

Abstract
Maribavir is an oral benzimidazole riboside for treatment of post‐transplant cytomegalovirus (CMV) infection/disease that is refractory to prior antiviral treatment (with or without resistance). Through competitive inhibition of adenosine triphosphate, maribavir prevents the phosphorylation actions of UL97 to inhibit CMV DNA replication, encapsidation, and nuclear egress. Maribavir is active against CMV strains with viral DNA polymerase mutations that confer resistance to other CMV antivirals. After oral administration, maribavir is rapidly and highly absorbed (fraction absorbed >90%). The approved dose of 400 mg twice daily (b.i.d.) achieves a steady‐state area under the curve per dosing interval of 128 h*μg/mL and trough concentration of 4.90 μg/mL (13.0 μM). Maribavir is highly bound to human plasma proteins (98%) with a small apparent volume of distribution of 27.3 L. Maribavir is primarily cleared by hepatic CYP3A4 metabolism; its major metabolite, VP44669 (pharmacologically inactive), is excreted in the urine and feces. There is no clinically relevant impact on maribavir pharmacokinetics by age, sex, race/ethnicity, body weight, transplant type, or hepatic/renal impairment status. In phase II dose‐ranging studies, maribavir showed similar rates of CMV viral clearance across 400, 800, or 1200 mg b.i.d. groups, ranging from 62.5–70% in study 202 (NCT01611974) and 74–83% in study 203 (EudraCT 2010–024247‐32). In the phase III SOLSTICE trial (NCT02931539), maribavir 400 mg b.i.d. demonstrated superior CMV viremia clearance at week 8 versus investigator‐assigned treatments, with lower treatment discontinuation rates. Dysgeusia, nausea, vomiting, and diarrhea were commonly experienced adverse events among patients treated with maribavir in clinical trials.
Clinical and Translational Card for Maribavir.
Mechanism of action: Competitive inhibitor of human CMV enzyme pUL97, a serine/threonine protein kinase that phosphorylates substrates involved in CMV DNA replication, capsid assembly, and nuclear egress.
Indication: Treatment of adults (and adolescents ≥12 years of age weighing ≥35 kg in US only) with post‐transplant CMV infection/disease that is refractory (with or without resistance) or intolerant (Australia and Taiwan only) to treatment with one or more prior antiviral therapies, including ganciclovir, valganciclovir, cidofovir, or foscarnet.
Dosage and administration: 400 mg (two 200 mg tablets) taken orally b.i.d. with or without food.
Metabolic pathways: Hepatic CYP3A4 (major); hepatic CYP1A2 (minor).
- Key pharmacokinetic characteristics:
-
⚬AUC0−tau, geometric mean (%CV), μg*h/mL: 128 (50.7%)a
-
⚬C max, geometric mean (%CV), μg/mL: 17.2 (39.3%)a
-
⚬T max, median, h: 1.0 to 3.0
-
⚬t 1/2, mean, h: 4.32b
-
⚬
aValues based on post hoc estimates from maribavir population pharmacokinetic model in transplant patients with CMV infection receiving 400 mg of maribavir b.i.d. with or without food.
bIn transplant patients.
Abbreviations: AUC0 −tau, area under the time concentration curve over a dosing interval; b.i.d., twice daily; C max, maximum concentration; CMV, cytomegalovirus; CV, coefficient of variation; DNA, deoxyribonucleic acid; T max, time to C max; t 1/2, terminal half life
INTRODUCTION
Cytomegalovirus (CMV) infection is common worldwide, and in healthy people is generally asymptomatic due to a robust immune response. 1 In immunocompromised transplant recipients, however, CMV infection—either transmitted from the donor tissue or re‐activated from latent infection in the recipient—can cause CMV‐related end‐organ disease, such as pneumonitis, retinitis, gastroenteritis, hepatitis, and esophagitis, as well as organ failure. 1 , 2
Maribavir is indicated for the treatment of adults (and in the United States only, adolescents 12 years of age or older weighing 35 kg or more) with post‐transplant CMV infection/disease that is refractory (or intolerant in Australia and Taiwan) to treatment (with or without resistance) with one or more prior antiviral therapies, including ganciclovir, valganciclovir, cidofovir, or foscarnet. 3 , 4 , 5 , 6
DRUG REGULATORY APPROVAL
Maribavir first gained regulatory approval in the United States (US Food and Drug Administration) in November 2021, 3 which was followed by approvals in Canada (Health Canada) in September 2022, 5 Australia (Therapeutic Goods Administration) in October 2022, 6 the European Union (European Commission/European Medical Agency) and United Kingdom (Medicines and Healthcare products Regulatory Agency) in November 2022, 4 and South Korea (Korean Ministry of Food and Drug Safety) in December 2022. Further regulatory reviews in other countries and regions are pending at the time of this review.
