

Abstract
Anaplastic lymphoma kinase (ALK) tyrosine kinase inhibitors (TKIs) show potent efficacy in several ALK-driven tumors, but the development of resistance limits their long-term clinical impact. Although resistance mechanisms have been studied extensively in ALK-driven non–small cell lung cancer, they are poorly understood in ALK-driven anaplastic large cell lymphoma (ALCL). Here, we identify a survival pathway supported by the tumor microenvironment that activates phosphatidylinositol 3-kinase γ (PI3K-γ) signaling through the C-C motif chemokine receptor 7 (CCR7). We found increased PI3K signaling in patients and ALCL cell lines resistant to ALK TKIs. PI3Kγ expression was predictive of a lack of response to ALK TKI in patients with ALCL. Expression of CCR7, PI3Kγ, and PI3Kδ were up-regulated during ALK or STAT3 inhibition or degradation and a constitutively active PI3Kγ isoform cooperated with oncogenic ALK to accelerate lymphomagenesis in mice. In a three-dimensional microfluidic chip, endothelial cells that produce the CCR7 ligands CCL19/CCL21 protected ALCL cells from apoptosis induced by crizotinib. The PI3Kγ/δ inhibitor duvelisib potentiated crizotinib activity against ALCL lines and patient-derived xenografts. Furthermore, genetic deletion of CCR7 blocked the central nervous system dissemination and perivascular growth of ALCL in mice treated with crizotinib. Thus, blockade of PI3Kγ or CCR7 signaling together with ALK TKI treatment reduces primary resistance and the survival of persister lymphoma cells in ALCL.
INTRODUCTION
Most anaplastic lymphoma kinase (ALK)–driven tumors respond well to treatment with ALK tyrosine kinase inhibitors (TKIs) (1). In ALK-rearranged anaplastic large cell lymphoma (ALCL), crizotinib, an ALK TKI, induces complete remissions that last for several years in most pediatric and adult patients with chemotherapy relapsed/refractory (R/R) disease (2–4). Because of this success, crizotinib received U.S. Food and Drug Administration approval in 2021. Despite this remarkable activity, resistance to crizotinib treatment appears within 3 months of therapy initiation in a subset of patients (5). Moreover, even patients with complete clinical and molecular remission of several years’ duration relapse after the discontinuation of TKI treatment (6–8). These data indicate that ALK+ lymphoma cells are not eradicated by TKI treatment and that therapy discontinuation allows the regrowth of rare persister cells. Resistant cells and persister cells share common mechanisms of tolerance to targeted TKI therapy (9). In epithelial tumors, resistance mechanisms include point mutations in the tyrosine kinase (TK) domain that abrogate drug binding, genomic amplification of the rearranged gene, and activation of bypass signaling pathways. These mechanisms can preexist or be acquired by drug-tolerant persister cells (9). In ALK+ non–small cell lung cancer (NSCLC), bypass mechanisms are provided by secondary activation of other receptor TKs (RTKs) expressed by the epithelial cells, such as mesenchymal-epithelial transition (MET) factor receptor and epidermal growth factor receptor (EGFR), that compensate for the blockade of ALK signaling (10, 11). In ALK+ ALCL, resistance to ALK TKIs is typically not associated with the acquisition of point mutations, suggesting the activation of bypass pathways (6, 11, 12). Because MET and EGFR are not expressed in ALCL, other yet-unknown bypass mechanisms are likely engaged. Blockade of con-comitant platelet-derived growth factor receptor (PDGFR) signaling potentiates the activity of ALK TKIs (13) and interleukin-10 receptor (IL-10R)–mediated cytokine signaling can contribute to primary resistance to crizotinib in ALK+ lymphoma cells (12). The downstream signals initiated by PDGFR and IL-10R converge into a bypass activation of Akt, signal transducer and activator of transcription 3 (STAT3), and mitogen-activated protein kinases (MAPKs), suggesting that the activation of these pathways, known to be essential for ALCL survival (14, 15), might be as well critical to compensate for the blockade of ALK signaling (12, 13). However, it is likely that more specific mechanisms originate from the tumor microenvironment to support the survival of ALK+ persister cells for years in the presence of ALK blockade. In the early phase of the disease, ALK+ ALCL cells tend to colonize the perivascular and intravascular spaces in lymph nodes, with a sinusoidal and perivascular distribution of tumor cells being a common pattern observed in ALK+ ALCL (16). Thus, the perivascular niche appears to be a favorable microenvironment for the homing of ALCL cells.
Here, we describe that expression of C-C motif chemokine receptor 7 (CCR7), phosphatidylinositol 3-kinase γ (PI3Kγ), and PI3Kδ is repressed by ALK activity in ALCL cells. ALK+ ALCL cells exploit the up-regulation of the CCR7/PI3Kγ axis to engage survival signals from perivascular niches during ALK blockade. Disruption of this survival axis through CCR7 deletion or PI3Kγ/δ blockade in vitro and in vivo increases the efficacy of ALK TKIs, supporting the use of these combination therapies for the eradication of ALCL persister cells.
RESULTS
The PI3K pathway is up-regulated in patients and ALCL cell lines resistant to ALK TKI treatment
To identify potential bypass mechanisms that lead to acquired resistance to crizotinib in ALK+ ALCL, we analyzed RNA sequencing (RNA-seq) data from primary tumors of two pediatric patients with ALK+ ALCL enrolled in the MAPPYACTS trial (NCT02613962) who developed resistance to ALK TKIs (fig. S1A) (12, 17). Gene Ontology (GO) analysis using pathway annotations from the Kyoto Encyclopedia of Genes and Genomes (KEGG) on genes that were differentially expressed in ALK TKI–resistant tumors versus ALK TKI–responsive tumors showed up-regulation of sets of genes involved in proliferation, survival, and cancer-associated pathways (fig. S1B). Genes in the PI3K-Akt pathway and in the Ras and MAPK signaling pathways were among the most up-regulated genes in ALK TKI–resistant tumors (Fig. 1A and fig. S1B). To corroborate these findings in experimental models, we created crizotinib-resistant (CR) ALK+ ALCL cell lines by exposing cells to increasing concentrations of crizotinib. After 6 months, seven out of the eight treated cell lines developed resistance to high concentrations of crizotinib (Fig. 1B and fig. S2, A and B), whereas the remaining line, SU-DHL1, did not develop resistance to crizotinib [median inhibitory concentration (IC50) remained <120 nM]. Sequencing of the ALK gene showed that most resistant cells had not acquired ALK mutations predicted to confer resistance to crizotinib, whereas, in two lines, ALK mutations were detected at a low frequency (table S1). We then performed RNA-seq on parental crizotinib-sensitive and CR ALCL cells and found 1525 differentially expressed genes (table S2). On the basis of analysis with the enrich R tool (18, 19), these genes included a number of genes associated with the KEGG PI3K-Akt signaling pathway. Specifically, 43 differentially expressed genes belonged to the set of 354 genes annotated in the KEGG PI3K-Akt pathway (fig. S2C and table S3). In this context, forkhead box O3 was down-regulated, but the mammalian target of rapamycin (mTOR) signaling was up-regulated in resistant cell lines, consistent with a function of mTOR in ALK+ ALCL survival (20). We concluded that an up-regulation of the PI3K pathway occurred in vivo and in vitro in ALCL cells that develop resistance to ALK TKI.
Fig. 1. Resistance to crizotinib is associated with PI3Kγ up-regulation in ALK+ ALCL.
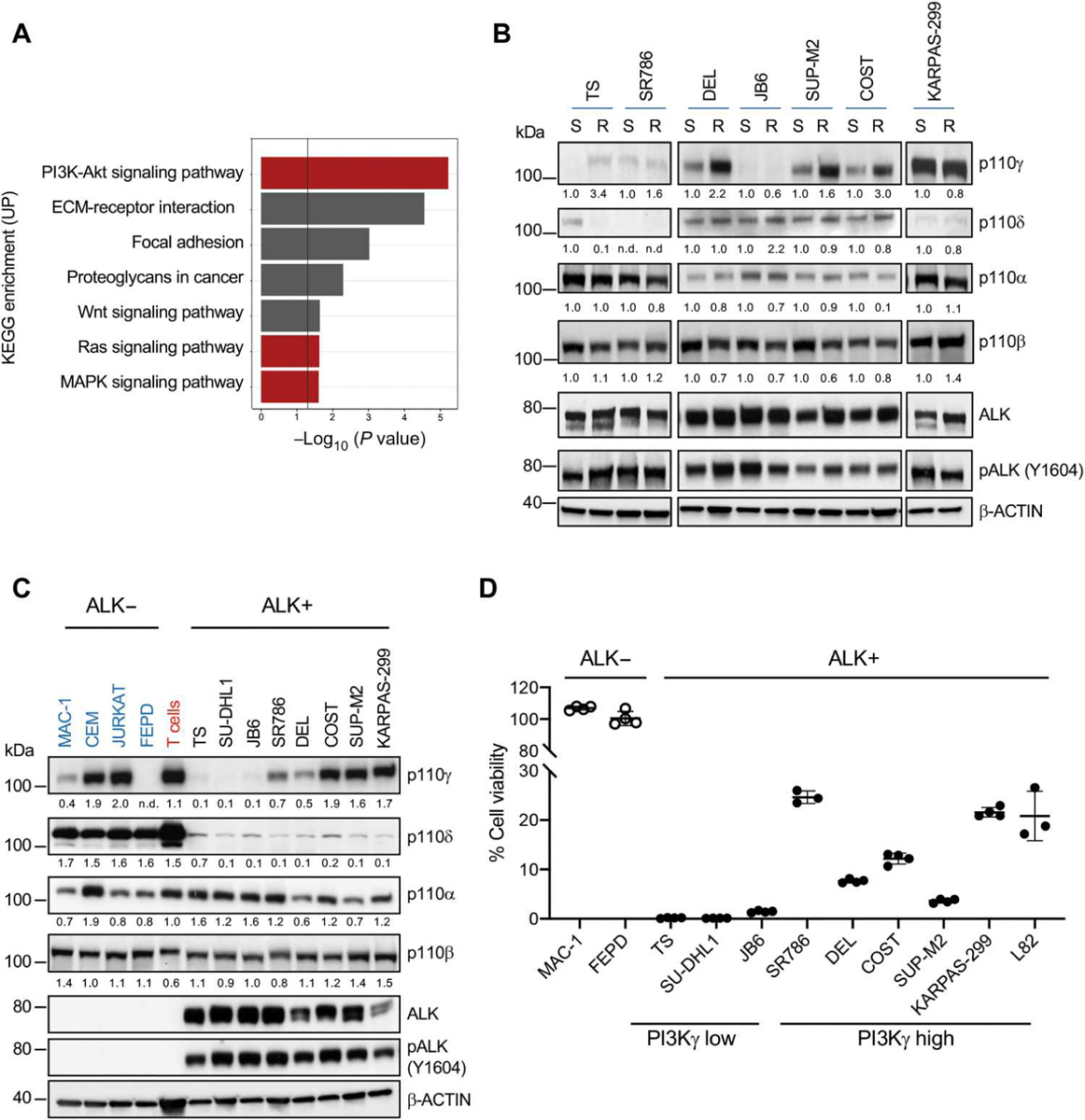
(A) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis for up-regulated genes identified by RNA-seq in samples from patients with ALK+ ALCL that relapsed on ALK inhibitor treatment (n = 2) versus sensitive to ALK inhibitor treatment (n = 2). The PI3K-Akt, Ras, and MAPK signaling pathways are highlighted in red. (B) Western blot analysis performed on paired crizotinib-sensitive (S) and resistant (R) ALK+ ALCL cell lines. (C) Western blot analysis performed on human ALK+ ALCL cell lines, ALK− T lymphoma lines, or normal T cells. Western blot bands were normalized on β-actin. (D) Cell viability measured using CellTiter-Glo of ALK+ and ALK− ALCL cell lines treated with crizotinib (100 nM) for 72 hours. n = 3 or 4 technical replicates. Data are shown as means ± SD. For Western blots, β-actin was used as a loading control, and two independent experiments with similar results were performed.
