

Abstract
PARP enzymes create ADP-ribose modifications to regulate multiple facets of human biology, and some prominent PARP family members are best known for the nucleic acid interactions that regulate their activities and functions. Recent structural studies have highlighted PARP interactions with nucleic acids, in particular for PARP enzymes that detect and respond to DNA strand break damage. These studies build on our understanding of how DNA break detection is linked to the catalysis of ADP-ribose modifications, provide insights into distinct modes of DNA interaction, and shed light on the mechanisms of PARP inhibitor action. PARP enzymes have several connections to RNA biology, including the detection of the genomes of RNA viruses, and recent structural work has highlighted how PARP13/ZAP specifically targets viral genomes enriched in CG dinucleotides.
INTRODUCTION
PARP enzymes are involved in multiple aspects of human biology, including genome maintenance, gene regulation, viral defense, protein homeostasis, and cell signaling and transport [1,2]. The ADP-ribosyltransferase fold typifies PARP enzymes and embodies the catalytic activity that creates ADP-ribose modifications using NAD+ [3,4]. The majority of PARP enzymes create mono-ADP-ribose modifications on proteins or DNA, and a subset of PARP enzymes can form poly(ADP-ribose), a polynucleotide with a distinctive structure and cell signaling capacity. Human PARP enzymes exhibit diverse domain organizations that dictate individual cellular tasks, usually through providing regulatory capacity or affording interaction with partner proteins or nucleic acids. Nucleic acids have long been appreciated as potent regulators of several PARP enzymes, particularly for those PARPs involved in DNA strand break detection and the cellular response to DNA damage. The structural basis for these nucleic acid interactions and their regulatory capacity have only fairly recently begun to be understood at a structural level. This review summarizes recent structural insights into human PARP structural biology with a focus on protein nucleic acid interactions.
PARP1 structural biology
PARP1 is the founding member of the PARP family and has prominent roles in gene regulation and genome maintenance [2]. With respect to genome maintenance, PARP1 rapidly detects DNA strand breaks, and PARP1 engagement of DNA breaks leads to a massive, allosteric stimulation of poly(ADP-ribose) production [5]. PARP1 achieves this sensitivity to DNA strand break damage through multiple domains that collectively interact with DNA and thereby allosterically enforce a catalytic domain conformation that is open and accessible to substrate NAD+ [6–11] (Figure 1 A–C). The most profound structural change in this activation process occurs in the PARP1 helical domain (HD), which blocks NAD+ binding to the catalytic active site when PARP1 is not engaged on a DNA break. A structure of multiple PARP1 domains bound to a DNA double-strand break suggested a mobile HD [7], and hydrogen-deuterium exchange mass spectrometry pinpointed specific HD helices that undergo massive unfolding transitions in the presence of a DNA single-strand break [8]; however, there were no high-resolution views of the PARP1 HD conformation that would permit NAD+ binding. A crystallographic study captured this open conformation by using a mutant version of PARP1 that favored the open HD conformation, and an NAD+ mimic was observed bound to the active site [12](Figure 1B,C). A surprising feature of this structure was a large-scale rotation of the ADP-ribosyltransferase (ART) fold relative to the rest of the complex, and this rotation appears necessary to fully open the path to NAD+ utilization. Importantly, this same study highlighted that the open configuration of the HD allows it to fully contribute to PARP1 affinity for DNA damage by adding to the multidomain assembly on a DNA break (Figure 1B).
Figure 1. Recent insights into PARP1 structural biology.
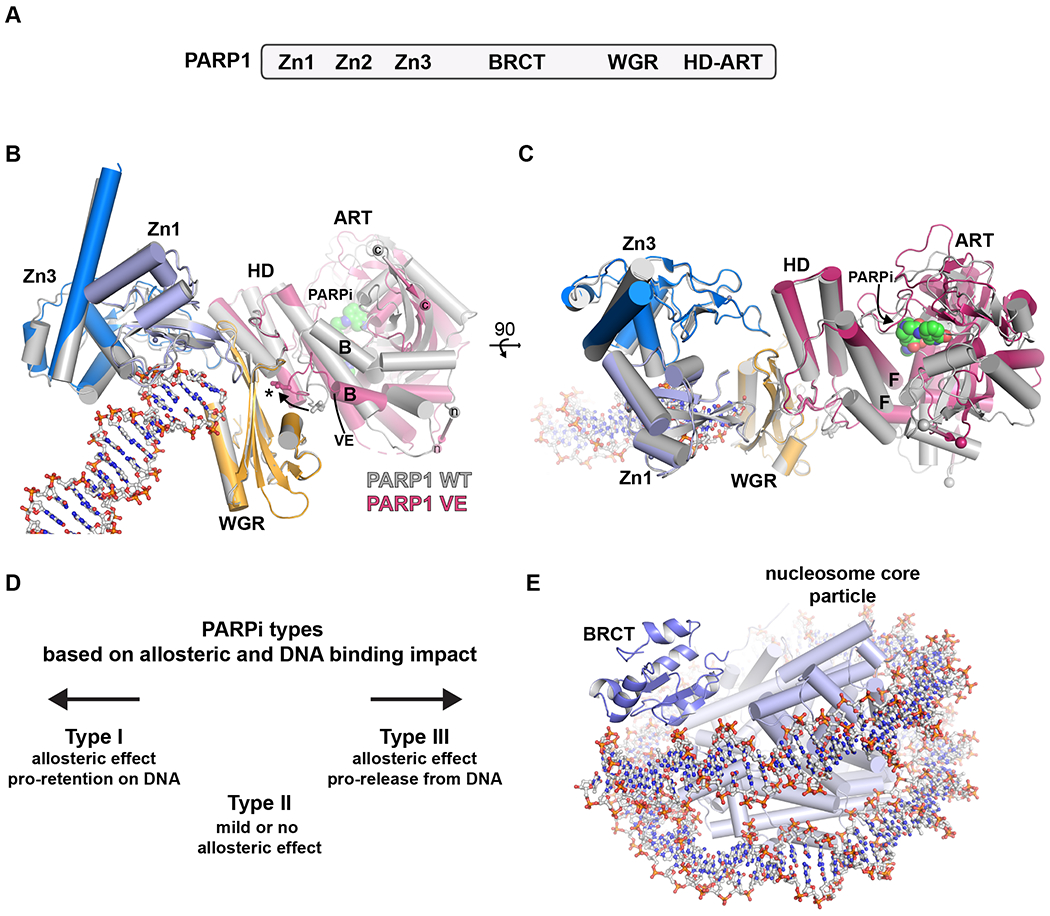
A. Human PARP1 domains. Zinc fingers Zn1 and Zn2 have similar structures and modes of engaging DNA damage; Zn1 also forms critical interdomain contacts. Zinc finger Zn3 has a distinct structure and is involved in DNA and interdomain contacts. The BRCT (BRCA1 C-terminus) fold is positioned within an extended unstructured region that bears the major sites of automodification. The WGR domain (Trp-Gly-Arg) is crucial for coupling the activities of the N-terminal DNA binding regions and the C-terminal catalytic region. The catalytic region is composed of a helical domain (HD) and the ADP-ribosyltransferase (ART) fold.
