

Abstract
Elimination of misfolded proteins by endoplasmic reticulum (ER)-associated protein degradation (ERAD) ensures that proteins proceeding through the secretory pathway are correctly folded and processed, which is critical to minimize ER stress. All ERAD pathways include a protein translocation process termed retrotranslocation, in which ubiquitinated misfolded substrates are extracted from the ER and degraded by the cytosolic 26S proteasome. Despite being integral to ERAD, the retrotranslocation process has been largely obscure. Recently, an explosion of discoveries has provided key mechanistic insights into this novel route of protein transport. These advances were facilitated by the development of in vitro and in vivo assays that utilize components from the yeast Saccharomyces cerevisiae. The assays permit detailed study of the distinct steps in ERAD-linked retrotranslocation, including ubiquitination of selected ERAD substrates, substrate removal from the ER, maintenance of cytosolic substrate solubility in the cytosol, and substrate degradation. Here we provide detailed protocols for these assays that pertain to work on retrotranslocation of integral membrane proteins (ERAD-M substrates), with the expectation that these approaches can be adapted for many related biochemical processes.
1. Introduction
Endoplasmic reticulum-associated protein degradation (ERAD) is a highly conserved quality control pathway that eliminates damaged or unassembled ER proteins (Foresti, Ruggiano, Hannibal-Bach, Ejsing, & Carvalho, 2013; Hampton & Garza, 2009). Defects in ERAD result in a toxic buildup of misfolded proteins and is associated with many pressing human maladies such as aging, neurodegenerative diseases, retinal degeneration, and diabetes (Chiti & Dobson, 2017; Hara et al., 2014; Zhao & Ackerman, 2006). ERAD occurs through the ubiquitin-proteasome pathway. ERAD substrates are poly-ubiquitinated, which marks them for degradation by the 26S proteasome. This ubiquitination is mediated by the sequential action of three enzymes. An E1 ubiquitin-activating enzyme uses ATP to create a high-energy thioester between ubiquitin and the E1 and then transfers the ubiquitin to the active-site cysteine of an E2 ubiquitin-conjugating (UBC) enzyme. Ubiquitin is then transferred from the E2 to the substrate or the growing ubiquitin chain by the action of an E3 ubiquitin ligase, resulting in a substrate-attached ubiquitin chain that is recognized and degraded by the proteasome (Bays, Wilhovsky, Goradia, Hodgkiss-Harlow, & Hampton, 2001; Hiller, Finger, Schweiger, & Wolf, 1996; Jarosch et al., 2002). Substrate specificity for a given pathway is largely determined by the E3 ligase.
In Saccharomyces cerevisiae, ERAD is mediated by the HRD (HMG-CoA Reductase Degradation) and DOA (Degradation of alpha2) pathways, both of which are conserved in virtually all eukaryotes (Carvalho, Goder, & Rapoport, 2006; Chen et al., 2006; Foresti et al., 2013; Hampton & Garza, 2009). In the HRD pathway, the Hrd1 E3 ligase targets a variety of substrates, including misfolded substrates with lesions within the membrane bilayer (ERAD-M), substrates with luminal lesions (ERAD-L), and some normal proteins such as the HMGR sterol synthetic enzyme (Bordallo, Plemper, Finger, & Wolf, 1998; Hampton, Gardner, & Rine, 1996). In the DOA pathway, the E3 ubiquitin ligase Doa10 ubiquitinates misfolded soluble and membrane proteins with lesions in their cytosolic domains (ERAD-C) but is also capable of targeting certain ERAD-M substrates (Wangeline, Vashistha, & Hampton, 2017).
Spatial separation of the various ERAD processes presents a major challenge for cells. In all examples of ERAD, there is a requirement to move substrates from their starting point within the ER lumen or ER membrane to the cytosol for proteasome-mediated destruction. This transport component of ERAD, broadly referred to as dislocation or retrotranslocation, has been known to occur since the earliest studies of ERAD when the cytosolic E2 ligase Ubc7 was identified to mediate the ubiquitin-dependent degradation of the luminal ERAD substrate CPY* (Hampton & Sommer, 2012; Hiller et al., 1996). Furthermore, many studies concluded that the Cdc48 ATPase (p97 in mammals) is required to drive the retrotranslocation process (Bays et al., 2001; Jarosch et al., 2002; Ye, Meyer, & Rapoport, 2003). The general requirement for retrotranslocation in all ERAD pathways is gradually revealing the mechanistic aspects of retrotranslocation, including the components required, the energetics, and the mechanism of transfer through the ER membrane. However, many open questions remain; to address these issues, we had previously developed an in vitro assay using the eight-transmembrane-spanning ER-resident substrate Hmg2, an isozyme of HMG-CoA reductase fused to GFP, which undergoes regulated ERAD mediated by the E3 ligase Hrd1 (Fig. 1A) (Garza, Sato, & Hampton, 2009; Garza, Tran, & Hampton, 2009; Hampton, 2005).
Fig. 1.
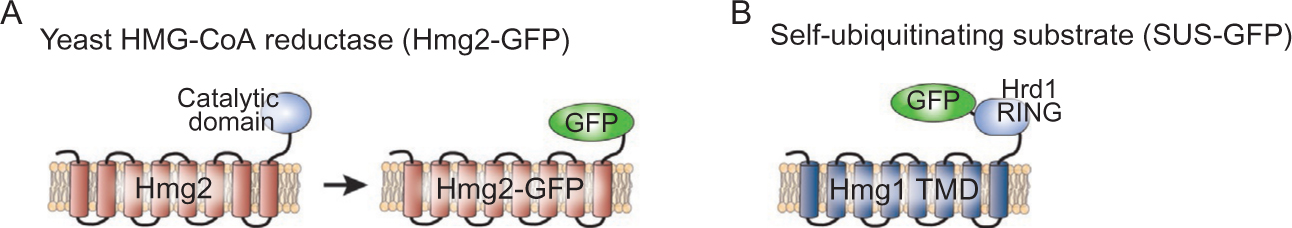
Fusion substrates used in the in vivo and in vitro retrotranslocation assays. (A) Hmg2 and the fluorescent fusion reporter Hmg2-GFP. Hmg2-GFP has the authentic catalytic domain of Hmg2 replaced by the GFP reporter and is able to undergo regulated degradation. (B) Fluorescent fusion retrotranslocation reporter, SUS-GFP. The transmembrane stable isoform of HMGR, Hmg1, is fused to the Hrd1 cytosolic RING domain along with the GFP at the C-terminus.
This assay has allowed several insights into ERAD-M retrotranslocation. First, it was demonstrated that the entire integral membrane Hmg2 protein is removed from the ER membrane and remains intact and soluble in the cytosol if proteasomal degradation is inhibited. Second, ATP hydrolysis by Cdc48 was found to be required for extraction of the tested integral membrane substrates. Third, the Hrd1 ligase was rate-limiting for retrotranslocation of these substrates (Garza, Sato, & Hampton, 2009; Garza, Tran, & Hampton, 2009).
It is thought that an aqueous protein channel allows the movement of substrates across the ER membrane. Channel candidates include the Derlins (Lilley & Ploegh, 2004; Sun et al., 2006), the Sec61 anterograde channel (Plemper, Böhmler, Bordallo, Sommer, & Wolf, 1997), or the multispanning domains of the ER ligases themselves (Baldridge & Rapoport, 2016; Plemper et al., 1997; Ravid, Kreft, & Hochstrasser, 2006). Recently, a strong case has been made for Hrd1 itself serving as an ERAD-L channel by the Rapoport group using in vitro reconstitution studies employing purified components from S. cerevisiae (Baldridge & Rapoport, 2016; Stein, Ruggiano, Carvalho, & Rapoport, 2014). This was further supported by a Cryo-EM structure of Hrd1, which revealed that the multispanning E3 ligase includes a channel that at least partially traverses the membrane (Schoebel et al., 2017). Finally, in vivo studies of Hrd1 led to a model in which autoubiquitination of Hrd1 opens the channel to catalyze retrotranslocation (Baldridge & Rapoport, 2016; Stein et al., 2014).
Although the studies above implicate Hrd1 as a channel for ERAD-L retrotranslocation, this factor did not appear to be involved in the retrotranslocation of multispanning integral membrane substrates of the ERAD-M class. The Hampton group used the in vitro retrotranslocation assay to examine the role of Hrd1 as a retrotranslocon for ERAD-M degradation (Garza, Sato, & Hampton, 2009). However, simply removing the ligase to test its role in retrotranslocation was not possible since Hrd1-dependent substrate ubiquitination is also a prerequisite for retrotranslocation. To uncouple E3 ligase activity from any potential channel activity, we devised a self-ubiquitinating substrate (SUS), which was composed of the multispanning membrane region fromHmg1(the stable HMGR isozyme) attached to the catalytic domain of Hrd1 (Fig. 1B). Using the Hrd1 catalytic domain, SUS catalyzes its own ubiquitination and degradation, with the appropriate dependencies including the attached Hrd1 E3RING domain, the Ubc7 E2 ligase, and Cdc48 (Garza, Sato, & Hampton, 2009). These studies showed that the integral membrane SUS protein does not require Hrd1 (or Doa10) for its degradation in vivo, despite the fact that it undergoes full retrotranslocation in the in vitro assay in a manner identical to bone fide ERAD-M substrates such as Hmg2. The behavior of the SUS substrate left open the question of how exactly it undergoes degradation and what the mechanism of retrotranslocation might be.
To complement the in vitro assay, we recently developed a physiologically relevant in vivo retrotranslocation assay (Neal, Mak, Bennett, & Hampton, 2017), inspired by earlier work from the Sommer group assaying in vivo ERAD-L retrotranslocation (Mehnert, Sommer, & Jarosch, 2013). The in vivo assay recapitulates two important features of ERAD that, for unknown reasons, were absent in the in vitro assay. First, Hmg2 examined in the in vivo assay undergoes normal, regulated degradation in response to the sterol pathway intermediate geranylgeranyl pyrophosphate (GGPP). Second, the effect of removing the proteasome adaptors Dsk2 and Rad23 in the in vivo assay placed them in the expected position along the HRD pathway: after retrotranslocation and before proteasomal degradation. The ease of the in vivo assay allowed us to quickly score candidate mutants—obtained from a yeast genetic screen—for retrotranslocation function. Use of this assay confirmed that SUS-GFP indeed undergoes retrotranslocation that is independent of Hrd1, leading to the genetic analysis that revealed the Dfm1 retrotranslocationfactorforERAD-Msubstrates(Nealetal.,2018). Theassay is described in detail below.
