

Abstract
Background
Leber’s hereditary optic neuropathy (LHON) is a mitochondrial disorder characterised by complex I defect leading to sudden degeneration of retinal ganglion cells. Although typically associated with pathogenic variants in mitochondrial DNA, LHON was recently described in patients carrying biallelic variants in nuclear genes DNAJC30, NDUFS2 and MCAT. MCAT is part of mitochondrial fatty acid synthesis (mtFAS), as also MECR, the mitochondrial trans-2-enoyl-CoA reductase. MECR mutations lead to a recessive childhood-onset syndromic disorder with dystonia, optic atrophy and basal ganglia abnormalities.
Methods
We studied through whole exome sequencing two sisters affected by sudden and painless visual loss at young age, with partial recovery and persistent central scotoma. We modelled the candidate variant in yeast and studied mitochondrial dysfunction in yeast and fibroblasts. We tested protein lipoylation and cell response to oxidative stress in yeast.
Results
Both sisters carried a homozygous pathogenic variant in MECR (p.Arg258Trp). In yeast, the MECR-R258W mutant showed an impaired oxidative growth, 30% reduction in oxygen consumption rate and 80% decrease in protein levels, pointing to structure destabilisation. Fibroblasts confirmed the reduced amount of MECR protein, but failed to reproduce the OXPHOS defect. Respiratory complexes assembly was normal. Finally, the yeast mutant lacked lipoylation of key metabolic enzymes and was more sensitive to H2O2 treatment. Lipoic Acid supplementation partially rescued the growth defect.
Conclusion
We report the first family with homozygous MECR variant causing an LHON-like optic neuropathy, which pairs the recent MCAT findings, reinforcing the impairment of mtFAS as novel pathogenic mechanism in LHON.
Keywords: Genetics, Medical; Neuromuscular Diseases; Ophthalmology
WHAT IS ALREADY KNOWN ON THIS TOPIC
Leber’s hereditary optic neuropathy (LHON) is a rare disease due to genetic defects affecting mitochondrial complex I function, most typically involving the mitochondrial genome but rarely also autosomal recessive. Recently, a single case was reported with MCAT variants and LHON-like phenotype, linking the mitochondrial fatty acid synthesis (mtFAS) pathways to LHON pathogenesis.
WHAT THIS STUDY ADDS
This study provides evidence that MECR is a new gene for autosomal recessive LHON, further supporting mtFAS as a novel pathway relevant for LHON and lipoic acid may be beneficial.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Further investigations are necessary to well characterise the novel link between mtFAS and LHON. However, MECR should be added to the molecular diagnostic pathway for LHON.
Background
Leber hereditary optic neuropathy (LHON (MIM: 535 000)) is a mitochondrial optic neuropathy characterised by sudden degeneration of retinal ganglion cells leading to subacute bilateral loss of central vision, with limited occurrence of spontaneous recovery.1 The disease typically shows incomplete penetrance, male prevalence and may be triggered by tobacco smoking.1
LHON is the most common disease caused by mtDNA pathogenic variants, with a minimum prevalence reaching 3.22 per 100 000.2 Three variants in respiratory complex I subunits are found in about 90% of pedigrees, m.3460G>A/MT-ND1, m.11778G>A/MT-ND4 and m.14484T>C/MT-ND6,1 while several rare mtDNA point mutations or combinations of rare polymorphisms account for some of the remaining 10%.3
Recently, an LHON-like phenotype with autosomal recessive inheritance has been associated with pathogenic variants in nuclear genes.4 The most relevant is DNAJC30, encoding a newly characterised mitochondrial chaperone that promotes complex I turnover.5 Several DNAJC30 pedigrees with a few pathogenic variants are described, all presenting with a mostly mild LHON phenotype (LHONA, MIM #619 382), except for a few cases with Leigh syndrome.6 A single case of isolated recessive LHON was also reported with compound heterozygous variants in NDUFS2 gene,7 while biallelic variants in this core subunit of complex I are usually associated with syndromic CI deficiency (MC1DN, MIM #618 228).8 Biallelic variants in another subunit of complex I, NDUFA12, were also recently associated with a phenotypic spectrum ranging from movement disorders (dystonia and/or spasticity) to isolated optic atrophy, besides a more typical presentation of Leigh syndrome.9 An evocative acute/subacute vision loss was reported, however, the ophthalmological description of these NDUFA12 optic atrophy cases was not detailed enough to draw firm conclusions about similarities with LHON. Finally, an LHON-like phenotype has been reported in one pedigree with pathogenic variants in MCAT, encoding the mitochondrial malonyl-CoA-acyl carrier protein transacylase.10 This is a key enzyme in mitochondrial fatty acid synthesis (mtFAS), which transfers malonyl-CoA to the mitochondrial acyl carrier protein NDUFAB1. Interestingly, the previous cases with homozygous variants in MCAT also presented with recessive optic atrophy, but with a progressive course, not obeying the LHON hallmark of subacute onset of visual loss.11
The MECR gene encodes the mitochondrial trans-2-enoyl-CoA reductase enzyme, also involved in mtFAS. Among the fatty acids synthesised by mtFAS, there is octanoic acid, the precursor of lipoic acid (LA), a cofactor involved in the activity of several mitochondrial enzymes, among which the pyruvate dehydrogenase, the α-ketoglutarate dehydrogenase, the branched-chain keto acid dehydrogenase, the 2-oxoadipate dehydrogenase and the glycine cleavage system.12
Pathogenic variants in MECR are associated with a recessive childhood-onset disorder characterised by dystonia, optic atrophy and basal ganglia abnormalities (DYTOAB, MIM: #617 282), also described with the acronym MEPAN (mitochondrial enoyl coA reductase protein-associated neurodegeneration). A total of eight families have been described with MECR-associated disease, all with a fairly uniform clinical presentation.13
We here describe the first family in which two siblings with homozygous MECR variants show an isolated optic neuropathy phenotype, with subacute onset. Thus, we propose MECR as another gene related to rare forms of LHON-like autosomal recessive optic neuropathy.