MECHANISM OF ACTION
The CMV virion comprises a double‐stranded DNA genome enclosed in a protein capsid, which is surrounded by a layer of tegument proteins and finally a lipid envelope. 7 As shown in Figure 1a, CMV virions enter host target cells either by fusion or by endocytosis in the case of host endothelial and epithelial cells. 1 The capsid and tegument proteins released into the cytoplasm move to the host cell nucleus and the CMV DNA transitions into the nucleus. 1 Following the replication of CMV DNA, the viral genome is encapsulated into capsids that exit the nucleus via nuclear egress. 2 , 8 The capsids form into mature virions in the cell cytoplasm by gaining tegument proteins and acquiring a glycoprotein envelope from the trans‐Golgi network. 2 , 8 Mature virions are then released into the extracellular space via exocytosis.
FIGURE 1.
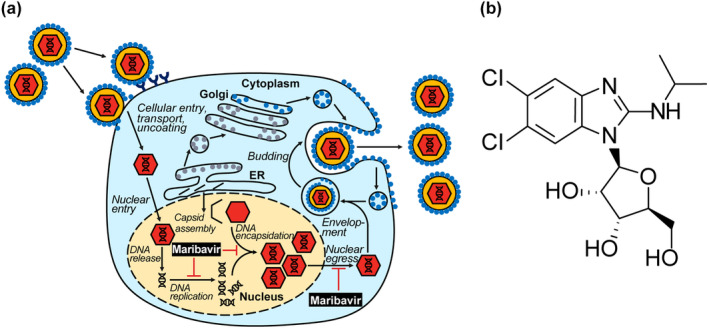
Mechanism of action of maribavir (a) and chemical structure (b). Maribavir is a benzimidazole riboside with a selective multimodal mechanism of action against human CMV. Maribavir is a competitive inhibitor of the CMV protein kinase UL97 at the adenosine triphosphate binding site and thus prevents the phosphorylation action of UL97, thereby inhibiting CMV DNA replication, CMV DNA encapsidation, and nuclear egress of viral capsids. CMV, cytomegalovirus; ER, endoplasmic reticulum.
Maribavir is a benzimidazole riboside antiviral agent (C15H19Cl2N3O4, molecular weight of 376.23 Da) 3 with a selective multimodal mechanism of action against human CMV (Figure 1a,b). 9 Maribavir targets UL97, a serine/threonine protein kinase that phosphorylates substrates involved in CMV DNA replication, capsid assembly, and nuclear egress. 10 , 11 Maribavir attaches to the UL97 adenosine triphosphate (ATP) binding site and competitively inhibits ATP (half maximal inhibitory concentration of 0.003 μM), thereby preventing phosphorylation required for CMV DNA replication, CMV DNA encapsidation, and nuclear egress of viral capsids. 3 , 9 , 10
Commonly utilized CMV antiviral agents, ganciclovir, valganciclovir, cidofovir, or foscarnet work by inhibiting CMV DNA polymerase UL54, and mutations in the UL54 gene confer CMV resistance to these agents. 12 Ganciclovir requires activation through phosphorylation by UL97, and UL97 mutations that decrease the phosphorylation of ganciclovir confer resistance to this drug. 12 Due to being an inhibitor of UL97 at the ATP‐binding site, maribavir is active against CMV strains with UL54 mutations and some of the UL97 mutations that confer resistance to ganciclovir. 9 In cell culture models, maribavir selectively inhibited CMV replication in yield reduction, DNA hybridization, and plaque reduction assays, with a mean half‐maximal effective concentration (EC50) of 0.11 μM, and EC50 range of 0.03 to 0.31 μM. 5 The cell culture antiviral activity of maribavir has also been evaluated against 10 CMV clinical isolates using DNA hybridization and plaque reduction assays, with median EC50 values of 0.1 μM (range, 0.03–0.13 μM) and 0.28 μM (range, 0.12–0.56 μM), respectively. 3 , 13 Maribavir has been shown to be effective at inhibiting replication in CMV variants that are resistant to ganciclovir, foscarnet, or cidofovir, as well as variants resistant to combinations of these drugs (EC50 range, 0.06–0.32 μM), 14 enabling maribavir to treat CMV strains that are resistant to conventional therapies.