Due to the unexpected solubility of multispanning membrane ERAD substrates following retrotranslocation, the factors or molecules facilitating the solubility of these hydrophobic proteins in the cytosol are of great interest. A strong suit of the in vivo assay is the ability to isolate the retrotranslocated intermediate from intact cells and to perform biophysical and proteomic analyses on retrotranslocated integral membrane substrates such as Hmg2. Using this assay has led to two important findings: (1) Retrotranslocated Hmg2 is not associated with lipids but is instead associated with proteins and (2) proteomic analysis of retrotranslocated Hmg2 identified Cdc48 as the major “retrochaperone” responsible for solubilizing full-length intact Hmg2 in the cytosol (Neal et al., 2017). Cdc48 was shown to be stochiometrically bound to retrotranslocated, ubiquitinated Hmg2 and is essential for preserving the solubility of this client protein on its way to the proteasome. Protocols for the in vivo assay that led to these discoveries are described in Section 3.
2. An in vitro assay for retrotranslocation of Hmg2-GFP
This section lists the yeast strains required to perform this assay, the detailed descriptions of the methods to monitor ubiquitination of Hmg2-GFP, and in vitro assay used to directly observe Hmg2-GFP retrotranslocation. In this assay, Hrd1 is expressed from the strong constitutive TDH3 promoter at levels sufficient to promote ERAD in the absence of other associated ERAD factors, includingHrd3, Usa1, and Yos9 (Carroll & Hampton, 2009; Carvalho et al., 2006; Mehnert et al., 2015; Vashistha, Neal, Singh, Carroll, & Hampton, 2016). (Notably, Hrd1 at native levels does not achieve optimal ubiquitination and degradation of Hmg2-GFP in the in vitro assay.) Accordingly, this approach defines the minimal components sufficient for successful retrotranslocation, providing the avenue for complete reconstitution. Hrd1 overexpressed at this level causes sufficient ubiquitination of Hmg2-GFP for the recovery and direct detection of the retrotranslocated substrate (see below). However, ectopic expression of Hrd1 in vivo alters Hmg2 ERAD, abrogating its physiological regulation by GGPP and changing the dependence of the Rad23/Dsk2 adaptors. Nevertheless, it has been very useful for a variety of studies (Bazirgan & Hampton, 2008; Garza, Sato, & Hampton, 2009; Sato, Schulz, Do, & Hampton, 2009).
2.1. Materials
2.1.1. Strains
All yeast strains are derived from the S288C background as described previously (Hampton & Rine, 1994). The initial genotype of the yeast strain used to create the strains used as cytosol and microsome donors is the following:
Matα ura3–52 ade2–101 met2 lys2–801 his3Δ200 trp1::hisG leu2Δ pep4Δ::HIS3 hmg2Δ::1myc-HMG2 hrd1Δ::kanMX ubc7Δ::LEU2 (Strain RHY4288).
The specific genetic insertions added to RHY4288 to create the respective microsome and donor strains are shown below. Mutations in other genes can then be introduced into one or the other strain.
| 1: Microsome donor strain(RHY2923): | ura3–52:: URA3 :: |
| TDH3PROM–HMG2–GFP | |
| trp1 :: hisG :: TRP1 :: | |
| TDH3PROM–HRD1–3HA | |
| 2. Cytosol donor strain(RHY4295): | trp1 :: hisG :: TRP1 :: |
| TDH3PROM–UBC7–2HA |
2.1.2. Microsome preparation
Microsome preparation from S. cerevisiae has been well documented (Garza, Sato, & Hampton, 2009; Neal et al., 2018, 2017) and is described in detail below with additional steps to ensure optimal yields. In most cases, microsome donor strains are grown at 30°C with aeration and vigorous shaking. With temperature-sensitive mutants, cells are grown at permissive temperature at 25°C and then shifted to the restrictive temperature at 37°C. To recapitulate the temperature-sensitive phenotype in the in vitro assay, the duration of growth at the restrictive temperature must be determined empirically.
The microsome donor strain is grown at the desired temperature in minimal medium (Garza, Sato, & Hampton, 2009) at an initial optical density at 600nm (OD600) of 0.1 OD until the culture reaches mid-log phase (OD600 of 0.3–0.5). 200mL of yeast are typically grown for microsome preparation. Note: We have observed that when microsome strains are grown in YPD, this condition induces loss of the overexpressed HRD1 gene. We do not observe this when strains are grown in minimal media.
30 OD units of cells are collected and prepared for bead lysis in a 50mL Falcon tube. Cell pellets are resuspended in 600μL of membrane fractionation (MF) buffer (20mM Tris–HCl pH 7.5, 100mM NaCl, 300mM sorbitol, with the following protease inhibitors (PIs): 1mM phenylmethylsulfonyl fluoride, 260μM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride, 100μM leupeptin hemisulfate, 76μM pepstatin A, 5mM aminocaproic acid, 5mM benzamidine, and 142μM N-tosyl-L-phenylalanine chloromethyl ketone). Note: phenylmethylsulphonyl fluoride is prepared fresh for each experiment.
Beads are added until they reach the meniscus of the cell suspension, and cells are lysed at 4°C using hand vortexing at top speed for 6 × 1-min intervals with 1-min intervals on ice between each vortexing. Lysis efficiency can be evaluated under the microscope using 20× magnification. Lysed cells are clearly distinguishable by their fragmented shape; for optimal yield it is critical to achieve ~80%–90% lysis efficiency. If below this range, continue to vortex for up to three more 1-min cycles at 4°C. We use either 0.5mm glass-based or silica-based beads for lysis (Biospec Products), but the lysis efficiency with silica-based beads appears to be somewhat higher. It is critical that each of the following steps is performed at 4°C.
The crude lysate is collected with a 1mL pipette and transferred to a 1.5mL Eppendorf tube. 200μL of cold MF buffer are used to rinse the beads, and this rinse fraction is added to the original lysate. The extract is centrifuged for 5s in a microcentrifuge (16,000 × g), and the resulting supernatant is transferred to a fresh 2mL Eppendorf tube. Centrifugation and transfer to fresh tubes is repeated until no pelleted cellular debris can be seen. This is typically repeated approximately six times.
Next, the microsomes are pelleted in a microcentrifuge at 20,000 × g for 30min. The pellets are resuspended in B88 buffer (20mM Hepes pH 6.8, 250mM sorbitol, 150mM KOAc, 5mM MgOAc, freshly made 1mM DTT, and the PIs listed above), to a final concentration of 0.3 OD equivalent units/μL of B88.
It is important to immediately use the microsome preparation for in vitro ubiquitination and retrotranslocation assays. In our experience, using fresh microsome preparations works best for in vitro ERAD assays; freezing these preparations does not reliably preserve function, although this avenue has not been intensely pursued.
2.1.3. Cytosol preparation
Cytosol is prepared from strains overexpressing Ubc7 in a hrd1 Δ ubc7 Δ double null strain in a manner similar to that described by Spang and Schekman (1998), using rapid freezing of cells with liquid nitrogen followed by mortar and pestle disruption of the frozen cells. Control cytosol is prepared in parallel from an otherwise identical ubc7 Δ strain (RHY4288). Smaller scale cytosol preparation can be performed with similar activity as described previously (Heck, Cheung, & Hampton, 2010). Again, for temperature-sensitive strains, duration for growth at non-permissive temperature must be validated empirically.
Cytosol donor strains are grown at the desired temperature in rich medium to mid-log phase (OD600 = ~1) at 30°C. 500mL of yeast are typically grown for cytosol preparation.
500 OD equivalents of cells are pelleted for 10min at room temperature at 5000 × g. The cells are rinsed once with water, once with B88 buffer, and resuspended in 500μL of B88 buffer (1 OD/μL B88 buffer) with the same amount of PIs as mentioned for microsomal preparation.
For cell lysis, the resuspended cells are transferred to a chilled mortar containing liquid nitrogen. The frozen cells are grounded with a pestle until a fine powder is achieved; this is usually achieved with approximately 15 strokes. During pestle treatment, it can be useful to keep a reservoir of liquid nitrogen to maintain freezing of the pellet during grinding. The frozen powder is transferred to a chilled 50mL Falcon tube and thawed on ice. It is critical that the each of the following steps is performed at 4°C. Note: the frozen powder may be stored at −80°C for later use.
ATP (Sigma) is added to the thawed cytosol from a 500mM stock solution to make a final added concentration of 1mM. Note: For AMP-PNP (Sigma) experiments, which test the role of ATP hydrolysis in ERAD, ATP is not added. ATP stock is typically made with deionized water, adjusted to pH 7.5, aliquoted and frozen at −80°C. An aliquot of ATP is thawed at the start of each in vitro ubiquitination experiment.
Once thawed, the crude cytosol lysate is centrifuged for 5min with a microcentrifuge (3000 × g) to remove debris, and then the supernatant is transferred to a new tube and centrifuged for 15 min with a microcentrifuge (20,000 × g).
The resulting supernatant is then ultracentrifuged at 100,000 × g for 1h using a Beckman TL-1000 tabletop ultracentrifuge along with a TLA 100.4rotor. After ultracentrifugation, the supernatant is removed carefully with a precision loading tip. The resulting cytosol preparation is measured for protein concentration with a NanoDrop (ThermoFisher) spectrophotometer [Absorbance at 280nm]. Cytosol protein concentrations are adjusted with B88 to 20–25mg/mL for ubiquitination and retrotranslocation assays, aliquoted and stored at −80°C. This concentration has worked best in the ubiquitination and retrotranslocation assays in the past as long as samples are not refrozen and thawed again.
2.2. In vitro ubiquitination of Hmg2-GFP
Prior to starting the in vitro ubiquitination assay, MG132 (benzyloxycarbonyl-Leu-Leu-aldehyde, Sigma) is added to a final concentration of 300μM to both microsome and cytosol preparations. Note: We found that the intensity of the ubiquitin signal for a membrane substrate is greater when proteasome inhibitor MG132 is added, suggesting that the 20S core proteasome activity causes a reduction in ubiquitination levels, either due to in vitro degradation of a fraction of the substrate by the proteasome or to increased activity of proteasome-associated ubiquitin-specific proteases that are sensitive to 20S proteasome inhibition (Garza, Sato, & Hampton, 2009; Neal et al., 2017). The schematic for the in vitro ubiquitination assay is depicted in Fig. 2 and described in detail below.
A typical in vitro ubiquitination reaction consists of a mixture of 10μL microsomes and 12μL cytosol.
To initiate the reaction, 1.3μL ATP (500mM stock) is added to each mixture to a final concentration of 30mM and the reactions are incubated for 1h at 30°C. From our experience, 30mM ATP and a reaction time of 1h give an optimal signal for Hmg2-GFP ubiquitination. For other substrates of interest, the optimal concentration of ATP and reaction time should be determined empirically. This is especially important since in a number of assays, there is an optimal ATP concentration that if exceed will begin to diminish ubiquitination signal intensity (Heck et al., 2010).