Methods
Clinical evaluation
The study adhered to the tenets of the Declaration of Helsinki. Informed consent was obtained from patients. Clinical assessments included best-corrected visual acuity by Snellen’s chart, colour vision tests (Ishihara test), slit-lamp biomicroscopy, Goldmann applanation tonometry, colour fundus photography, optical coherence tomography (OCT; Cirrus, Carl Zeiss Meditec, Dublin, California, USA; DRI Triton, Topcon, Tokyo, Japan), automated visual field test (Humphrey Field Analyzer, protocol Sita Standard 30-2; Zeiss, San Leandro, California). OCT protocols included the evaluation of peripapillary retinal nerve fibre layer (RNFL) thickness (3.4 acquisition protocol) and segmentation analysis of the macula (ganglion cell layer (GCL) between RNFL and the inner nuclear layer boundaries).
Genetic study
Whole exome sequencing (WES) was performed on DNA from patient 1 extracted from peripheral blood. The sample library was prepared and enriched using Truseq Rapid Exome Kit (Illumina), then sequenced with 150 bp paired-end reads on a NextSeq 500 instrument (Illumina). Bioinformatic analysis followed the GATK Best Practices workflow for germline variant discovery, aligning to reference genome GRCh37/hg19. We prioritised rare variants compatible with autosomal recessive inheritance in genes encoding mitochondrial proteins. Variants of interest were classified according to the America College of Human Genetics (ACMG) guidelines.14 Next Generation Sequencing (NGS) findings were confirmed by Sanger sequencing on a 3500 Dx Genetic Analyzer (Applied Biosystems). H3M2 tool was used for the identification of runs of homozygosity (ROHs) from WES alignments.15 We also checked the proband’s WES for pathogenic CNVs by combining four tools: ExomeDepth, CoNIFER, XHMM, cn.MOPS.16–19
Fibroblast model
Fibroblast cell lines derived from both patients were generated. The full description of cell lines and culture conditions, mitochondrial respiration evaluation, Western blot, antibodies and respiratory supercomplexes assembly by BN-Page and CI-in gel activity (CI-IGA) is available in online supplemental material.
jmg-2023-109340supp001.pdf (1.6MB, pdf)
Yeast model
The variant found in patients was studied in a yeast model disrupted in ETR1, the orthologous gene of MECR, and expressing human MECR. A detailed description of yeast model methods used for phenotypic analysis is available in the online supplemental material.
Statistical analysis
Statistical analysis was performed by one-way analysis of variance followed by a post hoc Bonferroni test for multiple comparisons or by t-test.
Results
Patient 1: case report
The proband is an early 50s woman who was first noticed for low vision at 6 years, detected during routine eye examinations at school. The past clinical charts showed at 22 years a reduction of visual acuity, 0.4 in her right eye (RE) and 0.5 in her left eye (LE), associated with a small central scotoma at visual fields.
Moreover, the patient reported two episodes of sudden and painless visual loss in RE at 28 and 44 years followed by recovery of visual function after 1 month. Visual fields at 29 years demonstrated a bilateral central scotoma (MD −4.29 in RE and MD −4.36 in LE) (online supplemental figure 1A).
Her medical history was remarkable for headache since the age of 12 years and mild sensorineural hearing impairment since she was 45 years. At 40 years of age, she reported cardiac arrhythmic problems (paroxysmal tachycardia) for which she is taking beta-blockers.
Patient 1: ophthalmological findings
We observed the patient 1 month after visual loss at 44 years and subsequent recovery of visual acuity. At ophthalmological evaluation, she had visual acuity of 0.32 in RE and 0.4 in LE . Defective colour vision was evident in both eyes (Ishihara test RE 6/12 and LE 8/12). Visual fields showed bilateral central scotoma (MD −6.09 in RE and MD −5.84 in LE) (online supplemental figure 1B). At fundus examination, a small optic disc with central small excavation and temporal pallor was observed. OCT showed a diffuse reduction of the RNFL (average thickness RE 79 µm and LE 73 µm) and GCL (average thickness OU 60 µm), associated with a small optic disc area (OD 1.57 mm2 and LE 1.50 mm2) (online supplemental figure 1C).
At the last examination, after a 9-year follow-up, the patient was stable, without significant changes in visual acuity and colour vision. Similarly, a stable central scotoma was noticed at visual fields (MD −3.18 in RE and MD −3.45 in LE) (figure 1A) and unchanged RNFL and GCL thinning at OCT were evident (figure 1A).
Figure 1.
