

Abstract
Limb-girdle muscular dystrophy 2E/R4 is caused by mutations in the β-sarcoglycan (SGCB) gene, leading to SGCB deficiency and consequent muscle loss. We developed a gene therapy approach based on functional replacement of the deficient SCB protein. Here we report interim results from a first-in-human, open-label, nonrandomized, phase 1/2 trial evaluating the safety and efficacy of bidridistrogene xeboparvovec, an adeno-associated virus-based gene therapy containing a codon-optimized, full-length human SGCB transgene. Patients aged 4–15 years with confirmed SGCB mutations at both alleles received one intravenous infusion of either 1.85 × 1013 vector genome copies kg−1 (Cohort 1, n = 3) or 7.41 × 1013 vector gene copies kg−1 (Cohort 2, n = 3). Primary endpoint was safety, and secondary endpoint was change in SGCB expression in skeletal muscle from baseline to Day 60. We report interim Year 2 results (trial ongoing). The most frequent treatment-related adverse events were vomiting (four of six patients) and gamma-glutamyl transferase increase (three of six patients). Serious adverse events resolved with standard therapies. Robust SGCB expression was observed: Day 60 mean (s.d.) percentage of normal expression 36.2% (2.7%) in Cohort 1 and 62.1% (8.7%) in Cohort 2. Post hoc exploratory analysis showed preliminary motor improvements using the North Star Assessment for Limb-girdle Type Muscular Dystrophies maintained through Year 2. The 2-year safety and efficacy of bidridistrogene xeboparvovec support clinical development advancement. Further studies are necessary to confirm the long-term safety and efficacy of this gene therapy. ClinicalTrials.gov registration: NCT03652259.
Subject terms: Targeted gene repair, Neuromuscular disease
Two-year interim data from an ongoing phase 1/2 trial of the AAV-based gene therapy bidridistrogene xeboparvovec in six patients with limb-girdle muscular dystrophy show a safe profile and preliminary increased expression of the β-sarcoglycan protein.
Main
Limb-girdle muscular dystrophies (LGMDs) are a clinically and genetically diverse group of autosomal inherited monogenic neuromuscular diseases characterized by muscle wasting and progressive weakness, mostly within proximal muscles around the pelvic and shoulder girdles1–3. Sarcoglycanopathies are among the most severe LGMDs, with patients typically showing rapidly progressive proximal muscle weakness, respiratory impairment and potential cardiac abnormalities. LGMD type 2E/R4 (LGMD2E/R4) is caused by mutations in the β-sarcoglycan gene (SGCB), leading to β-sarcoglycan (SGCB) protein deficiency and loss of sarcoglycan complex formation and preventing dystrophin-associated protein complex (DAPC) stabilization4,5. This loss of protein function manifests clinically in early childhood or late adolescence and progresses to loss of ambulation, potential respiratory insufficiency and often premature mortality6–8.
Globally, the incidence of LGMD2E/R4 is between one in 200,000 and one in 350,000 (ref. 9). Diagnosis typically occurs before the age of 10 years, with loss of ambulation by middle to late teens10–12. Patients present with elevated serum creatine kinase (CK), difficulty rising from the floor or walking and proximal muscle weakness. Joint contractures and kyphoscoliosis, combined with degenerating diaphragmatic muscle, lead to compromised pulmonary function13,14. Both the need for assisted ventilation with disease progression and cardiac involvement occurring in about 60% of patients contribute to poor prognosis and premature death6,13,15–18. There is an urgent need for a viable treatment option, because there are currently no approved therapies. Because LGMD2E/R4 is a monogenic disorder and its impact is largely restricted to skeletal and cardiac muscle, it is particularly suited for adeno-associated virus (AAV)-mediated gene transfer therapy.
Evaluation of any gene therapy platform must include a systematic and stepwise consideration of safety, transduction, expression, localization, cellular impact and clinical function19. The rAAVrh74.MHCK7.hSGCB construct, referred to as bidridistrogene xeboparvovec (SRP-9003), is a self-complementary AAV vector designed with these considerations in mind, containing a codon-optimized, full-length human SGCB (hSGCB) transgene, and driven by a muscle-specific promoter (MHCK7) to restore functional SGCB protein in muscle cells that leads to functional improvements in patients with LGMD2E/R4. AAVrh74 is a rhesus monkey-derived AAV vector that shows robust skeletal and cardiac muscle tissue tropism20,21. The MHCK7 promoter drives robust transgene expression in both skeletal and cardiac muscle and limits expression in off-target tissues. The addition of the promoter region of α-myosin heavy chain enhances cardiac expression20,22. The hSGCB transgene carries full-length human SGCB cDNA. The self-complementary nature of bidridistrogene xeboparvovec allows it to overcome the rate-limiting step for AAVs of second-strand synthesis, resulting in more efficient transduction of target cells with rapid and higher transgene expression23,24.
In preclinical studies, bidridistrogene xeboparvovec gene transfer provided complete restoration of human SGCB protein expression with concomitant improvements in muscle histopathology, kyphoscoliosis, physical activity and cardiac and diaphragmatic function in mice, and no safety issues observed; specifically, diaphragm force production increased by 94% (P < 0.05) and ambulation by 57% (P < 0.05) following treatment compared with SGCB−/− mice20.
This report presents preliminary safety, tolerability and efficacy data of the first-in-human application of bidridistrogene xeboparvovec systemic gene delivery in patients with LGMD2E/R4, including up to 2 years of follow-up. The study includes two dose cohorts who received bidridistrogene xeboparvovec doses of either 1.85 × 1013 vector genome copies (vg) kg−1 (Cohort 1) or 7.41 × 1013 vg kg−1 (Cohort 2), calculated based on a validated quantitative polymerase chain reaction (qPCR) method using a linear standard (Fig. 1a).
Fig. 1. SRP-9003-101 study design and study flow diagram.

a, Study design. b, Study flow diagram. EOS, end of study.
Results
Patient baseline demographics and clinical characteristics
A total of seven patients were assessed for eligibility; of the six patients who met eligibility criteria, three each were enrolled in Cohorts 1 and 2 (Fig. 1b). The first patient was enrolled on 26 October 2018 (Cohort 1) and the last on 17 January 2020 (Cohort 2); the trial is ongoing. Five patients remain on study, and one patient died in an accident unrelated to the study on Day 220. Baseline demographics and clinical characteristics are shown in Table 1. All SGCB mutations are homozygous; five patients have missense mutations in exons 3 and 4, which encode for the extracellular domain of SGCB protein, and one patient has a nonsense mutation in exon 1; all of these mutations lead to the absence of SGCB protein and a severe phenotype13. The mean age at baseline was 10.0 years in both cohorts. For Cohorts 1 and 2, respectively, at baseline, mean weight was 42.3 and 32.1 kg, mean body mass index (BMI) was 20.5 and 17.4 and mean CK levels were 16,575.3 and 7,042.7 U l−1. Mean (s.d.) North Star Assessment for Limb-girdle Type Muscular Dystrophies (NSAD) score at baseline was 43.0 (4.4) points in Cohort 1 and 39.3 (2.1) points in Cohort 2.
Table 1.
Baseline demographics and clinical characteristics
| Cohort 1 | Cohort 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Patient 1 | Patient 2 | Patient 3 | Totala (n = 3) | Patient 4 | Patient 5 | Patient 6 | Totala (n = 3) | |
| Age, years | 13 | 4 | 13 | 10.0 (5.2) | 11 | 11 | 8 | 10.0 (1.7) |
| Sexb | Female | Male | Female | NA | Male | Male | Female | NA |
| Height, cm | 155 | 103 | 157.5 | 138.5 (30.8) | 132 | 144 | 129.2 | 135.1 (7.9) |
| Weight, kg | 56.2 | 17.7 | 53.1 | 42.3 (21.4) | 29.4 | 39.5 | 27.3 | 32.1 (6.5) |
| BMI, kg m−2 | 23.4 | 16.7 | 21.4 | 20.5 (3.4) | 16.9 | 19 | 16.4 | 17.4 (1.4) |
| SGCB mutationc | Exon 3 c.341C>T; p.Ser114Phe Missense | Exon 4 c.452C>G; p.Thr151Arg Missense | Exon 3 c.341C>T; p.Ser114Phe Missense | NA | Exon 4 c.452C>G; p.Thr151Arg Missense | Exon 3 c.341C>T; p.Ser114Phe Missense | Exon 1 c.1_2delAT Deletion | NA |
| CKd, U l−1 | 10,727 | 28,014 | 10,985 | 16,575.3 (9907.0) | 6,447 | 8,938 | 5,743 | 7,042.7 (1678.7) |
| Functional measures | ||||||||
| NSAD score | 40 | 48 | 41 | 43.0 (4.4) | 41 | 37 | 40 | 39.3 (2.1) |
| Time to rise, s | 5 | 1.5 | 3.5 | 3.3 (1.8) | 3.3 | 3.5 | 5.7 | 4.2 (1.3) |
| Four-stair climb, s | 2.4 | 1.6 | 2.8 | 2.3 (0.6) | 3 | 3.1 | 3.1 | 3.1 (0.1) |
| 100-m, s | 52 | 35.1 | 48.8 | 45.3 (9.0) | 47.2 | 59.7 | 65.3 | 57.4 (9.3) |
| 10-m, s | 5 | 3.4 | 5.2 | 4.5 (1.0) | 4.9 | 5.8 | 6.1 | 5.6 (0.6) |
10-m, 10-m timed test; 100-m, 100-m timed test; BMI calculated as weight in kg divided by height in m2; NA, not applicable.
aData expressed as mean (s.d.).
bSex reported was determined based on self-report.
cPatients 1–5 had missense mutations; Patient 6 had a nonsense mutation; all mutations were homozygous.
dBaseline CK level defined as the maximum value of screening and Day −1 measurements; SI conversion factor: for conversion of U l−1 to μkat l−1, multiply by 0.0167.
