

Abstract
There is a need for better predictive models of the human immune system to evaluate safety and efficacy of immunomodulatory drugs and biologics for successful product development and regulatory approvals. Current in vitro models, which are often tested in two-dimensional (2D) tissue culture polystyrene, and preclinical animal models fail to fully recapitulate the function and physiology of the human immune system. Microphysiological systems (MPSs) that can model key microenvironment cues of the human immune system, as well as of specific organs and tissues, may be able to recapitulate specific features of the in vivo inflammatory response. This minireview provides an overview of MPS for modeling lymphatic tissues, immunity at tissue interfaces, inflammatory diseases, and the inflammatory tumor microenvironment in vitro and ex vivo. Broadly, these systems have utility in modeling how certain immunotherapies function in vivo, how dysfunctional immune responses can propagate diseases, and how our immune system can combat pathogens.
Keywords: Microphysiological systems, immunity, lymphatics, tissue interfaces, inflammatory diseases, tumor microenvironment
Impact Statement
This review discusses recent advancements in engineering microphysiological systems (MPSs) to model certain aspects of immunity. MPSs are increasingly able to model the function of vaccines in vitro and ex vivo by modeling lymphatic organs. Furthermore, MPS can recapitulate how tissue interfaces provide immunity to certain organs in our body. They may also serve a platform for modeling inflammatory diseases and provide a test bed for screening anti-inflammatory agents. Models of the tumor microenvironment may be predicative of the responses that drug and cellular immunotherapies have in patients.
Introduction
Model systems that recapitulate the biology of the human immune system are critical to the clinical translation and success of drugs and biologics. The safety of drugs and biologics depends on evaluating how these agents induce immunosuppressive, immunostimulatory, hypersensitivity, and/or autoimmune reactions in vivo.1,2 In addition to their safety profile, many drugs and biologics depend on the immune system for their mechanism of action and efficacy in treating diseases. These include vaccines and immunotherapies that boost the immune system’s natural ability to combat pathogens and cancers, as well as immunosuppressants that dampen the immune system during inflammatory and autoimmune diseases. Furthermore, inflammatory or anti-inflammatory drugs/biologics can have off-target effects in vivo, as the immune system is intimately linked with a variety of other biological processes in our body, such as the human gut microbiome 3 and wound healing. 4 Given the complexity of the immune system and its interdependence with other biological systems, predicting and measuring how drugs and biologics interact with the human immune system are a significant challenge for regulatory bodies and industries translating these therapies. In pursuing regulatory approval, drug developers typically evaluate safety and efficacy of new drugs and biologics using in vitro assays or animal models during the product development stage.
However, these animal models and in vitro assays have significant limitations in predicting how these drugs and biologics interact with the human immune system in vivo. Widely used murine models exhibit fundamental cellular and molecular differences in their inflammatory responses compared to the human in vivo immune system. 5 Humanized mice also demonstrate defects in their engrafted human immune system, which do not completely recapitulate human disease. 6 Furthermore, in vitro assays, which are most commonly performed in two-dimensional (2D) tissue culture polystyrene, fail to recapitulate in vivo cellular and chemical microenvironments of the human immune system and do not capture the complex interactions that molecular and cellular immune components have with different tissues and organ systems.
Microphysiological systems (MPSs) are microscale cell culture platforms that may recapitulate key features of the human immune system. These platforms, which may include monocultures, co-cultures of multiple cell types, tissue/organ explants, and/or organoids, 7 incorporate key features of a tissue’s or organ’s microenvironment, including chemical, physical, and cellular cues. In combination with biomaterials mimicking extracellular matrix (ECM) and microfluidic technologies, these systems can also control the spatial and temporal presentation of cues to model healthy and diseased tissues. Incorporation of molecular and cellular components of the immune system into these systems may potentially model human inflammatory processes with greater fidelity than animal models and traditional in vitro assays.
In this minireview, we provide an overview of the immunology of (1) lymphatic tissues, (2) immunity and inflammation at tissue interfaces, (3) inflammatory disorders and diseases, and (4) immunity in tumor microenvironments (TMEs), and then further discuss MPSs for modeling these immunological tissues and phenomena. For lymphatic tissues, we discuss how MPS systems can model lymphatic vessels and lymph node structures that are involved in generating adaptive immune responses with vaccines. Furthermore, we provide examples of the gastrointestinal (GI) mucosa barrier, the blood–brain barrier (BBB), and the placental barrier that MPS can model to assess inflammation and immunity at these tissue interfaces. We also discuss MPS models for recapitulating the pathology of atopic dermatitis (AD), arthritis, and cystic fibrosis (CF). Finally, we discuss approaches to model the TME and how these can be leveraged to evaluate chimeric antigen receptor (CAR) T-cells and immune checkpoint blockade therapies.
MPSs modeling lymphatic tissues
Vaccination
Vaccines, which provide acquired immunity to certain pathogens, are critical for reducing morbidity and mortality to many infectious diseases, including severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Between 2011 and 2020, there were over 600 clinical trials registered on National Institutes of Health’s (NIH) ClinicalTrials.gov investigating the clinical use of vaccines 8 and at least 46 clinical trials as of 2021 for SARS-CoV-2 vaccines. 9 Many types of vaccines are being explored preclinically and/or clinically, which include inactivated, attenuated, subunit, viral vector, nanoparticle, and RNA/DNA vaccines. 10 Furthermore, dendritic cell (DC) vaccines are being widely explored as cell-based vaccines for the treatment of various cancers, with over 130 on-going clinical trials as of 2021. 11 Vaccines against pathogens consist of antigens (or agents that produce antigens), which are components of the pathogen to which the body develops adaptive immunity. In addition to this, vaccines may contain adjuvants, which enhance the immune responses to antigens typically by activating pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), present on the surface and in the endosomes of antigen-presenting cells.
Vaccines are delivered to patients via several routes, including via intramuscular, subcutaneous, oral, and intranasal administration. Upon injection into the body, the antigen and adjuvant molecules are delivered to secondary lymphoid organs via three routes: (1) uptake by an antigen-presenting cell (i.e. DCs, macrophages, B-cells), (2) convection into draining lymph nodes via lymphatic vessels, and (3) diffusion into blood vasculature, where they are delivered to the spleen, liver, and kidneys. 12 The two arms of the adaptive immune system, consisting primarily of humoral immunity (B-cells) and cellular immunity (T-cells), are then activated in different ways. Activation of T-cell immunity typically begins with immature DCs that process antigen and mature via activation of their TLRs. 13 These mature DCs upregulate CCR7 and follow gradients of CCL21 into lymphatic vessels, where they eventually traffic to the lymph nodes in the T-cell zone. Here, DCs will present antigen to and activate naïve or memory T-cells, to induce a cell-mediated adaptive immune responses. Activation of B-cell humoral immunity is induced by antigens that are delivered to secondary lymph nodes without antigen-presenting cells. 13 These antigens, which can be in soluble form or displayed by DCs/macrophages, activate naïve B-cells that then process the antigen and present it to activated helper T-cells. The B-cells then migrate toward lymph node follicles, where they are activated by T follicular helper cells and form germinal centers. At these germinal centers, extensive B-cell proliferation, isotype switching, somatic hypermutation, affinity maturation, memory B-cell generation, and plasma cell generation occur. These plasma cells secrete large quantities of antibodies against pathogens. Given the complexity of these immunological processes, the safety and efficacy of vaccines are extensively tested preclinically in animal models. However, advancements in MPSs are enabling the study of these processes in vitro and ex vivo.
MPSs for evaluating vaccines
The migration of DCs toward gradients of CCL19 or CCL21 in biomaterials and/or microfluidic devices may provide insights into their migration toward lymphatics in response to vaccination. Previous work evaluating the migration of human mature dendritic cells (mDCs) in porous fibrillar collagen I scaffolds has revealed that mDCs adopt an amoeboid migration mode in response to CCL19 and CCL21 stimulation. 14 Microfluidic devices have also enabled the evaluation of mature DC migration in response to defined gradients of these cytokines, specifically in three-dimensional (3D) collagen I/Matrigel hydrogels 15 and under confinement under agarose gels; these studies have demonstrated that both exhausted and active mature DCs respond to gradients of CCL19 16 and that DCs may migrate more preferentially toward CCL21 than CCL19. 15 Laser-writing and chemokine immobilization technologies have further enabled the comparison of DC migration under soluble CCL19 versus immobilized gradients of CCL21. 17 Microfluidic devices have also been designed to simultaneously evaluate DC migration in response to CCL19 and T-cell activation by DCs. 18
MPS modeling the cellular and fluid flow microenvironments of lymphatics and lymph vessels have also been utilized to model immune cell trafficking to the lymph nodes. A 3D microfluidic model of lymphatic microvasculature with interstitial flow through a fibrin matrix has been shown to induce peripheral blood mononuclear cell (PBMC) recruitment with tumor necrosis factor (TNF)-α stimulation in a CCR7 and CXCR4–dependent manner. 19 Furthermore, a microfluidic perfusion model that was engineered to model the subcapsular sinus (Figure 1(A)) demonstrated that monocyte and metastatic cancer cell adhesion was dependent on E-selectin in the flow microenvironment. 20 Fluid flow has been shown to be critical to the expression of CCL21 by primary fibroblastic reticular cells (FRCs) cultured on 3D polyurethane hydrogel with collagen and Matrigel 21 This may be relevant to engineering MPS of the lymph node T-cell zone that can regulate T-cell and DC trafficking.
Figure 1.

Examples of lymphatic microphysiological systems that may be used to evaluate vaccines. (A) A divergent channel perfusion system is used to evaluate the effect of lymph node subcapsular sinus microenvironment-mimicking flow on cancer and immune cell adhesion in vitro. 20 (Source: Reproduced with permission, Copyright 2021 Elsevier.) (B) Murine lymph node slices cultured ex vivo can model cellular adaptive immune responses and antigen-specific responses. 27 (Source: Reproduced with permission, Copyright 2021 American Chemical Society.) (C) A lymph node follicle on a chip can induce ectopic lymph node follicle formation from primary B- and T-cells under flow to model lymph node germinal centers. In right confocal images, green denotes extracellular matrix fibers by second harmonic imaging and magenta denotes Hoechst staining of culture lymphocytes. 30 (Source: Reproduced with permission, an open-access journal printed by 2022 Wiley-VCH GmbH.)
Microfluidic devices and/or biomaterials can also be leveraged to study DC antigen-presentation to T-cells in a physiologically relevant manner. Microfluidic cell platforms have been utilized to evaluate early T-cell activation events between single DCs and T-cells 22 or small groups of compartmentalized DCs and T-cells. 23 DC activation of antigen-specific T-cells has also been evaluated in collagen I hydrogels, which mimic a key ECM protein found in lymph nodes.24,25 The effect of flow on antigen-presentation between DCs and T-cells in microfluidic devices has also been explored to evaluate the effect of interstitial flow observed in the in vivo lymph node microenvironment.25,26 Cultured lymph node slices from mice may also evaluate T-cells activation ex vivo in physiologically relevant microenvironments, as these slices presumably contain T-cells, DCs, and intact fibroblast reticular cell network (Figure 1(B)). 27 Stimulation of these slices with anti-CD3 to stimulate T-cells and/or R848 to stimulate the antigen-presenting cells resulted in upregulation of CD69 and cytokine production, demonstrating their ability to model activation of T-cells. 27
Several recent developments in germinal center modeling have also demonstrated the ability to test vaccine antibody production ex vivo. Polyethylene glycol (PEG) hydrogels functionalized with integrin binding REDV or RGD and seeded with naïve B-cells and 40LB stromal cells demonstrated ex vivo B-cell differentiation, as well as light-zone germinal center B-cell phenotype. Furthermore, the authors showed antigen-specific B-cell enrichment with first apoptosis signal FAS and B-cell receptor (BCR) stimulation in their 3D system. 28 This system was also leveraged to evaluate germinal center B-cell responses to novel designer conjugate vaccines in vitro. 29 In a two-channel microfluidic device, primary human B- and T-cells were shown to self-assemble into ectopic lymphoid follicles (LFs) when cultured in a collagen gel in one channel and with flow applied to the second channel (Figure 1(C)). When co-cultured with monocyte-derived DCs, these LFs induced plasma cell formation, B-cell class switching, and antibody production. 30 Organoids derived from human tonsils were able to similarly model key features of human germinal centers, in terms of antigen-specific antibody production, affinity maturation, plasma blast differentiation, and class-switching in response to rabies and SARS-COV-2 vaccines. 31 The MIMIC system was also developed, which consists of human umbilical vein endothelial cells (ECs) cultured on top of a collagen matrix with PMBCs; DCs, follicular DCs, T-cells, and B-cells were then sequentially added to the system to model the order of immune response in lymph nodes. 32 This system demonstrated antigen-specific antibody production in response to various vaccines. 32
While not directly relevant to the evaluation of vaccines, there have also been a number of MPSs modeling the physiology and function of healthy and diseased bone marrow, where all blood cells including immune cells are derived from. Microfluidic single-cell arrays have been developed to evaluate the behavior of hundreds of hematopoietic stem cells that are normal or exhibit chronic myeloid leukemia. 33 A recently developed bone marrow-on-a-chip utilizing human hematopoietic stem cells (HSCs), bone marrow stromal cells, and human umbilical vein ECs in a two-channel device with collagen and fibrin was able to support myeloerythroid differentiation, as well as model drug toxicity on erythrocytes and neutrophils. 34 Another model utilizing biphasic calcium phosphate–based scaffolds and human cell lines was able to model key features of the bone marrow niche and model bone marrow pathologies in response to disseminated cancer cells and leukemia. 35 A leukemia-on-a-chip modeling the perivascular and endosteal niches of the bone marrow was also able to model heterogeneous chemoresistance across various B-cell acute lymphoblastic leukemia cell lines. 36
Summary
MPSs modeling key features of lymphatic tissues, including lymphatic vessels, the lymph node subcapsular sinus, the lymph node T-cell zone/paracortex, and the lymph node B-cell zone/germinal center, are being developed with increasing complexity and physiological relevance for evaluating vaccine therapies. This is in part due to novel approaches for modeling key chemical and physical microenvironments of lymphatic tissues, such as chemokine gradients and fluid flow, as well as improved methods for engineering stromal and EC compartments of these lymphatic tissues through tissue engineering approaches and organoids. A current drawback of these systems is that they are currently evaluated as individual MPS systems, whereas vaccination and adaptive immune responses require a complex integration of various lymphatic tissues to fully model the entire cascade of T-/B-cell generation, antigen-presentation, adaptive immune cell activation, and immune cell migration. More accurate models of lymphatic tissues that fluidically link various lymphatic MPS systems will allow for more accurate modeling of the immune cell cascade for evaluating vaccines and immunotherapies.
