

Abstract
Accurate measurements of the molecular composition of single cells will be necessary for understanding the relationship between gene expression and function in diverse cell types. One of the most important phenotypes that differs between cells is their size, which was recently shown to be an important determinant of proteome composition in populations of similarly sized cells. We, therefore, sought to test if the effects of the cell size on protein concentrations were also evident in single-cell proteomics data. Using the relative concentrations of a set of reference proteins to estimate a cell’s DNA-to-cell volume ratio, we found that differences in the cell size explain a significant amount of cell-to-cell variance in two published single-cell proteome data sets.
Keywords: single cell proteomics, cell size, Histones, gene expression
Graphical Abstract
INTRODUCTION
Individual cells are the basis of life. It is therefore important to develop techniques that accurately quantify the molecular composition of single cells. Extensive progress examining mRNA composition has been achieved at single-cell resolution, helping to catalog diverse cell types in multicellular organisms.1 However, mRNA sequencing gives an incomplete measurement of the state of the cell because diverse post-transcriptional mechanisms also impact gene expression. For example, the correlation between mRNA and protein amounts is complicated by differing translation and degradation rates.2 Moreover, transcriptomic methods are blind to the diverse set of protein modifications that are often key to the activity and function. To address these limitations inherent to measuring only mRNA transcripts, single-cell proteomic methods have emerged.
Recent advances in single-cell proteomics are driven by low-volume sample preparation3–6 and an increase in measurement sensitivity from a new generation of mass spectrometers.7 Multiplexed peptide labeling approaches have also enabled the measurement of hundreds and sometimes thousands of proteins from single mammalian cells.5,6,8 Initial experiments have revealed that the proteomes of single cells may be influenced by the cell cycle phase,4,7 although it is unclear which other physiological features underlie cell-to-cell proteome heterogeneity. It is important to measure these and other quantifiable sources of proteome variation to better characterize features that are specific to the cell types and states.
We recently showed that cell size, or more precisely the DNA-to-cell volume ratio, is an important determinant of proteome content.9,10 Contrary to the assumption that most cellular components would remain at a constant concentration in cells of different sizes, we found widespread, size-dependent changes in the concentrations of individual proteins (Figure 1A)10.9 These changes in protein concentration likely reflect, to a large extent, the size-dependent changes in the cellular growth rate.11,10 Importantly, a recent proteome analysis of the NCI60 cancer lines revealed a similar pattern of size-dependent changes to the proteome.12 Thus, regardless of cell type, cell size has an important influence on proteome composition and, therefore, should contribute to cell-to-cell heterogeneity in the proteomes of single cells. Here, we used publicly available single-cell proteomic data sets to determine whether the size-dependent proteome changes we described previously are evident in single-cell measurements.
