

Abstract
A new, highly potent, selective, and water-soluble antagonist of the hA3 adenosine receptor was synthesized and tested in binding and functional assays. Compound 4 (5-[[(4-pyridyl)amino]carbonyl]amino-8-methyl-2-(2-furyl)-pyrazolo-[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine hydrochloride) displayed high water solubility (15 mM) and the highest affinity (Ki = 0.01 nM) and selectivity for the hA3 versus A1, A2A, and A2B receptors (>10000-fold) ever reported. A Schild analysis of the antagonism by 4 of agonist-induced inhibition of cAMP production in CHO cells expressing the hA3 receptor indicated a KB value of 0.20 nM.
Introduction.
It is well-known that adenosine regulates many physiological processes through the interaction with four known receptor subtypes classified as A1, A2A, A2B, and A3.1,2 In particular, the A3 adenosine receptor subtype, which is distributed in different organs (lung, liver, kidney, heart, and, with a lower density, the brain),3 exerts its action through the modulation of two second messenger systems: inhibition of adenylate cyclase4 and stimulation of phospholipases C5 and D.6 The potential therapeutic applications of activating or antagonizing this receptor subtype have been investigated in recent years. In particular, antagonists for the A3 receptor promise to be useful for the treatment of inflammation7 and in the regulation of cell growth.8,9 Consequently, much effort has been directed toward searching for potent and selective human A3 adenosine antagonists.10 Recently, Baraldi and co-workers reported a large series of pyrazolotriazolopyrimidines, bearing substituted phenylcarbamoyl residues at the amino group at the 5-position, as highly potent and selective antagonists of the human A3 adenosine receptor.11–13 In particular, 1 (5-[[(phenyl)-amino]carbonyl]amino-8-methyl-2-(2-furyl)-pyrazolo[4,3-e] 1,2,4-triazolo[1,5-c]pyrimidine (Chart 1) showed the most favorable binding affinity and selectivity for the human A3 adenosine receptor ever reported.13
Chart 1.
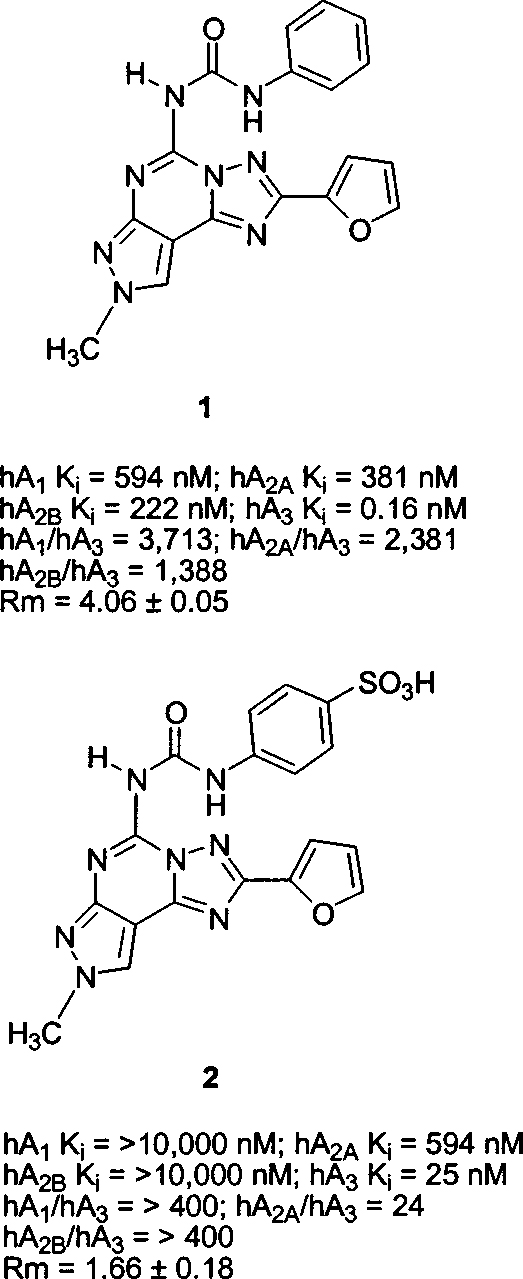
Structures, Biological Characterization, and Water Affinities of Reference Compounds
Unfortunately, a major problem within this class of compounds is the typical low water solubility, which has limited their use as pharmacological and diagnostic tools. The hydrophobicity of compound 1 is indicated by its high Rm value of 4.06. Previously an attempt for obtaining a water-soluble derivative has been made through the introduction of a sulfonic acid group at the para position of the phenyl ring (Chart 1) affording compound 2 (4-[3-(2-furan-2-yl-8-methyl-8H-pyrazolo-[4,3-e][1,2,4]triazolo[1,5-c]pyrimidin-5-yl)ureido]benzenesulfonic acid). As expected, a completely water-soluble derivative (Rm = 1.66, water solubility greater than 20 mM) was obtained; however, a significant loss of affinity (156-fold) and selectivity was observed.13 A hypothesis for the dramatic loss of affinity was provided through molecular modeling studies, which indicated that steric control seemed to be taking place around the para position of the phenyl ring in the putative A3 receptor binding site.13 Taking into account these observations and with the aim of obtaining derivatives with high affinity, selectivity, and water solubility, we synthesized a new derivative (Chart 2) by bioisosteric replacement of the phenyl ring with the 4-pyridyl moiety (3), thus providing higher water solubility while avoiding the steric hindrance of a substituent at the para position, which seemed to be responsible for the reduction of hA3 adenosine receptor affinity.