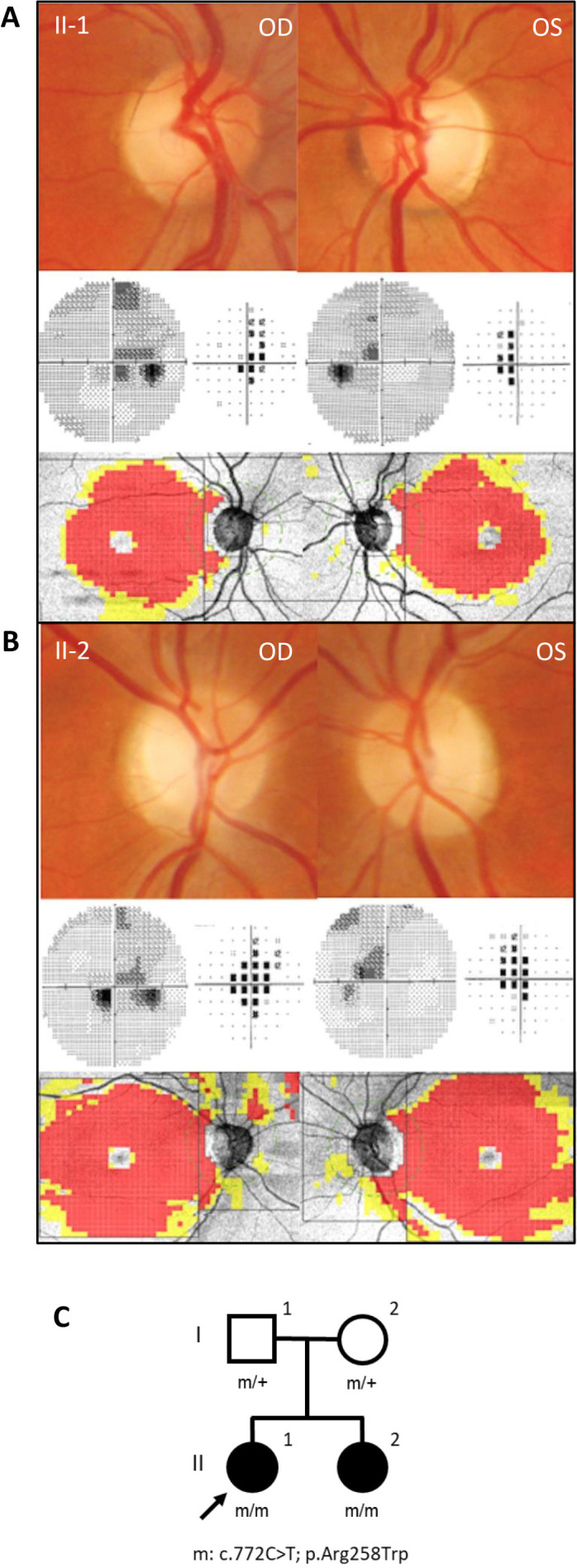
Clinical and pedigree data. Fundus pictures showing temporal optic disc pallor, visual field showing central scotoma, and optical coherence tomography revealing temporal retinal nerve fibre layer thinning and diffuse macular ganglion cell layerthinning for (A) patient II-1 and (B) patient II-2. (C) Family pedigree.
Patient 2: case report
This patient is the proband’s sister, now in the early 40s. She referred a sudden and painless visual loss in both eyes at 18 years during physical activity followed by slowly progressive recovery of vision. The past clinical charts demonstrated visual loss up to 0.05 in both eyes, associated with small central scotoma at visual fields. At 19 years, she had a bilateral central scotoma (MD −4.35 in RE and MD −3.39 in LE) (online supplemental figure 2A).
Her medical history was remarkable for headache since the age of 14 years and mild sensorineural hearing impairment since 38 years.
Patient 2: ophthalmological findings
The ophthalmological evaluation at 34 years of age disclosed visual acuity of 1.0 in RE and 0.8 in LE. Colour vision defect was evident in both eyes (Ishihara test RE 3/12 and LE 5/12). Visual fields showed a central scotoma (MD −3.39 in RE and MD −3.38 in LE) (online supplemental figure 2B). Ophthalmoscopy revealed a small optic disc with central small excavation and temporal pallor (online supplemental figure 2C). OCT showed a diffuse reduction of the RNFL associated with a small optic disc area (online supplemental figure 2C).
At the last examination, after a 7-year follow-up, the patient reported no further deterioration of visual function, the visual acuity and colour Ishihara test were unchanged. Visual fields revealed a stable central scotoma (MD −5.16 in RE and MD −3.51 in LE) (figure 1B). OCT did not show any change of RNFL and GCL thickness (figure 1B).
Genetic analysis
We performed WES on the proband (II-1, figure 1C) with a mean coverage of 79X, and 96% of target bases were above 20X. We detected a homozygous variant in the MECR gene (NM_016011.5), segregating also in the affected sister (II-1, figure 1C), while no likely pathogenic CNV was detected. The identified c.772C>T variant leads to the missense change p.Arg258Trp and is ultra-rare in GnomAD population database V.2.1.1,20 with max MAF 6.368E-5. This variant has been previously reported in two families with MECR dysfunction and typical DYTOABG/MEPAN phenotype, but in compound heterozygosity with loss-of-function variants,13 21 and accordingly to all reported evidence is classified as pathogenic according to ACMG guidelines.