The reaction is stopped by addition of 200μL of SUME buffer (1% SDS, 8 M urea, 10mM MOPS pH 6.8, 10mM EDTA) with PIs as above and 5mM N-ethylmaleimide (NEM, Sigma) to terminate the reactions and solubilize all proteins. Note: Ubiquitination is reversible and this modification can therefore easily be eliminated by deubiquitinases (DUBs). For this reason, it is essential to include DUB inhibitors such as NEM in the buffers used during the long incubation times used for immunoprecipitation in order to preserve the state of substrate ubiquitination (most DUBs are cysteine proteases that are inhibited by NEM).
Ubiquitination of Hmg2-GFP is measured by immunoprecipitation (IP) of Hmg2-GFP, resolution of the IP by SDS-PAGE, and immunoblotting for Hmg2-GFP and ubiquitin. For IP, 600μL IP buffer (IPB: 15mM Na2HPO4, 150mM NaCl, 2% Triton X-100, 0.1% SDS, 0.5% deoxycholate (DOC), 10mM EDTA, pH 7.5) is added to the 222μL of the solubilized ubiquitination reaction in step 4; PIs and NEM are added as before.
Insoluble material is removed from the IP reaction mix by centrifugation for 5min with a microcentrifuge (14,000 × g), and the resulting clarified supernatant is transferred to a fresh tube using a 1mL pipette. 15–25μL of rabbit polyclonal antiserum is added (the anti-GFP antiserum was prepared in collaboration with Scantibodies, Inc., Santee, CA). The IP incubation is carried out at 4°C for 12–16h.
100μL of protein A-Sepharose (GE Healthcare, Piscataway, NJ) equilibrated with IP buffer is added to the IP reactions. The tubes are then incubated for 2h at 4°C with gentle mixing. Protein A-Sepharose with bound protein is washed once with IPB and once with IPW (50mM NaCl, 10mM Tris, pH 7.5).
To prepare samples for loading, beads are aspirated to dryness and 55μL 2× urea sample buffer (USB: 8 M urea, 4% SDS, 10% ß-mercaptoethanol, 125mM Tris, pH 6.8) is added. The slurry is incubated at 50°C for 10min and centrifuged for 5min with a microcentrifuge (14,000 × g). The eluted proteins are removed to a new tube.
Eluted proteins are resolved by SDS-PAGE using 8% gels and are then transferred to nitrocellulose by electroblotting at 0.15 Amps for 2.5 h (4°C).
For anti-ubiquitin blots, it is important to note that ubiquitin is small and difficult to denature and the ubiquitin epitopes might not be accessible to antibodies due to insufficient denaturation during SDS-PAGE or renaturation on the membrane (Emmerich & Cohen, 2015). Therefore, after transfer to nitrocellulose membranes, the signal strength of anti-ubiquitin antibodies can frequently be enhanced significantly if the membrane is subjected to a denaturing treatment prior to blocking. Accordingly, the membranes are rinsed with water, sandwiched between sheets of Whatman paper, and placed in a glass dish. Deionized water is added to the dish and the membrane is boiled in a microwave oven at 3× 1-min intervals; periodically check in between intervals to ensure that the water has not evaporated. This brings about a remarkable increase in signal strength, presumably due to revealing of cryptic epitopes from the heat.
After denaturing treatment, the blocking and mouse anti-ubiquitin (Zymed Laboratories, South San Francisco, CA) incubations are carried out with Tris-buffered saline (TBS-T: 10 mM Tris–HCl, pH 8.0, 140 mM NaCl, 0.05% Tween 20) containing 20% heat-inactivated bovine serum. First the membrane is blocked by incubation in the serum-containing buffer prior to antibody treatment for 40min, followed by addition of anti-ubiquitin antibody (1: 4000 dilution) and overnight incubation at 4°C. On the next day, blots are washed three times for 10 min each with TBS-T. Note: From our experience, blocking and primary incubation with 20% inactivated bovine serum in TBS-T produces an enhanced signal for ubiquitin blots.
12. For anti-GFP or anti-HA blots, nitrocellulose membranes are blocked with 5% (w/v) nonfat dried milk in TBS-T and mouse anti-GFP (BD Biosciences, San Jose, CA) or anti-HA (Covance, Princeton, NJ) incubations are carried out in 2% non-fat dried milk in TBS-T. Goat anti-mouse conjugated with HRP (Jackson ImmunoResearch, West Grove, PA) is used to detect primary antibodies. Western Lightning chemiluminescence reagents (Perkin Elmer, Waltham, MA) are used for immunodetection and X-ray film is used for Western blot detection (Fig. 3). Signals on X-ray film are quantified by ImageJ (NIH). Band intensities were measured directly from films scanned in high resolution (600dpi) in TIFF file format.
Fig. 2.
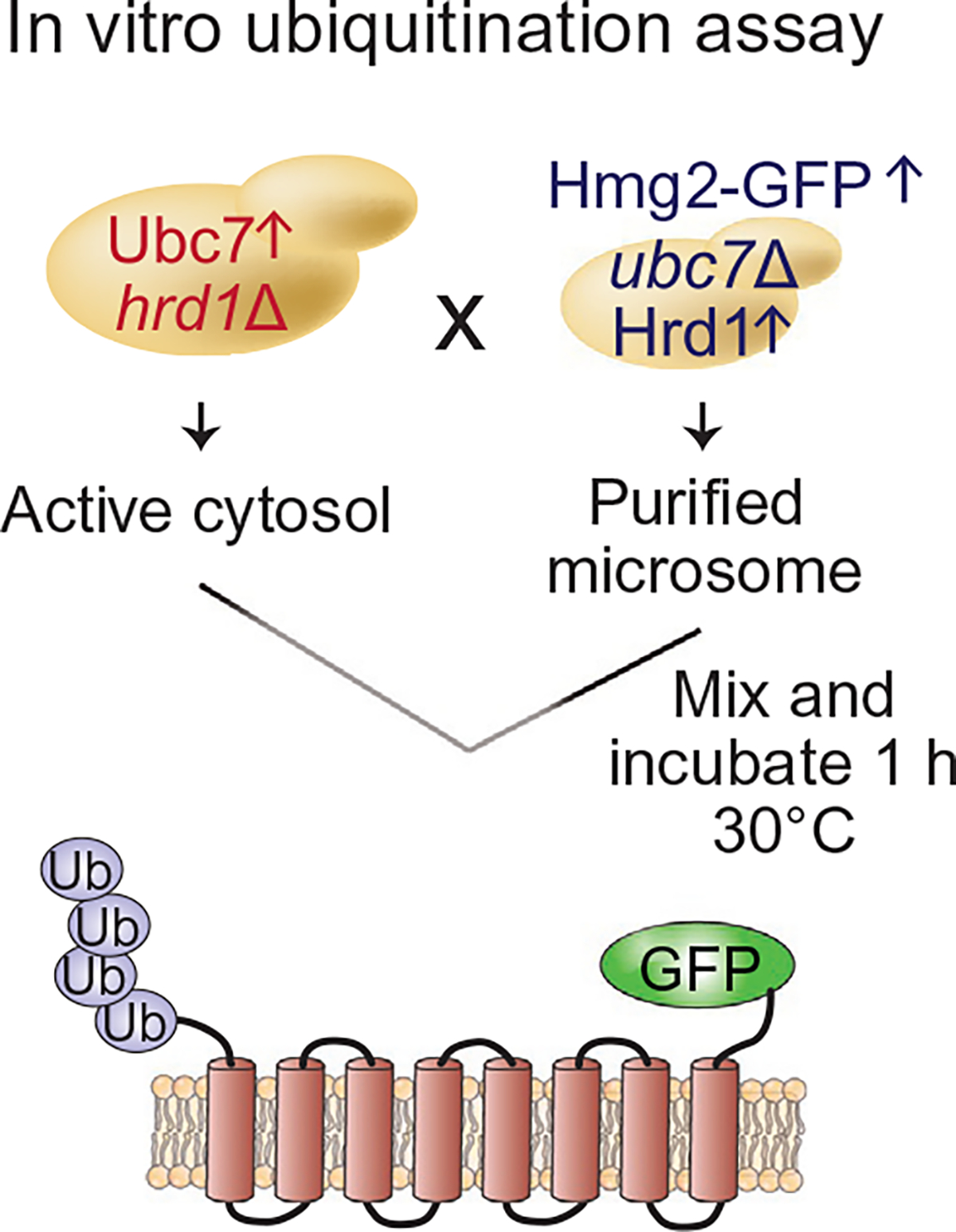
Schematic of in vitro ubiquitination of Hmg2-GFP. Microsomes were prepared from strains expressing TDH3prom-Hmg2-GFP with TDH3prom-Hrd1 and without ubc7 Δ (RHY2923). Cytosol was prepared from strains expressing TDH3prom-Ubc7-GFP (RHY4295) or devoid of Ubc7 (ubc7 Δ) (RHY4288) as a control for no ubiquitination of substrate. To initiate ubiquitination of Hmg2-GFP, microsomes and cytosol preparations were mixed and incubated for 1h at 30°C as indicated.
Fig. 3.
