

Editor’s Summary:
Most proteomic studies for Alzheimer’s disease (AD) are instrumental in identifying AD pathways but focus on single tissues and sporadic AD. Here, we present a large multi-tissue proteomic study, accessible through a web portal (omics.wustl.edu/proteomics). We obtained 1,305 proteins in brain, cerebrospinal fluid (CSF), and plasma from sporadic AD, TREM2 risk-variant carriers, autosomal dominant AD (ADAD), and healthy individuals. We identified 8 proteins in brain, 40 in CSF, and 9 in plasma that were altered by sporadic AD status in discovery and were replicated in several external studies. Our CSF-specific model provided higher prediction than the model with the gold standard biomarker CSF p-tau181/Aβ42 (area under the receiver operating characteristic curve, AUC = 0.90 vs. 0.81; P = 2.4×10-6). The plasma-specific model was comparable (AUC = 0.79 vs. 0.82; P > 0.05). We furthermore identified a proteomic signature that differentiated TREM2 variant carriers from both sporadic AD and healthy individuals with high predictive power (AUC range: 0.81–0.93). The proteins associated with sporadic AD were also altered in ADAD, but with greater effect size (1.36 times, P = 1.0×10-4). Brain derived proteins associated with ADAD were also replicated in CSF (P = 1.4×10-9). They enriched for several pathways including AD (Calcineurin, Apo E), Parkinson disease (α-Synuclein, LRRK2), and innate immune response (SHC1, ERK-1, SPP1). Our multi-tissue proteomic study contributes to the understanding of AD biology and to the creation of tissue-specific prediction models for individuals with specific genetic profiles, ultimately supporting its utility in creating individualized disease risk evaluation and treatment.
One Sentence Summary:
Multi-tissue proteomic profiling uncovers AD biology and enables tissue-specific prediction models for individuals with specific genetic profiles.
INTRODUCTION
Alzheimer’s disease (AD) is the most common cause of dementia, reducing the quality of life among patients and caregivers (1). AD is characterized by amyloid β (Aβ)-containing plaques and tau neurofibrillary tangles in the brain, resulting in neuronal loss, neuroinflammation, and memory decline (2). AD is genetically heterogeneous. Around 1–3% of cases are autosomal dominant AD (ADAD), carrying pathogenic variants in amyloid precursor protein (APP), presenilin-1 (PSEN1) and presenilin-1 (PSEN2), normally with onset before 65 years old (3). Except for ADAD, most AD cases are considered sporadic and manifest after 65 years old (4). We and others have identified several rare coding variants in triggering receptor expressed on myeloid cells 2 (TREM2) that increase risk of AD by almost two-fold, making TREM2 the second strongest genetic risk factor for sporadic AD after APOE (5, 6). However, despite these established genetically defined AD subtypes (ADAD or TREM2 risk variant carriers), no proteomic study systematically identifies dysregulated proteins across tissues under these conditions.
Timely diagnosis of AD is critical in clinical practice. neurofilament light chain (NfL), Aβ42, Aβ42/40 ratio, and phospho-tau181 (p-tau) are the gold standard of cerebrospinal fluid (CSF) protein biomarkers (7), and several noninvasive plasma biomarkers including Aβ42/40 ratio, p-tau217, p-tau231, and GFAP are also showing high performance (7–9). Identification of additional biomarker signatures in CSF and plasma is important, as they may be more effective disease-modifying targets. Even for clinical trials and therapies that target the already identified proteins, additional biomarkers that do not depend on the target protein are needed to monitor a treatment outcome. Therefore, it is important to develop prediction models that are independent of Aβ and tau pathology. Furthermore, all these fluid biomarkers only differentiate between sporadic AD cases and controls. Proteomic profiling of genetically defined AD subtypes, including individuals with AD-risk variants in TREM2 and ADAD cases, is important for fully understanding the biology of this heterogeneous disease and for identifying AD subtype-specific molecular biomarkers and therapeutic targets.
To identify proteomic profiles not only for sporadic AD, but also for genetically defined AD subtypes (carriers for TREM2 risk variants and ADAD individuals), we measured up to 1,305 proteins in brain, CSF, and plasma from the Knight Alzheimer Disease Research Center (Knight ADRC) and Dominantly Inherited Alzheimer Network (DIAN) (3, 10). We identified a set of proteins that were differentially altered in sporadic AD and genetically defined AD cases. Many of them were successfully validated through several independent cohorts using multiple orthogonal platforms. The replicated proteins were used to create tissue-specific prediction models and to identify the pathways leading to the disease. We also built a web portal ((omics.wustl.edu/proteomics) to support interactive visualization and exploration for the scientific community.
RESULTS
Multi-tissue Proteomic Signatures of Sporadic AD
To identify multi-tissue proteomic changes associated with sporadic AD, we quantified 1,305 proteins using a multiplexed, single-stranded DNA aptamer assay developed by SomaLogic (11). After a standard data processing, normalization, and quality control (Supplementary Materials and Methods), 1,092 proteins in brain, 713 in CSF, and 931 in plasma remained (Fig. 1). The correlation matrix of this proteomics data is in fig. S1. In each tissue, we performed differential abundance analysis comparing sporadic AD cases and cognitively normal individuals: 290 neuropathologically confirmed AD and 25 cognitively normal individuals with no brain pathology in brain; 176 clinical AD cases and 494 cognitively normal individuals in CSF; and 105 clinical AD cases and 254 cognitively normal individuals in plasma (Table 1). A web portal ((omics.wustl.edu/proteomics) was created to facilitate both exploration of our analysis and further investigation into individual proteins across disease status or sex (fig. S2). Specifically, we performed surrogate variable (SV) analysis (12) to remove batch effects and other unmeasured heterogeneity in each proteomic data. We then performed regression analysis with log-transformed protein abundance as a dependent variable and sporadic AD status as an independent variable, while including age, sex, and SVs as covariates. Results were not different when the technical batch was used instead of SVs (fig. S3-4). Instead of false discovery rate (FDR), we used a more stringent multiple test correction based on the number of independent proteins (see Supplementary Materials and Methods). Several publicly available datasets were downloaded and analyzed to replicate our findings in the discovery cohort (table S1, Fig. 1).
Fig. 1. Study outline.
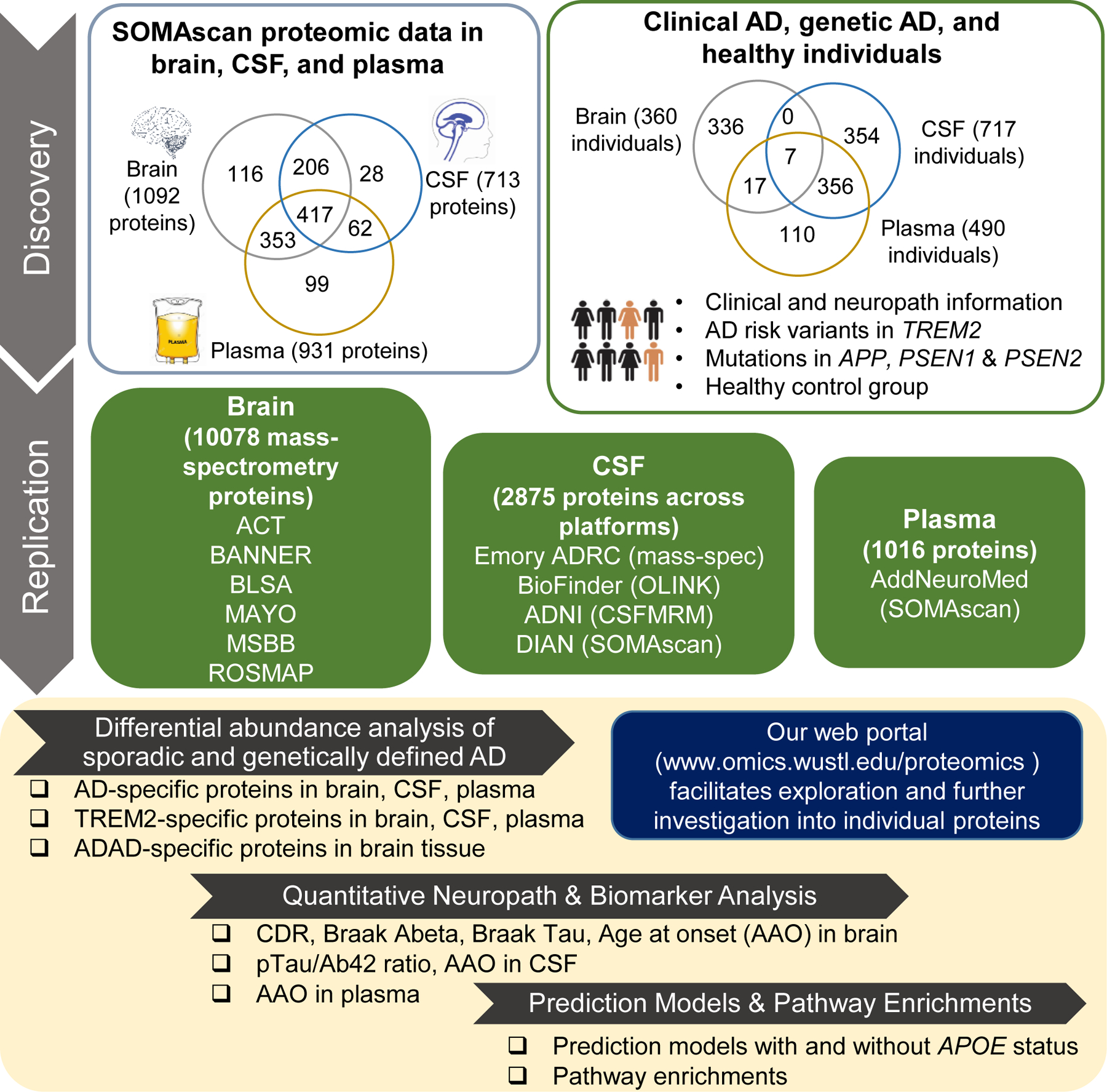
In the discovery stage, protein measures with SOMAscan targeting 1,305 proteins were obtained in brain, CSF, and plasma from well-characterized Knight ADRC and DIAN participants with comprehensive clinical information about AD pathology and cognition. This discovery cohort contained sporadic AD (290 in brain; 176 in CSF; 105 in plasma), TREM2 risk variant carriers (21 in brain; 47 in CSF; 131 in plasma), autosomal dominant AD (24 in brain), and healthy controls (25 in brain; 494 in CSF; 254 in plasma). Differential abundance analyses were performed for sporadic AD status, TREM2 risk variant carrier status, and autosomal dominant AD status. Several publicly available external proteomics data were then used to replicate our findings (details in Supplementary Materials and Methods). Finally, replicated proteins were used for creating tissue-specific prediction models and pathway enrichment analysis. In addition, we built a web portal (omics.wustl.edu/proteomics to support interactive visualization and exploration (fig. S2).
Table 1.