Figure 1.
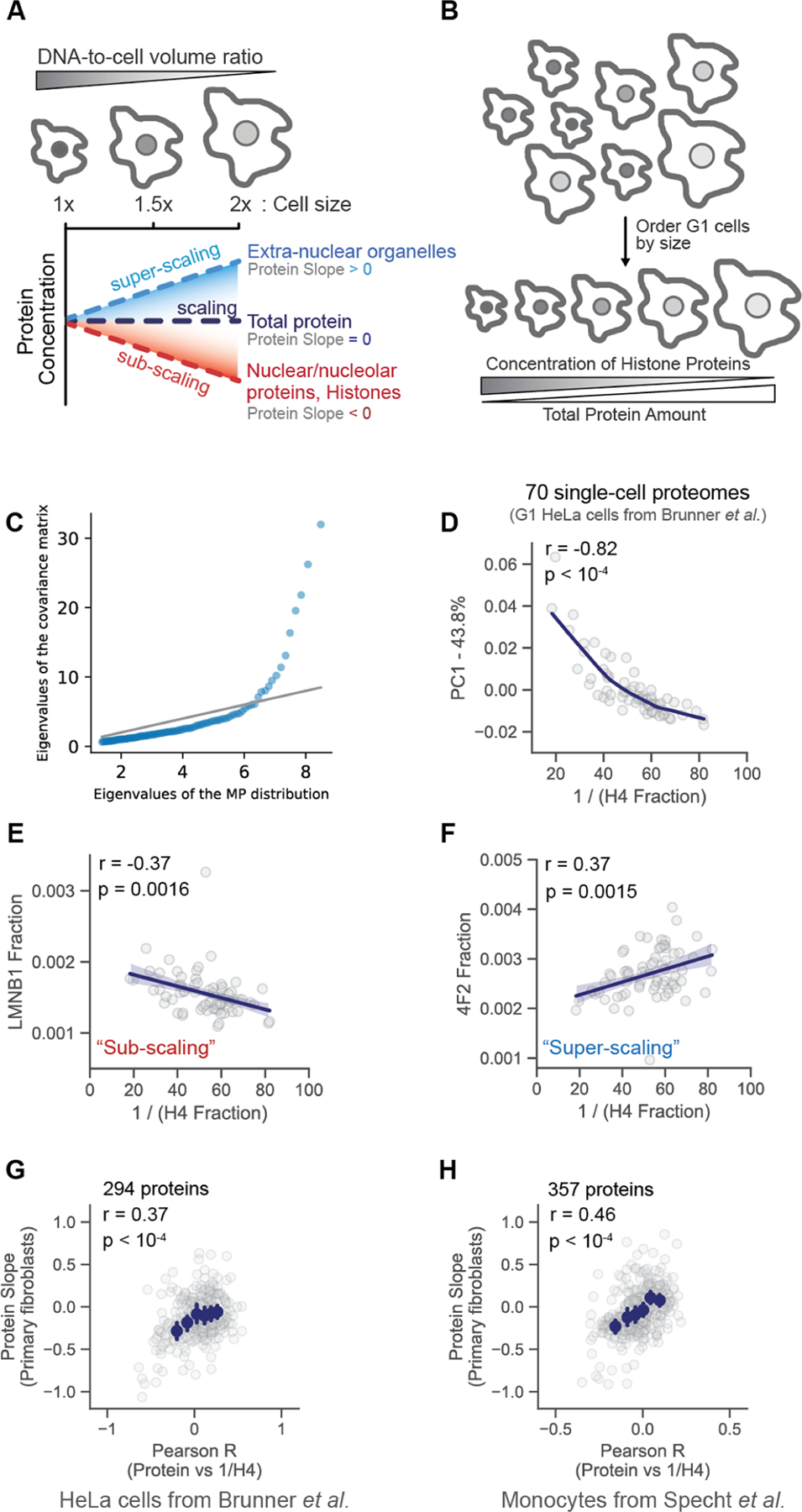
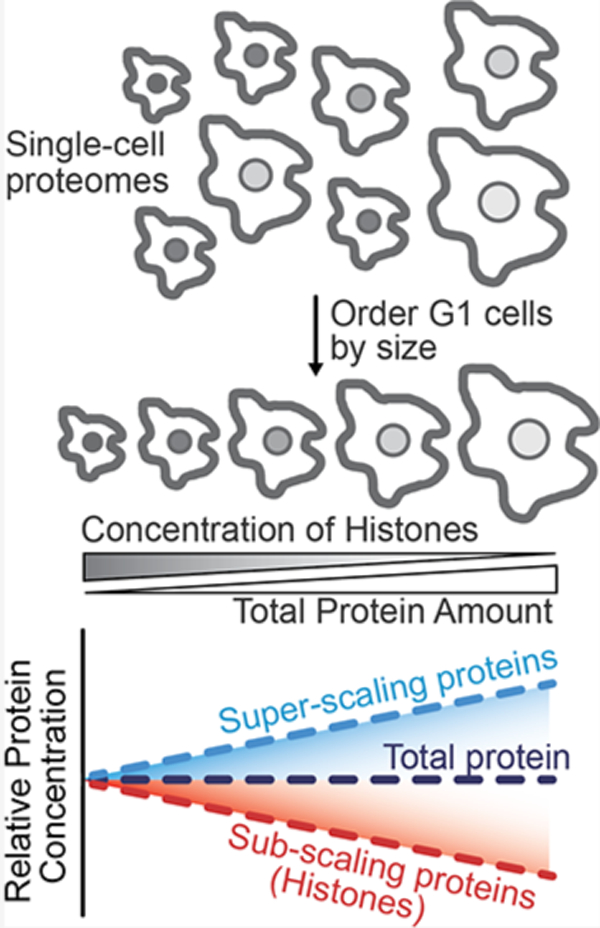
Cell size contributes to variation in the proteomes of single cells. (A) Proteomes vary with cell size. For example, the amount of histone proteins is maintained in proportion to the genome so that histone concentrations are inversely proportional to the cell size in G1 cells. The protein slope describes how the concentration of an individual protein scales with the cell size (Lanz et al.). Proteins with a slope of 0 maintain a constant cellular concentration regardless of cell volume (“scaling”). A slope value of 1 corresponds to an increase in concentration that is proportional to the increase in volume (“super-scaling”), and a slope of −1 corresponds to dilution (concentration ∼ 1/volume; “sub-scaling”). (B) Schematic illustrating how relative histone protein concentrations can be used as a proxy for the cell size in single cell proteomics data sets in which the cell size was not measured. (C) Quantile-quantile plot between the distribution of eigenvalues of the empirical covariance matrix and a sample of the Marcenko–Pastur distribution, which is the distribution expected from uncorrelated, normally distributed random variables. Eigenvalues above the gray identity line indicate the presence of an underlying signal. (D) Principal component analysis (PCA) analysis of 70 single cell proteomes. Each dot represents the proteome of a G1 cell from Brunner et al. The first principal component is plotted against a proxy for the G1 cell size (1/H4 Fraction). The fraction of the proteome represented by histone H4 is the H4 intensity/summed intensity of all other proteins. (E and F) correlation between the increasing G1 cell size (1/H4 Fraction) and the relative concentration (i.e., protein intensity/summed intensity of all other proteins) of two proteins previously found to (E) subscale and (F) superscale with the cell size (Lanz et al.). (G and H) A Pearson correlation coefficient was calculated by regressing the relative concentration of each individual protein against a proxy for each cell’s size (1/H4 concentration), as exemplified in (E) and (F). The r value for each protein from the (G) Brunner et al. and (H) Specht et al. data sets is plotted against the previously measured protein slope value.9 Histone H4 was excluded from the plot. Blue dots are x-binned data and error bars represent the 99% confidence interval. The plot in (H) was filtered to display the most abundant proteins. Figure S3B depicts an unfiltered version of this analysis.
METHODS
Data Curation
For Brunner et al., protein intensities for the individual G1 cells were obtained from PRIDE (ID: PXD024043). G1-labeled columns were extracted from the file named: “20210919_DIA-NN_SingleCellOutput.pg_matrix.tsv” (DIANN1.8 cell cycle folder). G1 cells with the fewest number of protein identifications were excluded until a shared set of ∼300 proteins was detected in each single cell. This resulted in the reanalysis of 70 of the 93 G1 cell proteomes (Table S1). For Specht et al., a dataframe containing relative protein concentrations for each single cell was downloaded from https://slavovlab.net (“Proteins-processed.csv”). Mock-treated monocytes were extracted from the “Proteins-processed.csv” dataframe using the “sdrf_scope2.tsv” table (Table S2).