Chart 2.
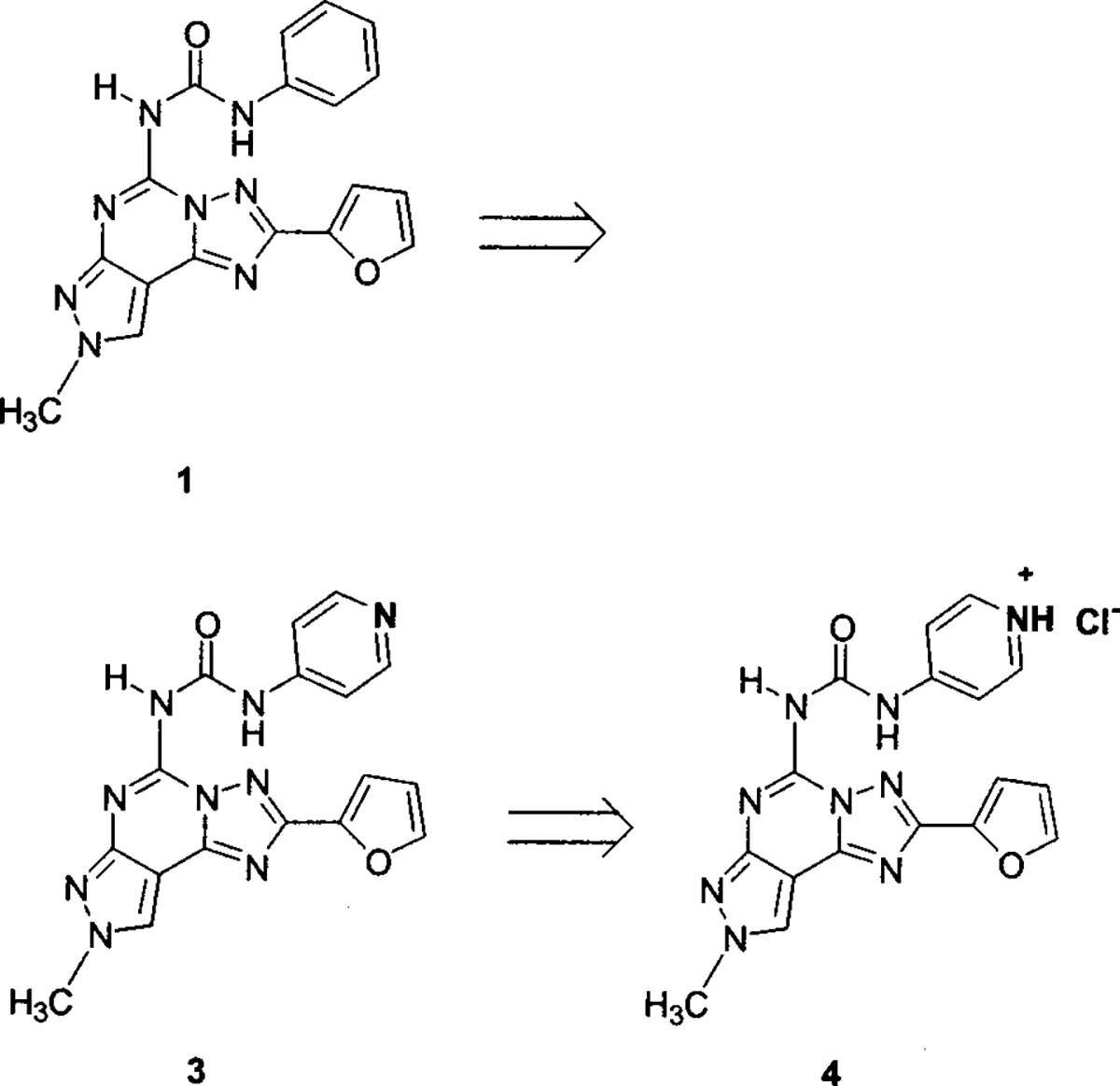
Rational Design of Water-Soluble hA3 Adenosine Receptor Antagonist
In fact, the introduction of a basic nitrogen was intended to further improve water solubility when protonated, i.e., in the form of the corresponding HCl salt (4).