Summary characteristics of participants with proteomic measures in the Knight ADRC and DIAN cohorts.
| Tissue | Status | Sample size (N) | % Female | Age (mean ± SD) |
|---|---|---|---|---|
| Brain | CO | 25 | 61.72 | 88.24 ± 8.85 |
| AD | 290 | 33.33 | 83.98 ± 8.83 | |
| ADAD | 24 | 76.00 | 55.67 ± 14.58 | |
| TREM2 | 21 | 57.14 | 82.57 ± 7.62 | |
|
| ||||
| CSF | CO | 494 | 55.26 | 73.15 ± 6.43 |
| AD | 176 | 46.02 | 74.60 ± 7.02 | |
| TREM2 | 47 | 44.68 | 74.00 ± 6.48 | |
|
| ||||
| Plasma | CO | 254 | 57.48 | 71.53 ± 7.31 |
| AD | 105 | 37.14 | 72.59 ± 7.67 | |
| TREM2 | 131 | 64.89 | 74.98 ± 8.17 | |
CO = healthy control; AD = sporadic AD cases; ADAD = Autosomal dominant AD; TREM2 = AD-risk variant (p.E151K, p.H157Y, p.L211P, p.R136Q, p.R163Q, p.R47H, p.R62H, p.T96K) carriers in TREM2; CSF = cerebrospinal fluid.
Brain proteomic profiles for sporadic AD
In the brain, 12 proteins showed significant association with AD status (Fig. 2A and 2B, see table S2 for p-values). All these proteins were also associated with other AD-related traits including age at onset and AD neuropathological characteristics [such as Braak scores and clinical dementia rating (CDR) (13) at death] at nominal P < 0.05 (table S3, fig. S5-6). To examine across-tissue consistency, we checked whether these proteins were also associated with AD risk or onset in CSF and plasma. Out of the 12 brain proteins associated with AD status, only six were found in both CSF and plasma data. Of these, five proteins (SMOC1, HGF, FSTL1, UBC9, and NET1) were associated with AD status or age at onset in both CSF and plasma data (P < 0.05, table S2), which represents a 333-fold enrichment (P = 5.8×10-13) to what would be expected by chance.
Fig. 2. Multi-tissue proteomics profiling of sporadic AD.
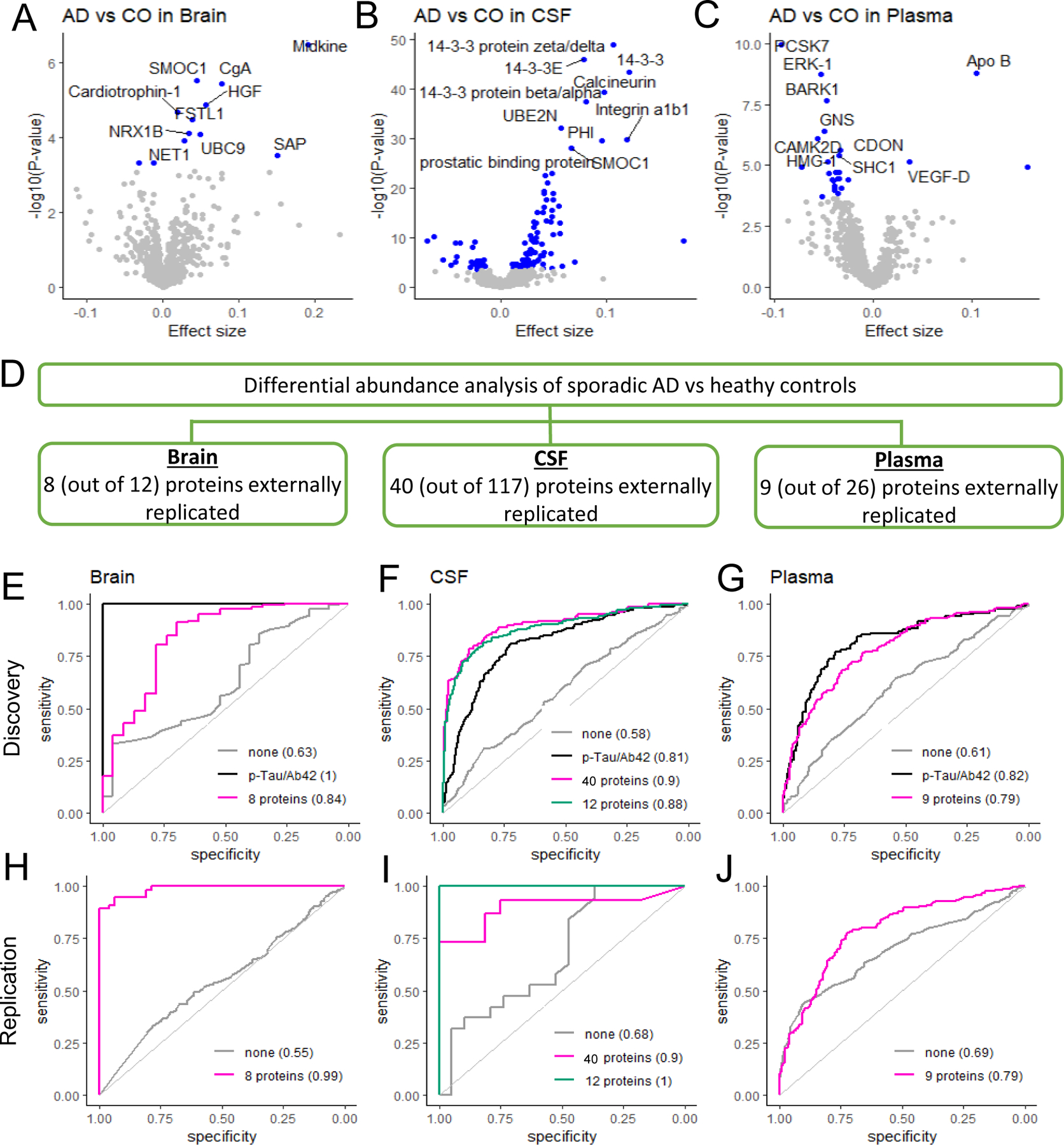
(A) Summary showing the number of identified and externally replicated proteins in three tissues. (B) Volcano plots for brain, CSF, and plasma in discovery analysis. Differential abundance between AD and healthy control (CO) groups is in x-axis and –log10(P-value) for statistical significance are in y-axis. The blue points show the proteins significant at the multiple testing-corrected threshold. While the top 10 proteins are labeled here, the volcano plots in the web portal ((omics.wustl.edu/proteomics) support interactive exploration for all proteins. (C) Tissue-specific prediction models based on the externally replicated proteins (eight in brain, 40 in CSF, and nine in plasma) for both discovery and replication data. Sex and age were included as covariates for all models. ‘None’ corresponds to the model including age and sex only, without any proteins or other biomarkers. Replication data were MassSpec Joint combining all mass-spec based cohorts in brain, Emory-ADRC mass-spec data in CSF, and AddNeuroMed in plasma.
To replicate our findings in the discovery cohort, we downloaded the mass-spectrometry data from the Adult Changes in Thought (ACT), Banner Sun Health Research Institute (BANNER), Baltimore longitudinal study of aging (BLSA), Mayo Clinic (MAYO), Mount Sinai Brain Bank (MSBB), and the Religious Orders Study and the Memory and Aging Project (ROSMAP). We integrated these mass-spectrometry data sets including 10,078 proteins from 415 AD patients and 194 controls (referred as MassSpec Joint) and subsequently performed differential abundance analysis for AD status (table S4). Of the nine proteins that were present in these datasets, eight replicated (Midkine, SMOC1, CgA, HGF, NRX1B, UBC9, NET1, and SAP) with the consistent direction at P < 0.05. This represents a 35-fold enrichment to what would be expected by chance (P = 1.3×10-12). In addition, to confirm that our results were not false positives due to the joint analysis that included six studies, we checked the published results of each individual study [Johnson et al (14), Higginbotham et al (15), and Wingo et al (16)]. Individual study analysis also provided 25–34-fold enrichments (table S1). Overall protein changes with AD status in our discovery data and the merged replication data, MassSpec Joint, were similar (P < 3.6×10-3; fig. S7A).
Here we (i) identified 12 proteins that were altered in sporadic AD brains from our discovery data; (ii) among six proteins available to test, five were validated across tissues; and (iii) among nine available to test, eight were replicated in the external replication data that had been generated with orthogonal proteomics platforms. Together, they indicate that our brain proteomic signature for sporadic AD is robust.
CSF proteomic profiles for sporadic AD
In CSF, 117 proteins were significantly associated with clinical AD status (Fig. 2A and 2B, see table S5 for p-values). Of these 117 proteins, 78 were found in brain and plasma data, and 27 proteins (including ERK-1 and LRRK2) were validated across tissues (138-fold enrichment, P = 3.3×10-50). For external replication, we obtained and analyzed Alzheimer’s Disease Neuroimaging Initiative (ADNI) multiple reaction monitoring (MRM) proteomic data. We also used results based on BioFinder OLINK data from Whelan et al (17) and Emory-ADRC mass-spectrometry data from Higginbotham et al (15). Of the 117 CSF proteins identified in the discovery cohort, 90 were present in external datasets (tables S5 and S6). In these external data sets, 40 proteins (including 14–3-3, Calcineurin, SMOC1, GFAP, SPP1, and Peroxiredoxin-1) were replicated at P < 0.05 and in the same direction (14–34-fold enrichments, P ≤ 4.4×10-5). Separately in the ADNI data (320 CSF samples), eight proteins were available among the 117 identified in discovery, and seven were replicated. In Higginbotham et al (15), there were 88 proteins among our identified proteins, and 34 were replicated. We speculate that a small sample size of this study (N=40) is a primary reason for a limited power in replicating the discovery findings. Correlation of altered protein changes between discovery and replication data was strong (r = 0.43–0.82, P < 3.4×10-7, fig. S7B). We therefore expect that more proteins would replicate in larger studies.
Plasma proteomic profiles for sporadic AD
In plasma, 26 proteins were associated with sporadic AD status after the multiple testing correction (Fig. 2A, table S7). Similar to previous analyses, we leveraged the multi-tissue data to replicate these findings. Of the 26 plasma proteins associated with AD status, 16 were found in brain and CSF, and seven proteins (including ERK-1, CDON, and SHC1) were replicated (175-fold enrichment, P= 6.8×10-15). For external replication, we downloaded and analyzed the AddNeuroMed SOMAscan 1.1K proteomic data that was processed and deposited by Sattlecker et al (18). Out of 26 associated proteins, we were able to test 19 in this data (table S8). Nine proteins (including CAMK2D and HMG-1) were replicated. This represented 18.9-fold enrichment (P = 2.8×10-10) to what would be expected by chance.
Here, we identified eight proteins in brain, 40 proteins in CSF, and nine proteins in plasma that were altered by AD status through a traditional discovery and replication strategy by leveraging several external proteomic datasets generated from orthogonal platforms. They included Apo E2 and SMOC1 and other previously identified proteins (table S9), related to amyloid/tau pathology. Several proteins identified in this study showed a very weak correlation with CSF Aβ42 (median correlation value for Aβ42 was 0.06 in brain, 0.002 in CSF, and 0.04 in plasma). While CSF proteins were modestly correlated with CSF p-Tau181 (median correlation = 0.35), proteins in brain and plasma were very weakly correlated (median correlation = −0.03 and 0.006, respectively). These proteins associated with AD, independent of amyloid/tau pathology, can be useful for potential therapeutics. Correlation plots for all these externally replicated proteins were in fig. S8.