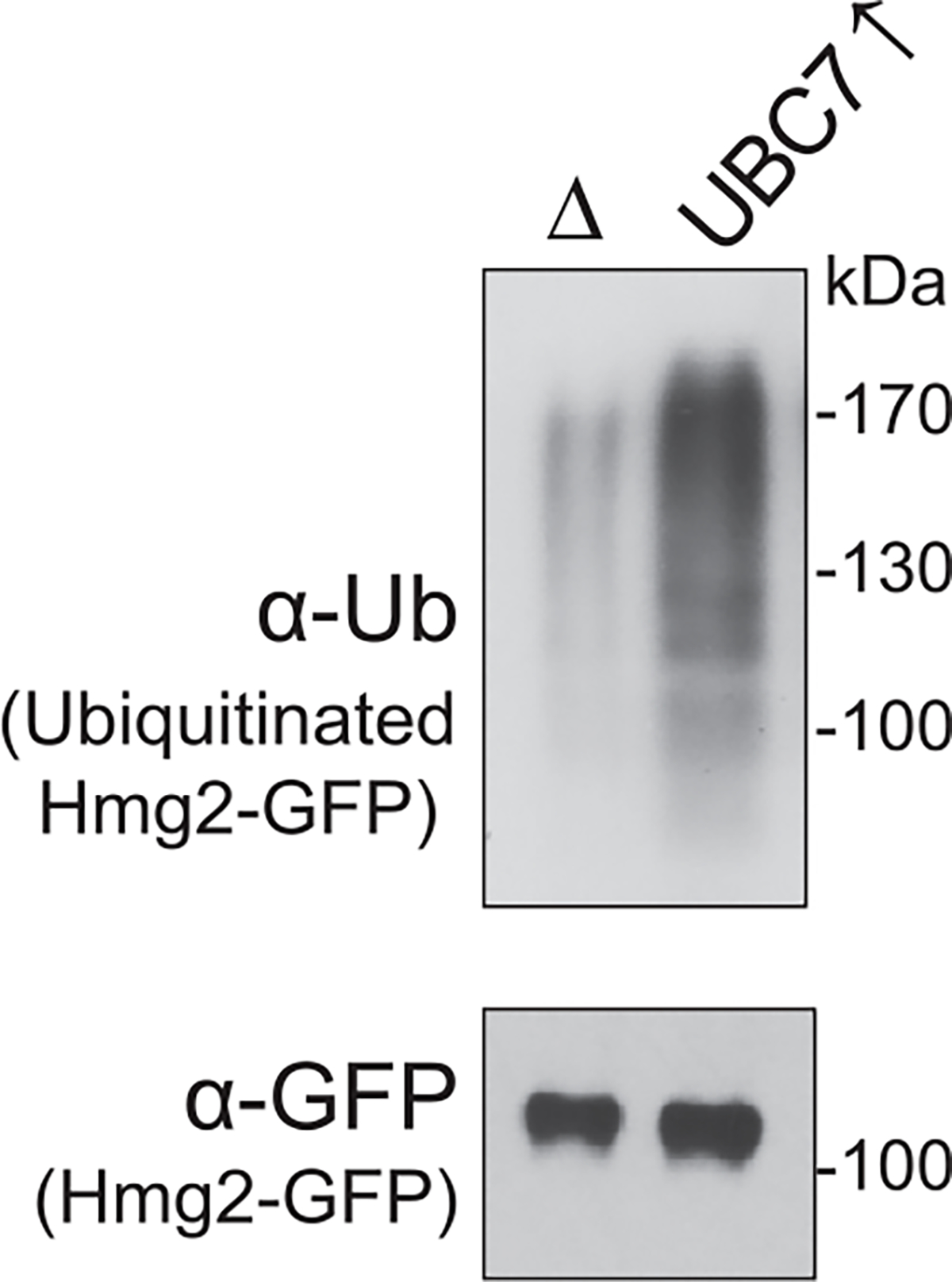
To discern for ubiquitination of the substrate, Hmg2-GFP is immunoprecipitated with anti-GFP, and immunoblotted for ubiquitin with anti-Ub or unmodified Hmg2-GFP with anti-GFP. Figure is taken from Neal, S., Mak, R., Bennett, E. J., & Hampton, R. (2017). A Cdc48 “retrochaperone” function is required for the solubility of retrotranslocated, integral membrane endoplasmic reticulum-associated degradation (ERAD-M) substrates. The Journal of Biological Chemistry 292, 3112–3128.
2.3. In vitro retrotranslocation assay of Hmg2-GFP
A schematic for the in vitro retrotranslocation assay is shown in Fig. 4, and the procedure is outlined below.
Fig. 4.
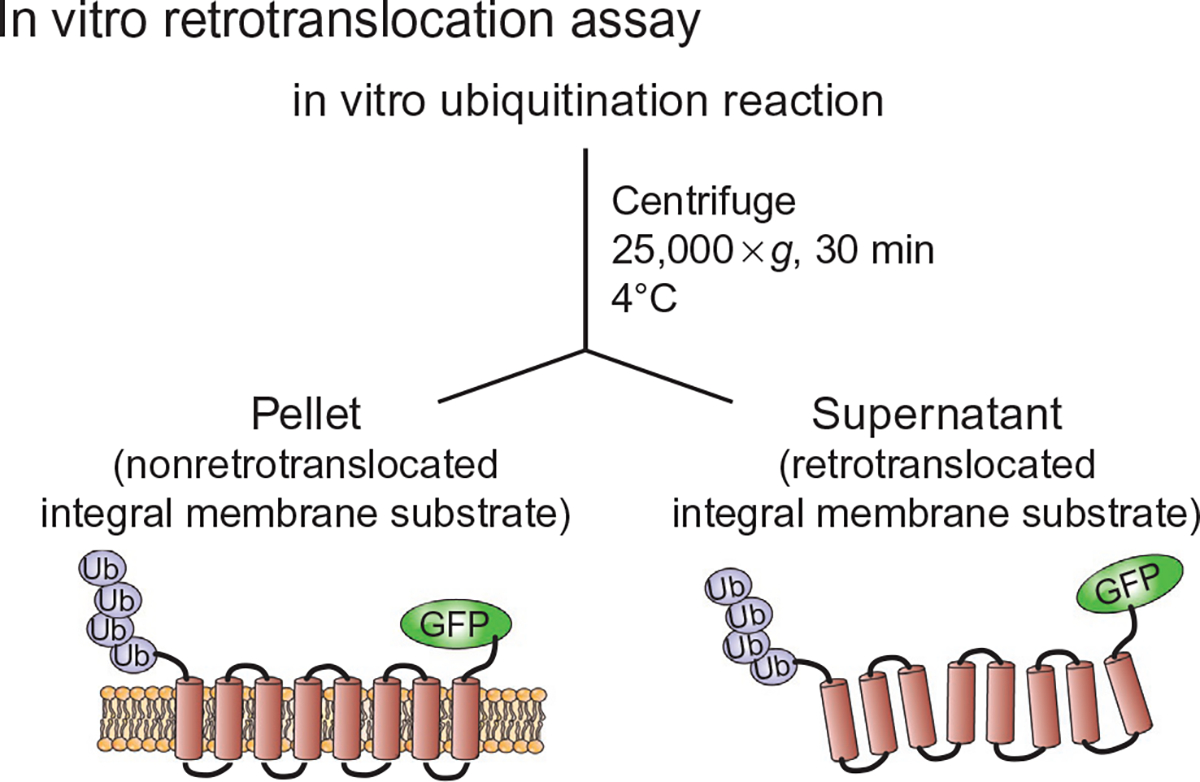
Schematic of in vitro retrotranslocation of Hmg2-GFP. In vitro ubiquitination reactions were carried out as described in Fig. 2, but with 3 × volume (72 μL). Reaction mixture containing in vitro ubiquitinated Hmg2-GFP is centrifuged at 4°C at 25,000 × g for 30 min to discern ubiquitinated Hmg2-GFP that either has been retrotranslocated into the supernatant cytosolic fraction (S) or remained in the microsome pellet fraction (P).
For each in vitro retrotranslocation assay, three samples are evaluated: total lysate, supernatant fraction, and pellet fraction. The ubiquitinated membrane protein that is moved from the pellet (membrane) fraction to the supernatants (cytosol) fraction is the portion that has been retrotranslocated. Typically, up to 50% of the ubiquitinated material can be found as solubilized, retrotranslocated supernatant immunoreactivity (Neal et al., 2017). The three fractions are typically derived from a vitro ubiquitination reaction that was scaled up three times. The in vitro ubiquitination reaction is run as described in the preceding section at 30°C for 1h and terminated by the addition of 1.5 μL of 50× (NEM) to a final concentration of 5 mM.
One reaction equivalent (typically 24 μL) is transferred to one tube designated as total (T) and another, equal reaction equivalent is transferred to a tube for centrifugation for 1h at 25,000 × g at 4°C.
The resulting supernatant (S) is carefully removed, and the resulting pellet (P) is resuspended with MF buffer plus PIs (same volume (24 μL) as the supernatant and total fractions). Each fraction is solubilized with SUME plus PIs and immunoprecipitated and detected as described above for the in vitro ubiquitination assay. The use of identical volumes allows for direct visual comparison of the immunoblotted proteins present in each fraction (Fig. 5).
Fig. 5.
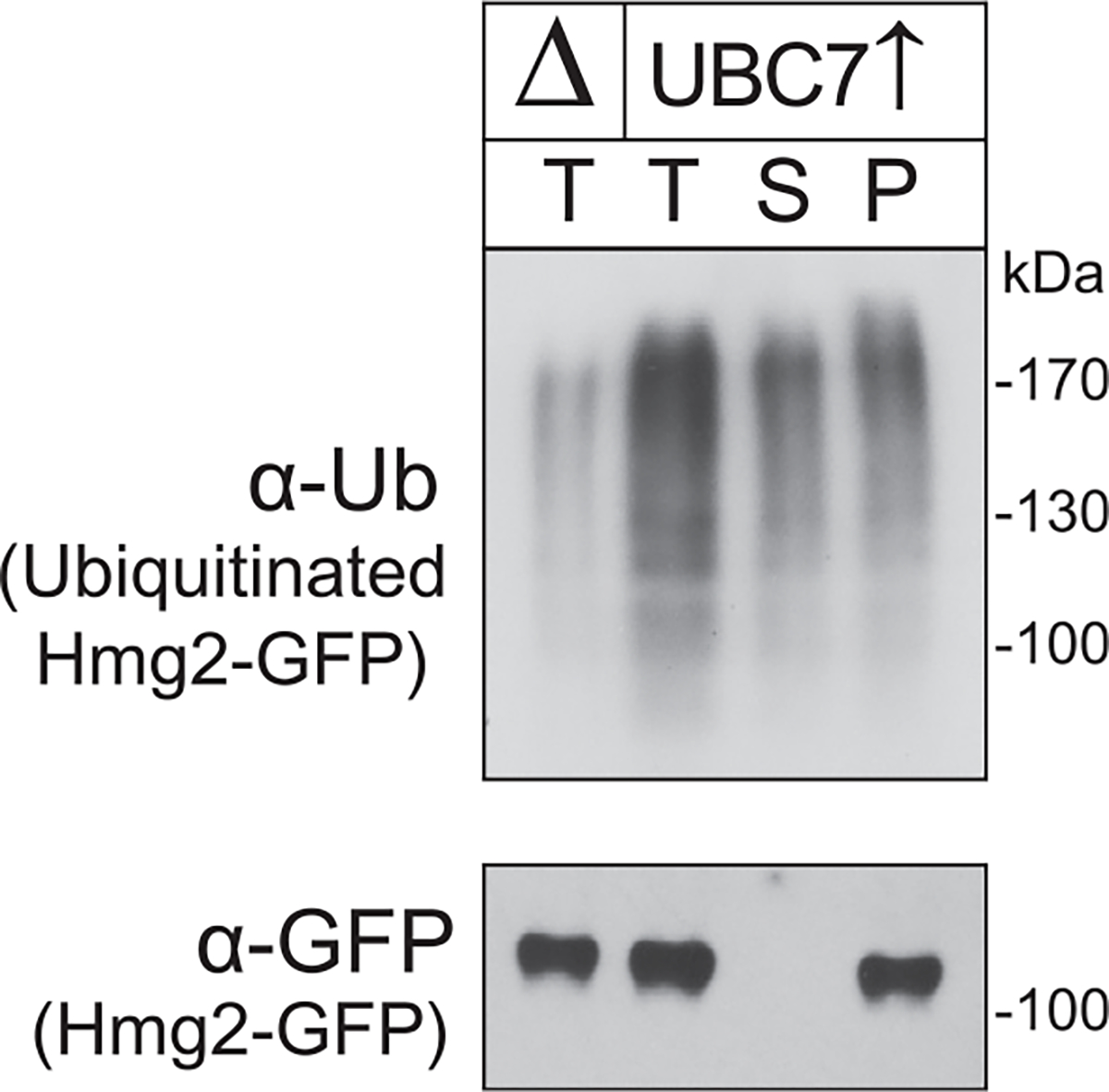
To discern for nonretrotranslocated and retrotranslocated ubiquitinated Hmg2-GFP, the substrate was immunoprecipitated from a 1× (24 μL) aliquot of the non-fractionated mixture (T) or from the supernatant (S) and pellet (P) of an identical volume (24 μL) of the same reaction mixture that was centrifuged, as described. Figure is taken from Neal, S., Mak, R., Bennett, E. J., & Hampton, R. (2017). A Cdc48 “retrochaperone” function is required for the solubility of retrotranslocated, integral membrane endoplasmic reticulum-associated degradation (ERAD-M) substrates. The Journal of Biological Chemistry 292, 3112–3128.