Safety and immunogenicity
The overall safety of bidridistrogene xeboparvovec was favorable across both cohorts at 2 years (Table 2 and Extended Data Table 1). In Cohort 1, two patients had treatment-related elevated γ-glutamyl transferase (GGT) levels that occurred either during or after tapering of oral steroids. One case was designated a serious adverse event (SAE) because the patient experienced AAV-related hepatitis and a transient increase in bilirubin. The SAE of hepatitis occurred on study Day 53, at the end of the steroid taper (patient was off steroids), and required short hospitalization; the event lasted 4 days and resolved with intravenous fluids and oral prednisone. In both cases, GGT levels returned to normal and symptoms were resolved within 30 days following supplemental steroid treatment. One patient experienced moderate upper gastrointestinal pain and mild vomiting that resolved within 1–2 days without steroid treatment. Other treatment-related adverse events were mild dizziness (n = 1) and mild decreased appetite (n = 1).
Table 2.
Primary outcome of treatment-related, treatment-emergent adverse events by system organ class and preferred term at 2 years
| System organ class preferred term | Cohort 1 (dose, 1.85 × 1013 vg kg−1)a (n = 3) | Cohort 2 (dose, 7.41 × 1013 vg kg−1)b (n = 3) | Total (n = 6) |
|---|---|---|---|
| Subjects with any treatment-related, treatment-emergent adverse event | 2 (66.7%) | 3 (100.0%) | 5 (83.3%) |
| Gastrointestinal disorders | 1 (33.3%) | 3 (100.0%) | 4 (66.7%) |
| Abdominal pain | 0 | 2 (66.7%) | 2 (33.3%) |
| Abdominal pain, upper | 1 (33.3%) | 1 (33.3%) | 2 (33.3%) |
| Nausea | 0 | 2 (66.7%) | 2 (33.3%) |
| Vomiting | 1 (33.3%) | 3 (100.0%) | 4 (66.7%) |
| General disorders and administration site conditions | 0 | 1 (33.3%) | 1 (16.7%) |
| Pyrexia | 0 | 1 (33.3%) | 1 (16.7%) |
| Hepatobiliary disorders | 1 (33.3%) | 0 | 1 (16.7%) |
| Hepatitis | 1 (33.3%)c | 0 | 1 (16.7%) |
| Hyperbilirubinemia | 1 (33.3%) | 0 | 1 (16.7%) |
| Investigations | 2 (66.7%) | 3 (100.0%) | 5 (83.3%) |
| GGT increased | 2 (66.7%) | 1 (33.3%) | 3 (50.0%) |
| Neutrophil count decreased | 0 | 1 (33.3%) | 1 (16.7%) |
| White blood cell count decreased | 0 | 2 (66.7%) | 2 (33.3%) |
| Metabolism and nutrition disorders | 1 (33.3%) | 1 (33.3%) | 2 (33.3%) |
| Decreased appetite | 1 (33.3%) | 0 | 1 (16.7%) |
| Dehydration | 0 | 1 (33.3%)c | 1 (16.7%) |
| Nervous system disorders | 1 (33.3%) | 0 | 1 (16.7%) |
| Dizziness | 1 (33.3%) | 0 | 1 (16.7%) |
Treatment-emergent adverse events are defined as all adverse events that started on or after the study drug administration date. Adverse events are coded using Medical Dictionary for Regulatory Activities v.22.0. Cutoff dates for safety data were 14 January 2021 (Cohort 1) and 18 January 2022 (Cohort 2).
a1.85 × 1013 vg kg−1 (linear standard qPCR).
b7.41 × 1013 vg kg−1 (linear standard qPCR).
cSAE.
Extended Data Table 1.
Primary outcome of treatment-emergent adverse events by system organ class and preferred term at 2 years
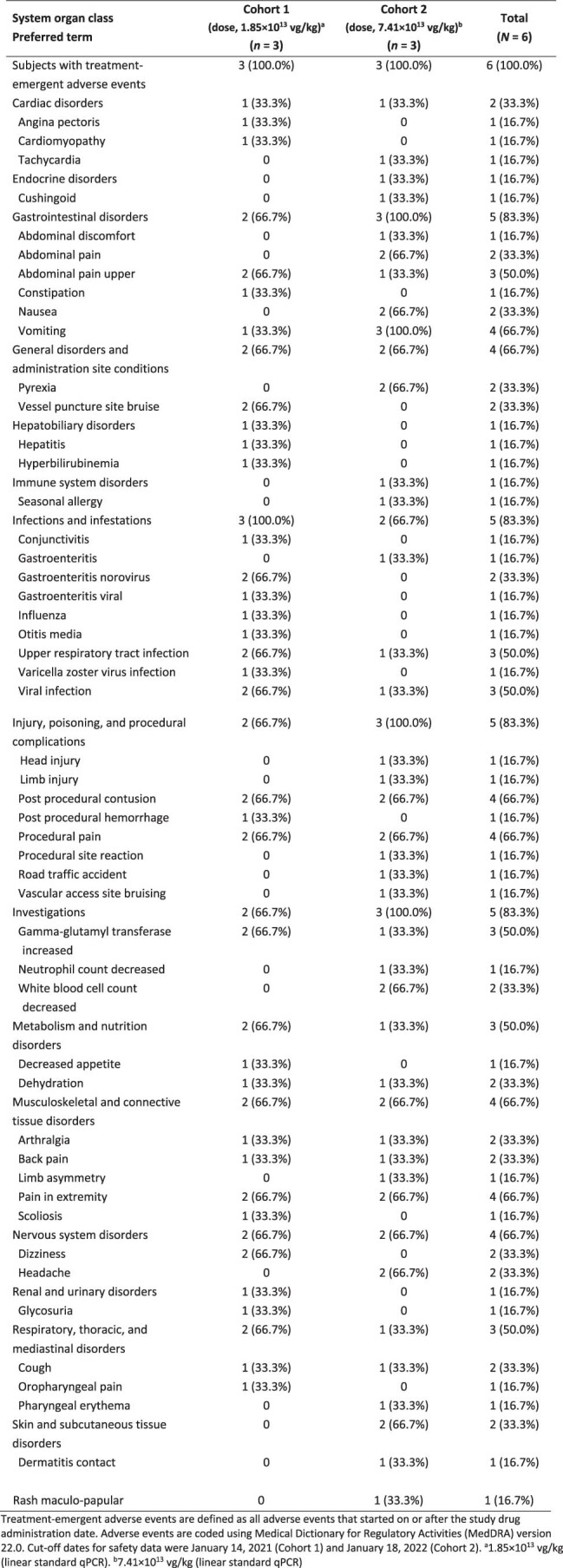
In Cohort 2, most patients experienced mild-to-moderate treatment-related gastrointestinal adverse events (for example, vomiting, nausea, abdominal pain), all of which resolved. One patient had mildly elevated GGT levels, which returned to normal while tapering steroids; the patient did not experience an increase following tapering. One patient experienced severe dehydration resulting from vomiting 3 days following infusion, which was classified as a treatment-related SAE and required brief hospitalization; symptoms resolved in 2 days with ondansetron, promethazine and intravenous fluids. There was one death due to a traffic accident unrelated to the study. None of the protocol-specified conditions for halting the study (for example, unexpected, serious or unacceptable risk to patients) occurred, and there were no discontinuations or deaths related to the study.
There were no adverse immune responses and no clinically meaningful B or T cell responses toward AAVrh74 or SGCB protein (ELISPots) or increases in anti-SGCB antibodies (ELISA assay) in any patient (Extended Data Fig. 1). There were expected increases in AAVrh74 antibodies in all six patients post dosing (Extended Data Fig. 1). T cell response toward AAVrh74 peptide pools (1–3) demonstrated increases in three patients, with the stronger response observed for peptide pool 2 (Extended Data Fig. 1). Elevations did not correspond to liver enzyme elevation or impact transgene expression. Notably, none of the adverse events were related to clinical complement activation.
Extended Data Fig. 1. Exploratory outcomes of individual immune responses to AAVrh74 capsid protein, AAV peptide pools, and hSGCB.

Exploratory outcomes of individual immune responses to (a) AAVrh74 capsid protein, (b) AAV peptide pools and (c) hSGCB. Footnote: Dotted line in ELISpot plots denotes threshold of 50 Spot Forming Colonies (SFC) per million cells. hSGCB, human β-sarcoglycan; IFN-γ SFC, gamma interferon spot-forming cells; PBMC, peripheral blood mononuclear cells. Missing datapoints are due to patient samples not collected at all or not collected at the scheduled timepoint (unscheduled data not shown). Asterisk (‘*’) denotes value <1/X. Hashtag (‘#’) denotes value >1/X.
No other laboratory abnormalities were noted that were suggestive of safety concerns. No decreases in platelet counts outside the normal range or electrocardiogram abnormalities were observed.
