

Abstract
Existing antiarrhythmic drugs to treat atrial fibrillation (AF) have incomplete efficacy, contraindications and adverse effects, including proarrhythmia. AP30663, an inhibitor of the KCa2 channel, has demonstrated AF efficacy in animals; however, its efficacy in humans with AF is unknown. Here we conducted a phase 2 trial in which patients with a current episode of AF lasting for 7 days or less were randomized to receive an intravenous infusion of 3 or 5 mg kg−1 AP30663 or placebo. The trial was prematurely discontinued because of slow enrollment during the coronavirus disease 2019 pandemic. The primary endpoint of the trial was cardioversion from AF to sinus rhythm within 90 min from the start of the infusion, analyzed with Bayesian statistics. Among 59 patients randomized and included in the efficacy analyses, the primary endpoint occurred in 42% (5 of 12), 55% (12 of 22) and 0% (0 of 25) of patients treated with 3 mg kg−1 AP30663, 5 mg kg−1 AP30663 or placebo, respectively. Both doses demonstrated more than 99.9% probability of superiority over placebo, surpassing the prespecified 95% threshold. The mean time to cardioversion, a secondary endpoint, was 47 (s.d. = 23) and 41 (s.d. = 24) minutes for 3 mg kg−1 and 5 mg kg−1 AP30663, respectively. AP30663 caused a transient increase in the QTcF interval, with a maximum mean effect of 37.7 ms for the 5 mg kg−1 dose. For both dose groups, no ventricular arrhythmias occurred and adverse event rates were comparable to the placebo group. AP30663 demonstrated AF cardioversion efficacy in patients with recent-onset AF episodes. KCa2 channel inhibition may be an attractive mechanism for rhythm control of AF that should be studied further in randomized trials. ClinicalTrials.gov registration: NCT04571385.
Subject terms: Atrial fibrillation, Drug development
In a phase 2 clinical trial, an inhibitor of the KCa2 potassium channel, which conducts a repolarizing current in the heart, met the primary endpoint of promoting cardioversion from atrial fibrillation to sinus rhythm within 90 minutes of drug administration.
Main
Atrial fibrillation (AF) is the most common cardiac arrhythmia and is associated with reduced quality of life and increased risk of stroke, heart failure and death1. Current treatment options for patients with AF episodes include pharmacological and electrical cardioversion as well as ‘wait and see’ approaches1–3. However, current pharmacological options for cardioversion have limited efficacy and a substantial risk of serious adverse effects, particularly an increased risk of causing potentially life-threatening ventricular arrhythmia, also known as proarrhythmia1,2. Consequently, use of these drugs are restricted in patients with left ventricular hypertrophy, ischemic heart disease and heart failure, resulting in barriers for prescribing the drugs and making many patients with AF ineligible for treatment. To avoid proarrhythmia, researchers have pursued drug targets with atrial but not ventricular effects, but have so far failed to demonstrate AF efficacy with these agents4–6.
The KCa2 ion (or SK) channel is a calcium-activated potassium channel that conducts a repolarizing current in the heart. It has the strongest association with AF in genome-wide association studies among genes encoding for ion channels7, with no association to the electrocardiogram (ECG) QT interval8. Inhibiting KCa2 results in prolongation of the atrial action potential duration in tissue from humans with AF9; KCa2 inhibitors have demonstrated efficacy in a range of animal models of AF without ventricular effects10–13. AP30663 is a new KCa2 inhibitor with demonstrated efficacy in animals14,15; it was well tolerated in a phase 1 trial, although with a finding of transient QTc prolongation (see Extended Data Fig. 1 for its chemical structure)16.
Extended Data Fig. 1. AP30663 chemical structure.
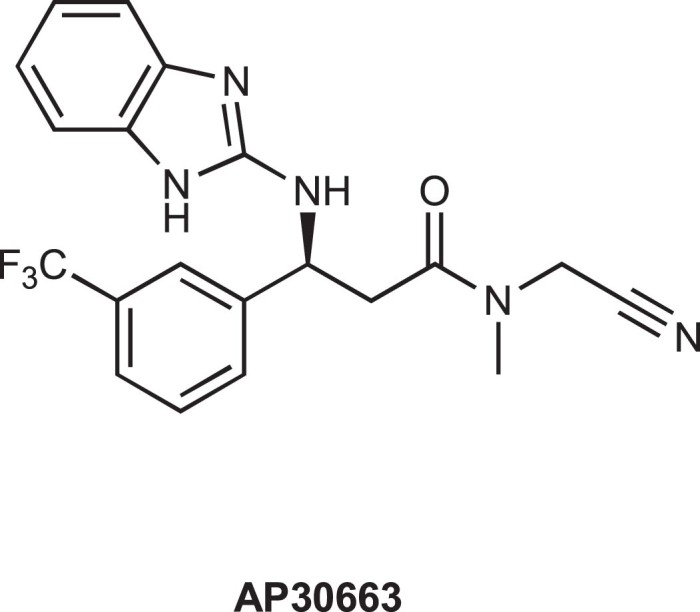
AP30663 chemical name: (3 R)-3-[(1H-1,3-benzodiazol-2-yl)amino]-N-(cyanomethyl)-N-methyl-3-[3-(trifluoromethyl)phenyl]propenamide.
The AF efficacy of KCa2 inhibition in humans has not previously been tested. In this phase 2 trial, we studied the cardioversion efficacy of a single intravenous infusion of AP30663 in patients with a recent-onset AF episode.
Results
Patient characteristics
We enrolled patients with a current episode of AF lasting 7 days or less and randomized them to receive an intravenous infusion of 3 or 5 mg kg−1 AP30663 or placebo. The trial was prematurely discontinued in December 2022 because of slow enrollment under the coronavirus disease 2019 pandemic. A total of 66 patients were randomized between 24 September 2019 and 9 December 2022. Of these, three patients did not receive the infusion and a further four were excluded from the full analysis set due to having atrial flutter instead of AF at randomization (n = 3) or undergoing direct current cardioversion within the 90 min during which the primary endpoint was assessed (n = 1). No patients discontinued the infusion and all patients receiving the infusion completed the 30 days follow-up visit (Fig. 1). The mean age in the three groups was between 64.3 and 65.5, 68.2–80.0% were male, 13.6–40.0% had heart disease and the mean AF episode duration was 59.9–88.0 h across groups, with a shorter mean duration in the placebo group (59.9 h) compared to the active groups (3 mg kg−1: 88.0; 5 mg kg−1: 87.9 h) (Table 1). No other meaningful differences were observed in baseline characteristics across the three groups.
Fig. 1. Patient flow diagram.