B. The DNA damage-dependent activity of PARP1 can be reconstituted with a minimal set of domains – Zn1, Zn3, WGR, and HD-ART. X-ray crystallographic analysis has captured these essential domains in complex with DNA duplexes mimicking DNA double-strand break damage. The domains collectively assemble on the DNA break, and induce a major increase in the conformational dynamics of the HD. The extent of HD structural change was captured in a recent crystal structure of a PARP1 VE mutant that favored the “open” conformation of the HD (PDB 7s6m[58]; shown with domains in non-grey colors). The mutant removes two residues (denoted VE) from a helix that is highly dynamic when PARP1 binds to DNA based on HXMS analysis (helix B, labeled ‘B’). The mutant structure featured a ~15 degree rotation of the ART domain relative to the rest of the complex, when compared to the wild-type protein (PDB 4dqy[7], all domains shown in grey). The rotation is highlighted by the change in position of the N- and C-terminal ends of the catalytic region (labeled ‘n’ and ‘c’). The ART rotation allowed the NAD+-mimic EB47 to access the active site (labeled as PARPi). The “open” conformation of the HD makes additional contributions to the multi-domain assembly on DNA (labeled with an asterisk and denoted by an arrow showing the change in position of a key leucine residue), thereby stabilizing the complex and increasing PARP1 affinity for DNA damage.
C. The same alignment of structures in panel B, but rotated 90 degrees to provide a second view. Helix F in the HD (labeled ‘F’) is sheared away from the ART to allow space for the NAD+ mimic.
D. PARP inhibitors can be classified into three types based on their impact on PARP1 allostery and DNA binding affinity. The left pointing arrow references the structures in panels B and C, in which a Type I inhibitor biases the HD to favor the “open” conformation that increases DNA binding affinity and leads to enhanced retention on DNA damage. The right pointing arrow references the structures in panels B and C, in which a Type III inhibitor engages the ART in a way that supports the HD in the closed conformation, and thereby decreases DNA binding affinity and favors release of PARP1 from DNA breaks.
E. A cryo-EM structure captured the BRCT domain of PARP1 in complex with the nucleosome particle (PDB 7scy[22]), illustrating the BRCT contacts made with continuous, undamaged DNA.
PARP inhibitors outcompete NAD+ binding to prevent ADP-ribose modifications from being formed. For PARP1, this prevents the creation of poly(ADP-ribose) and the roles that it plays in mediating the efficiency and speed of the DNA damage response. This deficiency is particularly toxic to cancer cells with deficiencies in homologous recombination repair of DNA damage [13–15]. PARP1 automodification with poly(ADP-ribose) weakens interaction with DNA breaks, and thereby contributes to the turnover and mobility of PARP1 at sites of DNA damage. Correspondingly, PARP inhibitors have the effect of modulating PARP1 residency time on DNA breaks by preventing poly(ADP-ribose) formation. The propensity of PARP inhibitors to “stall” PARP1 on DNA is proposed to be a major factor underlying cellular toxicity [16,17], and the inhibitors that best quench catalytic activity typically have the strongest effect of increasing PARP1 residence time on DNA breaks [18]. It has recently been appreciated that small molecules binding to the PARP1 active site have the potential to modulate the PARP1 allosteric activation mechanism in ways that influence affinity for DNA breaks [9,19]. For example, a non-hydrolyzable NAD+ mimic increased PARP1 affinity for DNA breaks by supporting the open HD conformation that contributes to DNA binding affinity [9]. A systematic analysis of clinical PARP inhibitor effects on PARP1 allostery and DNA break binding affinity indicated that none of the currently used inhibitors have a strong tendency to increase PARP1 affinity for DNA breaks [19]. Surprisingly, several clinical inhibitors indeed modulated PARP1 allostery, but in a manner that decreased PARP1 affinity for DNA breaks. This analysis led to a classification of PARP inhibitors into three types: type I, allosteric and pro-retention on DNA; type II, non-allosteric and no influence on DNA retention; type III, allosteric and pro-release from DNA [19](Figure 1D). The classification of PARP inhibitors toward PARP1 has also been observed with other structural approaches [20,21]. These findings open a new dimension of PARP inhibitor design, wherein the proximity of the NAD+ binding site to the HD permits inhibitor structures to be tailored in ways that engage the HD to influence allostery and PARP1 engagement on DNA.