In this family, the p.Arg258Trp was found inside a large ROH of 3.16 Mb, pointing to a possible parental relatedness. The main clinical and genetic findings of published cases with the additional family presented here are summarised in table 1.
Table 1.
MECR patients cohort
| Affected individual (ref) |
Origin | Nucleotide and protein variants | Age at onset of dystonia | Age at onset of optic atrophy | Intellect | MRI involvement | Age at last visit | Phenotype |
| 1 (Family A, II:1)13 | Ashkenazi Jewish | c.(695G>A);(855T>G) p.((Gly232Glu));(Tyr285Ter)] |
Early childhood | Mid-childhood | Preserved | Bilateral hyperintense T2 signal in the putamen | Late 40s | – |
| 2 (Family B, II:2)13 | Mixed Jewish | c.(695G>A);(830+2dup) p.((Gly232Glu));(?) |
1–5 y | NA | Preserved | Bilateral hyperintense pallidal T2 signal | <5 y | – |
| 3 (Family C, II:2)13 | Ashkenazi Jewish | c.(695G>A);(830+2dup) p.((Gly232Glu));(?) |
1–5 y | 5–10 y | Preserved | Bilateral hyperintense T2 signal in the dorsal striatum | 40s | – |
| 4 (Family C, II:8)13 | Ashkenazi Jewish | c.(695G>A);(830+2dup) p.((Gly232Glu));(?) |
1–5 y | 10–15 y | Preserved | NA | Late 20 s | – |
| 5 (Family D, II:1)13 | Tunisian | c.(854A>G);(854A>G) p.((Tyr285Cys));((Tyr285Cys)) |
5–10 y | 5–10 y | Relatively preserved (mild concentration difficulties) | Bilateral hyperintense T2 signal in the dorsal putamen | <10 y | – |
| 6 (Family E, II:1)13 30 | Anglo-Saxon | c.(772C>T);(247_250del) p.((Arg258Trp)); ((Asn83HisfsTer4)) |
1–5 y | 5–10 y | Deterioration of linguistic skills and executive functions to extremely low range at 9 y | Bilateral hyperintense pallidal T2 signal with cavitation and lactate peak on MRS | Late 10s | Choreoathetosis, retinopathy, cognitive decline (trans-2-enoyl-CoA reductase deficiency) |
| 7 (Family E, II:3)13 30 | Anglo-Saxon | c.(772C>T);(247_250del) p.((Arg258Trp)); ((Asn83HisfsTer4)) |
1–5 y | 1–5 y | Low average verbal comprehension with extremely low function on the other WISC IV indices | Bilateral hyperintense pallidal T2 signal with lactate peak on MRS | 10s | Choreoathetosis, retinopathy, cognitive decline (trans-2-enoyl-CoA reductase deficiency) |
| 8 (Family F, II:2)21 | Turkish | c.(772C>T);(1009C>T) p.((Arg258Trp));((Arg337Ter)) |
1–5 y | 1–5 y | NA | Hyperintense signal and cystic changes in the putamen, caudate nucleus and globus pallidus in addition to generalised mild cerebral atrophy | <5 y | – |
| 9 (Family G, II:1)31 | – | c.(830+2dup);(−39G>C) p.? |
<1 y | NA | – | – | NA | Delayed motor milestones and hypotonia, which evolved to include spasticity, an ataxic gait and progressive loss of motor skills |
| 10 (Family G, II:2)31 | – | c.(830+2dup);(−39G>C) p.? |
<1 y | NA | – | – | NA | Delayed motor milestones and hypotonia, which evolved to include spasticity, an ataxic gait and progressive loss of motor skills |
| 11 (Family H, II:1)32 | Chinese | c.(910G>T);(910G>T) p.((Asp304Tyr));((Asp304Tyr)) |
5–10 y | NA | Preserved | Bilateral hyperintense globus pallidus and cerebral peduncle T2 signals, with globus pallidus liquefaction and necrosis | 10s | – |
| 12 (Family I, II:1) (This study) | Italian | c.(772C>T);(772C>T) p.((Arg258Trp));((Arg258Trp)) |
– | 5–10 y | Preserved | – | 50s | Optic atrophy and mild sensorineural impairment |
| 13 (Family I, II:2) (this study) | Italian | c.(772C>T);(772C>T) p.((Arg258Trp));((Arg258Trp)) |
– | 15–20 y | Preserved | – | 30s | Optic atrophy and mild sensorineural impairment |
MECR coding DNA reference sequence NM_016011.5, MECR protein reference sequence NP_057095.4.
MRS, magnetic resonance spectroscopy; NA, not available; y, years.