MPSs modeling immunity and inflammation at tissue interfaces
Immunity and inflammation at the GI tract interface
The mucosal surface in the GI tract is the largest among all mucosal systems in the human body. The GI tract interface acts as a physical barrier to limit foreign substances, such as pathogens, dietary antigens, and toxins, from entering host internal tissue from the digestive lumen. The GI tract is made of specialized epithelial cells that form the wall of lumen tract, as well as mucins secreted by mucous and goblet cells on the luminal side, unstirred layer, and basement membrane on the abluminal side. The GI mucosal barrier dynamically regulates the interactions between the intestinal epithelial immune system and luminal microorganisms and toxins. The immune defense mechanisms include immunoglobulin A (IgA) secreted from B-cells, antimicrobial proteins, intraepithelial lymphocytes that secrete growth factors to repair compromised barrier and pro-inflammatory factors to fight against pathogens, and macrophages for “eating” pathogens that cross the epithelial layer. 37 Alterations in homeostasis due to the dysfunction of the GI barrier contribute to gut diseases. For example, Bradford et al. 38 showed that high TNF levels activated the mucosal immune system, which disrupt the gut barrier integrity. When interleukin (IL)-10 was knocked out, mice developed colitis accompanied with increased intestinal permeability. 39 It was also demonstrated that while myosin light-chain kinase (MLCK)-derived tight junction activation is insufficient to cause disease, it activates mucosal immune cells by increasing lamina propria T-cells, enhancing pro-inflammatory cytokine secretion such as interferon (IFN)-γ, TNF and IL-10, as well as relocating CD11c+ DCs to basement membrane. 40
MPS for modeling immunity at the GI interface
GI MPS models have been developed to study intestinal responses under physiological situations (i.e. relevant oxygen gradients, transepithelial gradient of morphogens),41,42 to customize gut-on-a-chips using biopsy tissue from patients 43 and to investigate the immune response under pathological conditions. For example, a microfluidic device was developed, featuring human Caco-2 intestinal epithelial cells on a 10-µm porous poly(dimethylsiloxane) (PDMS) matrix, flow rate of 30 µL/h, and specific mechanical deformations to promote formation of intestinal microvillus (Figure 2(A)). This model discovered an unknown contribution of IL-8, IL-6, IL-1β, and TNF-α to induce villus injury and compromise the barrier integrity after stimulation with immune cells and lipopolysaccharide (LPS) endotoxins together. 44 In 2018, Shi and Kim reported a colitis disease model induced by 2% dextran sodium sulfate (DSS) to demonstrate that direct contact between PBMCs and DSS-sensitized epithelial cells is necessary to cause epithelial oxidative stress and to produce pro-inflammatory cytokines. It was revealed that treatment efficacy by probiotic gut bacteria was significantly decreased when the gut barrier was compromised in their controllable models. 45 GI MPS can also be used to investigate the immune response after mucosal barriers are infected by parasites such as Toxoplasma gondii 46 or Helicobacter pylori. 47 These models displayed robust in vivo like signatures indicated by (1) decreased transcript levels of MKI67, LGR5, and AXIN2, (2) highly increased levels of VIL1, BEST4, and MUC2, 46 and (3) continuous expression of MUC5AC, MUC6, TFF1, and TFF2. 47 After infection, they found immune cells migrated to the infected sites to stimulate IFN-γ-dependent innate immune response 46 and to increase pro-inflammatory cytokines such as TNF-α to stimulate defense.46,47
Figure 2.
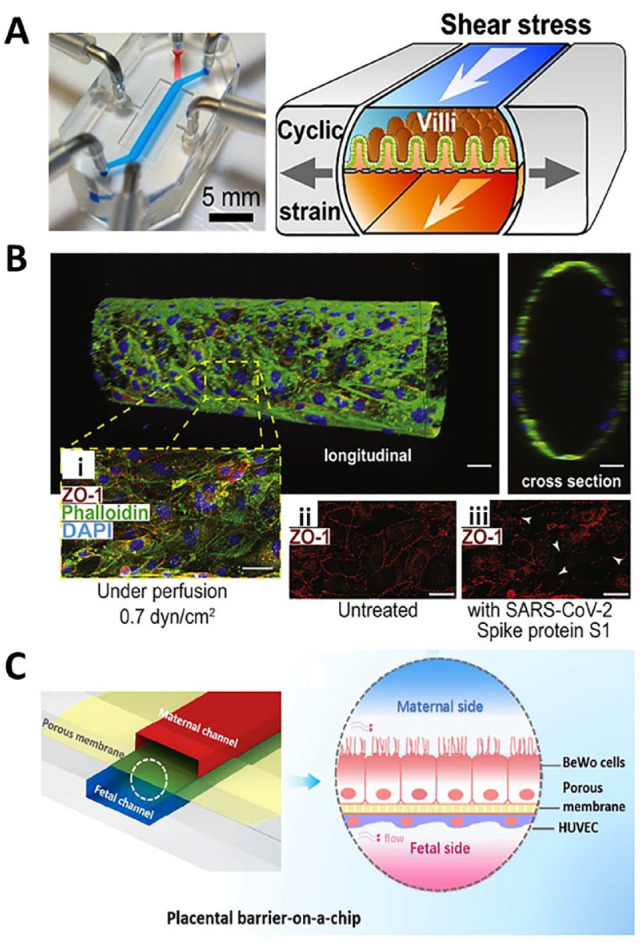
Examples of microphysiological systems to model tissue interface. (A) A GI-on-a-chip displays mechanical strain (gray arrows) and shear force (white arrows) contribute to the microvillous formation. 44 (Source: Reproduced with permission, Copyright 2015 National Academy of Sciences (NAS).) (B) A 3D human BBB model shows an endothelial monolayer at longitudinal and cross-sectional view, and that SARS-CoV-2 spike protein S1 disrupt tight junctions (white arrows). 58 (Source: Reproduced with permission, an open-access journal printed by 2020 Elsevier.) Scale bar: 20 μm. (C) A placenta-on-a-chip model shows the trophoblasts and endothelial cells on the opposite of a microporous membrane, and the flow mimic the physiological environment. 67 (Source: Reproduced with permission, Copyright 2018 American Chemical Society.)
Immunity and inflammation at the BBB
The BBB play a key role in maintaining sensitive brain homeostasis: the brain needs to keep an extra stable microenvironment for its precise neural connections. It includes a monolayer of ECs sealed by tight junctions to form the blood vessel wall, a dense brush-like glycocalyx on the luminal site of the ECs, a basement membrane surrounding the EC monolayer, pericytes embedded in the basement membrane, and astrocyte endfeet that wrap the pericyte processes and the basement membrane. The BBB also dynamically interacts with microglia and neurons, forming a “neurovascular unit” to maintain cerebral homeostasis. CNS parenchyma is described as an immune privileged site for two reasons: (1) DCs and other APCs are found beyond the CNS parenchyma in the brain, in regions such as meanings and circumventricular organs with incomplete BBB and (2) at healthy state, parenchymal intact BBB limits lymphocytes across into brain parenchyma from blood circulation.48–51 While microglia in the brain parenchyma play a primary role in immune surveillance for host defense, the central nervous system (CNS) is an immune privileged site: not only does it lack draining lymphatics and local antigen-presenting cells like DCs, but the BBB also prevents almost all lymphocytes from penetrating into the CNS from blood circulation. An exception to this is the migration of activated T-cells across the BBB, which is mediated by α4-integrins. 52 However, under a pathological status, resting T-cells can also pass through the BBB and invade the CNS due to upregulation of endothelial intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) on ECs. 52 There are several steps required for T-cells to cross the BBB. First, T-cells are captured by ICAM-1 and VCAM-1 present on the EC surface, followed by T-cell activation after attachment. The activated T-cells then arrest and start to crawl along the cerebral endothelium. Eventually, these T-cells cross the BBB through intact tight junctions, the paracellular pathway or the transcellular pathway. 53
MPS for modeling immunity at the BBB
Since the development of MPSs, various BBB models have been used to study the mechanisms of BBB inflammation such as T-cell penetration into the BBB under physiological flow. 54 These systems have also been used to investigate the immune response under neuroinflammation 55 pathogen infection conditions caused by the fungi Cryptococcus neoformans, 56 RNA viruses such as Zika virus (ZIKV) and Dengue virus, 57 or SARS-CoV-2 virus. 58 A neuroinflammation model was established by seeding brain microvessel EC on a physiologically relevant loose and porous ECM gel embedded with mural cells including brain pericytes and astrocytes, which play a key role in neuroprotection, in addition to secreting pro-inflammatory cytokines like IL-6 and IL-8 to regulate both the acute and late-stage immune responses and to trigger immune cell recruitment to injured sites. 55 A BBB model with microgravity-driven fluid flow demonstrated C. neoformans penetration-involved transcytosis by first attaching to the endothelial layer, then crossing the EC without changing EC morphology, and finally passing through the endothelium. 56 The brain-liver-on-a-chip was developed by connecting the BBB unit to a liver-on-a-chip unit and was used to study the neurotropism of C. neoformans to control microbial neurotropic behaviors, as well as to screen the genes that facilitate microorganisms cross BBB. 56 Bramley et al. 57 reported a 3D human BBB model by cultivating brain microvessel EC in a rotating wall vessel bioreactor to mimic in vivo microgravity and shear forces. This model has a similar transcriptional profiles to in vivo physiological states, including well-characterized BBB makers such as VE-cadherin, claudin-1, connecin-26, ZO-3, LRP1, Glut-1, IGFL1, and Na+/K+ ATPase-1. The model also demonstrated intact BBB resistance to viral infection by secreting antimicrobial factors like IFN-β, IFN-λ1, and IFN-λ2 as innate immune responses, while compromised BBB pretreated by TNF-α was susceptible to ZIKV infection. Many studies reported that SARS-CoV-2 virus can interact with the BBB by binding ACE2 receptor on cerebral EC to cause neurological symptoms, like headache, nausea, and dizziness. Using a gel-based cylindrical hollow space microfluidic system (Figure 2(B)), it was demonstrated that the virus-spike proteins S1 and S2 alone can activate the cerebral endothelial pro-inflammatory response by upregulating endothelial ICAM-1 and VCAM-1 for immune cell attachment. 58 Recently, a report showed SARS-CoV-2 infection also cause upregulation of inflammatory cytokines such as IL-6, IL-8, TNF-α, CXCL10, and CCL3 in glial cells. 59
Immunity and inflammation at the placental barrier
During pregnancy, the fetus must be protected from the immune system of the mother, while simultaneously permitting the delivery of oxygen and nutrients and the elimination of waste from the fetus. One strategy used by mammals is the formation of a placenta shortly after the embryo implants into the uterus. Initial formation of the human placenta is hemodichorial, having two layers. A single layer of multi-nucleated syncytiotrophoblasts (SYNTs) that are directly in touch with maternal blood are layered over a second layer of mononucleated cytotrophoblasts (CTBs). As the placenta continues to develop throughout the second and third trimesters, the CTB layer becomes progressively perforated and sparse. Eventually, the placenta becomes hemomonochorial, with only the SYNTs remaining as a barrier. The placenta offers a barrier not only to permit gas, nutrients, and waste exchange but also to provide an immune defense barrier to prevent maternal white blood cells and pathogens from entering the fetus while still allowing antibody transfer. The defense mechanisms include the following: (1) the TLRs on SYNT layer recognize pathogens and initiate innate immune responses; (2) the SYNTs form a dense brush-like structure at the maternal-facing surface to resist direct microbial infections such as Listeria monocytogenes and Toxoplasma gondii; 60 (3) the SYNTs constitutively secrete antiviral factors, such as type III IFNs (IFN-λ) and specific miroRNAs, to resist viral infections; 61 and (4) the SYNT express FcRn and FcγRIII receptors to actively transport protective IgG antibodies from the mother to the fetus. 62
Compromising the function of SYNT leads to more fetal infections. For example, it has been demonstrated that in IFN-λ deficient mouse models, the fetuses are more susceptible to ZIKV infection due to dysfunction of IFN-λ signaling. 61
MPS for modeling immunity at the placental barrier
Microfabrication combined with bioengineering technology enables the development of placental MPS to study drug transport, 63 placental defense mechanisms to resist ZIKV invasion, 64 the evaluation of common nanoparticles on the barrier, 65 and the immune response after the interface is insulted by parasites, 66 bacteria, 67 and viruses.64,66 First, it has been reported that a microengineered human placental barrier recaptures the efflux pump-mediated transport function for investigating the transport of glyburide (gestational diabetes drug) and diacetate (an antimicrobial drug) from the mother to the fetus.63,68 Second, the placental barrier functions as a physical barrier to resist virus invasion by different mechanisms, including the release of antiviral cytokines such as IFNs. Using an organoid-based human placental model, it was revealed that the constant release of IFN-λ1 and IFN-λ2 from SYNT plays a role in resistance to ZIKV during different trimesters of human gestation, which is comparable to the function of human chorionic villous explants. 64 Third, it has been demonstrated that the microengineered placental barrier could be used to evaluate the unknown and potentially serious effects of nanomedicine. For instance, a microengineered 3D placental model demonstrated that exposure to nanoparticles such as titanium dioxide nanoparticles (TiO2-NPs) compromises the integrity of the human placental barrier and significantly increases the number of maternal macrophages adhering to the trophoblastic cell layer. 65 Finally, several microengineered placental models have been developed to investigate how the placental barrier responds to pathogenic insults. McConkey et al. 66 constructed a 3D placental barrier with a rotating wall vessel bioreactor filled with dextran beads coated with collagen to serve as an ECM scaffold to induce the expression of SYNT-associated markers, such as BhCG, hPL, SYN, MFDS2, and PP13 in amounts comparable to primary human trophoblasts (PHTs). This model showed similar transcriptional profiles to PHT and was resistant to infection by vesicular stomatitis virus and Toxoplasma gondii. Escherichia coli is the one of the common inflammatory-causing bacteria found in fetuses. To investigate how the maternal-fetal interface responds to E. coli invasion, Zhu et al. 67 established a microengineered placental barrier with multicellular layers formation under flow. The porous matrix (0.4 µm) acted as the ECM, a key factor that enabled the trophoblasts and endothelium to assemble into a placental barrier with functional microvilli similar to that found in vivo (Figure 2(C)). By adding E. coli into this microsystem, an inflammatory placental model was established, as indicated by a high level of pro-inflammatory cytokines such as IL-1α, IL-1ß, IL-8, and TNF-α. Such inflammatory conditions activate maternal macrophages for epithelial attachment and maternal innate immunity. The microengineered placental barrier also allowed for the investigation of crosstalk in response to bacterial infection between placental surfaces and fetal vasculature through a microporous membrane. 67
