

Abstract
Obesity is a major cause of various metabolic disorders, including type 2 diabetes, nonalcoholic fatty liver disease (NAFLD) and cardiovascular diseases, in modern times. Fat tissue originally evolved as an organ to prepare for food shortages. However, when individuals consume excessive calories and engage in insufficient physical activity, it can lead to the excessive accumulation of lipids in white adipose tissue, potentially causing problems. In response to this excessive lipid accumulation extending to other tissues, insulin resistance is triggered in the body as a physiological response to prevent harmful effects. Additionally, in mammals, brown adipose tissue has evolved to generate energy and maintain body temperature. These inconspicuous defense mechanisms function coordinately to protect against systemic metabolic abnormalities affecting multiple organs. Understanding the dynamic nature of adipose tissues is now crucial for elucidating the details of the molecular abnormalities in obesity-associated metabolic diseases. This review outlines adipocyte plasticity and function with a focus on the physiological relevance and new pathways of insulin signaling.
Keywords: Type 2 diabetes, Adipocyte turnover, Insulin signaling, Insulin resistance, Therapeutic targets, Brown adipocyte
Introduction
In modern life, people tend to consume food and drink in excess while reducing daily physical activity, leading to an increase in body fat mass, referred to as “obesity.” Chronic overnutrition leads to obesity, with an increase in the abundance of visceral white adipose tissue (WAT) where excess energy is stored in the form of triglycerides (TGs). Obesity is associated with insulin resistance in various metabolic organs, including the liver, muscles, and adipose tissues, resulting in the development of type 2 diabetes (T2D) and metabolic syndrome [1]. There are other types of adipose tissue known as brown adipose tissue (BAT) and beige adipose tissue, which are rich in mitochondria. These tissues possess the potential for energy expenditure, which contributes to maintaining body temperature and reducing energy storage [2]. BAT is abundant in the interscapular region during infancy and adolescence but decreases significantly with aging [3]. Recent research has elucidated the significant metabolic advantages associated with active BAT, sparking interest in strategies to address obesity and T2D [4].
While studies have explored the anatomy, development, and secreted molecules associated with adipose tissues, there is still a need for further investigation into the molecular mechanisms governing adipocyte activation, turnover, and the soluble molecules regulating adipocyte maintenance and insulin sensitivity. Additionally, understanding how insulin signaling can modulate the functions of two distinct types of adipose tissues, that is, WAT for energy storage and BAT for energy expenditure, remains an intriguing area of research. This review discusses some of the aspects of the dynamic nature of adipose tissue and focuses on the newest aspects linking insulin signaling and adipose tissue to the control of systemic metabolism.
Adipose Tissues
There are three major types of adipose tissues found in mammals: WAT, beige or brite adipose tissue, and BAT [5]. WAT is involved in storing energy as TGs and releasing these TGs in response to changes in the body’s energy needs. In addition to its role in energy storage, WAT acts as an endocrine organ, releasing substances called adipokines with hormonal properties. These adipokines, including adiponectin related to insulin sensitivity and others such as resistin, retinol-binding protein 4 (RBP4), tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), interleukin-1 beta (IL-1β), and monocyte chemokine protein 1 (MCP1) related to insulin resistance and inflammation, influence metabolism [6]. Leptin acts on leptin receptors in the hypothalamus to suppress feeding and increase energy expenditure.
WAT is a heterogeneous tissue with diverse metabolic and regenerative plasticity and is composed of subcutaneous and visceral adipose tissues. It comprises lipid-filled adipocytes and nonadipocyte cells, including endothelial cells, uncharacterized stromal cells, preadipocytes, fibroblasts, and peripheral blood cells, which encompass immune-competent cells such as antigen-presenting cells, T cells, mononuclear cells, and macrophages.
In contrast to WAT, which contains a single large lipid droplet, BAT possesses multiple smaller lipid droplets. In adult humans, brown adipocytes are primarily found in regions such as the cervical, supraclavicular, paravertebral, and perirenal–adrenal areas [7]. Gene analysis of BAT from the supraclavicular region has revealed that it has characteristics of both traditional brown and beige adipocytes, indicating that human BAT is a diverse mixture of these cell types [8, 9]. These adipocytes have a high density of mitochondria and express uncoupling protein 1 (UCP1), which allows them to expend energy. Beige adipocytes, on the other hand, can transition from white to brown characteristics within WAT depots and share similarities with brown adipocytes. Studies have demonstrated that active BAT in humans has positive effects on metabolism. Higher BAT activity is associated with lower body mass index (BMI) and increased insulin sensitivity. Therefore, strategies aimed at boosting BAT mass and activity could hold promise for preventing obesity and its related health issues.