PARP1 contains a BRCT fold that is not required for interaction with DNA breaks or for catalytic activation by DNA breaks; however, the PARP1 BRCT fold was recently shown to interact with undamaged DNA and was proposed to contribute to how PARP1 navigates through the nucleus [22]. Hydrogen-deuterium exchange mass spectrometry experiments indicated that the BRCT domain of PARP1 interacted with undamaged DNA, and binding studies confirmed this interaction. A cryo-EM structure of the BRCT fold in complex with the nucleosome core particle provided insights into the mode of interaction and the residues likely to mediate the interaction (Figure 1E). The hydrogen-deuterium exchange mass spectrometry experiments also indicated that other domains of PARP1 interact with undamaged DNA, but these collective interactions do not lead to catalytic activation. PARP1 is an abundant nuclear protein and is likely to generally exist in the presence of more undamaged DNA than damaged DNA, so the interaction with undamaged DNA will be an important aspect of PARP1 to continue to decipher.
Insights into PARP1 mechanism have thus far come from individual domains or combinations of domains in complex with DNA, but a full-length structure at high resolution has yet to be captured. Negative stain analysis of full-length PARP1 in complex with a DNA single-strand break indicates that the composite structures are indeed representative of the full-length structure [23], and extending these studies to cryo-EM holds the promise of delivering the first high-resolution views of full-length PARP1.
PARP2 structural biology
PARP2 can be viewed as a more compact version of PARP1, with only the WGR domain and a short N-terminal extension to regulate its catalytic response to DNA strand break interaction (Figure 2A). Although there are conserved features of how PARP1 and PARP2 are allosterically activated by DNA breaks through WGR-HD contacts [24], recent structural analysis has indicated some sharp differences in how PARP1 and PARP2 utilize the WGR domain to engage DNA breaks. In crystal structures of the PARP2 WGR domain in complex with duplex DNA, the ends of DNA duplexes were juxtaposed in the crystal lattice and the WGR domain spanned the two DNA ends, suggesting that the protein might bring together DNA ends [25]. This binding mode is distinct from the WGR of PARP1, which collaborates with zinc fingers to effectively engage DNA breaks. A more recent structure of the WGR-CAT fragment of PARP2 also indicated that the WGR domain bridges DNA ends [26](Figure 2B). A curious feature of this structure was that the ART fold was substantially displaced from the WGR and HD, and the terminus of one DNA strand (not involved in a bridging interaction) was positioned near the active site. Although the exact positioning of the ART fold could be influenced by the crystal packing environment, it clearly highlights the mobility of the ART in the PARP2 activation process.
Figure 2. Recent insights into PARP2 structural biology.
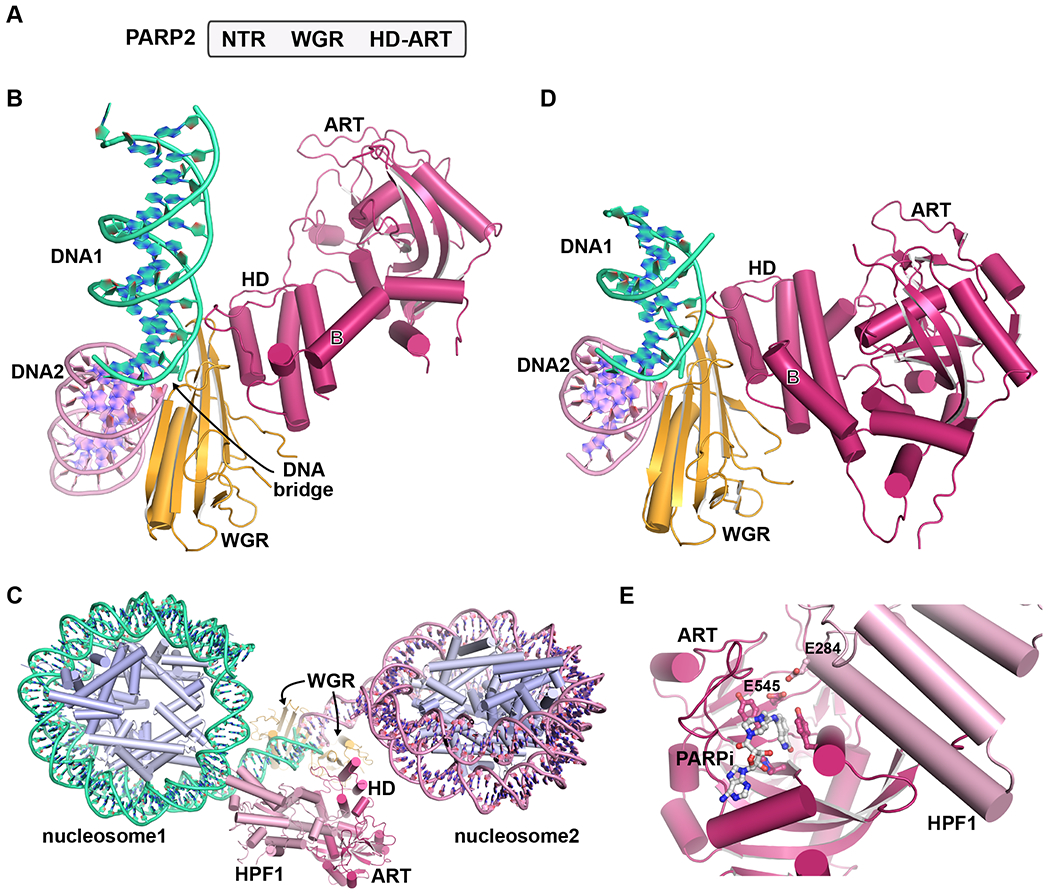
A. Human PARP2 domains. PARP2 bears WGR and HD-ART domains similar to PARP1. The WGR domain interacts with DNA strand breaks and communicates with the HD-ART. The N-terminal region (NTR) also contributes to overall DNA binding affinity.