Summary
Due to the complexity of tissue interfaces, mimicking the in vivo-like microenvironment is critical to ensuring that tissue barrier models function properly. The key factors for developing physiological barrier models include (1) ECM support with proper pore size to allow molecule diffusion and communication; for example, pore size of 40 nm creates physiologically relevant permeability; 54 (2) mechanical flexibility of ECM, as removal of mechanical distortions could cause outgrowth of bacteria; 44 (3) proper sheer forces applied to MPS;57,63,66 and (4) in vivo like structures. 46 As they feature these in vivo-like properties, MPS can be used to create defined conditions of specific barriers, such as an intestinal model with microvillus structures, a BBB model with specific tight junction and efflux pump formation, and a placental barrier model with the unique SYNT layer. Ultimately, mimicking these in vivo-like microenvironments may provide a system could potentially be useful in evaluating the safety and immunotoxicity of drugs and biologics at these tissue interfaces. Furthermore, they may serve as a platform for testing the efficacy of biologics and drugs for treating inflammatory disorders at these interfaces. While MPS for tissue interfaces bridges the gap between animal and clinical studies, however, these systems have several limitations. Tissue interface models normally contain more than one kind of cell. This brings challenges to optimize the culture medium for different cell growth in the same environment. Moreover, MPS is small scale. When scaling up the system, it might cause a different response from the microsystem. Although the tissue interface MPS includes more complexity than a 2D culture system, it still lacks dynamic, real-time regulation by signals and hormones from other parts of the body. In the tissue interface MPS field, there is ample room for work to conquer these limitations to establish more accurate systems.
MPSs for modeling inflammatory disorders and diseases
AD
AD or eczema is an inflammatory skin disease that usually occurs in infancy or early childhood. 69 AD causes the skin to become itchy, dry, red, and inflamed; patients with AD may develop subsequent conditions associated with other atopic disorders, such as allergic rhinitis and asthma.69,70 The treatment of AD can help control symptoms, and the condition improves with age. 71 However, in certain cases, AD can persist throughout adulthood, and it can even manifest in adults. 71 Pathogenesis of AD is strongly influenced by genetic, environmental, and immunological factors. 71 Skin barrier dysfunction is a major pathogenic factor of AD, and it has been shown that AD patients with loss-of-function mutations in the skin barrier protein filaggrin, encoded by the FLG gene, suffer severe forms of AD. 71 FLG is a key structural protein involved in the cornification of the outermost layer of the epidermis and epidermal differentiation, 70 and understanding the functions of FLG will help to explain the complex role of FLG in skin barrier disease. Furthermore, immunological pathologies of AD involve both resident and infiltrated immune cells, such as DCs, macrophages, neutrophils, and various subsets of T-cells. 71 These immune cells trigger immunological responses upon injury by increasing overproduction of Th2 molecular cues (i.e. thymic stromal lymphopoietin [TSLP] and cytokines [e.g. TNF-α, IFN-γ, IL-4, IL-13]) that further enhance Th2-type skin inflammation. 71 In addition, interactions among pathogens, allergens, toxins, irritants, and pollutants in the environment can trigger the production of antigen-specific IgE, hence contributing to exacerbations of AD. 72
MPS for modeling AD
Biofabricated 3D techniques can be used to generate skin tissues with various physiological complexities. These tissues have the potential to be used for drug screening, disease modeling, and personalized medicine. In a multiwell plate format, 3D-bioprinting techniques were employed to construct skin equivalent tissues including human epidermis, non-vascularized full-thickness (FTS) and vascularized full-thickness skin (VFTS), and disease models of AD induced by IL-4. 73 These 3D-fabricated models, which have different cell types in the dermal layer, a vascular feature, and pharmacological validity for drug screening, offer a more clinically relevant model to study human disease. 73 Furthermore, the high-throughput screening (HTS) multiwell format is compatible with other HTS devices, such as liquid handler and plate reader, allowing screening, quantitative measurement, biological testing, and evaluation. 3D immunocompetent skin models were also developed to investigate the interactions between the keratinocytes and T-cells to understand the pathophysiology of inflammatory skin diseases.74,75 The incorporation of T-cells, including Th1 and Th17 CD4+ T-cells, showed high expression of pro-inflammatory cytokines and chemokines, undifferentiated epidermal phenotypes, and T-cell migration that recapitulated inflammatory skin conditions.74,75 The immunocompetent models were validated with treatment of anti-inflammatory drugs and neutralizing antibodies that helped to reduce pro-inflammatory cytokine profiles.74,75 In addition, a 3D skin equivalent with a fibroblast-derived matrix allowed diseased phenotypes of inflammatory skin disorder such as AD and psoriasis to be developed. 76 The study described a fibroblast-derived matrix with an epidermis that was generated from primary keratinocytes with recombinant cytokine stimulation to induce pathological T-cell conditions. 76 Similarly, MPSs of skin models have been developed to test drugs and to study skin inflammation and edema (Figure 3(A) to (B)). 77 The microfluidic device of human skin-on-chip was created by co-culturing each cell type in a different layer with a porous membrane in between, to allow interlayer communication and mimic skin biology. Pro-inflammatory cytokines (i.e. IL-8, IL-6, and IL-1β) were found to be expressed in these inflammation and edema models. 77 Furthermore, a microphysiological full-thickness skin model demonstrated a stable structure and functionality of the skin, as well as an improvement in epidermal morphogenesis, differentiation, synthesis of basement membrane proteins, and skin barrier function in a controlled microenvironment. 78 In another microphysiological endothelial model that combined human dermal ECs, fibroblasts, and adipocytes, a model of microvasculature was used to study differentiation of protein behaviors, quantification of protein transport, and vascular absorption. 79
Figure 3.

Examples of microphysiological systems to model pathological inflammation. (A). A skin-on-a-chip microfluidic device presented a three-dimensional (3D) fluorescence image of the cross-section (A-A’). The 3D image showed four layers of three cell types that were uniformly stacked on two porous membranes. HaCaT cells, HS27 fibroblasts (Fbs), and HUVECs were stained in green, blue, and red, respectively. Scale bars: 300 μm. (B) Schematic of the skin edema model in a microfluidic device showed a control treatment, an inflammatory condition with TNF-α exposure, and a therapeutic treatment with TNF-α exposure after a dexamethasone (Dex) pretreatment on the top panel. On the bottom panel, immunofluorescence microscopy showed DAPI (blue) staining of HUVEC nuclei, and zonula occluden (ZO-1) (green) staining of HUVEC tight junctions. Gap junction was disrupted in the TNF-α-treat chip, and Dex prevented this TNF-α-induced disruption of the tight junctions. Scale bars: 100 μm. (Source: Reprinted from Figure 3(F) for Figure 3(A), Figure 7(B) for Figure 3(B), top panel, and Figure 6(G) to (I) for Figure 3(B), bottom panel from Wufuer et al. 77 Distributed under a Creative Commons Attribution 4.0 International License (CC BY 4.0) http://creativecommons.org/licenses/by/4.0/.) (C) A human joint-on-a-chip model displayed a co-culture system comprising chondral and fibroblasts-like synovial compartments for reciprocal inflammatory cross talk studies in arthritis research. 82 (Source: Reprinted from Figure 1(B) of Rothbauer et al. 82 with permission from the Royal Society of Chemistry. Distributed under a Creative Commons Attribution 3.0 Unported License (CC BY 3.0) https://creativecommons.org/licenses/by/3.0/.)
Rheumatoid arthritis
Rheumatoid arthritis (RA) is a chronic autoimmune disease that mostly affects the joints but can also affect the lung and vascular system. 80 The pathophysiology of RA is characterized by immune cell infiltration into the synovial membrane and joint cavity, as well as signaling network malfunction that impairs the tissue repair process, leading to organ damage. 80 Major histocompatibility complex (MHC) proteins have been identified as the strongest genetic risk factor for RA.80,81 The disease process of RA begins when a person is exposed to genetic and/or environmental factors that alter the homeostasis of the immune system. The onset of the autoimmune process causes production of autoantibodies, autoreactive T-cells and B-cells, and DNA instability. These conditions lead to T-cell maldifferentiation, the tissue’s inability to maintain tolerance, synoviocyte autoaggression, and the loss of tissue-protective macrophage populations which result in joint damage. 80 To further understand the disease process of RA, multiple 3D in vitro models were developed to mimic the disease process and gain insight into the therapeutic development.
MPS for modeling RA
Chondral organoids produced a high degree of cartilage physiology and architecture in an organoid-based joint-on-a-chip co-culture system comprised of chondral and synovial compartments, enabling reciprocal inflammatory crosstalk (Figure 3(C)). When compared to monocultures, these organoids showed higher level of cytokines such as IL-6, IL-8, and matrix metalloproteinase-13 (MMP-13). The study demonstrated a high level of microenvironmental and cellular control over chondrosynovial crosstalk on tissue-level behavior. 82
In addition, a microscale system was developed to mimic osteoarthritis using mechanical stimulation at a physiological and hyperphysiological compression (HPC) in conjunction with cytokine treatment. 83 Compression of HPC induced MMP-13 production and inflammatory gene expression (i.e. IL-8, IL-6), ECM deposition of collagen and aggrecan, resorption imbalance, and induction of osteoarthritic gene expression levels. 83 Furthermore, the model was used for screening and predicting anti-inflammatory drugs responses, which were comparable with previous preclinical and clinical reports. 83 In addition, a 3D model of synovial angiogenesis, in which RA fibroblast-like synoviocytes (RAFLSs) and ECs were combined into spheroids embedded in matrix, allowed the study of signaling pathways in distinct cell types and the testing of inhibitors. 84 To mimic RA disease progression, the spheroids were stimulated with RA synovial fluid (RASF) and growth factors to promote angiogenesis. The effect was blocked by an inhibitor that targeted the non-canonical nuclear factor-kB (NF-kB) pathway via NF-kB-inducing kinase (NIK). 84 Furthermore, a 3D in vitro model of cartilage destruction was developed using chondrocytes, synovial fibroblasts, and macrophages in a collagen and proteoglycan-enriched tri-culture system. 85 To obtain the pro-inflammatory phenotype in the disease model, macrophages were activated with LPS. The disease model showed increased apoptosis, overproduction of matrix degrading enzyme, upregulation of pro-inflammatory cytokines, and reduction of matrix components such as collagen and aggrecan. Furthermore, using celecoxib at an appropriate dose reduced cartilage injury and proteoglycan breakdown, as well as provided chondroprotective effects. 85 Another human cartilage-bone-synovium (CBS) model was developed in a co-culture system using cartilage (C), bone (B), and synovium (S) to study mechanical injury (INJ), and early disease progression in an inflammatory environment. 86 Both CBS co-cultures and CBS+ INJ models showed increased of chondrocyte death and induction of inflammatory cytokines (IL-1, IL-6, IL-8, and TNF-α) compared to the control, uninjured CB. 86 The study highlighted the role of inflammation and injury to the contribution of early-stage disease progression.