As in some scenarios it may not be possible to use independent datasets for replication, we tested whether protein identification across different tissues can serve as an alternative option for replication. An enrichment test showed that the proteins identified in all tissues were more likely to be replicated in external independent datasets than proteins identified in single tissues (15- to 40-fold enrichments, P ≤ 3.63×10-3, table S10). This suggests that multi-tissue proteomic data can be used as a viable replication strategy to support the findings.
Proteomic profiles of AD based on biomarkers
Most of AD biomarker studies in CSF and plasma published so far examined clinical AD status and not biomarker-based status (18). To allow for an easy comparison with these earlier studies, we also performed our analysis using clinical status as AD classifier. However, several studies indicated that up to 30% of cognitively normal elderly individuals could be pre-symptomatic for AD (19) and that other neurodegenerative diseases can clinically masquerade as AD dementia (20). It was shown that the CSF p-tau/Aβ42 ratio is a gold-standard biomarker not only for AD status but also for predicting AD progression from normal to dementia within 5 years (10). To examine whether our findings based on clinical status were robust, we subsequently performed differential analyses with both AT(N) classification and CSF p-tau/Aβ42 ratio. We had access to CSF p-tau/Aβ42 measures for 689 (out of 717) CSF samples and 393 (out of 490) plasma samples in the discovery study. Following the biomarker-based AT(N) classification (21), we obtained AT classification of amyloid/tau positivity in these CSF and plasma samples (Supplementary Materials and Methods). Of the 117 proteins associated with clinical AD status, 97 were significant for CSF p-tau/Aβ42 and 102 for the AT(N) classification (see table S11 for p-values). More importantly, there was a strong correlation between protein changes by clinical AD status and those by biomarker-based status (r = 0.86 and 0.88, respectively; P < 1.0×10-16; fig. S9). Similar results were found for plasma (fig. S9; table S12). This high correlation indicates that the results found by using clinical AD status can be interchangeable with those using biomarkers in this discovery study.
Tissue-specific Prediction Models
A prediction model (or predictive model) using protein biomarkers is critical for early diagnosis and monitoring disease progression. We created prediction models based on the eight, 40, and nine proteins that were detected in brain, CSF and plasma, respectively, and that were replicated in external datasets, while including sex and age as covariates (Fig. 2C). The prediction model based on the eight brain proteins provided high accuracy in distinguishing AD cases from cognitively normal individuals: an area under curve (AUC) of 0.84 in discovery and 0.99 in the independent MassSpec Joint data. The prediction model based on the 40 proteins detected in CSF provided an AUC of 0.89 in discovery and 0.90 in the Emory-ADRC mass-spec replication study. As this prediction model included too many proteins to translate into clinical practice, we performed a stepwise model selection and identified a panel of 12 proteins (table S13). These 12 proteins provided accuracy almost as high as all 40 proteins, leading to an AUC of 0.88 in discovery and 0.99 in replication data. This was significantly higher than the AUC based on the well-known CSF p-tau/Aβ42 ratio (AUC = 0.81; P = 2.4×10-6). Using the same approach for plasma, the prediction model based on the nine proteins led to an AUC of 0.79 in both discovery and AddNeuroMed replication data. This was not statistically different from the AUC with the CSF p-tau/Aβ42 ratio (AUC=0.82; P>0.05). The prediction model based on each protein was similar between the discovery and replication data (fig. S10).
Multi-tissue Proteomic Signatures of TREM2 Risk Variant Carriers
Several rare coding variants in TREM2 that increase risk of AD by almost two-fold have been identified (4). We therefore aimed to identify multi-tissue proteomic signatures of individuals carrying AD-risk variants in TREM2. Given the low frequency of occurrence of different TREM2 variants, all of them were combined into one group of TREM2 risk variant carriers. We generated brain, CSF, and plasma proteomic data for 21, 47, and 131 TREM2 variant carriers, respectively (Table 1) and compared their protein abundance with both cognitively normal individuals and individuals who were sporadic AD but did not carry any TREM2 variant. This is the first time a proteomic profile for TREM2 variant carriers has been generated.
In the brain, nine proteins (including α-Synuclein) were altered in TREM2 risk variant carriers compared to cognitively normal individuals at the multiple testing corrected threshold (Fig. 3A and 3B; table S14). In addition, 23 proteins (including LRRK2) were altered in TREM2 risk variant carriers when compared to other sporadic AD cases (table S15). Five proteins (ALT, HSP 70, HSP70 protein 8, PACAP-27, PSA1) were commonly found, indicating their distinct protein abundance across all three groups (TREM2 variant carriers vs. healthy controls vs. other sporadic AD cases). The external data included only four TREM2 variant carriers in MAYO, seven in MSBB, and eight in ROSMAP, which did not provide any statistical power to replicate our findings. As we demonstrated, our multi-tissue study design is a viable alternative approach to identify proteins that would replicate in external datasets, and we leveraged our data to identify those proteins that replicate across tissues. Out of these 27 TREM2-associated proteins found in the brain (combining nine and 23 proteins minus five overlaps), 11 were replicated only in CSF, six were replicated only in plasma, and five (ALT, α -Synuclein, MIS, LRRK2, and PAFAH beta subunit) were replicated in both tissues. This represents a 74-fold enrichment (P = 7.5×10-9) to what would be expected by chance.
Fig. 3. Multi-tissue proteomics profiling of TREM2 variant carrier status.
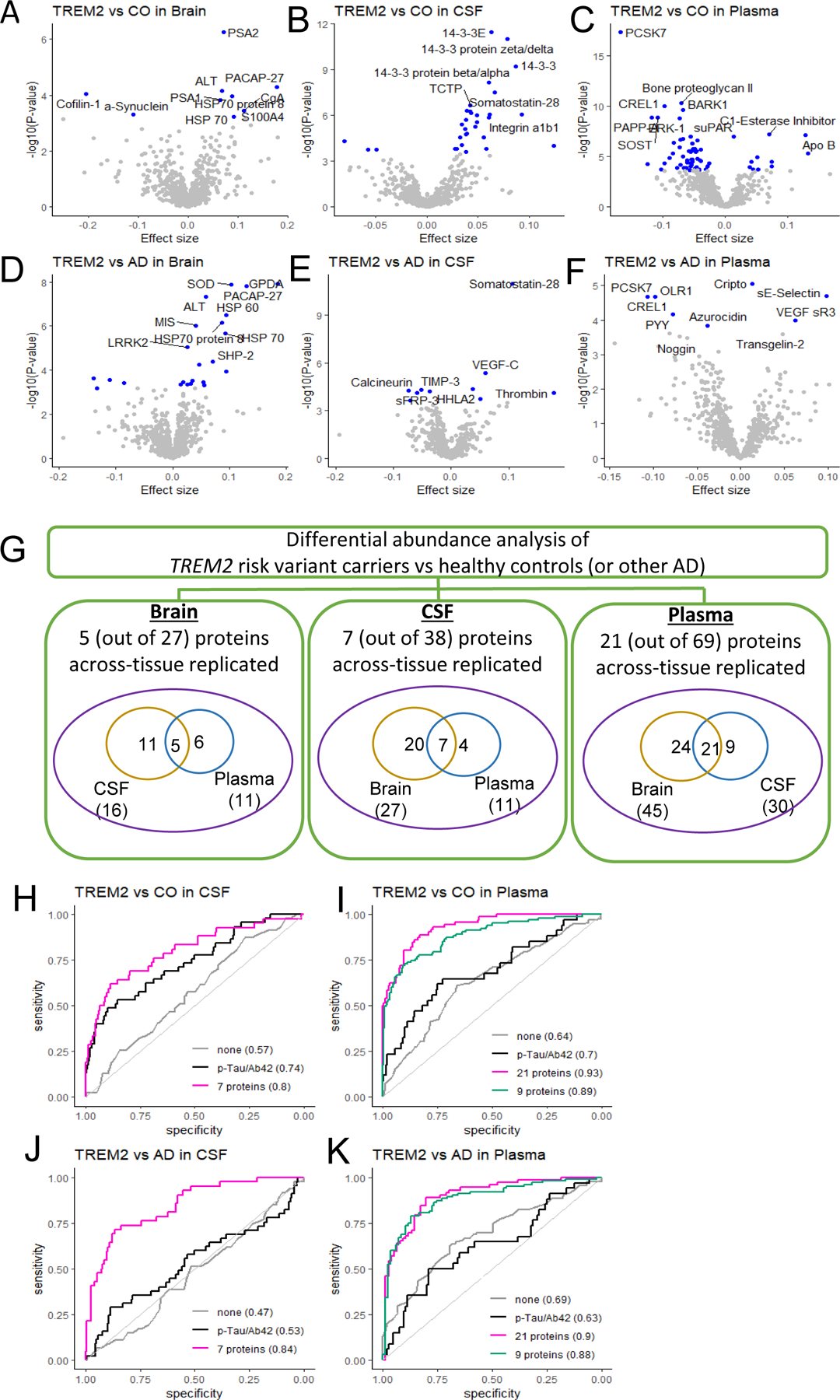
(A) Summary showing identified and across-tissue replicated proteins in three tissues. Multiple proteins showed differential abundance in TREM2 variant carriers (compared to controls or other sporadic AD cases) in at least one of the three tissues, several of which are replicated across tissue. (B) Volcano plots for brain, CSF, and plasma tissue. Differential abundance between TREM2 variant carriers and healthy control (CO) groups (at top panels) or other AD (at bottom panels) are in x-axis and –log10(P-value) for statistical significance are in y-axis. While the top 10 proteins are labeled here, the volcano plots in the web portal ((omics.wustl.edu/proteomics) support interactive exploration for all proteins. (C) Tissue-specific prediction models, including sex and age as covariates. ‘None’ corresponds to the model including age and sex only, without any proteins or other biomarkers.
In CSF, we identified 31 proteins altered in TREM2 variant carriers compared to healthy controls (table S16) and 10 proteins altered in TREM2 variant carriers compared to other sporadic AD cases (table S17). Three proteins (Nucleoside diphosphate kinase A, Somatostatin-28, and Thrombin) were common. Out of these 38 proteins (31 and 10 minus 3 overlaps), 20 were replicated in the brain, four were replicated in the plasma, and seven (14–3-3E, 14–3-3 protein zeta/delta, Somatostatin-28, SMOC1, Ubiquitin+1, QORL1, and Calcineurin) were replicated in both tissues. This represents a 73-fold enrichment (p=7.19×10-12) to what would be expected by chance. In plasma, we identified a total of 69 proteins: 65 proteins altered in TREM2 variant carriers compared to healthy controls (table S18); seven proteins altered in TREM2 variant carriers compared to other sporadic AD (table S19); and three overlaps (CREL1, Cripto, PCSK7). Among these 69 proteins, 24 proteins were present in the brain, nine proteins were replicated in the CSF, and 21 (including bone proteoglycan II, PAPP-A, ERK-1, suPAR and VCAM-1) were replicated, representing a 122-fold enrichment (p=5.47×10-38) to what would be expected by chance.