3. An in vivo assay for retrotranslocation of Hmg2-GFP
The in vitro assay is powerful and highly accessible for manipulation of reaction conditions, but the in vivo assay described here allows us to study ERAD in a more physiological context. Because the in vitro assay requires overexpressed Hrd1 to catalyze ERAD, both sterol pathway regulation and the absolute requirement for the Hrd1 partner Hrd3 observed in vivo are lost. Furthermore, the in vivo assay indicates that under normal conditions there is a detectable level of soluble retrotranslocated material en route to the proteasome. Consistent with this idea, addition of proteasome inhibitor MG132 causes a buildup of ubiquitinated Hmg2 in microsomal P and cytosol S fraction. The presence of retrotranslocated material in the absence of proteasome inhibitors supports the idea that full-length extraction and solubilization of multispanning membrane proteins is a part of normal ERAD-M, and amenable to observation with this approach. The in vivo assay also preserves sterol pathway regulation of Hmg2 stability. As shown previously, the 20-carbon sterol pathway molecule geranylgeranyl pyrophosphate (GGPP) is an endogenous regulator of Hmg2-GFP that stimulates increased Hmg2ubiquitinationand degradation(Garza, Tran, &Hampton,2009). We have demonstrated that addition of GGPP to cells along with proteasome inhibitors increases overall ubiquitination of Hmg2-GFP and increases levels of ubiquitinated Hmg2-GFP in the cytosolic fraction (Fig. 6). Together, these findings support the in vivo assay as a powerful tool for studying physiological retrotranslocation.
Fig. 6.
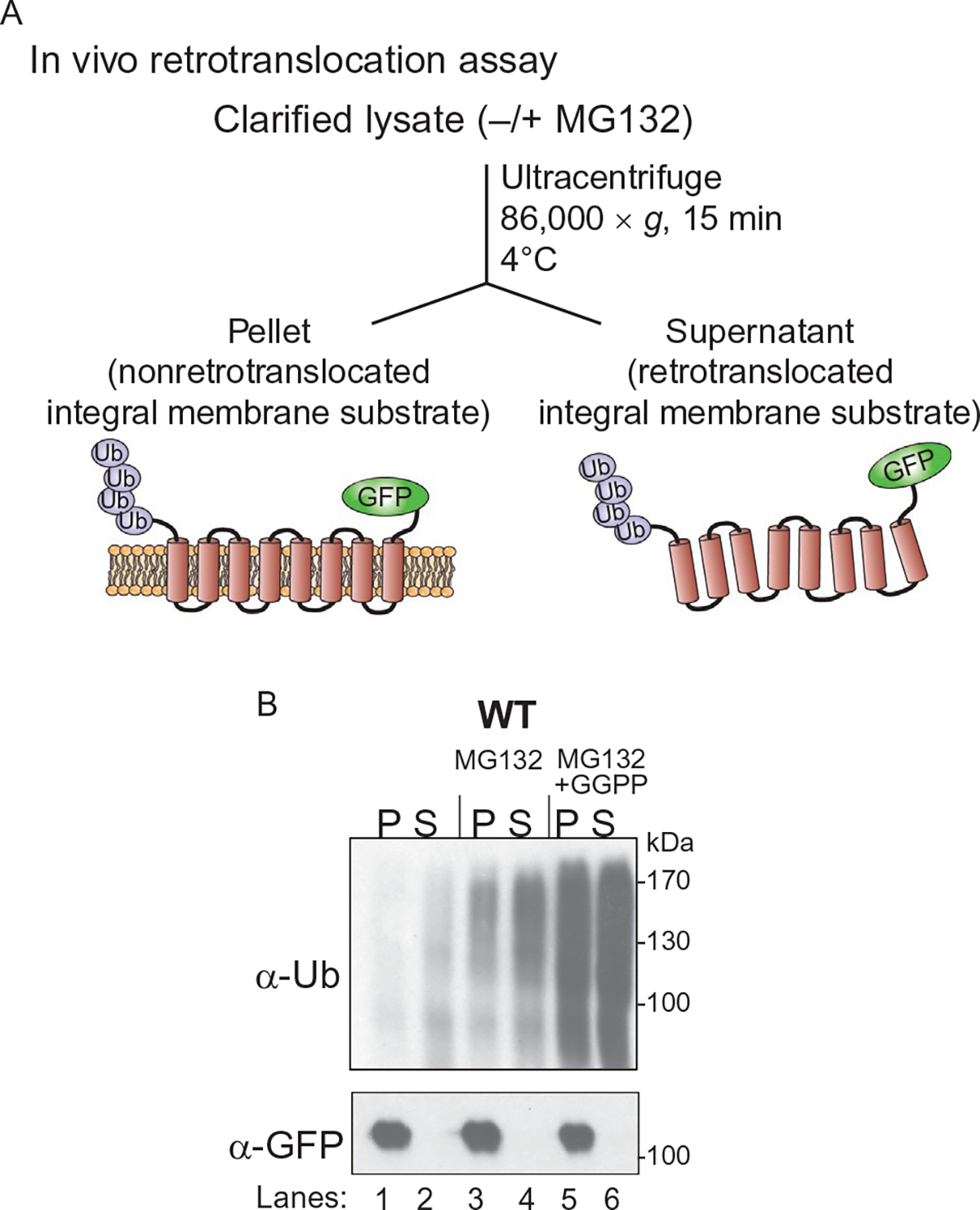
(A) Schematic of in vivo ubiquitination of Hmg2-GFP. Crude lysate was prepared from WT or various mutants defective in ERAD as indicated and ultracentrifuged to discern ubiquitinated Hmg2-GFP that either has been retrotranslocated into the soluble fraction (S) or remained in the ER membrane (P). (B) GGPP-induced ubiquitination and retrotranslocation of Hmg2-GFP. WT strains were grown to log phase and treated with different combinations of MG132 (25 μg/mL) and GGPP (11 μM). Crude lysate was prepared from each strain and ultracentrifuged to discern ubiquitinated Hmg2-GFP that either has been retrotranslocated into the soluble fraction (S) or remained in the membrane (P). Following fractionation, Hmg2-GFP was immunoprecipitated from both fractions, resolved on 8% SDS-PAGE, and immunoblotted for ubiquitin with anti-Ub or unmodified Hmg2-GFP with anti-GFP. Figure is taken from Neal, S., Mak, R., Bennett, E. J., & Hampton, R. (2017). A Cdc48 “retrochaperone” function is required for the solubility of retrotranslocated, integral membrane endoplasmic reticulum-associated degradation (ERAD-M) substrates. The Journal of Biological Chemistry 292, 3112–3128.
3.1. Materials
3.1.1. Strains
To generate a suitable strain for in vivo assays involving drug additions such as the proteasome inhibitor MG132, the multidrug resistance gene PDR5 is disrupted, since pdr5Δ null strains allow high potency inhibition of proteasomes of intact yeast with standard inhibitors such MG132 (Neal et al., 2017).
3.2. Assay of in vivo ubiquitination of Hmg2-GFP
Strains are grown from a low staring OD600 (typically 0.05) at the desired temperature in minimal medium and with vigorous shaking until the cultures reach a mid-log phase OD600 of 0.2–0.3. We typically grow 50mL of yeast culture.
For drug treatment, mid-log phase cultures are treated with MG132 at a final concentration of 25 μg/mL, added from a 25 mg/mL stock dissolved in DMSO for 2h at 30°C and/or final concentration of 11 μM of GGPP, added from a 2.2 mM stock of GGPP ammonium salt, for 1h at 30°C. We typically add GGPP after 1h of MG132 treatment. From our experience, these concentrations and the duration of drug treatments yield an optimal signal for ubiquitinated substrate. Importantly, only Hmg2 undergoes GGPP-stimulated ubiquitination. Neither unregulated Hmg2 variants, nor any other ERAD-M substrates, show any effect of added GGPP in this assay.
15 OD units of cells are collected and prepared for bead lysis in 50 mL Falcon tubes. Cell pellets are rinsed and resuspended in 400 μL MF buffer with the same amount of PIs as mentioned above for the in vitro assay.
Bead-based lysis is performed as described in Section 2.1.2, step 3. It is critical that subsequent steps are performed at 4°C.
The lysate is collected into a 2 mL Eppendorf tube, the glass beads are rinsed once with 400 μL of cold MF buffer, and added to the lysate. This combined crude lysate is centrifuged for 5min at 2500 × g. The resulting clarified lysate is transferred to a new chilled 2mL Eppendorf tube.
Ubiquitination of substrates is measured by IP of Hmg2-GFP, SDS-PAGE, and immunoblotting for Hmg2-GFP and ubiquitin. IP and immunoblot analysis were done as described above for the cell-free ubiquitination experiments.
3.3. In vivo retrotranslocation assay of Hmg2-GFP
The in vivo retrotranslocation assay involves ultracentrifugation of the clarified lysate from step 5, prior to addition of any detergents, into microsomal pellet and cytosolic supernatant fractions to assess the extent of retrotranslocation into the supernatant fraction.
200 μL of clarified lysate is ultracentrifuged at 86,000 × g for 15 min to yield microsomal pellet (P100) and cytosolic supernatant (S100) fractions.