Estimation of Cell Size
From the study of Brunner et al., we estimated the relative cell size for each of the single G1 cells using the “Histone H4 fraction”. To calculate the Histone H4 fraction, we divided the intensity value for Histone H4 (H4_HUMAN) by the summed intensity for all of the other proteins. To calculate this summed value, we considered only the ion intensity from proteins that were identified in all cells considered for our analysis. We chose a single histone protein, rather than the average of all histone proteins, to minimize missing values and therefore maximize the number of cells considered for our analysis. Histone H4 was chosen because there is a single H4 variant, and it was detected in all but 3 cells. The use of other core histone proteins or an averaged value produced similar results (Figure S1). From the study of Specht et al., the relative cell size was estimated using the relative concentration of Histone H4 (log2).
The approach with a single reference protein can be extended to several reference proteins. Namely, we select a small number of reference proteins known to scale significantly with cell size, i.e., the absolute value of the measured slope for those selected proteins is relatively large. We therefore construct a reference data set of protein fractions and its associated measured slope values and solve the following regression problem:
where is the vector of cell sizes we want to estimate, and the subscripts and refer to a specific protein or cell, respectively. In other words, the approach aims to find the cell sizes that, for the reference proteins selected, replicate as closely as possible the previously measured slopes.9 The estimated cell sizes allow the estimation of slopes for all of the proteins using standard linear regression.
Estimation of Proportion of Variance Attributable to the Cell Size
Subtracting the contribution of cell sizes for each protein in the data set yields a second data set whose total variance is expected to be lower. Indeed, if cell size is a contributor to cell-to-cell proteome variation, then removing its effect should decrease the total amount of variance. Denoting the sample covariance matrices of the original data set and the new data set with the effect of cell size removed by and , the total variances and are equal to the sum of the eigenvalues of their respective covariance matrices. Therefore, the amount of leftover variance after removal of the effect of slopes is given by . If the contribution of cell size is meaningful, we expect this ratio to be smaller than 1. We can compare this ratio to the leftover variance after removing the first principal component in the data set, i.e., , where is the maximum eigenvalue of the sample covariance matrix. The first principal component is the direction in the feature space that accounts for maximal variance. Comparing these two metrics reveals the amount of variance contributed by the cell size.