We also created prediction models that could distinguish TREM2 variant carriers from non-carriers in both sporadic AD cases and controls for CSF and plasma. In CSF, the prediction model based on the seven across-tissue replicated proteins provided an AUC of 0.79 for distinguishing TREM2 variant carriers from cognitively normal individuals (Fig. 3C). The same model showed an AUC of 0.84 for distinguishing TREM2 variant carriers from the other sporadic AD cases. While the CSF p-tau/Aβ42 ratio is a very good biomarker to distinguish AD cases from controls, no previous studies examined how the CSF p-tau/Aβ42 ratio provides prediction for TREM2 variant carriers. In this study, CSF p-tau/Aβ42 showed an AUC of 0.74 for TREM2 variant carriers vs. cognitively normal individuals and 0.53 for TREM2 variant carriers vs. other AD. Both AUC values from our TREM2-associated prediction model with seven proteins were significantly higher than the model based on the CSF p-tau/Aβ42 ratio (P < 1.6×10-5; Fig. 3C).
In plasma, our prediction model based on the 21 across-tissue replicated proteins provided an AUC of 0.93 in distinguishing TREM2 variant carriers from healthy individuals. Predictive performance was significantly higher than the model based on the CSF p-tau/Aβ42 ratio (AUC = 0.69; P = 1.1×10-3). Similarly, in distinguishing TREM2 risk variant carriers from other AD cases, the prediction model based on the same 21 proteins provided an AUC of 0.90, which is significantly higher (P = 1.5×10-4) than the AUC with the CSF p-tau/Aβ42 ratio (AUC=0.63). As the number of proteins is large, we performed a stepwise model selection and found a subset of nine proteins (table S13) that provided AUCs of 0.89 and 0.88 in distinguishing TREM2 variant carriers from cognitively normal individuals and from other sporadic AD cases, respectively (Fig. 3C). The prediction models including age, sex and APOE ε4 status as covariates provided similar performance (fig. S11).
Proteomic Signatures of Autosomal Dominant AD (ADAD)
To identify proteins associated with autosomal dominant AD status, we generated proteomic data from the parietal cortex of 24 individuals carrying pathogenic ADAD variants (19 individuals with PSEN1, one with PSEN2, and four with APP variants) recruited from the DIAN and the Knight ADRC studies. We identified 109 proteins altered in ADAD mutation carriers when compared to cognitively normal individuals with no brain pathology at the multiple testing-corrected threshold (Fig. 4A). As ADAD cases were much younger than the cognitively normal control group, age was not included in the model to avoid co-linearity. However, this could lead to false positives as some of the significant proteins could be associated with age rather than ADAD status. To address this, we identified 98 proteins associated with age in the control group at nominal significance (P < 0.05; Fig. 4B, table S20). Of these age-associated proteins, 17 (out of 109) were associated with ADAD status and excluded from any downstream analyses.
Fig. 4. Proteomic profiling of autosomal dominant AD (ADAD) status.
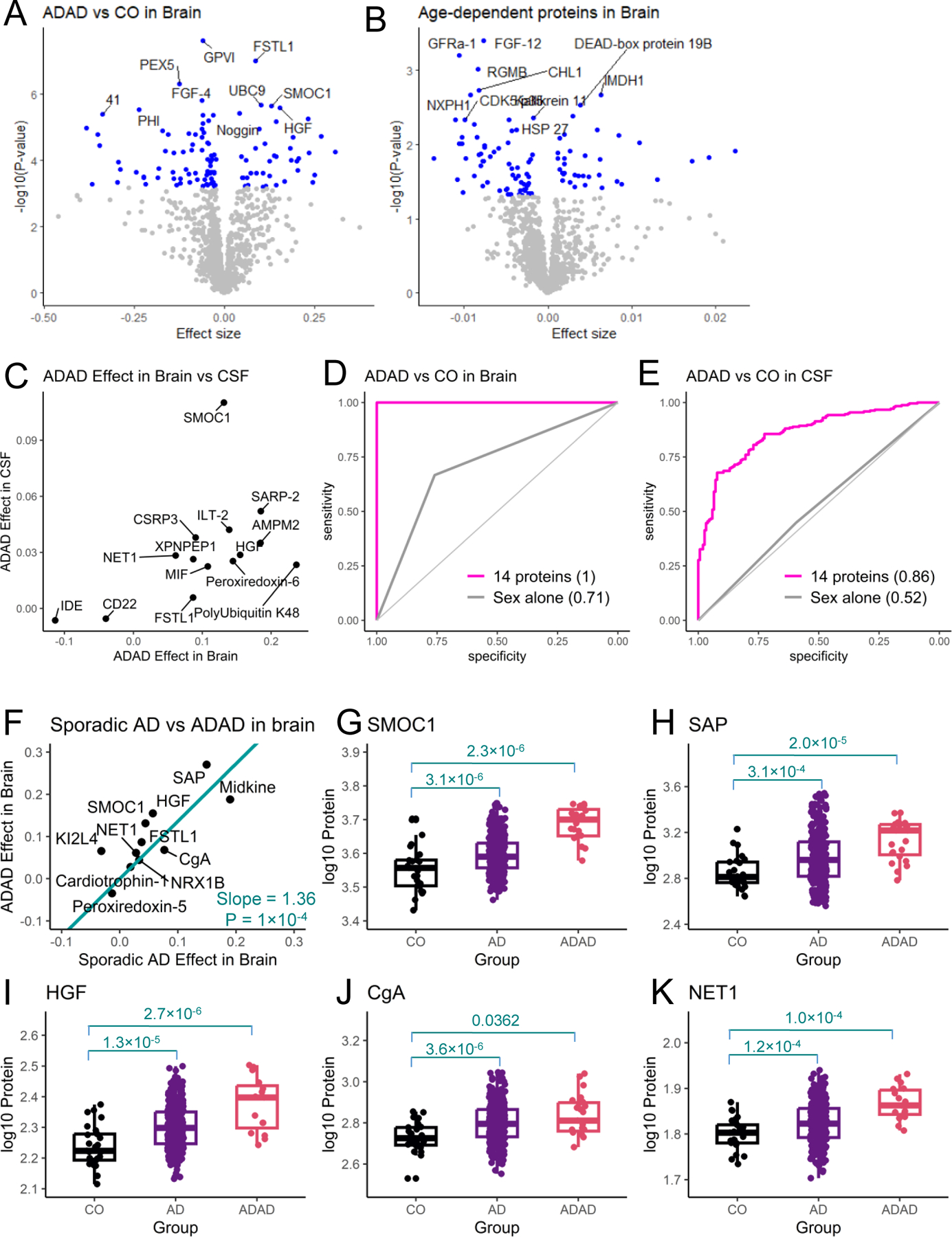
(A) Volcano plots of differential abundance analysis between individuals with ADAD and healthy control (CO) groups. (B) Volcano plots of differential abundance analysis of age dependent proteins in the brain. (C) The scatterplot of the 14 proteins replicated in CSF and their prediction models in brain and CSF. (D) The scatterplot of the 12 proteins associated with sporadic AD status. The effect of ADAD status on log-transformed protein abundance is in y-axis, and the effect of AD status is in x-axis. The box plots for the select 5 proteins are displayed.
In order to validate whether the remaining 92 proteins were also associated with ADAD status in CSF, we analyzed CSF proteins from 289 ADAD mutation carriers and 184 non-carriers from the DIAN study. Of the 92 proteins identified in brain, 89 passed QC in CSF proteomic data, and 14 were altered by ADAD status in CSF in the consistent direction (Fig. 4C, table S21), representing a 6.3-fold enrichment (p=4.88×10-8) to what would be expected by chance. We next leveraged these 14 proteins to create potential prediction models for distinguishing ADAD mutation carriers from non-carriers. A prediction model using these 14 proteins in the brain provided an AUC of 1, fully separating ADAD cases from the healthy controls. In CSF data, the same 14 proteins provided higher predictive performance than the model with sex alone (AUC = 0.86 vs 0.52, P < 2.2×10-16; Fig. 4C).
As presented earlier, we identified 12 proteins associated with sporadic AD status in brain tissue (Fig. 2A; table S2). We also sought to determine if the proteins altered by sporadic AD status showed similar alteration in ADAD individuals. We found that most of the proteins associated with sporadic AD brains displayed even stronger alteration in ADAD individuals compared to healthy individuals (table S22). The proteins associated with sporadic AD status showed 36% higher protein changes in ADAD brain samples on average (P = 1.0×10-4; Fig. 4D). For example, SMOC1 showed a significant association not only for sporadic AD status (protein change = 0.04: P = 3.1×10-6) but also for ADAD (protein change = 0.13; P = 2.3×10-6). As presented earlier, SMOC1 was also associated in sporadic AD status in both CSF (P = 8.4×10-29) and plasma (P = 0.002), suggesting that it could be used to create a new prediction model for AD.
Validation of Somalogic data
We recently demonstrated that Somalogic data correlated very well with classic enzyme linked immunosorbent assay (ELISA) for several well-known CSF biomarkers (22). We found excellent correlation (over 0.91) for NFL, Neurogranin, and VILIP-1. Prediction for AD status based on the CSF SomaScan proteins were also comparable to that based on the ELISA biomarkers.
To further validate our SomaScan data, we examined the 150 proteins we identified across different platforms in samples from the Emory, ADNI, and DIAN cohorts (table S23 and table S24). In the Emory cohort, the overall correlation for the overlapping proteins was 0.75 for CSF and 0.74 for plasma when comparing Somalogic vs. mass-spectrometry measures and 0.74 for both CSF and plasma when comparing Somalogic vs. Olink (Table S23). Many proteins showed higher correlation including SMOC1, a-Synuclein, 14–3-3E, TNFRSF1A, APOB, APCS, HSP90AA1, TGFBR3, VEGFC, CCDC80, CNTN5, and TFPI (fig. S12-S15). In ADNI, the overall correlation for the eight proteins that overlap was 0.72 (fig. S22). Of these proteins, GFAP, aldolase A (ALDOA), Peroxiredoxin-1 (PRDX1), Peroxiredoxin-6 (PRDX6), Polyubiquitin K48 (UBB, fig. S16) all with very high correlation, were replicated in CSF analysis using ADNI mass-spectrometry data (Table S17). APOB was identified in our plasma analyses. For the DIAN cohort, we found that the overall correlation for the overlapping 14 proteins were 0.79 (Table S23). These proteins included SMOC1 (identified in brain and CSF), Osteopontin (SPP1), PEBP1, ENO1, ENO2, YWHAZ and ALDOA (identified in CSF) (fig. S17).
