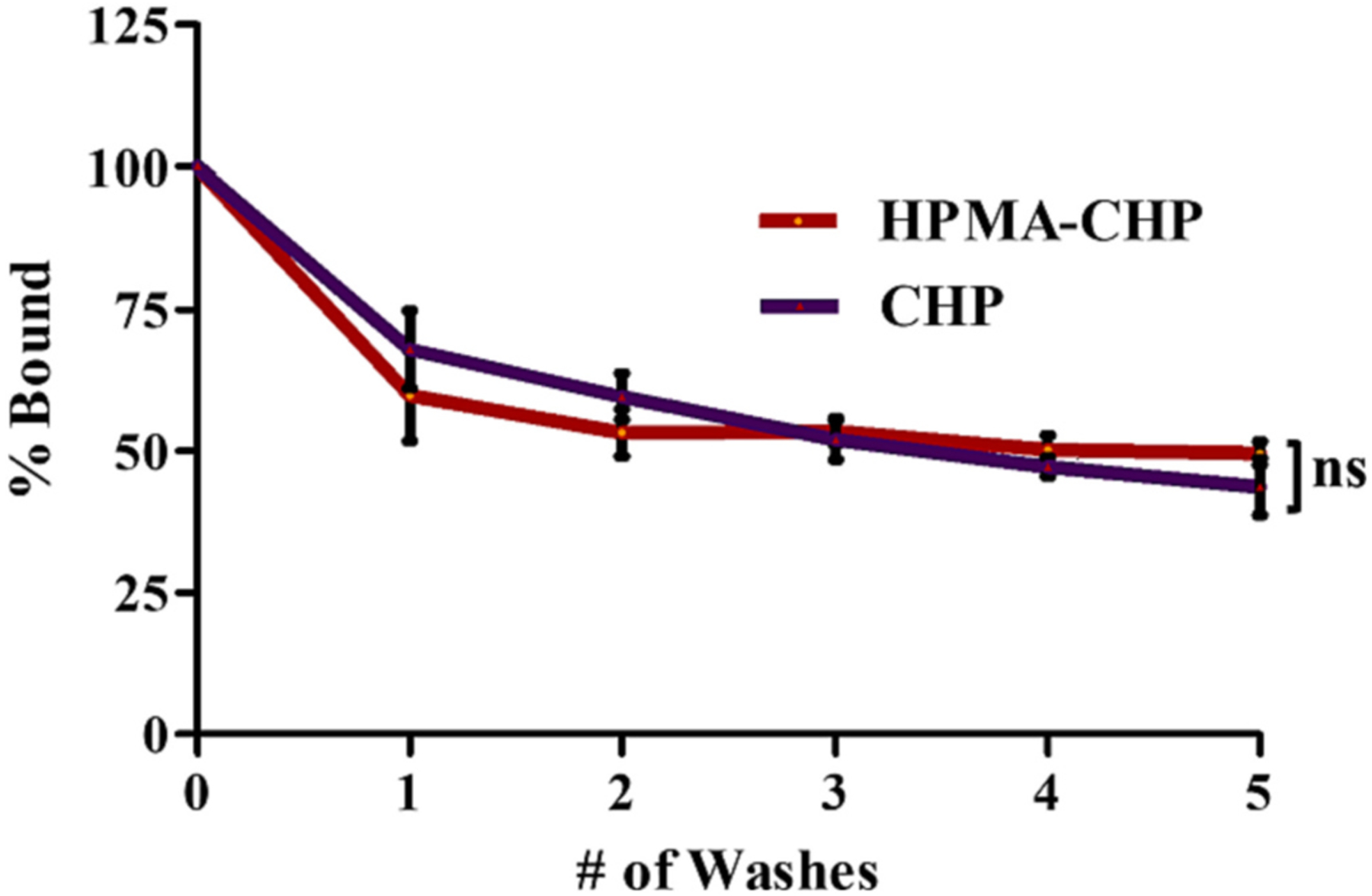
HPMA-CHP and CHP are similarly retained to gelatin-coated plates after washing with PBS with ~50% remaining after 5 washes. (Mean ± SD, N = 3, *p < 0.05, **p < 0.01, ***p < 0.001).
These results indicate that both HPMA-CHP and CHP show similar binding strength to collagen strands and that the strength of CHP binding is not reduced by conjugation to the HPMA backbone. The results also suggest that there was no multivalency-mediated enhancement of gelatin binding for HPMA-CHP, likely due to low CHP conjugation number (1.8 CHPs per HPMA polymer chain). In vivo, various factors such as local concentrations of the degraded collagens and HPMA-CHP conjugates, potential enzymatic degradation of peptides, and renal clearance may affect the relative sustained binding of HPMA-CHP and CHP.