Results and Discussion.
The synthesis of the desired compound has been performed following the general strategy depicted in Scheme 1. The major problem in the synthesis of derivative 3 involved the preparation of a 4-pyridyl isocyanate (5), which could not be synthesized by the usual method, i.e., reaction of the corresponding amine with phosgene or a phosgene equivalent, because of the reactivity and instability of pyridyl isocyanates.14
Scheme 1a.
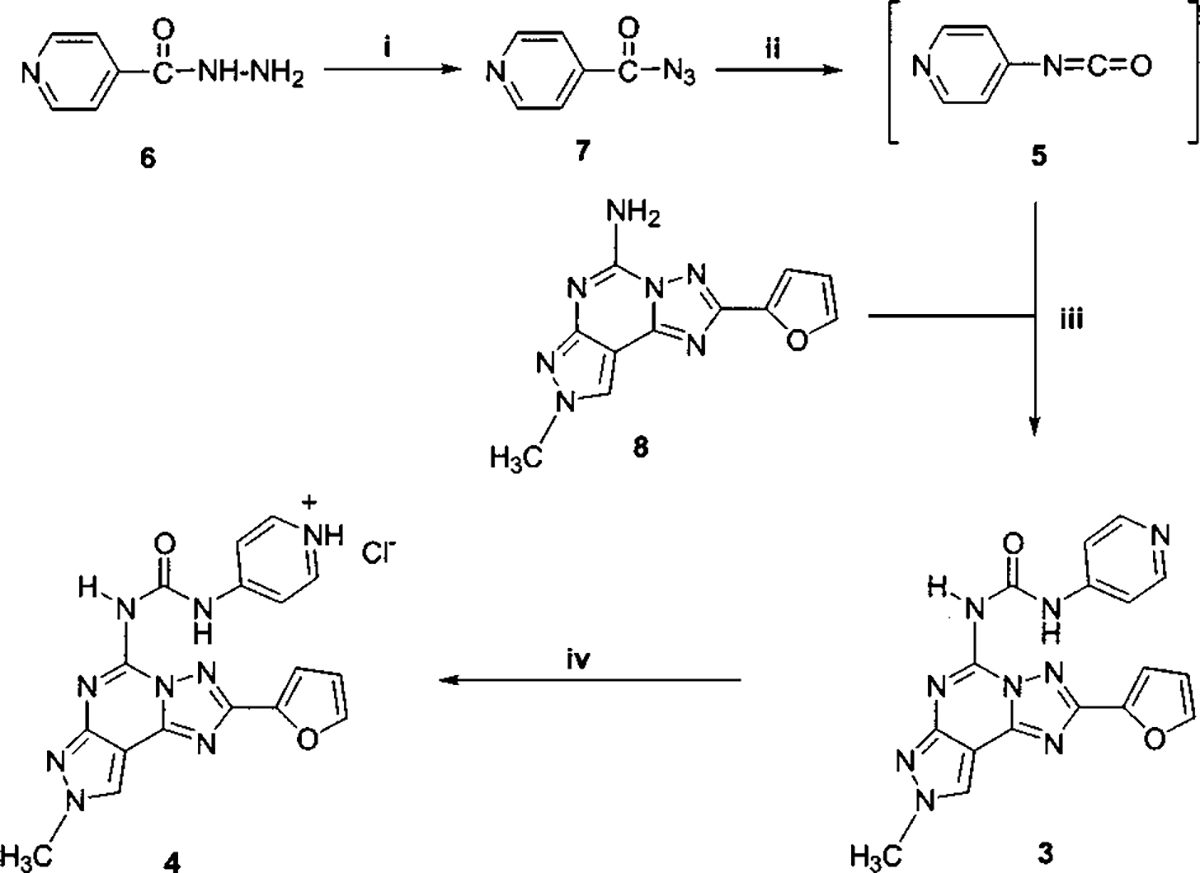
a (i) NaNO2, aqueous HCl, 0 °C, 1 h; (ii) benzene, reflux, 2 h; (iii) THF reflux overnight; (iv) HCl/MeOH, 0 °C, 30 min.