Bidridistrogene xeboparvovec transduction, SGCB expression and cell localization
Bidridistrogene xeboparvovec transduction and SGCB protein expression following systemic infusion were analyzed at two different timepoints (Day 60 and Year 2) and were detected by vector-specific droplet digital PCR (ddPCR), immunoblot and immunofluorescence (Table 3). Successful transduction in skeletal muscle (tibialis anterior and biceps) was confirmed in both cohorts at Day 60. In Cohort 1, data for vg per nucleus by ddPCR are not available; in Cohort 2, mean (s.d.) vg by ddPCR were 2.26 (0.92). Immunoblot analysis confirmed full-length SGCB protein expression in all patients, with a mean (s.d.) percentage of normal expression at Day 60 of 36.2% (2.7%) for Cohort 1 and 62.1% (8.7%) for Cohort 2. Representative immunoblot images of pretreatment (baseline), Day 60 and Year 2 are shown in Extended Data Fig. 2.
Table 3.
Secondary and exploratory outcomes of SGCB expression in muscle biopsies (tibialis anterior and biceps) at Day 60 and Year 2
| Cohort 1 (dose, 1.85 × 1013 vg kg−1)a (n = 3) | Cohort 2 (dose, 7.41 × 1013 vg kg−1)b (n = 3)c | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Timepoint | Immunoblot (% NC) | Percentage SGCB+ fibers (% NC) | SGCB intensity (% NC) | Vector copies per nucleus | Immunoblot (% NC) | Percentage SGCB+ fibers (% NC) | SGCB intensity (% NC) | Vector copies per nucleus | ||
| qPCR | ddPCR | qPCR | ddPCR | |||||||
| Baseline mean (s.d.) | NA | 8.9 (4.7) | 7.4 (6.0) | 0 | NA | 33.1 (4.8) | 5.9 (6.4) | 4.6 (5.7) | 0 | 0 |
| Median (range) | NA | 8.5 (4.4, 13.7) | 4.7 (3.3, 14.3) | 0 | NA | 35.0 (27.6, 36.6) | 5.0 (0, 12.8) | 2.5 (0.2, 11.1) | 0 | 0 |
| Day 60 mean (s.d.) | 36.2 (2.7) | 51.0 (10.6) | 47.4 (9.5) | 0.6 (0.4) | NA | 62.1 (8.7) | 72.3 (6.2) | 73.1 (21.8) | 4.2 (2.8) | 2.3 (0.9) |
| Median (range) | 34.7 (34.6, 39.3) | 48.9 (41.6, 62.5) | 47.4 (38.0, 56.9) | 0.4 (0.3, 1.1) | NA | 63.1 (53.0, 70.3) | 74.8 (65.3, 77.0) | 67.1 (54.9, 97.3) | 4.0 (1.6, 7.2) | 2.4 (1.3, 3.1) |
| Year 2 mean (s.d.) | 54.0 (16.1) | 46.8 (21.3) | 35.1 (22.9) | 0.1 (0.1) | 0.46 (0.36) | 60.3 (21.4) | 63.1 (21.6) | 43.5 (33.2) | 0.2 (0.1) | 0.5 (0.3) |
| Median (range) | 56.2 (37.0, 69.0) | 58.6 (22.3, 59.7) | 45.9 (8.8, 50.5) | 0.1 (0.1, 0.2) | 0.38 (0.2, 0.9) | 60.3 (45.1, 75.4) | 63.1 (47.9, 78.4) | 43.5 (20.0, 67.0) | 0.2 (0.2, 0.3) | 0.5 (0.3, 0.8) |
Values are mean (s.d.) and median (range); NC, normal control.
a1.85 × 1013 vg kg−1 (linear standard qPCR).
b7.41 × 1013 vg kg−1 (linear standard qPCR).
cn = 2 at Year 2.
Extended Data Fig. 2. Secondary and exploratory outcomes.

representative images of SGCB immunoblot analysis (a, b), immunoblot with α-actinin loading control (c, d), and γ2 sarcoglycan complex (e).
Immunofluorescence staining was used to determine the percentage of positive fibers expressing SGCB protein and its correct localization in the sarcolemma in muscle tissue. SGCB protein was correctly localized to the sarcolemma at Day 60 (Fig. 2a), resulting in a mean (s.d.) percentage of SGCB-positive fibers of 51% (10.6%) and 72% (6.2%) in Cohorts 1 and 2, respectively. Protein expression was also quantified by fluorescence intensity at Day 60 (Table 3), resulting in a mean (s.d.) intensity of 47% (9.5%) and 73% (21.8%) of normal for Cohorts 1 and 2, respectively.
Fig. 2. Secondary and exploratory outcomes of SGCB and sarcoglycan complex expression.

a, Representative immunofluorescence images of biopsied muscle sections stained for SGCB from each patient pre and post treatment (Day 60 and Year 2) compared with normal muscle. Biopsied tissue sections of tibialis anterior and biceps were processed and stained for detection of SGCB. Four ×20 images per tissue block were taken, with four per timepoint (that is, 16 images per patient per timepoint). Patient 5 died in an accident unrelated to the study and therefore the Year 2 biopsy timepoint is missing. b, Expression of sarcoglycan complex subunits in Cohorts 1 and 2 from muscle biopsies by immunofluorescence (mean percentage of positive fibers and mean intensity) at baseline, Day 60 and Year 2.
Bidridistrogene xeboparvovec vector genomes and protein expression in muscle tissue were durable, as seen in the analysis of muscle biopsies at Year 2 (Table 3). In Cohort 1, mean (s.d.) vg per nucleus by ddPCR were 0.46 (0.36); in Cohort 2, mean (s.d.) vg were 0.52 (0.33). SGCB protein expression by immunoblot was also sustained, with a mean (s.d.) percentage of normal expression at Year 2 of 54.0% (16.1%) for Cohort 1 and 60.3% (21.4%) for Cohort 2. At Year 2, mean (s.d.) percentage of SGCB-positive fibers was 47% (21.3%) and 63% (21.6%) in Cohorts 1 and 2, respectively, and mean (s.d.) intensity of normal expression was 35% (22.9%) and 44% (33.2%), respectively.
Exploratory endpoint of restoration of DAPC
Lack of SGCB protein expression results in the mislocalization and downregulation of the sarcoglycan protein complex (composed of α-, γ- and δ-sarcoglycan (SGCA, SGCG and SGCD, respectively)) because it is not able to assemble correctly at the membrane without SGCB.
Immunofluorescence staining of the sarcoglycan complex subunits at both baseline and Day 60 demonstrated that SGCB protein expression corresponded to a clear improvement in the colocalization of SGCA, SGCG and SGCD subunits and rescue of the sarcoglycan complex at the membrane following treatment in both cohorts (Fig. 2b). At Day 60, both the percentage of fibers staining positive and fluorescence intensity increased for SGCA, SGCG and SGCD in patients of both cohorts, but increases were higher in Cohort 2 patients. At Year 2, the expression of sarcoglycan complex subunits remained higher than baseline in all patients and was generally maintained at similar expression levels compared with Day 60 (Fig. 2b). Individual patient data are shown in Supplementary Tables 1 and 2.
Exploratory endpoint of CK reduction
Creatine kinase in serum was markedly elevated in patients with LGMD2E/R4 as a consequence of muscle fiber breakdown. At 60 days post treatment, serum CK levels were reduced in all patients (Extended Data Table 2); mean (s.d.) percentage change from baseline to Day 60 was −92.4% (9.8%) for Cohort 1 and −94.9% (4.5%) for Cohort 2. At 2 years post treatment, all patients in Cohorts 1 and 2 maintained reductions in serum CK levels (Extended Data Table 2); mean (s.d.) percentage change from baseline was −83.4% (4.7%) and −67.3% (18.2%), respectively.
Extended Data Table 2.
Exploratory outcome of serum creatine kinase levels (U/L) throughout the study

Exploratory endpoints of functional outcomes
At 1 year post treatment, both cohorts showed improvements in functional outcomes (Table 4, Extended Data Fig. 3 and Extended Data Table 3). Motor performance was assessed using the NSAD scale; NSAD consists of 29 items, including running, squatting down and jumping (score range 0–54), with higher scores indicating higher levels of function. At 1 year, mean (s.d.) change from baseline in NSAD score was an increase of 5.7 (1.5) in Cohort 1 and an increase of 4.0 (1.4) in Cohort 2 (Extended Data Fig. 4). Timed function tests also improved at 1 year, with mean (s.d.) changes from baseline indicating shorter times for time to rise (Cohort 1, −0.8 s (0.4); Cohort 2, −1.1 s (1.1)); four-stair climb (Cohort 1, −0.5 s (0.3); Cohort 2, −0.4 s (0)); 100-m walk test (Cohort 1, −5.3 s (3.2); Cohort 2, −7.9 (5.4)); and 10-m walk test (Cohort 1, −0.6 s (0.2); Cohort 2, −0.6 s (0.2)).
Table 4.