3.5. HPMA-CHP exhibited higher accumulation and longer retention in murine breast tumors compared to HPMA-SCHP as a result of denatured collagen binding
An orthotopic breast cancer model, induced through injecting MDA-MB-231 cells into the mammary fat pad of NOD/scid mice, was employed to provide a representative tumor and collagen microenvironment. Tumor-bearing mice were administered HPMA-CHP, HPMA-SCHP, or CHP via tail-vein injection, and fluorescence was measured at t = 0 (pre-injection), 0.2 (post-injection), 6, 12, 24, 48, 72, and 168 h using an IVIS Spectrum (Caliper LifeSciences). Mice within each experimental group (HPMA-CHP, HPMA-SCHP, and CHP) were given identifiers prior to injection of experimental compounds [#1–5, from left to right in images shown in supplemental data (Figs. S1–S3)] and animal #3 in each group was chosen for all representative images shown. Fluorescent signal in mice over time showed accumulation at the breast tumor indicated by a black dashed oval in pre-injection mouse (Fig. 6). Radiant efficiency over time at the tumor site showed that HPMA-CHP has a higher accumulation compared to HPMA-SCHP at each timepoint throughout the 168 h, and a higher area under the curve (AUC) (Fig. 7a and b). The half-lives were not statistically different between HPMA-CHP and HPMA-SCHP. Semi-quantitative pharmacokinetic data (non-compartmental analysis) at the tumor as a function of fluorescent signal indicated that HPMA-CHP and HPMA-SCHP had areas under the curve (AUC) of 1.68 × 1013 and 9.09 × 1012 radiant efficiency units (rad eff) × hr, respectively, and maximum concentrations (Cmax) of 1.74 × 1011 and 8.80 × 1010 rad eff units, respectively (Table 1). Both had a time to maximum concentration (tmax) of 24 h. The tumor half-lives (t1/2) of HPMA-CHP and HPMA-SCHP were 74.2 and 66.5 h, respectively.
Fig. 6.
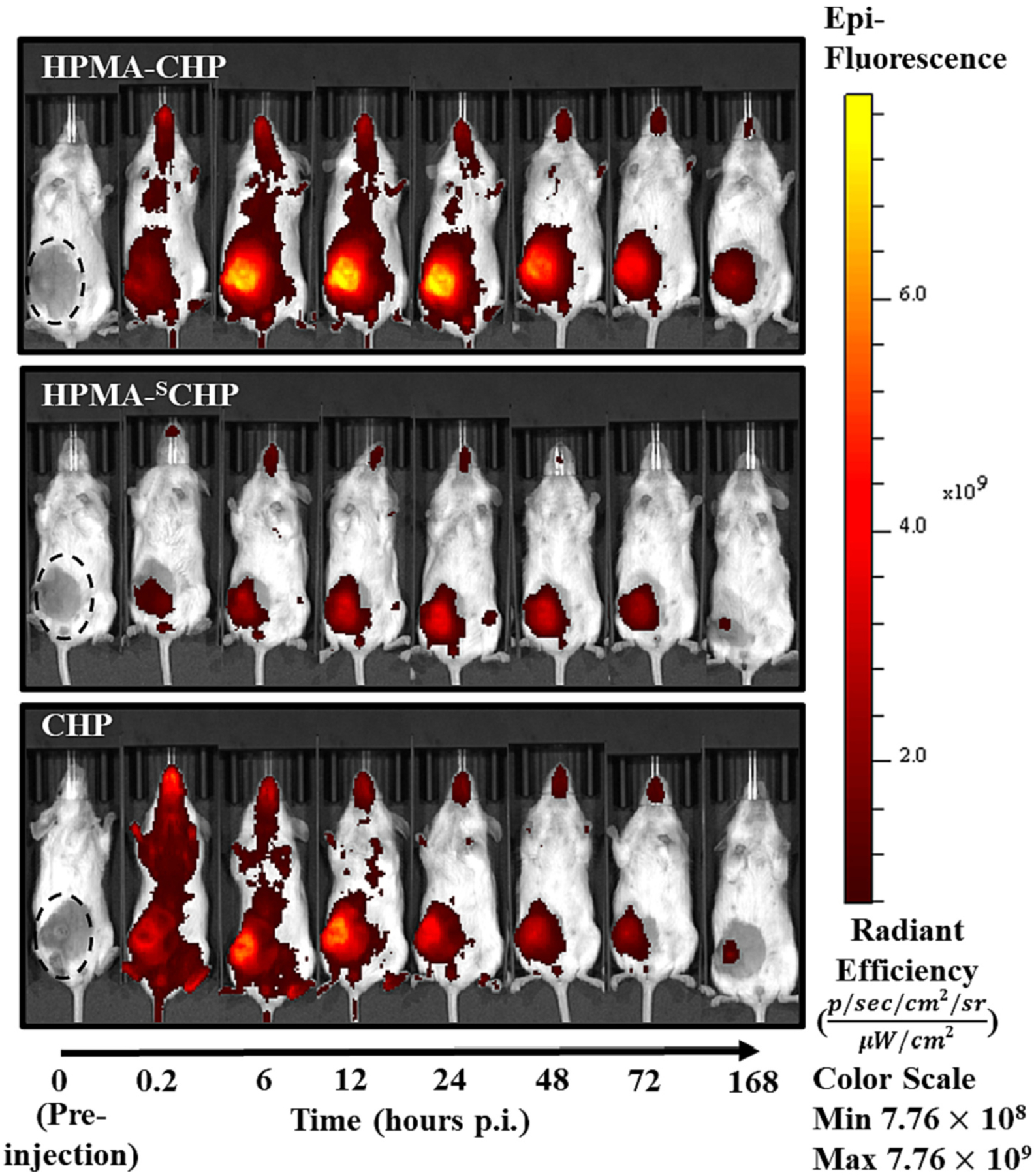
In vivo near-IR fluorescence images of breast tumor-bearing mice at t = 0 (pre-injection), 0.2, 6, 12, 24, 48, 72, and 168 h after tail-vein injection of Cy5.5-labeled HPMA-CHP, HPMA-SCHP, or CHP. Black dashed oval indicates location of breast tumor.
Fig. 7.