For this reason, 5 was prepared as reported in the literature starting from the commercially available isonicotinoyl hydrazide (6), which after reaction with sodium nitrite under acid conditions afforded the corresponding acyl azide 7.15 The latter was in turn converted into 5 upon Curtius rearrangement induced by heating 7 at reflux in dry benzene for 2 h.16 The crude isocyanate was refluxed overnight in dry THF with the precursor containing the tricyclic system bearing a methyl group at the N8 position (8). The desired product 3 was purified using flash chromatography and eluted with a MeOH/EtOAc gradient of 0–30%. The corresponding hydrochloride 4 was obtained by treatment of 3 for 30 min at 0 °C with methanol saturated with HCl gas. The hydrophobicity of the newly synthesized substances was measured in reverse-phase TLC experiments and reported as Rm values17 (Rm = log(1/Rf − 1) as shown in Table 1. As expected, both derivatives showed increased hydrophilicity with respect to reference 1, but most importantly, the hydrochloride salt 4 freely dissolved in water to a maximum concentration of 15 mM. Table 1 also compares the receptor binding affinities of 3 and 4 determined at human A1,18 A2A,19 A2B,20 and A320 receptors expressed in CHO (A1, A2A, A3) and HEK-293 (A2B) cells.
Table 1.
Binding Affinity at hA1, hA2A, hA2B, and hA3 Adenosine Receptors and Water Solubility of Synthesized Compounds
| compd | Rm(0)a | hA1 Ki (nM)b | hA2A Ki (nM)c | hA2B Ki (nM)d | hA3 Ki (nM)e | hA1/hA3 | hA2A/hA3 | hA2B/hA3 |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| 3 | 3.06 ± 0.06 | 250 ± 23 | 60 ± 10 | 200 ± 16 | 0.04 ± 0.009 | 6250 | 1500 | 5000 |
| 4 | 2.29 ± 0.05 | 350 ± 22 | 100 ± 12 | 250 ± 24 | 0.01 ± 0.005 | 35000 | 10000 | 25000 |
The Rm values of 3 and 4 were measured with a mobile phase of different concentrations of MeOH/H2O. Rm values are reported as theoretical at 0% organic solvent in the mobile phase (Rm(0)).
Displacement of specific [3H]DPCPX26 binding at human A2A receptors expressed in CHO cells (n = 3–6).
Displacement of specific [3H]ZM 24138526 binding at human A2A receptors expressed in HEK-293 cells.
Displacement of specific [3H]DPCPX binding at human A2B receptors expressed in HEK-29326 cells (n = 3–6).
Displacement of specific [3H]MRE3008-F2 026 binding at human A3 receptors expressed in HEK-293 cells. Data are expressed as Ki ± SEM (n = 3–6). The affinity of 4 at the rat adenosine receptor in RBL-2H3 cells was also determined using previously reported methods.21 Ki values (n = 3) were 226 ± 50 nM (A1), 97.6 ± 26.2 nM (A2A), and >1 μM (A3).
Surprisingly, both substances showed very high affinity at the human A3 adenosine receptor subtype, with Ki values in the picomolar range (10–40 pM) and with high levels of selectivity. In particular, the hydrochloride salt 4 showed increased affinity and selectivity with respect to the reference compound bearing the phenylcarbamoyl moiety 1. The Ki value of 4 at the hA3 receptor was 0.01 nM, thus indicating high selectivity versus other subtypes: hA1/hA3 = 35 000, hA2A/hA3 = 10 000, hA2B/hA3 = 25 000. These values were more favorable than the selectivity ratios for compound 1, for which the Ki value at the hA3 was reported to be 0.16 nM: hA1/hA3 = 3700, hA2A/hA3 = 2400, hA2B/hA3 = 1400. Moreover, this class of compounds, as previously demonstrated, proved to be inactive in a rat model with Ki values at the rA3 typically greater than 1 μM.11 4 at 1 μM displaced only 35% of the specific binding of [125I]I-AB-MECA at A3 receptors in membranes of rat basophilic RBL-2H3 cells.21b Concerning the affinity differences observed between the salt and neutral species, we speculate that a difference in their relative dissociation/association rates during the biological assay could be a reasonable explanation. These results not only represent the first example of a highly potent, selective, and water-soluble human A3 adenosine antagonist but strongly suggest an involvement of the pyridine nitrogen in the receptor recognition. Using a homology modeling approach based on the crystal structure of the rhodopsin22 as a template, we have built an improved model of the transmembrane helical domains (TMs) of the human A3 receptor,23 which can be considered a further refinement of the hypothetical binding site for A3 receptor antagonists already proposed.13,22,24 As shown in Figure 1, after the Monte Carlo/annealing sampling, we propose that the hypothetical binding site of 4 is surrounded by TMs 3, 5, 6, and 7, with the furan ring pointing toward the extracellular environment. Similar to conformational results already described for other pyrazolotriazolopyrimidines,12,13,22 the lowest energy conformation of 4 featured the carbamoyl moiety in the 5-position surrounded by four polar amino acids: Ser242 (TM6), Ser271 (TM7), His274, and Ser275 (TM7).
Figure 1.