B. A crystal structure of human PARP2 was determined in complex with duplex DNA (PDB 7aeo[26]). The WGR domain was positioned at the junction of two DNA molecules (DNA1 in light grey, DNA2 in dark grey), effectively forming a bridge across the discontinuity in the backbone of the juxtaposed DNA ends. For clarity, a WGR domain bound to the opposite side of the DNA junction is not shown. The HD forms contacts with the WGR, and the ART is translated away from the HD relative to catalytic domain structures in the absence of DNA. For reference to PARP1 in Figure 1, helix B is labeled.
C. In a recent cryo-EM study, the human PARP2/HFP1 complex was captured bridging the DNA ends extending from two different nucleosomes (PDB 6x0n[27]). In the most complete map/model produced from the study, a complete PARP2/HPF1 complex is positioned on one DNA junction (see panel D), and the WGR alone is positioned on the other DNA junction.
D. Focused refinement in the same cryo-EM study also yielded a map/model for PARP2 bridging two DNA ends (PDB 6x0l[27]). The WGR contacts across the DNA ends agrees with the X-ray structure in panel B. The ART domain remains in close contact with the HD, in contrast to the X-ray structure in panel B. The second WGR domain is not shown for clarity.
E. The ART domain of PARP2 was crystallized in complex with HPF1 (PDB 6tx3[59]). HPF1 completes the PARP2 (and PARP1) active site by contributing E284 to allow deprotonation of Serine residues, permitting their modification with ADP-ribose.
A cryo-EM study analyzed PARP2 interaction with DNA ends extending from two different nucleosome core particles [27], giving a visually stunning representation of the DNA bridge mediated by the PARP2 WGR domain (Figure 2C,D). In this complex, the ART domain remained in close contact with the HD (Figure 2D), in contrast to the crystal structure described in the last section. The model deposited for the PARP2 complex with DNA ends (Figure 2D) did not exhibit the ART domain rotation described in the previous section for PARP1. It could be that only one dominant conformation of the catalytic region was captured, or that the domain rotation is not conserved in PARP2 activation. The PARP2 structure did not contain substrate or substrate mimics in the active site, so it is unclear whether the modeled conformation represents the active form of PARP2. A different cryo-EM study also captured PARP2 bridging DNA ends extending from nucleosomes, but only the WGR domain was modeled in this case [28].
Several structures have thus captured PARP2 spanning two DNA ends, suggesting a potential role for PARP2-mediated DNA bridging in repair processes. PARP2 DNA bridges were recently studied using a magnetic tweezer-based assay, and the results indicated a remarkably stable bridging interaction [29]. PARP1 did not exhibit the capacity to tether DNA ends in the same experimental setup. Further studies are needed to better understand the potential conditions under which PARP2 might play this end-bridging role in the cell.
Even though PARP1/2/3 are generally considered conserved in the WGR-CAT regions, the recent structures and results described here indicate interesting specialization between PARP1 and PARP2 in terms of DNA break engagement. High-resolution structures of PARP3 in complex with DNA might indicate other possibilities for the WGR domain to engage DNA breaks. It is also notable that PARP inhibitors have different allosteric effects on PARP1 and PARP2. None of the current clinical PARPi exhibit type I behavior toward PARP1 [19]. In contrast, several clinical PARPi exhibit type I behavior toward PARP2, thereby strongly increasing PARP2 affinity for DNA breaks [30]. Thus, we continue to learn more about interesting variations even among highly related members of the PARP family.
HPF1 regulation of PARP1/2
Histone PARylation factor 1 (HPF1) has only recently emerged as a central regulator of PARP1 and PARP2 in the DNA damage response [31–34]. HPF1 permits PARP1 and PARP2 to modify serine residues, notably those located on histone tails, in addition to the glutamate and aspartate residues that are the typical sites of modification [32]. Indeed, the discovery of HPF1 has exposed an underappreciated prevalence of serine-ADP-ribose modifications [35–37]. Several structural studies, including the PARP2/nucleosome complex highlighted above, have now demonstrated that HFP1 “completes” the PARP1/PARP2 ART fold, in particular contributing a glutamic acid residue to a joint PARP/HPF1 active site [27,38,39](Figure 2E). The glutamic acid residue is expected to deprotonate serine residues and make them chemically competent for modification. The discovery of HPF1 opens the exciting possibility of other PARP family members working with core regulatory factors.
PARP interactions with RNA
There is a growing appreciation of how PARP enzymes are connected to RNA biology [40,41], and several PARP enzymes contain known RNA binding modules, including PARP10, PARP12, PARP13, and PARP14 [1]. However, there are limited structural insights into RNA-mediated regulation of PARP enzymes. There are reports of RNA structures that stimulate PARP1 and PARP2 activity [42–44]. The level of activation by RNA is generally much lower than the level of activation seen with DNA breaks, and perhaps this is expected for normal functions in unstressed cells that might require more modest levels of PARP activity, compared to the “firehose” levels of PAR production in response to DNA breaks. Structural biology will clearly play an important role in establishing the basis for these RNA interactions with PARP1 and PARP2 and suggesting mechanisms of regulation.
Studies of PARP13 have provided the first insights into PARP-RNA interactions. PARP13 also goes by the name zinc finger antiviral protein or ZAP [45–48]. PARP13/ZAP detects and promotes the degradation of viral RNA. PARP13 preferentially interacts with RNA containing CG dinucleotides, which steers its RNA-binding activity toward viral genomes enriched in CG dinucleotides (and some viral genomes with suppressed CG dinucleotide content are less susceptible to PARP13). RNA interaction is mediated by four N-terminal CCCH-type zinc fingers (Figure 3A). Structures of the N-terminal zinc fingers of PARP13 bound to RNA oligonucleotides have illustrated the specificity of PARP13 for CG dinucleotides in RNA [49,50]. The structure of human PARP13 N-terminal zinc fingers indicated that the specificity comes largely from the second zinc finger (Figure 3B). The mouse PARP13 structure captured the same mode of CG dinucleotide detection, and also indicated how other zinc fingers can contribute to RNA interactions (Figure 3C). It is anticipated that multiple PARP13 molecules will bind to the large segments of viral RNA presenting CG dinucleotides, perhaps forming a scaffolding structure that could initiate RNA degradation in concert with other cellular factors.