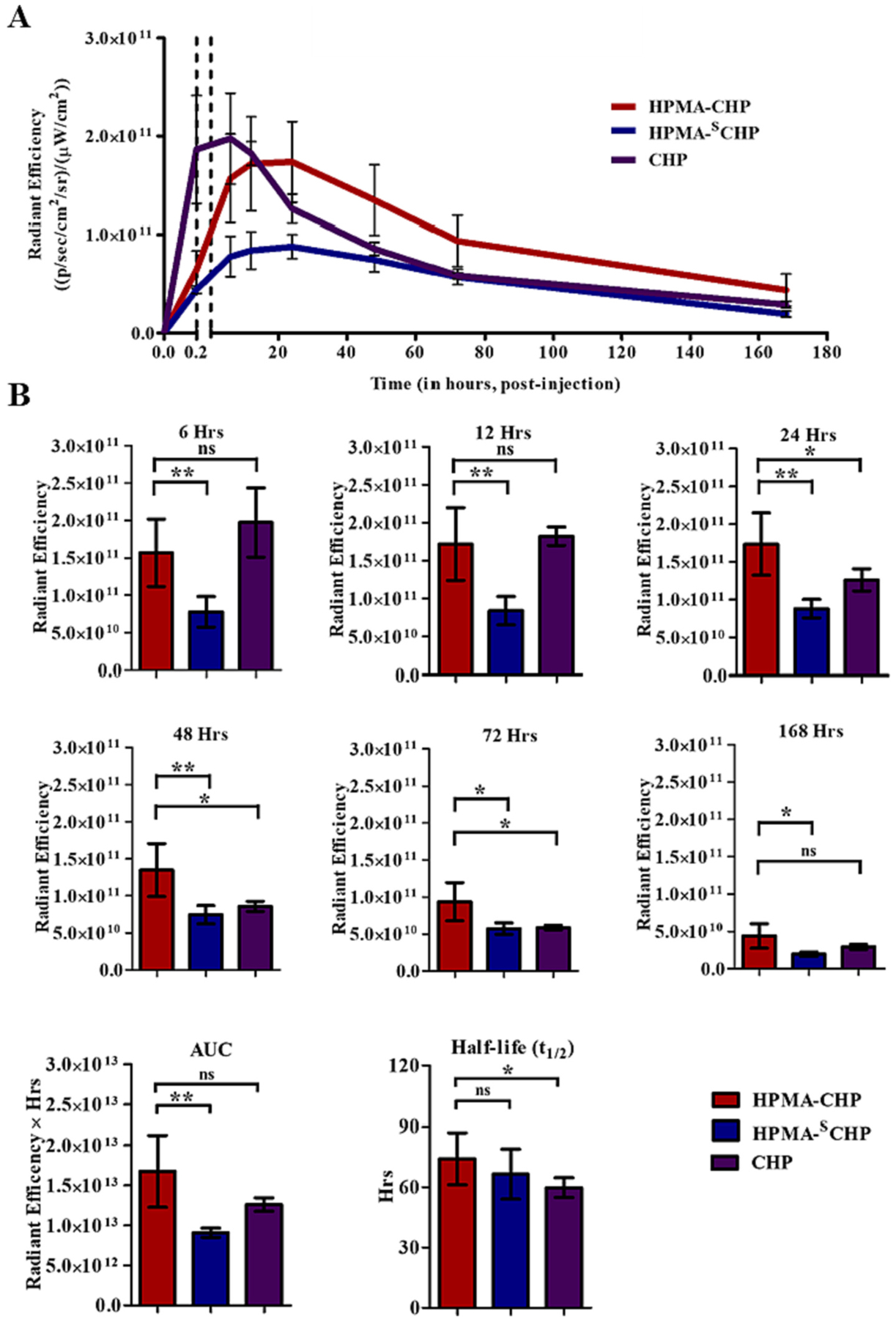
(A) Radiant efficiency at breast tumor over time of Cy5.5-labeled HPMA-CHP, HPMA-SCHP, and CHP administered to breast tumor-bearing mice over 168 h, and (B) comparison of breast tumor accumulation at timepoints t = 6, 12, 24, 48, 72, and 168 h, areas under the curve (AUC), and half-lives. (Mean ± SD, N = 5, *p < 0.05, **p < 0.01, ***p < 0.001).
Table 1.
Non-compartmental pharmacokinetic analysis of HPMA-CHP, HPMA-SCHP, and CHP at the tumor, showing area under the curve (AUC), maximum concentration (Cmax), time to maximum concentration (tmax), and half-life (t1/2), determined using fluorescent signal in radiant efficiency (rad eff) units.
| HPMA-CHP | HPMA-SCHP | CHP | |
|---|---|---|---|
| AUC (rad eff × hr) | 1.68 × 1013 | 9.09 × 1012 | 1.26 × 1013 |
| Cmax (rad eff) | 1.74 × 1011 | 8.80 × 1010 | 1.97 × 1011 |
| tmax (hr) | 24 | 24 | 6 |
| t1/2 (hr) | 74.2 | 66.5 | 59.8 |
The AUC for HPMA-CHP was 1.8 times that of HPMA-SCHP, and the Cmax of HPMA-CHP was higher than that of HPMA-SCHP, suggesting that if a drug molecule was conjugated to the HPMA copolymer, HPMA-CHP would allow 1.8 times the total drug exposure (compared to HPMA without CHP) at the tumor over 168 h and potentially a greater therapeutic effect of a given dose of drug with fewer side effects. The greater concentration of HPMA-CHP compared to HPMA-SCHP at every point shows potential for utility of this carrier to improve efficacy and reduce systemic toxicity of therapeutics.
3.6. HPMA-CHP exhibited higher retention in murine breast tumors compared to CHP without polymer backbone
While CHP and HPMA-CHP showed similar Cmax in the tumor, with time to maximum level (tmax) of ~6 h for CHP and ~ 24 h for HPMA-CHP, CHP was eliminated faster. While at the 168 h timepoint, the difference between HPMA-CHP and CHP was not significant, HPMACHP showed greater accumulation than CHP up to the 72 h timepoint, indicating greater retention. This is also corroborated by the longer tumor half-life of HPMA-CHP compared to CHP. The tumor half-life of the targeted peptide was 59.8 h compared to 74.2 h for the targeted polymer.