Principal Component Analysis
A dataframe was created that contained individual proteins as rows with columns corresponding to single G1 cell proteomes. Principal component analysis (PCA) analysis was performed in Python by using the sklearn package. The results of the PCA analysis were visualized with Seaborn’s scatterplot.
Pearson r Correlation Analysis
From the study of Brunner et al., a data frame containing intensity values of 295 proteins (row) for 70 single cells (column) was converted to intensity fractions. For each cell, the intensity of each protein was divided by the summed intensity of all proteins to calculate each protein’s proteome fraction, an estimate of a protein’s relative concentration. A Pearson correlation (python’s scipy package) was calculated by regressing the relative concentration of each individual protein against a proxy for each cell’s size (exemplified in Figure 1E, F). For Specht et al., we used the log2 ratio values published by the authors, so the r value was derived from a regression between the relative protein concentration (log2) versus the relative Histone H4 concentration (log2). Only the most abundant ∼350 proteins were considered for Figure 1H (filtered by peptide detections in our own data set). Our analysis of the entire Specht et al. study’s data set can be found in Figure S3 and Table S2.
RESULTS
To investigate whether cell size can explain cell-to-cell variations in proteome content, we reanalyzed data from two recently published single-cell proteome data sets5,7 (Tables S1 and S2). One of these utilized Bruker’s ultrahigh-sensitivity timsTOF SCP (label-free DIA) to measure the proteomes of single HeLa cells that were proceeding through the cell cycle after being synchronized.7 The authors distinguished the cell cycle phase of single cells based on their measurements. To disentangle cell size and cell-cycle-related effects, we only considered the proteomes of G1-enriched single cells for our analysis. An eigenvalue analysis of this set of G1 cell proteomes found that the top eigenvalues of the covariance matrix deviate significantly from the Marcenko–Pastur distribution,13 i.e., the distribution of eigenvalues for data sets with no latent variables (Figure 1C). This means that there is a significant signal in our data despite the noisy nature of single-cell proteomics data. To crudely approximate the relative size of each cell, we used the histone proteins because their amount is proportional to the amount of DNA.9,14 Smaller cells, therefore, possess proportionally higher concentrations of histone proteins than larger cells (Figure 1B). We used the fraction of total ion intensity represented by Histone H4 as representative of the inverse of the cell volume (a proxy for cell size). We chose Histone H4 because it has only one variant and was detected in nearly every G1 cell in the Brunner et al. data set. We performed PCA on 70 G1-enriched single-cell proteomes from Brunner et al., reasoning that proteins with cell size-dependent abundances could help explain the variance in these cells. The fraction of total ion signal attributable to histone H4 significantly correlated with the first principal component, indicating the importance of cell size in contributing to cell-to-cell proteome variation (Figure 1D). Other core histone proteins produced similar results (Figure S1). In contrast, substituting a histone protein for a common housekeeping enzyme, PGK1, whose concentration is expected to be independent of the cell size,9 did not produce a significant correlation (Figure S2).
To further explore the relationship between single-cell proteome variation and cell size, we calculated Pearson coefficients (r) for each protein from the correlation between its relative protein concentration and Histone H4, a proxy for cell size (Figure 1E,F). We then correlated these r values with the protein concentration size dependence previously reported by Lanz et al. (Figure 1G). Concentration size dependence was calculated as a protein slope. In brief, the protein slope is calculated from a linear regression between the log2 of an individual protein’s concentration and the log2 of the cell volume. Thus, a protein slope value of 0 describes proteins for which concentration does not change with cell volume (scaling), a protein slope value of 1 describes proteins for which concentrations increase proportionally to cell volume (super-scaling), and a protein slope value of −1 describes proteins that are perfectly diluted by cell growth such that their concentration is inversely proportional to cell volume (subscaling). The Pearson r value correlating concentration and histone H4 derived from single cells was correlated with the previously published protein slope values (Figure 1G). Having established that cell size influences variation in one single-cell proteomics data set, we next sought to examine the robustness of this result in a second data set. To do this, we repeated this analysis on a second data set generated using a different single-cell proteomic platform.5 Using SCoPE2, Specht et al. distinguished individual monocytes that were or were not differentiated into macrophages. Like the HeLa cells measured by Brunner et al. (Figure 1G), Pearson regression analysis of single monocyte and macrophage proteomes from Specht et al. produced r values, which significantly correlated with the protein slope values (Figures 1H and S3). Taken together, these correlations strongly support the hypothesis that variations in cell size measurably contribute to single-cell proteome variation.