Figure 3. Recent insights into PARP13/ZAP structural biology.
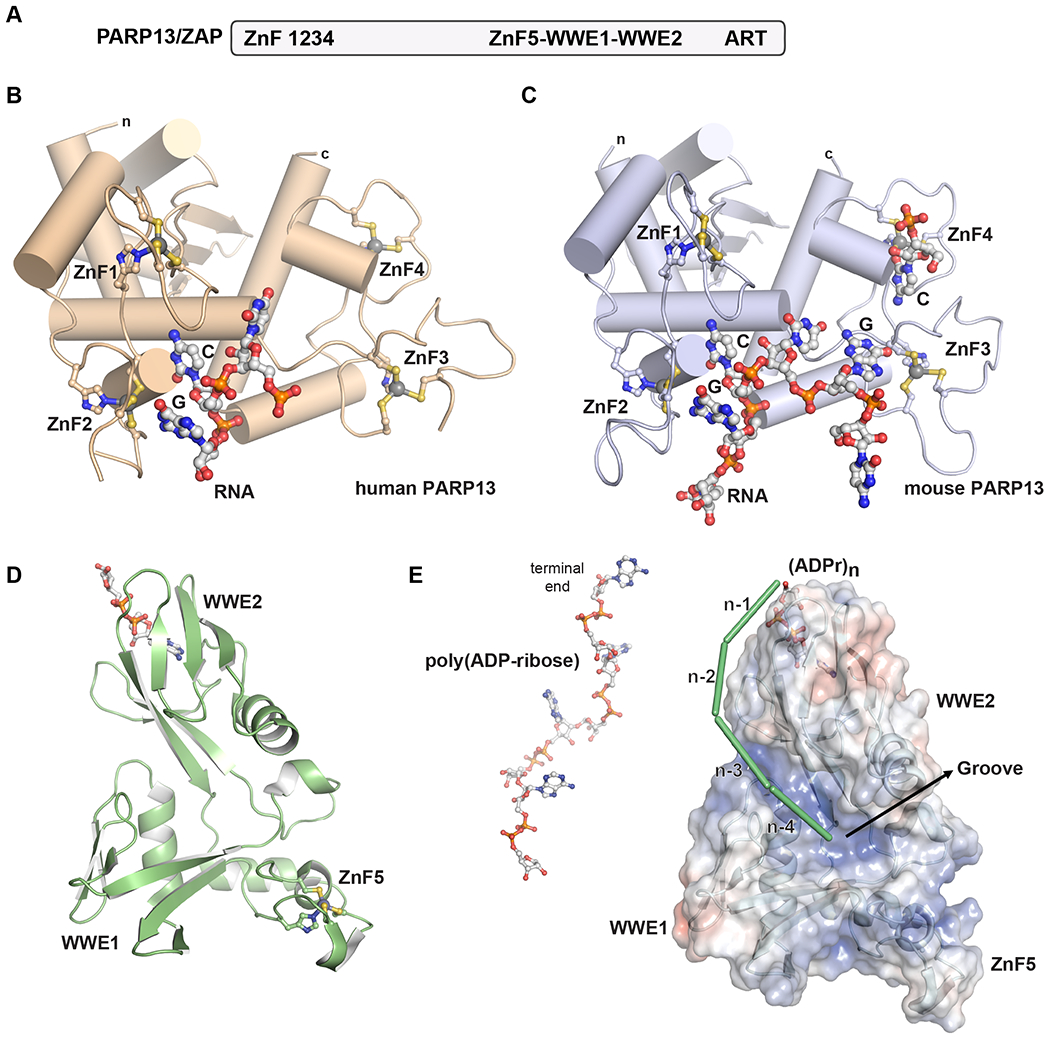
A. Human PARP13/ZAP domains. Four CCCH zinc fingers (ZnF) are located at the N-terminus, followed by an extended unstructured region. A central structured region is composed of a fifth zinc finger (ZnF5) and two WWE domains: ZnF5-WWE1-WWE2. The ART fold is located at the C-terminus and lacks the residues required for catalytic activity.
B. A crystal structure of human PARP13 in complex with an RNA oligonucleotide demonstrates how specificity toward CG dinucleotides is achieved primarily within ZnF2 (PDB 6uej[49]).
C. A crystal structure of mouse PARP13 in complex with an RNA oligonucleotide also capture the specific interactions made with a CG dinucleotide in ZnF2, and also specific contacts made with a G nucleotide in ZnF3, and a C nucleotide in ZnF4 (PDB 6l1w[50]). The C nucleotide in ZnF4 comes from the terminus of the RNA chain bound to another PARP13 molecule.
D. Crystal structure of the ZnF5-WWE1-WWE2 region of human PARP13 in complex with ADP-ribose (PDB 7tgq[60]). Only the WWE2 domain has a functional binding site. The three domains collectively assemble into a single module.
E. Structures, mutagenesis, and binding analysis support a model for PARP13 interaction with poly(ADP-ribose) in which binding is anchored at one end of the poly(ADP-ribose) chain, and an electropositive groove formed by the collective ZnF5-WWE1-WWE2 structure supports binding of polymers of a certain length [56]. A model of four units of a poly(ADP-ribose) chain are shown on the left for comparison to the binding model.
Interestingly, the PARP13 ART fold at the C-terminus lacks the residues required for catalytic activity, and some PARP13 isoforms lack the ART fold [51,52]. The central region of PARP13 has a CCCH zinc finger and tandem WWE domains (Figure 3A). This central region interacts with poly(ADP-ribose) and the interaction contributes to the antiviral properties of PARP13 [53–55]. Recent structures have provided first insights into how this region of PARP13 engages poly(ADP-ribose), using the collective fold of the zinc finger and tandem WWEs [55,56](Figure 3D,E). More mechanistic work is needed to understand how the RNA-binding, poly(ADP-ribose) binding, and ART fold collectively operate to mediate PARP13 antiviral functions [57].