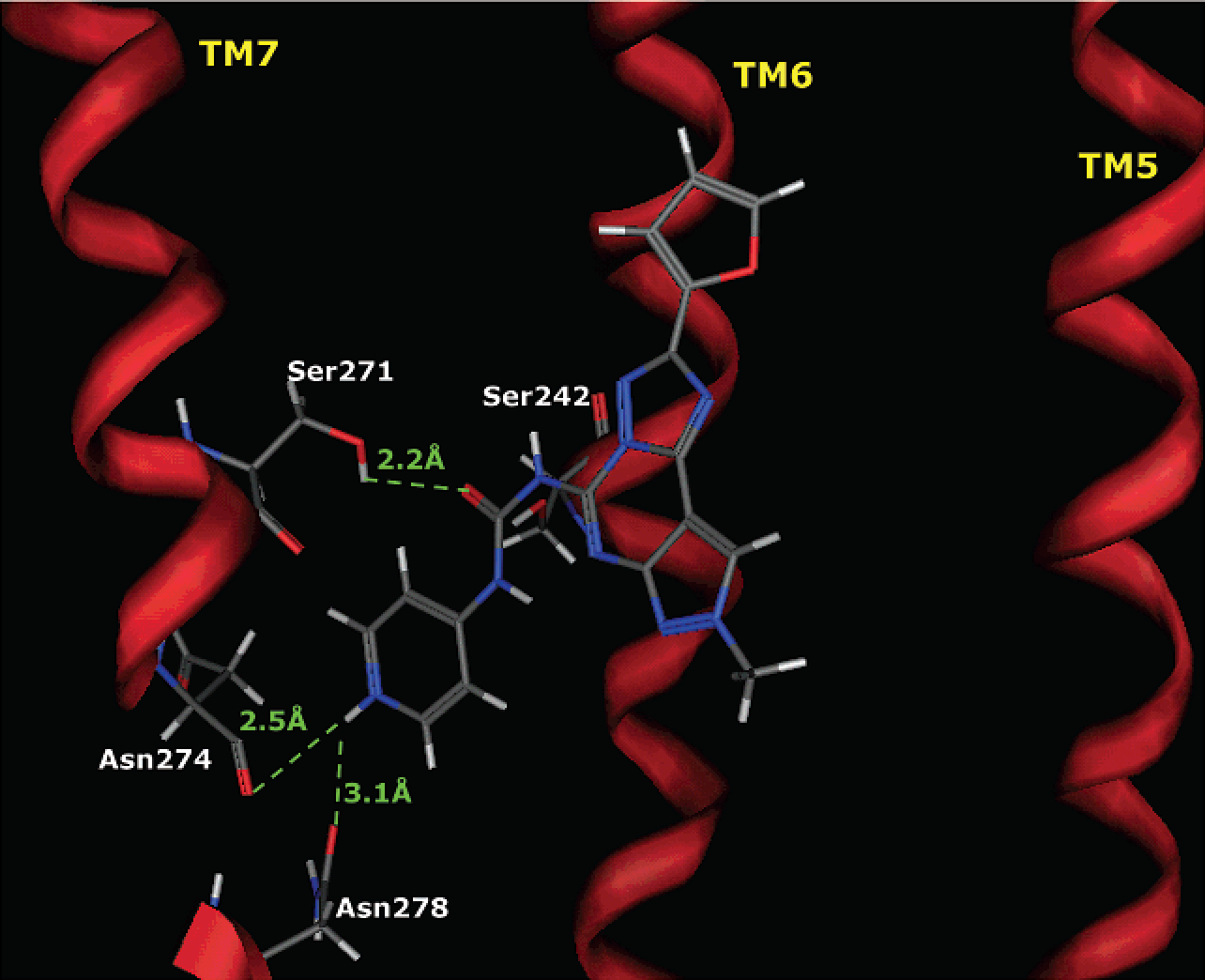
Side view of the human A3-4 complex model. The side chains of the important residues in proximity to the docked 4 molecule are highlighted and labeled.
Accordingly, this region seemed to be critical for the recognition of this class of antagonists. Moreover, additional strong electrostatic interactions appeared to occur between the positively charged pyridinium moiety of 4 and the carbonyl oxygen atoms of Asn274 (N+H····OC distance = 2.5 Å) and Asn278 (N+H····OC distance = 3.1 Å), both located on TM7. These electrostatic interactions might be responsible for the increase of the affinity in the protonated form, i.e., the hydrochloride derivative 4. Interestingly, these two asparagine residues (Asn274 and Asn278) are largely conserved among a number of GPCRs. It should be noted, however, that this human A3 receptor model, based on the TM region of the receptor, is not able to clearly explain the corresponding increase of selectivity for this subtype even taking into account which of the important TM residues are different in the other receptor subtpyes. However, as already reported by Jacobson and co-workers, multiple regions of the adenosine receptors, including a segment of the second extracellular loop, are involved in ligand recognition.25 Other investigations are in progress in our lab to better describe the role of the extracellular domain on the ligand recognition process. For confirmation of the high potency of this compound in a functional assay, the inhibition of cAMP26 generation by Cl-IB-MECA in membranes of CHO cells stably transfected with the human A3 receptor was evaluated.