Insulin/IGF1 Receptor Signaling in Adipocytes
Insulin and IGF1 control growth and metabolism by binding to insulin receptor (IR) and IGF1 receptor (IGF1R) on cell surfaces. Because IR and IGF1R are highly homologous heterodimers, they share pathways, leading to similar responses [10]. When IR and IGF1R are activated by their respective ligands, IR and IGF1R undergo conformational changes, which induce Tyr phosphorylation of their β-subunits. This leads to the recruitment of protein substrates such as insulin substrate 1 and 2 (IRS-1 and IRS-2), and src homology and collagen (Shc). The following reactions activate phosphoinositide 3-kinase (PI3K) and Ras/mitogen-activated protein kinase (MAPK) signaling. Downstream of PI3K, a set of kinases, including phosphoinositide-dependent kinase 1 (PDK-1), Akt, protein kinase C (PKC), mechanism target of rapamycin (mTOR), S6 ribosomal kinase (S6K) and glycogen synthase kinase 3 (GSK3), transmit the signal. These kinases regulate transcription factors such as forkhead box O1 (FoxO1), peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC1α) and sterol regulatory element-binding protein (SREBP), thereby influencing glucose, lipids, and mitochondrial function, as well as cell growth, differentiation, and programmed cell death [10, 11].
IR and IGF1R functionally belong to the receptor tyrosine kinase family. Despite their high degree of homology and shared downstream signaling, their activation leads to distinct physiological outcomes. IR primarily regulates metabolic functions, while IGF1R regulates mitogenesis and growth. The receptor-specific effects in adipocytes depend on differences in the extracellular and intracellular domains and the varying affinities for different intracellular substrates. IR prefers the phosphorylation of IRS proteins, whereas IGF1R prefers the phosphorylation of Shc, a transforming protein containing src homology 2 (SH2) domains. Indeed, the substitution of amino acids in the juxtamembrane region of IR and IGF1R reveals another significant sequence difference near the NPEY motif of the juxtamembrane region. This residue, positioned at 973, is leucine in IR and phenylalanine in IGF1R. Substituting leucine-973 in the juxtamembrane region of IR with phenylalanine decreases binding to IRS-1 and increases binding to Shc [12] (Fig. 1).
Fig. 1.

Leucine 973 was identified as a key residue that differentiates IR signaling from IGF1R signaling. Substituting leucine-973 in the juxtamembrane region of IR with phenylalanine has been shown to decrease binding to IRS-1 and increase binding to Shc and activation of the ERK1/2 and p70S6K-S6 pathways
Role of IR/IGF1R Signaling in Mature Adipose Tissue
The initial fat-specific insulin receptor knockout (FIRKO) mice, generated using the aP2-Cre gene promoter, displayed a partial defect in adipocyte development, increased insulin sensitivity and enhanced longevity [13]. To further investigate the effect of insulin sensitivity in mature adipocytes in adult mice, we created mice with inducible knockout of IR and/or IGF1R in adipocytes (Ai-IRKO, Ai-IGFRKO and Ai-DKO) using tamoxifen-inducible adiponectin-CreERT2 to drive recombination [14]. Genetic disruption of IR or IGF1R in mature adipocytes in adult mice leads to different phenotypes. Models with an inducible fat-specific knockout of IR using tamoxifen-regulated adiponectin-Cre (Ai-IRKO) rapidly developed severe lipodystrophy, cold intolerance and systemic insulin resistance accompanied by NAFLD and pancreatic beta-cell hyperplasia. However, loss of IGF1R alone (Ai-IGFRKO) produced only a modest reduction in WAT and no significant effect on glucose metabolism [14]. Despite the modest role of IGFR1R alone, combined knockout of IR and IGF1R (Ai-DKO) resulted in a more severe phenotype than knockout of IR alone. Ai-DKO mice displayed greater reductions in BAT mass, increased hyperglycemia and profound cold intolerance, indicating a strong interaction between the IR and IGF1R complementary pathways in BAT maintenance and function [14].