Figure shows patient flow through the trial and reasons for exclusion. The full analysis set was used for all efficacy analyses including for the primary endpoint.
Table 1.
Patient demographics and baseline characteristics in the safety analysis set
| Characteristic | Placebo (n = 26) | AP30663 3 mg kg−1 (n = 15) | AP30663 5 mg kg−1 (n = 22) |
|---|---|---|---|
| Age, years | 64.3 ± 9.23 | 65.4 ± 8.48 | 65.5 ± 10.38 |
| Male sex, n (%) | 18 (69.2) | 12 (80.0) | 15 (68.2) |
| Weight, kg | 88.6 ± 14.6 | 90.3 ± 11.9 | 85.4 ± 12.0 |
| Duration of current AF episode, h | 59.9 ± 43.0 | 88.0 ± 24.6 | 87.9 ± 45.5 |
| Prior diagnosis of AF, n (%) | 6 (23.1) | 5 (33.3) | 4 (18.2) |
| Time since first AF diagnosis, years | 4.3 ± 2.8 | 2.4 ± 2.7 | 6.7 ± 12.3 |
| Heart rate during AF, bpm | 98.0 ± 24.1 | 102.0 ± 28.7 | 93.9 ± 19.2 |
| ECG QTcF interval, ms | 408.0 ± 17.4 | 418.8 ± 27.2 | 409.9 ± 25.9 |
| Left ventricular ejection fraction, % | 60.3 ± 8.4 | 57.8 ± 9.7 | 57.3 ± 10.2 |
| Left atrial dimension and diameter (anterior–posterior, end systolic), mm | 47.5 ± 9.2 | 51.9 ± 9.5 | 45.2 ± 7.9 |
| Heart disease, n (%) | 6 (23.1) | 6 (40.0) | 3 (13.6) |
| Ischemic heart disease, n (%) | 4 (15.4) | 6 (40.0) | 2 (9.1) |
| Heart failure, n (%) | 1 (3.8) | 0 | 2 (9.1) |
| Valvular heart disease, n (%) | 1 (3.8) | 0 | 0 |
| Diabetes, n (%) | 8 (30.8) | 8 (53.3) | 4 (18.2) |
| Oral anticoagulant drug use, n (%) | 18 (69.2) | 11 (73.3) | 13 (59.1) |
| Rate control drug use, n (%)a | 20 (76.9) | 13 (86.7) | 9 (40.9) |
| Betaxolol, n (mean daily dose) | 0 | 1 (10 mg) | 1 (40 mg) |
| Bisoprolol, n (mean daily dose) | 13 (6 mg) | 5 (8 mg) | 6 (4 mg) |
| Carvedilol, n (mean daily dose) | 2 (38 mg) | 3 (83 mg) | 0 |
| Metoprolol, n (mean daily dose) | 4 (213 mg) | 3 (134 mg) | 0 |
| Nebivolol, n (mean daily dose) | 1 (10 mg) | 2 (5 mg) | 1 (5 mg) |
| Digoxin, n (mean daily dose) | 0 | 0 | 1 (500 µg) |
| Prior AF ablation, n (%) | 1 (3.8) | 1 (6.7) | 1 (4.5) |
aDefined as digoxin or beta-blocker use. There was no use of verapamil in the trial.
The ± values represent the mean ± s.d. unless otherwise indicated.
Efficacy endpoints
The primary endpoint of cardioversion within 90 min occurred in 42% (5 of 12) and 55% (12 of 22) of patients receiving AP30663 at 3 mg kg−1 and 5 mg kg−1, respectively, and in 0% (0 of 25) of patients receiving placebo (Fig. 2 and Table 2). The posterior probability of superiority to placebo was greater than 99.9% for both doses, thereby exceeding the prespecified level of 95% and confirming superiority for both doses versus placebo. Secondary endpoints all numerically favored the active treatment over placebo: mean time to cardioversion was 47 min (s.d. ± 23) for AP30663 3 mg kg−1 and 41 min (s.d. ± 24) for AP30663 5 mg kg−1. Patients who did not convert within the 90 min after the start of the infusion underwent a direct current cardioversion; a post hoc analysis of this showed 100% success in cardioversion to sinus rhythm for both AP30663 dose groups versus 88% (22 of 25) for those receiving placebo. All patients treated with AP30663 3 mg kg−1 and 5 mg kg−1 were in sinus rhythm 24 h after the start of the infusion, compared with 76% (19 of 25) of patients receiving placebo. One relapse of AF within 5 min after pharmacological or direct current cardioversion was seen, occurring after a direct current cardioversion in the placebo group.
Fig. 2. Primary endpoint of cardioversion.

a, Cardioversion rates in the full analysis set. The bar heights show the percentage of patients with cardioversion; the error bars show the 95% confidence intervals (CIs). AP30663 3 mg kg−1, n = 12 patients; AP30663 5 mg kg−1, n = 22 patients; placebo, n = 25 patients. b, Time to cardioversion in the full analysis set.
Table 2.
Efficacy endpoints in the full analysis set
| Placebo (n = 25) | AP30663 3 mg kg−1 (n = 12) | AP30663 5 mg kg−1 (n = 22) | |
|---|---|---|---|
| Primary endpoint | |||
| Cardioversion within 90 min, n (%) | 0/25 (0) | 5/12 (42) | 12/22 (55) |
| Within-arm 95% CI, % | 0–14% | 15–72% | 32–76% |
| Posterior probability of superiority versus placebo, % | NA | >99.9% | >99.9% |
| Secondary endpoints | |||
| Time to cardioversion, min ± s.d. | NA | 47 ± 23 | 41 ± 24 |
| P for pairwise comparison to placebo | NA | 0.001 | <0.0001 |
| Relapse of AF within 5 min after pharmacological or direct current cardioversion, n (%) | 1/25 (4.0) | 0 | 0 |
| Sinus rhythm 3 h after the start of the infusion, n (%) | 21/25 (84.0) | 11/11 (100) | 20/21 (95.2) |
| Sinus rhythm 24 h after the start of the infusion, n (%) | 19/25 (76) | 11/11 (100) | 21/21 (100) |
| Sinus rhythm 30 days after the start of the infusion, n (%) | 16/25 (64.0) | 9/10 (90.0) | 15/21 (71.4) |
| Patients with direct current cardioversion, n (%) | 25/25 (100) | 7/12 (58) | 10/22 (45) |
| Successful direct current cardioversion, n (%)a | 22/25 (88) | 7/7 (100%) | 10/10 (100) |
aPost hoc analysis.