Larger molecular weight of the conjugates leading to reduced rate of elimination from the tumor and potential multivalency may account for some of the differences observed. Though the HPMA-CHP and CHP exhibit similar accumulations at the 168 h timepoint, the conjugate offers advantages over the peptide alone. In addition to the increased retention provided by the HPMA-CHP compared to CHP, the polymer offers opportunities for multifunctionality through incorporation of drugs and imaging agents in the side chains for sustained and targeted delivery of ECM-acting therapeutics, combination drug delivery, as well as for diagnostic and theranostic applications.
3.7. Organ distribution showed differential accumulation at 168 h
To better assess biodistribution, mice were euthanized after 168 h, and lung (lg), heart (h), liver (lv), kidneys (k), spleen (s), stomach and intestines (s&i), and breast tumor (tumor) were collected to measure fluorescent signal (Fig. 8a). Fluorescence images of all organs are shown in supplemental data (Figs. S4–S6). Percentage of signal in each organ or tumor out of total signal in all organs/tumor indicated that HPMA-CHP and HPMA-SCHP have a higher proportion of the signal in the liver and stomach, while CHP showed a higher proportion in the lungs and kidneys (Fig. 8b). The percentage of total signal in tumors is similar for all three. Comparing the total signal in each organ or tumor between the three groups (Fig. 8c), there was more accumulation in the tumor for the targeted polymer compared to the non-targeted polymer. There was also more accumulation of HPMA-CHP in the lungs, heart, kidneys, spleen, and liver compared to HPMA-SCHP. The difference was not significant for the stomach and intestines. HPMA-CHP and CHP had similar accumulation in the tumor at this timepoint. HPMA-CHP accumulated more in the spleen, liver, and stomach and intestines compared to CHP, while CHP accumulated more in the lungs and kidneys compared to HPMA-CHP. The difference in accumulation in the heart for these was not significant.
Fig. 8.
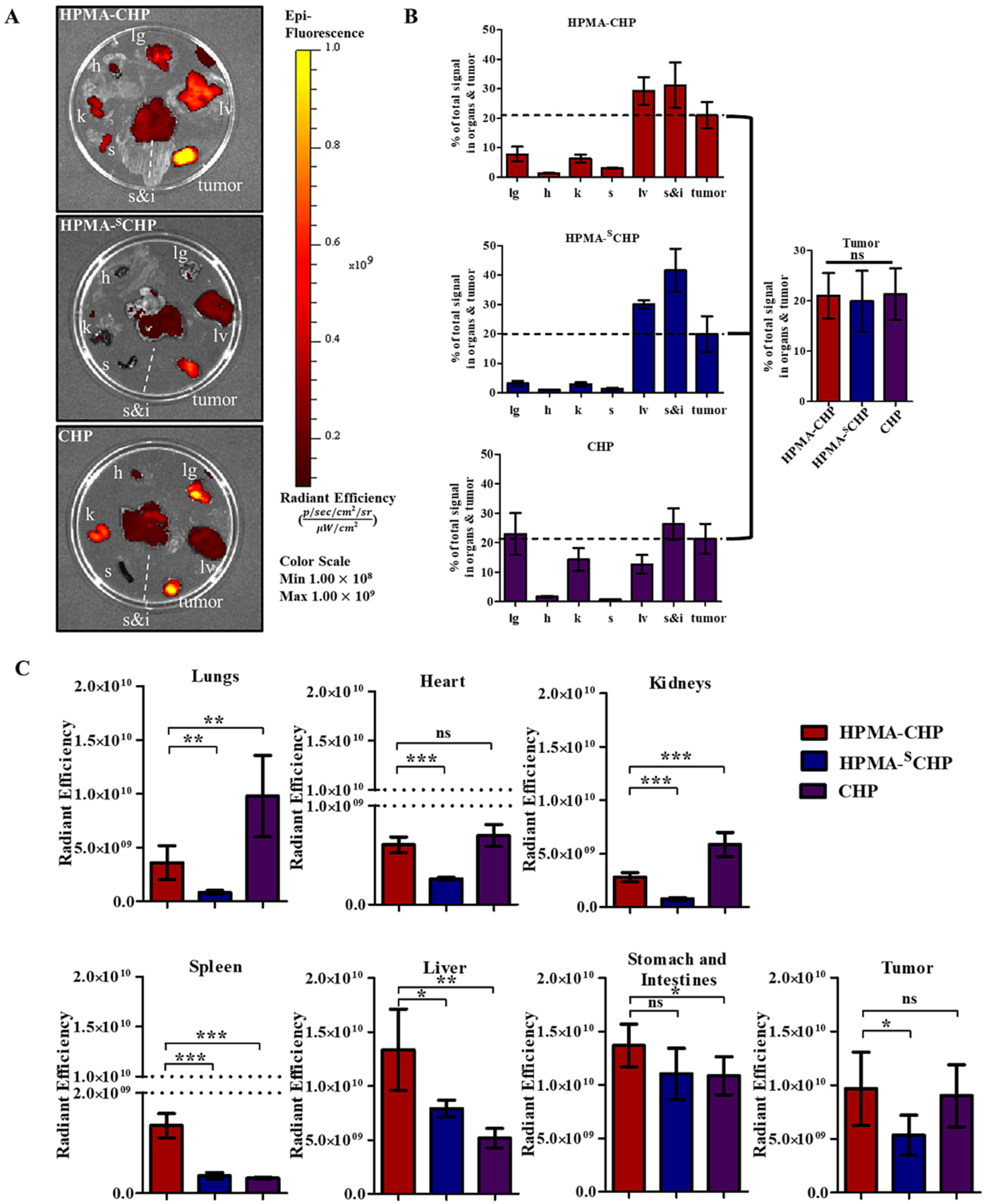
(A) Fluorescence images of harvested lung (lg), heart (h), liver (lv), kidneys (k), spleen (s), stomach and intestines (s&i), and breast tumor (tumor) from mice 168 h after tail-vein injection of Cy5.5-labeled HPMA-CHP, HPMA-SCHP, or CHP, (B) percent of signal in each organ or tumor out of total signal in organs and tumor at 168 h, with comparison between percentage in tumors between the three groups, and (C) fluorescent signal in radiant efficiency units in each organ or tumor in HPMA-CHP, HPMA-SCHP, and CHP. (Mean ± SD, N = 5, *p < 0.05, **p < 0.01, ***p < 0.001).