Some strains of CMV show resistance toward maribavir, in cell culture and in patients. In cell culture, CMV variants with amino‐acid substitutions clustered around the ATP‐binding site region of the UL97 gene, including L337M, V353A, L397R, T409M, and H411L/N/Y, have been shown to confer reduced susceptibility to maribavir in a range from 3.5‐fold to greater than 200‐fold. Substitutions identified in the UL27 gene have been shown to reduce susceptibility by 1.7‐ to 4.8‐fold. Treatment‐emergent maribavir‐resistant strains associated with substitutions in UL97 (F342Y, T409M, H411L/Y, and C480F) have been identified in clinical trials. 3 In the phase III SOLSTICE study (ClinicalTrials.gov NCT02931539), treatment‐emergent maribavir resistance mutations were detected in 26% of patients randomized to maribavir, mostly in nonresponders, including in those with a viral load rebound while on therapy. The most common mutations were UL97 T409M, H411Y, and C480F, and mutations were first detected 26 to 130 (median 56) days after starting maribavir. The majority of patients who developed maribavir resistance went on to achieve viremia clearance after receiving alternative therapy. 15
The availability of antiviral agents with differing mechanistic targets raises the possibility of combination therapy to overcome viral resistance. Maribavir has additive interactions against wild‐type CMV and CMV variants with letermovir, foscarnet, and cidofovir; but a strong antagonism with ganciclovir, because ganciclovir activity is dependent on phosphorylation by UL97. 16
PHARMACOKINETIC AND PHARMACODYNAMIC CHARACTERISTICS
Absorption, distribution, metabolism, and excretion properties
Most drugs for treatment of CMV infection, such as ganciclovir, cidofovir, or foscarnet, require intravenous (i.v.) administration; valganciclovir, a prodrug of ganciclovir, has moderate bioavailability after in vivo bioconversion to ganciclovir. 17 To minimize the need for i.v. infusions, as well as enzymatic conversion to active agent, maribavir was developed for oral dosing with a direct inhibitory mechanism on CMV.
After oral administration, maribavir is rapidly absorbed, with a median time to reach maximum plasma concentration of 1–3 h postdose 3 , 4 , 18 ; the fraction absorbed is greater than 90%. In healthy volunteers, maribavir demonstrated dose‐proportional pharmacokinetics up to 2400 mg. 3 , 18 The area under the curve (AUC) of maribavir, which is most relevant to the antiviral efficacy, is not impacted by co‐administration with food, although a high‐fat diet decreases the peak concentration in plasma (C max) of maribavir by ~28% relative to the fasted state. 4 , 18 The oral absorption of maribavir is not significantly affected by crushing the tablet or by increases in gastric pH. 4 , 18
Maribavir has a relatively small volume of distribution (mean apparent steady‐state volume of distribution of 27.3 L). 3 , 4 It is highly bound to human plasma proteins (98%) in both healthy subjects and transplant recipients. 3 , 4 , 18 Based on nonclinical studies, maribavir may cross the blood–brain barrier, but penetration into the central nervous system is expected to be low compared with plasma levels. 4 Maribavir has been shown to cross the blood–retinal barrier, with a vitreous human‐to‐plasma ratio of up to 0.28. 18
Maribavir is primarily cleared through hepatic metabolism, with renal clearance accounting for less than 2% of the total clearance. 3 , 18 Maribavir is metabolized by CYP3A4 (70%–85% of hepatic microsomal CYP pathways) and CYP1A2 (15%–30%) to form the major de‐alkylated metabolite, VP44469, which is inactive against CMV UL97; VP44469 is excreted in urine and feces. 3 , 4 , 18 , 19 Direct glucuronidation of maribavir is a minor metabolic pathway. 4 , 19 The mean observed half‐life of maribavir in transplant recipients is ~4.3 h, which is similar to that in healthy volunteers (3.9 h). 3 , 4 , 20
Following 400 mg twice‐daily (b.i.d.) doses in transplant patients with CMV infections, steady‐state exposure of maribavir was reached within 2 days of dosing. The geometric mean steady‐state (% coefficient of variation for geometric mean) values for AUC during the dosing interval (AUC0−τ), C max, and observed plasma concentration at the end of a dosing interval were 128 (50.7%) μg*h/mL, 17.2 (39.3%) μg/mL, and 4.90 (89.7%) μg/mL (13.0 μM), respectively. 3 , 4 Accumulation was low with b.i.d. dosing, with predicted accumulation ratios of 1.47 and 1.37 for AUC and C max, respectively. 3 , 4
Drug–drug interactions
Effect of other drugs on maribavir
As the primary metabolic pathway of maribavir is through CYP3A, co‐administration of maribavir with CYP3A4 inhibitors or inducers may affect its clearance and exposure. 4 Although maribavir exposure may be increased with co‐administration of CYP3A inhibitors, dose adjustment is not needed given maribavir's wide therapeutic window (i.e., similar anti‐CMV efficacy at doses 400–1200 mg b.i.d. and similar rate of dysgeusia [the most common adverse event; AE] at less than or equal to 1200 mg b.i.d.). 4 , 21 , 22 In contrast, without dose adjustment, the decreased exposure of maribavir from concomitant administration of strong or moderate CYP3A4 inducers may reduce antiviral efficacy. 4 For this reason, co‐administration of maribavir with rifampicin, rifabutin, or St. John's wort is not recommended, and co‐administration with other common CYP3A4 inducers requires a maribavir dose increase to 800 mg b.i.d. (for carbamazepine) or 1200 mg b.i.d. (for efavirenz, phenobarbital, or phenytoin). 3 , 4
Effect of maribavir on other drugs