The primary endpoint was analyzed with Bayesian statistics and the prior probability of success at a dose was modeled with a uniform Beta (1,1) prior. AP30663 was considered superior to placebo if the posterior probability was greater than 0.95. Time to cardioversion was analyzed using Kaplan–Meier curves and two-sided P values are reported for pairwise comparison to placebo. No adjustment for multiple testing was done. NA, not applicable.
None of the three individuals (one allocated to placebo and two allocated to AP30663 3 mg kg−1) who were excluded from the full analysis set because of having atrial flutter at randomization met the primary endpoint.
Safety endpoints
Adverse events were reported in 50% (13 of 26) of patients for the AP30663 5 mg kg−1 group, 27% (4 of 15) for AP30663 3 mg kg−1 and 50% (11 of 22) for placebo (Table 3). No deaths occurred and all four serious adverse events were observed in the placebo group. All four serious adverse events were recurrence of AF that led to hospitalization. Changes in systolic blood pressure were 1.2 mmHg (s.d. = 10.1) for the AP30663 5 mg kg−1 group, 3.4 mmHg (s.d. = 10.3) for AP30663 3 mg kg−1 and 1.7 mmHg (s.d. = 11.7) for placebo, all measured during the infusion. Changes in diastolic blood pressure were 2.2 mmHg (s.d. = 8.3) for the AP30663 5 mg kg−1 group, −2.7 mmHg (s.d. = 7.6) for the AP30663 3 mg kg−1 group and −0.5 mmHg (s.d. = 7.1) for the placebo group, all measured during the infusion (Extended Data Table 1).
Table 3.
Adverse events in the safety analysis set
| Event | Placebo (n = 26) | AP30663 3 mg kg−1 (n = 15) | AP30663 5 mg kg−1 (n = 22) |
|---|---|---|---|
| Adverse event | 13 (50.0) 19 | 4 (26.7) 6 | 11 (50.0) 19 |
| Serious adverse eventa | 4 (15.4) 4 | 0 | 0 |
| Leading to death | 0 | 0 | 0 |
| Adverse event leading to discontinuation of infusion | 0 | 0 | 0 |
| Adverse event according to organ class and preferred term | |||
| Cardiac disorders | 13 (50.0) 15 | 2 (13.3) 3 | 9 (40.9) 13 |
| AF | 11 (42.3) 12 | 1 (6.7) 1 | 7 (31.8) 7 |
| Atrial flutter | 1 (3.8) 1 | 0 | 3 (13.6) 3 |
| Atrioventricular block (first-degree) | 1 (3.8) 1 | 2 (13.3) 2 | 0 |
| Left bundle branch block | 0 | 0 | 1 (4.5) 1 |
| Right bundle branch block | 0 | 0 | 1 (4.5) 1 |
| Left ventricular failure | 0 | 0 | 1 (4.5) 1 |
| Supraventricular tachycardia | 1 (3.8) 1 | 0 | 0 |
| Vascular disorders | 2 (7.7) 2 | 2 (13.3) 2 | 1 (4.5) 1 |
| Hypotension | 0 | 1 (6.7) 1 | 1 (4.5) 1 |
| Phlebitis | 2 (7.7) 2 | 0 | 0 |
| Hypertension | 0 | 1 (6.7) 1 | 0 |
| Investigations | 0 | 1 (6.7) 1 | 1 (4.5) 1 |
| Increased blood bilirubin | 0 | 0 | 1 (4.5) 1 |
| Electrocardiogram prolonged QT | 0 | 1 (6.7) 1 | 0 |
| Metabolism and nutrition disorders | 1 (3.8) 1 | 0 | 1 (4.5) 1 |
| Diabetes mellitus | 0 | 0 | 1 (4.5) 1 |
| Hypokalemia | 1 (3.8) 1 | 0 | 0 |
| Renal and urinary disorders | 0 | 0 | 2 (9.1) 2 |
| Hematuria | 0 | 0 | 2 (9.1) 2 |
| Nervous system disorders | 0 | 0 | 1 (4.5) 1 |
| Postural dizziness | 0 | 0 | 1 (4.5) 1 |
| Psychiatric disorders | 1 (3.8) 1 | 0 | 0 |
| Insomnia | 1 (3.8) 1 | 0 | 0 |
All data are based on investigator-reported adverse events. Data reported are the number of patients (%) and number of events (n). aA serious adverse event was defined as death, a life-threatening episode, hospitalization or prolongation of existing hospitalization, a persistent or substantial disability or incapacity, or an event otherwise considered to be an important medical event.
Extended Data Table 1.
Changes in blood pressure in the safety analysis set

A mean decrease in heart rate was seen for all groups. This occurred earlier for the active groups compared to the placebo group, coinciding with the timing of conversions from AF to sinus rhythm (Extended Data Fig. 2).
Extended Data Fig. 2. Changes in heart rate in the safety analysis set.

The plots show least squares mean and 90% CI based on a linear mixed-effects model. AP30663 3 mg/kg n = 15 patients, AP30663 5 mg/kg n = 22 patients, Placebo n = 26 patients.
A transient difference in change in QTcF was observed (Fig. 3), with an estimated maximum least squares mean effect of +37.7 ms (s.e.m. = 3.5) at 45 min for the AP30663 5 mg kg−1 group, compared to +19.4 ms (s.e.m. = 4.3) for the 3 mg kg−1 group and −1.3 ms (s.e.m. = 3.21) for the placebo group (Extended Data Table 2). QTcF changes for all groups, including the placebo group, remained at more than 10 ms at 24 h. No other meaningful differences in ECG markers were observed.
Fig. 3. Change in QTcF from baseline in the safety analysis set.

Plot showing the least squares mean and 90% CIs based on a linear mixed-effects model at the indicated time points. AP30663 3 mg kg−1, n = 15 patients; AP30663 5 mg kg−1, n = 22 patients; placebo, n = 26 patients.
Extended Data Table 2.
Changes in ECG QTcF interval in the safety analysis set

*In protocol version 4.0, Holter monitoring was shortened from 24 to 8 hours post-infusion.
Based on a linear mixed-effects model
With Holter monitoring, the most important ventricular finding was episodes of nonsustained ventricular tachycardia seen in both the active and placebo groups with the longest episodes being four beats (Extended Data Table 3).
Extended Data Table 3.
Ventricular tachyarrhythmia on Holter in the safety analysis set

No apparent effect of AP30663 was observed on laboratory parameters (Extended Data Table 4).
Extended Data Table 4.