Yeast model
The Arg258 residue is conserved in fungi and animals (figure 2A), and we confirmed that human MECR wild-type allele (MECRwt ) can partially complement the oxidative growth of the etr1Δ S. cerevisiae strain, as already reported (figure 2B).13 When the strain was transformed with MECR harbouring the Arg258Trp variant (MECRR258W ), the mutant showed a reduced oxidative growth compared with the strain harbouring MECRwt on a medium supplemented with 2% ethanol, indicating the variant is associated with an oxidative phenotype (figure 2B). This result was confirmed by the oxygen consumption rate (OCR), which was similar to that of the etr1Δ strain transformed with the empty plasmid and decreased by 30% compared with the strain transformed with MECRwt (figure 2C). By measuring the steady-state levels of MECR protein, we observed two bands, whose size is compatible with the unprocessed MECR protein and with the functional MECR protein in which the mitochondrial localisation signal was removed: in the MECRR258W strain, the MECR levels were decreased by 80%, suggesting that reduced levels of mutant MECR contributed to the phenotypic defects (figure 2D).
Figure 2.

Yeast modelling of MECRR258W variant. (A) Alignment of MECR proteins from different organisms, including Mus musculus (mammals), Gallus gallus (birds), Chelonia mydas (reptiles), Xenopus laevis (amphibians), Danio rerio (bony fish), Scyliorhinus canicula (cartilaginous fish), Asterias rubens (echinoderms), Drosophila melanogaster (arthropods), Ophiocordyceps sinensis (mollusks), Caenorhabditis elegans (roundworms), Schizosaccharomyces pombe and Saccaromyces cerevisiae (fungi). (B) Spot assay of W303-1B etr1Δ strain transformed with pFL38ETR1, empty YEplac112TEToff and YEPlac112TEToffMECR, either wild-type or harbouring R258W mutation, on YP medium supplemented with either 2% glucose or 2% ethanol. Pictures were taken after 3 days. (C) Oxygen consumption rate (OCR) on strains transformed with MECR wt or MECRR258W . OCR was measured on four independent clones for each strain and normalised to the OCR of the strain transformed with MECRwt and reported as mean±SD. Statistical analysis was performed by one-way analysis of variance followed by a post hoc Bonferroni test. ***p<0.001. (D) Representative image of a Western blot on the same strains reported in panel C for MECR and Por1 as loading control, with MECR/Por1 levels normalised to the strain harbouring MECRwt .
Fibroblasts analysis
To gain further insights into the consequences of the MECRR258W mutation, we investigated fibroblasts derived from the two patients carrying homozygous MECR variant here reported.
Western blot analysis on total cellular lysates indicated that MECR protein levels were significantly decreased in mutants compared with controls. As observed in the yeast model, these results confirm that MECRR258W mutation causes protein reduction (figure 3A,B).
Figure 3.

Characterisation of patient-derived fibroblasts carrying MECRR258W variant. (A) Western blot of MECR; GAPDH was used as loading control. A representative blot of three independent experiments (biological replicates) is shown. (B) Densitometry of MECR content. All data are means and SD Statistical analysis was performed by t-test. **: p<0.01. (C) OCR expressed as picomoles O2/min, normalised for protein content, under basal conditions and after injection of oligomycin (O), carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP; F), rotenone (R) and antimycin A (A). Data are expressed as means ± SD of three independent experiments (biological replicates). (D) Basal, ATP-linked, maximal and CI respiration. All values are means and SD of three independent experiments (biological replicates). (E) Western blot of OXPHOS subunits; GAPDH was used as loading control. A representative blot of three independent experiments is shown for each protein. (F) Densitometry of OXPHOS subunits of three independent experiments (biological replicates). All data are means and SD. (G) CI-IGA of SCs from mitoplasts obtained from control and mutant cells. One representative experiment of three is shown. IGA, in gel activity.
Since the MECRR258W mutation in yeast showed an oxidative phosphorylation (OXPHOS) defect, we investigated the OCR after having incubated fibroblasts in a galactose-containing medium for 48 hours, thus forcing cells to rely on OXPHOS to produce ATP. Contrary to the yeast model, the MECRR258W mutant cell lines showed no differences in basal, ATP-linked and maximal OCR compared with controls. Moreover, the analysis showed a comparable CI-driven OCR between mutants and controls (figure 3C,D).
The steady-state expression of representative complexes subunits of respiratory chain complexes explored by Western blotting showed a similar abundance of NDUFB8 (CI), SDHB (CII), UQCRC2 (CIII), COII (CIV) and ATP5A (CV) in the MECRR258W mutant and control cell lines (figure 3E,F). Moreover, we examined the supramolecular organisation of the respiratory chain complexes (SCs) by BN-PAGE. According to previous results, the SCs containing active CI were detected in all cell lines, as shown in the CI-in gel activity (CI-IGA) panel (figure 3G), suggesting that the mutant form of MECR protein does not interfere with the formation of SCs in fibroblasts.
Rescue of the phenotype by LA in yeast
It was previously reported that deletion of ETR1 is associated with undetectable levels of lipoylated Lat1 and Kgd2, the dihydrolipoamide acetyltransferase subunit and the dihydrolipoyl trans-succinylase subunit, respectively, of the pyruvate dehydrogenase and of the α-ketoglutarate dehydrogenase, due to the lack of LA biosynthesis.13 22 We observed lipoylated Lat1 and Kgd2 in the MECRwt strain, whereas the two proteins are undetectable in the MECRR258W strain (figure 4A), suggesting that the variant deeply affected the ability of the protein to synthesise the LA precursor. LA is also a potent ROS scavenger, and it was previously demonstrated that a decrease in its level due to variants in genes involved in its synthesis is associated with increased sensitivity to oxidant agents such as H2O2.23 By measuring the sensitivity of the MECRwt and MECRR258W strain to H2O2, we observed that the presence of the variant is associated with a decreased growth (figure 4B) and a decreased viability (figure 4C).