For pharmacodynamic‐based drug–drug interactions (DDIs), co‐administration of valganciclovir or ganciclovir with maribavir is contraindicated, as the antiviral activity of ganciclovir is dependent on phosphorylation by the CMV UL97 protein kinase and so may be antagonized by maribavir's inhibition of UL97. 4 For pharmacokinetic‐based DDIs, maribavir has been shown to potentially inhibit the P‐gp transporter and the BCRP transporter at clinically relevant concentrations. Plasma levels of digoxin, a P‐gp transporter substrate, increased when co‐administered with maribavir (AUC and C max ratios 1.21 and 1.25, respectively); caution should be exercised when maribavir and sensitive P‐gp substrates, such as digoxin and dabigatran, are taken together. Co‐administration of maribavir with BCRP substrates, such as rosuvastatin, might increase their exposure and patients should be closely monitored for occurrences of myopathy and rhabdomyolysis. 4 In another DDI study, the AUC and C max of tacrolimus increased by 51% and 38%, respectively, following co‐administration with maribavir, possibly due to inhibition of CYP3A4 activity and/or P‐glycoprotein. 23 When co‐administered with maribavir, levels of tacrolimus and other immunosuppressants, such as cyclosporine, everolimus, or sirolimus, should be frequently monitored. 4
In vitro, maribavir was an inducer of CYP1A2; therefore, in the absence of data from clinical studies, co‐administration of maribavir with CYP1A2 substrates with a narrow therapeutic window, such as tizanidine and theophylline, should be avoided. 4 In vitro, maribavir inhibited the OAT3 and MATE1 transporters. Plasma concentrations of sensitive substrates of OAT3 (such as ciprofloxacin, imipenem, and cilastin) or MATE1 (metformin) may be increased. 4
Population pharmacokinetics
A population pharmacokinetic (PopPK) model for maribavir was developed using nonlinear mixed‐effects modeling to describe its plasma concentration time course after oral administration. Pharmacokinetic data were collated from participants enrolled in nine phase I studies, two phase II studies, and one phase III study, which included healthy volunteers and recipients of hematopoietic stem cell transplant (HSCT) or solid organ transplant (SOT) with CMV infection. 24 , 25 The pharmacokinetic profiles of maribavir were adequately described by a two‐compartment model with first‐order absorption and elimination, and an absorption lag time. Covariates included body weight for clearance and volume terms, “transplant recipients with CMV” and moderate/strong CYP3A4 inhibitor or inducer as categorical covariates for apparent oral clearance (CL/F), and maribavir dose for the rate of absorption. 24 The parameter estimates of the PopPK model are presented in Table S1.
Overall, the PopPK model showed that CL/F was 24% lower for transplant patients with CMV infection compared with all other participants, and AUC and C max were 27% and 5% higher, respectively, in transplant patients with CMV infection than in healthy volunteers. 24 , 25 These differences are not considered clinically relevant. No clinically relevant impact on maribavir pharmacokinetics related to age (18–79 years), sex, race (White, Black, Asian, or others), ethnicity (Hispanic/Latino, or non‐Hispanic/Latino), or body weight was identified (Figure S1). Transplant types (i.e., HSCT vs. SOT; or among SOT types of liver, lungs, kidneys, or heart) (Figure S2), graft‐versus‐host disease, or diarrhea did not impact the pharmacokinetics of maribavir. 25
Pharmacokinetics in special populations
In the United States, maribavir is approved for the treatment of CMV infection in adolescents (aged 12 years or older and weighing 35 kg or more) based on evidence from clinical trials in adults as well as PopPK modeling and simulation. As age and body weight showed no clinically meaningful effect on maribavir exposures, simulations projected similar maribavir exposure between adults and adolescents aged 12 years or older and weighing 35 kg or more. 3 , 24
In addition, no dosage adjustment is required for individuals with mild (Child‐Pugh Class A) or moderate (Child‐Pugh Class B) hepatic impairment; mild, moderate, or severe renal impairment; or in patients greater than 65 years, based on pharmacokinetic studies in these populations, as well as the lack of effect of age in the PopPK model. Maribavir has not been studied in patients with severe hepatic impairment or end‐stage renal disease. 3 , 4
Exposure–response relationship on safety and anti‐CMV efficacy
Logistic regression models were used to evaluate associations between steady‐state pharmacokinetic exposure of maribavir and efficacy endpoints and safety parameters using data from 231 HSCT/SOT recipients in the phase III SOLSTICE study (ClinicalTrials.gov NCT02931539). For the efficacy analysis, the primary end point of confirmed CMV viremia clearance at the end of week 8 and the key secondary efficacy end point of confirmed CMV viremia clearance and symptom control at week 8 maintained through week 16 were evaluated. Key treatment‐emergent adverse events (AEs) based on frequency (>10% of patients), severity, or special interest data obtained during the 8‐week treatment phase were also selected for exposure–safety analysis: dysgeusia, nausea, neutropenia, diarrhea, vomiting, viral infections, anemia, fatigue, pyrexia, renal disorder, increase in immunosuppressant drug concentration level, and thrombocytopenia. These analyses indicated that there were no clinically meaningful relationships between interpatient maribavir exposure and the probability of achieving primary or key secondary efficacy end points or the probability of dysgeusia, fatigue, increase in immunosuppressant drug concentration, or serious AEs. 25
KEY CLINICAL TRIALS
Four clinical studies, including two dose‐ranging safety and efficacy phase II studies (study 202, NCT01611974; and study 203, EUDRACT 2010–024247‐32) and two active‐controlled phase III studies (SOLSTICE, NCT02931539; and AURORA, NCT02927067) have been conducted to evaluate maribavir in SOT and/or HSCT recipients with documented CMV infection. 21 , 22 , 26 , 27 Study details are provided in Table 1. In addition, a phase III open‐label, single‐arm, repeated‐dose study to evaluate the safety and tolerability, pharmacokinetics, and antiviral activity of maribavir for the treatment of CMV infection in children and adolescents who have received an HSCT or SOT (study 2004, NCT05319353) is planned. 28
TABLE 1.