Changes in biochemistry parameters in the safety analysis set
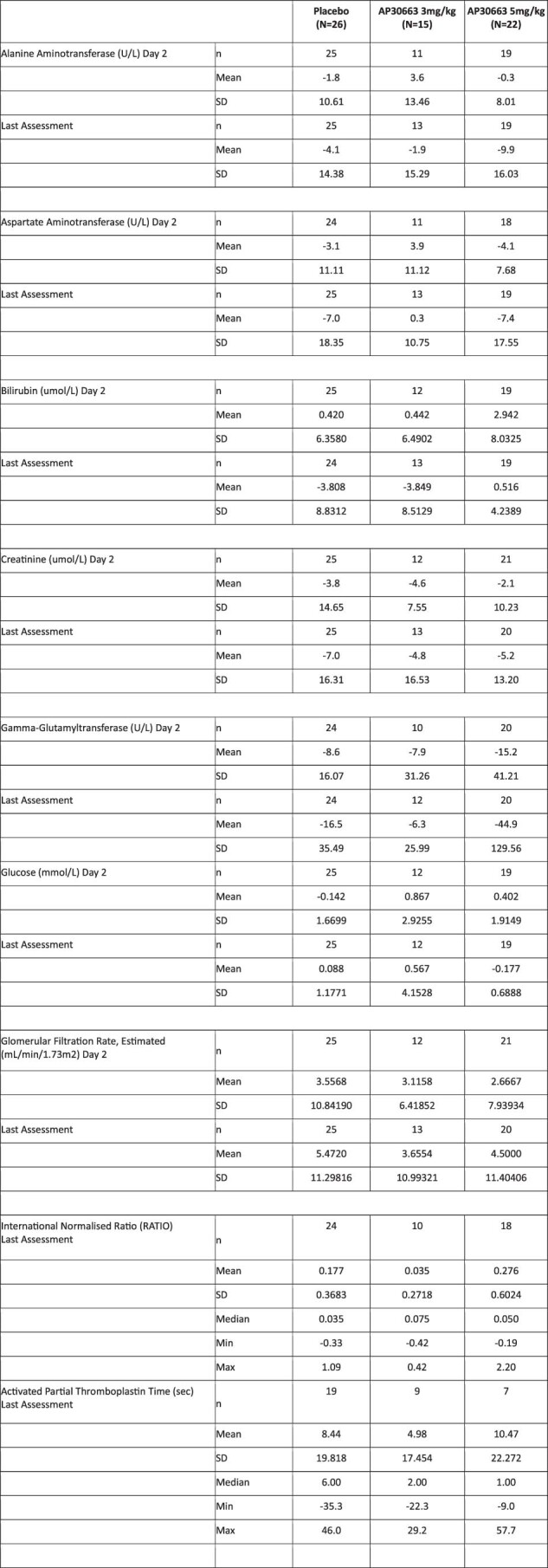
Last Assessment is Day 30 or early discontinuation visit.
Pharmacokinetic endpoint
The plasma concentration over time is shown in Extended Data Fig. 3.
Extended Data Fig. 3. AP30663 mean plasma concentration in the pharmacokinetic analysis set.

H: hours. Error bars show the standard deviation. AP30663 3 mg/kg n = 15 patients, AP30663 5 mg/kg n = 22 patients.
Discussion
In this phase 2 clinical trial, AP30663, a new KCa2 ion channel inhibitor, demonstrated efficacy in cardioverting AF to sinus rhythm compared to placebo in patients with a recent-onset AF episode. There was a numerical dose–response between the two doses tested and results from all secondary endpoints supported the efficacy of AP30663.
Among currently existing drugs for pharmacological AF cardioversion, vernakalant and flecainide are the most efficacious1. Most clinical trial data are available for vernakalant, which is a multichannel blocker that targets cardiac sodium channels and the atrium-specific potassium channel KV1.5, but not KCa2 (refs. 17,18). Vernakalant has demonstrated superior efficacy to ibutilide and amiodarone in randomized trials19,20. In a meta-analysis that included placebo-controlled trials enrolling a very similar population to ours, vernakalant had a 48% cardioversion rate within 90 min versus 6% with placebo, resulting in a 42% placebo-adjusted cardioversion rate21. In our trial, AP30663, at the highest dose tested, demonstrated a 55% placebo-adjusted cardioversion rate, supporting a competitive efficacy of AP30663.
The clinical efficacy of AP30663 aligns with the effects observed in animal models, where AP30663 showed a pronounced effect on the atrial effective refractory period in one pig model, and successfully cardioverted AF and prevented its reinduction in another pig model where vernakalant did not cardiovert any longer14. Other KCa2 inhibitors have shown similar AF antiarrhythmic effects in rats, guinea pigs, rabbits, pigs, dogs, goats and horses; however, none of these have been tested in humans10–13,22.
AP30663 inhibits the KCa2 channel through negative allosteric modulation that decreases the calcium sensitivity of the channel. A recent study found that the KCa2 current is upregulated and is the dominant repolarizing atrial current in patients with AF because of increased Ca2+ sensitivity and increased trafficking of KCa2 to the cell membrane9. This upregulation of the KCa2 current in patients with AF contributes to the action potential shortening characteristic seen in AF; the AP30663 mechanism of action can be hypothesized to directly counteract this effect.
The safety profile of AP30663 was consistent with results from the phase 1 trial, with the exception that infusion site reactions were reported in the phase 1 trial whereas none were reported in the current trial16. Inhibition of the KCa2 channel in animal studies has been associated with central nervous system adverse effects in the form of tremors and ataxia at high doses; this was also seen with AP30663 in the safety animal studies16. However, these effects have not been observed with AP30663 in any of the clinical trials, including the current one. Although AP30663 caused a transient increase in the QTcF interval, no clinically relevant ventricular arrhythmias were observed in any treatment group. The increase in QTcF remained above 10 ms for all groups including the placebo group at 24 h, highlighting the difficulties in QTc assessment when comparing baseline values assessed while patients are in AF with post-baseline values assessed once patients converted to sinus rhythm, with resulting changes in heart rate. These findings suggest that the maximum increase in QTcF seen at 45 min is probably overestimated for the active groups because some patients had converted to sinus rhythm at this time point. In genome-wide association studies, no association between the genes encoding for the KCa2 channels and the QTc interval has been found8. Additionally, while AP30663 potently inhibits KCa2, it also inhibits the Kv11.1 channel (KCNH2 or hERG gene) to a lesser degree16. Kv11.1 inhibition is the most common cause of drug-related QTc prolongation23; collectively, these findings suggest that the QTc increase is caused by an off-target inhibition of the Kv11.1 channel.
Existing drugs with different mechanisms of action, when effective in AF cardioversion, also show efficacy in sinus rhythm maintenance (prevention of AF recurrence)1,2,24–26. Our demonstration of human cardioversion efficacy through inhibition of KCa2 suggests that this may also be a promising drug target for maintaining sinus rhythm.