Collagen is ubiquitous in the body, and a basal level of remodeling occurs as a part of normal tissue maintenance [1,19]. The higher signal in all organs, except the stomach and intestines, of HPMA-CHP compared to HPMA-SCHP may be due to binding and retention at the remodeling collagen at these sites. For the stomach and intestines, the organs were harvested together and the total signal quantified, and we believe that the signal primarily comes from the intestines rather than the stomach. The similar accumulation between the targeted and non-targeted polymers, and the difference in accumulation between targeted polymer and peptide, suggests that localization in the intestines may be due to excretion through hepatic route followed by biliary clearance [48,49], rather than the result of collagen binding. The higher accumulation of the CHP compared to HPMA-CHP in the kidneys supports renal excretion as the excretion route of the CHP, likely due to the smaller size of CHP. However, as CHP is expected to be eliminated more quickly due to its smaller size, the continued presence in the kidneys at this timepoint may suggest binding to collagen, rather than solely clearance route. The higher accumulation of HPMA-CHP compared to HPMA-SCHP in the kidneys further supports HPMA-CHP and CHP binding to basal collagen remodeling known to be present in the kidneys [50]. Additionally, the copolymers contain a significant amount of terminal COOH groups in the side chains, which ionize to COO– in vivo, and are thus highly negatively charged. Kidney accumulation of HPMA copolymers with high COO- content in the side chains has been observed, and negative charge has been proposed as a potential cause [51,52], and this may have also contributed to the overall kidney accumulation of both copolymers in our study. Polymer charge has been reported to influence biodistribution, due to its influence on glomerular permeability and clearance [53,54], which may have contributed to the observed organ accumulations. Further studies are needed to investigate the mechanisms of kidney accumulation of these constructs.
The higher HPMA-CHP accumulation in the spleen, liver, and stomach and intestines, compared to CHP may be due to the increased size of the polymeric constructs compared to free peptides and contribution of macrophage uptake [55]. Accumulation of HPMA-CHP in the liver and spleen may be due to leakiness in the endothelium and basement membranes in the blood vessels of these organs, as has been observed in the past for other HPMA copolymers [56]. Additionally, the difference in hepatic extraction ratios between peptides and polymers may also play a role. Although there is a higher signal of HPMA-CHP compared to HPMA-SCHP at the heart, potentially reflecting a higher level remaining in circulation, the total level at the heart is very low (>1 order of magnitude lower) compared to other sites. It is possible that the HPMA-SCHP was excreted earlier due to less retention at the tumor and other sites, and that the HPMA-CHP and CHP were retained in the body longer due to collagen binding. The signal from the harvested tumor corroborates the result seen in the in vivo imaging at the 168 h timepoint. In the lungs, both the accumulation of CHP and the difference between HPMA-CHP and HPMA-SCHP suggest a role of binding to collagen, present in the alveolar interstitium [57]. Previous studies have compared HPMA copolymers bound to two different targeting peptides (RGD4C and RGDfK) with different binding affinities to the same receptor and found that when bound to HPMA copolymers the overall biodistributions and tumor accumulations were similar, indicating a greater influence of the polymer [58]. In this study, accumulation at nontarget organs may have been caused by the CHP and the high prevalence of collagen throughout the body to which CHP can bind.
4. Summary and further discussion
The ECM is intricately involved in cell life through complex soluble and insoluble signaling interactions [3,5]. In the tumor-associated ECM, these signaling mechanisms are appropriated to promote tumor growth and metastasis [59]. This results in more deposition of collagen in the tumor ECM by the cancer cells, resulting in a dense matrix that is difficult for chemotherapeutics to penetrate, which limits the efficacy of cancer targeting strategies that involve binding to cell surface receptors [21,22]. Targeting the surrounding collagen matrix provides a strategy that potentially circumvents this problem. One of the changes in the ECM during tumor progression is an increased expression of ECM degradative enzymes, specifically cathepsins and MMPs, which alter the tumor ECM and its signaling dynamics by remodeling the collagens [60]. The increased availability of remodeling collagen was used in this work as a target for tumor selectivity of our polymer delivery vehicle.
We developed HPMA copolymer-CHP conjugates that can bind selectively to denatured collagen and demonstrated their targeted and sustained localization in breast tumor tissue in a mouse model. Targeted conjugates demonstrated an advantage over both non-targeted polymers and collagen hybridizing peptides alone. Compared to CHP, for tumor targeting, the incorporation of the polymer alters pharmacokinetics. We observed an increased AUC for HPMA-CHP compared to HPMA-SCHP, and an increased tumor half-life for HPMA-CHP compared to CHP. These characteristics would allow for greater drug exposure when utilizing HPMA-CHP for drug delivery.