Consistent with its binding affinity, 4 showed an IC50 value of 0.7 ± 0.06 nM, compared to an IC50 of 2.10 ± 0.21 nM for 1. A Schild analysis of the antagonism of the effects of 4 on Cl-IB-MECA26-induced inhibition of forskolin-stimulated cAMP was carried out (Figure 2).19 A KB value of 0.20 ± 0.03 nM was calculated, thus demonstrating 4 to be the most potent antagonist of the human A3 receptor ever reported.
Figure 2.
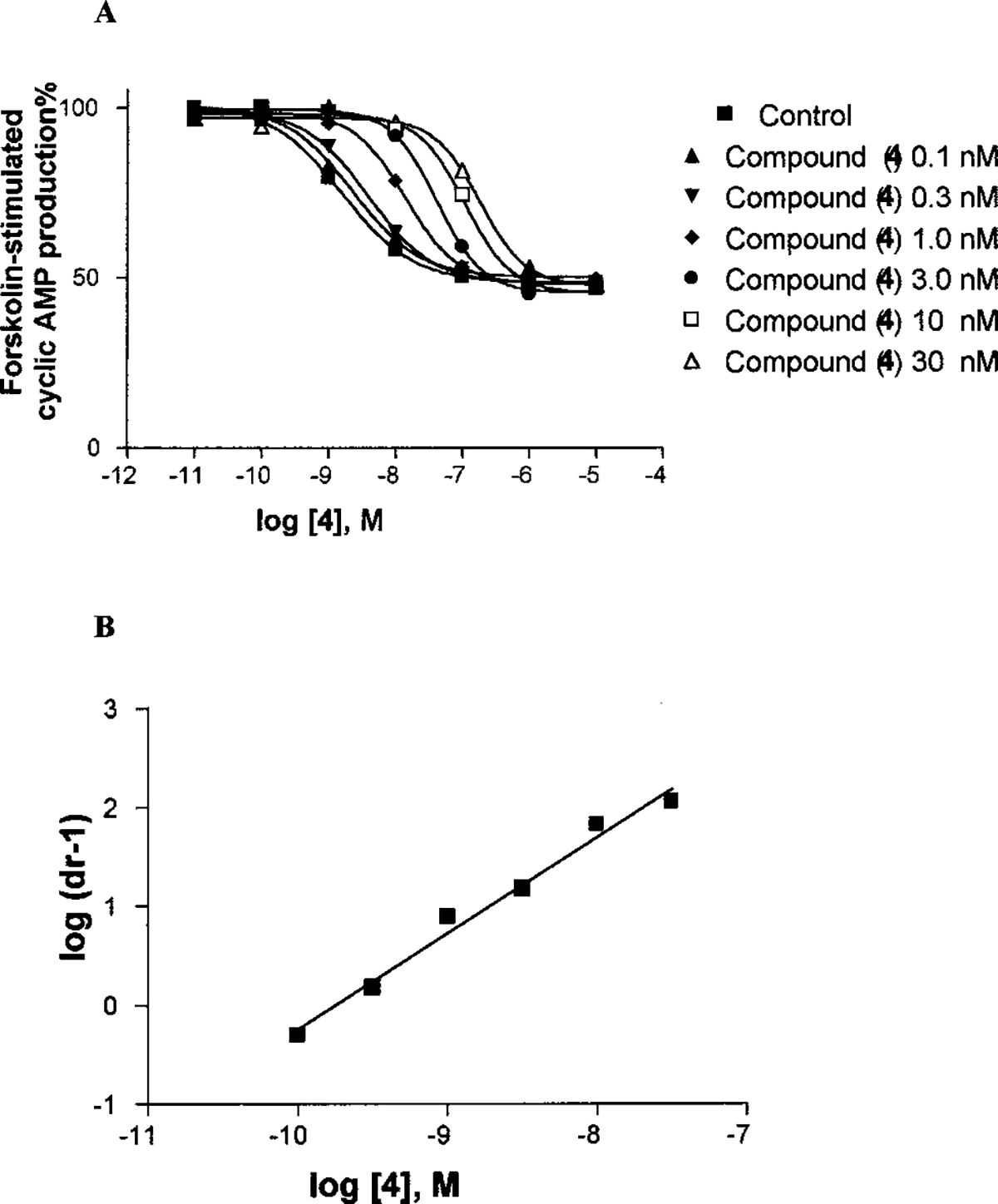
Effects of 4 on the inhibition of cyclic AMP production induced by the agonist Cl-IB-MECA in human A3 adenosine receptor-expressing CHO cells (A) and Schild analysis (B) of the data. The procedures used are described in Supporting Information. The data shown in panel A were derived from one experiment performed in duplicate and are typical of three independent experiments giving similar results. The KB value for antagonism by 4 was calculated from three independent experiments.