Exploratory outcomes of change from baseline in functional outcomes
| Cohort 1 (dose, 1.85 × 1013 vg kg−1)a (n = 3) | Cohort 2 (dose, 7.41 × 1013 vg kg−1)b (n = 3) | Natural history cohort (n = 5) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Month 6 (n = 3) | Year 1 (n = 3) | Year 2 (n = 3) | Month 6 (n = 3) | Year 1 (n = 2) | Year 2 (n = 2) | Month 6 (n = 5) | Year 1 (n = 5) | Year 2 (n = 5) | |
| NSAD score | |||||||||
| Mean (s.d.) | 3.0 (1.7) | 5.7 (1.5) | 5.7 (1.5) | 3.7 (3.5) | 4.0 (1.4) | 0.5 (0.7) | –2.2 (3.7) | –2.0 (2.1) | –5.3 (3.1) |
| Median (range) | 4.0 (1.0, 4.0) | 6.0 (4.0, 7.0) | 6.0 (4.0, 7.0) | 4.0 (0, 7.0) | 4.0 (3.0, 5.0) | 0.5 (0, 1.0) | −1.0 (−7.0, 2.0) | −2.0 (−4.0, 1.0) | −6.0 (−8.0, −2.0) |
| Time to rise, s | – | – | – | ||||||
| Mean (s.d.) | −0.2 (0.8) | −0.8 (0.4) | −0.6 (0.2) | −1.3 (0.9) | −1.1 (1.1) | −0.7 (0.4) | |||
| Median (range) | −0.6 (−0.7, 0.8) | −0.6 (−1.2, −0.5) | −0.6 (−0.7, −0.4) | −0.8 (−2.3, −0.7) | −1.1 (−1.8, −0.3) | −0.7 (−1.0, −0.4) | |||
| Four-stair climb, s | – | – | – | ||||||
| Mean (s.d.) | −0.5 (0.4) | −0.5 (0.3) | −0.3 (0.4) | −0.4 (0.3) | −0.4 (0) | −0.3 (0.3) | |||
| Median (range) | −0.4 (−0.9, −0.1) | −0.5 (−0.8, −0.2) | −0.5 (−0.6, 0.2) | −0.4 (−0.7, −0.1) | −0.4 (−0.4, −0.4) | −0.3 (−0.5, −0.1) | |||
| 100-m walk, s | |||||||||
| Mean (s.d.) | −3.8 (2.9) | −5.3 (3.2) | −2.8 (6.4) | −6.3 (6.7) | −7.9 (5.4) | −2.9 (9.7) | −0.8 (5.6) | 1.6 (6.8) | 5.2 (10.2) |
| Median (range) | −4.6 (−6.2, 0.6) | −3.6 (−8.9, −3.3) | −4.4 (−8.2, 4.2) | −2.8 (−14.0, −2.1) | −7.9 (−11.7, −4.1) | −2.9 (−9.7, 4.0) | 1.2 (−7.5, 5.5) | 3.3 (−8.1, 7.7) | 9.0 (−6.4, 13.0) |
| 10-m walk, s | |||||||||
| Mean (s.d.) | −0.6 (0.3) | −0.6 (0.2) | −0.2 (0.5) | −0.6 (0.6) | −0.6 (0.2) | −0.3 (0.9) | −0.7 (0.6) | −0.2 (0.7) | −0.1 (0.3) |
| Median (range) | −0.6 (−0.9, −0.3) | −0.5 (−0.9, −0.5) | −0.3 (−0.6, 0.3) | −0.3 (−1.3, −0.2) | −0.6 (−0.7, −0.4) | −0.3 (−0.9, 0.4) | −0.7 (−1.5, 0) | −0.1 (−1.0, 0.6) | −0.1 (−0.3, 0.1) |
A positive (+) change in NSAD and a negative (−) change in timed tests indicate improvement in function.
a1.85 × 1013 vg kg−1 (linear standard qPCR).
b7.41 × 1013 vg kg−1 (linear standard qPCR).
Extended Data Fig. 3.

Exploratory outcome of changes in functional outcomes throughout the study and post hoc comparison with natural history cohort presented as scores of individual patients for (a) Time to rise, (b) 4-stair climb, (c) 100m, (d) 10m.
Extended Data Table 3.
Exploratory outcomes of additional functional outcome measures assessed throughout the study: Cohort 1 and Cohort 2

Extended Data Fig. 4. Exploratory outcome of changes in NSAD score throughout the study and post hoc comparison with natural history cohort.

a–c, Changes in NSAD score in Cohort 1 (a), Cohort 2 (b), and SRP-9003-101 study population (Cohorts 1 and 2; n = 6) versus natural history cohort (n = 5) (c) presented as total NSAD score of individual patients (left panel) and mean (s.d.) change from baseline by cohort (right panel). The NSAD scale ranges from 0 to 54, with higher scores indicating a higher level of function. Footnote: aMMRM analysis included fixed effects for treatment arm, visit, and treatment arm by visit interaction, and baseline NSAD, baseline 100m, and baseline 10m as continuous covariates; the first-order autoregressive structure was used for variance-covariance matrix of within-patient errors. Functional data for Patient 5 is available until month 6 due to a recreational accident unrelated to the study. LS, least square; MMRM, mixed model repeated measures; NSAD, North Star Assessment for Limb Girdle Type Muscular Dystrophies.
Improvements over baseline in functional outcomes were generally sustained at 2 years in both cohorts (Table 4, Extended Data Fig. 3 and Extended Data Table 3). Mean (s.d.) change from baseline in NSAD score at 2 years was 5.7 (1.5) in Cohort 1 and 0.5 (0.7) in Cohort 2 (Extended Data Fig. 4). Mean (s.d.) changes from baseline in timed function tests at 2 years also indicated sustained improvements: time to rise (Cohort 1, −0.6 s (0.2); Cohort 2, −0.7 s (0.4)); four-stair climb (Cohort 1, −0.3 s (0.4); Cohort 2, −0.3 s (0.3)); 100-m test (Cohort 1, −2.8 s (6.4); Cohort 2, −2.9 s (9.7)); and 10-m test (Cohort 1, −0.2 s (0.5); Cohort 2, −0.3 s (0.9)). Hand-held dynamometry assessments showed sustained improvements at 2 years in six of six muscle groups in Cohort 1 and in four of six muscle groups in Cohort 2 (Extended Data Table 3).
Post hoc comparison with natural history cohort
An exploratory comparison of functional outcomes in the SRP-9003-101 study population of bidridistrogene xeboparvovec-treated patients (Cohorts 1 and 2, n = 6) versus an untreated natural history cohort was also performed. The natural history cohort (n = 5) was identified from the Nationwide Children’s Hospital (NCH) natural history dataset using the same key study inclusion criteria as in SRP-9003-101 (further details provided in Methods). Whereas age and sex were well balanced between cohorts, the NCH natural history cohort had better functions as measured by NSAD, 100-m test and 10-m test at baseline (Extended Data Table 4). Alternative matching criteria were explored but were unsuccessful in achieving balanced baseline functions due to the limited sample size. A mixed-model, repeated-measures analysis was thus used to adjust for differences in baseline functions. The mixed-model, repeated-measures model included fixed effects for treatment arm, visit and treatment arm by visit interaction, as well as baseline NSAD, baseline 100-m test and baseline 10-m test as continuous covariates; the first-order autoregressive structure was used for variance–covariance matrix of within-patient errors; the Kenward–Roger approximation was used to estimate the denominator degrees of freedom. Individual patient-level data for the matched natural history cohort are shown in Extended Data Table 5.
Extended Data Table 4.
Post hoc exploratory baseline comparison of bidridistrogene xeboparvovec–treated patients versus natural history cohort

Extended Data Table 5.
Exploratory functional outcome measures: individual patient data for the matched natural history cohort

In this exploratory comparison, patients treated with bidridistrogene xeboparvovec consistently showed higher NSAD change from baseline scores compared with the natural history cohort over 2 years of follow-up (Extended Data Fig. 4); least-squares mean difference in the change in NSAD score from baseline for the bidridistrogene xeboparvovec-treated cohort versus the natural history cohort at Year 2 was 7.3 (95% confidence interval (CI), 0.7–13.9). Timed function tests also showed improvements, with least-squares mean differences between cohorts in change from baseline of −6.2 s (95% CI, −19.6 to 7.1) for 100-m test and −0.8 s (95% CI, −2.2 to 0.6) for 10-m test. In addition, descriptive statistics are provided in Extended Data Table 6. The unadjusted mean difference in the change in NSAD score from baseline for the bidridistrogene xeboparvovec-treated cohort versus the natural history cohort at Year 2 was 8.9 (95% CI, 3.5–14.4). Timed function tests also showed improvements, with unadjusted mean differences between cohorts in change from baseline of −8.1 s (95% CI, −22.3 to 6.2) for 100-m test and −0.1 s (95% CI, −1.2 to 1.0) for 10-m test.
Extended Data Table 6.
Post hoc exploratory analysis – descriptive summary of functional outcomes between the treated cohort and the matched natural history cohort

Discussion
Interim results of this open-label trial provide biological and clinical evidence for the safety and efficacy of systemic gene therapy with bidridistrogene xeboparvovec, at both low and high doses, in patients with LGMD2E/R4.
Efficient transduction with widespread and robust full-length SGCB expression in muscle biopsies was shown by vector genome counts, immunofluorescence and immunoblot in all six patients at Day 60 post treatment, demonstrating success of the targeted delivery of the transgene to muscle tissue. Greater effects on transduction and SGCB expression were observed in the higher-dose cohort, indicating a dose–response in biological activity between low and high doses of bidridistrogene xeboparvovec. Immunofluorescence analysis showed that SGCB expression was associated with upregulation of SGCA, SGCG and SGCD expression, suggesting that bidridistrogene xeboparvovec can restore the sarcoglycan complex at the sarcolemma, which is required for DAPC stabilization. SGCB gene transduction and protein expression were also analyzed 2 years post treatment. We observed a reduction in SGCB genome copies compared with Day 60, which could be related to a higher amount of episomal DNA in the nucleus in the first months post gene transference. The initial overestimation of copy number was corrected by episomal DNA stabilization, which in turn provided a path for maintaining transgene expression and protein production at high levels, as measured by immunoblot and the percentage of SGCB-positive fibers. We observed a reduction in immunofluorescence intensity of SGCB at year 2 that was more evident in Cohort 1, but expression levels were comparable with Day 60.