Compared to cell surface-targeted HPMA copolymer conjugates reported in the literature, such as αvβ3 integrin-targeting [41,56,61,62], HER2-targeting [63], HS receptor GRP78-targeting [44], and others, targeting the ECM collagen is not limited by the tumor cell heterogeneity [64], and might be less limited by diffusion across the stroma [21]. The involvement of collagen in many cancers and the flexibility to attach different drugs and imaging agents makes this polymer a versatile delivery platform.
In addition to increasing tumor localization for drug delivery, targeting and anchoring at the extracellular matrix opens opportunities for delivery of drugs that specifically act on the ECM. The involvement of the ECM in cancer progression has led to many therapeutic strategies, such as MMP inhibitors, that focus on suppressing ECM signaling activity [4], which has a critical role in instigating metastasis [65]. While MMP inhibitors (e.g., batimastat, marimastat), have undergone clinical trials, they failed due to delivery limitations (including water insolubility) and side effects (such as peritonitis when delivered intraperitoneally and joint pain) [66,67]. A water-soluble polymeric carrier that anchors in the tumor ECM may improve efficacy and reduce toxicity of such drugs, which may enable successful clinical use, as well as a new theranostic approach through incorporation of imaging agents and drugs. Further, pursuing extracellular targets could enable a combination therapy, simultaneously treating both tumor cells and the environmental contribution of dynamic remodeling.
5. Conclusion
This study demonstrates the utility of a collagen-targeted water-soluble copolymer conjugate for targeted accumulation in a model of breast cancer. The copolymer-peptide conjugate exhibited an increased accumulation at the tumor compared to the non-targeted conjugate as well as an increase in retention over the peptide alone. In future studies, comparison of HPMA copolymers targeting cell surface receptors against collagen-targeting HPMA copolymers could elucidate advantages of extracellular matrix targeting, limitations imposed by stromal diffusion, and potential reduction of off-site toxicities. The important next step in this study is to explore the use of this targeting strategy in other desmoplastic cancer models, as well as to utilize it as a carrier for the delivery of ECM-acting agents and other therapeutics.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Shawn Owen for numerous discussions and guidance. This work was funded by the Ruth L. Kirschstein National Institutes of Health (NIH) National Research Service Award (NRSA) awarded to N.S. (5F31CA213901) and NIH R21OD026618 awarded to S.M.Y. The authors would also like to acknowledge the Preclinical Research Resource (PRR) at the Huntsman Cancer Institute (HCI), supported by the National Cancer Institute (NCI) of the NIH under Award Number P30CA042014, for their assistance with in vivo experiments. Figures were made using http://BioRender.com.
Footnotes
Credit author statement
N.S.: substantial contribution to the conceptualization, investigation, formal analysis, funding acquisition, and writing - original draft of this work. B.Y. and J.K.: contribution to the investigation of this work. S.M.Y. and H.G.: conceptualization, analysis, funding acquisition, resources, and writing - review and editing of this work. All authors have read and agreed to the published version of the manuscript.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jconrel.2022.10.017.
Data availability
Data will be made available on request.
References
- [1].Frantz C, Stewart KM, Weaver VM, The extracellular matrix at a glance, J. Cell Sci 123 (2010) 4195–4200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bonnans C, Chou J, Werb Z, Remodelling the extracellular matrix in development and disease, Nat. Rev. Mol. Cell Biol 15 (2014) 786–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Insua-Rodriguez J, Oskarsson T, The extracellular matrix in breast cancer, Adv. Drug Deliv. Rev 97 (2016) 41–55. [DOI] [PubMed] [Google Scholar]
- [4].Subrahmanyam N, Ghandehari H, Harnessing extracellular matrix biology for tumor drug delivery, J. Pers. Med 11 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK, Extracellular matrix structure, Adv. Drug Deliv. Rev 97 (2016) 4–27. [DOI] [PubMed] [Google Scholar]
- [6].Muncie JM, Weaver VM, The physical and biochemical properties of the extracellular matrix regulate cell fate, Curr. Top. Dev. Biol 130 (2018) 1–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Walker C, Mojares E, Del Río Hernández A, Role of extracellular matrix in development and cancer progression, Int. J. Mol. Sci 19 (2018) 3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Filipe EC, Chitty JL, Cox TR, Charting the unexplored extracellular matrix in cancer, Int. J. Exp. Pathol 99 (2018) 58–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Humphrey JD, Dufresne ER, Schwartz MA, Mechanotransduction and extracellular matrix homeostasis, Nat. Rev. Mol. Cell Biol 15 (2014) 802–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Cox TR, The matrix in cancer, Nat. Rev. Cancer 21 (4) (2021) 217–238. [DOI] [PubMed] [Google Scholar]
- [11].Ribeiro Franco PI, Rodrigues AP, de Menezes LB, Pacheco Miguel M, Tumor microenvironment components: allies of cancer progression, Pathol. Res. Pract 216 (1) (2020), 152729. [DOI] [PubMed] [Google Scholar]
- [12].Multhaupt HAB, Leitinger B, Gullberg D, Couchman JR, Extracellular matrix component signaling in cancer, Adv. Drug Deliv. Rev 97 (2016) 28–40. [DOI] [PubMed] [Google Scholar]