Conclusion
The structures highlighted in this review represent some of the first views of PARP nucleic acid interactions, but we can likely expect more structures as our understanding of PARP biology continues to mature. Poly(ADP-ribose) is itself a special type of polynucleotide. Detailed analysis of interactions with poly(ADP-ribose) are generally lacking, thus it will be interesting to see this area of PARP biology further develop, as there are interesting opportunities for DNA/RNA binding surfaces to be co-opted for interaction with poly(ADP-ribose), and perhaps thereby exert regulatory functions.
ACKNOWLEDGMENTS
Work in the Pascal laboratory is support by the Canadian Institutes of Health Research (PJT153295 and PJT173370) and the National Cancer Institute of the NIH (CA259037 and CA92584).
Footnotes
DECLARATION OF INTERESTS
The author is a cofounder of Hysplex with interests in PARPi development.
REFERENCES
* of special interest
** of outstanding interest
- 1.Lüscher B, Ahel I, Altmeyer M, Ashworth A, Bai P, Chang P, Cohen M, Corda D, Dantzer F, Daugherty MD, et al. : ADP-ribosyltransferases, an update on function and nomenclature. FEBS J 2021, doi: 10.1111/febs.16142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupte R, Liu Z, Kraus WL: PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev 2017, 31:101–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barkauskaite E, Jankevicius G, Ahel I: Structures and Mechanisms of Enzymes Employed in the Synthesis and Degradation of PARP-Dependent Protein ADP-Ribosylation. Mol Cell 2015, 58:935–946. [DOI] [PubMed] [Google Scholar]
- 4.Karlberg T, Langelier M-F, Pascal JM, Schüler H: Structural biology of the writers, readers, and erasers in mono- and poly(ADP-ribose) mediated signaling. Mol Aspects Med 2013, 34:1088–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pandey N, Black BE: Rapid Detection and Signaling of DNA Damage by PARP-1. Trends Biochem Sci 2021, 46:744–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Langelier M-FM-F, Pascal JMJM: PARP-1 mechanism for coupling DNA damage detection to poly(ADP-ribose) synthesis. Curr Opin Struct Biol 2013, 23:134–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langelier M, Planck JL, Roy S, Pascal JM: Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science 2012, 336:728–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dawicki-McKenna JM, Langelier M-F, DeNizio JE, Riccio AA, Cao CD, Karch KR, McCauley M, Steffen amin D, Black BE, Pascal JM: PARP-1 Activation Requires Local Unfolding of an Autoinhibitory Domain. Mol Cell 2015, 60:755–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Langelier M-F, Zandarashvili L, Aguiar PM, Black BE, Pascal JM: NAD+ analog reveals PARP-1 substrate-blocking mechanism and allosteric communication from catalytic center to DNA-binding domains. Nat Commun 2018, 9:844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eustermann S, Wu W-F, Langelier M-F, Yang J-C, Easton LE, Riccio AA, Pascal JM, Neuhaus D: Structural Basis of Detection and Signaling of DNA Single-Strand Breaks by Human PARP-1. Mol Cell 2015, 60:742–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogden TEH, Yang JC, Schimpl M, Easton LE, Underwood E, Rawlins PB, McCauley MM, Langelier MF, Pascal JM, Embrey KJ, et al. : Dynamics of the HD regulatory subdomain of PARP-1; substrate access and allostery in PARP activation and inhibition. Nucleic Acids Res 2021, 49:2266–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.*.Rouleau-Turcotte É, Krastev DB, Pettitt SJ, Lord CJ, Pascal JM: Captured snapshots of PARP1 in the active state reveal the mechanics of PARP1 allostery. Mol Cell 2022, 82:2939–2951.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]; X-ray structure of a PARP1 mutant that favors the conformation that interacts with NAD+. An NAD+ mimic is co-crystallized with the assembled domains of PARP1 bound to a DNA double strand break. The contribution of the HD to DNA binding affinity is quantified.
- 13.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, Helleday T: Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434:913–7. [DOI] [PubMed] [Google Scholar]
- 14.Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. : Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434:917–21. [DOI] [PubMed] [Google Scholar]
- 15.Lord CJ, Ashworth A: PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355:1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pommier Y, O’Connor MJ, de Bono J: Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 2016, 8:362ps17. [DOI] [PubMed] [Google Scholar]
- 17.Murai J, Huang SN, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y: Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res 2012, 72:5588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hopkins TA, Shi Y, Rodriguez LE, Solomon LR, Donawho CK, DiGiammarino EL, Panchal SC, Wilsbacher JL, Gao W, Olson AM, et al. : Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol Cancer Res 2015, 13:1465–1477. [DOI] [PubMed] [Google Scholar]
- 19.**.Zandarashvili L, Langelier M-F, Velagapudi UK, Hancock MA, Steffen JD, Billur R, Hannan ZM, Wicks AJ, Krastev DB, Pettitt SJ, et al. : Structural basis for allosteric PARP-1 retention on DNA breaks. Science 2020, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]; PARP inhibitors are classified into three types based on their impact on PARP1 allostery and DNA binding affinity using hydrogen-deuterium exchange mass spectrometry and biochemical analysis. Chemical modification to a clinical PARP inhibitor converts between classes and leads to higher toxicity in cultured cancer cells.