CF
CF is a genetic condition that is caused by mutations in the CF transmembrane conductance regulator (CFTR) gene. 87 The severity of CF is determined by which organs are affected, and lung CF is the leading cause of morbidity and mortality in patients due to progressive loss of lung function and persistent inflammation. 88 A defect in CFTR function aggravates lung infection and stimulates the innate immune system, causing an imbalance in the release of anti-/pro-inflammatory cytokines and the infiltration of neutrophils, macrophages, and epithelial cells. 89
In addition, the immune system in influenced by hyperinflammatory responses resulting from dysregulation of TLR and suppression of T-cell-mediated host response caused by myeloid-derived suppressor cells. 89 Therefore, a better understanding of the innate immune system, pathways, mechanisms involved, and appropriate study models will help in the evaluation and identification of potential treatments for CF.
MPS for modeling CF
Advances in microphysiological lung-on-a-chip devices have provided more physiologically relevant platforms to test the efficacy of these cells in the lung tissues. These systems offer a new opportunity for disease modeling and drug testing. Human airway-on-a-chip models of asthma and lung inflammation, utilizing isolated epithelial cells from healthy individuals and patients with chronic obstructive pulmonary disease (COPD), demonstrated hallmarks of overproduction of secreted cytokines, increased neutrophil migration, and greater susceptibility to viral and bacterial infection. 90 Furthermore, IL-13 treatment of small airways resulted in an increase in goblet cells, overproduction of inflammatory cytokines such as granulocyte colony-stimulating factor (G-CSF) and granulocyte macrophage-colony-stimulating factor (GM-CSF) in the vascular channel, and a decrease in cilia.
These disease phenotypes were rescued by tofacitinib, a potent inhibitor of the Janus kinases/signal transducer and activator of transcription protein (JAK/STAT) pathway for IL-13 signaling. 90 Similarly, another human lung airway model offered a better understanding of lung obstructive disease by conducting a mechanistic study of epithelial–airway smooth muscle (ASM) interactions in a controlled environment through biochemical and mechanical transduction, allowing for the study of airway contraction, positive–negative feedback of epithelial-ASM, and intercellular signaling interactions. 91 Furthermore, a lung MPS in a high-throughput multiwell format was established to study fibrotic disease and was further expanded to study cancer and CF by incorporating of microvascular into airway. 92 The vascular co-culture system allowed the study of epithelial–fibroblast interaction and epithelium resembling CF on neutrophil migration in the 3D structure. 92
Neutrophils were incorporated into the system to evaluate the role of neutrophilic inflammation in the lung, how epithelial cells affected the neutrophil migration in the diseased lung compared to the control model, and how the innate immune system was regulated in the healthy and disease lung epithelium. 92 In a study that combined three complementary MPS, the systems were used to address the intricate mechanisms of lung disease and the effects of cigarette smoke. 93 The three MPS included (1) transwell system for co-culture of immune and epithelial cells, (2) precision-cut lung slides, and (3) lung-on-a-chip model. 93 These complimentary MPS allowed to model the lung airways and study crosstalk between different lung cell types and disease progression. 93 Together, these lung MPS provide a platform for studying lung disease, monitoring disease progression, and evaluating therapeutic responses.
Summary
Overall, MPSs provide promising platforms to study and to further our understanding of the pathogenesis of inflammatory diseases, including AD, CF, and RA, as they are physiologically relevant and reproducible. With the complex 3D microenvironment, these MPS models offer the opportunity to investigate targets of various therapeutics such as integrity of barrier function, crosstalks of different cell types in the tissues, incorporation of immune cells, integration of mechanical/biological injury, and the possibility to mimic the vascular and immunological/molecular environment. The physiological complexity of MPS helps to overcome the weakness of 2D models, as they provide an alternative platform for a deeper understanding of the signaling processes in these inflammatory diseases and in vitro tools for disease modeling, drug discovery, and therapy targeting the maintenance and repair of the barrier function, as well as modeling inflammatory disorder. However, with the advantages of MPS, there are several limitations to note. For example, due to physiological complexity, it might not be possible to increase large-scale production and maintenance of these MPS. In addition, the complexity of these MPS involves multiple cell sources that might be hard to get access and requires optimization including media composition and duration of the culture. Although these MPS help to bridge the gap between in vitro and in vivo models, they need to be validated to confirm their efficiency and reproducibility, thus requiring further studies to be able to utilize for therapeutic approaches.
MPSs for evaluating cancer immunotherapies
Immune cells in the TME
The TME is the cellular and extracellular environment within which cancer exists. The TME is often complex and comprises cytokines, hormones, growth factors, ECM, vasculature (blood and lymphatic), and various cell types that include tumor, stromal, immune, endothelial, and fibroblasts (Figure 4(A)). 94 Given its complex composition, the TME represents a unique environment that develops during tumor progression, where TME interactions with the surrounding host environment are orchestrated by the tumor. 95 Within the TME, these various components interact with each other to initiate, protect, and promote the tumor via various mechanisms of immune evasion. 94 Mechanisms of tumor escape from immune intervention have been previously reviewed. 96
Figure 4.

How microphysical tumor models are being used to understand the tumor microenvironment and possible cancer therapeutics. (A) Illustration of the complexity of the tumor microenvironment and some of its main components and how drug transport interacts with this environment 97 . (Source: Reproduced with permission, Copyright 2021 WILEY – VCH VERLAG GMBH & CO. KGAA). (B) The top image illustrates HUVEC-derived vasculature created in a microfluidic device used to assess permeability and shows how a tumor spheroid influences vascular network density within the in vitro model. The bottom three images show different cancer types modeled and vascularized within a microfluidic device. The image on the bottom left illustrates HUVEC-derived vasculature within ovarian carcinoma, where the white arrow indicating a ring of fibroblast taken at day 7. The middle and right most images show lung adenocarcinoma fixed at day 7. (Scale bar 200 microns). 97 (Source: Reproduced with permission, Copyright 2021 WILEY – VCH VERLAG GMBH & CO. KGAA.) (C) Illustrates the process by which T-cell therapeutics are developed and used in a microfluidics to assess their anticancer ability. 108 (Source: Reproduced with permission, Copyright 2021 American Society for Clinical Investigation.) (D) Illustrates the immunotherapeutic high-throughput observation chamber (iHOC) system which uses tumor spheroids as a microphysical model for the exploration of PDL-1 in T-cell infiltration of the tumor environment. Also included is an illustration showing PD-1’s role in T-cell immunosuppression and how this effects tumor infiltration within the tumor microenvironment. 113 (Source: Edited and reproduced with permission, Copyright 2020 WILEY VCH.)
The TME includes both adaptive and innate immune cells including T-cells, and natural killer (NK) cells and macrophages, respectively. T-cells directly interact with tumor cells, as well as contribute to the chemokine and cytokine signaling within and around the TME. There are many different types of T-cells which are critical to the immune response through actions such as killing infected cells, recruiting other immune cells through cytokine signaling, and regulating the immune response (to prevent chronic inflammation). 94 T-cells within the TME are usually either Th1, Th2, or Treg T-cells, which produce pro-inflammatory or anti-inflammatory cytokines. 94 The proportion and characteristics of T-cells within the microenvironment can influence tumor progression. NK cells are circulatory lymphoid cells which have cytotoxic effector functions; their main goal is to kill circulating tumor cells. 95 However, tumor cells can inhibit NK cells and/or avoid detection. 95 Macrophage functions include wound healing, tissue repair, pathogen phagocytosis, antigen-presentation to T-cells, and release of cytokines to induce an immune response. In the TME, there are also tumor-associated macrophages (TAMs) that are known to be immunosuppressive through reprogramming by factors in the microenvironment. These TAMs help in the promotion of the TME by releasing growth factors that promote growth, angiogenesis, movement, and invasion of cancer. 94 All three of these immune cell types can be targeted therapeutically to reduce cancer progression. However, the TME is complex and difficult to replicate in a lab setting. MPSs are promising research tools as they can be used to model various aspects of the complex TME, leading to physiologically relevant conditions for testing the effectiveness of and troubleshooting therapeutic strategies.
MPS for modeling the TME
MPS platforms may be leveraged to recapitulate major constituents of the TME for targeted evaluation of tumor progression toward improving mechanistic understanding and identifying therapeutic approaches. Indeed, targeting the TME may provide a viable strategy for inhibiting cancer progression and potentially overcoming antitumor drug resistance. 3D vascularized tumor models built using MPS tools offer physiologically relevant microenvironments that may have more predictive value in preclinical drug–response studies over simple 2D cultures. One major constituent that is often modeled using MPS involves the tumor vascular niche and how tumor-associated ECs influence response to anti-tumor agents. Recent work evaluated the influence of two different tumor types, an ovarian cancer model (Skov3) and lung adenocarcinoma model (A549), on vascular network formation within a custom MPS and found that tumor cells affected localized microvascular density differently depending on the cancer type (Figure 4(B)). 97 Such differences in localized tumor-induced vascular dysregulation were associated with reduced chemotherapeutic delivery to tumor spheroids within this MPS and may model a potential mechanism of tumor-acquired drug resistance. 97 While some research approaches utilize bespoke MPS with customized designs for in-depth mechanistic studies of tumor vascular niches, others have leveraged enhanced-throughput platforms to engineer tumor-induced vascular formations at higher scales for drug-screening applications. One example involved the fabrication of an injection-molded platform that is amenable to the evaluation of nine different tumor cell lines on vasculature formation in a standardizable and user-friendly fashion. 98 Within the higher-throughput injection-molded platform, it was observed that the morphology of microvascular network formations for each tumor co-culture condition was characteristic of the tumor-type evaluated. 98 The ability to model tumor-induced vasculatures with higher-throughput using MPS enables the preclinical screening of many different anti-tumor drugs that target the cancer-associated vasculature. While vascular elements are commonly integrated into TME models to investigate metastasis and evaluate vasculature-targeting therapies, several studies using non-vascular TME models have also been reported, as discussed later. In recognition of the utility of standardizable MPS platforms as preclinical tools for studying anticancer drugs, significant effort is being invested into evaluating the reproducibility of MPS models between laboratories and demonstrating the enhanced predictive value of MPS models over traditional 2D cell cultures. 99
Another constituent of the TME that is often recapitulated using MPS are non-malignant cancer–associated fibroblasts (CAFs) and their crosstalk with cancer cells. MPS feature mechanisms for spatiotemporal control over co-culture configuration of multiple different cell types which can be manipulated to study cell–cell communication. As an example, one study utilized a multichannel setup to separately co-culture pancreatic stellate cells (PSCs), which include pancreatic ductal adenocarcinoma stroma-derived cancer-associated fibroblasts, with human pancreatic cancer cells without initial cell–cell contact. 100 It was found that the PSCs exhibited morphological changes and activation, which induced epithelial–mesenchymal transition–related changes in co-cultured pancreatic cancer cells that imparted drug resistance to chemotherapeutic drugs. Modeling non-malignant elements of the TME using MPS could offer a valuable method for studying mechanisms of drug resistance and therefore lead to the identification of novel targets for anticancer treatment.
In addition, MPSs have also been used to model immune elements of the TME. Given that macrophage polarization within the TME is thought to play a significant role in influencing tumor progression, several 3D organotypic models have been leveraged for the study of TAMs. In one model based on a transwell-plate format, it was shown that human squamous cell carcinoma cells co-cultured with TAMs induced M2 macrophage polarization over prolonged periods without the need for exogenous IL-4, which led to increased tumor cell invasion in the model. 101 To better study the role of TAMs in tumor progression, recent efforts have leveraged the advantages of microfluidic platforms to incorporate TAMs into engineered TMEs involving human lung adenocarcinoma (A549) cell aggregates. 102 It was found that M1 and M2b subtypes of TAMs induced the greatest degree of tumor cell dispersion within the studied co-culture model. Another study worked on recapitulating the TME using cancer spheroids suspended in alginate gels. They found that by encapsulating single cells of CAFs ThP-1 or PBM, they were able to create tumor spheroids positive for common markers, such as N-cadherin and vimentin in addition to the appropriate architecture and accumulation of ECM components such as collagen type I, collagen type IV, and fibronectin. 103 They were able to use their model to activate monocytes into a TAM-associated phenotype without the addition of external factors – providing greater immunological relevance. These spheroids were then used to assess the effects of chemo- and immunotherapies such as Cisplatin and Paclitaxel, where they observed the repolarization of M2-like macrophages into M1-phenotypes. 103 As an example of a non-vascular TME model for studying the cytotoxic effects of NK cells, one group reported a proof-of-concept immunocompetent MPS that comprised of the co-culture of HCT116 colorectal tumor microtissueis, cardiac microtissues, and human-derived NK cells within a customized commercial platform. 104 To compensate for the lack of vasculature within the immunocompetent MPS, gravity-driven perfusion was employed by cyclically tilting the culture device over a tiling angle of 5° each way. Utilizing this non-vascular TME model to study various cell–cell interactions, it was reported that the NK cells exhibited specific cytotoxic activity against the tumor spheroids while sparing non-tumor cardiac tissues to an extent. 104 Other non-vascular immunocompetent MPS models of the TME have alternatively evaluated the role of lymphatic structures in cancer metastasis. For example, recent endeavors have explored the development of an in vitro head and neck cancer (HNC) TME by incorporating HNC tumor spheroids and lymphatic vessels within a bespoke MPS platform. 105 The authors reported the novel secretion of pro-inflammatory macrophage migration inhibitory factor by lymphatic ECs that have been exposed to HNC cells within the MPS, suggesting that lymphatic structures may be an important element to consider when building immunocompetent TME models. Understanding the behavior of these various TME constituents may therefore prove to be important for the development of new cancer immunotherapies.