Conclusions.
The present study revealed a novel, potent, selective, and most importantly, water-soluble hA3 adenosine receptor antagonist. This derivative featured a basic 4-pyridylcarbamoyl moiety at the N5 position of the pyrazolotriazolopyrimidine nucleus, and the corresponding hydrochloride salt 4 displayed a Ki value of 0.01 nM at the hA3 and selectivities versus the other adenosine receptor subtypes ranging from 10 000 to 35 000. This increase of affinity compared to neutral arylcarbamate derivatives could be attributed in receptor modeling to strong electrostatic interactions between the pyridinium moiety of 4 and the side chain carbonyl oxygen atoms of Asn274 and Asn278, both located on TM7. In view of the potency, selectivity, and water solubility, 4 could be an ideal candidate for pharmacological and clinical investigation of the hA3 adenosine receptor subtype.
Supplementary Material
Acknowledgment.
We thank the Regione Friuli Venezia Giulia and the University of Trieste for financial support.
Footnotes
Supporting Information Available: Experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Fredholm BB; Ijzerman AP; Jacobson KA; Klotz KN; Linden J International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001, 53, 527–552. [PMC free article] [PubMed] [Google Scholar]
- (2).Ralevic V; Burnstock G Receptors for purines and pyrimidines. Pharmacol. Rev. 1998, 50, 413–492. [PubMed] [Google Scholar]
- (3).Salvatore CA; Jacobson MA; Taylor HE; Linden J; Johnson RG Molecular cloning and characterization of the human A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 10365–10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jacobson KA; Suzuki F Recent developments in selective agonists and antagonists acting at purine and pyrimidine receptors. Drug. Dev. Res. 1996, 39, 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Abbracchio MP; Brambilla R; Kim HO; von Lubitz DKJE; Jacobson KA; Cattabeni F G-protein-dependent activation of phospholipase-C by adenosine A3 receptor in rat brain. Mol. Pharmacol. 1995, 48, 1038–1045. [PubMed] [Google Scholar]
- (6).Ali H; Choi OH; Fraundorfer PF; Yamada K; Gonzaga HMS; Beaven MA Sustained activation of phospholipase-D via adenosine A3 receptors is associated with enhancement of antigen-ionophore-induced and Ca2+-ionophore-induced secretion in a rat mast-cell line. J. Pharmacol. Exp. Ther. 1996, 276, 837–845. [PubMed] [Google Scholar]
- (7).Ramkumar V; Stiles GL; Beaven MA; Ali H The A3 adenosine receptors is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J. Biol. Chem. 1993, 268, 16887–16890. [PubMed] [Google Scholar]
- (8).Jacobson KA; Moro S; Kim YC; Li AH A3 adenosine receptors: protective vs. damaging effects identified using novel agonists and antagonists. Drug Dev. Res. 1998, 45, 113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Brambilla R; Cattabeni F; Ceruti S; Barbieri D; Franceschi C; Kim Y; Jacobson KA; Klotz KN; Lohse MJ; Abbracchio MP Activation of the A3 adenosine receptor effects cell cycle progression and cell growth. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2000, 361, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Baraldi PG; Cacciari B; Romagnoli R; Merighi S; Varani K; Borea PA; Spalluto G A3 Adenosine receptor ligands; history and perspectives. Med. Res. Rev. 2000, 20, 103–128. [DOI] [PubMed] [Google Scholar]
- (11).Baraldi PG; Cacciari B; Romagnoli R; Spalluto G; Klotz K-N; Leung E; Varani K; Gessi S; Merighi S; Borea PA Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as highly potent and selective human A3 adenosine receptor antagonists. J. Med. Chem. 1999, 42, 4473–4478. [DOI] [PubMed] [Google Scholar]
- (12).Baraldi PG; Cacciari B; Romagnoli R; Spalluto G; Moro S; Klotz KN; Leung E; Varani K; Gessi S; Merighi S; Borea PA Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as highly potent and selective human A3 adenosine receptor antagonists: Influence of the chain at N8 pyrazole nitrogen. J. Med. Chem. 2000, 43, 4768–4780. [DOI] [PubMed] [Google Scholar]