Increased PI3Kγ expression induces resistance to crizotinib
To elucidate the mechanism of PI3K pathway activation, we analyzed the expression of all PI3K class I isoforms. We found that four out of seven CR cell lines displayed increased mRNA and protein abundance of PI3Kγ, whereas no increase was seen in PI3Kα, PI3Kβ, or PI3Kδ (Fig. 1B and fig. S3, A to C). We also observed the up-regulation of PI3Kγ expression in vivo in ALCL xenografts that had become resistant to lorlatinib (fig. S3D) (21), suggesting that increased PI3Kγ expression could contribute to ALK TKI resistance.
We next extended the characterization of PI3K isoforms to a larger panel of ALK+ and ALK− lymphoma cell lines. In contrast to normal T cells or other T cell lymphoma or leukemia cell lines, all ALK+ ALCL lines displayed low PI3Kδ expression. PI3Kγ showed variable expression in ALK+ ALCL lines ranging from very-low to intermediate-high expression that was comparable to normal peripheral blood T cells (Fig. 1C and fig. S3E). By fluorescence in situ hybridization (FISH) analysis, the ALK+ ALCL cell line, L82, showed amplification (>five copies) of the PIK3CG locus consistent with a previous report based on CNV analysis (22), whereas all other lines contained two PIK3CG copies (fig. S3F). Cell lines with higher PI3Kγ expression showed greater resistance to crizotinib than cell lines with low PI3Kγ (Fig. 1D). The positive correlation between PI3Kγ expression and increased IC50 was also seen with other ALK TKIs (brigatinib, ceritinib, alectinib, and lorlatinib) (fig. S4).
To determine the expression of PI3Kγ in treatment-naïve primary ALCL, we developed an immunohistochemistry staining assay with an antibody whose specificity in formalin-fixed samples was validated in cells transduced with a PI3Kγ-encoding retrovirus (fig. S5A). Consistent with the heterogeneity found in cell lines, primary ALK+ ALCL showed variable PI3Kγ expression, as did primary ALK− ALCL (Fig. 2, A and B). In contrast, other T cell lymphomas, such as peripheral T cell lymphoma–not otherwise specified (PTCL-NOS) and angioimmunoblastic T cell lymphoma, showed more consistent PI3Kγ expression like that seen in reactive T cells (Fig. 2, A and B, and fig. S5B). No gains or amplification of PIK3CG was detected by FISH in 41 primary ALCL samples (fig. S3G). Thus, PI3Kγ mRNA and protein abundance is heterogeneous in both primary ALCL and ALK+ ALCL cell lines.
Fig. 2. High PI3Kγ expression induces spontaneous resistance to crizotinib in patients with ALK+ ALCL.
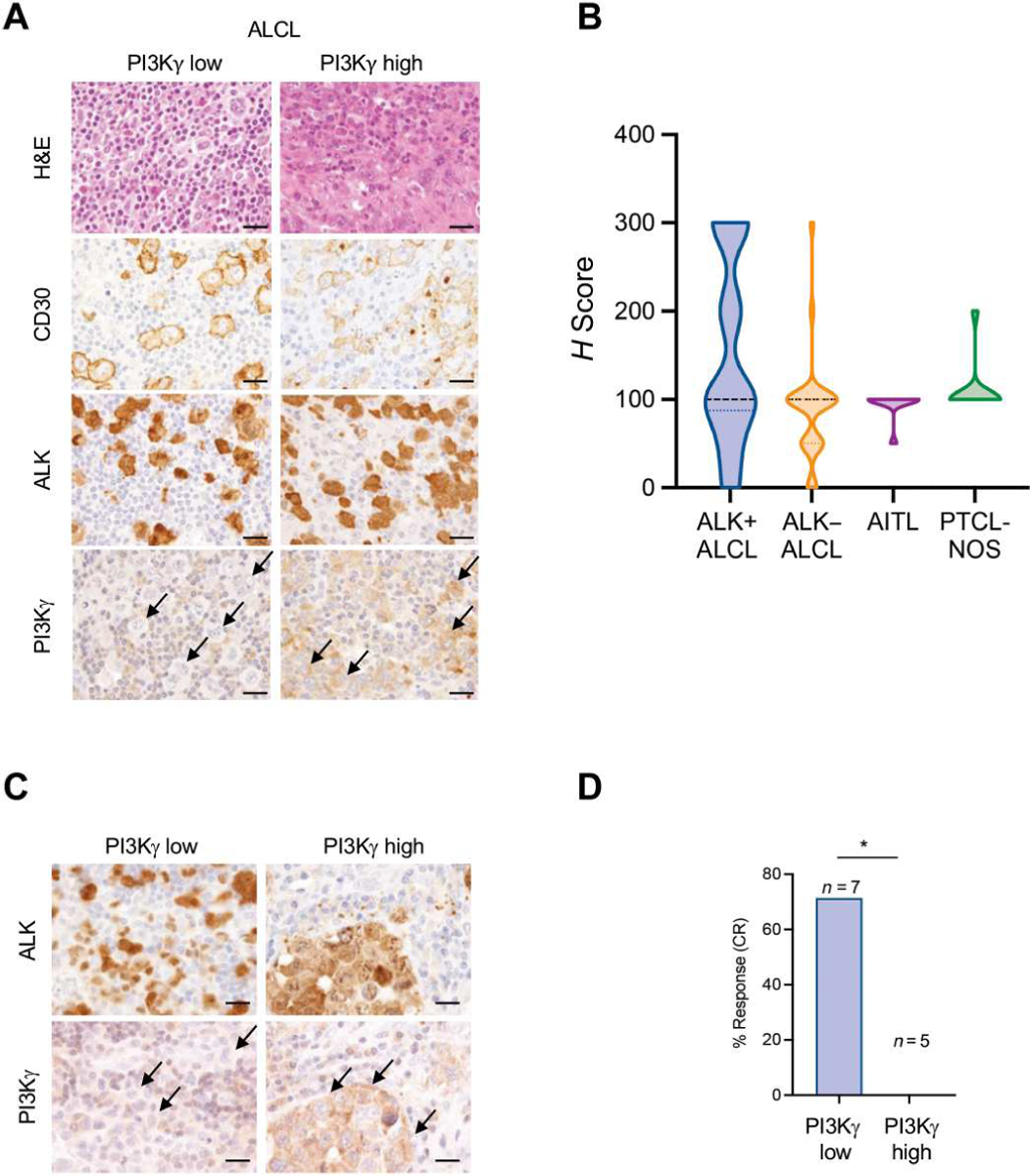
(A) Photomicrographs of representative hematoxylin and eosin (H&E) staining and immunohistochemical staining performed with the indicated antibodies in primary ALK+ lymphoma samples with low or high expression of PI3Kγ. PI3Kγ antibody was validated in formalin-fixed samples of cells transduced with a PI3Kγ-encoding retrovirus (fig. S5A). Black arrows indicate PI3Kγ expression in lymphoma cells. Scale bars, 100 μm. (B) PI3Kγ staining in human T cell lymphoma subtypes. Expression across the violin plot is shown. ALK+ ALCL, n = 41; ALK− ALCL, n = 29; angioimmunoblastic T cell lymphoma (AITL), n = 10; PTCL-NOS n = 11. The number of patient samples is indicated for each lymphoma subtype. PI3Kγ expression was quantified by immunostaining, and an H score was assigned. (C) Immunohistochemical staining of ALK and PI3Kγ in primary samples of ALK+ ALCL. Black arrows indicate PI3Kγ expression in lymphoma cells. Scale bars, 100 μm. (D) Percentage of response to crizotinib treatment of patients with ALK+ ALCL expressing low (H score < 100; n = 7) or high (H score > 100; n = 5) PI3Kγ. C Scale bars, 50 μm. *P < 0.05. Significance was determined by unpaired, two-tailed Student’s t test.
Because the availability of patients in the MAPPYACTS trial was limited, we investigated samples from another series of patients with R/R ALK+ ALCL who had been treated with crizotinib and for whom clinical response was known and histology was available (NCT02419287 and EudraCT 2010-022978-14) (2, 4). We previously demonstrated that crizotinib could induce long-lasting clinical responses in ALK+ ALCL (23), although some patients responded poorly and relapsed during crizotinib treatment (5). In this patient cohort, none of the patients with high expression of PI3Kγ (H score > 100) responded to crizotinib (zero of five), in contrast to five out of seven (71%) patients with low expression of PI3Kγ (H score < 100) who responded to crizotinib (Fig. 2, C and D).
To test the hypothesis that PI3Kγ confers resistance to ALK TKI, we overexpressed PI3Kγ in two ALCL cell lines, TS and SU-DHL1, with low baseline PI3Kγ expression (fig. S6A). Overexpression of PI3Kγ was sufficient to decrease crizotinib sensitivity in ALCL cells (fig. S6B). Likewise, the overexpression of a constitutively active form of PI3Kγ (PI3KγCAAX) caused resistance to crizotinib (fig. S6, C and D). In a complementary experiment, we observed that the knockdown of PI3Kγ with two different short hairpin RNAs (fig. S6E) decreased cell viability after crizotinib treatment in all ALCL lines tested, although less profoundly than the knockdown of PI3Kα that is known to be essential for ALK signaling in ALCL (fig. S6, F and G) (24). Overall, these data support the conclusion that PI3Kγ expression is both necessary and sufficient to confer partial resistance to ALK TKIs in ALK+ ALCL.