This trial has some limitations. We enrolled patients with an AF episode lasting less than 7 days. Other drugs have shown decreased efficacy with longer AF durations27–29, and efficacy should not be generalized to patients with longer AF episodes. The trial had a limited sample size. The early termination of the trial because of slow enrollment, which prevented testing doses higher than 5 mg kg−1. Underrepresentation of female patients in the trial was another limitation. Collectively, the results of this phase 2 trial should be considered hypothesis-generating; the efficacy and safety, in particular the potential consequences of QT prolongation, need to be studied in a larger phase 3 trial.
In conclusion, the KCa2 inhibitor AP30663 demonstrated superior AF cardioversion efficacy compared with placebo in patients with recent-onset AF episodes. AP30663 caused a transient increase of the QTc interval, but no ventricular arrhythmias were observed. KCa2 inhibition may be an attractive pathway for rhythm control of AF and should be studied in future randomized trials.
Methods
Trial design
We conducted a phase 2 randomized, double-blind, placebo-controlled, parallel-group trial with an adaptive design with the potential to test doses between 2 and 6 mg kg−1. An independent data monitoring committee reviewed unblinded safety and efficacy data during the trial and was responsible for providing recommendations to the sponsor regarding dose changes according to the adaptive design (see the ‘Statistical analyses’ section of the Methods). See Supplementary Note 1 for a list of data monitoring committee members.
The maximum number of individuals that could be enrolled based on the adaptive design was 108. Patients wore 12-lead Holter monitors to assess both efficacy and safety for at least 8 h after the infusion; the collected data were analyzed by a core laboratory for arrhythmias and semiautomated measurement of ECG intervals based on triplicate ECG extracts at prespecified time points. Patients were followed until day 30 after receiving the infusion. Please refer to the protocol in Supplementary Note 2 for further information.
In December 2022, the sponsor decided to stop the trial early because of slow enrollment, mainly due to the coronavirus disease 2019 pandemic, at a time when the sample size in the 5 mg kg−1 dose allowed for sufficient statistical power to evaluate this dose. At that point in the adaptive design, subsequent interim analysis could have allowed testing of a 4 or 6 mg kg−1 dose.
The trial protocol was approved by the following ethics committees: Den Videnskabsetiske Komité, Region Sjælland, Denmark and Egészségügyi Tudományos Tanács Klinikai Farmakológiai Etikai Bizottsága, Budapest, Hungary.
All participants provided written informed consent. The trial was sponsored and funded by Acesion Pharma.
ClinicalTrials.gov registration: NCT04571385. EudraCT registration: 2018-004445-17.
Trial population
Male and female patients aged 18–80 years with a body weight of 50–110 kg and a current episode of symptomatic AF lasting between 3 h and 7 days were deemed eligible. Patients were required to be treated with anticoagulation according to current guidelines. Patients were allowed to have stable ischemic heart disease and heart failure (New York Heart Association classes I and II, left ventricular ejection fraction 40% or higher). Exclusion criteria included recent cardioversion, current or recent use of antiarrhythmic drug classes I or III (including amiodarone), QTcF greater than 450 ms or previous torsade de pointes episodes. The complete list of inclusion and exclusion criteria can be found in the next sections.
Inclusion criteria
The inclusion criteria were: provision of written informed consent; clinical indication for cardioversion of AF; current episode of symptomatic AF lasting between 3 h and 7 days inclusive at randomization; adequate anticoagulation according to international or national guidelines; body weight 50–110 kg inclusive (with clothes, without shoes); and male patients and postmenopausal women aged 18–80 years inclusive.
Exclusion criteria
The exclusion criteria were: significant clinical illness or surgical procedure within 4 weeks before the screening visit; present renal dysfunction (estimated glomerular filtration rate less than 30 ml min−1), hepatic dysfunction (alanine aminotransferase or aspartate aminotransferase higher than three times the upper limit of normal) or uncontrolled hyperthyroidism or hypothyroidism; a history of significant mental, renal or hepatic disorder, chronic obstructive pulmonary disease or other significant disease, as judged by the investigator; any cardioversion attempt of AF or atrial flutter within 1 week before randomization. Previous failed attempt (no conversion) of pharmacological or direct current cardioversion of previous or current AF episode; failure to find a large antecubital (or equivalent) vein for the infusion; any of the following events, or any other significant cardiovascular event as judged by the investigator, during the last 6 weeks before randomization: myocardial infarction, unstable angina pectoris or other signs of myocardial ischemia, stroke or transient ischemic attack, myocardial revascularization (percutaneous coronary intervention, coronary artery bypass graft) or other revascularization procedure; hemodynamically unstable condition as judged by the investigator; systolic blood pressure lower than 90 mmHg or higher than 180 mmHg, or diastolic blood pressure higher than 105 mmHg at randomization; blood hemoglobin lower than 100 g l−1 at screening; congestive heart failure according to New York Heart Association class III or IV; left ventricular ejection fraction lower than 40% on echocardiography or other clinically significant abnormality on the echocardiogram (not older than 6 months), as judged by the investigator; known hypertrophic cardiomyopathy or significant left ventricular hypertrophy (free wall or septal thickness greater than 13 mm); any clinically significant valvular heart disease; a history or previous signs of sinus nodal disease; pacemaker or implantable cardioverter defibrillator therapy; a personal or family history of torsades de pointes, any other polymorphic ventricular tachycardia, sustained ventricular tachycardia, long QT syndrome or Brugada syndrome; QTc (QTcF) interval greater than 450 ms at randomization. When measured during AF, the mean heart rate should be 50–100 bpm. The QTcF should be calculated at AF as the mean of at least five consecutive RR intervals with consecutive QT intervals; QRS complex duration longer than 120 ms at randomization; known atrioventricular (AV) block I (prolonged PQ interval longer than 220 ms), AV block II, AV block III or complete bundle branch block; potassium in serum below 3.5 or above 5.3 mmol l−1 at randomization. Patients with low potassium levels at screening may be appropriately supplemented with potassium before baseline, according to local standards. Retesting of the potassium level is required and the patient can be randomized after potassium has returned to the reference range; anticipated change in dose or initiation of loop diuretic from screening to the end of the infusion; use of any antiarrhythmic drug class I or III within 7 days or, for amiodarone specifically, 12 weeks before randomization; use of QT-prolonging drug or drug that inhibits cytochrome P450 3A4, as well as St John’s wort within 10 days before randomization; administration of an investigational drug within the preceding 3 months before randomization; administration of AP30663 at any time before randomization; a history of drug addiction or alcohol abuse within the last 12 months, at the discretion of the investigator; blood or plasma donation within the preceding 4 weeks before randomization; any suspected or manifested clinically significant infection, as judged by the investigator; involvement in the planning and conduct of the study (applies to Acesion Pharma staff, Syneos Health staff and staff at the investigational site); clinical judgment by the investigator that the patient should not participate in the study; any malignant cancer within 3 years (except for successfully treated in situ nonmelanoma skin cancer and in situ cervical cancer) of signing the informed consent form.