Completed key clinical trials investigating the efficacy and safety of maribavir in the treatment of cytomegalovirus infection in adults and adolescents.
| Study | Trial design | Treatment duration/follow‐up period | Dose(s) | Study population | Number of subjects |
|---|---|---|---|---|---|
| NCT01611974 21 (Study 202) | Phase II, randomized, double‐blind, multicenter, dose‐ranging | Treatment up to 24 weeks | Maribavir 400, 800, or 1200 mg b.i.d. | HSCT/SOT recipients, ≥12 years of age a with R/R CMV infection and plasma CMV DNA ≥1000 copies/mL |
N = 120 randomized; 47 HCT and 73 SOT
|
| EUDRACT 2010–024247‐32 22 (Study 203) | Phase II, randomized, open‐label, dose‐ranging, dose‐blind, multicenter, active‐controlled | Treatment up to 12 weeks and post‐treatment follow‐up of 12 weeks |
Maribavir 400, 800, or 1200 mg b.i.d. Valganciclovir 900 mg b.i.d. weeks 1–3 and q.d. after 3 weeks |
HSCT/SOT recipients, ≥18 years of age, with CMV reactivation and plasma CMV DNA 1000–100,000 copies/mL |
N = 161 randomized
N = 159 received treatment; 82 HCT and 77 SOT
|
| NCT02931539 26 (SOLSTICE) | Phase III, randomized, open‐label, multicenter, active‐controlled | 8‐weeks treatment and post‐treatment follow‐up of 12 weeks |
Maribavir: 400 mg b.i.d. IAT (foscarnet, ganciclovir, valganciclovir, cidofovir, foscarnet/valganciclovir, foscarnet/ganciclovir) |
HSCT and SOT recipients, aged ≥12 years a with refractory CMV infection with or without resistance and plasma CMV DNA at screening of ≥910 IU/mL |
N = 352 randomized; 141 HCT and 211 SOT
|
|
(AURORA) |
Phase III, randomized, double‐blind, double‐dummy, multicenter, active‐controlled | 8‐weeks treatment and post‐treatment follow‐up of 12 weeks |
Maribavir 400 mg b.i.d. Valganciclovir 900 mg b.i.d. Placebo matched to either maribavir or valganciclovir b.i.d. |
HSCT recipients aged ≥16 years with first post‐transplant CMV infection with plasma CMV DNA at screening of ≥455 IU/mL to ≤91,000 IU/mL |
N = 533 recipients randomized
|
Abbreviations: b.i.d., twice daily; CMV, cytomegalovirus; HSCT, hematopoietic stem cell transplant; IAT, investigator‐assigned treatment; q.d., once daily; R/R, refractory/resistant; SOT, solid organ transplant.
Although entry criteria permitted enrollment of children (≥12 years of age), only adults (≥18 years of age) were enrolled into these studies.
In the two phase II, dose‐ranging studies, patients were treated with maribavir 400 mg, 800 mg or 1200 mg b.i.d. for up to 24 weeks (study 202) 21 or up to 12 weeks with a 12‐week follow‐up (study 203). 22 In the phase III SOLSTICE and AURORA trials, patients were treated with maribavir 400 mg b.i.d. for 8 weeks with a 12‐week follow‐up. 26 , 27 Three studies had active comparators, valganciclovir in study 203 22 and AURORA 27 and investigator‐assigned treatment (IAT; ganciclovir, valganciclovir, foscarnet, or cidofovir, as mono or combination therapy, at the investigators' discretion) in SOLSTICE. 26
Primary efficacy end points in studies 202 and 203 were proportions of patients with a response to treatment defined as undetectable levels of CMV DNA in the plasma within 3 22 or 6 21 , 22 weeks after the start of treatment. Secondary efficacy analyses included the time to the first undetectable plasma CMV DNA, proportion of patients with CMV recurrence, and time to first CMV recurrence. 21 , 22 For SOLSTICE and AURORA, the primary end point was percentage of patients with confirmed CMV viremia clearance at the end of week 8, and the key secondary end point was a composite of confirmed CMV viremia clearance and symptom control at the end of week 8, maintained through week 16. 26 , 27
CLINICAL EFFICACY AND SAFETY
An overview of the efficacy and safety outcomes from three published clinical trials (study 202, study 203, and SOLSTICE) evaluating maribavir as a treatment for CMV infection in SOT and/or HSCT recipients is provided in Tables 2 and 3, respectively. Study results from AURORA have yet to be published at the time of writing this review.