PI3Kγ and PI3Kδ expression are repressed by ALK oncogenic activity in ALCL
The unexpected finding that PI3Kδ expression was low in ALK+ ALCL prompted us to investigate the regulation of PI3Kδ expression in ALCL. We and others previously reported that, in lymphoma cells, ALK signaling suppresses the expression of several genes that mediate T cell receptor (TCR) signaling, including CD3, lymphocyte cell–specific protein-tyrosine kinase (LCK) and ZAP70 (25, 26). Inhibition of ALK activity by crizotinib or by treatment with the ALK degrader TL13–112 (27) up-regulated PI3Kδ expression (Fig. 3, A and B, and fig. S7A). Conversely, the ectopic expression of nucleophosmin (NPM)–ALK in ALK− ALCL resulted in decreased PI3Kδ expression (fig. S7, B and C). Furthermore, treatment with the STAT3 degrader SD36 (28) increased PI3Kδ mRNA and protein abundance, indicating that ALK-induced suppression of PI3Kδ was transcriptionally regulated through STAT3 (Fig. 3, C and D). Degradation of STAT3 also increased PI3Kγ mRNA and protein expression (Fig. 3D and fig. S7D). However, the overexpression of PI3Kδ, either wild type (WT) or the constitutively active form PI3KδE1021K, did not increase resistance to crizotinib (fig. S7, E and F). Thus, ALK and STAT3 activities both repress PI3Kδ expression, whereas PI3Kγ is repressed by STAT3 in ALK+ ALCL.
Fig. 3. PI3Kδ is repressed by ALK in ALK+ ALCL cell lines and constitutive activation of PI3Kγ accelerates ALK-dependent lymphomagenesis.
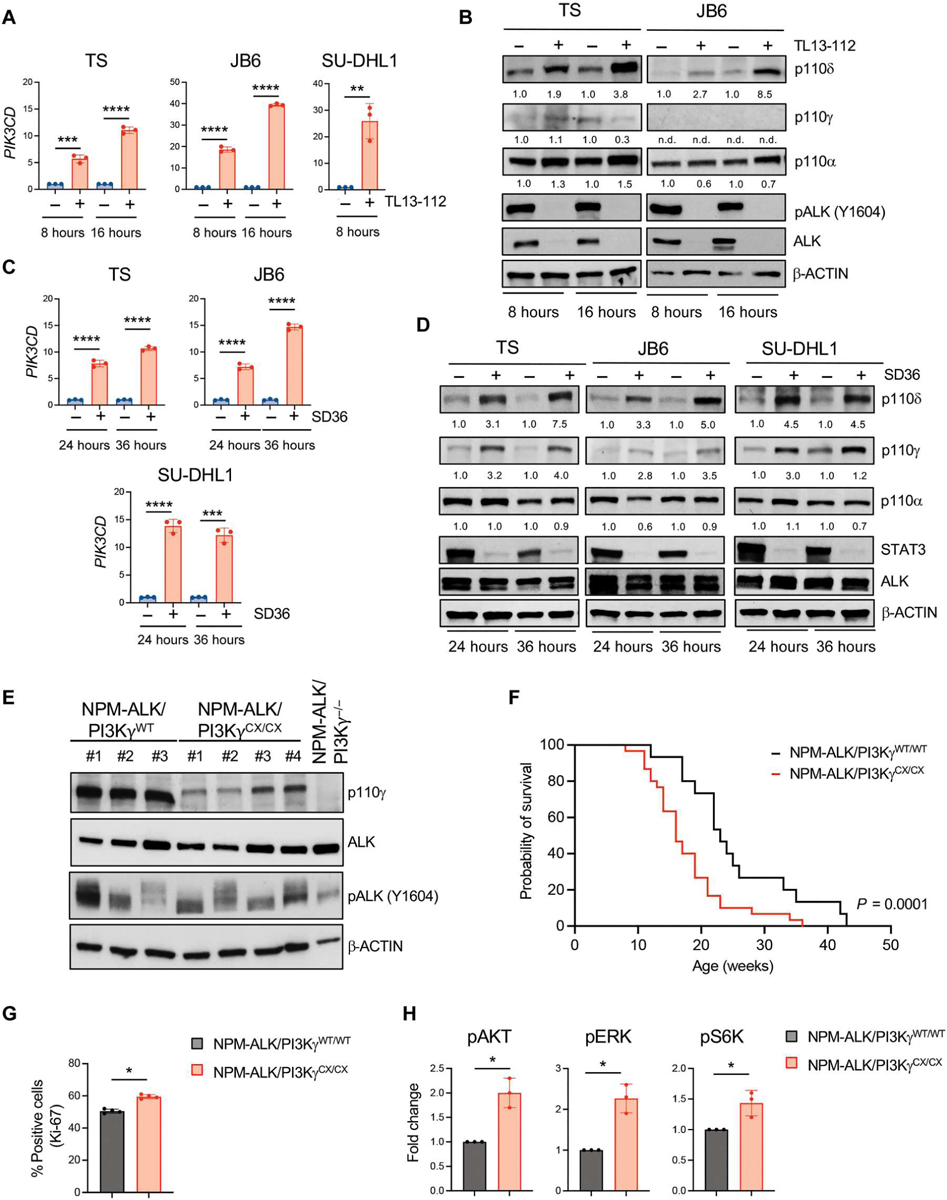
(A) qRT-PCR analysis of PIK3CD mRNA expression performed on TS, JB6, and SU-DHL1 cell lines treated with TL134–112 (100 nM). n = 3 technical replicates. (B) Western blot analysis on TS and JB6 cell lines treated with TL13–112 (100 nM). (C) qRT-PCR analysis of PIK3CD mRNA expression performed on ALK+ ALCL cell lines treated with SD36 (1 μM). n = 3 technical replicates. (D) Western blot analysis on ALK+ ALCL human cell lines treated with SD36 (1 μM). (E) Western blot analysis of lymphomas obtained from C57BL/6 mice with the indicated genotypes. (F) Kaplan-Meier survival analysis of NPM-ALK transgenic mice crossed with mice expressing an active form of PI3Kγ (PI3KγCX/CX) (black, NPM-ALK/PI3KγWT/WT, n = 30 mice; red, NPM-ALK/PI3KγCX/CX, n = 30 mice). ****P < 0.0001. Significance was determined by log-rank (Mantel-Cox) test. (G) Quantification of Ki-67–positive cells in sections of primary lymphomas (n = 4) with the indicated genotypes. (H) Amount of phosphorylated Akt, ERK, and S6K measured in murine primary tumors with the indicated genotypes by Bio-Rad Bio-Plex. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Significance was determined by unpaired, two-tailed Student’s t test. Data are shown as means ± SD. For Western blots, β-actin was used as a loading control, and two independent experiments with similar results were performed.
Constitutive activation of PI3Kγ in mice accelerates NPM-ALK-driven lymphomagenesis
We then investigated the contribution of PI3Kγ in ALK-driven lymphoma by crossing NPM-ALK transgenic mice (29) with PI3Kγ (PI3Kγ−/− mice) knockout mice (30) and mice expressing a constitutively active form of PI3Kγ (PI3KγCX/CX mice) in T cells (Fig. 3E) (31). PI3Kγ deficiency did not affect lymphoma onset or survival (fig. S8A), indicating that PI3Kγ is not required for T cell transformation by NPM-ALK. In contrast, the presence of homozygous PI3KγCX alleles significantly accelerated NPM-ALK lymphoma onset (P < 0.0001; Fig. 3F). Lymphomas from NPM-ALK/PI3KγCX/CX mice showed higher proliferation rates in vivo than those from NPM-ALK/PI3KγWT/WT mice (P = 0.0008; Fig. 3G). Activation of Akt, extracellular signal–regulated kinase 1/2 (ERK1/2), and S6K was higher in NPM-ALK/PI3KγCX/CX than in NPM-ALK/PI3KγWT/WT lymphomas (Fig. 3H). Thus, constitutive activation of PI3Kγ accelerates NPM-ALK–driven lymphoma by contributing to the activation of pathways that promote cell proliferation.
Duvelisib cooperates with crizotinib to block ALCL cell growth
When used as single agents, the PI3Kγ/δ dual inhibitor duvelisib and other PI3K inhibitors had limited effects on ALK+ ALCL growth and apoptosis in vitro and in vivo (Fig. 4, A and B, and fig. S8, B and C). This is consistent with our previously published report (32) and ongoing data from the PRIMO trial in patients with T cell lymphoma (NCT03372057). Thus, we tested the combined treatment of crizotinib at low concentration with duvelisib in ALCL lines (KARPAS-299, L82, and COST) that highly express PI3Kγ and are partially resistant to ALK inhibition. We demonstrated that duvelisib potentiated the activity of crizotinib in vitro (fig. S8B). Moreover, combination therapy with crizotinib and duvelisib in KARPAS-299 and L82 xenografts reduced cell proliferation and increased apoptosis of ALCL cells compared with crizotinib alone (Fig. 4, A and B). Last, we tested the combination of crizotinib and duvelisib in an ALK+ ALCL patient-derived xenograft (PDX). Non-obese diabetic mice (NOD)/severe combined immunodeficient (SCID) IL-2Rγ (IL-2Rγ)–null (NSG) mice were injected subcutaneously with PDXs. PDX tumors grew rapidly when injected subcutaneously and were insensitive to duvelisib alone but responded to crizotinib and to the combined treatment of crizotinib with duvelisib. The combined treatment induced more durable responses than crizotinib alone (Fig. 4, C and D). Histology of tumors showed a reduction of residual ALK+ lymphoma cells in PDXs from mice receiving crizotinib in combination with duvelisib compared with either duvelisib or crizotinib alone (Fig. 4E and fig. S8D). Ex vivo analysis of tumor lysates demonstrated that the PDX tumors moderately expressed PI3Kγ and PI3Kδ, and up-regulated expression of PI3Kγ upon prolonged treatment with crizotinib (Fig. 4F and fig. S8E). In addition, PDX tumors showed a greater reduction of ERK1/2 and Akt phosphorylation with the combination of crizotinib and duvelisib than crizotinib or duvelisib alone (Fig. 4F and fig. S8F). Thus, we concluded that, although PI3Kγ inhibitors are ineffective as single agents in ALK+ ALCL, they can potentiate the therapeutic activity of ALK TKIs.
Fig. 4. Duvelisib potentiates crizotinib treatment in ALK+ ALCL xenografts.