CAR T-cells
CAR T-cell therapy is a new and promising cancer therapy that has gained traction in recent years. CAR T is one of the three approaches for adoptive T-cell transfer which include tumor-infiltrating lymphocytes, T-cell receptor (TCR) T-cells, and CAR T-cells. 106 CARs are the synthetic receptors that consist of the following main components: an extracellular target antigen-binding domain, a hinge region, a transmembrane domain, and one or more intracellular signaling domains. 106 The transmembrane domain regulates functions such as expression level, stability, signaling and/or synapse formation, and dimerization of endogenous signaling molecules. 106 CAR T has been successful in binding to target antigens on cell surface that are independent from MHC receptor, which increases T-cell activation, thereby inducing an antitumor response and preventing tumor cells from evading the immune system. 107 Success of these approaches has led to the US Food and Drug Administration (FDA) approval of anti-CD19 and anti-B-cell maturation antigen (BCMA) CAR T-cell therapy against B-cell malignancies. 107 Despite great success, there are still many limitations of CAR T-cell therapy. Some limitations include antigen escape, on-target off-tumor effects, CAR T-cell trafficking and tumor infiltration, immunosuppressive microenvironment, and CAR T-cell-associated toxicities. 107 To further expand CAR T-cell therapy success, these limitations must be explored. However, many models are insufficient in replicating the in vivo microenvironment. MPSs are the closest in vitro models to replicating the physiological system of the TME. Using these systems, the limitations listed before can be explored and broken through.
MPS for evaluating CAR T-cells
Analytical platforms based on MPS offer a promising screening modality for the enhanced-throughput assessment of CAR T-cell functionality and bioactivity (i.e. potency) in physiologically relevant contexts. The development of such advanced analytical tools is also important for understanding lot-to-lot variability of CAR T-cells that inherently arise from the manufacturing and processing of complex biologics. For example, organotypic platforms that can also incorporate physical and immunological barriers of the TME have been utilized for the study of limited TCR or CAR T-cell efficacy against solid tumors, which represents a major obstacle in adoptive T-cell therapy. One study utilized a compartmentalized microfluidic device to investigate the anti-tumor effects of TCR T-cells against HepG2 human liver carcinoma cell aggregates within various TME conditions. 108 Using this model, researchers discerned that differences in cytotoxic abilities between two different TCR T-cell preparations may be due to differences in T-cell motility and dependency on IL-2. Such nuances are difficult to replicate using traditional 2D models. Another group developed a 3D microphysiological tumor model comprising non-small cell lung cancer cells or triple-negative breast cancer cells that also allowed for the introduction of therapeutic CAR T-cells via flow. 109 When CAR T-cells engineered to target receptor tyrosine kinase-like orphan receptor 1 (ROR1) were introduced into the tumor model, it was found that ROR1-CAR T-cells (derived from multiple different donors) infiltrated the tumor aggregates and exhibited significant cytolytic activity. One other example of an organotypic model for assessing CAR T-cell function involves a microdevice platform that incorporates a 3D solid tumor section cultured in an engineered hypoxic gradient microenvironment with neighboring microfluidic channels for CAR T-cell delivery (Figure 4(C)). 110 It was found that CAR T-induced cytotoxicity can be spatially modulated by hypoxia and that the inhibition of PD-L1 expression by hypoxic cancer cells did not enhance CAR T-killing effects within the model. While not performed in a self-enclosed MPS, such studies performed using an organotypic model support the value of incorporating physiologically relevant MPS platforms into analytical tools for evaluating cell therapies.
Immune checkpoint blockade
The immune checkpoint is a safety mechanism that prevents hyperactivation of immune cells and helps the immune system to function properly. The checkpoint molecules are receptors present on the surface of the immune cells, and when engaged with their ligands, initiate activating or inhibitory signaling. A major role of immune checkpoints is to identify and eliminate cancer cells to prevent further cancer growth. However, cancer cells have developed mechanisms to evade these checkpoints through the loss or mutation of immunogenic tumor antigens. 111 This prevents the activation of the immune cells (T-cells, lymphocytes) from recognizing, and thereby destroying cancer cells. For example, some cancer cells present programmed death ligand 1 (PD-L1) which then targets the programmed death 1 (PD-1) (receptor) molecule that is present on activated T-cells. When PD-L1 and PD-1 interact, it prevents the T-cell from attacking, and therefore, the cancer cell survives. 112 To surmount cancer mutations that confer immune evasion, researchers are developing checkpoint blockade immunotherapy which is intended to overcome the PD-1 activation mechanism. For example, scientists have developed drugs that prevent the interaction between PD-1 and PD-L1. This prevents the inactivation of T-cells by cancer cells and leads to destruction of the cancer cell.
MPS for evaluating checkpoint blockade therapies
MPS technologies may be used to model and monitor checkpoints in cancer–immune interactions within the TME in response to various immunotherapies. To provide a system for evaluating cancer immunotherapies based on immune checkpoint blockade, one approach involved a high-throughput MPS platform that modeled interactions between MDA-MB-231 breast cancer cell aggregates and Jurkat T-cells when immune checkpoint inhibitors are introduced (Figure 4(D)). 113 Investigators were able to observe how the PD-1 checkpoint could be modulated to adjust T-cell proliferation, cytokine secretion, and antitumor activity. Indeed, MPS methodologies enable the ex vivo interrogation of various animal-derived and patient-derived tumors to immune checkpoint blockade. In one study, various murine- and patient-derived organotypic tumor spheroids containing tumor-infiltrating lymphoid and myeloid subpopulations were cultured within collagen hydrogels in a microfluidic culture system and treated with various combination therapies targeting PD-1 blockade. 114 The researchers were able to recapitulate in vivo-like responses of the various tumors to PD-1 blockade via cytokine profiling toward the identification of methods to combat drug resistance. Another case study investigated the effectiveness of PD-1 checkpoint immunotherapy in a glioblastoma MPS model. 115 Various patient-specific glioblastoma subtypes were co-cultured in a hyaluronan-rich Matrigel hydrogel with human brain microvascular ECs, TAMs, and CD8+ T-cells within a compartmentalized MPS to model the brain TME. It was found that T-cell and TAM phenotype were dependent on glioblastoma subtype, where the effectiveness of PD-1 blockade was enhanced when combined with the colony-stimulating factor-1 receptor (CSF-1R) inhibition of immunosuppressive M2 TAMs in the model. Together, these examples illustrate the utility of MPS as platforms for broadly interrogating various tumor responses to immune checkpoint blockade, as well as for personalized screening of immunotherapies under patient-specific contexts.
Summary
MPS complexity provides physiological relevance, allowing for easier translation into human trials of many different cancer immunotherapies. Modeling the TME using MPS has greatly advanced research by recapitulating the 3D microenvironment of in vivo conditions, creating a model that more accurately illustrates the effects of many immunotherapies. This provides great insight into the mechanisms of action within the TME that are unable to be explored in 2D conditions. Many different aspects of immunotherapy ranging from immune cell interactions to immunotherapy drugs can be assessed with these types of models for different types of cancer in addition to different specificities of these therapies. However, these MPS systems are still evolving, and there are many improvements that have yet to be accomplished. As of now, these MPS systems do recapitulate complexity of the TME systems better than their 2D counterparts; however, they are unable to include every aspect of the in vivo environment. In addition, many of these MPS models require extensive time, money, and skill to be performed making them inaccessible to many. Nevertheless, MPS systems are promising new alternatives to in vivo models that provide comprehensive assessments of various cancer immunotherapies.
Future directions and considerations
A critical consideration in further developing MPSs for modeling immunity is the type and source of immune cells incorporated into the devices. Broadly, the immune cells incorporated into these devices may consist of cells from the innate immune system, which include monocytes, neutrophils, macrophages, DCs, and NK cells that provide rapid, non-specific immunity, as well as cells from the adaptive immune system, which include T- and B-cells that provide antigen-specific immune responses and immunological memory. In MPSs for modeling immunity/inflammation at tissue interfaces and inflammatory disorders/disease, the models discussed in this review mostly focus on incorporating cells from the innate immune system (Table 1), potentially due to the decreased complexity of modeling these cellular responses. MPSs modeling lymphatic tissues and cancer immunotherapies have for the most part incorporated both innate and adaptive immune systems into their models (Table 1), due to the critical importance of the adaptive immune system in these tissues and conditions. However, many these MPS fail to properly model antigen-specific T- and B-cells responses, often studying polyclonal B- and T-cells responses in these systems.
Table 1.
Microphysiological systems utilizing innate and/or adaptive immune cells.
| MPS category | Innate immune cells | Adaptive immune cells | Innate and adaptive immune cells |
|---|---|---|---|
| Modeling lymphatic tissues | Dendritic cells14–17
Monocytes 20 |
B-cells28,29
T-cells and B-cells 30 |
Dendritic cells and T-cells18,22,24–26
Dendritic cells, T-cells, and B-cells27,32 PMBCs19,31 |
| Modeling immunity and inflammation at tissue interfaces | Neutrophils
46
Natural kill cells 46 Macrophages 65 |
T-cells 54 | PBMCs44–47 |
| Modeling inflammatory disorders and diseases | Macrophages
85
Neutrophils90,92 |
T-cells74,75 | |
| Evaluating cancer immunotherapies | Nonocytes, macrophages101–103
Natural killer cells 104 |
T-cells108–110,113 | Tumor-infiltrating myeloid and lymphoid cells114,115 |
MPS: microphysiological system.
Modeling the adaptive immune system remains a critical challenge in modeling the breadth of antigen-specific responses, as well as recapitulating self versus non-self immunity. The theoretical diversity of TCRs and BCRs in the human body is believed to be in the range of 1012–1018 different TCRs/BCRs;116,117 both capturing this diversity and eliciting antigen-specific responses in lymphatic organ MPS systems to all potential antigens remains a challenge. Assuming this diversity can be recapitulated, there is also the challenge in modeling tolerance (i.e. deletion of self-reactive T-cells), as well as self versus non-self immunity that arises from differences in human leukocyte antigen (HLA) antigens on cells from different donors. Taking this diversity into account requires either HLA matching different human cell types that are incorporated into a single MPS or deriving all cells in an MPS from a single donor. The later approach would benefit from methods of generating cells from induced pluripotent stem cells (iPSCs), in order to capture patient-to-patient variability. In terms of generating iPSC-derived immune cells, there has been success generating iPSC-derived macrophages, NK cells, DCs, and T-cells for immunotherapies, 118 as well as eosinophils, mast cells, neutrophils, and B-cells.119–122 However, it remains to be seen whether these iPSC-derived immune cells can capture the diversity of function and phenotype of the innate immune system, as well as antigen-diversity of T- and B-cells.
In addition to challenges in modeling the cellular immune components in MPS, emerging advancements in combining organoids and organ-on-a-chip, which have traditionally been recognized as two distinct model systems, have yielded better approaches to model the structural components of tissues. Organoids, 3D multicellular tissues created by self-organizing stem and progenitor cells have been shown to recapitulate the development process and early structural features of a variety organs, including kidneys, 123 intestines, 124 and tonsil tissues, 31 These organoids typically capture critical cell–cell interactions between different cell types, as well as cellular heterogeneities exhibited in these developing progenitor and stem cells. However, these approaches often fail to recapitulate other key microenvironmental cues. On the contrary, organ-on-a-chip systems, which commonly utilize microfluidic devices or microfabrication technologies, focus on incorporating biochemical and mechanical cues to cells in devices, often in the form of fluid flow, ECM hydrogels, and/or mechanical actuation. However, these technologies often fail to accurately model critical cell–cell contacts and cell heterogeneity found in these organs. Recently, the line between these two distinct technologies has blurred, with efforts to combine these two distinct technologies for engineering intestines 125 and vascularized kidneys; 126 these technologies have also been leveraged for evaluating immunological phenomena, in the context of macrophage-intestine crosstalk and T-cell specific antibodies in kidneys, respectively.125,126 The integration of these approaches will yield more physiologically relevant approaches for modeling immunity in vitro.