- (13).Baraldi PG; Cacciari B; Moro S; Spalluto G; Pastorin G; Da Ros T; Klotz K-N; Varani K; Gessi S; Borea PA Synthesis, Biological Activity, and Molecular Modeling Investigation of New Pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine Derivatives as Human A3 Adenosine Receptor Antagonists. J. Med. Chem. 2002, 45, 770–780. [DOI] [PubMed] [Google Scholar]
- (14).Singha CN; Dixit N; Sathyanarayana DN 1H and 13C NMR spectra of some unsymmetric N,N′-dipyridyl ureas: spectral assignments and molecular conformations. J. Chem. Soc., Perkin Trans. 2 1997, 157–162. [Google Scholar]
- (15).Curtius T; Mohr E Transformation of nicotinic acid to beta-amidopyridine. Ber. 1898, 31, 2493–2495. [Google Scholar]
- (16).Hyden S; Wilbert G Pyridine isocyanates. Chem. Ind. (London) 1967, 33, 1406–1407. [Google Scholar]
- (17).Biagi GL; Barbaro AM; Sapone A; Borea PA; Varani K; Recanatini M Study of liphophilic character of serotoninergic ligands. J. Chromatogr., A 1996, 723, 135–143. [Google Scholar]
- (18).Lohse MJ; Klotz K-N; Lindernborn-Fotinos J; Reddington M; Schwabe U; Olsson RA 8-Cyclopentyl 1,3-dipropylxanthine DPCPX a selective high affinity antagonist radioligand for A1 adenosine receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1987, 336, 204–210. [DOI] [PubMed] [Google Scholar]
- (19).Ongini E; Dionisotti S; Gessi S; Irenius E; Fredholm BB Comparison of CGS 15943 and SCH 58261 as antagonist at human A3 adenosine receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1999, 359, 7–10. [DOI] [PubMed] [Google Scholar]
- (20).Varani K; Merighi S; Gessi S; Klotz KN; Leung E; Baraldi PG; Cacciari B; Spalluto G; Borea PA [3H]MRE3008-F20: a novel antagonist radioligand for the pharmacological and biochemical characterization of human A3 adenosine receptors. Mol. Pharmacol. 2000, 57, 968–975. [PubMed] [Google Scholar]
- (21).(a) Jacobson KA; Gallo-Rodriguez C; Melman N; Fischer B; Maillard M; van Bergen A; van Galen PJM; Karton Y Structure-activity relationships of 8-styrylxanthines as A2-selective adenosine antagonists. J. Med. Chem. 1993, 36, 1333–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ji X-D; Gallo-Rodriguez C; Jacobson KA A selective affinity label for A3 adenosine receptors. Biochem. Biophys. Res. Commun. 1994, 203, 570–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Baraldi PG; Caccairi B; Romagnoli R; Moro S; Ji X-D; Jacobson KA; Gessi S; Borea PA; Spalluto G Fluorosulfonyl- and bis-(β-chloroethyl)amino-phenyl functionalized pyrazolo[4,3-e]1,2,4-triazolo[1,5-c]pyrimidine derivatives as irreversible antagonists at the human A3 adenosine receptor: molecular modeling studies. J. Med. Chem. 2001, 44, 2735–2742. [DOI] [PubMed] [Google Scholar]
- (23).Palczewski K; Kumasaka T; Hori T; Behnke CA; Motoshima H; Fox BA; Trong IL; Teller DC; Okada T; Stenkamp RE; Yamamoto M; Miyano M Crystal structure of rhodopsin: a G protein-coupled receptor. Science 2000, 289, 739–745. [DOI] [PubMed] [Google Scholar]
- (24).Moro S; Li AH; Jacobson KA Molecular modeling studies of human A3 adenosine antagonists: structural homology and receptor docking. J. Chem. Inf. Comput. Sci. 1998, 38, 1239–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Olah ME; Jacobson KA; Stiles GL Role of the second extracellular loop of adenosine receptors in agonist and antagonist binding. Analysis of chimeric A1/A3 adenosine receptors. J. Biol. Chem. 1994, 269, 24692–24698. [PMC free article] [PubMed] [Google Scholar]
- (26).Abbreviations: DPCPX,8-cyclopentyl-1,3-dipropylxanthine;[125I]-AB-MECA, [125I]-1-[6-[[(4-amino-3-iodophenyl)methyl]amino]-9H-purin-9-yl]-1-deoxy-N-methyl-β-D-ribofuranuronamide; THF, tetrahydrofuran; CHO, Chinese hamster ovary; HEK, human embryonic kidney; MRE3008-F20, 5-[[(4-methoxyphenyl)amino]-carbonyl]amino-8-propyl-2-(2-furyl)-pyrazolo[4,3-e]-1,2,4-triazolo-[1,5-c]pyrimidine; ZM 241385, 5-amino-7-(2-phenylethyl)-2-(2-furyl)pyrazolo[4,3-e]-1,2,4-triazolo[1,5-c]pyrimidine; Cl-IB-MECA, 2-chloro-3-iodobenzyl-5′-(N-methylcarbamoyl)adenosine; cAMP, cyclic adenosine-5′-monophosphate′.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.