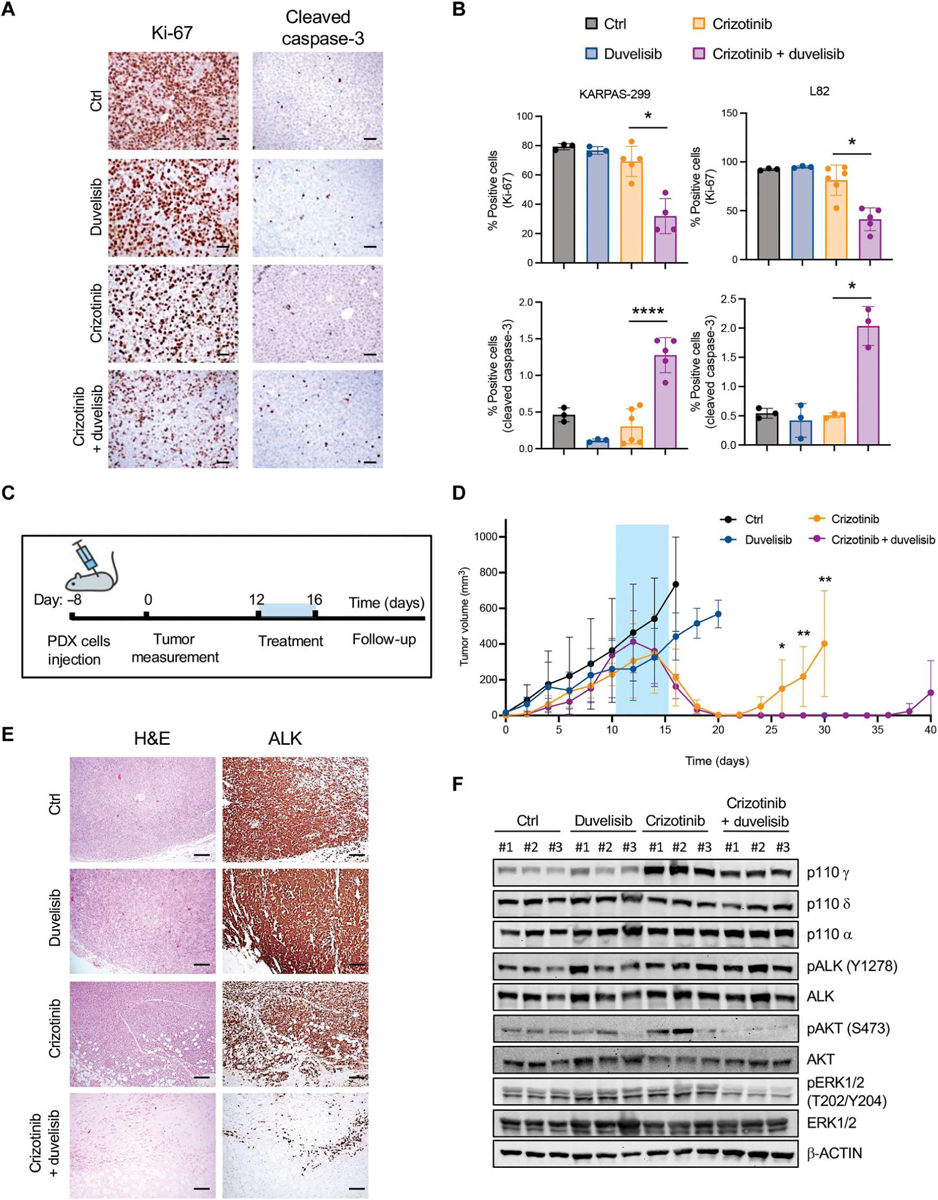
(A) Representative immunohistochemistry (IHC) for Ki-67 and cleaved caspase-3 of xenografted tumors collected from NSG mice injected subcutis with ALK+ ALCL cells and treated when tumors reached 300 to 500 mm3. Mice were treated with a vehicle, duvelisib (10 mg/kg), crizotinib (30 mg/kg), or duvelisib and crizotinib for 6 days. Scale bars, 50 μm. (B) Quantification of Ki-67 (top) and cleaved caspase-3–positive (bottom) cells in ALK+ ALCL xenograft lymphoma as in (A) (n = 3 to 6 tumors). (C) Schematic representation of the in vivo experiment with ALK+ ALCL PDXs. PDXs were injected subcutis. Treatment started when tumors reached 300 to 500 mm3. Mice were treated for 5 days and monitored for follow-up until day 40 after treatment. (D) Tumor growth of PDXs treated with a vehicle (n = 9 tumors), duvelisib (10 mg/kg; n = 6 tumors), crizotinib (30 mg/kg; n = 8 tumors), or duvelisib and crizotinib (n = 8 tumors) for 5 days. (E) Representative H&E staining and IHC for ALK of PDX tumors treated as indicated in (C) and (D). Tumors were collected as follows: control mice, day 16; duvelisib, day 20; crizotinib, day 30; crizotinib and duvelisib, day 40. Scale bars, 100 μm. (F) Western blot analysis on ALK+ ALCL PDX tumor samples treated and collected as indicated in (E). *P < 0.05, ** P < 0.01, and ****P < 0.0001. Significance was determined by unpaired, two-tailed Student’s t test. Data are shown as means ± SD. For Western blots, β-actin was used as a loading control, and two independent experiments with similar results were performed.
PI3Kγ signaling activates the MAPK pathway through the CCL19/CCL21-CCR7 chemokine receptor axis
PI3Kγ is a class IB PI3K typically activated by G protein–coupled receptors (GPCRs) (33, 34). We analyzed RNA-seq data for the expression of GPCRs known to activate PI3Kγ signaling and found that most ALK+ ALCL lines, primary tumors, and PDXs expressed CCR7 and C-X-C motif chemokine receptor 4 (CXCR4) (fig. S9, A to C), in accordance with previous studies on primary ALCL (35). Flow cytometry confirmed that ALK+ ALCL cell lines had moderate to high expression of CCR7, approaching the expression in normal T cells (fig. S9D). To further characterize CCR7 expression by ALK+ ALCL cells, we performed single-cell RNA-seq (scRNA-seq) analysis of two primary ALK+ ALCLs and one PDX. Both the primary cases and the PDX demonstrated CCR7 expression in the ALK+ lymphoma cells (Fig. 5, A and B, and fig. S10, A to C). The same population of ALK+ cells showed low to undetectable expression of PI3Kγ and PI3Kδ (fig. S10, D and E). Collectively, these data indicate that CCR7 is consistently expressed by ALK+ ALCL cells. In addition, inhibition of ALK by crizotinib or degradation of ALK with TL13–112 induced an increase in CCR7 mRNA expression and a twofold increase of CCR7 surface expression (Fig. 5, C to F, and fig. S11). Available chromatin immunoprecipitation sequencing (ChIP-seq) data (36) showed two distinct STAT3 binding sites in the CCR7 gene or 3′ downstream that were strongly reduced upon ALK inhibition (fig. S12A), prompting us to investigate the role of STAT3 in CCR7 regulation. Degradation of STAT3 by SD36 in ALK+ ALCL cell lines (COST, DEL, KARPAS-299, and SU-DHL1) resulted in increased CCR7 mRNA and protein expression (fig. S12, B to I), demonstrating that CCR7 expression is repressed in ALCL cells by ALK and STAT3 signaling.
Fig. 5. CCR7 is specifically expressed in ALK+ cells in primary ALCL and derepressed by ALK degradation.
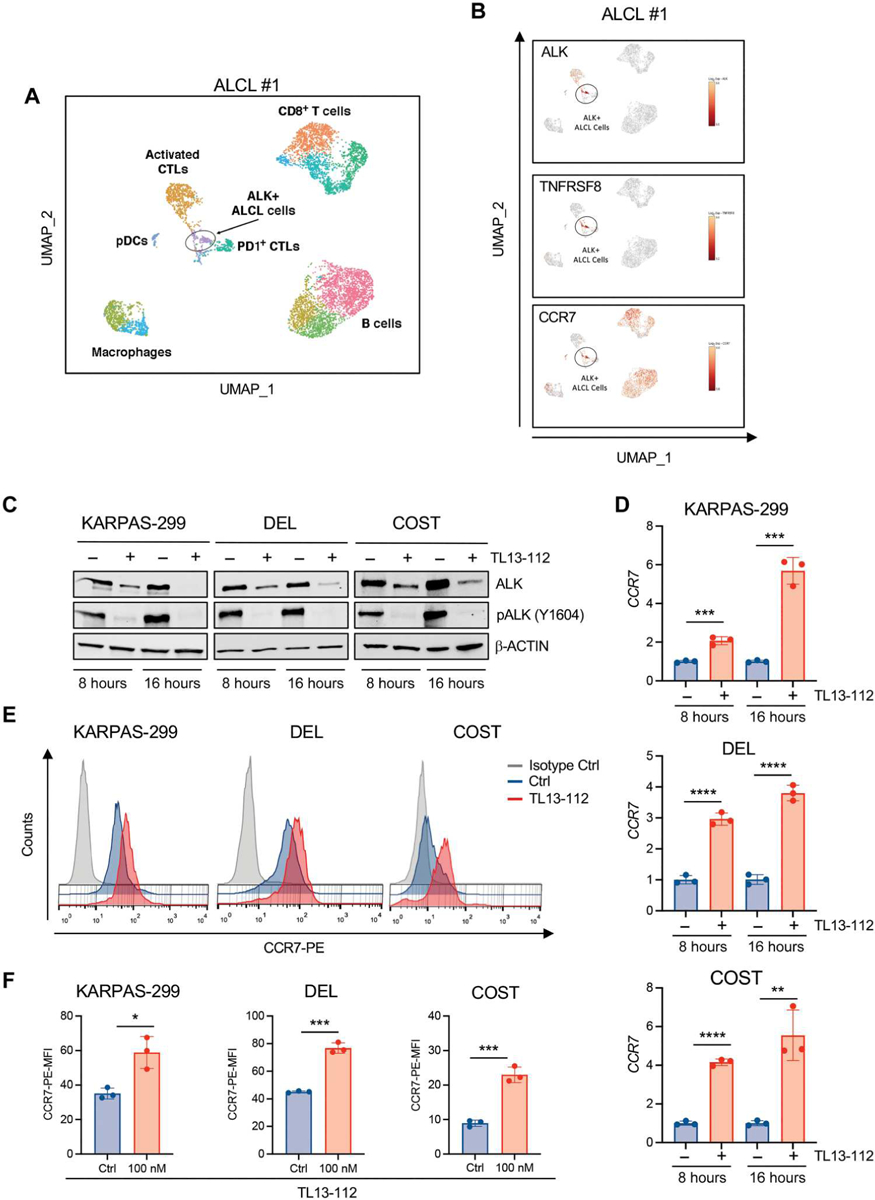
(A) Uniform Manifold Approximation and Projection (UMAP) plot of scRNA-seq data for CD45+ cells from a primary lymph node of a patient with ALK+ ALCL, color-coded by the main group of cell type. (B) UMAP plots [CD45+ cell positioning as shown in (A)] normalized for expression of selected genes. (C) Western blot analysis of high-PI3Kγ–expressing ALK+ ALCL cells treated with TL13–112 (100 nM). (D) qRT-PCR analysis of CCR7 mRNA expression performed on ALK+ ALCL cell lines treated with TL13–112 (100 nM). n = 3 technical replicates. (E) CCR7 cell surface expression intensity measured by flow cytometry in ALK+ ALCL cell lines treated with dimethyl sulfoxide or TL13–112 (100 nM). KARPAS-299 cells were treated for 24 hours and DEL and COST cells for 16 hours. (F) Histograms show mean fluorescence intensity (MFI) of CCR7 cell surface expression in ALK+ ALCL cells treated as in (E). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Significance was determined by unpaired, two-tailed Student’s t test. Data are shown as means ± SD. For Western blots, β-actin was used as a loading control. Blots are representative of two independent experiments with similar results.