Functional Pathways
Finally, we examined functional pathways for the proteins identified in our study using Enrichr (23). As expected, the identified proteins were enriched for the AD pathway (FDR = 1.9×10-2, Fig. 5A, table S25), including Apo E (APOE), Calcineurin (PPP3R1 and PPP3CA), and ERK-1 (MAPK3) (Fig. 5B, fig. S19). This finding supports our study design to identify and validate proteins for AD. In addition to the AD pathway, the identified proteins altered in sporadic AD and TREM2 variant carriers were enriched for the Parkinson disease (PD) pathway (FDR = 2.1×10-12 for sporadic AD; FDR = 1.4×10-4 for TREM2 variant carriers; table S25). Among the proteins in the PD pathway, α-synuclein (SNCA) was altered in both sporadic AD (P = 9.3×10-8) and TREM2 carriers (P = 5.0×10-4), compared to controls. Similarly, LRRK2 was altered in sporadic AD (P = 8.3×10-9) and TREM2 carriers (P = 9.3×10-6). We also found that identified proteins in sporadic AD and TREM2 carriers were enriched for the innate immune system pathway (FDR = 2.0×10-3 for sporadic AD; FDR =8.4×10-3 for TREM2 carriers). In particular, proteins in sporadic AD were enriched for the neutrophil mediated immunity (FDR = 1.6×10-6). The proteins in this pathway include Osteopontin (SPP1) and Integrin a1b1 (ITGB1), among others.
Fig. 5. Pathway enrichments for sporadic and genetically defined AD.
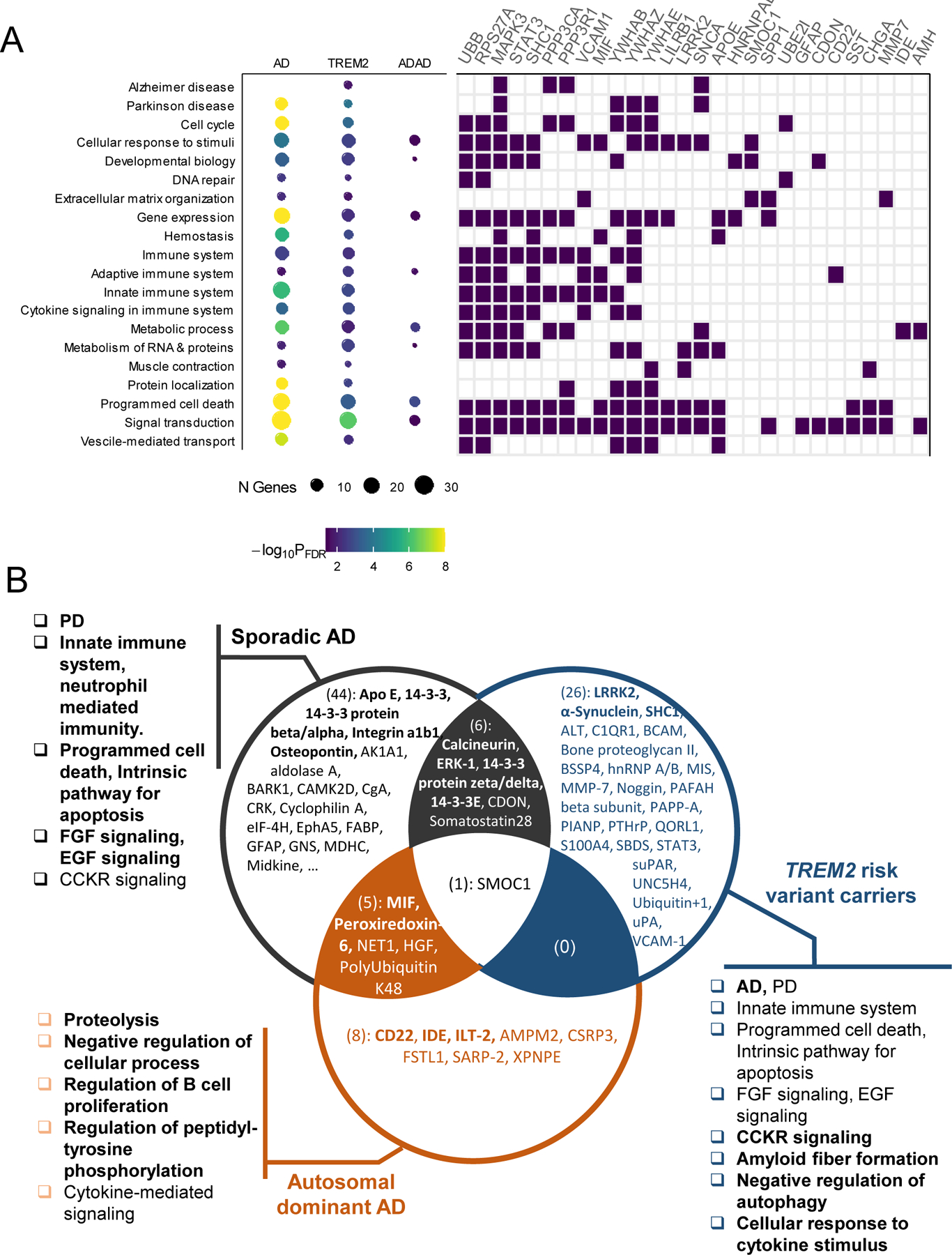
(A) The dot chart (on the left, size corresponding to the number of identified genes and color corresponding to the FDR corrected significance, table S25). The tile plot (on the right) show genes that belong to the specific pathway. A full list of 81 genes is shown in fig. S12. (B) The Venn diagram shows overlap of identified proteins across the three groups: 56 externally replicated proteins for sporadic AD, 33 across-tissue replicated proteins for TREM2, 14 proteins replicated in DIAN CSF data for ADAD. Proteins mentioned in Pathway section are in boldface (for example, Apo E). The pathways highlighted in Pathway section are included. The most enriched pathways across 3 groups are in boldface.
The most strongly enriched pathways for sporadic AD were the programmed cell death (FDR = 8.6×10-9) and intrinsic pathway for apoptosis (FDR = 7.0×10-11), that included several 14–3-3 proteins (YWHAE, YWHAZ). This suggests that some of the cell death associated with AD pathogenesis is regulated by apoptosis and not due to necrosis, entosis, ferroptosis, or lysosomal dependent cell death. Signaling pathways, including FGF signaling pathway (FDR = 9.0×10-9) and EGF receptor signaling pathway (FDR = 8.7×10-10), were also among the top pathways for sporadic AD.
The most strongly enriched pathway by TREM2-specific proteins was cholecystokinin (CCK) receptor signaling (FDR = 5.4×10-7). This pathway includes ITGB1, PPP3CA, YWHAB, and MAPK3. CCK is a satiety hormone that is highly expressed in the brain, including hippocampus. It has been shown that CSF CCK was related to memory scores, higher CSF tau and p-tau values in ADNI cohort (24). Other notable pathways related to TREM2-specific proteins were amyloid fiber formation (FDR = 2.5×10-3), negative regulation of autophagy (FDR = 5.3×10-3), and cellular response to cytokine stimulus (FDR = 5.7×10-3).
Pathways enriched by ADAD-specific proteins also pointed to biological processes involved in this AD subtype. Notable pathways were proteolysis (FDR = 3.2×10-3), negative regulation of cellular process (FDR = 4.0×10-2), regulation of B cell proliferation (FDR = 2.7×10-2), and regulation of peptidyl-tyrosine phosphorylation (FDR = 3.2×10-3). They include IDE, MIF, a pro-inflammatory cytokine involved in the innate immune response, and ILT-2 (LILRB1) and CD22. IDE is involved in the cellular breakdown of insulin and reported to be involved in the degradation and clearance of naturally secreted amyloid beta-protein by neurons and microglia (25). These proteins were also enriched for cytokine-mediated signaling pathway (FDR = 4.1×10-2), indicating that inflammation also plays a role in autosomal dominant AD.
DISCUSSION
This is the first large-scale, multi-tissue proteomic characterization of sporadic and genetically defined AD sub-types. Particularly, we obtained proteomic measures from Knight ADRC and DIAN cohorts and identified proteomic profiles for sporadic AD, TREM2 variant carriers, and autosomal dominant AD cases in three tissues. These proteomic profiles, replicated in independent datasets and across tissues, were used to create tissue-specific prediction models and to identify functional pathways. Our CSF-specific model for sporadic AD provided higher predictive performance than that with CSF p-tau/Aβ42 values (AUC = 0.88–0.90 vs. 0.81, Fig. 2). The predictive performance using plasma proteins was similar to that of CSF p-tau/Aβ42. These models were replicated in independent datasets with different protein quantification, indicating that our model is robust, reproducible, and reliable.
In addition, we demonstrated that (i) SomaScan measurements were in good agreements with mass-spec and OLINK measurements, with an overall correlation coefficient of around 0.74 (table S23, fig. S18); (ii) many proteins identified here showed very strong correlation including SMOC1, α-Synuclein (SNCA), 14–3-3E (YWHAE), GFAP and ALDOA (correlation >0.81, fig. S12-S18); and (iii) the correlation of these proteins between Somalogic vs mass-spectrometry or Olink were remarkably consistent regardless of cohorts and tissue. Similar findings have been reported recently by Dammer at al (26). The fact that these proteins replicated in multiple mass spectrometry data and that show a very high correlation across platform not only strengthens our findings but also indicates that the Somalogic measurements are robust. Taken together, all these validations demonstrate that the aptamer-based Somalogic approach is as accurate as any other platforms including with classic ELISA, Olink proximity extension assay, and multiple mass spectrometry data.
We created the prediction models for clinical AD status instead of the biomarker-based status because the latter was unavailable in most of the replication cohorts. The biomarker-based AT(N) status provides classification based on core AD pathophysiological features: the Aβ pathway (A), aggregated tau pathophysiology (T), and neuronal injury and neurodegeneration (N) (21). The AT(N) classification identifies individuals in pre-symptomatic stage. Because not all AT(N)-positive individuals develop cognitive impairment, as some may carry protective factors (27), our prediction models are optimized for symptomatic AD. AT(N)-based models can identify individuals with AD pathology, not those with clinical symptoms. We excluded any samples showing symptoms outside AD. Because our controls may include pre-symptomatic individuals, we also performed analyses using CSF p-tau/Aβ42 and AT(N) as an outcome. The top proteins from these analyses [with clinical status or CSF p-tau/Aβ42 or AT(N)] were the same, and the effect size for all proteins were highly correlated in both CSF and plasma (tables S11-S12, fig. S8), indicating that in the Knight-ADRC using clinical status or biomarker-defined status would lead to the same results.