Since single-cell proteomics measurements are noisy, we anticipate that there is significant noise in our estimate of cell size using only histone H4. We therefore sought to derive a more robust method for measuring the size-dependent proteome variation in single cell proteome data sets. To do this, we decided to use more than one reference protein to approximate a cell’s size. We selected a subset of reference proteins and reconstructed cell sizes based on a least-squared-error heuristic (Figure 2A and Table S3). Under this framework, the estimated cell size distribution is the one that most closely reproduces the set of measured protein slopes for this subset of proteins. Assuming that protein-to-protein noise is uncorrelated, using more than one reference protein reduces measurement noise. The reference proteins were chosen based on their (i) large absolute measured slope value and (ii) large median correlation coefficients with other reference proteins. These criteria ensure that the reference proteins encode meaningful variations from which a signal can be extracted.
Figure 2.
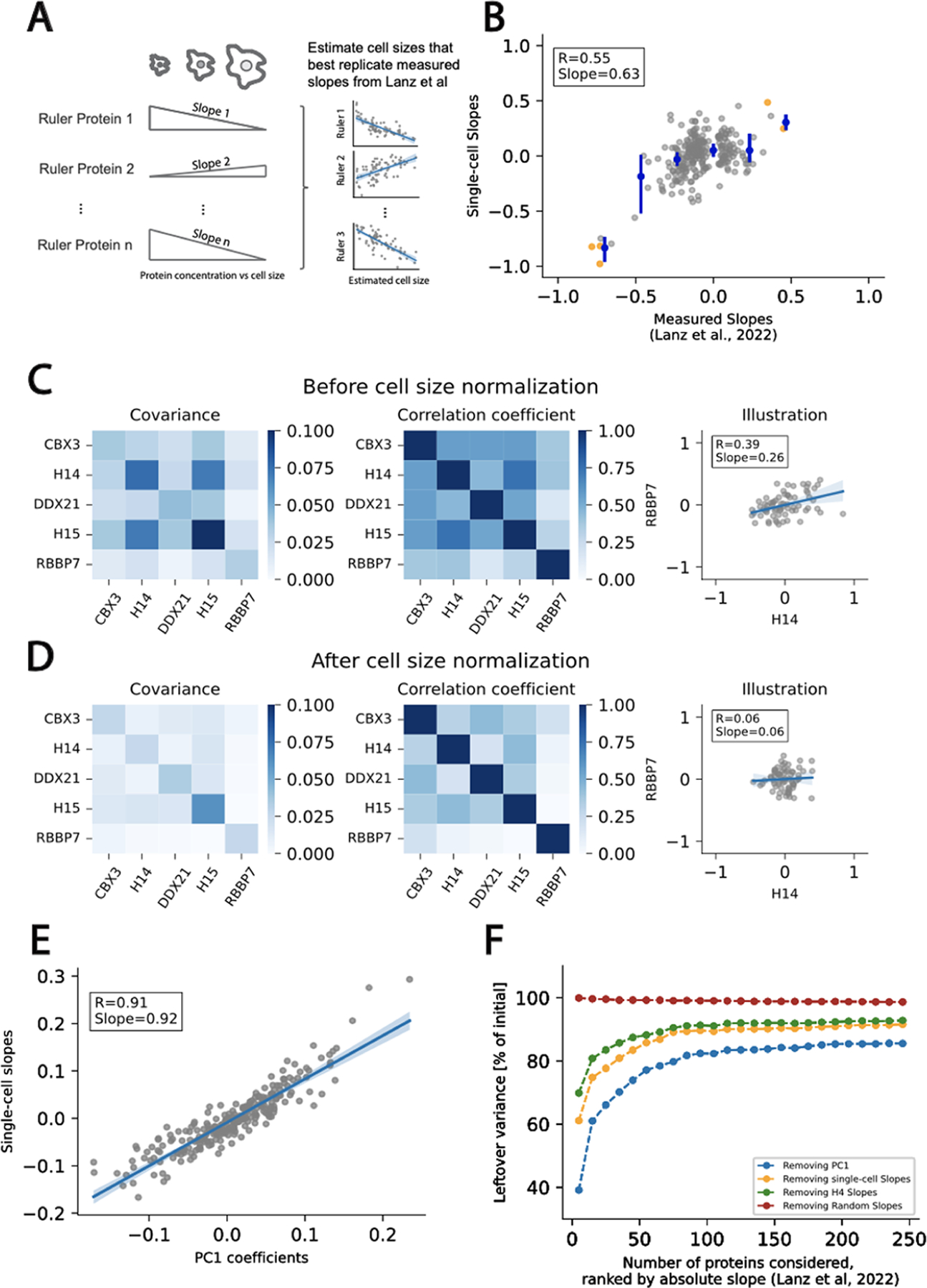
Robust estimation of cell size used to determine the size-dependent variance in single-cell proteomics data. (A) Schematic illustrating the methodology to estimate cell size. We first select a small subset of reference proteins, like histone H4, whose concentrations were shown to be strongly size-dependent (Lanz et al.). Using these reference proteins and their corresponding protein slope values, we performed a least-squares regression to estimate the size of each single cell. (B) Having estimated the size of each cell, we then calculate a size slope for each protein in our single cell proteomics data sets (“Single-cell Slopes”). Plot depicts a comparison of slopes estimated from the single cell proteomics data and those measured previously.9 Orange dots denote reference proteins and blue dots with error bars denote x-binned values. (C and D) Comparison of protein concentration covariance and correlation in the initial data set (C) and after removing the estimated effect of cell size (D) for a set of 5 proteins with large absolute measured slopes. Removing the estimated effect of the cell size reduces the covariance and the correlation coefficient between protein pairs. We illustrate this effect with a given protein pair. (E) Relationship between the estimated slopes and the coefficients of the first principal component. Both quantities are very close to each other, indicating the estimated slopes approximate the direction of maximum variance in the data set. (F) Amount of variance leftover after removing the first principal component (blue), the single cell slopes (orange), the effect of H4 only (green), and the estimated effect of the cell size from random slopes (red). The number of proteins included in the analysis (x-axis) was gradually increased based on protein absolute slopes. For example, if 50 proteins were included in the analysis, this set contains the 50 proteins with the highest absolute slopes. The maximum amount of removable variance is bounded by the first principal component (PC1 blue).