CCR7 is a GPCR that is essential for the activation, migration, and homing of B and T lymphocytes in various tissues and engages the PI3K-Akt signaling pathway (37–39). CCR7 is activated by chemokine (C-C motif) ligand 19 (CCL19) and 21 (CCL21), and PI3Kγ is required for the CCL19-induced chemotactic responses in T cells (40). Thus, we hypothesized that PI3Kγ mediates signaling resulting from stimulation of CCR7 by CCL19/21 in ALK+ ALCL. First, we tested cell lines from lymphomas arising in NPM-ALK/PI3KγWT/WT, NPM-ALK/PI3KγCX/CX (expressing active PI3KγCAAX), and NPM-ALK/PI3Kγ−/− mice (Fig. 6A). When treated with CCL19/21, NPM-ALK/PI3KγWT/WT and NPM-ALK/PI3KγCX/CX lymphoma cells showed an increase in ERK1/2 phosphorylation, whereas pALK, pAkt, and pSTAT3 remained unchanged (Fig. 6A) (41). By contrast, increased ERK1/2 phosphorylation was reduced in NPM-ALK/PI3Kγ−/− cells (Fig. 6A). The activation of ERK1/2 induced by CCL19/21 in NPM-ALK/PI3KγWT/WT and NPM-ALK/PI3KγCX/CX lymphoma cells was almost completely blocked by treating cells with duvelisib, further supporting the conclusion that MAPK activation induced by CCR7 engagement is mediated by PI3Kγ in ALK+ murine lymphomas (Fig. 6B and fig. S13A). Next, we stimulated human ALK+ ALCL lines expressing either intermediate-high PI3Kγ (DEL, COST, KARPAS-229, and SUP-M2) or low PI3Kγ (TS and JB6) with CCL19/21. CCL19/21 increased ERK1/2 and Akt phosphorylation in cells with high PI3Kγ even when cells were treated with crizotinib to abrogate the effects of ALK signaling (Fig. 6C and fig. S13B). Furthermore, as in mouse cells, duvelisib blocked the activation of ERK1/2 induced by CCL19/21 in both treatment-naïve and TKI-resistant human ALK+ ALCL cells (Fig. 6D and fig. S13C).
Fig. 6. PI3Kγ signaling activates MAPK pathway through CCR7.
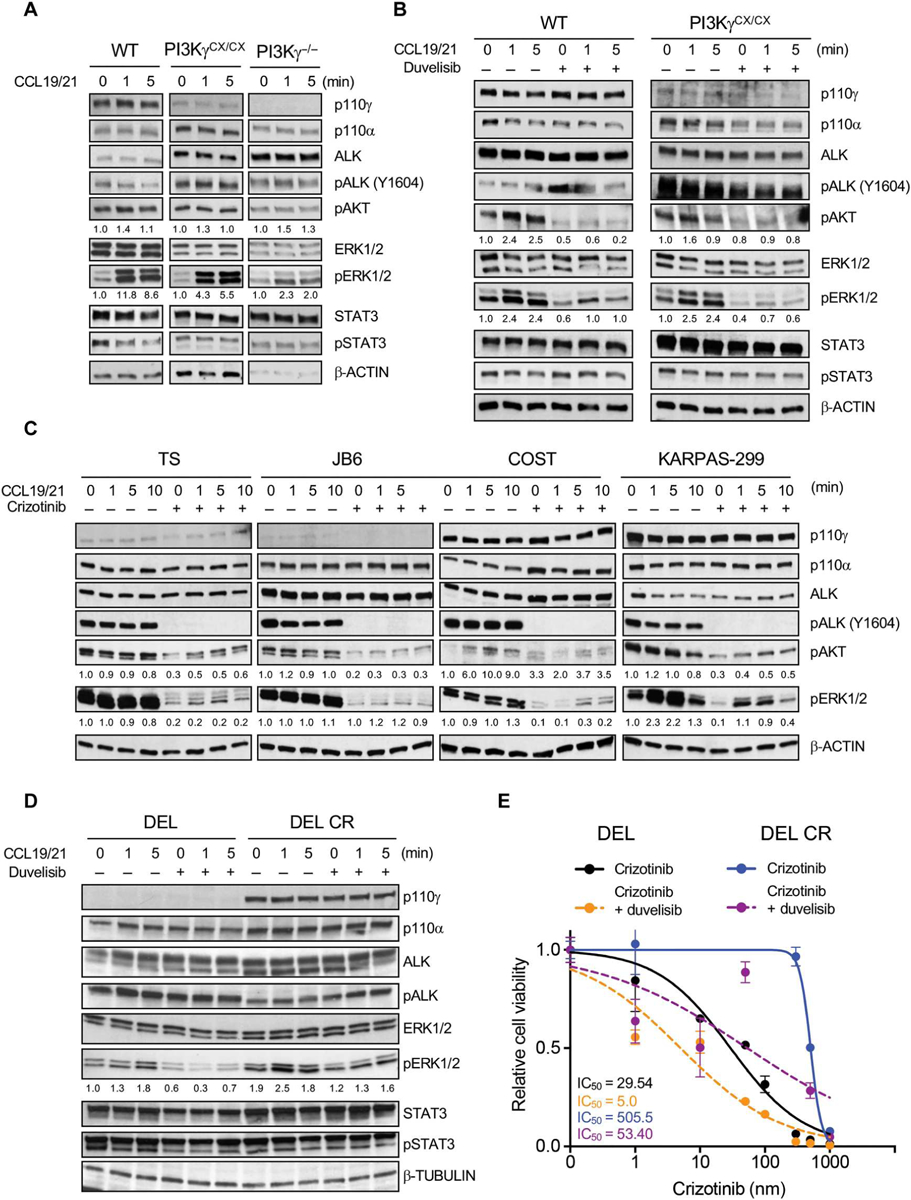
(A) Western blot analysis performed on of NPM-ALK+ lymphoma cells derived from primary tumors collected in mice with the indicated genotypes and stimulated with CCL19/21 (100 ng/ml) ex vivo. (B) Western blot analysis of NPM-ALK+ lymphoma cells derived from primary tumors collected in mice with the indicated genotypes. Cells were stimulated with CCL19/21 (100 ng/ml) and treated with duvelisib (1 μM) for 3 hours ex vivo. (C) Western blot analysis performed on human ALK+ ALCL cell lines. Cells were stimulated with CCL19/21 (100 ng/ml) and left untreated or treated with crizotinib (300 nM) for 3 hours ex vivo. (D) Western blot analysis of a crizotinib-sensitive and -resistant (CR) ALK+ ALCL cell lines stimulated with CCL19/21 (100 ng/ml) in the presence or absence of duvelisib (1 μM). β-Tubulin was used as a loading control. (E) Dose-response curves of crizotinib-sensitive (DEL) and CR (DEL-CR) ALK+ ALCL cells treated with increasing concentrations of crizotinib in single or in combination with duvelisib (10 μM). n = 3 technical replicates. Data are shown as means ± SD. For Western blots, β-actin was used as a loading control. Blots are representative of two independent experiments with similar results.
Duvelisib also partially restored the sensitivity to crizotinib or lorlatinib in cells that had up-regulated PI3Kγ expression while developing resistance to ALK TKI (Fig. 6E and fig. S13, D and E). Thus, CCR7 engagement results in PI3Kγ-dependent activation of the MAPK pathway in ALK+ ALCL and may convey TKI resistance.
The perivascular niche protects lymphoma cells from the effects of crizotinib treatment
ALK+ ALCL cells can secrete cytokines, such as IL-10 and IL-17, that support cell survival through an autocrine loop (42–45), but we did not detect CCL19/21 expression in ALK+ ALCL cell lines (fig. S9A) or ALK+ ALCL cells from two primary tumors and a PDX (fig. S14, A to C). In contrast, CCL19/21 was expressed in the tumor microenvironment by vascular and stromal cells (fig. S14D), suggesting that ALK+ ALCL might engage CCR7 through CCL19/21 produced in the microenvironment. ALK+ ALCL cells tend to preferentially colonize the perivascular and intravascular spaces (Fig. 7A) (16). In addition, rare ALK+ persister cells are found in the proximity of vessels in bone marrow biopsies obtained from patients with ALK+ ALCL in clinical remission (Fig. 7B). Thus, we reasoned that the CCL19/21 secreted by endothelial cells (ECs) may sustain MAPK activation through an engagement of the CCR7-PI3Kγ axis to confer resistance to crizotinib. First, we generated three CCR7 knockout ALCL cell lines by deleting the CCR7 gene using CRISPR-Cas9. The effective deletion was confirmed by two independent anti-CCR7 antibodies (fig. S15, A and B). Then, we used a three-dimensional (3D) ALCL-vascular microfluidic chip (46) to study the interaction of ALCL cells with ECs (Fig. 7C). The 3D vascular model consisted of a well-formed and perfusable macrovessel with ALCL flowing inside and interacting with the ECs (Fig. 7C). In this model, we investigated whether the presence of human umbilical vein ECs (HUVECs) that produce CCL19 and CCL21 (fig. S14E) would protect ALCL cells from the apoptotic effects induced by crizotinib through an engagement of the CCR7 receptor. In the absence of HUVEC cells, crizotinib treatment had the same effect on cell viability in CCR7WT or CCR7KO ALK+ ALCL cells, consistent with a lack of a CCL19/21-CCR7–mediated autocrine loop (Fig 7, D and E, and fig. S15, C and D). In contrast, the presence of HUVEC cells increased resistance to crizotinib, and the addition of duvelisib abrogated the protective effect provided by ECs (Fig. 7D and fig. S15, C and D). Deletion of CCR7 in ALK+ ALCL cells reduced, but did not completely abrogate, the protective effect of HUVEC cells (Fig. 7E and fig. S15, E and F). Last, we leveraged the 3D ALCL-vascular microfluidic chip to test a larger series of PI3K inhibitors. ALK+ ALCL cells were cultivated in the presence of HUVEC cells and treated with the PI3Kγ-specific inhibitor eganelisib and two PI3Kδ-selective inhibitors, acalisib and idelalisib. In these experimental settings, only duvelisib and eganelisib potentiated the effect of crizotinib, whereas acalisib and idelalisib did not show a significant effect (fig. S15G). Overall, these data confirm that PI3Kγ inhibition overcomes the microenvironment-mediated resistance to ALK TKIs and supports the use of eganelisib as an alternative to duvelisib for the treatment of patients with ALK+ ALCL in combination with an ALK TKI.