Retrotranslocation of substrates is measured by comparison of immunoprecipitation of Hmg2-GFP from the P100 and S100 fractions, with the S100 material representing retrotranslocation. P100 is resuspended in 200 μL of SUME with PIs and 5 mM NEM followed by addition of 600 μL of IPB with PIs and NEM. The S100 supernatant is added directly to 600 μL IPB with PIs and NEM. Subsequent steps for IP and immunoblot analysis of retrotranslocated Hmg2-GFP are carried out as described above for the in vitro ubiquitination assay (Fig. 7). Note that employing the same final volumes for the P100 and S100 allows facile visual assessment of the fraction of ubiquitinated substrate that has been moved to the cytosol by retrotranslocation.
Fig. 7.
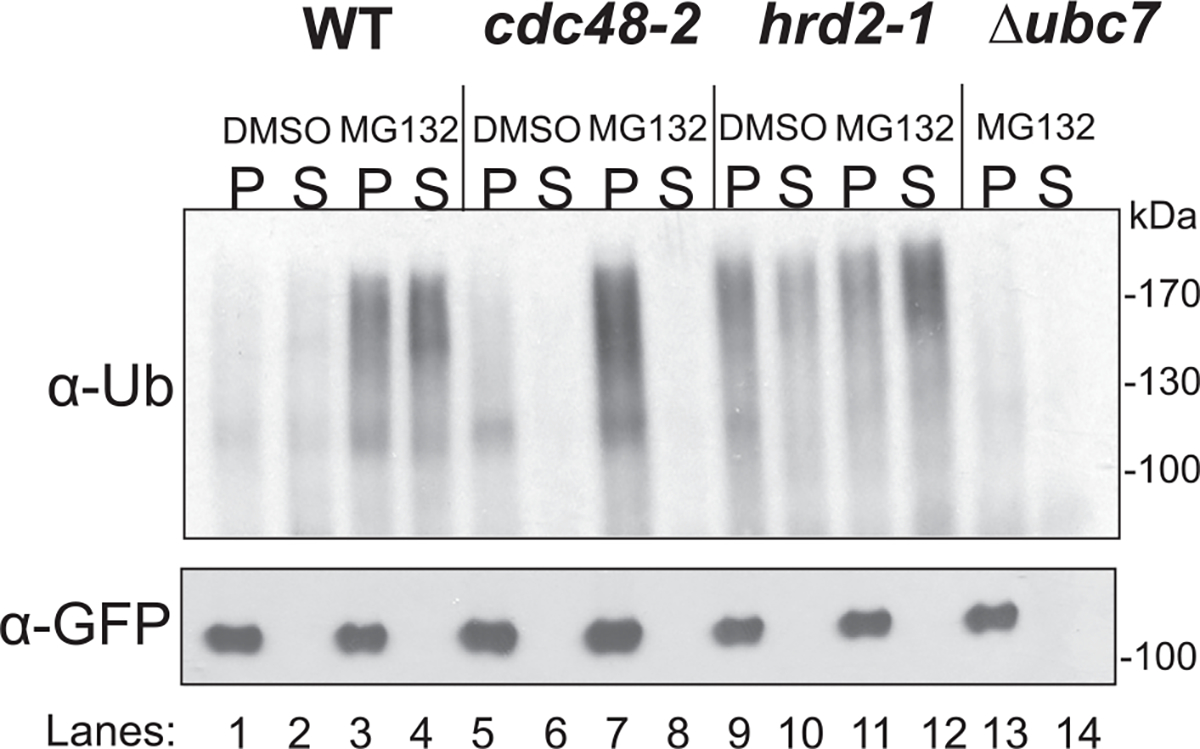
in vivo retrotranslocation of Hmg2-GFP requires Cdc48. in vivo retrotranslocation assay was performed with WT, cdc48–2, proteasome mutant hrd2–1, and ubc7 Δ cells and treated with vehicle or MG132 (25 μg/mL). cdc48–2 was used as a positive control for block in retrotranslocation (lanes 5–8). The ERAD mutant hrd2–1 is an allele of the 26S proteasome base subunit Rpn1 that is strikingly deficient in Hmg2 degradation. As expected in an hrd2–1 strain, there was an increase in total ubiquitination of Hmg2-GFP and more accumulation in the soluble fraction compared with WT. Moreover, the signal intensity in each fraction was greater still when MG132 was added to the hrd2–1 strain due to further inhibition of the proteasome (lanes 9–12). In cells lacking Ubc7, the principal HRD pathway E2, Hmg2-GFP was not ubiquitinated, and so no retrotranslocated Hmg2-GFP was generated (lanes 13 and 14). Figure is taken from Neal, S., Mak, R., Bennett, E. J., & Hampton, R. (2017). A Cdc48 “retrochaperone” function is required for the solubility of retrotranslocated, integral membrane endoplasmic reticulum-associated degradation (ERAD-M) substrates. The Journal of Biological Chemistry 292, 3112–3128.
4. Characterizing the nature of retrotranslocated Hmg2-GFP
Both in vitro and in vivo retrotranslocation assays allow for the isolation and direct examination of the retrotranslocated intermediate. In this section, the use of biophysical and proteomic analyses to understand the physical state of retrotranslocated Hmg2-GFP is described.
4.1. Ubiquitin cleavage from retrotranslocated Hmg2-GFP
Several experiments indicate that the retrotranslocated protein is soluble, intact and full-length; the most striking result shows that full-length Hmg2 can be recovered from the cytosolic supernatant fraction by cleaving off the attached ubiquitin with Usp2, purified, constitutively active ubiquitin protease (Garza, Sato, & Hampton, 2009; Neal et al., 2017). These studies showed that the intact 8-TMD-containing Hmg2 is in a soluble form after retrotranslocation. The procedure for ubiquitin removal from retrotranslocated Hmg2-GFP is outlined here.
As starting material, a supernatant fraction containing either in vivo and in vitro retrotranslocated Hmg2-GFP is used, prepared as above, except that the employed retrotranslocation assay is carried out without added leupeptin or NEM in any of the buffers since these compounds both inhibit ubiquitin protease activity and so prevent removal of the ubiquitin chains from the substrate.
For removing ubiquitin from in vitro retrotranslocated Hmg2-GFP, seven separate 24 μL in vitro ubiquitination reactions are incubated for 1h at 30°C and then centrifuged at 25,000 × g. Note: 7× separate reactions must be performed. Scaling this reaction has been problematic and did not result in optimal ubiquitination of Hmg2-GFP. The resulting supernatant fractions are pooled to yield a sufficient volume for analysis. For in vivo retrotranslocated Hmg2-GFP, supernatant from 15 OD equivalents of MG132 treated cells was enough for analysis. Note: seven separate in vitro ubiquitination reactions are used to recover and detect a robust signal for Hmg2-GFP once the polyubiquitination chain is cleaved.
Ubiquitin removal is accomplished with the broadly active Usp2 ubiquitin protease (human recombinant Usp2Core) that either can be purified from Escherichia coli that express the recombinant enzyme (a gift from Rohan Baker, Australian National University) or purchased commercially (LifeSensors Inc., Malvern, PA). 50 μL of supernatant containing retrotranslocated Hmg2-GFP is incubated with 10 μL of Usp2Core (5 μg) for 1h at 37°C. In our experience, this concentration and incubation time is optimal for complete ubiquitin removal.
The reaction is quenched with 200 μL of SUME with PIs and retrotranslocated Hmg2-GFP is immunoprecipitated as described above for the in vitro ubiquitination reactions. The final volume of eluted proteins is 55 μL.
20 μL of the immunoprecipitate is used for detection with anti-GFP and anti-ubiquitin antibodies (Fig. 8).
Fig. 8.
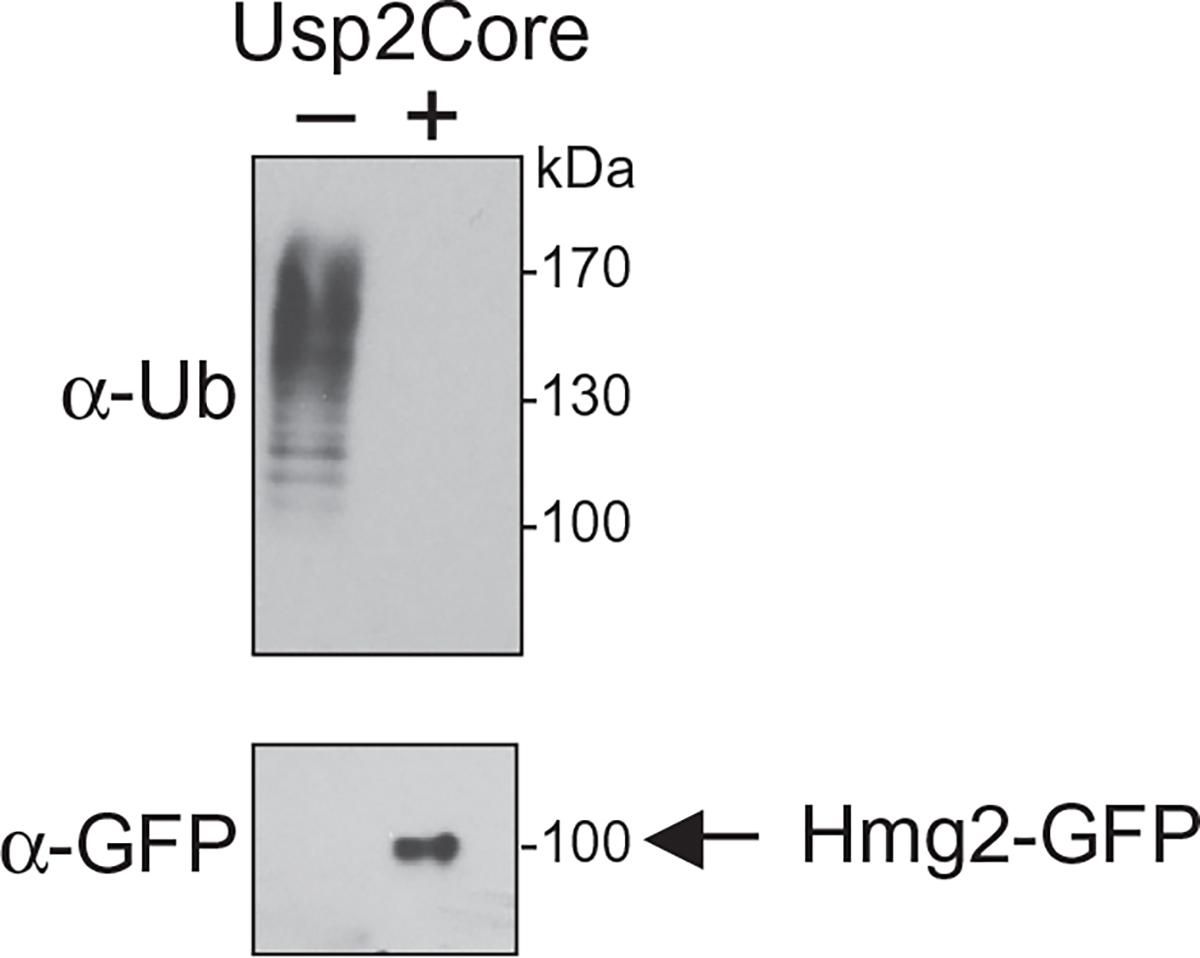
Full-length Hmg2-GFP retrotranslocates into soluble fraction in vivo. Supernatant fraction containing retrotranslocated Hmg2-GFP was isolated from in vivo retrotranslocation assay and incubated in the presence or absence of Usp2Core for 1h at 37°C. Full-length Hmg2-GFP was immunoprecipitated and immunoblotted with antiUbi to verify that the ubiquitin has been cleaved from the degradation intermediate or with anti-GFP to blot for recovery of full-length retrotranslocated Hmg2-GFP. Figure is taken from Neal, S., Mak, R., Bennett, E. J., & Hampton, R. (2017). A Cdc48 “retrochaperone” function is required for the solubility of retrotranslocated, integral membrane endoplasmic reticulum-associated degradation (ERAD-M) substrates. The Journal of Biological Chemistry 292, 3112–3128.