The trial was conducted at ten sites in Hungary and Denmark.
Trial intervention
The trial ended up testing 3 mg kg−1 and 5 mg kg−1 doses of AP30663 versus placebo; participants were randomized to AP30663 or placebo in a 1:1 ratio for part 1 of the trial testing the 3 mg kg−1 dose and subsequently in a 2:1 ratio. Randomization was performed using a computer-generated random sequence and interactive voice and Web response system. The intravenous infusion was prepared by an unblinded team and handed over to a blinded team for administration over 30 min. Use of other antiarrhythmic class I or III drugs (including amiodarone) was prohibited until the day after receiving the infusion.
If patients did not cardiovert within the 90 min after the start of the infusion, they were to undergo an electrical (direct current) cardioversion.
Trial endpoints
The primary endpoint was the proportion of patients converting from AF to sinus rhythm within 90 min from the start of the infusion and who subsequently had no AF recurrence within 1 min of conversion. Key secondary efficacy endpoints included time to conversion from AF, the proportion of patients with relapse of AF within 5 min after pharmacological (AP30663) or direct current cardioversion and the proportion of patients in sinus rhythm at 3 and 24 h after the start of the infusion. Key secondary safety endpoints included adverse events and change in QT interval corrected using the Fridericia formula (QTcF). Pharmacokinetics were also assessed as a secondary endpoint.
Exploratory endpoints addressing the potential for treatment effect interactions with baseline variables and pharmacokinetic parameters were specified in the protocol; however, due to the limited sample size, these analyses were severely underpowered and are not reported in this article.
Statistical analyses
To ensure an efficient adaptive trial design, Bayesian statistics were used. In part 1 of the trial, a dose of 3 mg kg−1 was tested; if deemed safe, a dose of 5 mg kg−1 as well as potentially a dose of 2 mg kg−1 were to be tested based on the results from an interim analysis. Dose testing according to the adaptive trial design was guided by achieving an AF cardioversion rate greater than 0.65 at the interim analysis; if the posterior probability of this was 0.90 or greater, the current dose would be closed for sufficient efficacy and a dose one level below would be opened, if available. If a posterior probability of this was less than 0.10, the current dose would be closed and a higher dose opened, if available. Interim analyses were done after enrolling 32 individuals in the 3 mg kg−1 dose, which led to further testing of the 5 mg kg−1 dose only. An interim analysis after enrolling 18 individuals in the 5 mg kg−1 dose did not result in dose change. The prior probability of success at a dose was modeled with a uniform Beta (1,1) prior; the posterior distribution was modeled for each dose independently using a Beta posterior distribution. This method was also applied to the final analysis of the primary endpoint because the primary analysis and each AP30663 dose were considered superior to placebo if the posterior probability was greater than 0.95. Time to conversion was analyzed using Kaplan–Meier curves. No type I error rate control was implemented across the interim analyses or at the final analysis because this was a phase 2 non-confirmatory trial. In all analyses, individuals given placebo were pooled.
ECG parameters were analyzed based on a linear mixed-effects model; a least squares mean with 90% CIs was reported for these.
The safety analysis set was prespecified to be used for all safety analyses and consisted of all randomized participants who were administered double-blind trial treatment. The full analysis set was prespecified to be the primary one for all efficacy analyses and consisted of all randomized participants who were administered double-blind trial treatment and had an evaluable AF conversion status within 90 min from the start of the infusion.
The sample size was selected based on a similar previous trial30; no formal sample size calculations were made. Please refer to the Statistical Analysis Plan in Supplementary Note 3 for further information.
Data analyses were done by the contract research organization Syneos Health and by the sponsor. All authors had access to the data. SAS v.9.4 or higher and R v.3.5.2 or higher were used for the analyses. Data were collected through the electronic data capture system Medidata Classic Rave (2019–2023).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41591-023-02679-9.
Supplementary information
Supplementary Notes 1–3. Supplementary Note 1 contains the list of investigators and DMC members. Supplementary Note 2 contains the protocol. Supplementary Note 3 contains the SAP.
Acknowledgements
We thank the patients who participated in the trial and the site investigators and staff. Grant funding for the present study was received from Wellcome Trust (Translational Award, 200450/Z/16/Z) and Innovation Fund Denmark (8064-00074B).
Extended data
Author contributions
N.E., M.G., U.S.S., J.G.D. and B.H.B. designed the trial. B.V. and A.G.H. amended the trial design based on input from J.T., among others. P.D., S.H.H. and D.L.B. served on the data monitoring committee. A.G.H. and B.V. monitored the trial for the sponsor while in the conduct phase. A.G.H. performed the supplementary statistical analyses. A.G.H. wrote the first draft of the paper and all authors revised it. All authors vouch for the accuracy and completeness of the reported data.
Peer review
Peer review information
Nature Medicine thanks Jonathan Piccini and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Michael Basson, in collaboration with the Nature Medicine team.
Data availability
Data originating from the trial are considered commercially sensitive; as such, they are not publicly available. To the extent that current legislation allows it, the authors will provide access to individual deidentified participant-level data that underlie the data presented in this article to researchers who provide a methodologically sound proposal for academic purposes to interpret, verify and extend research in the article that does not violate intellectual property or confidentiality obligations, beginning 12 months after article publication. Researchers should contact the corresponding author when applying for data access. Use of data will be restricted to the agreed purpose.