- 20.Xue H, Bhardwaj A, Yin Y, Fijen C, Ephstein A, Zhang L, Ding X, Pascal JM, VanArsdale TL, Rothenberg E: A two-step mechanism governing PARP1-DNA retention by PARP inhibitors. Sci Adv 2022, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sefer A, Kallis E, Eilert T, Röcker C, Kolesnikova O, Neuhaus D, Eustermann S, Michaelis J: Structural dynamics of DNA strand break sensing by PARP-1 at a single-molecule level. Nat Commun 2022, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rudolph J, Muthurajan UM, Palacio M, Mahadevan J, Roberts G, Erbse AH, Dyer PN, Luger K: The BRCT domain of PARP1 binds intact DNA and mediates intrastrand transfer. Mol Cell 2021, 81:4994–5006.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Langelier MF, Billur R, Sverzhinsky A, Black BE, Pascal JM: HPF1 dynamically controls the PARP1/2 balance between initiating and elongating ADP-ribose modifications. Nat Commun 2021, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langelier M-FM-F, Riccio AA, Pascal JM: PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res 2014, 42:7762–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Obaji E, Haikarainen T, Lehtiö L: Structural basis for DNA break recognition by ARTD2/PARP2. Nucleic Acids Res 2018, 46:12154–12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.*. Obaji E, Maksimainen MM, Galera-Prat A, Lehtiö L: Activation of PARP2/ARTD2 by DNA damage induces conformational changes relieving enzyme autoinhibition. Nat Commun 2021, 12. X-ray structure of human PARP2 in complex with DNA double-strand break damage captures the WGR domain bridging the ends of DNA duplexes and highlights conformational changes associated with DNA damage detection.
- 27.*.Bilokapic S, Suskiewicz MJ, Ahel I, Halic M: Bridging of DNA breaks activates PARP2–HPF1 to modify chromatin. Nature 2020, 585:609–613. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cryo-EM study of the PARP2/HPF1 complex bound to two nucleosomes, with PARP2 serving as a bridge between the two nucleosomes. The bridging interaction is shown to specifically stimulate PARP2 activity.
- 28.Gaullier G, Roberts G, Muthurajan UM, Bowerman S, Rudolph J, Mahadevan J, Jha A, Rae PS, Luger K: Bridging of nucleosome-proximal DNA double-strand breaks by PARP2 enhances its interaction with HPF1. PLoS One 2020, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bell NAW, Molloy JE: Single-molecule force spectroscopy reveals binding and bridging dynamics of PARP1 and PARP2 at DNA double-strand breaks. Proc Natl Acad Sci U S A 2023, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langelier M-F, Lin X, Zha S, Pascal JM: Clinical PARP inhibitors allosterically induce PARP2 retention on DNA. Sci Adv 2023, 9:eadf7175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gibbs-Seymour I, Fontana P, Rack JGM, Ahel I: HPF1/C4orf27 Is a PARP-1-Interacting Protein that Regulates PARP-1 ADP-Ribosylation Activity. Mol Cell 2016, 62:432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonfiglio JJ, Fontana P, Zhang Q, Colby T, Gibbs-Seymour I, Atanassov I, Bartlett E, Zaja R, Ahel I, Matic I: Serine ADP-Ribosylation Depends on HPF1. Mol Cell 2017, 65:932–940.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Palazzo L, Leidecker O, Prokhorova E, Dauben H, Matic I, Ahel I: Serine is the major residue for ADP-ribosylation upon DNA damage. Elife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prokhorova E, Zobel F, Smith R, Zentout S, Gibbs-Seymour I, Schützenhofer K, Peters A, Groslambert J, Zorzini V, Agnew T, et al. : Serine-linked PARP1 auto-modification controls PARP inhibitor response. Nat Commun 2021, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larsen SC, Hendriks IA, Lyon D, Jensen LJ, Nielsen ML: Systems-wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep 2018, 24:2493–2505.e4. [DOI] [PubMed] [Google Scholar]
- 36.Longarini EJ, Matic I: The fast-growing business of Serine ADP-ribosylation. DNA Repair (Amst) 2022, 118. [DOI] [PubMed] [Google Scholar]
- 37.Leidecker O, Bonfiglio JJ, Colby T, Zhang Q, Atanassov I, Zaja R, Palazzo L, Stockum A, Ahel I, Matic I: Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat Chem Biol 2016, 12:998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.**.Suskiewicz MJ, Zobel F, Ogden TEH, Fontana P, Ariza A, Yang J-C, Zhu K, Bracken L, Hawthorne WJ, Ahel D, et al. : HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 2020, 579:598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]; X-ray crystal structure of the human PARP2 ART domain in complex with HPF1 identifies a composite active site formed between the two proteins. Biochemical analysis indicates that HPF1 interaction requires PARP1/2 to adopt the active HD conformation.
- 39.*.Sun FH, Zhao P, Zhang N, Kong LL, Wong CCL, Yun CH: HPF1 remodels the active site of PARP1 to enable the serine ADP-ribosylation of histones. Nat Commun 2021, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]; X-ray crystallographic analysis of the ART domain of PARP1 in complex with HPF1 demonstrates the composite active site formed between the two proteins.
- 40.Bock FJ, Todorova TT, Chang P: RNA Regulation by Poly(ADP-Ribose) Polymerases. Mol Cell 2015, 58:959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim DS, Challa S, Jones A, Kraus WL: PARPs and ADP-ribosylation in RNA biology: From RNA expression and processing to protein translation and proteostasis. Genes Dev 2020, 34:302–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Léger K, Bär D, Savić N, Santoro R, Hottiger MO: ARTD2 activity is stimulated by RNA. Nucleic Acids Res 2014, 42:5072–5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim DS, Camacho CV., Nagari A, Malladi VS, Challa S, Kraus WL: Activation of PARP-1 by snoRNAs Controls Ribosome Biogenesis and Cell Growth via the RNA Helicase DDX21. Mol Cell 2019, 75:1270–1285.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laspata N, Kaur P, Mersaoui SY, Muoio D, Liu ZS, Bannister MH, Nguyen HD, Curry C, Pascal JM, Poirier GG, et al. : PARP1 associates with R-loops to promote their resolution and genome stability. Nucleic Acids Res 2023, 51:2215–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gao G, Guo X, Goff SP: Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science (80- ) 2002, 297. [DOI] [PubMed] [Google Scholar]
- 46.Guo X, Ma J, Sun J, Gao G: The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc Natl Acad Sci U S A 2007, 104:151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Y, Gao G: ZAP-mediated mRNA degradation. RNA Biol 2008, 5. [DOI] [PubMed] [Google Scholar]
- 48.Takata MA, Gonçalves-Carneiro D, Zang TM, Soll SJ, York A, Blanco-Melo D, Bieniasz PD: CG dinucleotide suppression enables antiviral defence targeting non-self RNA. Nature 2017, 550:124–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.*.Meagher JL, Takata M, Gonçalves-Carneiro D, Keane SC, Rebendenne A, Ong H, Orr VK, MacDonald MR, Stuckey JA, Bieniasz PD, et al. : Structure of the zinc-finger antiviral protein in complex with RNA reveals a mechanism for selective targeting of CG-rich viral sequences. Proc Natl Acad Sci U S A 2019, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]; An X-ray structure of the four N-terminal zinc fingers of human PARP13/ZAP in complex with single-stranded RNA illustrates the selectivity towards CG dinucleotides.