TABLE 2.
Summary of key efficacy results from phase II and III studies evaluating maribavir for treatment of cytomegalovirus infection.
| Study | Primary efficacy result | Secondary efficacy results |
|---|---|---|
| Phase II | ||
| NCT01611974 21 (study 202) | Patients with confirmed undetectable plasma CMV DNA within 6 weeks, maribavir all doses combined % [95% CI], (n/N)
|
Patients with CMV recurrence, n/N (%; 95% CI)
Probability of CMV recurrence
|
| EUDRACT 2010–024247‐32 22 (study 203) |
Patients with confirmed undetectable plasma CMV DNA within 3 weeks, maribavir all doses combined vs. valganciclovir % [95% CI], (n/N)
Patients with confirmed undetectable plasma CMV DNA within 6 weeks, maribavir all doses combined vs. valganciclovir % [95% CI], (n/N)
|
Median (95% CI) time to the first undetectable plasma CMV DNA within 6 weeks, days
Patients with CMV recurrence, maribavir all doses combined vs. valganciclovir, n/N (%; 95% CI)
Median time from undetectable plasma CMV DNA to first CMV recurrence, days
|
| Phase III | ||
| NCT02931539 26 (SOLSTICE) | Patients with confirmed CMV viremia clearance at 8 weeks, % (n/N)
|
Key secondary end point: Confirmed CMV viremia clearance and symptom control at the end of week 8, maintained through week 16, %
|
Abbreviations: CI, confidence interval; CMV, cytomegalovirus; IAT, investigator‐assigned treatment.
TABLE 3.
Summary of key safety results from phase II and phase III studies evaluating maribavir for treatment of cytomegalovirus infection.
| Study | Safety | |||
|---|---|---|---|---|
| Phase II | ||||
| NCT01611974 21 (study 202) a | Maribavir 400, 800, 1200 mg doses combined (N = 120) | |||
| Discontinuation due to AE 34.2% (n = 41) | ≥1 SAE 67.5% (n = 81) | Deaths 26.7% (n = 32) | Most common AEs (≥20%):
|
|
| EUDRACT 2010–024247‐32 22 (Study 203) a | Maribavir 400, 800, 1200 mg doses combined (N = 119) and valganciclovir (N = 40) | |||
|
Discontinuation due to AE
|
SAE
|
Deaths
|
Most common AEs (≥20%), maribavir vs. valganciclovir:
|
|
| Phase III | ||||
| NCT02931539 26 (SOLSTICE) b | Maribavir 400 mg (N = 234) and IAT (N = 116) | |||
Discontinuation due to AE
|
SAE
|
Most common AEs (≥15%), maribavir vs. IAT:
|
||
Abbreviations: AE, adverse event; CMV, cytomegalovirus; IAT, investigator‐assigned treatment; SAE, serious adverse event.
AEs and SAEs that occurred or worsened during treatment were defined as those that occurred during the period from the start of study treatment through 7 days after the last dose.
AEs and SAEs during on‐treatment observation period, which started at the time of study‐assigned treatment initiation through 7 days after the last dose of study‐assigned treatment or through 21 days if cidofovir was used, or until the maribavir rescue treatment initiation or until the non‐study CMV treatment initiation, whichever was earlier.
Deaths during overall study period including death due to relapse of Hodgkin's disease that occurred 3 days prior to initiation of study treatment.
Deaths during overall study period including one patient who had onset of fatal AE on day 3 of maribavir rescue therapy.
In study 202, maribavir showed similar clinical efficacy in terms of CMV viral clearance across the maribavir dose groups with 70%, 62.5%, and 67.5% of patients treated with maribavir 400, 800, or 1200 mg b.i.d., respectively, having undetectable plasma CMV DNA at week 6. 21 These data were consistent with the high rates of CMV clearance at week 6 in study 203, which again were similar across the maribavir 400, 800, or 1200 mg b.i.d. dose groups at 79%, 83%, and 74%, respectively. The rates of viral clearance at 6 weeks observed for maribavir were all numerically higher than those observed for valganciclovir at 67%. 22
Based on the safety and efficacy results from the dose‐finding phase II studies, maribavir 400 mg b.i.d. was selected for further efficacy and safety evaluation in the phase III studies. The SOLSTICE trial included HSCT/SOT recipients with refractory CMV infection (with or without resistance to CMV antiviral agents) and compared maribavir 400 mg b.i.d. versus IAT (ganciclovir, valganciclovir, foscarnet, or cidofovir). A significantly higher proportion of patients treated with maribavir achieved confirmed CMV viremia clearance at week 8 than in the IAT group (55.7% [131/235] vs. 23.9% [28/117]; adjusted difference: 32.8%; p < 0.001), demonstrating superiority of maribavir to IAT in viremia clearance at this timepoint. 26 In the second phase III study (AURORA), maribavir was compared against valganciclovir as a pre‐emptive treatment for CMV infection in HSCT recipients. Results from this study have not yet been published.