Another advantage of using clinical status is the ability to compare our model with CSF p-tau/Aβ42 based models (tables S11-S12, fig. S8). We demonstrated that our model provided higher performance than p-tau/Aβ42 in CSF and comparable performance in plasma. Nakamura et al (28) presented blood-based biomarkers APP669–711/Aβ42 and Aβ40/Aβ42 for predicting Aβ positivity (AUC: 0.88–0.96). This and other Aβ-based blood assays are emerging as powerful biomarkers. However, as several FDA-approved targets are developed, non Aβ-based biomarkers are needed to monitor target engagement and treatment outcome. Zhang et al (29) identified 11 plasma proteins for predicting Aβ positivity (AUC = 0.73), two for p-tau positivity (AUC = 0.67), and seven for AT classification (AUC = 0.77) in one cohort. Our model based on the nine plasma proteins provided higher predictive performance (AUC = 0.79) in both discovery and replication cohorts. Palmqvist et al (30) created prediction models with plasma p-tau217, NFL, APOE genotype, MRI, and cognitive test, providing an AUC of 0.92 in discovery and 0.86 in replication. The AUC for p-tau217 alone was 0.72 in discovery and 0.78 in replication for predicting clinical AD. The prediction power of plasma p-tau217 was similar to those using CSF tau and Aβ42/Aβ40 (7–9). Our plasma models including only basic demographic information (age, sex) along with identified proteins provided comparable AUC (0.82–0.79) as those using blood p-tau217 (30). Similar to Palmqvist et al (30) and others (7), we considered independent replication cohorts to validate the model. Better prediction models could be constructed by combining our proteomics profiles with MRI and cognitive test, as demonstrated in previous studies.
In this study, we identified Apo E (APOE), Calcineurin (PPP3R1 and PPP3CA), and ERK-1 (MAPK3) associated with sporadic AD. APOE is the strongest and most common genetic risk factor for AD (31), and individuals with the APOE ε4 allele have lower CSF Aβ42 values (31) and lower Aβ42 clearance in brain (32). Genetic variants in PPP3R1 have been associated with higher CSF p-tau values and earlier age at onset (33). MAPK3 was also reported to be involved in AD pathology (34), likely by affecting tau phosphorylation. Calcineurin and ERK-1 were recently reported as part of the causal AD pathway by pQTL and Mendelian randomization analyses (35). In addition, we identified α-synuclein and LRRK2 associated with AD for the first time. On autopsy, around 30% of AD cases, including autosomal dominant AD, present with Lewy bodies, which are deposits of α-synuclein (36). With the discovery of pathogenic mutations in LRRK2 causing autosomal dominant PD (37), LRRK2 has emerged as a promising target for PD (38). Those reports, together with our analyses, support for pathological events and proteins shared across neurodegenerative diseases, specifically in this case for AD and PD. These findings could also help to develop fluid biomarkers to identify AD cases with PD pathology without waiting for autopsy.
The most strongly enriched pathways for sporadic AD were programmed cell death (FDR = 8.6×10-9), intrinsic pathway for apoptosis (FDR = 7.0×10-11), EGF receptor signaling pathway (FDR = 8.7×10-10), and the neutrophil mediated immunity (FDR = 1.6×10-6), among others. The neutrophil mediated immunity includes Osteopontin (SPP1) and Integrin a1b1 (ITGB1). SPP1 was recently implicated in microglia activation and AD (39). ITGB1 is a microglia gene and shown to be differentially expressed in the hippocampus and peripheral blood mononuclear cells (PBMC) in AD (40), important in microglia activation (41), and part of the causal AD pathway (42). Consistent with recent findings that meningeal lymphatics and endothelial-specific proteins affect microglia and AD risk (43), we found several endothelial-specific proteins (ERK-1, SHC1, and BCAM). This may be useful for fully understanding how changes in brain endothelial cells and in the blood-brain barrier contribute the disease. The EGF receptor signaling pathway regulates growth, survival, proliferation, and differentiation in mammalian cells. Dysregulation of this pathway is associated with white matter injury (44) and downstream to Aβ oligomers (45) and prevents APOE4 and Aβ-induced cognitive and cerebrovascular deficits (46), suggesting that developing strategies targeting this pathway may be particularly efficacious once Aβ pathology is present.
To our knowledge, this is the first study that identified proteins altered in TREM2 risk variant carriers and autosomal dominant AD cases. TREM2 is a microglia gene and activation of microglia results in their production of pro-inflammatory cytokines such as IL-1, IL-6, and TNF-α. Multiple pro-inflammatory (CCL21, CSF1R, IL24) and anti-inflammatory (IL1RN and ANXA1) proteins are a part of the cellular response to cytokine stimulus, extending the number of proteins that are part of the pathological pathways in TREM2 carriers. Once individuals with specific genetic profiles are identified, it is possible to create customized prediction models, an instrumental step toward individualized, specific disease risk evaluation and treatment. Integrating proteomic data with other omics (including genomics, transcriptomics, and metabolomics) can help better understanding the complex etiology of AD, further improving the discovery of AD biomarkers and their use in clinical management, as reviewed by Aerqin et al (47). In addition, there may be different downstream, and potentially causal, pathways leading to disease in these individuals. For example, in TREM2 variant carriers, we found a higher enrichment of the negative regulation of autophagy pathway (FDR = 5.3×10-3), which is also involved in cell death and seems to play a major role in AD pathogenesis in TREM2 variant carriers based on own analyses. Other unique pathway was cholecystokinin (CCK) receptor signaling (FDR = 5.4×10-7) for TREM2 risk variant carriers. For autosomal dominant AD cases, proteolysis (FDR = 3.2×10-3) and regulation of peptidyl-tyrosine phosphorylation (FDR = 3.2×10-3). These findings highlight the role of these biological processes and their relationship to TREM2 and ADAD downstream pathological events, which may represent new therapeutic targets.
The strength of this study is relatively large sample sizes, replication of proteins associated with sporadic AD cases in the multiple external data generated using orthogonal proteomics methods, and uniquely identifying proteins associated with the genetically defined AD cases. However, there are several limitations. First, several AD biomarkers in CSF and plasma including Aβ42/Aβ40 ratio, p-tau217, and p-tau231 that provide good performance were not available in our samples. Therefore, we were not able to compare our prediction models with the models based on these biomarkers. Second, to replicate our findings, we sought multiple external datasets (Supplementary Materials and Methods). As the proteomic data available in these studies were generated using a different platform (OLINK and mass-spectrometry), not all proteins identified in discovery were assayed. In addition, several replication datasets had smaller sample sizes than our discovery data, providing limited power. The replicated proteins therefore likely represent only a subset of proteins associated with AD status. Third, genetically defined AD cases (TREM2 variant carriers and autosomal dominant AD individuals) are extremely rare, even in AD cases. There were not enough carriers (four TREM2 variant carriers in MAYO, seven in MSBB, eight in ROSMAP, and no ADAD cases) in public datasets, and we were unable to pursue replication of the proteins dysregulated in these genetically defined AD cases. Fourth, we performed very stringent QC (Supplementary Materials and Methods). Because of this, not all proteins passed QC across all three tissues. This limited across-tissue replications and should be accounted for future studies.
In summary, we identified new proteins and pathways implicated in sporadic and genetically defined AD. We also demonstrated that, by leveraging proteins associated with AD status commonly across tissues, one could validate and confirm potential new biomarkers, which has important implications for the validation of AD biomarkers in future studies. A web portal ((omics.wustl.edu/proteomics) is available for further investigation. While additional validation of some of our findings will be needed, these results highlight the need for multi-tissue proteomics to fully understand the biology of AD and create prediction models for individuals with specific genetic profiles, supporting its utility in generating clinically useful biomarker arrays.
MATERIALS AND METHODS
Study Design
The goal of this study is to identify tissue-specific (brain, CSF and plasma) proteomic profiles for sporadic AD as well as genetically-defined AD cases (Fig. 1). This study included the brain (N=360), CSF (N=717), and plasma (N=490) data from the Knight ADRC (10) and the Dominantly Inherited Alzheimer Network (DIAN) (3) cohorts. The recruited individuals were evaluated by Clinical Core personnel of the Knight ADRC (see Supplementary Materials and Methods). Brain samples were obtained from 290 autopsy-confirmed AD cases, 21 TREM2 risk variant carriers, and 25 cognitively normal individuals with no brain pathology (Table 1). CSF samples were from 176 individuals with a clinical diagnosis of AD, 47 TREM2 risk variant carriers, and 494 cognitively normal individuals. Plasma samples were from 105 individuals with a clinical diagnosis of AD, 131 TREM2 risk variant carriers, and 254 cognitively normal individuals. In CSF and plasma data, AD cases had a diagnosis of dementia of the Alzheimer’s type (DAT) using criteria equivalent to the National Institute of Neurological and Communication Disorders and Stroke-Alzheimer’s Disease and Related Disorders Association for probable AD (48). In addition, we obtained the brain samples from 24 individuals carrying ADAD mutations, out of which 18 were from the DIAN cohort. Among these ADAD individuals, 19, one, and four carried pathogenic mutations in PSEN1, PSEN2, and APP, respectively. The Institutional Review Board of Washington University School of Medicine in St. Louis approved the study and research was performed in accordance with the approved protocols.
Proteomics Data
For omics characterization in brain, CSF, and plasma tissues, we quantified 1,305 proteins using a multiplexed, single-stranded DNA aptamer assay developed by SomaLogic (11). The assay covers a dynamic range of 108 and measures all three major categories: secreted, membrane, and intracellular proteins. The proteins cover a wide range of molecular functions and include proteins known to be relevant to human disease. Aliquots of gray matter homogenate (150 μl) of tissue were provided to the Genome Technology Access Center at Washington University in St. Louis for protein measurement. As previously described (11), modified single-stranded DNA aptamers are used to bind specific protein targets, which are then quantified by a DNA microarray. Protein concentrations are quantified as relative fluorescent units (RFU) of intensity in this DNA microarray. We performed a standard data processing, normalization, and extensive quality control (see Supplementary Materials and Methods),
Validation of Somalogic data
To further validate our SomaScan data, we examined the consistencies of protein abundance of the 150 proteins we identified across different platforms in samples from the Emory, ADNI, and DIAN cohorts. In the Emory cohort, 35 samples had CSF and plasma proteomic data measured in SomaScan, Olink, mass spectrometry (2). ADNI and DIAN cohorts had 110 and 457 samples, respectively, who were measured in both SomaScan and mass spectrometry in CSF (table S23).
Pathway Enrichments
Functional enrichment analysis was performed with Enrichr (23). Within each AD subtype, we merged all proteins identified and validated across tissues. The genes that target these merged proteins were used as an input for enrichment analysis. This corresponded 62 genes (targeting 56 externally replicated proteins) for sporadic AD, 37 genes (targeting 33 across-tissue replicated proteins) for TREM2 variant carriers, and 14 genes (targeting 14 proteins replicated in independent DIAN CSF data) for ADAD. Among multiple gene-set libraries, KEGG, Reactome, Panther pathways and GO biological process were considered. The significance of functional enrichment was reported as the p-value of Fisher’s exact test, followed by Benjamini–Hochberg adjustment for false discovery rates (FDR) in testing multiple hypotheses. We considered results with FDR < 0.05 as significant and included them for creating the dot chart and tile plots to graphically display our findings.
Statistical Analysis
We performed multiple rigorous statistical analyses including differential abundance analysis in both discovery and external replication cohorts, and analysis for neuropathological characteristics using Braak neurofibrillary tangle scores and CDR at death in brain data. See Supplementary Materials and Methods for more details.