Insulin Signaling-Dependent Regulation of Transcription Factors
One of the actions of Akt is to phosphorylate members of the FoxO family of forkhead transcription factors, including FoxO1, FoxO3, and FoxO4. This phosphorylation event results in the exclusion of FoxOs from the nucleus, effectively inhibiting their transcriptional activity. Over the past decade, extensive research has illuminated the crucial role of deactivating FoxOs, particularly FoxO1, in the regulation of insulin action and the control of whole-body energy metabolism. FoxO1 plays a pivotal role in regulating adipocyte differentiation and in mediating insulin-controlled protein degradation in muscle tissue. Since insulin functions to counteract the actions of FoxOs by preventing their nuclear localization, genetic knockout experiments targeting FoxO1 in the liver or FoxO-1, -3, and -4 in muscle or adipocytes have resulted in the reversal of the consequences of insulin receptor/IGF1R loss, including its effects on gene expression and metabolism in these tissues [15–17].
We searched for IR/IGF1R-binding proteins in brown preadipocytes using a proteomics approach. We have identified two members of the FoxK family of Forkhead transcription factors, forkhead box K1 (FoxK1) and forkhead box K2 (FoxK2), as new targets of insulin action.
In contrast to FoxO1, which is turned off by insulin, these transcription factors with a FOX domain, FoxK1 and FoxK2, were found to translocate from the cytoplasm to the nucleus following insulin stimulation, suggesting a potential positive role in insulin signaling [18] (Fig. 2). Phosphoproteomics analysis revealed that FoxK1/2 phosphorylation sites included clusters at the N-terminus (FoxK1: S225, S229, and T233) that increased and clusters at the C-terminus (FoxK1: S402, S406, S454, and S458; FoxK2: S415 and S419) that decreased upon insulin stimulation. These motifs were identified as phosphorylation targets of GSK3, specifically -SxxxS-. In the basal state, GSK3 phosphorylates FoxK1/K2, leading to increased interaction with 14-3-3 proteins and nuclear exclusion. On the other hand, nuclear translocation is regulated by Akt-mTOR signaling by insulin. FoxK1/K2 regulate genes involved in the cell cycle, apoptosis and lipid metabolism in hepatocytes, while in adipocytes and muscle, FoxK1/K2 regulate glycolytic and mitochondrial metabolism [18, 19].
Fig. 2.

Regulation of FoxK1/K2 by insulin signaling. FoxK1/K2 translocate from the cytoplasm to the nucleus reciprocally with the translocation of FoxO1
Maintenance of Insulin Sensitivity of Adipocytes
The restoration of insulin sensitivity in adipose tissue is important for the regulation of adipose tissue metabolism. The interaction between the insulin receptor and insulin itself inevitably relies on processes such as receptor desensitization and dephosphorylation for the recovery of receptor sensitivity. Protein tyrosine phosphatases, such as protein tyrosine phosphatase 1B (PTP1B), have been identified as negative regulators of insulin action by dephosphorylating tyrosine residues on activated IR, IGF1R, and IRS1 proteins, thereby attenuating their activity [20]. PTP1B plays an essential role in insulin action, and studies using PTP1B knockout (KO) mice have shown enhanced insulin sensitivity, increased insulin receptor phosphorylation, and resistance to obesity and insulin resistance [21, 22].
In contrast, the activities of downstream protein kinases such as Akt, PKC, S6K, and extracellular signal-regulated kinase (ERK) are regulated by serine/threonine protein phosphatase 2A (PP2A), which constitutes approximately 80% of serine/threonine phosphatase activity in cells [23]. However, the molecular mechanism of how the tyrosine-dephosphorylating enzyme PTP1B and the serine/threonine-dephosphorylating enzyme PP2A coordinate their activities to control IR signaling remains to be elucidated. Recently, it has been discovered that α4, a protector within the catalytic subunit of PP2A, plays a role in controlling the transcription factor Y-box protein 1 (YBX1) associated with PTP1B. This mechanism establishes a feedback loop that regulates insulin receptor signaling from downstream to upstream tyrosine phosphorylation status. Notably, adipocyte-specific knockout of α4 in mice impaired insulin-induced Akt-mediated serine/threonine phosphorylation and reduced insulin-induced insulin receptor tyrosine phosphorylation. This effect is attributed to a decreased association between α4 and YBX1, leading to an increase in the expression of the tyrosine phosphatase PTP1B [24] (Fig. 3).
Fig. 3.