In terms of tolerability, discontinuation due to AEs ranged from 23% to 34% of patients for all maribavir doses combined in the phase II studies, and was 13.2% among patients treated with maribavir 400 mg b.i.d. in SOLSTICE. 21 , 22 , 26 Patients in the maribavir group in SOLSTICE had a lower treatment discontinuation rate (13.2%) than those treated with IAT (31.9%). In SOLSTICE, maribavir had a lower frequency of neutropenia than valganciclovir/ganciclovir (maribavir 1.7% vs. valganciclovir/ganciclovir 25.0%) and acute kidney injury versus foscarnet (maribavir 1.7% vs. foscarnet 19.1%), which are treatment‐limiting toxicities frequently associated with these therapies. 26
Dysgeusia was common among patients treated with maribavir, with 26–60% of patients treated with the recommended maribavir dose of 400 mg b.i.d. experiencing this AE across trials; but it rarely led to treatment discontinuation. Gastrointestinal AEs of nausea, vomiting, and diarrhea were also frequently reported AEs with maribavir treatment (Table 3). 21 , 22 , 26
In summary, maribavir has demonstrated clinically meaningful efficacy against CMV infection in clinical trials, with significantly higher rates of CMV viral clearance in HSCT and SOT recipients with refractory CMV infection (with or without resistance) than conventional CMV therapies. 26 The most common AEs reported in patients receiving maribavir in clinical trials were dysgeusia, nausea, vomiting, and diarrhea. Maribavir was associated with lower rates of neutropenia and acute kidney injury than valganciclovir/ganciclovir or foscarnet, respectively.
Main clinical pearls for a clinician to consider when prescribing maribavir:
Maribavir is dosed orally for patient convenience, associated with a lower frequency of neutropenia than ganciclovir/valganciclovir, and active against CMV strains that are resistant to other CMV antivirals.
Dysgeusia, the most frequently reported AE in patients receiving maribavir in clinical trials, was mostly reported as mild and usually resolved either on or shortly after treatment.
Co‐administration of maribavir with ganciclovir or valganciclovir is not recommended.
When co‐administered with maribavir, levels of tacrolimus and other immunosuppressants, such as cyclosporine, everolimus, or sirolimus, should be frequently monitored.
Co‐administration of maribavir with rifampin or St. John's wort is not recommended. When maribavir is co‐administered with other strong or moderate CYP3A4 inducers, maribavir dosage should be increased to up to 1200 mg b.i.d.
Please refer to local prescribing information when co‐administering maribavir with other drugs.
FUNDING INFORMATION
This work was supported by Takeda Development Center Americas, Inc., Lexington, MA, USA.
CONFLICT OF INTEREST STATEMENT
All authors are employees of Takeda Development Center Americas, Inc., and receive stock (and stock options) from Takeda.
Supporting information
Data S1
ACKNOWLEDGMENTS
Under the direction of the authors, medical writing support was provided by Joanne Vaughan, Excel Medical Affairs (Fairfield, CT, USA), and was funded by Takeda Development Center Americas, Inc., Lexington, MA, USA.