4.2. Sucrose gradient flotation assay of retrotranslocated Hmg2-GFP
We suspected that specific proteins, which we term “retrochaperones” (Neal et al., 2017), were required for the high solubility of retrotranslocated full-length Hmg2-GFP, as has been shown for the Ste6* (also called Ste6–166) Doa10 pathway substrate (Nakatsukasa et al., 2016). Nevertheless, it remained formally possible that lipids are involved in solubilizing Hmg2 by formation of micelles and/or lipid droplet-like particles (Jo, Hartman, & DeBose-Boyd, 2013). To address this, in vivo retrotranslocated Hmg2-GFP was isolated and analyzed by a sucrose density flotation assay to evaluate its association with lipids: protein complexes “sink” to the lower fractions of a sucrose gradient, while lipid-based complexes such as lipid droplets rise to the top of such gradients due to the buoyancy of such molecular species. This flotation assay is adapted from (Zhang et al., 2001) and described in detail below.
Supernatant fraction containing in vivo retrotranslocated Hmg2-GFP is isolated as described above except MSB buffer (50 mM HEPES, 150mM NaCl, 5mM EDTA, pH 7.6) was used instead of MF buffer.
100μL of supernatant fraction containing ubiquitinated soluble Hmg2-GFP is mixed with 300 μL of dense sucrose solution (2.4 M sucrose in MSB buffer) in a 1mL ultracentrifuge tube (Beckman Coulter) on ice.
Subsequent steps include the overlaying of sucrose solutions of progressively lower densities. Specifically, MSB supplemented with 1.5 M sucrose (500μL) and then 0.25 M sucrose (400 μL) are successively layered into the centrifuge tube containing the dense sucrose solution containing soluble ubiquitinated Hmg2-GFP. The layering must be done without causing any turbulence that could mix the step concentrations and compromise the buoyancy experiment.
The resulting sucrose-layered tube is centrifuged in a Beckman SW55 rotor at 100,00 × g for 16h at 4°C.
Fractions of 100 μL are gently removed from the top of the gradient and transferred to Eppendorf tubes, allowing analysis of the position of proteins in the gradients.
For analysis of retrotranslocated Hmg2-GFP, each fraction is immunoprecipitated with anti-GFP antibodies and immunoblotted as described above in the in vitro ubiquitination assay.
Various control proteins are also evaluated in the gradients (Fig. 9) to ensure successful partition based on biophysical properties. The soluble cytoplasmic enzyme phosphoglycerate kinase (PGK) remains in the lower fractions (7–13) closer to the bottom of the tube, whereas the membrane-associated ER-resident integral membrane protein Sec61 floats to higher fractions (4–10). The retrotranslocated Hmg2-GFP shows mobility coincident with PGK, suggesting that the in vivo retrotranslocated Hmg2-GFP is not associated with lipids; rather, it appeared to be fully proteinaceous.
Fig. 9.
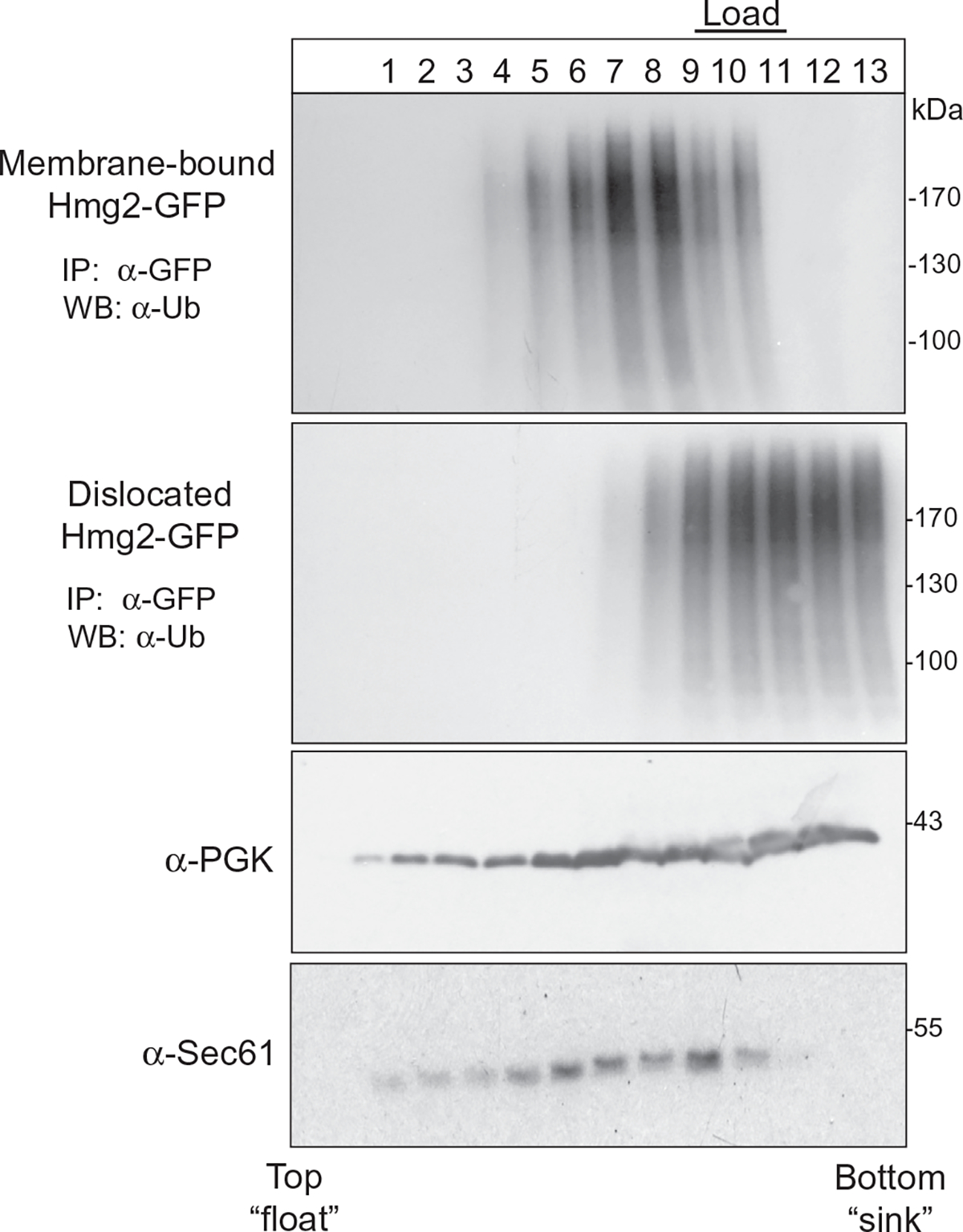
In vivo retrotranslocated Hmg2 does not associate with lipids. Supernatant fraction containing retrotranslocated Hmg2-GFP was isolated and prepared, layered at the bottom of a centrifuge tube, and subjected to ultracentrifugation as described. Aliquots were removed from the top to the bottom of the sucrose gradient, and each fraction was either directly immunoblotted for Sec61 and PGK with anti-Sec61 or anti-PGK, respectively, or immunoprecipitated with anti-GFP and immunoblotted for ubiquitin and Hmg2-GFP. Figure is taken from Neal, S., Mak, R., Bennett, E. J., & Hampton, R. (2017). A Cdc48 “retrochaperone” function is required for the solubility of retrotranslocated, integral membrane endoplasmic reticulum-associated degradation (ERAD-M) substrates. The Journal of Biological Chemistry 292, 3112–3128.
4.3. Proteomic analysis of retrotranslocated Hmg2-GFP and associated retrochaperones
Our observation that retrotranslocated Hmg2-GFP was proteinacious demanded analysis of the associated proteins that would allow an eight-transmembrane domain ERAD-M substrate to exist in a soluble state. To identify these retrochaperones, we used an unbiased proteomics approach (Neal et al., 2017). We maximized the amount of retrotranslocated material by modifying and scaling up the in vivo retrotranslocation assay. Accordingly, the in vivo retrotranslocation assay supernatant fraction was derived from simultaneous proteasome inhibitor MG132 and ubiquitin GGPP treatment of a large sample—30 OD units—of cells. Furthermore, the ultracentrifuged S fraction is subjected to preparative anti-GFP co-IP using GFP-Trap® agarose (Chromotek) in the absence of detergent to capture retrotranslocated, ubiquitinated Hmg2-GFP and any accompanying proteins. As controls, Co-IPs are performed on supernatant fractions from otherwise identical strains expressing cytosolic GFP. This Co-IP procedure for proteomic analysis of retrotranslocated Hmg2-GFP is outlined below.
Strains are grown from a low starting OD (~0.05) at the desired temperature in minimal medium with vigorous shaking until the cultures reach a mid-log phase OD600 of 0.2–0.3. We typically grow 200 mL of yeast culture.
For drug treatment, mid-log phase cultures are treated with MG132 at a final concentration of 25 μg/mL (added from a 25 mg/mL stock) for 2h at 30°C and GGPP ammonium salt at a final concentration of 11 μM (added from a 2.2mM stock) for 1h at 30°C. GGPP is added after the first hour of MG132 treatment.
30 OD units of cells are collected and prepared for bead lysis in 50 mL Falcon tubes. Cell pellets are rinsed and resuspended in ice cold 800 μL MF buffer with PIs.
Bead-based lysis is performed as described above. It is critical that the following steps are performed at 4°C.