Competing interests
A.G.H., B.V., U.S.S., J.G.D., M.G. and B.H.B. are employees of Acesion Pharma and hold shares or warrants in the company. N.E. is a consultant to Acesion Pharma. P.D. received consulting fees and honoraria from Acesion Pharma. D.L.B. has served on the Advisory Boards of Angiowave, Bayer, Boehringer Ingelheim, Cardax, CellProthera, Cereno Scientific, Elsevier Practice Update Cardiology, High Enroll, Janssen, Level Ex, McKinsey, Medscape Cardiology, Merck, MyoKardia, NirvaMed, Novo Nordisk, PhaseBio, PLx Pharma, Regado Biosciences and Stasys. He has served on the Board of Directors of Angiowave (stock options), Boston VA Research Institute, Bristol Myers Squibb (stock), DRS.LINQ (stock options), High Enroll (stock), the Society of Cardiovascular Patient Care and TobeSoft. He has been the Inaugural Chair of the American Heart Association Quality Oversight Committee. He has been a consultant for Broadview Ventures and Hims. He has served on the Data Monitoring Committees of Acesion Pharma, Assistance Publique-Hôpitaux de Paris, the Baim Institute for Clinical Research (formerly the Harvard Clinical Research Institute, for the PORTICO trial, funded by St. Jude Medical, now Abbott), Boston Scientific (Chair, PEITHO trial), the Cleveland Clinic (including for the ExCEED trial, funded by Edwards), Contego Medical (Chair, PERFORMANCE 2), the Duke Clinical Research Institute, the Mayo Clinic, the Mount Sinai School of Medicine (for the ENVISAGE trial, funded by Daiichi Sankyo, and for the ABILITY-DM trial, funded by Concept Medical), Novartis, Population Health Research Institute and Rutgers University (for the National Institutes of Health-funded MINT trial). He has received honoraria from the American College of Cardiology (Senior Associate Editor, Clinical Trials and News, ACC.org; Chair, ACC Accreditation Oversight Committee), Arnold and Porter law firm (work related to Sanofi and Bristol Myers Squibb clopidogrel litigation), Baim Institute for Clinical Research (formerly the Harvard Clinical Research Institute; RE-DUAL PCI clinical trial steering committee funded by Boehringer Ingelheim; AEGIS-II executive committee funded by CSL Behring), Belvoir Publications (Editor in Chief, Harvard Heart Letter), Canadian Medical and Surgical Knowledge Translation Research Group (clinical trial steering committees), CSL Behring (AHA lecture), Cowen and Company, Duke Clinical Research Institute (clinical trial steering committees, including for the PRONOUNCE trial, funded by Ferring Pharmaceuticals), HMP Global (Editor in Chief, Journal of Invasive Cardiology), Journal of the American College of Cardiology (Guest Editor; Associate Editor), K2P (Co-chair, interdisciplinary curriculum), Level Ex, Medtelligence/ReachMD (CME steering committee), MJH Life Sciences, Oakstone CME (Course Director, Comprehensive Review of Interventional Cardiology), Piper Sandler, Population Health Research Institute (for the COMPASS operations committee, publications committee, steering committee and USA national co-leader, funded by Bayer), Slack Publications (Chief Medical Editor, Cardiology Today’s Intervention), Society of Cardiovascular Patient Care (Secretary and Treasurer), WebMD (CME steering committee), Wiley (steering committee), Clinical Cardiology (Deputy Editor), NCDR-ACTION Registry Steering Committee (Chair) and VA CART Research and Publications Committee (Chair). He is named on a patent for sotagliflozin (assigned to the Brigham and Women’s Hospital who assigned it to Lexicon; neither P.D. nor the Brigham and Women’s Hospital receive any income from this patent). He has received research funding from Abbott, Acesion Pharma, Afimmune, Aker Biomarine, Alnylam, Amarin, Amgen, AstraZeneca, Bayer, Beren, Boehringer Ingelheim, Boston Scientific, Bristol Myers Squibb, Cardax, CellProthera, Cereno Scientific, Chiesi, CinCor, Cleerly, CSL Behring, Eisai, Ethicon, Faraday Pharmaceuticals, Ferring Pharmaceuticals, Forest Laboratories, Fractyl, Garmin, HLS Therapeutics, Idorsia, Ironwood, Ischemix, Janssen, Javelin, Lexicon, Lilly, Medtronic, Merck, Moderna, MyoKardia, NirvaMed, Novartis, Novo Nordisk, Otsuka, Owkin, Pfizer, PhaseBio, PLx Pharma, Recardio, Regeneron, the Reid Hoffman Foundation, Roche, Sanofi, Stasys, Synaptic, The Medicines Company, Youngene and 89Bio. He has received royalties from Elsevier (Editor, Braunwald’s Heart Disease). He has been a site co-investigator for Abbott, Biotronik, Boston Scientific, CSI, Endotronix, St. Jude Medical (now Abbott), Philips, SpectraWAVE, Svelte and Vascular Solutions. He is a trustee of the American College of Cardiology. He has carried out unfunded research for FlowCo. S.H.H. and J.T. declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
is available for this paper at 10.1038/s41591-023-02679-9.
Supplementary information
The online version contains supplementary material available at 10.1038/s41591-023-02679-9.
References
- 1.Hindricks G, et al. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC). Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021;42:373–498. doi: 10.1093/eurheartj/ehaa612. [DOI] [PubMed] [Google Scholar]
- 2.January CT, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130:e199–e267. doi: 10.1161/CIR.0000000000000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pluymaekers NAHA, et al. Early or delayed cardioversion in recent-onset atrial fibrillation. N. Engl. J. Med. 2019;380:1499–1508. doi: 10.1056/NEJMoa1900353. [DOI] [PubMed] [Google Scholar]
- 4.Shunmugam SR, et al. A double-blind, randomised, placebo-controlled, cross-over study assessing the use of XEN-D0103 in patients with paroxysmal atrial fibrillation and implanted pacemakers allowing continuous beat-to-beat monitoring of drug efficacy. J. Interv. Card. Electrophysiol. 2018;51:191–197. doi: 10.1007/s10840-018-0318-2. [DOI] [PubMed] [Google Scholar]
- 5.Podd SJ, Freemantle N, Furniss SS, Sulke N. First clinical trial of specific IKACh blocker shows no reduction in atrial fibrillation burden in patients with paroxysmal atrial fibrillation: pacemaker assessment of BMS 914392 in patients with paroxysmal atrial fibrillation. Europace. 2016;18:340–346. doi: 10.1093/europace/euv263. [DOI] [PubMed] [Google Scholar]
- 6.Walfridsson H, et al. Is the acetylcholine-regulated inwardly rectifying potassium current a viable antiarrhythmic target? Translational discrepancies of AZD2927 and A7071 in dogs and humans. Europace. 2015;17:473–482. doi: 10.1093/europace/euu192. [DOI] [PubMed] [Google Scholar]