Fig. 7. The perivascular niche protects ALK+ ALCL cells from crizotinib–induced apoptosis.
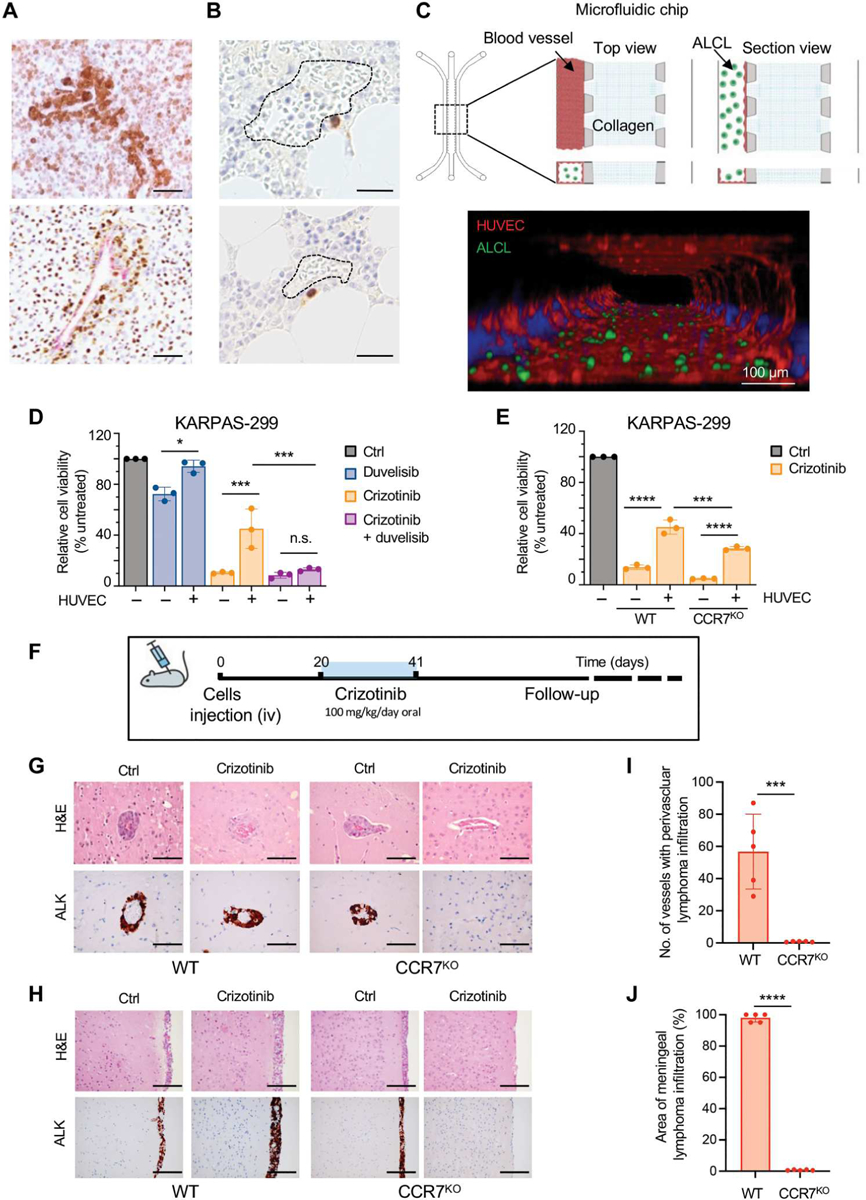
(A) Representative IHC for ALK in primary ALK+ ALCL lymph nodes showing perivascular colonization of ALK+ cells. Scale bars, 100 μm. (B) Perivascular distribution of rare persister ALK+ cells (stained in brown) detected by anti-ALK IHC in bone marrow biopsies obtained from patients with ALK+ ALCL in clinical remission. Dotted line: bone marrow vascular space. Scale bars, 100 μm. (C) Top: Schematic representation of the microfluidic chip (left) and schematic of microphysio-logical model of ALCL-vascular interaction and blood vessel formation in a 3D microfluidic model (right). A commercial microfluidic chip with a central collagen hydrogel channel (collagen), flanked by two fluidic media channels, was used (blood vessels). ALCL cells inside the channel (ALCL). Bottom: Confocal microscope imaging of macrovessels (F-actin, red) and ALK+ ALCL (GFP, green) in the microfluidic model. ALK+ ALCL cells were transduced with a lentivirus-expressing enhanced green fluorescent protein. Scale bar, 100 μm. (D) Cell viability of KARPAS-299CCR7WT and KARPAS-299CCR7KO in coculture with or without blood vessels (HUVEC) treated with crizotinib (300 nM) or duvelisib (10 μM) or in combination. (E) Cell viability KARPAS-299CCR7WT and KARPAS-299CCR7KO in coculture with or without HUVEC treated with crizotinib (300 nM) for 72 hours. n = 3 biological replicates. *P < 0.05, ***P < 0.001, and ****P < 0.0001, n.s., not significant. Significance was determined by one-way ANOVA. (F) Schematic representation of the in vivo experiment. COSTCCR7WT and COSTCCR7KO were injected intravenously (iv) into NSG mice, and mice were treated with crizotinib (100 mg/kg/die). (G and H) Representative H&E staining (top) and IHC for ALK (bottom) of the central nervous system of mice injected with COSTCCR7WT and COSTCCR7KO without or with crizotinib treatment. Perivascular [(G), scale bars, 100 μm] or meningeal [(H), scale bars, 200 μm] infiltration of ALK+ lymphoma cells. (I and J) Histograms of lymphoma cell infiltration in the brain perivascular vessels (I) or meninges (J) of mice inoculated with COSTCCR7WT (n = 5) and COSTCCR7KO (n = 5) and treated with crizotinib as in (F). ***P < 0.001 and ****P < 0.0001. Significance was determined by unpaired, two-tailed Student’s t test. Data are shown as means ± SD.
These data demonstrate that resistance to ALK TKI is enhanced by the presence of ECs in the tumor microenvironment at least partially through an engagement of the CCR7 receptor. To study whether the CCR7 signaling would support persistence of ALK+ ALCL cells in vivo, we evaluated whether CCR7 would increase TKI resistance in a mouse experimental model designed to mimic the relapse of perivascular lymphoma upon crizotinib suspension (Fig. 7F). NSG mice engrafted with CCR7WT or CCR7KO ALK+ ALCL cells (KARPAS-299) developed systemic disease involving several organs. including the brain, wherein ALK+ ALCL cells disseminated to the perivascular space and to the vascularized meningeal space (Fig. 7, G and H), allowing for a histologic characterization of the perivascular niche (Fig. 7G). Although the perivascular and meningeal growth of CCR7WT ALK+ ALCL was not impaired by treatment with crizotinib, the same treatment eradicated CCR7KO ALK+ ALCL cells from the perivascular and meningeal spaces (Fig. 7, G to J, and table S4). We conclude that an engagement of the CCR7 receptor may lead to resistance of ALK+ ALCL to ALK TKIs by preventing the eradication of the perivascular persister lymphoma cells.
DISCUSSION
In this study, we identified an actionable bypass mechanism of TKI resistance in ALK+ ALCL that is mediated by the CCR7-PI3Kγ signaling axis to sustain prosurvival signaling during ALK blockade. This mechanism is different from those exploited by ALK+ NSCLC, which typically rely on the activation of compensatory RTKs, such as MET and EGFR, to sustain oncogenic signaling including MAPK activation after ALK blockade (10). Although CCR7 is also expressed on epithelial tumors where it is implicated in the metastatic spread of NSCLC (47), MET and EGFR are not substantially expressed by ALCL cells, indicating a tumor-specific modality of activating survival signals during ALK inhibition. In addition, we found that ALK represses the CCR7-PI3Kγ axis and that ALK inhibition alleviates this repression leading to the up-regulation of PI3Kγ and CCR7. Thus, we speculate that ALCL cells exploit the plasticity of PI3Kγ and CCR7 up-regulation to activate rescue signals from the microenvironment during ALK blockade. These signals might contribute not only to upfront TKI resistance seen in a fraction of patients with ALK+ ALCL (5, 23) but also to the long-term persistence of ALCL cells in patients treated with ALK TKIs (6, 8).
The ALK-mediated repression of PI3Kγ and CCR7 requires STAT3, a master signaling mediator activated by oncogenic ALK (15, 48). ALK signaling through STAT3 also repressed the expression of PI3Kδ, another PI3K enzyme with an important role in the normal biology of T cells (40, 49, 50). PI3Kδ is required for optimal maturation (30) and potent TCR activation in T cells (51), whereas PI3Kγ contributes to optimal TCR activation by antigen-presenting cells by regulating F-actin polarization (52). Thus, repression of PI3Kγ and PI3Kδ in the presence of active ALK and STAT3 is consistent with the global repression of molecules involved in TCR signaling seen in ALK+ ALCL (25, 53). The suppression of the expression of the TCR and TCR-related downstream molecules in ALCL increases the biological fitness of cells that are driven by the potent and broad oncogenic activity of ALK (25, 26, 53). Likewise, the repression of PI3Kγ, PI3Kδ, and CCR7 in the presence of an active ALK signaling could represent a modality exploited by ALCL cells to prevent the hyperactivation of critical pathways that could result in a toxic excess of signaling (54, 55). We recently published that reduced sensitivity to crizotinib in ALCL can be partially mediated by an autocrine loop that activates IL-10 receptor signaling (12), whereas p110δ activates IL-10 production (56). Therefore, it is possible that the increased expression of PI3Kδ during ALK inhibition might also contribute to TKI resistance.