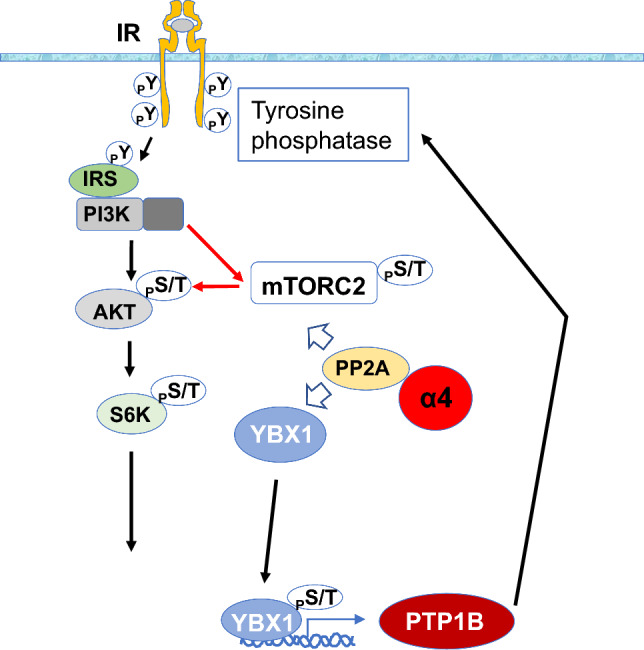
α4 plays a crucial role in the regulation of insulin signaling by stabilizing PP2A and facilitating the dephosphorylation of mTORC2. Additionally, α4 interacts with Y-box protein 1 (YBX1), which acts as a transcription factor for the Tyr phosphatase PTP1B. α4 prevents YBX1 from translocating to the nucleus and inhibits the expression of PTP1B. This feedback mechanism enhances signaling through the insulin receptor
Furthermore, the knockout of α4 in adipocytes impaired adipogenesis and altered mitochondrial oxidation, leading to increased inflammation, systemic insulin resistance, hepatosteatosis, islet hyperplasia, and impaired thermogenesis [24]. In adipose tissue of the knockout mice, macrophage infiltration, inflammation, and cell death are induced, underscoring the importance of α4 in adipose tissue maintenance through the regulation of insulin signaling. Overall, these findings elucidated the intricate interplay between PTP1B and PP2A through the modulatory role of α4, revealing a feedback mechanism that impacts insulin receptor signaling and the functionality of adipose tissues.
Adipose Tissue Regeneration
Until recently, it was generally believed that fat cells were mainly generated during childhood and adolescence, and therefore, fat tissue mass was established before adulthood. However, recent studies have reported evidence that supports the potential for the generation of fat cells in adulthood. Several methods have been used to estimate the turnover rate of adipocytes in mice and humans. One method that involves measuring the concentration of 14C in fat cells, utilizing the changes in atmospheric 14C levels released from nuclear bomb tests, reported that approximately 8.4% of fat cell turnover occurs annually in both lean and obese subjects [25]. The average age of triglycerides in fat cells is approximately 1.6 years [26]. Other studies using long-term labeling with 2H have shown that the half-life of TGs was estimated to be approximately 6 months [27], suggesting a higher turnover rate for fat cells and TGs.
Furthermore, studies using 2H labeling suggested that 0.16–0.29% of fat cells may turnover daily [28]. The basal turnover of adipocytes is very low in rodents but can be accelerated by a high-fat diet (HFD). The turnover differs between visceral and subcutaneous fat, with lineage tracing studies indicating an increase in fat generation in visceral fat within 4 weeks of HFD feeding [29]. The regenerative capacity of adipose tissue has been validated in models known for acute adipocyte removal, such as FAT-ATTAC mice (mice in which fat tissue apoptosis is induced by caspase-8 activation), mice with adipocyte-specific inducible knockout of IR and IGF1R and mice with adipocyte-specific inducible knockout of α4 [14, 24, 30]. These models lead to rapid fat loss, followed by the rapid proliferation and differentiation of preadipocyte cells, resulting in the generation of new BAT and WAT to recover fat tissues and an improvement in metabolic syndrome within 30 days. These results suggest a robust homeostatic mechanism that induces the recovery and regeneration of fat tissue.