- 50.*.Luo X, Wang X, Gao Y, Zhu J, Liu S, Gao G, Gao P: Molecular Mechanism of RNA Recognition by Zinc-Finger Antiviral Protein. Cell Rep 2020, 30. [DOI] [PubMed] [Google Scholar]; X-ray structure of the four N-terminal zinc fingers of mouse PARP13/ZAP in complex with CG dinucleotide-containing RNA. Binding analysis indicates preferred sequence: C(n7)G(n)CG, where n represents any nucleotide.
- 51.Karlberg T, Klepsch M, Thorsell AG, Andersson CD, Linusson A, Schüler H: Structural basis for lack of ADP-ribosyltransferase activity in poly(ADP-ribose) polymerase-13/zinc finger antiviral protein. J Biol Chem 2015, 290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li MMH, Aguilar EG, Michailidis E, Pabon J, Park P, Wu X, de Jong YP, Schneider WM, Molina H, Rice CM, et al. : Characterization of Novel Splice Variants of Zinc Finger Antiviral Protein (ZAP). J Virol 2019, 93:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dasovich M, Beckett MQ, Bailey S, Ong SE, Greenberg MM, Leung AKL: Identifying Poly(ADP-ribose)-Binding Proteins with Photoaffinity-Based Proteomics. J Am Chem Soc 2021, 143:3037–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kliza KW, Liu Q, Roosenboom LWM, Jansen PWTC, Filippov DV, Vermeulen M: Reading ADP-ribosylation signaling using chemical biology and interaction proteomics. Mol Cell 2021, 81:4552--4567.e8. [DOI] [PubMed] [Google Scholar]
- 55.*.Xue G, Braczyk K, Gonçalves-Carneiro D, Dawidziak DM, Sanchez K, Ong H, Wan Y, Zadrozny KK, Ganser-Pornillos BK, Bieniasz PD, et al. : Poly(ADP-ribose) potentiates ZAP antiviral activity. PLoS Pathog 2022, 18:e1009202. [DOI] [PMC free article] [PubMed] [Google Scholar]; X-ray crystallographic analysis of the ZnF5-WWE1-WWE2 region of human PARP13/ZAP in complex with ADP-ribose. Structure-based mutagenesis and analysis of viral infectivity indicates a role for poly(ADP-ribose) in potentiating the antiviral function of PARP13/ZAP.
- 56.*.Kuttiyatveetil JRA, Soufari H, Dasovich M, Uribe IR, Mirhasan M, Cheng SJ, Leung AKL, Pascal JM: Crystal structures and functional analysis of the ZnF5-WWE1-WWE2 region of PARP13/ZAP define a distinctive mode of engaging poly(ADP-ribose). Cell Rep 2022, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]; X-ray crystallographic analysis of the ZnF5-WWE1-WWE2 region of mouse PARP13/ZAP in complex with ADP-ribose and in complex with ATP. Binding studies with defined poly(ADP-ribose) lengths and termini support a new model of interaction with poly(ADP-ribose) that anchors on the end of the poly(ADP-ribose) chain.
- 57.Ficarelli M, Neil SJD, Swanson CM: Targeted Restriction of Viral Gene Expression and Replication by the ZAP Antiviral System. Annu Rev Virol 2021, 8:265–283. [DOI] [PubMed] [Google Scholar]
- 58.*.Rouleau-Turcotte É, Krastev DB, Pettitt SJ, Lord CJ, Pascal JM: Captured snapshots of PARP1 in the active state reveal the mechanics of PARP1 allostery. Mol Cell 2022, 82:2939–2951.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]; X-ray structure of a PARP1 mutant that favors the conformation that interacts with NAD+. An NAD+ mimic is co-crystallized with the assembled domains of PARP1 bound to a DNA double strand break. The contribution of the HD to DNA binding affinity is quantified.
- 59.**.Suskiewicz MJ, Zobel F, Ogden TEH, Fontana P, Ariza A, Yang JC, Zhu K, Bracken L, Hawthorne WJ, Ahel D, et al. : HPF1 completes the PARP active site for DNA damage-induced ADP-ribosylation. Nature 2020, 579:598–602. [DOI] [PMC free article] [PubMed] [Google Scholar]; X-ray crystal structure of the human PARP2 ART domain in complex with HPF1 identifies a composite active site formed between the two proteins. Biochemical analysis indicates that HPF1 interaction requires PARP1/2 to adopt the active HD conformation.
- 60.Xue G, Braczyk K, Gonçalves-Carneiro D, Dawidziak DM, Zawada K, Ong H, Wan Y, Zadrozny KK, Ganser-Pornillos BK, Bieniasz PD, et al. : Poly(ADP-ribose) potentiates ZAP antiviral activity. bioRxiv 2020, doi: 10.1101/2020.12.17.423219. [DOI] [PMC free article] [PubMed] [Google Scholar]