ALCL cells have a propensity to colonize the perivascular niches of blood and lymphatic vessels (Fig. 7A) (16). In 3D microfluidic chip models of the vascular microenvironment, the presence of ECs was sufficient to increase the resistance of ALCL cells to crizotinib. In these models, the protective effect of the ECs was reduced by inhibition of PI3Kγ with duvelisib or eganelisib, as well as genetic deletion of CCR7. CCR7 is specifically activated by the chemokines CCL19 and CCL21 that are secreted by ECs (57) and is critical for T cell homing and positioning within the lymph node (39, 58). CCR7 signaling is important for other hematologic malignancies, such as the homing to the lymph node and the survival of chronic lymphocytic leukemia (CLL) cells (59, 60) and the homing to the central nervous system of T cell acute lymphoblastic leukemia (T-ALL) cells (38). When engaged by its ligands, CCR7 activated the MAPK and PI3K-Akt pathways in ALK+ ALCL cells. This activation was blunted by the genetic deletion of PI3Kγ or by treatment with duvelisib, indicating that an engagement of CCR7 can sustain the activation of MAPK and PI3K-Akt through PI3Kγ signaling, two of the pathways with central roles in the pathophysiology of ALCL (14, 15). Together these findings suggest that the perivascular niche supports preferential homing but also the survival of ALCL persister cells.
Our study has several limitations. The focus of this manuscript is largely on p110γ and on its role in sustaining resistance to ALK TKI inhibition in ALK+ ALCL, but we cannot exclude that this might be one of the multiple avenues that ALCL cells can exploit to survive during TKI inhibition. In addition, p110δ might also have a role in mediating TKI resistance in ALCL, likely in other contexts or through different signaling pathways than the CCR7 receptor; therefore, further investigation is required by studying a large series of patients that develop resistance to ALK TKIs.
Our study has several potential therapeutic implications. First, PI3Kγ and CCR7 expressions are potential biomarkers for predicting the response of ALK+ ALCL to ALK TKIs. In the small cohort of patients analyzed and in ALCL cell lines, PI3Kγ expression positively correlated with resistance to crizotinib and other ALK TKIs. Overexpression of PI3Kγ or a constitutively active form of PI3Kγ (PI3KγCAAX) was sufficient to impart crizotinib resistance to ALCL cells with low baseline PI3Kγ expression. Although most patients undergo complete remission with crizotinib, a subset responds poorly and shows disease progression within the first 3 months of treatment (2). Additional analysis of PI3Kγ and CCR7 expression in a large cohort of patients with ALK+ ALCL treated with ALK TKIs will be needed to determine their utility as biomarkers. Second, PI3Kγ can be inhibited by the PI3Kγ/δ dual inhibitor duvelisib, which has shown efficacy as a single agent in treatment of T cell lymphoma (32) and currently is being tested in a phase 2 clinical trial of patients with R/R PTCL (NCT03372057) (61). Although duvelisib alone has very limited effects on ALK+ ALCL, likely because of the dominant signaling initiated by ALK, we demonstrated that duvelisib can potentiate the antitumor effects of crizotinib in vitro and in vivo. Although PI3Kα is also involved in ALK-mediated signaling in ALCL (24), the identification of PI3Kγ as the main PI3K that contributes to the activation of the PI3K pathways during ALK TKI resistance supports the use of the selective PI3Kγ/δ inhibitors despite their immune-mediated toxicities that can be challenging for patients (62) or of less-toxic PI3K inhibitors such as the PI3Kγ-selective eganelisib or the pan-PI3K inhibitor copanlisib.
CCR7/PI3Kγ signaling could also be targeted by blockade of the CCR7 receptor. CCR7 antibodies showed promising preclinical results in CLL (63, 64) and are now in a phase 1 clinical trial for refractory CLL (NCT04704323). CCR7 allosteric antagonists have also been identified (65). CCR7 blocking antibodies alter homing of CLL cells in lymph nodes (66). Thus, targeting CCR7 in ALK+ ALCL could not only block the survival signaling but also displace persister lymphoma cells from the perivascular niche during TKI inhibition. In addition, blockade of CCR7 in combination with ALK TKIs could prevent or reduce the central nervous system dissemination of ALCL in patients, a severe complication that is currently treated with central nervous system prophylaxis in patients at risk (67). Because part of the CCR7 signaling is independent of PI3Kγ, a combination of CCR7 blocking antibodies with duvelisib could generate a broader vertical inhibition of this axis to further increase sensitivity to ALK TKIs. Although single-cell analysis of ALCL samples showed that ALK+ cells distinctly express CCR7 and ECs produce CCL19/21, the microenvironment of ALK+ ALCL is made up of several other cell types, such as fibroblasts or macrophages that produce CCL19 or CCL21. Furthermore, experiments with 3D microfluidic chips demonstrated that protection from apoptosis induced by ECs was still partially observed in CCR7 knockout cells, suggesting that additional signaling pathways mediated by the endothelium might also contribute to TKI resistance. For example, we recently showed that IL-10 mediates resistance to ALK TKI (12) and a subset of ALK+ ALCL express CXCR4, a receptor involved in the formation of a vascular niche where residual T-ALL cells survive and contribute to leukemia progression (68).
Last, the concept of displacing lymphoma cells with blockade of the CCR7-PI3Kγ signaling axis could be extended to other T cell lymphomas. For example, ALK– ALCL also express CCR7 (35) and may also be driven by Janus kinase (JAK)/STAT activation or TK fusions (69, 70) that can be targeted by specific TKIs (71). Primary ALK– ALCL also shows the heterogeneity of PI3Kγ expression (Fig. 2B and fig. S5B) and high expression of CCR7 (fig. S9C). Thus, it is intriguing to predict that ALK– ALCLs or other T cell lymphoma would also benefit from therapies that combine duvelisib or CCR7 blockade with TKIs. Overall, our data suggest that combined treatments should be further evaluated in patients with TKI refractory lymphomas to determine whether they could delay relapses and possibly eradicate long-term persister cells.
MATERIALS AND METHODS
Study design
The primary objective of this study was to investigate mechanisms of resistance and persistence in ALK+ ALCL during treatment with ALK TKIs that contribute to relapse or treatment failure. To achieve this objective, we first analyzed RNA-seq data previously generated from primary lymphoma of patients with ALCL enrolled in the MAPPYACTS trial (ClinicalTrials.gov identifier: NCT02613962). The MAPPYACTS trial protocol, amendments, and informed consent were approved by the ethics committee and complied with local regulations and the Declaration of Helsinki (no. 2015-A00464–45) (12). We also generated in vitro ALK+ ALCL cell lines resistant to crizotinib and performed RNA-seq on both parental cells sensitive to crizotinib and resistant cells. Once we identified the involvement of the CCR7-PI3Kγ signaling as a potential mechanism of resistance, we used human ALCL cell lines and murine lymphoma cell lines derived from NPM-ALK transgenic mouse models, NPM-ALK transgenic mouse models, PI3Kγ knockout (PI3Kγ−/−) mice and transgenic mice expressing constitutive active PI3KγCAAX (PI3KγCX/CX), patients’ primary lymphoma samples, and ALCL PDXs.
Human and murine cell lines were used for in vitro studies, including Western blot, reverse transcription quantitative polymerase chain reaction (RT-qPCR), FISH, IC50 experiments, ChIP-seq analysis, and 3D lymph node perivascular niche modeling. Patients′ primary lymphoma samples obtained with informed consent under Dana-Farber/Harvard Cancer Center Institutional Review Board (IRB) protocol 14–076, and ALCL PDXs were used for scRNA-seq analysis. Patients’ primary samples for histological and immunohistochemical staining were enrolled in the phase 2 studies from 2010 to 2019 (NCT02419287 and EudraCT 2010-022978-14) (4).
Human ALCL cell lines and ALCL PDXs were also used for in vivo studies. All procedures that involved animals were approved by the Institutional Animal Care and Use Committee of University of Torino. Animal experiments were performed under protocols approved by the Italian Ministry of Health (no. 2542017-PR). In all experiments, mice of similar age and size were used across all groups. None of the mice was excluded from the analysis and no randomization or blinding method was used. The number of mice is presented in the figure legends. All experiments were performed independently at least three times except where noted. No data were excluded from the analysis. Histopathological evaluation was performed in a double-blind manner. Sample sizes, experimental replicates, and statistics are specified in the figures, figure legends, methods, and data files. Primary data are reported in data file S1.
Reagents and cell lines
Human ALK+ ALCL cell lines (TS, SU-DHL1, SUP-M2, JB6, SR-786, KARPAS-299, DEL, and L82) and ALK− cell lines (MAC-1, FEPD, CEM, and JURKAT) were obtained from DSMZ (German collection of microorganisms and cell cultures). Cells were passed for fewer than 6 months after receipt and resuscitation. The ALK+ ALCL cell line, COST, was provided by L. Lamant (Institut Universitaire du Cancer Toulouse Oncopole, Toulouse, France). Human embryonic kidney (HEK) 293FT packaging cells (HEK-293T and 293 Phoenix packaging cells) were used for lentivirus production and obtained from American Type Culture Collection. Doxycycline-inducible MAC1 Tet ON cells were previously described (36, 54). Cell lines were maintained in RPMI 1640 medium (Lonza) or Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS), 2% penicillin, streptomycin (5 mg/ml; Gibco), and 1% glutamine (Gibco). Cell lines were grown at 37°C in a humidified atmosphere with 5% CO2. All cell lines were routinely tested for mycoplasma contamination and always tested negative.
Murine cell lines were obtained from primary tumors from transgenic mice with corresponding genotypes and were maintained in RPMI 1640 medium containing 10% fetal calf serum as previously described (36). Frozen tissues of murine primary ALK+ lymphomas were treated as previously described (29).
Crizotinib and lorlatinib were provided by Pfizer and dissolved in dimethyl sulfoxide for in vitro experiments. ALK TKIs, brigatinib, ceritinib, and alectinib were purchased from Selleckchem. The dual inhibitor PI3Kγ/δ duvelisib; the PI3Kγ-specific inhibitor eganelisib (IPI-549/); and two PI3Kδ-selective inhibitors, acalisib and idelalisib, were purchased from Selleckchem. The selective ALK degrader TL13–112 and STAT3 degrader SD36 were purchased by MedChemExpress. All drugs were dissolved according to the manufacturer’s instructions.