The utility of MPSs for evaluating immunity depends on the analytical chemistries and bioassays that are utilized to probe cellular behavior on these systems. Depending on the biomaterial utilized in the MPS, extraction of cells from the device may be technically challenging and may impair cell viability upon extraction. Assays including quantitative polymerase chain reaction (PCR), western blot, enzyme-linked immunosorbent assays (ELISAs), mass spectrometry, flow/mass spectrometry, and RNA-seq may have limited utility in analyzing these systems, due to the intrinsic small volumes and cell numbers incorporated into these systems. Single-cell RNA-seq, in particular, will have tremendous utility in analyzing these models, due to both the low required cell numbers for the analysis and insights into cellular heterogeneity in these systems. 127 The further development of multiplex assays that can analyze multiple analytes, proteins, and/or genes in small volumes of samples will improve our ability to analyze these systems and evaluate their ability to model immunity in vitro.128,129
A long-term goal for modeling immunity in vitro is to engineer MPSs representing different organs and tissues that are linked fluidically by channels or tubing that models blood vessel and lymphatic vessel flow between organs. These fluidically linked MPS systems would be able to essentially model a “body-on-a-chip” and systemic circulation, as well as potentially recapitulate the pharmacokinetics of various drugs and biologics. Notable works have linked intestine, liver, kidney, bone marrow heart, lung, skin, BBB, and/or brain chips via vascular channels to model the pharmacokinetics of drugs.128,129 Similar work linking heart, liver, bone, and skin chips via vascular channels also modeled the pharmacokinetics of doxorubicin and modeled clinically observed miRNA responses. 130 Relevant to immunity, fluidically linking cultured lymph node slices with tumor slices was able to model immunosuppressive function of tumors on lymphatic organs. 131 We envision that this approach could long term be applied to fluidically linked primary and secondary lymphoid organs, to induce the differentiation, proliferation, and activation of various immune cells in our body. For example, a fluidically linked bone marrow, thymus, spleen, and lymph node chip could model the generation, differentiation, and activation of T- and B-cells.
Conclusions
MPSs have demonstrated the ability to recapitulate certain immunological functions and features of lymphatic tissues, tissue interfaces, inflammatory diseases, and TMEs. Broadly, these systems have exhibited the ability to model how certain immunotherapies function in our body, how dysfunctional immune responses can propagate disease, and how our immune system can provide immunity and protection from pathogens. However, these systems are still limited by their inability to model the complex cellular and molecular interactions of the in vivo immune system. Despite these limitations, MPSs for modeling immunity may have long-term utility in predicting specific effects (but not complete responses) that drugs and biologics have on the human immune system and in certain immune-related diseases. However, these systems will need to be qualified and validated in order for regulatory bodies to accept them as predictive models. While MPSs may not currently replace the usage of in vivo models for modeling the complexity of the immune system, they may significantly reduce time and resources spent while increasing the efficiency and speed of product development in preclinical stage by potentially reducing the need for animal testing.
Acknowledgments
The authors acknowledge Nirjal Bhattarai and Ronit Mazor for their review for this manuscript.
Footnotes
Authors’ Contributions: All authors participated in the design, interpretation of the studies, writing, and review of the manuscript.
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by BJK’s appointment to the Research Participation Program at CBER administered by the Oak Ridge Institute for Science and Education through the US Department of Energy and the US Food and Drug Administration. This work was also supported by BJK’s appointment to the Interagency Oncology Task Force Fellowship through the National Cancer Institute and the US Food and Drug Administration. This work was partially supported by the Perinatal Health Center of Excellence, and research funds from the Division of Cellular and Gene Therapies in CBER/FDA. This work also was supported by X-XN’s appointment to the Translational Science Interagency Fellowship through the National Center for Advancing Translational Sciences (NCATS) and the US Food and Drug Administration.
ORCID iD: Brian J Kwee  https://orcid.org/0000-0002-4426-1363
https://orcid.org/0000-0002-4426-1363
References
- 1. Descotes JJ. Methods of evaluating immunotoxicity. Expert Opin Drug Metab Toxicol 2006;2:249–59 [DOI] [PubMed] [Google Scholar]
- 2. Pineda CC, Castañeda Hernández G, Jacobs IA, Alvarez DF, Carini C. Assessing the immunogenicity of biopharmaceuticals. Biodrugs 2016;30:195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cerf-Bensussan N, Gaboriau-Routhiau V. The immune system and the gut microbiota: friends or foes? Nat Rev Immunol 2010;10:735–44 [DOI] [PubMed] [Google Scholar]
- 4. Park JE, Barbul A. Understanding the role of immune regulation in wound healing. Am J Surg 2004;187:11S–6 [DOI] [PubMed] [Google Scholar]
- 5. Zschaler JJ, Schlorke D, Arnhold J. Differences in innate immune response between man and mouse. Crit Rev Immunol 2014;34:433–54 [PubMed] [Google Scholar]
- 6. Brehm MA, Shultz LD, Luban J, Greiner DL. Overcoming current limitations in humanized mouse research. J Infect Dis 2013;208:S125–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Advancing alternative methods at FDA, https://www.fda.gov/science-research/about-science-research-fda/advancing-alternative-methods-fda (accessed 1 December 2022).
- 8. Flores LE, Frontera WR, Andrasik MP, Del Rio C, Mondríguez-González A, Price SA, Krantz EM, Pergam SA, Silver JK. Assessment of the inclusion of racial/ethnic minority, female, and older individuals in vaccine clinical trials. JAMA Netw Open 2021;4:e2037640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Korang SK, von Rohden E, Veroniki AA, Ong G, Ngalamika O, Siddiqui F, Juul S, Nielsen EE, Feinberg JB, Petersen JJ, Legart C, Kokogho A, Maagaard M, Klingenberg S, Thabane L, Bardach A, Ciapponi A, Thomsen AR, Jakobsen JC, Gluud C. Vaccines to prevent COVID-19: a living systematic review with trial sequential analysis and network meta-analysis of randomized clinical trials. PLoS ONE 2022;17:e0260733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brisse MM, Vrba SM, Kirk N, Liang Y, Ly H. Emerging concepts and technologies in vaccine development. Front Immunol 2020;11:583077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wang LLW, Janes ME, Kumbhojkar N, Kapate N, Clegg JR, Prakash S, Heavey MK, Zhao Z, Anselmo AC, Mitragotri S. Cell therapies in the clinic. Bioeng Trans Med 2021;6:e10214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Irvine DJ, Aung A, Silva M. Controlling timing and location in vaccines. Adv Drug Deliv Rev 2020;158:91–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Abbas AK, Lichtman AH, Pillai S. Cellular and molecular immunology E-book. Amsterdam: Elsevier, 2014 [Google Scholar]
- 14. Cougoule C, Lastrucci C, Guiet R, Mascarau R, Meunier E, Lugo-Villarino G, Neyrolles O, Poincloux R, Maridonneau-Parini I. Podosomes, but not the maturation status, determine the protease-dependent 3D migration in human dendritic cells. Front Immunol 2018;9:846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Haessler U, Pisano M, Wu M, Swartz MA. Dendritic cell chemotaxis in 3D under defined chemokine gradients reveals differential response to ligands CCL21 and CCL19. Proc Nat Acad Sci 2011;108:5614–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Choi Y, Sunkara V, Lee Y, Cho Y-K. Exhausted mature dendritic cells exhibit a slower and less persistent random motility but retain chemotaxis against CCL19. Lab Chip 2022;22:377–86 [DOI] [PubMed] [Google Scholar]
- 17. Schwarz J, Bierbaum V, Merrin J, Frank T, Hauschild R, Bollenbach T, Tay S, Sixt M, Mehling M. A microfluidic device for measuring cell migration towards substrate-bound and soluble chemokine gradients. Sci Rep 2016;6:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mitra B, Jindal R, Lee S, Dong DX, Li L, Sharma N, Maguire T, Schloss R, Yarmush ML. Microdevice integrating innate and adaptive immune responses associated with antigen presentation by dendritic cells. RSC Advances 2013;3:16002–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Serrano JC, Gillrie MR, Li R, Ishamuddin SH, Kamm RD. On-chip engineered human lymphatic microvasculature for physio-/pathological transport phenomena studies. 2022, https://www.biorxiv.org/content/10.1101/2022.03.06.483122v1
- 20. Birmingham KG, O’Melia MJ, Bordy S, Aguilar DR, El-Reyas B, Lesinski G, Thomas SN. Lymph node subcapsular sinus microenvironment-on-a-chip modeling shear flow relevant to lymphatic metastasis and immune cell homing. Iscience 2020;23:101751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tomei AA, Siegert S, Britschgi MR, Luther SA, Swartz MA. Fluid flow regulates stromal cell organization and CCL21 expression in a tissue-engineered lymph node microenvironment. J Immunol 2009;183:4273–83 [DOI] [PubMed] [Google Scholar]
- 22. Dura B, Dougan SK, Barisa M, Hoehl MM, Lo CT, Ploegh HL, Voldman J. Profiling lymphocyte interactions at the single-cell level by microfluidic cell pairing. Nat Commun 2015;6:1–13 [DOI] [PubMed] [Google Scholar]
- 23. Faley S, Seale K, Hughey J, Schaffer DK, VanCompernolle S, McKinney B, Baudenbacher F, Unutmaz D, Wikswo JP. Microfluidic platform for real-time signaling analysis of multiple single T cells in parallel. Lab Chip 2008;8:1700–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gunzer M, Schäfer A, Borgmann S, Grabbe S, Zänker KS, Bröcker EB, Kämpgen E, Friedl P. Antigen presentation in extracellular matrix: interactions of T cells with dendritic cells are dynamic, short lived, and sequential. Immunity 2000;13:323–32 [DOI] [PubMed] [Google Scholar]
- 25. Shanti A, Samara B, Abdullah A, Hallfors N, Accoto D, Sapudom J, Alatoom A, Teo J, Danti S, Stefanini C. Multi-compartment 3D-cultured organ-on-a-chip: towards a biomimetic lymph node for drug development. Pharmaceutics 2020;12:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rosa PM, Gopalakrishnan N, Ibrahim H, Haug Halaas MØ. The intercell dynamics of T cells and dendritic cells in a lymph node-on-a-chip flow device. Lab Chip 2016;16:3728–40 [DOI] [PubMed] [Google Scholar]
- 27. Belanger MC, Ball AG, Catterton MA, Kinman AW, Anbaei P, Groff BD, Melchor SJ, Lukens JR, Ross AE, Pompano RR. Acute lymph node slices are a functional model system to study immunity ex vivo. ACS Pharmacol Trans Sci 2021;4:128–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Purwada A, Shah SB, Béguelin W, August A, Melnick AM, Singh A. Ex vivo synthetic immune tissues with T cell signals for differentiating antigen-specific, high affinity germinal center B cells. Biomaterials 2019;198:27–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moeller TD, Shah SB, Lai K, Lopez-Barbosa N, Desai P, Wang W, Zhong Z, Redmond D, Singh A, DeLisa MP. Profiling germinal center-like B cell responses to conjugate vaccines using synthetic immune organoids. ACS Cent Sci 2023;9:787–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Goyal G, Prabhala P, Mahajan G, Bausk B, Gilboa T, Xie L, Zhai Y, Lazarovits R, Mansour A, Kim MS, Patil A, Curran D, Long JM, Sharma S, Junaid A, Cohen L, Ferrante TC, Levy O, Prantil-Baun R, Walt DR, Ingber DE. Ectopic lymphoid follicle formation and human seasonal influenza vaccination responses recapitulated in an organ-on-a-chip. Adv Sci 2022;9:e2103241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wagar LE, Salahudeen A, Constantz CM, Wendel BS, Lyons MM, Mallajosyula V, Jatt LP, Adamska JZ, Blum LK, Gupta N, Jackson KJL, Yang F, Röltgen K, Roskin KM, Blaine KM, Meister KD, Ahmad IN, Cortese M, Dora EG, Tucker SN, Sperling AI, Jain A, Davies DH, Felgner PL, Hammer GB, Kim PS, Robinson WH, Boyd SD, Kuo CJ, Davis MM. Modeling human adaptive immune responses with tonsil organoids. Nat Med 2021;27:125–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Higbee RG, Byers AM, Dhir V, Drake D, Fahlenkamp HG, Gangur J, Kachurin A, Kachurina O, Leistritz D, Ma Y, Mehta R, Mishkin E, Moser J, Mosquera L, Nguyen M, Parkhill R, Pawar S, Poisson L, Sanchez-Schmitz G, Schanen B, Singh I, Song H, Tapia T, Warren W, Wittman V. An immunologic model for rapid vaccine assessment – a clinical trial in a test tube. Altern Lab Anim 2009;37:19–27 [DOI] [PubMed] [Google Scholar]