- [13].Li Y, Foss CA, Summerfield DD, Doyle JJ, Torok CM, Dietz HC, Pomper MG, Yu SM, Targeting collagen strands by photo-triggered triple-helix hybridization, Proc. Natl. Acad. Sci. U. S. A 109 (2012) 14767–14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Li Y, Yu SM, Targeting and mimicking collagens via triple helical peptide assembly, Curr. Opin. Chem. Biol 17 (2013) 968–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shoulders MD, Raines RT, Collagen structure and stability, Annu. Rev. Biochem 78 (2009) 929–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ricard-Blum S, The collagen family, Cold Spring Harb. Perspect. Biol 3 (2011), a004978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hwang J, Huang Y, Burwell TJ, Peterson NC, Connor J, Weiss SJ, Yu SM, Li Y, In situ imaging of tissue remodeling with collagen hybridizing peptides, ACS Nano 11 (2017) 9825–9835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Jabłońska-Trypuć A, Matejczyk M, Rosochacki S, Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs, J. Enzyme Inhib. Med. Chem 31 (2016) 177–183. [DOI] [PubMed] [Google Scholar]
- [19].Fields GB, Interstitial collagen catabolism, J. Biol. Chem 288 (2013) 8785–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Walker RA, The complexities of breast cancer desmoplasia, Breast Cancer Res. 3 (2001) 143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jain RK, Transport of molecules in the tumor interstitium: a review, Cancer Res. 47 (1987) 3039–3051. [PubMed] [Google Scholar]
- [22].Jain RK, Stylianopoulos T, Delivering nanomedicine to solid tumors, Nat. Rev. Clin. Oncol 7 (2010) 653–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Stylianopoulos T, Wong C, Bawendi MG, Jain RK, Fukumura D, Chapter six - multistage nanoparticles for improved delivery into tumor tissue, in: Düzgüneş N (Ed.), Methods in Enzymology, Academic Press, 2012, pp. 109–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA, The pancreas cancer microenvironment, Clin. Cancer Res 18 (2012) 4266–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Valmiki S, Aid MA, Chaitou AR, Zahid M, Valmiki M, Fawzy P, Khan S, Extracellular matrix: a treasure trove in ovarian cancer dissemination and chemotherapeutic resistance, Cureus 13 (2021), e13864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Trenevska I, Li D, Banham AH, Therapeutic antibodies against intracellular tumor antigens, Front. Immunol 8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cathcart J, Pulkoski-Gross A, Cao J, Targeting matrix metalloproteinases in cancer: bringing new life to old ideas, Genes Dis. 2 (2015) 26–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ughachukwu P, Unekwe P, Efflux pump-mediated resistance in chemotherapy, Annal. Med. Health Sci. Res 2 (2012) 191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wahyudi H, Reynolds AA, Li Y, Owen SC, Yu SM, Targeting collagen for diagnostic imaging and therapeutic delivery, J. Control. Release 240 (2016) 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].An B, Lin YS, Brodsky B, Collagen interactions: drug design and delivery, Adv. Drug Deliv. Rev 97 (2016) 69–84. [DOI] [PubMed] [Google Scholar]
- [31].Järveläinen H, Sainio A, Koulu M, Wight TN, Penttinen R, Extracellular matrix molecules: potential targets in pharmacotherapy, Pharmacol. Rev 61 (2009) 198–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Bennink LL, Li Y, Kim B, Shin IJ, San BH, Zangari M, Yoon D, Yu SM, Visualizing collagen proteolysis by peptide hybridization: from 3D cell culture to in vivo imaging, Biomaterials 183 (2018) 67–76. [DOI] [PubMed] [Google Scholar]
- [33].Hwang J, San BH, Turner NJ, White LJ, Faulk DM, Badylak SF, Li Y, Yu SM, Molecular assessment of collagen denaturation in decellularized tissues using a collagen hybridizing peptide, Acta Biomater. 53 (2017) 268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shoulders MD, Kamer KJ, Raines RT, Origin of the stability conferred upon collagen by fluorination, Bioorg. Med. Chem. Lett 19 (2009) 3859–3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kessler JL, Li Y, Fornetti J, Welm AL, Yu SM, Enrichment of collagen fragments using dimeric collagen hybridizing peptide for urinary Collagenomics, J. Proteome Res 19 (2020) 2926–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Yang J, Kopeček J, Design of smart HPMA copolymer-based nanomedicines, J. Control. Release 240 (2016) 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yang J, Kopeček J, The light at the end of the tunnel-second generation HPMA conjugates for cancer treatment, Curr. Opin. Colloid Interface Sci 31 (2017) 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Buckway B, Wang Y, Ray A, Ghandehari H, Overcoming the stromal barrier for targeted delivery of HPMA copolymers to pancreatic tumors, Int. J. Pharm 456 (2013) 202–211. [DOI] [PubMed] [Google Scholar]
- [39].Frazier N, Payne A, de Bever J, Dillon C, Panda A, Subrahmanyam N, Ghandehari H, High intensity focused ultrasound hyperthermia for enhanced macromolecular delivery, J. Control. Release 241 (2016) 186–193. [DOI] [PubMed] [Google Scholar]
- [40].Pike DB, Ghandehari H, HPMA copolymer–cyclic RGD conjugates for tumor targeting, Adv. Drug Deliv. Rev 62 (2010) 167–183. [DOI] [PubMed] [Google Scholar]
- [41].Borgman MP, Aras O, Geyser-Stoops S, Sausville EA, Ghandehari H, BiodistributionofHPMAcopolymer-aminohexylgeldanamycin-RGDfKconjugatesforprostatecancerdrug delivery, Mol. Pharm 6 (2009) 1836–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Maeda H, Wu J, Sawa T, Matsumura Y, Hori K, Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review, J. Control. Release 65 (2000) 271–284. [DOI] [PubMed] [Google Scholar]