These findings follow reports of delandistrogene moxeparvovec gene transfer therapy in Duchenne muscular dystrophy25,26. The construct used in the current study contains the same vector and promoter used in delandistrogene moxeparvovec, demonstrating consistent ability to drive transgene expression and consequent functional benefit in multiple disease states. Thereby, efficient and targeted production of native SGCB protein in the current trial resulted in the functional benefits observed.
Systemic administration of bidridistrogene xeboparvovec was well tolerated, with minimal SAE findings and no unexpected immunological responses. Some patients in both cohorts had elevated liver enzymes, but no associated clinical manifestations were observed. One SAE was observed in each cohort; in both cases, symptoms resolved with standard therapies and no stopping/discontinuation rules were triggered by these adverse events. Importantly, clinically relevant complement activation reported in gene transfer programs using other vectors (that is, non-AAVrh74) was not observed, which is in line with other AAVrh74-based programs in development.
This study has the inherent limitations of a small, open-label trial with an interim, short-term analysis that limits the ability to draw inferences from secondary, exploratory and subgroup analyses, and highlights the need for longer follow-up to assess the safety and efficacy of the therapy. Moreover, variability associated with the immunofluorescence technique, and the use of an external comparator (natural history cohort) that was matched by age with the treatment cohorts but not by baseline NSAD, pose additional limitations. Despite these limitations, patients in both cohorts treated with bidridistrogene xeboparvovec demonstrated improvement or stabilization of NSAD scores from baseline up to 2 years. Functional gains were accompanied by sustained reductions in serum CK levels, indicating less muscle damage, although early reductions were probably due to prophylactic treatment of prednisone that was continued post treatment for at least 30 and 60 days for Cohorts 1 and 2, respectively. Exploratory post hoc analyses further showed that bidridistrogene xeboparvovec-treated patients had clinically important improvements in functional outcomes compared with a natural history cohort over 2 years of follow-up. A recent observational, retrospective study of a large cohort of patients with sarcoglycanopathies identified residual mutated protein expression levels <30% as an independent risk factor for loss of ambulation before 18 years of age27. In our LGMD2E/R4 trial, both low- and high-dose cohorts showed full wild-type protein production with a mean percentage of normal expression of 36.2 and 62.1, respectively, at Day 60, suggesting that sufficient SGCB expression levels were achieved and predictive of improved functional outcomes.
An important concern for gene transfer therapy is the potential for waning of effects over time. Until there is broader and longer-term clinical experience with gene therapies for multiple disease-state applications, we can only speculate on long-term efficacy. Long-term follow-up of two clinical trials involving 12 patients with spinal muscular atrophy type 1 (up to 7 years) and four patients with Duchenne muscular dystrophy (up to 4 years), treated with a dose range of AAV-based vectors similar to that in the present study, showed therapeutic efficacy sustained over time26,28. Assessment of long-term safety and efficacy from a small cohort of patients with LGMD2E/R4 is challenging, but the present trial yielded encouraging results for bidridistrogene xeboparvovec for at least 2 years; extended follow-up of patients is currently ongoing, with the final study analysis planned at 5 years.
Methods
Study design and participants
This first-in-human, single-center, nonrandomized, open-label, phase 1/2 systemic gene delivery trial (SRP-9003-101) evaluated the safety, tolerability and efficacy of bidridistrogene xeboparvovec in patients with LGMD2E/R4 (ClinicalTrials.gov identifier: NCT03652259; Fig. 1a). This trial was conducted at NCH (Columbus, OH), preregistered on 28 August 2018 (NCT03652259) and began on 26 October 2018. Although the duration of follow-up is 5 years, the protocol is currently amended to extend the study for an additional 2 years with a total of 7 years. This study is being conducted in accordance with the principles of the Declaration of Helsinki and the International Council for Harmonisation Good Clinical Practice guidelines29. The study protocol was approved by the NCH Institutional Review Board. Parents or legal guardians of all patients provided written informed consent before study participation and genetic testing and consented to disclosure of their demographics for publication purposes. There was no compensation for participation in the study other than covering for meals and travel-related expenses.
Vector description
SRP-9003 is a self-complementary, nonreplicating, recombinant AAVrh74 vector containing codon-optimized, full-length SGCB cDNA under the control of the myosin heavy-chain/muscle CK (MHCK7) promoter (the full sequence is given in Supplementary Information)20,30. The vector cassette contains minimal elements required for gene expression, including AAV2 inverted terminal repeats, hSGCB, SV40 chimeric intron and synthetic polyadenylation site, all under the control of the MHCK7 promoter (Supplementary Fig. 1). The amino acid sequence of the SGCB protein produced from the SRP-9003 transgene is identical to that of normal wild-type hSGCB protein.
Outcomes
The primary endpoint was to evaluate the safety of bidridistrogene xeboparvovec. The secondary endpoint was the change in quantity of SGCB protein in skeletal muscle from baseline to Day 60 as assessed by immunoblot and immunofluorescence staining analysis (fiber intensity and percentage of SGCB-positive fibers). Exploratory endpoints included functional outcomes (NSAD score and time function tests), skeletal muscle strength, serum CK levels, immunogenicity of bidridistrogene xeboparvovec and durability of SGCB expression in skeletal muscle. Because patients are now beyond the post-dosing interval where safety events are most likely to be attributed to gene therapy, and all protocol-defined muscle biopsies used to evaluate SCGB expression have been completed, this report was generated to provide preliminary results for the primary and secondary endpoints. A subsequent clinical report of the final 5-year data will be warranted after final study completion.
Eligible patients were aged 4–15 years (inclusive) with confirmed SGCB gene mutations for SGCB deficiency at both alleles. Additional inclusion criteria were demonstrable muscle weakness and ≥40% of normal 100-m test predicted for age-, height- and weight-matched healthy controls at the screening visit31. Sex or gender was not considered in the study design. Sex was self-reported by participants. Full eligibility criteria are as follows.
Subject inclusion criteria:
Subjects aged 4–15 years, inclusive
Males or females of any ethnic group
SGCB DNA gene mutations at both alleles (if genetic testing was completed at a laboratory that is not Clinical Laboratory Improvement Amendments certified, the testing may be repeated at the discretion of the principal investigator)
Weakness demonstrated, based on history of difficulty running, jumping and climbing stairs
100-meter walk/run test result: ≥40% of that predicted for age-, height-, gender- and weight-matched healthy controls at the screening visit
Ability to cooperate with muscle testing
Willingness of sexually active subjects with reproductive capacity to practice reliable method of contraception (if appropriate) during the first 6 months following gene therapy
Subject exclusion criteria:
Active viral infection based on clinical observations
Cardiac MRI-determined left ventricular ejection fraction <40%
Serological evidence of infection by human immunodeficiency virus, hepatitis B or hepatitis C
Diagnosis of (or ongoing treatment for) an autoimmune disease
Abnormal laboratory values considered clinically significant
Concomitant illness or requirement for chronic drug treatment that, in the opinion of the principal investigator, creates unnecessary risks for gene transfer
Pregnancy
With AAVrh74-binding antibody titers >1:400 as determined by ELISA; if endpoint titer is positive at screening, testing may be repeated in 1 month
Has a medical condition or circumstance that could compromise the protocol compliance or compromise safety
Severe infection (for example, pneumonia, pyelonephritis or meningitis) within 4 weeks preceding gene transfer visit (enrollment may be postponed)
Family does not want to disclose subject’s study participation with primary care physician or other medical providers
Study treatment
Enrolled patients received a single systemic infusion of bidridistrogene xeboparvovec via a peripheral vein over approximately 1–2 h. Twenty-four hours preceding gene delivery, prophylactic oral prednisone (1 mg kg−1) or comparable glucocorticoid was administered and continued daily for 30 days for Cohort 1. In response to a treatment-related SAE of hepatitis in one patient, the data safety monitoring board approved a protocol amendment to specify administration of immunosuppression for at least 60 days for Cohort 2. In both cohorts this was followed by a tapering regimen based on the individual’s immune response to gene transfer as assessed by both ELISpot and liver function monitoring of GGT. Patients in Cohort 1 were treated with bidridistrogene xeboparvovec at a dose of 1.85 × 1013 vg kg−1 (calculated by qPCR using a linear reference standard). This dose was selected with the goal of achieving ≥20% of normal expression levels extrapolated from preclinical studies correlating expression and function20. Muscle biopsies at Day 60 determined the dosage level for Cohort 2. According to the protocol adopted, because the number of muscle fibers expressing SGCB was <50% of normal control intensity in one or more of the treated patients, the bidridistrogene xeboparvovec dose was escalated to 7.41 × 1013 vg kg−1 by linear qPCR (standard equivalent of 2 × 1014 vg kg−1) following review and endorsement by the Data Safety Monitoring Board. Patients in Cohort 2 received bidridistrogene xeboparvovec at least 4 weeks post biopsy of Cohort 1.