Supplementary Material
Acknowledgements
We thank all the participants and their families, as well as the many involved institutions and their staff.
Funding:
This work was supported by grants from the National Institutes of Health [RF1AG074007 (to Y.J.S.), R01AG044546 (to C.C.), P01AG003991 (to C.C. and J.C.M.), RF1AG053303 (to C.C.), RF1AG058501 (to C.C.), and U01AG058922 (to C.C.)], and the Chan Zuckerberg Initiative (CZI).
The recruitment and clinical characterization of research participants at Washington University were supported by NIH P30AG066444 (to J.C.M.), P01AG03991 (to J.C.M.), and P01AG026276 (to J.C.M.).
This work was supported by access to equipment made possible by the Hope Center for Neurological Disorders, the Neurogenomics and Informatics Center (NGI: https://neurogenomics.wustl.edu/)and the Departments of Neurology and Psychiatry at Washington University School of Medicine.
DIAN: Data collection and sharing for this project was supported by The Dominantly Inherited Alzheimer Network (DIAN, U19AG032438) funded by the National Institute on Aging (NIA), the Alzheimer’s Association (SG-20–690363-DIAN), the German Center for Neurodegenerative Diseases (DZNE), Raul Carrea Institute for Neurological Research (FLENI), Partial support by the Research and Development Grants for Dementia from Japan Agency for Medical Research and Development, AMED, the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), Spanish Institute of Health Carlos III (ISCIII), Canadian Institutes of Health Research (CIHR), Canadian Consortium of Neurodegeneration and Aging, Brain Canada Foundation, and Fonds de Recherche du Québec Santé. This manuscript has been reviewed by DIAN Study investigators for scientific content and consistency of data interpretation with previous DIAN Study publications. We acknowledge the altruism of the participants and their families and contributions of the DIAN research and support staff at each of the participating sites for their contributions to this study.
ADNI: Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH-12–2-0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (http://www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
AMP-AD: The results published here are in whole or in part based on data obtained from the AD Knowledge Portal (https://adknowledgeportal.org). Study data were provided by the Baltimore Longitudinal Study of Aging (BLSA), the Banner Sun Health Research Institute (Banner), the Mount Sinai School of Medicine Brain Bank (MSSB), the Adult Changes in Thought Study (ACT), the Mayo Clinic Brain Bank, and the Religious Orders Study (ROS) and the Memory and Aging Project (MAP).
Footnotes
Overline: NEURODEGENERATIVE DISEASE
Competing interests: CC has received research support from: GSK and EISAI. The funders of the study had no role in the collection, analysis, or interpretation of data; in the writing of the report; or in the decision to submit the paper for publication. CC is a member of the advisory board of Vivid Genomics and Circular Genomics and owns stocks.
Dr. Fagan has received research funding from the National Institute on Aging of the National Institutes of Health, Biogen, Centene, Fujirebio and Roche Diagnostics. She is a member of the scientific advisory boards for Roche Diagnostics and Genentech and also consults for Diadem, DiamiR and Siemens Healthcare Diagnostics Inc. There are no conflicts. Dr. Seyfried is co-founder of Emtherapro Corp. There are no conflicts.
Data and materials availability: All Data associated with this study are present in the paper or the supplementary Materials. Proteomic data from the Knight ADRC participants are available at the NIAGADS and can be accessed at https://www.niagads.org/Knight ADRC-collection; under accession no. NG00102: https://www.niagads.org/datasets/ng00102.
The summary results using these data are also available to the scientific community through a public web browser: (omics.wustl.edu/proteomics. Data generated from the DIAN cohort can be be obtained by applying through the DIAN website at https://dian.wustl.edu/our-research/for-investigators/diantu-investigator-resources/dian-tu-biospecimen-request-form/.
Please provide availability information about the human samples used.
Please deposit code used in a public data base like Zenodo.
References
- 1.A. s. Association, 2020 Alzheimer’s disease facts and figures. Alzheimers Dementia, (2020); published online EpubMar 10 ( 10.1002/alz.12068). [DOI] [PubMed] [Google Scholar]
- 2.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM, Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34, 939–944 (1984); published online EpubJul ( 10.1212/wnl.34.7.939). [DOI] [PubMed] [Google Scholar]
- 3.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC, N. Dominantly Inherited Alzheimer, Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367, 795–804 (2012); published online EpubAug 30 ( 10.1056/NEJMoa1202753). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ulrich JD, Holtzman DM, TREM2 Function in Alzheimer’s Disease and Neurodegeneration. ACS Chem Neurosci 7, 420–427 (2016); published online EpubApr 20 ( 10.1021/acschemneuro.5b00313). [DOI] [PubMed] [Google Scholar]
- 5.Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J, Alzheimer Genetic Analysis G, TREM2 variants in Alzheimer’s disease. N Engl J Med 368, 117–127 (2013); published online EpubJan 10 ( 10.1056/NEJMoa1211851). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benitez BA, Cooper B, Pastor P, Jin SC, Lorenzo E, Cervantes S, Cruchaga C, TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol Aging 34, 1711 e1715–1717 (2013); published online EpubJun ( 10.1016/j.neurobiolaging.2012.12.018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olsson B, Lautner R, Andreasson U, Ohrfelt A, Portelius E, Bjerke M, Holtta M, Rosen C, Olsson C, Strobel G, Wu E, Dakin K, Petzold M, Blennow K, Zetterberg H, CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: a systematic review and meta-analysis. Lancet Neurol 15, 673–684 (2016); published online EpubJun ( 10.1016/S1474-4422(16)00070-3). [DOI] [PubMed] [Google Scholar]
- 8.Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Li Y, Gordon BA, Holtzman DM, Morris JC, Benzinger TLS, Xiong C, Fagan AM, Bateman RJ, High-precision plasma beta-amyloid 42/40 predicts current and future brain amyloidosis. Neurology 93, e1647–e1659 (2019); published online EpubOct 22 ( 10.1212/WNL.0000000000008081). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li Y, Schindler SE, Bollinger JG, Ovod V, Mawuenyega KG, Weiner MW, Leslie SM, Masters CL, Fowler CJ, Trojanowski JQ, Korecka M, Martins RN, Janelidze S, Hansson O, Bateman RJ, Validation of Plasma Amyloid-beta 42/40 for Detecting Alzheimer Disease Amyloid Plaques. Neurology, (2021); published online EpubDec 14 ( 10.1212/WNL.0000000000013211). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM, Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol 64, 343–349 (2007); published online EpubMar ( 10.1001/archneur.64.3.noc60123). [DOI] [PubMed] [Google Scholar]
- 11.Gold L, Ayers D, Bertino J, Bock C, Bock A, Brody EN, Carter J, Dalby AB, Eaton BE, Fitzwater T, Flather D, Forbes A, Foreman T, Fowler C, Gawande B, Goss M, Gunn M, Gupta S, Halladay D, Heil J, Heilig J, Hicke B, Husar G, Janjic N, Jarvis T, Jennings S, Katilius E, Keeney TR, Kim N, Koch TH, Kraemer S, Kroiss L, Le N, Levine D, Lindsey W, Lollo B, Mayfield W, Mehan M, Mehler R, Nelson SK, Nelson M, Nieuwlandt D, Nikrad M, Ochsner U, Ostroff RM, Otis M, Parker T, Pietrasiewicz S, Resnicow DI, Rohloff J, Sanders G, Sattin S, Schneider D, Singer B, Stanton M, Sterkel A, Stewart A, Stratford S, Vaught JD, Vrkljan M, Walker JJ, Watrobka M, Waugh S, Weiss A, Wilcox SK, Wolfson A, Wolk SK, Zhang C, Zichi D, Aptamer-based multiplexed proteomic technology for biomarker discovery. PLoS One 5, e15004 (2010); published online EpubDec 7 ( 10.1371/journal.pone.0015004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD, The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28, 882–883 (2012); published online EpubMar 15 ( 10.1093/bioinformatics/bts034). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morris JC, The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 43, 2412–2414 (1993); published online EpubNov ( [DOI] [PubMed] [Google Scholar]
- 14.Johnson ECB, Dammer EB, Duong DM, Ping L, Zhou M, Yin L, Higginbotham LA, Guajardo A, White B, Troncoso JC, Thambisetty M, Montine TJ, Lee EB, Trojanowski JQ, Beach TG, Reiman EM, Haroutunian V, Wang M, Schadt E, Zhang B, Dickson DW, Ertekin-Taner N, Golde TE, Petyuk VA, De Jager PL, Bennett DA, Wingo TS, Rangaraju S, Hajjar I, Shulman JM, Lah JJ, Levey AI, Seyfried NT, Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Nat Med 26, 769–780 (2020); published online EpubMay ( 10.1038/s41591-020-0815-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Higginbotham L, Ping L, Dammer EB, Duong DM, Zhou M, Gearing M, Hurst C, Glass JD, Factor SA, Johnson ECB, Hajjar I, Lah JJ, Levey AI, Seyfried NT, Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci Adv 6, (2020); published online EpubOct ( 10.1126/sciadv.aaz9360). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wingo AP, Fan W, Duong DM, Gerasimov ES, Dammer EB, Liu Y, Harerimana NV, White B, Thambisetty M, Troncoso JC, Kim N, Schneider JA, Hajjar IM, Lah JJ, Bennett DA, Seyfried NT, Levey AI, Wingo TS, Shared proteomic effects of cerebral atherosclerosis and Alzheimer’s disease on the human brain. Nat Neurosci 23, 696–700 (2020); published online EpubJun ( 10.1038/s41593-020-0635-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whelan CD, Mattsson N, Nagle MW, Vijayaraghavan S, Hyde C, Janelidze S, Stomrud E, Lee J, Fitz L, Samad TA, Ramaswamy G, Margolin RA, Malarstig A, Hansson O, Multiplex proteomics identifies novel CSF and plasma biomarkers of early Alzheimer’s disease. Acta Neuropathol Commun 7, 169 (2019); published online EpubNov 6 ( 10.1186/s40478-019-0795-2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sattlecker M, Kiddle SJ, Newhouse S, Proitsi P, Nelson S, Williams S, Johnston C, Killick R, Simmons A, Westman E, Hodges A, Soininen H, Kloszewska I, Mecocci P, Tsolaki M, Vellas B, Lovestone S, AddNeuroMed C, Dobson RJ, Alzheimer’s disease biomarker discovery using SOMAscan multiplexed protein technology. Alzheimers Dement 10, 724–734 (2014); published online EpubNov ( 10.1016/j.jalz.2013.09.016). [DOI] [PubMed] [Google Scholar]