The lysate is collected and pooled with one bead rinse using 800 μL of cold MF buffer to give crude lysate. The crude lysate is centrifuged for 5min at 2500 × g. The resulting clarified lysate is transferred to an ultracentrifuge tube.
The clarified lysate is ultracentrifuged at 86,000 × g for 15min to yield a cytosolic supernatant fraction.
-
While the lysate is being centrifuged, GFP-Trap®-agarose beads are equilibrated for co-IP of retrotranslocated Hmg2-GFP.
GFP-Trap®-agarose stock tube is gently vortexed and 35 μL of agarose slurry is transferred to a chilled Eppendorf tube to which 500 μL of ice cold non-detergent IP buffer (15mM Na2HPO4, 150mM NaCl, 10mM EDTA, pH 7.5) with PIs is added.
The agarose slurry is centrifuged at 2500 × g for 2min at 4°C and the supernatant is discarded.
GFP-Trap®-agarose is resuspended in 600μL of ice cold non-detergent IP buffer with PIs.
The agarose slurry is divided into three fresh chilled Eppendorf tubes (200μL per tube).
All three tube are centrifuged at 2500 × g for 2min at 4°C and the supernatant is discarded from all three tubes.
Each agarose pellet is resuspended in 600μL of ice cold non-detergent IP buffer with PIs.
Once ultracentrifugation of the yeast lysate is completed, 600 μL of the supernatant is divided equally into the three tubes (200 μL/tube) containing equilibrated GFP-Trap agarose as prepared above. IP incubation is carried out overnight at 4°C with gentle agitation.
Next day: All three IPs are centrifuged at room temperature at 2500 × g for 2min. Supernatants are removed and saved for analysis.
GFP-Trap bound protein from all three tubes is resuspended in non-detergent IP buffer. The three IP samples are combined into one Eppendorf tube and washed in 2× 1mL with non-detergent IP buffer.
For elution of proteins bound to GFP-Trap agarose, beads are resuspended in 50 μL of 0.2 M glycine pH 2.5 and incubated for 30s under constant mixing.
Tube is centrifuged at room temperature at 20,000 × g for 5min.
Supernatant is transferred to new tube and 5 μL of 1 M Tris base pH 10.4 is added for neutralization.
Neutralized sample is then subjected for LC–MS/MS mass spectrometry analysis.
Acknowledgments
We thank the Hampton lab members for in depth discussions and technical assistance. These studies were supported by NIH grant 5R37DK051996–18 (to R.Y.H.). S.E.N was funded by the postdoctoral NIH grant 1F32GM111024–01 and Burroughs Wellcome Fund 1013987. S.D. was supported from funding by the CRI Irvington Postdoctoral Fellowship.
References
- Baldridge RD, & Rapoport TA (2016). Autoubiquitination of the Hrd1 ligase triggers protein Retrotranslocation in ERAD. Cell, 166, 394–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bays NW, Wilhovsky SK, Goradia A, Hodgkiss-Harlow K, & Hampton RY (2001). HRD4/NPL4 is required for the proteasomal processing of ubiquitinated ER proteins. Molecular Biology of the Cell, 12, 4114–4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazirgan OA, & Hampton RY (2008). Cue1p is an activator of Ubc7p E2 activity in vitro and in vivo. The Journal of Biological Chemistry, 283, 12797–12810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordallo J, Plemper RK, Finger A, & Wolf DH (1998). Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Molecular Biology of the Cell, 9, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SM, & Hampton RY (2009). Usa1p is required for optimal function and regulation of the Hrd1p endoplasmic reticulum-associated degradation ubiquitin ligase. The Journal of Biological Chemistry, 285(8), 5146–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho P, Goder V, & Rapoport TA (2006). Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell, 126, 361–373. [DOI] [PubMed] [Google Scholar]
- Chen B, Mariano J, Tsai YC, Chan AH, Cohen M, & Weissman AM (2006). The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proceedings of the National Academy of Sciences of the United States of America, 103, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, & Dobson CM (2017). Protein Misfolding, amyloid formation, and human disease: A summary of Progress over the last decade. Annual Review of Biochemistry, 86, 27–68. [DOI] [PubMed] [Google Scholar]
- Emmerich CH, & Cohen P (2015). Optimising methods for the preservation, capture and identification of ubiquitin chains and ubiquitylated proteins by immunoblotting. Biochemical and Biophysical Research Communications, 466, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foresti O, Ruggiano A, Hannibal-Bach HK, Ejsing CS, & Carvalho P (2013). Sterol homeostasis requires regulated degradation of squalene monooxygenase by the ubiquitin ligase Doa10/Teb4. eLife, 2, e00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza RM, Sato BK, & Hampton RY (2009). In vitro analysis of Hrd1p-mediated retrotranslocation of its multispanning membrane substrate 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase. The Journal of Biological Chemistry, 284, 14710–14722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garza RM, Tran PN, & Hampton RY (2009). Geranylgeranyl pyrophosphate (GGPP) is a potent regulator of HRD-dependent HMG-CoA reductase degradation in yeast. The Journal of Biological Chemistry, 284(51), 35368–35380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY (2005). Fusion-based strategies to identify genes involved in degradation of a specific substrate. Methods in Enzymology, 399, 310–323. [DOI] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, & Rine J (1996). Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Molecular Biology of the Cell, 7, 2029–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, & Garza RM (2009). Protein quality control as a strategy for cellular regulation: Lessons from ubiquitin-mediated regulation of the sterol pathway. Chemical Reviews, 109, 1561–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, & Rine J (1994). Regulated degradation of HMG-CoA reductase, an integral membrane protein of the endoplasmic reticulum, in yeast. The Journal of Cell Biology, 125, 299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, & Sommer T (2012). Finding the will and the way of ERAD substrate retrotranslocation. Current Opinion in Cell Biology, 24, 460–466. [DOI] [PubMed] [Google Scholar]
- Hara T, Hashimoto Y, Akuzawa T, Hirai R, Kobayashi H, & Sato K (2014). Rer1 and calnexin regulate endoplasmic reticulum retention of a peripheral myelin protein 22 mutant that causes type 1A Charcot-Marie-tooth disease. Scientific Reports, 4, 6992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck JW, Cheung SK, & Hampton RY (2010). Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proceedings of the National Academy of Sciences of the United States of America, 107, 1106–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller MM, Finger A, Schweiger M, & Wolf DH (1996). ER degradation of a misfolded luminal protein by the cytosolic ubiquitin-proteasome pathway. Science, 273, 1725–1728. [DOI] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, et al. (2002). Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nature Cell Biology, 4, 134–139. [DOI] [PubMed] [Google Scholar]
- Jo Y, Hartman IZ, & DeBose-Boyd RA (2013). Ancient ubiquitous protein-1 mediates sterol-induced ubiquitination of 3-hydroxy-3-methylglutaryl CoA reductase in lipid droplet-associated endoplasmic reticulum membranes. Molecular Biology of the Cell, 24, 169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilley BN, & Ploegh HL (2004). A membrane protein required for dislocation of misfolded proteins from the ER. Nature, 429, 834–840. [DOI] [PubMed] [Google Scholar]
- Mehnert M, Sommer T, & Jarosch E (2013). Der1 promotes movement of misfolded proteins through the endoplasmic reticulum membrane. Nature Cell Biology, 16, 77–86. [DOI] [PubMed] [Google Scholar]
- Mehnert M, Sommermeyer F, Berger M, Kumar Lakshmipathy S, Gauss R, Aebi M, et al. (2015). The interplay of Hrd3 and the molecular chaperone system ensures efficient degradation of malfolded secretory proteins. Molecular Biology of the Cell, 26, 185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Kamura T, Bagola K, Mehnert M, Jarosch E, Sommer T, et al. (2016). Subcellular Fractionation Analysis of the Extraction of Ubiquitinated Polytopic Membrane Substrate during ER-Associated Degradation. PLoS One, 11 e0148327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S, Jaeger PA, Duttke SH, Benner CK, Glass C, Ideker T, et al. (2018). The Dfm1 Derlin is required for ERAD Retrotranslocation of integral membrane proteins. Molecular Cell, 69, 306–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neal S, Mak R, Bennett EJ, & Hampton R (2017). A Cdc48 “retrochaperone” function is required for the solubility of retrotranslocated, integral membrane endoplasmic reticulum-associated degradation (ERAD-M) substrates. The Journal of Biological Chemistry, 292, 3112–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemper RK, Böhmler S, Bordallo J, Sommer T, & Wolf DH (1997). Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature, 388, 891–895. [DOI] [PubMed] [Google Scholar]
- Ravid T, Kreft SG, & Hochstrasser M (2006). Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. The EMBO Journal, 25, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato BK, Schulz D, Do PH, & Hampton RY (2009). Misfolded membrane proteins are specifically recognized by the transmembrane domain of the Hrd1p ubiquitin ligase. Molecular Cell, 34, 212–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoebel S, Mi W, Stein A, Ovchinnikov S, Pavlovicz R, DiMaio F, et al. (2017). Cryo-EM structure of the protein-conducting ERAD channel Hrd1 in complex with Hrd3. Nature, 548, 352–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spang A, & Schekman R (1998). Reconstitution of retrograde transport from the Golgi to the ER in vitro. The Journal of Cell Biology, 143, 589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein A, Ruggiano A, Carvalho P, & Rapoport TA (2014). Key steps in ERAD of luminal ER proteins reconstituted with purified components. Cell, 158, 1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Zhang R, Gong X, Geng X, Drain PF, & Frizzell RA (2006). Derlin-1 promotes the efficient degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. The Journal of Biological Chemistry, 281, 36856–36863. [DOI] [PubMed] [Google Scholar]
- Vashistha N, Neal SE, Singh A, Carroll SM, & Hampton RY (2016). Direct and essential function for Hrd3 in ER-associated degradation. Proceedings of the National Academy of Sciences of the United States of America, 113, 5934–5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangeline MA, Vashistha N, & Hampton RY (2017). Proteostatic tactics in the strategy of sterol regulation. Annual Review of Cell and Developmental Biology, 33, 467–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, & Rapoport TA (2003). Function of the p97-Ufd1-Npl4 complex in retrotranslocation from the ER to the cytosol: Dual recognition of nonubiquitinated polypeptide segments and polyubiquitin chains. The Journal of Cell Biology, 77114, 21–9525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nijbroek G, Sullivan ML, McCracken AA, Watkins SC, Michaelis S, et al. (2001). NNNN70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Molecular Biology of the Cell, 12, 1303–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, & Ackerman SL (2006). Endoplasmic reticulum stress in health and disease. Current Opinion in Cell Biology, 18, 444–452. [DOI] [PubMed] [Google Scholar]