- 33. Faley SL, Copland M, Wlodkowic D, Kolch W, Seale KT, Wikswo JP, Cooper JM. Microfluidic single cell arrays to interrogate signalling dynamics of individual, patient-derived hematopoietic stem cells. Lab Chip 2009;9:2659–64 [DOI] [PubMed] [Google Scholar]
- 34. Chou DB, Frismantas V, Milton Y, David R, Pop-Damkov P, Ferguson D, MacDonald Vargel Bölükbaşı AÖ, Joyce CE, Moreira Teixeira LS, Rech A, Jiang A, Calamari E, Jalili-Firoozinezhad S, Furlong BA, O’Sullivan LR, Ng CF, Choe Y, Marquez S, Myers KC, Weinberg OK, Hasserjian RP, Novak R, Levy O, Prantil-Baun R, Novina CD, Shimamura A, Ewart L, Ingber DE. On-chip recapitulation of clinical bone marrow toxicities and patient-specific pathophysiology. Nat Biomed Eng 2020;4:394–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Voeltzel T, Fossard G, Degaud M, Geistlich K, Gadot N, Jeanpierre S, Mikaelian I, Brevet M, Anginot A, Le Bousse-Kerdilès MC, Trichet V, Lefort S, Maguer-Satta V. A minimal standardized human bone marrow microphysiological system to assess resident cell behavior during normal and pathological processes. Biomater Sci 2022;10:485–98 [DOI] [PubMed] [Google Scholar]
- 36. Ma C, Witkowski MT, Harris J, Dolgalev I, Sreeram S, Qian W, Tong J, Chen X, Aifantis I, Chen W. Leukemia-on-a-chip: dissecting the chemoresistance mechanisms in B cell acute lymphoblastic leukemia bone marrow niche. Sci Adv 2020;6:eaba5536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duerkop BA, Vaishnava S, Hooper LV. Immune responses to the microbiota at the intestinal mucosal surface. Immunity 2009;31:368–76 [DOI] [PubMed] [Google Scholar]
- 38. Bradford EM, Ryu SH, Singh AP, Lee G, Goretsky T, Sinh P, Williams DB, Cloud AL, Gounaris E, Patel V. Epithelial TNF receptor signaling promotes mucosal repair in inflammatory bowel disease. J Immunol 2017;199:1886–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Papoutsopoulou S, Pollock L, Walker C, Tench W, Samad SS, Bergey F, Lenzi L, Sheibani-Tezerji R, Rosenstiel P, Alam MT, Martins Dos Santos VAP, Müller W, Campbell BJ. Impact of interleukin 10 deficiency on intestinal epithelium responses to inflammatory signals. Front Immunol 2021;12:690817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Su L, Shen L, Clayburgh DR, Nalle SC, Sullivan EA, Meddings JB, Abraham C, Turner JR. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology 2009;136:551–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jalili-Firoozinezhad S, Gazzaniga FS, Calamari EL, Camacho DM, Fadel CW, Bein A, Swenor B, Nestor B, Cronce MJ, Tovaglieri A. A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nat Biomed Eng 2019;3:520–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shin W, Hinojosa CD, Ingber DE, Kim HJ. Human intestinal morphogenesis controlled by transepithelial morphogen gradient and flow-dependent physical cues in a microengineered gut-on-a-chip. Iscience 2019;15:391–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shin YC, Shin W, Koh D, Wu A, Ambrosini YM, Min S, Eckhardt SG, Fleming RD, Kim S, Park S. Three-dimensional regeneration of patient-derived intestinal organoid epithelium in a physiodynamic mucosal interface-on-a-chip. Micromachines 2020;11:663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kim HJ, Li H, Collins JJ, Ingber DE. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc Nat Acad Sci 2016;113:E7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shin W, Kim HJ. Intestinal barrier dysfunction orchestrates the onset of inflammatory host–microbiome cross-talk in a human gut inflammation-on-a-chip. Proc Nat Acad Sci 2018;115:E10539–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Humayun M, Ayuso JM, Park KY, Martorelli Di, Genova B, Skala MC, Kerr SC, Knoll LJ, Beebe DJ. Innate immune cell response to host-parasite interaction in a human intestinal tissue microphysiological system. Sci Adv 2022;8:eabm8012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jeong H-J, Park J-H, Kang JH, Kong S-H, Park T-E. Organoid-based human stomach micro-physiological system to recapitulate the dynamic mucosal defense mechanism. 2022, https://www.biorxiv.org/content/10.1101/2022.03.02.482603v1 [DOI] [PMC free article] [PubMed]
- 48. Gallizioli M, Miró-Mur F, Otxoa-de-Amezaga A, Cugota R, Salas-Perdomo A, Justicia C, Brait VH, Ruiz-Jaén F, Arbaizar-Rovirosa M, Pedragosa J, Bonfill-Teixidor E, Gelderblom M, Magnus T, Cano E, Del Fresno C, Sancho D, Planas AM. Dendritic cells and microglia have non-redundant functions in the inflamed brain with protective effects of type 1 cDCs. Cell Rep 2020;33:108291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol 2012;12:623–35 [DOI] [PubMed] [Google Scholar]
- 50. D’Agostino PM, Gottfried-Blackmore A, Anandasabapathy N, Bulloch K. Brain dendritic cells: biology and pathology. Acta Neuropathol 2012;124:599–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mapunda JA, Tibar H, Regragui W, Engelhardt B. How does the immune system enter the brain. Front Immunol 2022;13:805657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Nishihara H, Soldati S, Mossu A, Rosito M, Rudolph H, Muller WA, Latorre D, Sallusto F, Sospedra M, Martin R. Human CD4+ T cell subsets differ in their abilities to cross endothelial and epithelial brain barriers in vitro. Fluid Barrier CNS 2020;17:1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood–brain barriers. Trends Immunol 2012;33:579–89 [DOI] [PubMed] [Google Scholar]
- 54. Mossu A, Rosito M, Khire T, Li Chung H, Nishihara H, Gruber I, Luke E, Dehouck L, Sallusto F, Gosselet F, McGrath JL, Engelhardt B. A silicon nanomembrane platform for the visualization of immune cell trafficking across the human blood–brain barrier under flow. J Cereb Blood Flow Metab 2019;39:395–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Herland A, van der Meer AD, FitzGerald EA, Park TE, Sleeboom JJ, Ingber DE. Distinct contributions of astrocytes and pericytes to neuroinflammation identified in a 3D human blood-brain barrier on a chip. PLoS ONE 2016;11:e0150360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kim J, Lee KT, Lee JS, Shin J, Cui B, Yang K, Choi YS, Choi N, Lee SH, Lee JH, Bahn YS, Cho SW. Fungal brain infection modelled in a human-neurovascular-unit-on-a-chip with a functional blood–brain barrier. Nat Biomed Eng 2021;5:830–46 [DOI] [PubMed] [Google Scholar]
- 57. Bramley JC, Drummond CG, Lennemann NJ, Good CA, Kim KS, Coyne CB. A three-dimensional cell culture system to model RNA virus infections at the blood-brain barrier. mSphere 2017;2:e00206–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Buzhdygan TP, DeOre BJ, Baldwin-Leclair A, Bullock TA, McGary HM, Khan JA, Razmpour R, Hale JF, Galie PA, Potula R. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood–brain barrier. Neurobiol Dis 2020;146:105131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang P, Jin L, Zhang M, Wu Y, Duan Z, Guo Y, Wang C, Chen W, Liao Z, Wang Y, Lai R, Lee LP, Qin J. Blood-brain barrier injury and neuroinflammation induced by SARS-CoV-2 in a lung-brain microphysiological system. Nat Biomed Eng. Epub ahead of print 22 June 2023. DOI: 10.1038/s41551-023-01054-w [DOI] [PubMed] [Google Scholar]
- 60. Robbins JR, Skrzypczynska KM, Zeldovich VB, Kapidzic M, Bakardjiev AI. Placental syncytiotrophoblast constitutes a major barrier to vertical transmission of Listeria monocytogenes. PLoS Pathogens 2010;6:e1000732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jagger BW, Miner JJ, Cao B, Arora N, Smith AM, Kovacs A, Mysorekar IU, Coyne CB, Diamond MS. Gestational stage and IFN-λ signaling regulate ZIKV infection in utero. Cell Host Microbe 2017;22:366e3–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ander SE, Diamond MS, Coyne CB. Immune responses at the maternal-fetal interface. Sci Immunol 2019;4:eaat6114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Blundell C, Yi YS, Ma L, Tess ER, Farrell MJ, Georgescu A, Aleksunes LM, Huh D. Placental drug transport-on-a-chip: a microengineered in vitro model of transporter-mediated drug efflux in the human placental barrier. Adv Healthc Mater 2018;7:1700786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Corry J, Arora N, Good CA, Sadovsky Y, Coyne CB. Organotypic models of type III interferon-mediated protection from Zika virus infections at the maternal–fetal interface. Proc Nat Acad Sci 2017;114:9433–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yin F, Zhu Y, Zhang M, Yu H, Chen W, Qin J. A 3D human placenta-on-a-chip model to probe nanoparticle exposure at the placental barrier. Toxicol in Vitro 2019;54:105–13 [DOI] [PubMed] [Google Scholar]
- 66. McConkey CA, Delorme-Axford E, Nickerson CA, Kim KS, Sadovsky Y, Boyle JP, Coyne CB. A three-dimensional culture system recapitulates placental syncytiotrophoblast development and microbial resistance. Sci Adv 2016;2:e1501462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhu Y, Yin F, Wang H, Wang L, Yuan J, Qin J. Placental barrier-on-a-chip: modeling placental inflammatory responses to bacterial infection. ACS Biomater Sci Eng 2018;4:3356–63 [DOI] [PubMed] [Google Scholar]
- 68. Rabussier G, Bünter I, Bouwhuis J, Soragni C, van Zijp T, Ng CP, Domansky K, de Windt LJ, Vulto P, Murdoch CE, Bircsak KM, Lanz HL. Healthy and diseased placental barrier on-a-chip models suitable for standardized studies. Acta Biomater 2023;164:363–76 [DOI] [PubMed] [Google Scholar]
- 69. Bieber T. Atopic dermatitis. N Engl J Med 2008;358:1483–94 [DOI] [PubMed] [Google Scholar]
- 70. Bantz SK, Zhu Z, Zheng T. The atopic march: progression from atopic dermatitis to allergic rhinitis and asthma. J Clin Cell Immunol 2014;5:202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Peng W, Novak N. Pathogenesis of atopic dermatitis. Clin Exp Allergy 2015;45:566–74 [DOI] [PubMed] [Google Scholar]
- 72. Cork MJ, Robinson DA, Vasilopoulos Y, Ferguson A, Moustafa M, MacGowan A, Duff GW, Ward SJ, Tazi-Ahnini R. New perspectives on epidermal barrier dysfunction in atopic dermatitis: gene-environment interactions. J Allergy Clin Immunol 2006;118:3–2122 [DOI] [PubMed] [Google Scholar]
- 73. Liu X, Michael S, Bharti K, Ferrer M, Song MJ. A biofabricated vascularized skin model of atopic dermatitis for preclinical studies. Biofabrication 2020;12:035002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. van den Bogaard EH, Tjabringa GS, Joosten I, Vonk-Bergers M, van Rijssen E, Tijssen HJ, Erkens M, Schalkwijk J, Koenen HJPM. Crosstalk between keratinocytes and T cells in a 3D microenvironment: a model to study inflammatory skin diseases. J Invest Dermatol 2014;134:719–27 [DOI] [PubMed] [Google Scholar]
- 75. Shin JU, Abaci HE, Herron L, Guo Z, Sallee B, Pappalardo A, Jackow J, Wang EHC, Doucet Y, Christiano AM. Recapitulating T cell infiltration in 3D psoriatic skin models for patient-specific drug testing. Sci Rep 2020;10:4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Morgner B, Tittelbach J, Wiegand C. Induction of psoriasis- and atopic dermatitis-like phenotypes in 3D skin equivalents with a fibroblast-derived matrix. Sci Rep 2023;13:1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wufuer M, Lee G, Hur W, Jeon B, Kim BJ, Choi TH, Lee S. Skin-on-a-chip model simulating inflammation, edema and drug-based treatment. Sci Rep 2016;6:37471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sriram Alberti GM, Dancik Y, Wu B, Wu R, Feng ZJ, Ramasamy S, Bigliardi PL, Bigliardi-Qi M, Wang Z. Full-thickness human skin-on-chip with enhanced epidermal morphogenesis and barrier function. Materials Today 2017;21:326–40 [Google Scholar]
- 79. Offeddu GS, Serrano JC, Wan Z, Bryniarski MA, Humphreys SC, Chen SW, Dhoolypala H, Conner K, Kamm RD. Microphysiological endothelial models to characterize subcutaneous drug absorption. ALTEX 2023;40:299313. [DOI] [PubMed] [Google Scholar]
- 80. Weyand CM, Goronzy JJ. The immunology of rheumatoid arthritis. Nat Immunol 2021;22:10–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Damerau A, Gaber T. Modeling rheumatoid arthritis in vitro: from experimental feasibility to physiological proximity. Int J Mol Sci 2020;21:7916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rothbauer M, Byrne RA, Schobesberger S, Olmos Calvo I, Fischer A, Reihs EI, Spitz S, Bachmann B, Sevelda F, Holinka J, Holnthoner W, Redl H, Toegel S, Windhager R, Kiener HP, Ertl P. Establishment of a human three-dimensional chip-based chondro-synovial coculture joint model for reciprocal cross talk studies in arthritis research. Lab Chip 2021;21:4128–43 [DOI] [PubMed] [Google Scholar]