Sun K, Fournier M, Sundberg AK, Song IH. Maribavir: Mechanism of action, clinical, and translational science. Clin Transl Sci. 2024;17:e13696. doi: 10.1111/cts.13696
REFERENCES
- 1. Griffiths P, Reeves M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat Rev Microbiol. 2021;19:759‐773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Steingruber M, Marschall M. The cytomegalovirus protein kinase pUL97: host interactions, regulatory mechanisms and antiviral drug targeting. Microorganisms. 2020;8:515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takeda Pharmaceuticals America Inc . LIVTENCITY US prescribing information. Accessed May 24, 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/215596lbl.pdf
- 4. Takeda Pharmaceuticals International AG Ireland Branch . LIVTENCITY summary of product characteristics (EMA). Accessed May 24, 2023. https://www.ema.europa.eu/en/documents/product‐information/livtencity‐epar‐product‐information_en.pdf
- 5. Takeda Canada Inc . LIVTENCITY product monograph including patient medication information. Accessed May 24, 2023. https://assets‐dam.takeda.com/raw/upload/v1666348510/legacy‐dotcom/siteassets/en‐ca/home/what‐we‐do/our‐medicines/product‐monographs/livtencity/LIVTENCITY‐PM‐Eng.pdf
- 6. Takeda Pharmaceuticals Australia Pty Ltd . LIVTENCITY Australia product information. Accessed May 24, 2023. https://www.tga.gov.au/resources/artg/380132
- 7. Kalejta RF. Tegument proteins of human cytomegalovirus. Microbiol Mol Biol Rev. 2008;72:249‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Draganova EB, Thorsen MK, Heldwein EE. Nuclear egress. Curr Issues Mol Biol. 2021;41:125‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Halpern‐Cohen V, Blumberg EA. New perspectives on antimicrobial agents: maribavir. Antimicrob Agents Chemother. 2022;66:e0240521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shannon‐Lowe CD, Emery VC. The effects of maribavir on the autophosphorylation of ganciclovir resistant mutants of the cytomegalovirus UL97 protein. Herpesviridae. 2010;1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Prichard MN. Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir. Rev Med Virol. 2009;19:215‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chou S. Approach to drug‐resistant cytomegalovirus in transplant recipients. Curr Opin Infect Dis. 2015;28:293‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Biron KK, Harvey RJ, Chamberlain SC, et al. Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a benzimidazole L‐riboside with a unique mode of action. Antimicrob Agents Chemother. 2002;46:2365‐2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Drew WL, Miner RC, Marousek GI, Chou S. Maribavir sensitivity of cytomegalovirus isolates resistant to ganciclovir, cidofovir or foscarnet. J Clin Virol. 2006;37:124‐127. [DOI] [PubMed] [Google Scholar]
- 15. Chou S, Alain S, Cervera C, et al. Drug resistance assessed in a phase 3 clinical trial of maribavir therapy for refractory or resistant cytomegalovirus infection in transplant recipients. J Infect Dis. 2023:jiad293. 10.1093/infdis/jiad293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chou S, Ercolani RJ, Derakhchan K. Antiviral activity of maribavir in combination with other drugs active against human cytomegalovirus. Antivir Res. 2018;157:128‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Genentech USA, Inc . Valcyte US prescribing information. Accessed May 24, 2023. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/021304s012,022257s007lbl.pdf
- 18. Song I, Ilic K, Sun K, Martin P. Clinical pharmacology of maribavir (SHP620): a comprehensive overview. Biol Blood Marrow Transplant. 2019;25:S342. [Google Scholar]
- 19. Song I, Sun K, Ilic K, Martin P. Summary of maribavir (SHP620) drug–drug interactions based on accumulated clinical and nonclinical data. Biol Blood Marrow Transplant. 2019;25:S370‐S371. [Google Scholar]
- 20. Wang LH, Peck RW, Yin Y, Allanson J, Wiggs R, Wire MB. Phase I safety and pharmacokinetic trials of 1263 W94, a novel oral anti‐human cytomegalovirus agent, in healthy and human immunodeficiency virus‐infected subjects. Antimicrob Agents Chemother. 2003;47:1334‐1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Papanicolaou GA, Silveira FP, Langston AA, et al. Maribavir for refractory or resistant cytomegalovirus infections in hematopoietic‐cell or solid‐organ transplant recipients: a randomized, dose‐ranging, double‐blind, phase 2 study. Clin Infect Dis. 2019;68:1255‐1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maertens J, Cordonnier C, Jaksch P, et al. Maribavir for preemptive treatment of cytomegalovirus reactivation. N Engl J Med. 2019;381:1136‐1147. [DOI] [PubMed] [Google Scholar]
- 23. Pescovitz MD, Bloom R, Pirsch J, Johnson J, Gelone S, Villano SA. A randomized, double‐blind, pharmacokinetic study of oral maribavir with tacrolimus in stable renal transplant recipients. Am J Transplant. 2009;9:2324‐2330. [DOI] [PubMed] [Google Scholar]
- 24. Sun K, Hayes S, Farrell C, Song IH. Population pharmacokinetic modeling and simulation of maribavir to support dose selection and regulatory approval in adolescents with posttransplant refractory cytomegalovirus. CPT Pharmacometrics Syst Pharmacol. 2023;12:719‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Song I, Chen G, Hayes S, et al. Population pharmacokinetics and exposure–response relationships of maribavir in transplant recipients with cytomegalovirus infection. Poster Presented at Transplantation & Cellular Therapy Meetings of ASTCT and CIBMTR. April 23–26, 2022, Salt Lake City, UT . Transplant Cell Ther. 2022;28: S368‐S369. [Google Scholar]
- 26. Avery RK, Alain S, Alexander BD, et al. Maribavir for refractory cytomegalovirus infections with or without resistance post‐transplant: results from a phase 3 randomized clinical trial. Clin Infect Dis. 2022;75:690‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. ClinicalTrials.gov . A study of maribavir compared to valganciclovir to treat cytomegalovirus infections in people who have received stem cell transplants. Accessed April, 2023. https://clinicaltrials.gov/ct2/show/results/NCT02927067
- 28. ClinicalTrials.gov . A study to evaluate the safety and tolerability, pharmacokinetics, and antiviral activity of maribavir for the treatment of children and teenage transplant recipients with CMV infection. https://clinicaltrials.gov/ct2/show/NCT05319353. Accessed April, 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1