Figure 4.

Oxidative stress and LA supplementation on yeast model. (A) Representative image of a Western blot of lipoylated Lat1, Kgd2 and Por1 as loading control on the same proteins extract as in figure 2, panel D. (B) Growth phenotype of the same strains as in panel A on YPD medium without or with 2 mM H2O2. Pictures were taken after 3 days. (C) Viability test assay after treatment with different concentrations of H 2 O 2 for different times on MECRwt and MECRR258W strains. Viability was measured on three independent clones for each strain, at least 5000 cells plated for each clone, and reported as mean±SD. **p<0.01; ***p<0.001. (D) Cell concentration of the same strains as in panel C, after 48-hour, 72-hour and 96-hour growth in liquid YP medium supplemented with 2% ethanol and, for the MECRR258W strain, without or with 40 µg/mL LA. Cell concentration was measured on three independent replicates for each strain/condition and reported as mean±SD. *p<0.05; ***p<0.001. (E) Representative image of a Western blot of lipoylated Lat1 and Kgd2 proteins extracted from untreated MECRwt strain and untreated or LA-supplemented MECRR258W strain. (F) Viability test assay after treatment with 1 mM H2O2 for 2 hours and rescue with different concentrations of LA on MECRR258W strain. Viability was measured on three independent replicates for each condition, at least 5000 cells plated for each replicate, and reported as mean±SD. ***p<0.001. LA, lipoic acid.
The yeast model allowed evaluation of whether the supplementation with LA could ameliorate the detrimental phenotype induced by the variant. The growth rate of the MECRR258W strain in liquid YP medium supplemented with ethanol was lower compared with the MECRwt strain. When the MECRR258W strain was supplemented with LA at a high concentration (40 µg/mL), a slight but significant increase in the growth rate was observed after 72 and 96 hours (figure 4D). By measuring the levels of lipoylated Lat1 or Kgd2 in the mutant strain, no proteins were detectable even at a high concentration of LA (figure 4E), indicating that the rescue of the growth was not due to lipoylation of such proteins.
To evaluate whether the improved oxidative growth was due to the intrinsic antioxidant characteristics of LA, we measured the effect of pretreatment with LA at different concentrations after exposure of the mutant strain to 1 mM H2O2 for 2 hours. Pretreatment with at least 4 µg/mL LA increased the viability by approximately 20% (figure 4F): this value was slightly lower compared with that of MECRwt strain exposed to H2O2 without LA pretreatment. Thus, we confirm the beneficial effects of LA supplementation to reduce oxidative stress.
Discussion
We described two sisters presenting with optic atrophy: the proband apparently had a childhood onset, with subsequent subacute episodes, whereas the younger sister showed a subacute onset resembling LHON. The co-existence of childhood onset and subacute loss of central vision at 18 years of age in the same family summarises the known spectrum of LHON.1 Nonetheless, the multiple subacute episodes reported in the proband with childhood onset are a remarkable feature, not typically seen in LHON. The only extraocular feature was a mild sensorineural hearing impairment, not standard in LHON, even though the involvement of the acoustic nerve has been reported also in LHON.24
The genetic investigations identified a known recessive pathogenic MECR variant, previously found in compound heterozygosity with null alleles and associated with the phenotype of DYTOABG/MEPAN. This same variant in homozygous state was here associated with the novel phenotype of LHON-like isolated optic neuropathy and mild sensorineural hearing impairment. The p.Arg258Trp variant strongly decreased the MECR protein levels, both in patients' fibroblasts and in yeast, suggesting that instability of the mutant protein, rather than a decrease in its activity, is the main cause of the pathological phenotype. Despite a childhood onset of the disease in one case, like the syndromic published cases, the two affected siblings in our family did not show any sign of dystonia or MRI abnormalities, even after 40 years of age. It is possible that the residual quantity of MECR protein observed in mutant fibroblasts is sufficient for sustaining some biochemical activity, thus explaining a less severe phenotype compared with the described paediatric cases.
The damaging effect of p.Arg258Trp variant was confirmed through yeast modelling, where the reduction of MECR protein levels is paired with no detectable lipoylation in mitochondrial metabolic enzymes such as Lat1 or Kgd2. This defect translates into impaired oxidative growth and OCR, as well as reduced protection from oxidative stress induced by H2O2. We then supplemented LA to growth medium and observed a partial rescue of growth phenotype, especially when administered after exposure to H2O2, confirming that the beneficial effect of LA may be primarily driven by its antioxidant function, as previously observed.25 Thus, LA could be a viable therapeutical option for MECR patients, even if it does not compensate directly for the lack of protein lipoylation typical of mtFAS dysfunction. Appropriate clinical trials titrating LA in patients are needed.