- 19.Vlassenko AG, McCue L, Jasielec MS, Su Y, Gordon BA, Xiong C, Holtzman DM, Benzinger TL, Morris JC, Fagan AM, Imaging and cerebrospinal fluid biomarkers in early preclinical alzheimer disease. Ann Neurol 80, 379–387 (2016); published online EpubSep ( 10.1002/ana.24719). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guo T, Shaw LM, Trojanowski JQ, Jagust WJ, Landau SM, I. Alzheimer’s Disease Neuroimaging, Association of CSF Abeta, amyloid PET, and cognition in cognitively unimpaired elderly adults. Neurology 95, e2075–e2085 (2020); published online EpubOct 13 ( 10.1212/WNL.0000000000010596). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jack CR Jr., Bennett DA, Blennow K, Carrillo MC, Feldman HH, Frisoni GB, Hampel H, Jagust WJ, Johnson KA, Knopman DS, Petersen RC, Scheltens P, Sperling RA, Dubois B, A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87, 539–547 (2016); published online EpubAug 2 ( 10.1212/WNL.0000000000002923). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Timsina J, Gomez-Fonseca D, Wang L, Do A, Western D, Alvarez I, Aguilar M, Pastor P, Henson RL, Herries E, Xiong C, Schindler SE, Fagan AM, Bateman RJ, Farlow M, Morris JC, Perrin R, Moulder K, Hassenstab J, Chhatwal J, Mori H, I. Alzheimer’s Disease Neuroimaging, C. Dominantly Inherited Alzheimer Network, Sung YJ, Cruchaga C, Comparative Analysis of Alzheimer’s Disease Cerebrospinal Fluid Biomarkers Measurement by Multiplex SOMAscan Platform and Immunoassay-Based Approach. J Alzheimers Dis 89, 193–207 (2022) 10.3233/JAD-220399). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A, Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013); published online EpubApr 15 ( 10.1186/1471-2105-14-128). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Plagman A, Hoscheidt S, McLimans KE, Klinedinst B, Pappas C, Anantharam V, Kanthasamy A, Willette AA, I. Alzheimer’s Disease Neuroimaging, Cholecystokinin and Alzheimer’s disease: a biomarker of metabolic function, neural integrity, and cognitive performance. Neurobiol Aging 76, 201–207 (2019); published online EpubApr ( 10.1016/j.neurobiolaging.2019.01.002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu H, Liu B, Li L, Lemere CA, Microglia Do Not Take Up Soluble Amyloid-beta Peptides, But Partially Degrade Them by Secreting Insulin-degrading Enzyme. Neuroscience 443, 30–43 (2020); published online EpubSep 1 ( 10.1016/j.neuroscience.2020.07.020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dammer EB, Ping L, Duong DM, Modeste ES, Seyfried NT, Lah JJ, Levey AI, Johnson ECB, Multi-Platform Proteomic Analysis of Alzheimer’s Disease Cerebrospinal Fluid and Plasma Reveals Network Biomarkers Associated with Proteostasis and the Matrisome. bioRxiv, 2022.2006.2020.494087 (2022) 10.1101/2022.06.20.494087). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE, Cummings J, van der Flier WM, Alzheimer’s disease. Lancet 397, 1577–1590 (2021); published online EpubApr 24 ( 10.1016/S0140-6736(20)32205-4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nakamura A, Kaneko N, Villemagne VL, Kato T, Doecke J, Dore V, Fowler C, Li QX, Martins R, Rowe C, Tomita T, Matsuzaki K, Ishii K, Ishii K, Arahata Y, Iwamoto S, Ito K, Tanaka K, Masters CL, Yanagisawa K, High performance plasma amyloid-beta biomarkers for Alzheimer’s disease. Nature 554, 249–254 (2018); published online EpubFeb 8 ( 10.1038/nature25456). [DOI] [PubMed] [Google Scholar]
- 29.Zhang Y, Ghose U, Buckley NJ, Engelborghs S, Sleegers K, Frisoni GB, Wallin A, Lleo A, Popp J, Martinez-Lage P, Legido-Quigley C, Barkhof F, Zetterberg H, Visser PJ, Bertram L, Lovestone S, Nevado-Holgado AJ, Shi L, Predicting AT(N) pathologies in Alzheimer’s disease from blood-based proteomic data using neural networks. Front Aging Neurosci 14, 1040001 (2022) 10.3389/fnagi.2022.1040001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Palmqvist S, Tideman P, Cullen N, Zetterberg H, Blennow K, I. Alzheimer’s Disease Neuroimaging, Dage JL, Stomrud E, Janelidze S, Mattsson-Carlgren N, Hansson O, Prediction of future Alzheimer’s disease dementia using plasma phospho-tau combined with other accessible measures. Nat Med 27, 1034–1042 (2021); published online EpubJun ( 10.1038/s41591-021-01348-z). [DOI] [PubMed] [Google Scholar]
- 31.Cruchaga C, Kauwe JS, Nowotny P, Bales K, Pickering EH, Mayo K, Bertelsen S, Hinrichs A, I. Alzheimer’s Disease Neuroimaging, Fagan AM, Holtzman DM, Morris JC, Goate AM, Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet 21, 4558–4571 (2012); published online EpubOct 15 ( 10.1093/hmg/dds296). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM, Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med 3, 89ra57 (2011); published online EpubJun 29 ( 10.1126/scitranslmed.3002156). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cruchaga C, Kauwe JS, Mayo K, Spiegel N, Bertelsen S, Nowotny P, Shah AR, Abraham R, Hollingworth P, Harold D, Owen MM, Williams J, Lovestone S, Peskind ER, Li G, Leverenz JB, Galasko D, I. Alzheimer’s Disease Neuroimaging, Morris JC, Fagan AM, Holtzman DM, Goate AM, SNPs associated with cerebrospinal fluid phospho-tau levels influence rate of decline in Alzheimer’s disease. PLoS Genet 6, (2010); published online EpubSep ( 10.1371/journal.pgen.1001101). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhao WQ, Ravindranath L, Mohamed AS, Zohar O, Chen GH, Lyketsos CG, Etcheberrigaray R, Alkon DL, MAP kinase signaling cascade dysfunction specific to Alzheimer’s disease in fibroblasts. Neurobiol Dis 11, 166–183 (2002); published online EpubOct ( 10.1006/nbdi.2002.0520). [DOI] [PubMed] [Google Scholar]
- 35.Yang C, Farias FHG, Ibanez L, Suhy A, Sadler B, Fernandez MV, Wang F, Bradley JL, Eiffert B, Bahena JA, Budde JP, Li Z, Dube U, Sung YJ, Mihindukulasuriya KA, Morris JC, Fagan AM, Perrin RJ, Benitez BA, Rhinn H, Harari O, Cruchaga C, Genomic atlas of the proteome from brain, CSF and plasma prioritizes proteins implicated in neurological disorders. Nat Neurosci, (2021); published online EpubJul ( 10.1038/s41593-021-00886-6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ringman JM, Monsell S, Ng DW, Zhou Y, Nguyen A, Coppola G, Van Berlo V, Mendez MF, Tung S, Weintraub S, Mesulam MM, Bigio EH, Gitelman DR, Fisher-Hubbard AO, Albin RL, Vinters HV, Neuropathology of Autosomal Dominant Alzheimer Disease in the National Alzheimer Coordinating Center Database. J Neuropathol Exp Neurol 75, 284–290 (2016); published online EpubMar ( 10.1093/jnen/nlv028). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, Wszolek ZK, Gasser T, Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44, 601–607 (2004); published online EpubNov 18 ( 10.1016/j.neuron.2004.11.005). [DOI] [PubMed] [Google Scholar]
- 38.Tolosa E, Vila M, Klein C, Rascol O, LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol 16, 97–107 (2020); published online EpubFeb ( 10.1038/s41582-019-0301-2). [DOI] [PubMed] [Google Scholar]
- 39.Thrupp N, Sala Frigerio C, Wolfs L, Skene NG, Fattorelli N, Poovathingal S, Fourne Y, Matthews PM, Theys T, Mancuso R, de Strooper B, Fiers M, Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep 32, 108189 (2020); published online EpubSep 29 ( 10.1016/j.celrep.2020.108189). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ma G, Liu M, Du K, Zhong X, Gong S, Jiao L, Wei M, Differential Expression of mRNAs in the Brain Tissues of Patients with Alzheimer’s Disease Based on GEO Expression Profile and Its Clinical Significance. Biomed Res Int 2019, 8179145 (2019) 10.1155/2019/8179145). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pan XD, Zhu YG, Lin N, Zhang J, Ye QY, Huang HP, Chen XC, Microglial phagocytosis induced by fibrillar beta-amyloid is attenuated by oligomeric beta-amyloid: implications for Alzheimer’s disease. Mol Neurodegener 6, 45 (2011); published online EpubJun 30 ( 10.1186/1750-1326-6-45). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, Xiong Z, Manor LC, Cao H, Li T, Integrative computational evaluation of genetic markers for Alzheimer’s disease. Saudi J Biol Sci 25, 996–1002 (2018); published online EpubJul ( 10.1016/j.sjbs.2018.05.019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Da Mesquita S, Papadopoulos Z, Dykstra T, Brase L, Farias FG, Wall M, Jiang H, Kodira CD, de Lima KA, Herz J, Louveau A, Goldman DH, Salvador AF, Onengut-Gumuscu S, Farber E, Dabhi N, Kennedy T, Milam MG, Baker W, Smirnov I, Rich SS, N. Dominantly Inherited Alzheimer, Benitez BA, Karch CM, Perrin RJ, Farlow M, Chhatwal JP, Holtzman DM, Cruchaga C, Harari O, Kipnis J, Meningeal lymphatics affect microglia responses and anti-Abeta immunotherapy. Nature 593, 255–260 (2021); published online EpubMay ( 10.1038/s41586-021-03489-0). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scafidi J, Hammond TR, Scafidi S, Ritter J, Jablonska B, Roncal M, Szigeti-Buck K, Coman D, Huang Y, McCarter RJ Jr., Hyder F, Horvath TL, Gallo V, Intranasal epidermal growth factor treatment rescues neonatal brain injury. Nature 506, 230–234 (2014); published online EpubFeb 13 ( 10.1038/nature12880). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang L, Chiang HC, Wu W, Liang B, Xie Z, Yao X, Ma W, Du S, Zhong Y, Epidermal growth factor receptor is a preferred target for treating amyloid-beta-induced memory loss. Proc Natl Acad Sci U S A 109, 16743–16748 (2012); published online EpubOct 9 ( 10.1073/pnas.1208011109). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thomas R, Zuchowska P, Morris AW, Marottoli FM, Sunny S, Deaton R, Gann PH, Tai LM, Epidermal growth factor prevents APOE4 and amyloid-beta-induced cognitive and cerebrovascular deficits in female mice. Acta Neuropathol Commun 4, 111 (2016); published online EpubOct 27 ( 10.1186/s40478-016-0387-3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aerqin Q, Wang ZT, Wu KM, He XY, Dong Q, Yu JT, Omics-based biomarkers discovery for Alzheimer’s disease. Cell Mol Life Sci 79, 585 (2022); published online EpubNov 8 ( 10.1007/s00018-022-04614-6). [DOI] [PubMed] [Google Scholar]
- 48.Berg L, McKeel DW Jr., Miller JP, Storandt M, Rubin EH, Morris JC, Baty J, Coats M, Norton J, Goate AM, Price JL, Gearing M, Mirra SS, Saunders AM, Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol 55, 326–335 (1998); published online EpubMar ( [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.