Safety assessments
Safety was assessed throughout the study by monitoring of vital signs, physical examinations, electrocardiograms, incidence of treatment-emergent adverse events and treatment-emergent SAEs and select laboratory assessments including hematological analysis, serum chemistry panels (liver function tests and CK level) and urinalysis. Patients were assessed at baseline and monitored on Days 1, 7, 14, 30, 45, 60, 90, 180 and 270 and Years 1, 1.5 (Month 18), 2, 2.5 (Month 30), 3, 4 and 5.
Immunological response was assessed by measurement of antibodies (ELISA) and T cell responses (ELISpot) to rAAVrh74 and/or SGCB (ELISA) at baseline, Days 7, 14, 30, 60, 90, 180 and 270 and Years 1, 1.5 and 2.
Biological assessments
Muscle biopsies were performed at baseline, Day 60 and Year 2 on the tibialis anterior and biceps muscles, with appropriate anesthesia. Biopsies were performed by an interventional radiologist guided by ultrasound and using a 10-g Vacora biopsy probe32. Immunoblot assays were executed according to validated methodology adapted from Charleston et al.33. Muscle biopsy tissue was mounted with minimal optimal cutting temperature compound, gum tragacanth medium and/or Cryo-Gel, cryosectioned and allocated for analysis by immunoblot, qPCR, ddPCR and immunofluorescence. qPCR was used initially for analysis of vg numbers before switching to ddPCR as the preferred method. SGCB levels of samples were calculated from a five-point standard curve using full-length recombinant SGCB protein ranging from 125 to 2,000 pg. The protocol was modified before Cohort 2 sample analysis, expanding the standard curve range from 125 to 3,000 pg and ranging from 5 to 80%. Reported SGCB levels were the average value of both biological replicates and two technical gel replicates for each sample result. Mean values for each patient were obtained from four tissue blocks. Recombinant protein was used to generate a standard curve for quantitative analysis. BIOQUANT automated software was used to quantify the level of SGCB expression, and ImageJ NIH software34 to determine the number of muscle fibers expressing SGCB.
ELISpot assay
Whole-blood samples were collected at intervals as defined by the adopted protocol. Peripheral blood mononuclear cells (PBMCs) were separated from whole blood by density-gradient centrifugation. Briefly, fresh blood samples were spun in a tabletop centrifuge (Sorvall Legend RT, Marshall Scientific) and the top plasma layer was removed and stored at −80 °C. The remaining blood was mixed with PBS−/− (Invitrogen) underlayed with Ficoll-Paque (Cytiva) and spun for density-gradient separation. The middle layer containing PBMCs was removed and subjected to various wash–spin cycles before final resuspension in human AIM V medium (Invitrogen) supplemented with human antibody serum. PBMCs were then plated in IP filter plates (MilliporeSigma) at a density of 2 × 105 cells per well, except for the positive control wells that were plated at 2.5 × 104 cells per well. IP plate wells were precoated with monoclonal antibody provided with the human interferon-γ ELISpot kit (U-Cytech Biosciences). Three peptide pools were used for the AAVrh74 capsid protein (Genemed Synthesis) containing 34–36 peptides, each 18 amino acids in length and overlapping by 11 residues. One peptide pool encompassed the SGCB transgene (Genemed Synthesis) containing 44 peptides, each 18 amino acids in length and overlapping by 11 residues. Peptides were added directly to wells at a final concentration of 40 μg ml−1. Following the addition of PBMCs and peptides, the plate was incubated at 37 °C in 5–7% CO2 and 100% humidity for 36–48 h and then developed according to the manufacturer’s protocol. Interferon-γ spot formation was counted using Immunospot software (S6 Universal, Cellular Technology Ltd).
ELISA
Serum samples were diluted from 1:25 to 1:26,214,400 in blocking solution (5% nonfat dry milk, 1% normal goat serum (Invitrogen) in PBS−/−) and added to wells for 1 h at room temperature. Sera from previously screened positive and negative human samples were used for assay controls. Goat anti-human HRP IgG-FC conjugated (Bethyl Laboratories) was used as the secondary antibody, at 1:10,000 dilution for 30 min at room temperature, followed by exposure with Ultra TMB-ELISA solution (Thermo Fisher Scientific). Absorbance of wells at optical density 450 nm was measured and used to calculate the titer of positive reaction; the lower limit of detection is 1:25.
Immunoblot
Immunoblot was performed according to methods adapted from Pozsgai et al.20. SGCB levels of treatment-blinded samples were calculated from a five-point standard curve ranging from 125 to either 2,000 pg (Cohort 1, Day 60) or 3,000 pg (Cohort 1, Year 2; Cohort 2, Day 60 and Year 2) and were loaded in the same gel as the normal control pool and samples.
qPCR
TaqMan qPCR was performed to quantify the number of vg present in targeted muscle biopsies, as previously described26. Biodistribution analysis was performed on tissue samples collected from three vector-dosed mdx animals per dose level. Tissues were harvested at necropsy, and vector-specific primer probe sets specific for sequences of the MHCK7 promoter were utilized. A vector-specific primer probe set was used to amplify a sequence of the intronic region directly downstream from the MHCK7 promoter that is unique and located within the SRP-9003 cassette. Copy number is reported as vg number per nucleus.
ddPCR
The ddPCR assay was used to quantify vector genome copies from human muscle tissue biopsies. The human Myogenin gene was used as a reference to report vg per diploid human genome (also referred to as vectors per nucleus). The assay was performed using ddPCR Supermix for Probes (No dUTP) (Bio-Rad Laboratories) on a QX200 ddPCR System (Bio-Rad Laboratories) according to the manufacturer’s instructions, with a thermocycler ramp rate of 2 °C s−1; 95 °C for 10 min; 40 cycles of (94 °C for 30 s; 60 °C for 1 min); 98 °C for 10 min; 4 °C hold. Primers and probes (Table 1) were synthesized from Integrated DNA Technologies. Data were analyzed using QuantaSoft-Regulatory Edition v.1.7.4 (Bio-Rad Laboratories). Primers and probes used for the ddPCR assay were: vg forward primer 5′ GCAACAGACCTTTCATGGGCAAACC 3′; vg reverse primer 5′ TATAACCAGGCATCTCGGGTGTCCC 3′; human genome forward primer 5′ CTCATTGGAACTCCAGCAGGCCT 3′; human genome reverse primer 5′ GCTCTCACAGGACACCAGCTTAAAGG 3′; vg probe 5′ /56-FAM/CCCTGCTGT/ZEN/CTAGCATGCCCCACTACG/3IABkFQ/ 3′; and human genome probe 5′ /5HEX/AGCGACCTT/ZEN/CACTGGGCCACAAAATCTG/3IABkFQ/ 3′.
Immunofluorescence
Immunostaining was conducted according to Good Clinical Laboratory Practice using qualified equipment and validated methods and software. Standard operating procedures and associated forms were utilized for related laboratory activities.
Frozen muscle samples mounted on wooden blocks were cut using a Thermo Scientific HM525NX Cryostat at 10-µm thickness. Frozen sections were mounted onto slides (Fisherbrand Superfrost Plus, catalog no. 12-550-15) and stored at −40 °C until staining. Slides were warmed to ambient temperature and fixed briefly with cold acetone (Fisher Scientific, catalog no. A18-4) and allowed to dry in air. Once dry, a hydrophobic boundary was created around frozen sections using a Pap Pen (BioGenex, catalog no. XT001PP). A no-primary slide for each antibody was stained in parallel to assess nonspecific binding of primary antibodies. A 10% goat serum blocking buffer was prepared using 1× TBS (Fisher Scientific, catalog no. BP24721) and goat serum (Gibco, catalog no. 16210064). A 1% goat serum wash solution was prepared (1× TBS, goat serum).
All steps of the staining procedure were conducted in a humidity chamber with a lid. Slides were incubated in 10% blocking buffer for 20 min, after which excess blocking buffer was removed from slides.
Primary antibody SGCD (mouse monoclonal, Leica Biosystems, catalog no. NCL-d-SARC) was diluted in 1× TBS and prepared at a dilution of 1:25. Slides were incubated for 1 h then 1× TBS was applied to the no-primary slide in place of diluted primary antibody.
Primary antibodies SGCB (mouse monoclonal, Leica Biosystems, catalog no. NCL-L-b-SARC) and SGCA (mouse monoclonal, Leica Biosystems, catalog no. NCL-L-a-SARC) were each diluted in 1× TBS and prepared at a dilution of 1:50. Slides were incubated for 1 h then 1× TBS was applied to the no-primary slide in place of diluted primary antibody.
Primary antibody SGCG (mouse monoclonal, Leica Biosystems, catalog no. NCL-g-SARC) was diluted in 1× TBS and prepared at a dilution of 1:100. Slides were incubated for 1 h then 1× TBS was applied to the no-primary slide in place of diluted primary antibody.
Excess primary antibody and 1× TBS were removed from slides after 1 h. Sections were washed three times with 1% goat serum wash solution, each for 5 min. Excess wash solution was removed from slides between each wash and after the final wash. Secondary antibody Alexa Fluor 594 (goat anti-mouse IgG Highly Cross-Adsorbed Secondary Antibody, Invitrogen, catalog no. A11032) was diluted 1:300 in 1× TBS. Slides were incubated for 1 h, and excess secondary antibody removed from slides after one further hour. Sections were washed three times with 1% goat serum wash solution, each for 5 min. Excess wash solution was removed from slides between each wash and after the final wash. Slides were then coverslipped using Vectashield antifade mounting medium (Vector Labs, catalog no. H-100010) and glass coverslips (Gold Seal Coverslips, Electron Microscopy Sciences, catalog no. 6376910) and stored at −40 °C.