Remarkably, this report on MECR pairs the recent observation of an LHON-like phenotype associated with biallelic variants in MCAT, also involved in the mtFAS pathway. The knockdown in human cells of either MCAT or MECR leads to mitochondrial dysfunction characterised by impaired assembly of respiratory complexes, mainly due to the loss of acyl-NDUFAB1 and its interaction with LYRM proteins.26 Moreover, pathogenic variants in both genes have been associated with a severe childhood-onset oxidative phosphorylation defect, also characterised by reduced levels of intact complexes/supercomplexes in the MCAT described case.27 Conversely, we failed to observe such a drastic downstream consequence of mutant MECR on OXPHOS complexes and respiratory function in our patient-derived primary cultures of fibroblasts. As mentioned, the residual activity of MECR in these two patients may be sufficient to maintain a basal mitochondrial respiration, which might well be defective in neurons like the retinal ganglion cells. Consistently, the phenotype of our patients is mild, limited to optic atrophy, whereas the full-blown encephalopathy described in the other reported patients broadly belongs to the spectrum of Leigh syndrome.13 27 Indeed, the dual clinical presentation of Leigh syndrome and subacute optic neuropathy is now described for the complex I-related genes DNAJC30 and NDUFS2 and possibly NDUFA12, as well as the mtFAS-related MCAT and MECR, suggesting that this phenotypic spectrum could be common to different defects in mitochondrial enzymes. On the contrary, even the common mtDNA mutations associated with LHON may occasionally lead to Leigh syndrome, remarking the mechanistic link between these two clinical phenotypes of mitochondrial disease.28 29
Overall, the dysfunctional mtFAS pathway emerges as a previously overlooked pathogenic mechanism reflecting on mitochondrial function, in addition to the already proposed protein lipoylation defect, which possibly contributes to the mitochondrial respiratory impairment typical of mtFAS-linked disease.13 In conclusion, mtFAS impairment is a new pathogenic mechanism leading to LHON-like recessive optic neuropathy, in addition to the previously described cases with more severe Leigh-like encephalopathic involvement. Further studies on patient-derived neuronal cells are needed to clarify the consequences of MECR mutations associated with the LHON-like phenotype in the cells targeted by this disease. However, the beneficial effect of LA on oxidative stress suggests that this condition is possibly amenable to treatment.
Acknowledgments
The authors are deeply grateful to the patients who participated to this work.
Footnotes
CF, AD, MLC and CVT contributed equally.
AM, PB, EB and LC contributed equally.
Contributors: Conceptualisation: LC, EB, PB, VC, CF. Data curation: CF, AD, MLC, CVT. Formal analysis: CF, AD, MLC, CVT, CLM, MB, DO, FP, MC, FB. Funding acquisition: EB, LC. Investigation and methodology: all authors. Software: AT, SEH, LA, JL, DK, FC, JME. Writing—original draft: CF, AD, MLC, CVT, CLM, VC, AM, PB, EB, LC. Guarantor: LC. Writing—review and editing: all authors.
Funding: The research activity was supported by the Italian Ministry of Health grant GR-2016-02361449 to LC and ‘Ricerca Corrente’ funding to VC and AM.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
This study involves human participants and was approved by Comitato Etico di Area Vasta Emilia Centro della Regione Emilia-Romagna (CE-AVEC), #211/2018/SPER/AUSLBO. Participants gave informed consent to participate in the study before taking part.
References
- 1. Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res 2004;23:53–89. 10.1016/j.preteyeres.2003.10.003 [DOI] [PubMed] [Google Scholar]
- 2. Yu-Wai-Man P, Griffiths PG, Brown DT, et al. The epidemiology of Leber hereditary optic neuropathy in the North east of England. Am J Hum Genet 2003;72:333–9. 10.1086/346066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caporali L, Iommarini L, La Morgia C, et al. Peculiar combinations of individually non-pathogenic missense mitochondrial DNA variants cause low penetrance Leber’s hereditary optic neuropathy. PLoS Genet 2018;14:e1007210. 10.1371/journal.pgen.1007210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Newman NJ, Yu-Wai-Man P, Biousse V, et al. Understanding the molecular basis and pathogenesis of hereditary optic neuropathies: towards improved diagnosis and management. Lancet Neurol 2023;22:172–88. 10.1016/S1474-4422(22)00174-0 [DOI] [PubMed] [Google Scholar]
- 5. Stenton SL, Sheremet NL, Catarino CB, et al. Impaired complex I repair causes recessive Leber’s hereditary optic neuropathy. J Clin Invest 2021;131:e138267. 10.1172/JCI138267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stenton SL, Tesarova M, Sheremet NL, et al. DNAJC30 defect: a frequent cause of recessive Leber hereditary optic neuropathy and Leigh syndrome. Brain 2022;145:1624–31. 10.1093/brain/awac052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gerber S, Ding MG, Gérard X, et al. Compound heterozygosity for severe and hypomorphic NDUFS2 mutations cause non-syndromic LHON-like optic neuropathy. J Med Genet 2017;54:346–56. 10.1136/jmedgenet-2016-104212 [DOI] [PubMed] [Google Scholar]
- 8. Pagniez-Mammeri H, Loublier S, Legrand A, et al. Mitochondrial complex I deficiency of nuclear origin. Mol Genet Metab 2012;105:163–72. 10.1016/j.ymgme.2011.11.188 [DOI] [PubMed] [Google Scholar]