Based on our single reference protein analysis in Figure 1, the scaling behavior of proteins is expected to be qualitatively conserved across cell types. Using the reconstructed cell size distribution and the single-cell proteome measurements, we compute single-cell slopes for each of the remaining proteins in the data set, which were well correlated with the previously measured slopes (Figure 2B). These results are qualitatively similar to the analysis shown in Figure 1, but quantitatively more robust due to the use of five reference proteins to estimate the cell size. Indeed, a smaller number of rulers yields larger variations in the estimated single-cell slopes and cell size distributions (Figure S4). The correlation between estimated and measured slopes does not vary significantly when more than five rulers are used (Figure S5).
Having found that cell size contributes to single-cell proteome variation, we next sought to quantify the extent of this contribution to the total variation in the single-cell proteomics data. To explore this contribution, we first subtracted the estimated contribution of cell size to each protein concentration from our data. This resulted in a significant decrease in the covariance and correlation coefficient among a set of proteins whose concentrations varied with cell size (Figure 2C,D). To assess whether the estimated cell size distribution is the primary source of variance in the data set, we compared the first principal component coefficients with the imputed single-cell slopes (Figure 2E). The strong correlation demonstrates that the estimated slopes closely align with the direction of the maximum variance. Consistent with this observation, no correlation is observed with the second PC coefficients (Figure S6). Finally, the imputed cell size distribution allowed us to estimate the proportion of variance in the data set that is attributable to cell size (see the Methods section; Figure 2F). The variance due to cell size differences is comparable to the variance attributed to the first principal component, which sets the upper bound for the removable variance by a single linear transformation. While using histone H4 as a single reference protein does remove some variance in the data set, using more reference proteins increases the amount of variance that can be accounted for. We note that using protein slopes derived from the measurement of another cell type9 yields generally similar results (Figure S7). In contrast, using a set of randomly generated protein slopes does not remove any variance in the data set.