- [43].Matsumura Y, Maeda H, A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs, Cancer Res. 46 (1986) 6387–6392. [PubMed] [Google Scholar]
- [44].Frazier N, Payne A, Dillon C, Subrahmanyam N, Ghandehari H, Enhanced efficacy of combination heat shock targeted polymer therapeutics with high intensity focused ultrasound, Nanomedicine 13 (2017) 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Strohalm J, Kopeček J, Poly[N-(2-hydroxypropyl)methacrylamide]. IV. Heterogeneous polymerization, Die Angew. Makromol. Chem 70 (1978) 109–118. [Google Scholar]
- [46].Li Y, Mo X, Kim D, Yu SM, Template-tethered collagen mimetic peptides for studying heterotrimeric triple-helical interactions, Biopolymers 95 (2011) 94–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wang AY, Foss CA, Leong S, Mo X, Pomper MG, Yu SM, Spatio-temporal modification of collagen scaffolds mediated by triple helical propensity, Biomacromolecules 9 (2008) 1755–1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Noguchi Y, Wu J, Duncan R, Strohalm J, Ulbrich K, Akaike T, Maeda H, Early phase tumor accumulation of macromolecules: a great difference in clearance rate between tumor and normal tissues, Jpn. J. Cancer Res 89 (1998) 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kostka L, Etrych T, High-molecular-weight HPMA-based polymer drug carriers for delivery to tumor, Physiol. Res 65 (2016) S179–s190. [DOI] [PubMed] [Google Scholar]
- [50].Alexakis C, Maxwell P, Bou-Gharios G, Organ-specific collagen expression: implications for renal disease, Nephron Exp. Nephrol 102 (2006) e71–e75. [DOI] [PubMed] [Google Scholar]
- [51].Borgman MP, Coleman T, Kolhatkar RB, Geyser-Stoops S, Line BR, Ghandehari H, Tumor-targeted HPMA copolymer-(RGDfK)-(CHX-A-DTPA) conjugates show increased kidney accumulation, J. Control. Release 132 (2008) 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Liu GW, Prossnitz AN, Eng DG, Cheng Y, Subrahmanyam N, Pippin JW, Lamm RJ, Ngambenjawong C, Ghandehari H, Shankland SJ, Pun SH, Glomerular disease augments kidney accumulation of synthetic anionic polymers, Biomaterials 178 (2018) 317–325. [DOI] [PubMed] [Google Scholar]
- [53].Etrych T, Šubr V, Strohalm J, Šírová M, Ríhová B, Ulbrich K, HPMA copolymer-doxorubicin conjugates: the effects of molecular weight and architecture on biodistribution and in vivo activity, J. Control. Release 164 (2012) 346–354. [DOI] [PubMed] [Google Scholar]
- [54].Rennke HG, Venkatachalam MA, Glomerular permeability of macromolecules. Effect of molecular configuration on the fractional clearance of uncharged dextran and neutral horseradish peroxidase in the rat, J. Clin. Invest 63 (1979) 713–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Blanco E, Shen H, Ferrari M, Principles of nanoparticle design for overcoming biological barriers to drug delivery, Nat. Biotechnol 33 (2015) 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mitra A, Nan A, Papadimitriou JC, Ghandehari H, Line BR, Polymer-peptide conjugates for angiogenesis targeted tumor radiotherapy, Nucl. Med. Biol 33 (2006) 43–52. [DOI] [PubMed] [Google Scholar]
- [57].Bradley KH, McConnell SD, Crystal RG, Lung collagen composition and synthesis. Characterization and changes with age, J. Biol. Chem 249 (1974) 2674–2683. [PubMed] [Google Scholar]
- [58].Mitra A, Coleman T, Borgman M, Nan A, Ghandehari H, Line BR, Polymeric conjugates of mono- and bi-cyclic αVβ3 binding peptides for tumor targeting, J. Control. Release 114 (2006) 175–183. [DOI] [PubMed] [Google Scholar]
- [59].Pickup MW, Mouw JK, Weaver VM, The extracellular matrix modulates the hallmarks of cancer, EMBO Rep. 15 (2014) 1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kai F, Drain AP, Weaver VM, The extracellular matrix modulates the metastatic journey, Dev. Cell 49 (2019) 332–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Zarabi B, Borgman MP, Zhuo J, Gullapalli R, Ghandehari H, Noninvasive monitoring of HPMA copolymer-RGDfK conjugates by magnetic resonance imaging, Pharm. Res 26 (2009) 1121–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ray A, Larson N, Pike DB, Grüner M, Naik S, Bauer H, Malugin A, Greish K, Ghandehari H, Comparison of active and passive targeting of docetaxel for prostate cancer therapy by HPMA copolymer-RGDfK conjugates, Mol. Pharm 8 (2011) 1090–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Radford DC, Yang J, Doan MC, Li L, Dixon AS, Owen SC, Kopeček J, Multivalent HER2-binding polymer conjugates facilitate rapid endocytosis and enhance intracellular drug delivery, J. Control. Release 319 (2020) 285–299. [DOI] [PubMed] [Google Scholar]
- [64].Crucitta S, Cucchiara F, Mathijssen R, Mateo J, Jager A, Joosse A, Passaro A, Attili I, Petrini I, van Schaik R, Danesi R, Del Re M, Treatment-driven tumour heterogeneity and drug resistance: lessons from solid tumours, Cancer Treat. Rev 104 (2022), 102340. [DOI] [PubMed] [Google Scholar]
- [65].Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ, Collagen reorganization at the tumor-stromal interface facilitates local invasion, BMC Med. 4 (2006) 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Winer A, Adams S, Mignatti P, Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes, Mol. Cancer Ther 17 (2018) 1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Wojtowicz-Praga SM, Dickson RB, Hawkins MJ, Matrix metalloproteinase inhibitors, Investig. New Drugs 15 (1997) 61–75. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.