Depending on the time of patient enrollment and biopsy timepoints, images were captured using either an Axioskop microscope with AxioVision Rel software (Zeiss) or an ECLIPSE Ni-U microscope (Nikon) fitted with a PROKYON camera equipped with GRYPHAX software (Jenoptik). All fresh-frozen muscle samples (four blocks per patient per timepoint) included four nonoverlapping images of immunofluorescence-labeled tissue sections, each at ×20 objective. Positive and negative control tissues per batch run were used to set an optimum exposure time. All images were captured in an identical manner and with the same exposure time for each batch session. Proper storage conditions of the tissue samples and stained slides protected stability for optimal fluorophore signal intensity without reduction of expression. Images were saved for analysis in .tiff format using eight bits per channel for fluorochrome images captured for image analysis. For imaging analysis, four ×20 images per tissue block were taken, with four per timepoint (that is, 16 images per patient per timepoint).
BIOQUANT image analysis
Images (×20) were measured using BIOQUANT software v.2019. To determine the upper and lower thresholds for analysis, the normal and negative control images were used by selecting the stained membrane, including the various shades of intensity within the run. Threshold settings for the dataset were obtained from two trained technical analysts independently. Threshold values obtained for red, green and blue channels were averaged, with the average value for each channel taken as the consensus threshold for that dataset. A sequence of images was analyzed by selecting the field measurement tool. Values for Fluorescence Quantitation (D6) and Red Fluorescence Intensity (D3) BIOQUANT measurement algorithms were recorded. An average value was calculated for expression percentage of normal controls for the dataset, which was normalized as 100%.
Percentage positive fiber analysis
Images (×20) were assessed for the relative number of positive fibers compared with total fibers based on visual examination of digital images. SGCB-positive fiber established criteria were defined as positive if the membrane contained approximately 30% or greater additive staining of the fiber perimeter (membrane) above the low-intensity levels of the run, including controls. The total percentage of the fiber perimeter did not need to be continuous and was subjectively determined by consciously combining all segments of perimeter positivity to determine a total perimeter intensity percentage. Fibers counted were defined by the structural appearance of their cross-section. To facilitate scoring, NIH ImageJ with the Cell Counter plugin was used to count total fibers. By convention, a ʻType #ʼ and color was selected to score/mark positive fibers. The Cell Counter tracked counts as fibers were selected. Positive fibers were scored based on the original image exposure; there was no adjustment to the brightness or contrast of any image during the positive-image scoring process. Once positive fiber selections were completed, the brightness and contrast of the image were adjusted to visualize negative fibers more easily. No additional positive fibers were scored once brightness/contrast was adjusted. The remaining fibers were scored as negative using a different Type # and color for the score/mark. Once scoring was complete, ʻExport Imageʼ was selected from the Cell Counter workspace and this image was saved separately as an annotated .jpeg image. Total fiber counts were determined after all fibers were counted.
Functional assessments
Functional efficacy was assessed using NSAD, a 29-item activity-level scale for measurement of motor performance, with a score range of 0–54 (higher scores indicate a higher level of function)35–38. Other functional outcomes included time to rise, four-stair climb, 100-m test, 10-m test and hand-held dynamometry for knee extensors and flexors and elbow flexors.
NCH natural history cohort
The NCH natural history study is a prospective, observational, longitudinal cohort study assessing the natural history of LGMD2A and LGMD2E (ClinicalTrials.gov identifier: NCT03488784)37. To select individuals in the untreated NCH natural history cohort for comparison with treated patients in the SRP-9003-101 trial, the predefined inclusion criteria for SRP-9003-101 were also applied to the natural history dataset, as follows:
LGMD2E/R4 ambulatory subjects only; for the natural history cohort, this was defined as those with nonmissing 10-m test value
Baseline age 4–15 years, inclusive
100-m test result: ≥40% of that predicted for age-, height-, gender- and weight-matched healthy controls
Supplementary Fig. 2 shows the derivation of the natural history comparator group. From an overall dataset of 35 subjects, five control subjects who met these criteria were identified. The matched patients were originally consented to NCT03488784 from November 2016 to August 2019.
Statistics and reproducibility
Sample size was based on enrollment feasibility; there was no formal determination based on statistical considerations. Analyses were primarily descriptive in nature and no formal statistical tests were performed. Descriptive statistics are presented for all endpoints and included number of subjects (n), mean, s.d., minimum and maximum for continuous variables and number and percentage for categorical variables. Baseline and demographic characteristics, safety, biopsy and functional data were summarized using descriptive statistics. No sex- or gender-based analysis was performed owing to the small sample size. SAS v.9.4 (SAS Institute Inc.) was used for demographics and safety data, and Prism v.5 (GraphPad Software) for biopsy data. For the NCH natural history cohort, baseline was defined as the first timepoint where both the 100-m test and NSAD were nonmissing.
For each patient and timepoint, four biopsies were collected. Each biopsy was analyzed with two technical replicates using a validated immunoblot method. The immunoblot experiment was not repeated after obtaining valid results from two technical replicates. The average coefficient of variation of technical replicates was 10.17%. During validation of the immunoblot method, the following parameters were assessed and deemed suitable for the analysis of study samples: accuracy, precision, specificity, sensitivity, linearity and measurement of range and robustness. Validation was performed based on Food and Drug Administration guidelines.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41591-023-02730-9.
Supplementary information
Supplementary Tables 1 and 2 and Figs. 1–3.
Source data
Unprocessed immunoblots used in Extended Data Fig. 2.
Acknowledgements
This study (no. NCT03652259) was funded by Sarepta Therapeutics, Inc. We thank the patients and their families for their participation in the study. We also thank A. S. Meadows and E. Koenig for their assistance with the study as former employees of Sarepta Therapeutics, Inc. Medical writing and editorial support was provided by L. Quintana-Gallardo of Sarepta Therapeutics, Inc. and P. Briassouli of Eloquent Scientific Solutions, and was funded by Sarepta Therapeutics, Inc.
Extended data
Author contributions
J.R.M. designed and was the principal investigator on the clinical trial, recruited patients, managed safety issues and followed patients for outcome measures. E.R.P. designed experiments, analyzed and reviewed data and contributed to manuscript drafting. S.L. analyzed and reviewed data and contributed to manuscript drafting. D.A.G. analyzed and reviewed data and contributed to manuscript drafting. K.J.L. designed clinical trials, was a subinvestigator on the clinical trial, recruited patients for trial, managed safety issues and followed patients for outcome measures. K.C. is Program Manager in the Center for Gene Therapy, oversaw the CRN/CRC team and worked with internal/external stakeholders to operationalize the trial. N.F.R. collected data as a clinical evaluator for the trial data and natural history comparative data, analyzed and reviewed data and contributed to manuscript drafting. M.A.I. collected data as a clinical evaluator for the trial, analyzed and reviewed data and contributed to manuscript drafting. L.N.A. contributed to study design and data collection, served as a coinvestigator for the comparative natural history data study and assisted with data interpretation and editing of the manuscript. B.S. served as a clinical evaluator. L.P.L. contributed to study design and data collection, served as principal investigator for the comparative natural history data study and assisted with data interpretation and editing of the manuscript. S.N. is a Sponsor Medical Director/Study Lead, internal study management, and assisted with data interpretation and editing of the manuscript. R.P. analyzed and reviewed data and contributed to manuscript drafting. X.L. analyzed and reviewed data and contributed to manuscript drafting. H.S. analyzed and reviewed data and contributed to manuscript drafting. L.R.R.-K. conceived the study, designed experiments, analyzed data and wrote the paper.
Peer review
Peer review information
Nature Medicine thanks James Dowling, Rong Lu, Simone Spuler and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Data availability
Individual data for all patients are presented in the paper but, for any further information, qualified researchers may reach out to Sarepta Therapeutics, Inc. by contacting medinfo@sarepta.com. Data requests will be fulfilled within 90 days. Because the study is ongoing, access to the data is limited to those that support the findings of this study. Source data are provided with this paper.
Competing interests
J.R.M. received financial support from Sarepta Therapeutics, Inc. while at NCH at the time of the study and currently is an employee of Sarepta Therapeutics, Inc. E.R.P., S.L., D.A.G., S.N., R.P., X.L., H.S. and L.R.R.-K. are, or have been, employees of Sarepta Therapeutics, Inc. and may own stock/options in the company. L.P.L. and L.N.A. received fees from Sarepta Therapeutics, Inc. for licensure of the LGMD natural history dataset and served on the LGMD advisory board. L.P.L., L.N.A. and N.F.R. perform outcome measures training. B.S., K.J.L., K.C., N.F.R. and M.A.I. have no competing interests to declare and J.R.M with L.R.R.-K., are coinventors of bidridistrogene xeboparvovec.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
is available for this paper at 10.1038/s41591-023-02730-9.
Supplementary information
The online version contains supplementary material available at 10.1038/s41591-023-02730-9.