DISCUSSION
Our goal here was to test for effects of cell size in single-cell proteomics data. Ideally, we would have directly measured cell size and cell cycle phase (e.g., using DNA content or a FUCCI reporter) for each cell in a single-cell proteomics data set. However, since we currently do not have the ability to do this, we instead re-analyzed publicly available data sets. Because our only goal was to see whether the effect of the cell size is measurable, we used internal protein proxies for the cell size such as 1/histone or a set of “ruler” proteins. We chose these proxies because we knew they would reflect the cell size based on our previous work examining bulk populations of cells.9 The results presented here definitively show that cell size is important and must be considered when analyzing single-cell proteomes. While our simple methods are sufficient to prove this overall concept, we anticipate that future researchers will choose to accurately measure the cell size prior to preparing cells for single-cell mass spectrometry using emerging technology platforms.3,4,6
To properly correct for the proteome effects of the cell size (i.e., DNA-to-cell volume ratio), both cell size and cell cycle information must be accounted for. This is because the larger cells in a proliferating population are typically in late S or G2 phase and have thus duplicated their genome. This confounding factor can be corrected for as long as cell cycle information is collected alongside the cell size measurement. By collecting both cell cycle and size information for each cell, we expect that proteome heterogeneity due to differences in cell size can be accounted for and subtracted from the data to better isolate other cell biological features of interest. Our attempts to perform such a normalization using internal protein proxies for size (e.g., 1/Histone) are confounded by the noisy nature of single-cell proteomes. However, once accurate size and cell cycle normalization are performed, other biological differences between cells should become more apparent in single-cell proteomes.
While our data demonstrate that cell size is a major contributor to variation in single-cell proteomes extracted from cells of the same type, it is important to note that the relationship may be more complex when data sets contain very different types of cells, as was recently demonstrated in a melanoma cell line.4 Nevertheless, even in these complex assemblies of cells, we still anticipate there to be some size-dependent signal because several different cell lines exhibit similar size scaling across their proteomes.9,12 We also note that differences in ploidy across cell types may have a large effect on the proteome. For example, a comparison of the proteomes of 2N, 4N, and 8N cells revealed that they were highly similar despite the near-4-fold increase in cell size.9 Thus, what we refer to as size scaling is more accurately attributed to changes in the DNA-to-cell volume ratio.
In summary, we reanalyzed the proteome heterogeneity in single-cell data sets reported by two independent groups using different single-cell preparation and measurement platforms. In both cases, we found that differences in cell size substantially contribute to the variance in single-cell proteomes. Remarkably, the effects of cell size trended in agreement with a recent report of cell size-dependent changes to the proteome that were measured in bulk for different types of cells.9 Taken together, these analyses support the conclusion that differences in cell size will account for a significant amount of proteome heterogeneity in single cells. We therefore recommend accounting for differences in cell size in future analyses of single-cell proteomes.
Supplementary Material
ACKNOWLEDGMENTS
We thank Martin Wühr and members of the Skotheim and Elias laboratories for feedback and suggestions on the manuscript. We also thank the authors of Brunner et al. and Specht et al. for collecting and curating their single-cell data in a manner that facilitated our reanalysis.
Funding
Chan Zuckerberg Biohub (collaborative postdoctoral fellowship M.C.L.; investigator award J.M.S.); NIH P01 CA254867.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jproteome.3c00441.
Histone H4 data and analysis for single cells from Brunner et al. (XLSX)
Histone H4 data and analysis for single cells from Specht et al. (XLSX)
Data and analysis related to Figure 2 (XLSX) Supporting Information (PDF)
The authors declare no competing financial interest.
Contributor Information
Michael C. Lanz, Department of Biology, Stanford University, Stanford, California 94305, United States; Chan Zuckerberg Biohub, Stanford, California 94305, United States
Lucas Fuentes Valenzuela, Department of Biology, Stanford University, Stanford, California 94305, United States.
Joshua E. Elias, Chan Zuckerberg Biohub, Stanford, California 94305, United States
Jan M. Skotheim, Department of Biology, Stanford University, Stanford, California 94305, United States; Chan Zuckerberg Biohub, Stanford, California 94305, United States
Data Availability Statement
This study analyzed previously published data.