- 9. Magrinelli F, Cali E, Braga VL, et al. Biallelic loss-of-function NDUFA12 variants cause a wide phenotypic spectrum from Leigh/Leigh-like syndrome to isolated optic atrophy. Mov Disord Clin Pract 2022;9:218–28. 10.1002/mdc3.13398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gerber S, Orssaud C, Kaplan J, et al. MCAT mutations cause nuclear LHON-like optic neuropathy. Genes (Basel) 2021;12:521. 10.3390/genes12040521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H, Yuan S, Minegishi Y, et al. Novel mutations in malonyl-Coa-Acyl carrier protein transacylase provoke autosomal recessive optic neuropathy. Hum Mol Genet 2020;29:444–58. 10.1093/hmg/ddz311 [DOI] [PubMed] [Google Scholar]
- 12. Hiltunen JK, Autio KJ, Schonauer MS, et al. Mitochondrial fatty acid synthesis and respiration. Biochim Biophys Acta 2010;1797:1195–202. 10.1016/j.bbabio.2010.03.006 [DOI] [PubMed] [Google Scholar]
- 13. Heimer G, Kerätär JM, Riley LG, et al. MECR mutations cause childhood-onset dystonia and optic atrophy, a mitochondrial fatty acid synthesis disorder. Am J Hum Genet 2016;99:1229–44. 10.1016/j.ajhg.2016.09.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the Association for molecular pathology. Genet Med 2015;17:405–24. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Magi A, Tattini L, Palombo F, et al. H3M2: detection of runs of Homozygosity from whole-exome sequencing data. Bioinformatics 2014;30:2852–9. 10.1093/bioinformatics/btu401 [DOI] [PubMed] [Google Scholar]
- 16. Plagnol V, Curtis J, Epstein M, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics 2012;28:2747–54. 10.1093/bioinformatics/bts526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krumm N, Sudmant PH, Ko A, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res 2012;22:1525–32. 10.1101/gr.138115.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fromer M, Purcell SM. Using XHMM software to detect copy number variation in whole-exome sequencing data. Curr Protoc Hum Genet 2014;81:7. 10.1002/0471142905.hg0723s81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Klambauer G, Schwarzbauer K, Mayr A, et al. Cn.MOPS: mixture of Poissons for discovering copy number variations in next-generation sequencing data with a low false discovery rate. Nucleic Acids Res 2012;40:e69. 10.1093/nar/gks003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gorukmez O, Gorukmez O, Havalı C. Novel MECR Mutation in childhood-onset dystonia, optic atrophy, and basal ganglia signal abnormalities. Neuropediatrics 2019;50:336–7. 10.1055/s-0039-1688767 [DOI] [PubMed] [Google Scholar]
- 22. Kursu VAS, Pietikäinen LP, Fontanesi F, et al. Defects in mitochondrial fatty acid synthesis result in failure of multiple aspects of mitochondrial biogenesis in Saccharomyces cerevisiae. Mol Microbiol 2013;90:824–40. 10.1111/mmi.12402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brown JA, Sherlock G, Myers CL, et al. Global analysis of gene function in yeast by quantitative phenotypic profiling. Mol Syst Biol 2006;2:2006.0001. 10.1038/msb4100043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rance G, Kearns LS, Tan J, et al. Auditory function in individuals within Leber’s hereditary optic neuropathy pedigrees. J Neurol 2012;259:542–50. 10.1007/s00415-011-6230-7 [DOI] [PubMed] [Google Scholar]
- 25. Feng D, Witkowski A, Smith S. Down-regulation of mitochondrial acyl carrier protein in mammalian cells compromises protein lipoylation and respiratory complex I and results in cell death. J Biol Chem 2009;284:11436–45. 10.1074/jbc.M806991200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nowinski SM, Solmonson A, Rusin SF, et al. Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. Elife 2020;9:e58041. 10.7554/eLife.58041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Webb BD, Nowinski SM, Solmonson A, et al. Recessive pathogenic variants in MCAT cause combined oxidative phosphorylation deficiency. Elife 2023;12:e68047. 10.7554/eLife.68047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Funalot B, Reynier P, Vighetto A, et al. Leigh-like encephalopathy complicating Leber’s hereditary optic neuropathy. Ann Neurol 2002;52:374–7. 10.1002/ana.10299 [DOI] [PubMed] [Google Scholar]
- 29. Fruhman G, Landsverk ML, Lotze TE, et al. Atypical presentation of Leigh syndrome associated with a Leber hereditary optic neuropathy primary mitochondrial DNA mutation. Mol Genet Metab 2011;103:153–60. 10.1016/j.ymgme.2011.02.014 [DOI] [PubMed] [Google Scholar]
- 30. Riley LG, Cowley MJ, Gayevskiy V, et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet Med 2020;22:1254–61. 10.1038/s41436-020-0793-6 [DOI] [PubMed] [Google Scholar]
- 31. Frésard L, Smail C, Ferraro NM, et al. Identification of rare-disease genes using blood transcriptome sequencing and large control cohorts. Nat Med 2019;25:911–9. 10.1038/s41591-019-0457-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu Z, Shimura M, Zhang L, et al. Whole exome sequencing identifies a novel homozygous MECR mutation in a Chinese patient with childhood-onset dystonia and basal ganglia abnormalities, without optic atrophy. Mitochondrion 2021;57:222–9. 10.1016/j.mito.2020.12.014 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jmg-2023-109340supp001.pdf (1.6MB, pdf)
Data Availability Statement
Data are available upon reasonable request.