- 7.Miyazawa K, et al. Cross-ancestry genome-wide analysis of atrial fibrillation unveils disease biology and enables cardioembolic risk prediction. Nat. Genet. 2023;55:187–197. doi: 10.1038/s41588-022-01284-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sollis E, et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 2023;51:D977–D985. doi: 10.1093/nar/gkac1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heijman J, et al. Enhanced Ca2+-dependent SK-channel gating and membrane trafficking in human atrial fibrillation. Circ. Res. 2023;132:e116–e133. doi: 10.1161/CIRCRESAHA.122.321858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Diness JG, et al. Inhibition of small-conductance Ca2+-activated K+ channels terminates and protects against atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2010;3:380–390. doi: 10.1161/CIRCEP.110.957407. [DOI] [PubMed] [Google Scholar]
- 11.Qi X-Y, et al. Role of small-conductance calcium-activated potassium channels in atrial electrophysiology and fibrillation in the dog. Circulation. 2014;129:430–440. doi: 10.1161/CIRCULATIONAHA.113.003019. [DOI] [PubMed] [Google Scholar]
- 12.Haugaard MM, et al. Pharmacologic inhibition of small-conductance calcium-activated potassium (SK) channels by NS8593 reveals atrial antiarrhythmic potential in horses. Heart Rhythm. 2015;12:825–835. doi: 10.1016/j.hrthm.2014.12.028. [DOI] [PubMed] [Google Scholar]
- 13.Gatta G, et al. Effective termination of atrial fibrillation by SK channel inhibition is associated with a sudden organization of fibrillatory conduction. Europace. 2021;23:1847–1859. doi: 10.1093/europace/euab125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diness JG, et al. The KCa2 channel inhibitor AP30663 selectively increases atrial refractoriness, converts vernakalant-resistant atrial fibrillation and prevents its reinduction in conscious pigs. Front. Pharmacol. 2020;11:159. doi: 10.3389/fphar.2020.00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bentzen BH, et al. Mechanisms of action of the KCa2-negative modulator AP30663, a novel compound in development for treatment of atrial fibrillation in man. Front. Pharmacol. 2020;11:610. doi: 10.3389/fphar.2020.00610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gal P, et al. First clinical study with AP30663—a KCa2 channel inhibitor in development for conversion of atrial fibrillation. Clin. Transl. Sci. 2020;13:1336–1344. doi: 10.1111/cts.12835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fedida D. Vernakalant (RSD1235): a novel, atrial-selective antifibrillatory agent. Expert Opin. Investig. Drugs. 2007;16:519–532. doi: 10.1517/13543784.16.4.519. [DOI] [PubMed] [Google Scholar]
- 18.Simó-Vicens R, Sauter DRP, Grunnet M, Diness JG, Bentzen BH. Effect of antiarrhythmic drugs on small conductance calcium-activated potassium channels. Eur. J. Pharmacol. 2017;803:118–123. doi: 10.1016/j.ejphar.2017.03.039. [DOI] [PubMed] [Google Scholar]
- 19.Simon A, et al. Vernakalant is superior to ibutilide for achieving sinus rhythm in patients with recent-onset atrial fibrillation: a randomized controlled trial at the emergency department. Europace. 2017;19:233–240. doi: 10.1093/europace/euw052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Camm AJ, et al. A randomized active-controlled study comparing the efficacy and safety of vernakalant to amiodarone in recent-onset atrial fibrillation. J. Am. Coll. Cardiol. 2011;57:313–321. doi: 10.1016/j.jacc.2010.07.046. [DOI] [PubMed] [Google Scholar]
- 21.McIntyre WF, et al. Vernakalant for cardioversion of recent-onset atrial fibrillation: a systematic review and meta-analysis. Europace. 2019;21:1159–1166. doi: 10.1093/europace/euz175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diness JG, et al. Termination of vernakalant-resistant atrial fibrillation by inhibition of small-conductance Ca2+-activated K+ channels in pigs. Circ. Arrhythm. Electrophysiol. 2017;10:e005125. doi: 10.1161/CIRCEP.117.005125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tisdale JE, et al. Drug-induced arrhythmias: a scientific statement from the American Heart Association. Circulation. 2020;142:e214–e233.. doi: 10.1161/CIR.0000000000000905. [DOI] [PubMed] [Google Scholar]
- 24.Falk RH, Pollak A, Singh SN, Friedrich T. Intravenous dofetilide, a class III antiarrhythmic agent, for the termination of sustained atrial fibrillation or flutter. J. Am. Coll. Cardiol. 1997;29:385–390. doi: 10.1016/S0735-1097(96)00506-2. [DOI] [PubMed] [Google Scholar]
- 25.Reisinger J, et al. Prospective comparison of flecainide versus sotalol for immediate cardioversion of atrial fibrillation. Am. J. Cardiol. 1998;81:1450–1454. doi: 10.1016/S0002-9149(98)00223-9. [DOI] [PubMed] [Google Scholar]
- 26.Torp-Pedersen C, et al. A randomized, placebo-controlled study of vernakalant (oral) for the prevention of atrial fibrillation recurrence after cardioversion. Circ. Arrhythm. Electrophysiol. 2011;4:637–643. doi: 10.1161/CIRCEP.111.962340. [DOI] [PubMed] [Google Scholar]
- 27.Stambler BS, et al. Efficacy and safety of repeated intravenous doses of ibutilide for rapid conversion of atrial flutter or fibrillation. Circulation. 1996;94:1613–1621. doi: 10.1161/01.CIR.94.7.1613. [DOI] [PubMed] [Google Scholar]
- 28.Pratt CM, et al. Usefulness of vernakalant hydrochloride injection for rapid conversion of atrial fibrillation. Am. J. Cardiol. 2010;106:1277–1283. doi: 10.1016/j.amjcard.2010.06.054. [DOI] [PubMed] [Google Scholar]
- 29.Roy D, et al. Vernakalant hydrochloride for rapid conversion of atrial fibrillation: a phase 3, randomized, placebo-controlled trial. Circulation. 2008;117:1518–1525. doi: 10.1161/CIRCULATIONAHA.107.723866. [DOI] [PubMed] [Google Scholar]
- 30.Roy D, et al. A randomized, controlled trial of RSD1235, a novel anti-arrhythmic agent, in the treatment of recent onset atrial fibrillation. J. Am. Coll. Cardiol. 2004;44:2355–2361. doi: 10.1016/j.jacc.2004.09.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Notes 1–3. Supplementary Note 1 contains the list of investigators and DMC members. Supplementary Note 2 contains the protocol. Supplementary Note 3 contains the SAP.
Data Availability Statement
Data originating from the trial are considered commercially sensitive; as such, they are not publicly available. To the extent that current legislation allows it, the authors will provide access to individual deidentified participant-level data that underlie the data presented in this article to researchers who provide a methodologically sound proposal for academic purposes to interpret, verify and extend research in the article that does not violate intellectual property or confidentiality obligations, beginning 12 months after article publication. Researchers should contact the corresponding author when applying for data access. Use of data will be restricted to the agreed purpose.