Future Perspectives
Brown adipocytes were discovered in 1551 [31], but their thermogenic potential was not revealed until 1961 [32]. The application of fluorodeoxyglucose (18F-FDG) positron-emission tomographic and computed tomographic (PET–CT) scans confirmed the presence of BAT in adults in 2009 [2]. The discovery has reignited research attention, with a focus on the effects of BAT on metabolism and its potential applications in obesity treatment. As BAT mass decreases with age, increasing the abundance and activity of thermogenic adipose tissue holds promise as a strategy to counter metabolic diseases and obesity. Pharmacological or technological interventions are expected to lead to the development of a physiologically valid approach through the activation of BAT to increase energy expenditure, leading to a reduction in visceral fat and the alleviation of obesity. Furthermore, it is essential to elucidate the role of adipocyte turnover in the pathogenesis of insulin resistance and T2D.
Acknowledgements
I sincerely thank all the contributors to the work conducted in the laboratory of Emeritus Professor Eiichi Araki at Kumamoto University and Professor C. Ronald Kahn’s laboratory at the Joslin Diabetes Center, Harvard University, Boston, USA. A summary of this review was presented at the Lilly Award Lecture at the 66th Annual Meeting of the Japan Diabetes Society, Kagoshima, Japan. I would like to express my sincere gratitude to Emeritus Professor Eiichi Araki and Professor C. Ronald Kahn for their mentoring and support. The author was supported by a grant from the Astellas Foundation for Research on Metabolic Disorders and JSPS KAKENHI (JP 18K16208, JP 21K08532).
Declarations
Conflict of Interest
MS declares that he has no conflicts of interest associated with this research.
Human or Animal Rights
This article does not include data collected from any studies involving human or animal subjects.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kahn CR, Wang G, Lee KY. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J Clin Invest. 2019;129(10):3990–4000. doi: 10.1172/JCI129187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cypess AM, Kahn CR. The role and importance of brown adipose tissue in energy homeostasis. Curr Opin Pediatr. 2010;22(4):478–484. doi: 10.1097/MOP.0b013e32833a8d6e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sakers A, De Siqueira MK, Seale P, Villanueva CJ. Adipose-tissue plasticity in health and disease. Cell. 2022;185(3):419–446. doi: 10.1016/j.cell.2021.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harms M, Seale P. Brown and beige fat: development, function and therapeutic potential. Nat Med. 2013;19(10):1252–1263. doi: 10.1038/nm.3361. [DOI] [PubMed] [Google Scholar]
- 5.Cohen P, Kajimura S. The cellular and functional complexity of thermogenic fat. Nat Rev Mol Cell Biol. 2021;22(6):393–409. doi: 10.1038/s41580-021-00350-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Funcke JB, Scherer PE. Beyond adiponectin and leptin: adipose tissue-derived mediators of inter-organ communication. J Lipid Res. 2019;60(10):1648–1684. doi: 10.1194/jlr.R094060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leitner BP, Huang S, Brychta RJ, Duckworth CJ, Baskin AS, McGehee S, et al. Mapping of human brown adipose tissue in lean and obese young men. Proc Natl Acad Sci U S A. 2017;114(32):8649–8654. doi: 10.1073/pnas.1705287114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jespersen NZ, Larsen TJ, Peijs L, Daugaard S, Homoe P, Loft A, et al. A classical brown adipose tissue mRNA signature partly overlaps with brite in the supraclavicular region of adult humans. Cell Metab. 2013;17(5):798–805. doi: 10.1016/j.cmet.2013.04.011. [DOI] [PubMed] [Google Scholar]
- 9.Lidell ME, Betz MJ, Dahlqvist Leinhard O, Heglind M, Elander L, Slawik M, et al. Evidence for two types of brown adipose tissue in humans. Nat Med. 2013;19(5):631–634. doi: 10.1038/nm.3017. [DOI] [PubMed] [Google Scholar]