References
- 1.Murphy AP, Straub V. The classification, natural history and treatment of the limb girdle muscular dystrophies. J. Neuromuscul. Dis. 2015;2:S7–S19. doi: 10.3233/JND-150105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wicklund MP. The limb-girdle muscular dystrophies. Continuum (Minneap. Minn.) 2019;25:1599–1618. doi: 10.1212/CON.0000000000000809. [DOI] [PubMed] [Google Scholar]
- 3.Thompson R, Straub V. Limb-girdle muscular dystrophies – international collaborations for translational research. Nat. Rev. Neurol. 2016;12:294–309. doi: 10.1038/nrneurol.2016.35. [DOI] [PubMed] [Google Scholar]
- 4.Straub V, Murphy A, Udd B. Limb girdle muscular dystrophies – nomenclature and reformed classification. Neuromuscul. Disord. 2018;28:702–710. doi: 10.1016/j.nmd.2018.05.007. [DOI] [PubMed] [Google Scholar]
- 5.Angelini C, Tasca E, Nascimbeni AC, Fanin M. Muscle fatigue, nNOS and muscle fiber atrophy in limb girdle muscular dystrophy. Acta Myol. 2014;33:119–126. [PMC free article] [PubMed] [Google Scholar]
- 6.Albuquerque MA, et al. Clinical aspects of patients with sarcoglycanopathies under steroids therapy. Arq. Neuropsiquiatr. 2014;72:768–772. doi: 10.1590/0004-282X20140126. [DOI] [PubMed] [Google Scholar]
- 7.Winckler PB, et al. Clinicogenetic lessons from 370 patients with autosomal recessive limb-girdle muscular dystrophy. Clin. Genet. 2019;96:341–353. doi: 10.1111/cge.13597. [DOI] [PubMed] [Google Scholar]
- 8.Fayssoil A, et al. Natural history of cardiac and respiratory involvement, prognosis and predictive factors for long-term survival in adult patients with limb girdle muscular dystrophies type 2C and 2D. PLoS ONE. 2016;11:e0153095. doi: 10.1371/journal.pone.0153095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore SA, et al. Limb-girdle muscular dystrophy in the United States. J. Neuropathol. Exp. Neurol. 2006;65:995–1003. doi: 10.1097/01.jnen.0000235854.77716.6c. [DOI] [PubMed] [Google Scholar]
- 10.Sandonà D, Betto R. Sarcoglycanopathies: molecular pathogenesis and therapeutic prospects. Expert Rev. Mol. Med. 2009;11:e28. doi: 10.1017/S1462399409001203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Angelini C, et al. The clinical spectrum of sarcoglycanopathies. Neurology. 1999;52:176–179. doi: 10.1212/WNL.52.1.176. [DOI] [PubMed] [Google Scholar]
- 12.Bönnemann CG, et al. Beta-sarcoglycan (A3b) mutations cause autosomal recessive muscular dystrophy with loss of the sarcoglycan complex. Nat. Genet. 1995;11:266–273. doi: 10.1038/ng1195-266. [DOI] [PubMed] [Google Scholar]
- 13.Semplicini C, et al. Clinical and genetic spectrum in limb-girdle muscular dystrophy type 2E. Neurology. 2015;84:1772–1781. doi: 10.1212/WNL.0000000000001519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Politano L, et al. Evaluation of cardiac and respiratory involvement in sarcoglycanopathies. Neuromuscul. Disord. 2001;11:178–185. doi: 10.1016/S0960-8966(00)00174-7. [DOI] [PubMed] [Google Scholar]
- 15.Sveen ML, Thune JJ, Køber L, Vissing J. Cardiac involvement in patients with limb-girdle muscular dystrophy type 2 and Becker muscular dystrophy. Arch. Neurol. 2008;65:1196–1201. doi: 10.1001/archneur.65.9.1196. [DOI] [PubMed] [Google Scholar]
- 16.Fanin M, Melacini P, Boito C, Pegoraro E, Angelini C. LGMD2E patients risk developing dilated cardiomyopathy. Neuromuscul. Disord. 2003;13:303–309. doi: 10.1016/S0960-8966(02)00280-8. [DOI] [PubMed] [Google Scholar]
- 17.Melacini P, et al. Heart involvement in muscular dystrophies due to sarcoglycan gene mutations. Muscle Nerve. 1999;22:473–479. doi: 10.1002/(SICI)1097-4598(199904)22:4<473::AID-MUS8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 18.Carotti M, Fecchio C, Sandonà D. Emerging therapeutic strategies for sarcoglycanopathy. Expert Opin. Orphan Drugs. 2017;5:381–396. doi: 10.1080/21678707.2017.1307731. [DOI] [Google Scholar]
- 19.Asher DR, et al. Clinical development on the frontier: gene therapy for Duchenne muscular dystrophy. Expert Opin. Biol. Ther. 2020;20:263–274. doi: 10.1080/14712598.2020.1725469. [DOI] [PubMed] [Google Scholar]
- 20.Pozsgai ER, Griffin DA, Heller KN, Mendell JR, Rodino-Klapac LR. Systemic AAV-mediated β-sarcoglycan delivery targeting cardiac and skeletal muscle ameliorates histological and functional deficits in LGMD2E mice. Mol. Ther. 2017;25:855–869. doi: 10.1016/j.ymthe.2017.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zygmunt DA, Crowe KE, Flanigan KM, Martin PT. Comparison of serum rAAV serotype-specific antibodies in patients with Duchenne muscular dystrophy, Becker muscular dystrophy, inclusion body myositis, or GNE myopathy. Hum. Gene Ther. 2017;28:737–746. doi: 10.1089/hum.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salva MZ, et al. Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol. Ther. 2007;15:320–329. doi: 10.1038/sj.mt.6300027. [DOI] [PubMed] [Google Scholar]
- 23.Raj D, Davidoff AM, Nathwani AC. Self-complementary adeno-associated viral vectors for gene therapy of hemophilia B: progress and challenges. Expert Rev. Hematol. 2011;4:539–549. doi: 10.1586/ehm.11.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCarty DM. Self-complementary AAV vectors; advances and applications. Mol. Ther. 2008;16:1648–1656. doi: 10.1038/mt.2008.171. [DOI] [PubMed] [Google Scholar]
- 25.Mendell JR, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 26.Mendell JR, et al. Assessment of systemic delivery of rAAVrh74.MHCK7.micro-dystrophin in children with Duchenne muscular dystrophy: a nonrandomized controlled trial. JAMA Neurol. 2020;77:1122–1131. doi: 10.1001/jamaneurol.2020.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alonso-Pérez J, et al. New genotype-phenotype correlations in a large European cohort of patients with sarcoglycanopathy. Brain. 2020;143:2696–2708. doi: 10.1093/brain/awaa228. [DOI] [PubMed] [Google Scholar]
- 28.Al-Zaidy SA, et al. AVXS-101 (onasemnogene abeparvovec) for SMA1: comparative study with a prospective natural history cohort. J. Neuromuscul. Dis. 2019;6:307–317. doi: 10.3233/JND-190403. [DOI] [PubMed] [Google Scholar]
- 29.Dixon JR., Jr. The International Conference on Harmonization Good Clinical Practice guideline. Qual. Assur. 1998;6:65–74. doi: 10.1080/105294199277860. [DOI] [PubMed] [Google Scholar]
- 30.Pozsgai ER, Griffin DA, Heller KN, Mendell JR, Rodino-Klapac LR. beta-Sarcoglycan gene transfer decreases fibrosis and restores force in LGMD2E mice. Gene Ther. 2016;23:57–66. doi: 10.1038/gt.2015.80. [DOI] [PubMed] [Google Scholar]
- 31.Alfano LN, et al. The 100-meter timed test: normative data in healthy males and comparative pilot outcome data for use in Duchenne muscular dystrophy clinical trials. Neuromuscul. Disord. 2017;27:452–457. doi: 10.1016/j.nmd.2017.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Tarnopolsky MA, Pearce E, Smith K, Lach B. Suction-modified Bergström muscle biopsy technique: experience with 13,500 procedures. Muscle Nerve. 2011;43:717–725. doi: 10.1002/mus.21945. [DOI] [PubMed] [Google Scholar]
- 33.Charleston JS, et al. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology. 2018;90:e2146–e2154. doi: 10.1212/WNL.0000000000005680. [DOI] [PubMed] [Google Scholar]
- 34.Rasband, W. S. ImageJ (1997–2018) version 1.54F; https://imagej.nih.gov/ij/
- 35.Jacobs MB, et al. Assessing dysferlinopathy patients over three years with a new motor scale. Ann. Neurol. 2021;89:967–978. doi: 10.1002/ana.26044. [DOI] [PubMed] [Google Scholar]
- 36.James M, et al. North Star Assessment for dysferlinopathy: longitudinal performance in the clinical outcome study of dysferlinopathy. Neuromuscul. Disord. 2017;27:S145. [Google Scholar]
- 37.Miller N, et al. Limb girdle muscular dystrophies: P.143 measuring motor function in LGMD2E: determining which functional outcomes are the most responsive to change in a 1-year study. Neuromuscul. Disord. 2020;30:S88. doi: 10.1016/j.nmd.2020.08.146. [DOI] [Google Scholar]
- 38.Moore U, et al. Assessment of disease progression in dysferlinopathy: a 1-year cohort study. Neurology. 2019;92:e461–e474. doi: 10.1212/WNL.0000000000006858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Tables 1 and 2 and Figs. 1–3.
Unprocessed immunoblots used in Extended Data Fig. 2.
Data Availability Statement
Individual data for all patients are presented in the paper but, for any further information, qualified researchers may reach out to Sarepta Therapeutics, Inc. by contacting medinfo@sarepta.com. Data requests will be fulfilled within 90 days. Because the study is ongoing, access to the data is limited to those that support the findings of this study. Source data are provided with this paper.