REFERENCES
- (1).(a) Tabula Sapiens C; Jones RC; Karkanias J; Krasnow MA; Pisco AO; Quake SR; Salzman J; Yosef N; Bulthaup B; Brown P; et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 2022, 376 (6594), No. eabl4896. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Regev A; Teichmann SA; Lander ES; Amit I; Benoist C; Birney E; Bodenmiller B; Campbell P; Carninci P; Clatworthy M; et al. The Human Cell Atlas. Elife 2017, 6, No. e27041. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wu AR; Neff NF; Kalisky T; Dalerba P; Treutlein B; Rothenberg ME; Mburu FM; Mantalas GL; Sim S; Clarke MF; et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat. Methods 2014, 11 (1), 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Liu Y; Beyer A; Aebersold R On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165 (3), 535–550. [DOI] [PubMed] [Google Scholar]
- (3).Matzinger M; Muller E; Durnberger G; Pichler P; Mechtler K Robust and Easy-to-Use One-Pot Workflow for Label-Free Single-Cell Proteomics. Anal. Chem 2023, 95 (9), 4435–4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Leduc A; Huffman RG; Cantlon J; Khan S; Slavov N Exploring functional protein covariation across single cells using nPOP. Genome Biol 2022, 23 (1), 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Specht H; Emmott E; Petelski AA; Huffman RG; Perlman DH; Serra M; Kharchenko P; Koller A; Slavov N Single-cell proteomic and transcriptomic analysis of macrophage heterogeneity using SCoPE2. Genome Biol 2021, 22 (1), 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Petelski AA; Emmott E; Leduc A; Huffman RG; Specht H; Perlman DH; Slavov N Multiplexed single-cell proteomics using SCoPE2. Nat. Protoc 2021, 16 (12), 5398–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Brunner AD; Thielert M; Vasilopoulou C; Ammar C; Coscia F; Mund A; Hoerning OB; Bache N; Apalategui A; Lubeck M; et al. Ultra-high sensitivity mass spectrometry quantifies single-cell proteome changes upon perturbation. Mol. Syst. Biol 2022, 18 (3), No. e10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Budnik B; Levy E; Harmange G; Slavov N SCoPE-MS: mass spectrometry of single mammalian cells quantifies proteome heterogeneity during cell differentiation. Genome Biol 2018, 19 (1), 161. [DOI] [PMC free article] [PubMed] [Google Scholar]; Derks J; Leduc A; Wallmann G; Huffman RG; Willetts M; Khan S; Specht H; Ralser M; Demichev V; Slavov N Increasing the throughput of sensitive proteomics by plexDIA. Nat. Biotechnol 2022, 41, 50 DOI: 10.1038/s41587-022-01389-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lanz MC; Zatulovskiy E; Swaffer MP; Zhang L; Ilerten I; Zhang S; You DS; Marinov G; McAlpine P; Elias JE; et al. Increasing cell size remodels the proteome and promotes senescence. Mol. Cell 2022, 82 (17), 3255–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Lanz MC; Zhang S; Swaffer MP; Ziv I; Hernández Götz L; McCarty F; Jarosz DF; Elias JE; Skotheim JM Genome dilution by cell growth drives starvation-like proteome remodeling in mammalian and yeast cells. bioRxiv 2023, DOI: 10.1101/2023.10.16.562558. [DOI] [PubMed] [Google Scholar]
- (11).Zatulovskiy E; Lanz MC; Zhang S; McCarthy F; Elias JE; Skotheim JM Delineation of proteome changes driven by cell size and growth rate. Front. Cell Dev. Biol 2022, 10, No. 980721. [DOI] [PMC free article] [PubMed] [Google Scholar]; Liu X; Yan J; Kirschner MW Beyond G1/S regulation: how cell size homeostasis is tightly controlled throughout the cell cycle? bioRxiv 2022, DOI: 10.1101/2022.02.03.478996. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cadart C; Monnier S; Grilli J; Saez PJ; Srivastava N; Attia R; Terriac E; Baum B; Cosentino-Lagomarsino M; Piel M Size control in mammalian cells involves modulation of both growth rate and cell cycle duration. Nat. Commun 2018, 9 (1), 3275. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ginzberg MB; Chang N; D’Souza H; Patel N; Kafri R; Kirschner MW Cell size sensing in animal cells coordinates anabolic growth rates and cell cycle progression to maintain cell size uniformity. Elife 2018, 7, No. e26957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cheng L; Chen J; Kong Y; Tan C; Kafri R; Björklund M Size-scaling promotes senescence-like changes in proteome and organelle content. bioRxiv 2021, No. 455193. [Google Scholar]
- (13).Johnstone IM On the distribution of the largest eigenvalue in principal components analysis. Ann. Stat 2001, 29 (2), 295–327. [Google Scholar]
- (14).Wisniewski JR; Hein MY; Cox J; Mann MA ″proteomic ruler″ for protein copy number and concentration estimation without spike-in standards. Mol. Cell Proteomics 2014, 13 (12), 3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]; Swaffer MP; Kim J; Chandler-Brown D; Langhinrichs M; Marinov GK; Greenleaf WJ; Kundaje A; Schmoller KM; Skotheim JM Transcriptional and chromatin-based partitioning mechanisms uncouple protein scaling from cell size. Mol. Cell 2021, 81 (23), 4861–4875. [DOI] [PMC free article] [PubMed] [Google Scholar]; Claude KL; Bureik D; Chatzitheodoridou D; Adarska P; Singh A; Schmoller KM Transcription coordinates histone amounts and genome content. Nat. Commun 2021, 12 (1), 4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study analyzed previously published data.