- 83. Occhetta P, Mainardi A, Votta E, Vallmajo-Martin Q, Ehrbar M, Martin I, Barbero A, Rasponi M. Hyperphysiological compression of articular cartilage induces an osteoarthritic phenotype in a cartilage-on-a-chip model. Nat Biomed Eng 2019;3:545–57 [DOI] [PubMed] [Google Scholar]
- 84. Maracle CX, Kucharzewska P, Helder B, van der Horst C, Correa de, Sampaio P, Noort AR, van Zoest K, Griffioen AW, Olsson H, Tas SW. Targeting non-canonical nuclear factor-kappaB signalling attenuates neovascularization in a novel 3D model of rheumatoid arthritis synovial angiogenesis. Rheumatology 2017;56:294–302 [DOI] [PubMed] [Google Scholar]
- 85. Peck Y, Leom LT, Low PFP, Wang DA. Establishment of an in vitro three-dimensional model for cartilage damage in rheumatoid arthritis. J Tissue Eng Regen Med 2018;12:e237–49 [DOI] [PubMed] [Google Scholar]
- 86. Dwivedi G, Flaman L, Alaybeyoglu B, Struglics A, Frank EH, Chubinskya S, Trippel SB, Rosen V, Cirit M, Grodzinsky AJ. Inflammatory cytokines and mechanical injury induce post-traumatic osteoarthritis-like changes in a human cartilage-bone-synovium microphysiological system. Arthritis Res Ther 2022;24:198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, Rozmahel R, Cole JL, Kennedy D, Hidaka N, Zsiga M, Buchwald M, Riordan JR, Tsui L-C, Collins FS. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989;245:1059–65 [DOI] [PubMed] [Google Scholar]
- 88. Heijerman HH. Infection and inflammation in cystic fibrosis: a short review. J Cyst Fibros 2005;4:3–5 [DOI] [PubMed] [Google Scholar]
- 89. Rieber NN, Hector A, Carevic M, Hartl D. Current concepts of immune dysregulation in cystic fibrosis. Int J Biochem Cell Biol 2014;52:108–12 [DOI] [PubMed] [Google Scholar]
- 90. Benam KH, Villenave R, Lucchesi C, Varone A, Hubeau C, Lee HH, Alves SE, Salmon M, Ferrante TC, Weaver JC, Bahinski A, Hamilton GA, Ingber DE. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat Methods 2016;13:151–7 [DOI] [PubMed] [Google Scholar]
- 91. Kilic O, Yoon A, Shah SR, Yong HM, Ruiz-Valls A, Chang H, Panettieri RA, Jr, Liggett SB, Quiñones-Hinojosa A, An SS, Levchenko A. A microphysiological model of the bronchial airways reveals the interplay of mechanical and biochemical signals in bronchospasm. Nat Biomed Eng 2019;3:532–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Mejias JC, Nelson MR, Liseth O, Roy K. A 96-well format microvascularized human lung-on-a-chip platform for microphysiological modeling of fibrotic diseases. Lab Chip 2020;20:3601–11 [DOI] [PubMed] [Google Scholar]
- 93. Lagowala DA, Wally A, Wilmsen K, Kim B, Yeung-Luk B, Choi J, Swaby C, Luk M, Feller L, Ghosh B, Niedrkofler A, Tieng E, Sherman E, Chen D, Upadya N, Zhang R, Kim DH, Sidhaye V. Microphysiological models of lung epithelium-alveolar macrophage co-cultures to study chronic lung disease. Adv Biol 2023;2023:e2300165 [DOI] [PubMed] [Google Scholar]
- 94. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Letters 2017;387:61–8 [DOI] [PubMed] [Google Scholar]
- 95. Whiteside T. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008;27:5904–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Whiteside TL. Immune suppression in cancer: effects on immune cells, mechanisms and future therapeutic intervention. Semin Cancer Biol 2006;16:3–15 [DOI] [PubMed] [Google Scholar]
- 97. Haase K, Offeddu GS, Gillrie MR, Kamm RD. Endothelial regulation of drug transport in a 3D vascularized tumor model. Adv Funct Mater 2020;30:2002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lee S, Lim J, Yu J, Ahn J, Lee Y, Jeon NL. Engineering tumor vasculature on an injection-molded plastic array 3D culture (IMPACT) platform. Lab Chip 2019;19:2071–80 [DOI] [PubMed] [Google Scholar]
- 99. Liu Y, Sakolish C, Chen Z, Phan DT, Bender RHF, Hughes CC, Rusyn I. Human in vitro vascularized micro-organ and micro-tumor models are reproducible organ-on-a-chip platforms for studies of anticancer drugs. Toxicology 2020;445:152601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Lee J-H, Kim S-K, Khawar IA, Jeong S-Y, Chung S, Kuh H-J. Microfluidic co-culture of pancreatic tumor spheroids with stellate cells as a novel 3D model for investigation of stroma-mediated cell motility and drug resistance. J Exp Clin Cancer Res 2018;37:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Linde N, Gutschalk CM, Hoffmann C, Yilmaz D, Mueller MM. Integrating macrophages into organotypic co-cultures: a 3D in vitro model to study tumor-associated macrophages. PLoS ONE 2012;7:e40058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Bai J, Adriani G, Dang T-M, Tu H-XL, Penny T-Y, Wong S-C, Kamm RD, Thiery J-P. Contact-dependent carcinoma aggregate dispersion by M2a macrophages via ICAM-1 and β2 integrin interactions. Oncotarget 2015;6:25295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Rebelo SP, Pinto C, Martins TR, Harrer N, Estrada MF, Loza-Alvarez P, Cabeçadas J, Alves PM, Gualda EJ, Sommergruber W, Brito C. 3D-3-culture: a tool to unveil macrophage plasticity in the tumour microenvironment. Biomaterials 2018;163:185–97 [DOI] [PubMed] [Google Scholar]
- 104. Nguyen OTP, Misun PM, Lohasz C, Lee J, Wang W, Schroeder T, Hierlemann A. An immunocompetent microphysiological system to simultaneously investigate effects of anti-tumor natural killer cells on tumor and cardiac microtissues. Front Immunol 2021;12:781337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Yada RC, Desa DE, Gillette AA, Bartels E, Harari PM, Skala MC, Beebe DJ, Kerr SC. Microphysiological head and neck cancer model identifies novel role of lymphatically secreted monocyte migration inhibitory factor in cancer cell migration and metabolism. Biomaterials 2023;298:122136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science 2018;359:1361–5 [DOI] [PubMed] [Google Scholar]
- 107. Sterner RC, Sterner RM. CAR-T cell therapy: current limitations and potential strategies. Blood Cancer Journal 2021;11:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Pavesi A, Tan AT, Koh S, Chia A, Colombo M, Antonecchia E, Miccolis C, Ceccarello E, Adriani G, Raimondi MT. A 3D microfluidic model for preclinical evaluation of TCR-engineered T cells against solid tumors. JCI Insight 2017;2:e89762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wallstabe L, Göttlich C, Nelke LC, Kühnemundt J, Schwarz T, Nerreter T, Einsele H, Walles H, Dandekar G, Nietzer SL. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 2019;4:e126345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Ando Y, Siegler EL, Ta HP, Cinay GE, Zhou H, Gorrell KA, Au H, Jarvis BM, Wang P, Shen K. Evaluating CAR-T cell therapy in a hypoxic 3D tumor model. Adv Healthc Mater 2019;8:e1900001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Zhang J, Tao A. Antigenicity, immunogenicity, allergenicity. Aller Bioinf 2015;8:175–86 [Google Scholar]
- 112. Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res 2015;21:687–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Jiang X, Ren L, Tebon P, Wang C, Zhou X, Qu M, Zhu J, Ling H, Zhang S, Xue Y, Wu Q, Bandaru P, Lee J, Kim HJ, Ahadian S, Ashammakhi N, Dokmeci MR, Wu J, Gu Z, Sun W, Khademhosseini A. Cancer-on-a-chip for modeling immune checkpoint inhibitor and tumor interactions. Small 2021;17:e2004282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D. Ex vivo profiling of PD-1 blockade using organotypic tumor spheroidsex vivo profiling of immune checkpoint blockade. Cancer Discover 2018;8:196–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Cui X, Ma C, Vasudevaraja V, Serrano J, Tong J, Peng Y, Delorenzo M, Shen G, Frenster R-TT, Morales J. Dissecting the immunosuppressive tumor microenvironments in Glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. Elife 2020;9:e52253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Arstila TP, Casrouge A, Baron V, Even J, Kanellopoulos J, Kourilsky P. A direct estimate of the human alphabeta T cell receptor diversity. Science 1999;286:958–61 [DOI] [PubMed] [Google Scholar]
- 117. Imkeller K, Wardemann H. Assessing human B cell repertoire diversity and convergence. Immunol Rev 2018;284:51–66 [DOI] [PubMed] [Google Scholar]
- 118. Xue D, Lu S, Zhang H, Zhang L, Dai Z, Kaufman DS, Zhang J. Induced pluripotent stem cell-derived engineered T cells, natural killer cells, macrophages, and dendritic cells in immunotherapy. Trends Biotechnol 2023;41:907–22 [DOI] [PubMed] [Google Scholar]
- 119. Lai W, Xie H, Liu Y, Zheng F, Zhang Y, Lei Q, Lv L, Dong J, Song J, Gao X, Yin M, Wang C, Deng H. Human pluripotent stem cell-derived eosinophils reveal potent cytotoxicity against solid tumors. Stem Cell Report 2021;16:1697–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Igarashi A, Ebihara Y, Kumagai T, Hirai H, Nagata K, Tsuji K. Mast cells derived from human induced pluripotent stem cells are useful for allergen tests. Allergol Int 2018;67:234–42 [DOI] [PubMed] [Google Scholar]
- 121. Trump LR, Nayak RC, Singh AK, Emberesh S, Wellendorf AM, Lutzko CM, Cancelas JA. Neutrophils derived from genetically modified human induced pluripotent stem cells circulate and phagocytose bacteria in vivo. Stem Cells Transl Med 2019;8:557–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. French A, Yang CT, Taylor S, Watt SM, Carpenter L. Human induced pluripotent stem cell-derived B lymphocytes express sIgM and can be generated via a hemogenic endothelium intermediate. Stem Cells Dev 2015;24:1082–95 [DOI] [PubMed] [Google Scholar]
- 123. Morizane R, Lam AQ, Freedman BS, Kishi S, Valerius MT, Bonventre JV. Nephron organoids derived from human pluripotent stem cells model kidney development and injury. Nat Biotechnol 2015;33:1193–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, McLaren-Douglas A, Blokker J, Jaksani S, Bartfeld S, Volckman R, van Sluis P, Li VS, Seepo S, Sekhar Pedamallu C, Cibulskis K, Carter SL, McKenna A, Lawrence MS, Lichtenstein L, Stewart C, Koster J, Versteeg R, van Oudenaarden A, Saez-Rodriguez J, Vries RG, Getz G, Wessels L, Stratton MR, McDermott U, Meyerson M, Garnett MJ, Clevers H. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell 2015;161:933–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Nikolaev M, Mitrofanova O, Broguiere N, Geraldo S, Dutta D, Tabata Y, Elci B, Brandenberg N, Kolotuev I, Gjorevski N, Clevers H, Lutolf MP. Homeostatic mini-intestines through scaffold-guided organoid morphogenesis. Nature 2020;585:574–8 [DOI] [PubMed] [Google Scholar]
- 126. Kroll KT, Mata MM, Homan KA, Micallef V, Carpy A, Hiratsuka K, Morizane R, Moisan A, Gubler M, Walz AC, Marrer-Berger E, Lewis JA. Immune-infiltrated kidney organoid-on-chip model for assessing T cell bispecific antibodies. Proc Natl Acad Sci U S A 2023;120:e2305322120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Svensson V, Vento-Tormo R, Teichmann SA. Exponential scaling of single-cell RNA-seq in the past decade. Nat Protoc 2018;13:599–604 [DOI] [PubMed] [Google Scholar]
- 128. Herland A, Maoz BM, Das D, Somayaji MR, Prantil-Baun R, Novak R, Cronce M, Huffstater T, Jeanty SSF, Ingram M, Chalkiadaki A, Benson Chou D, Marquez S, Delahanty A, Jalili-Firoozinezhad S, Milton Y, Sontheimer-Phelps A, Swenor B, Levy O, Parker KK, Przekwas A, Ingber DE. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat Biomed Eng 2020;4:421–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Novak R, Ingram M, Marquez S, Das D, Delahanty A, Herland A, Maoz BM, Jeanty SSF, Somayaji MR, Burt M, Calamari E, Chalkiadaki A, Cho A, Choe Y, Chou DB, Cronce M, Dauth S, Divic T, Fernandez-Alcon J, Ferrante T, Ferrier J, FitzGerald EA, Fleming R, Jalili-Firoozinezhad S, Grevesse T, Goss JA, Hamkins-Indik T, Henry O, Hinojosa C, Huffstater T, Jang KJ, Kujala V, Leng L, Mannix R, Milton Y, Nawroth J, Nestor BA, Ng CF, O’Connor B, Park TE, Sanchez H, Sliz J, Sontheimer-Phelps A, Swenor B, Thompson G, 2nd, Touloumes GJ, Tranchemontagne Z, Wen N, Yadid M, Bahinski A, Hamilton GA, Levner D, Levy O, Przekwas A, Prantil-Baun R, Parker KK, Ingber DE. Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat Biomed Eng 2020;4:407–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Ronaldson-Bouchard K, Teles D, Yeager K, Tavakol DN, Zhao Y, Chramiec A, Tagore S, Summers M, Stylianos S, Tamargo M, Lee BM, Halligan SP, Abaci EH, Guo Z, Jacków J, Pappalardo A, Shih J, Soni RK, Sonar S, German C, Christiano AM, Califano A, Hirschi KK, Chen CS, Przekwas A, Vunjak-Novakovic G. A multi-organ chip with matured tissue niches linked by vascular flow. Nat Biomed Eng 2022;6:351–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Shim S, Belanger MC, Harris AR, Munson JM, Pompano RR. Two-way communication between ex vivo tissues on a microfluidic chip: application to tumor-lymph node interaction. Lab Chip 2019;19:1013–26 [DOI] [PMC free article] [PubMed] [Google Scholar]