- 10.Boucher J, Kleinridders A, Kahn CR. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol. 2014 doi: 10.1101/cshperspect.a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batista TM, Haider N, Kahn CR. Defining the underlying defect in insulin action in type 2 diabetes. Diabetologia. 2021;64(5):994–1006. doi: 10.1007/s00125-021-05415-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai W, Sakaguchi M, Kleinridders A, Gonzalez-Del Pino G, Dreyfuss JM, O'Neill BT, et al. Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat Commun. 2017;8:14892. doi: 10.1038/ncomms14892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299(5606):572–574. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 14.Sakaguchi M, Fujisaka S, Cai W, Winnay JN, Konishi M, O'Neill BT, et al. Adipocyte dynamics and reversible metabolic syndrome in mice with an inducible adipocyte-specific deletion of the insulin receptor. Cell Metab. 2017;25(2):448–462. doi: 10.1016/j.cmet.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O'Neill BT, Lee KY, Klaus K, Softic S, Krumpoch MT, Fentz J, et al. Insulin and IGF-1 receptors regulate FoxO-mediated signaling in muscle proteostasis. J Clin Invest. 2016;126(9):3433–3446. doi: 10.1172/JCI86522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Os I, Zhang W, Wasserman DH, Liew CW, Liu J, Paik J, et al. FoxO1 integrates direct and indirect effects of insulin on hepatic glucose production and glucose utilization. Nat Commun. 2015 doi: 10.1038/ncomms8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Homan EP, Brandao BB, Softic S, El Ouaamari A, O'Neill BT, Kulkarni RN, et al. Differential roles of FOXO transcription factors on insulin action in brown and white adipose tissue. J Clin Invest. 2021 doi: 10.1172/JCI143328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakaguchi M, Cai W, Wang CH, Cederquist CT, Damasio M, Homan EP, et al. FoxK1 and FoxK2 in insulin regulation of cellular and mitochondrial metabolism. Nat Commun. 2019;10(1):1582. doi: 10.1038/s41467-019-09418-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sukonina V, Ma H, Zhang W, Bartesaghi S, Subhash S, Heglind M, et al. FOXK1 and FOXK2 regulate aerobic glycolysis. Nature. 2019;566(7743):279–283. doi: 10.1038/s41586-019-0900-5. [DOI] [PubMed] [Google Scholar]
- 20.Goldstein BJ, Ahmad F, Ding W, Li PM, Zhang WR. Regulation of the insulin signalling pathway by cellular protein-tyrosine phosphatases. Mol Cell Biochem. 1998;182(1–2):91–99. doi: 10.1023/A:1006812218502. [DOI] [PubMed] [Google Scholar]
- 21.Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, et al. Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science. 1999;283(5407):1544–1548. doi: 10.1126/science.283.5407.1544. [DOI] [PubMed] [Google Scholar]
- 22.Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, et al. Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol. 2000;20(15):5479–5489. doi: 10.1128/MCB.20.15.5479-5489.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24(5):186–191. doi: 10.1016/S0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- 24.Sakaguchi M, Okagawa S, Okubo Y, Otsuka Y, Fukuda K, Igata M, et al. Phosphatase protector alpha4 (alpha4) is involved in adipocyte maintenance and mitochondrial homeostasis through regulation of insulin signaling. Nat Commun. 2022;13(1):6092. doi: 10.1038/s41467-022-33842-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, et al. Dynamics of fat cell turnover in humans. Nature. 2008;453(7196):783–787. doi: 10.1038/nature06902. [DOI] [PubMed] [Google Scholar]
- 26.Arner P, Bernard S, Salehpour M, Possnert G, Liebl J, Steier P, et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature. 2011;478(7367):110–113. doi: 10.1038/nature10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Strawford A, Antelo F, Christiansen M, Hellerstein MK. Adipose tissue triglyceride turnover, de novo lipogenesis, and cell proliferation in humans measured with 2H2O. Am J Physiol Endocrinol Metab. 2004;286(4):E577–E588. doi: 10.1152/ajpendo.00093.2003. [DOI] [PubMed] [Google Scholar]
- 28.Neese RA, Misell LM, Turner S, Chu A, Kim J, Cesar D, et al. Measurement in vivo of proliferation rates of slow turnover cells by 2H2O labeling of the deoxyribose moiety of DNA. Proc Natl Acad Sci U S A. 2002;99(24):15345–15350. doi: 10.1073/pnas.232551499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang QA, Tao C, Gupta RK, Scherer PE. Tracking adipogenesis during white adipose tissue development, expansion and regeneration. Nat Med. 2013;19(10):1338–1344. doi: 10.1038/nm.3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, et al. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med. 2005;11(7):797–803. doi: 10.1038/nm1262. [DOI] [PubMed] [Google Scholar]
- 31.Gesner C. Conradi gesneri medici tigurini historiae animalium: Lib 1—de quadrupedibus viviparis. Zürich, 1551. p. 842. https://www.e-rara.ch/zuz/doi/10.3931/e-rara-1927.
- 32.Smith RE. Thermogenic activity of hibernating gland in the cold-acclimated rat. Physiologist. 1961;4:113. [Google Scholar]