Mice and in vivo experiments with xenografts and ALK+ ALCL PDXs
CD4-NPM-ALK, PI3Kγ−/−, and PI3KγCX/CX transgenic mice were described elsewhere (29, 31). Briefly, NSG mice were purchased from Charles River Laboratories.
For subcutaneous (sc) xenografts, ALK+ ALCL cell lines (5 × 106 cells) were injected into both flanks of NSG mice. Treatment started when tumors reached a volume ranging between 300 and 500 mm3. Mice were treated with duvelisib (10 mg/kg), crizotinib (30 mg/kg), or a combination of both by oral gavage once a day for a week. Control mice were treated with a vehicle. Tumor size was measured with a caliper every 2 days. Both drugs were dissolved in 0.5% methylcellulose + 0.05% Tween 80. Mice were euthanized at a humane endpoint.
For intravenous injection, NSG mice were inoculated intravenously (iv) with ALK+ ALCL cell lines (5 × 106 cells) in 0.2 ml of phosphate-buffered saline (PBS). According to our previous experience, treatment started 15 days after cell injection. Mice were treated with crizotinib, monitored during follow-up, and euthanized at a humane endpoint under anesthesia. All organs were isolated and immediately fixed in formalin solution for histopathological examination as previously described (72).
ALK+ ALCL PDX was kindly provided by G. Inghirami (Weill Cornell Medical College, NY, USA) (73). ALK+ ALCL PDX was generated from a patient with crizotinib-naïve ALCL. Cells were injected in both flanks of NSG mice. Treatment started when tumors reached a volume ranging between 300 and 500 mm3. Mice were treated with duvelisib (10 mg/kg), crizotinib (30 mg/kg), or a combination of the two inhibitors by oral gavage as described above. Mice were euthanized at a humane endpoint.
Animals were housed and maintained in the Animal Facility of the Department of Molecular Biotechnology and Health Sciences of University of Torino. Animal experiments were performed under protocols approved by the Italian Ministry of Health for University of Torino (approval no. 2542017-PR). None of the mice was excluded from the analysis, and no randomization or blinding method was used.
In vitro generation of resistant cell lines
To generate CR ALK+ ALCL cell lines, we added crizotinib to the medium at a starting concentration of 10 nM. The medium was replaced with fresh RPMI 1640 supplemented with crizotinib every 48 or 72 hours, and cell viability was assessed by flow cytometry after staining with 200 nM tetramethylrhodamine, methyl ester. After two or three passages at the same crizotinib concentration, a higher dose of crizotinib was added to the medium until a final concentration of 1 μM was achieved. The resulting pool of resistant cells (designated CR) was maintained in RPMI 1640 with 10% FBS containing 0.5 to 1 μM crizotinib.
CCR7 knockout by CRISPR-Cas9 gene editing
CCR7 was knocked out with a CRISPR-Cas9 system. Two (74) single-guide RNAs (sgRNA) targeting exon 3 of human CCR7 gene were designed using deskgene tool (www.deskgen.com) [CCR7 exon 3 sgRNA 1: GAGGTCACGGACGATTACAT; PAM CGG; cut site: chr17 (+40,555,783: −40,555,783); GC%: 50; activity score: 63; off-target score: 99. CCR7 exon 3 sgRNA 2: CGACCAGCCCATTGCCCAGT; PAM AGG; cut site: chr17 (+40,555,657: −40,555,657); GC%: 65; activity score: 56; off-target score: 94] and cloned into LentiCRISPRv2 puro (Addgene, plasmid #52961). Lentiviral particles were produced in HEK-293T cells, and viral supernatant was used to transduce ALCL cell lines (5 × 106 cells/ml) that were then infected with LentiCRISPRv2 puro containing CCR7 sgRNA #1 or CCR7 sgRNA #2, according to the protocol described above. After infection and 3 days of puromycin selection, cells were grown for 10 additional days to allow gene editing. CCR7 expression was assessed by flow cytometry and two different antibodies. Because of the CCR7 expression heterogeneity within each cell line, CCR7-negative populations were sorted using BD FACSA-ria II (BD Biosciences), and single-cell clones were obtained through sequential dilutions. CCR7 expression was then reassessed in each single-cell clone. To confirm the biallelic deletion of the clones (biallelic knockout), the PCR products were cloned and sequenced.
Human studies
Clinical and molecular data from patients with ALK+ ALCL relapsed on ALK inhibitor treatment (n = 2) or sensitive to ALK inhibitor treatment (n = 2) were obtained from both male (n = 3) and female (n = 1) pediatric participants included in the MAPPYACTS trial (ClinicalTrials.gov identifier: NCT02613962; https://clinicaltrials.gov/ct2/show/NCT02613962) with informed consent. The MAPPYACTS trial protocol, amendments, and informed consent were approved by the ethics committee and complied with local regulations and the Declaration of Helsinki (no. 2015-A00464–45). Additional fresh ALK+ lymphoma tissue for scRNA-seq experiments was obtained with informed consent under Dana-Farber/Harvard Cancer Center IRB protocol 14–076. For histological and immunohistochemical staining, primary samples were collected from patients enrolled in the phase 2 studies from 2010 to 2019 (NCT02419287 and EudraCT 2010-022978-14).
mRNA-seq data analysis of human samples
Libraries were prepared with the TruSeq Stranded mRNA kit (Illumina) according to supplier recommendations. Each transcriptome library was sequenced on an Illumina NextSeq500 as paired-end 75–base pair sequencing mode. Data were provided under the MAPPYACTS protocol and have been published (12). Raw paired-end FASTQ files were mapped against the human reference genome (GRCh38.p12) and GENCODE transcriptome annotation version v29 using Salmon (version 0.14.0) with the default parameters (75, 76). Strand-specific transcript counts were converted into gene counts with the tximport package in R (77). Differential gene expression analysis and normalization were performed using the edge R package in R (78).
GO analysis
GO analysis was performed with the topGO package (Alexa A, Rahnenführer J. topGO: Enrichment Analysis for GO. doi:10.18129/B9.bioc.topGO) in R using pathway annotations from the KEGG (79).
3D ALCL-vascular microfluidic chip
The 3D ALCL-vascular microfluidic chip models were developed using a commercial microfluidic chip “3-D cell culture chip” (DAX-1, AIM Biotech) as previously described (80). To generate the 3D microfluidic chip model, a mixture of collagen hydrogel was prepared by dissolving collagen I rat tail into 10× PBS with phenol red (Sigma-Aldrich, 114537–5G) followed by pH adjustment by NaOH solution. A pH of 7.0 to 7.5 was confirmed using Panpeha Whatman paper (Sigma-Aldrich). The collagen hydrogel (final concentration of 2.5 mg/ml) was injected into the center gel region of the 3D microfluidic chamber (10 to 12 μl per microfluidic chamber). Before injection, collagen hydrogel was kept on ice. After incubation for 40 min at 37°C in sterile humidity chambers, collagen hydrogel channels were hydrated with RPMI 1640. Then, all the side walls of one flanked channel (media channel) were coated with a collagen solution (150 μg/ml) in PBS to allow better adhesion of ECs to the channel. After 15 min, the channel was washed once with a medium. To create the 3D macrovessel, a 50-μl cell suspension of HUVECs (3 × 106 cells/ml; Lonza, C2519AS) cultured in VascuLife medium (Lifeline) was injected in the media channel coated with collagen. The microfluidic chip was rotated twice to create the 3D macrovessel with a confluent hollow lumen. To allow the cells to attach to the media-gel interface to form a monolayer, the chip was placed face down for 15 min to let cells adhere. After that, a 50-μl cell suspension was reinjected, and the chip was faced upside down to cover the upper part of the 3D vascular channel. After 90 min of incubation in the humidity chamber in an incubator at 37°C, cell culture medium was gently added in both media channels. Chips were placed in an incubator to form a confluent monolayer. The day after, ALCL cells (2 × 105 cells/ml) (COST, KARPAS-299, and DEL), WT cells, or CCR7KO cells were added into the 3D macrovessel. The medium was supplemented with 300 nM crizotinib, 10 μM duvelisib, or their combination. Microfluidic chips were cultured for 72 hours, and cell proliferation analysis was performed using CellTiter-Glo Luminescent Cell Viability Assay (Promega). ALCL cells were collected from the microfluidic device, plated in a 96-well plate, mixed 1:1 with a CellTiterGlo (CTG) solution, and analyzed using the GloMax-Multi Detection System.
Statistical analysis
All experiments were performed at least two times. Results are presented as means ± SD or SEM. Statistical significance (P) was calculated using the nonparametric two-tailed Mann-Whitney U test, Student’s t test, and one- and two-way analysis of variance (ANOVA), Mantel-Cox and Kaplan-Meier tests when appropriate (GraphPad Software, La Jolla, CA, USA). All statistical analysis was performed using GraphPad Prism. The numbers of samples and independent biological experimental repeats are indicated in the figures or figure legends. P < 0.05 was considered statistically significant, where *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. The statistical analysis was performed using GraphPad Prism 9.0. software.
Supplementary Material
Acknowledgments:
We would like to thank M. Stella Scalzo, R. Dall’Olio, S. Germani, B. Mussolin, P. Gugliotta, and G. Lomazzo for technical assistance for immunohistochemical stains and FISH analysis and Q. Wang for analyzing data in fig. S5B.
Funding:
The work has been supported by grants R01 CA196703-01 to R. Chiarle; Leukemia & Lymphoma Society (LLS) SCOR grant to D.M.W., R. Chiarle, G.G.I., and J.C.A.; European Union Horizon 2020 Marie Sklodowska-Curie Innovative Training Network (ITN-ETN) grant award no. 675712 for the European Research Initiative for ALK-Related Malignancies (ERIA) to N.P., S.D.T., I.M., C.G.-P., and R. Chiarle; the FANTOM Marie Sklodowska-Curie Innovative Training Network (ITN-ETN) within the Horizon Europe - the Framework Programme for Research and Innovation (2021–2027) to S.D.T., C.G.-P., C.V., and R. Chiarle; the project National Institute for Cancer Research (Programme EXCELES, ID project no. LX22NPO5102) - Funded by the European Union - Next Generation EU to S.D.T.; the MAPPYACTS trial by the Institut National du Cancer grant PHRC-K14-175 to B.G.; the Foundation ARC grant MAPY201501241 to B.G.; the Association Imagine for Margo to B.G.; MFAG 9479 to C.A.; grant nos. CA229086, CA229100, CA195568, and LLS 7011-16 to G.G.I.; AIRC IG-21875 and MIUR PRIN 2017 to E.H.; AIRC IG-24828 to L. Mologni.; AIRC IG 2019 - ID. 23146 to C.V. M.C. was supported by the MIT-POLITO grant (BIOMODE - Compagnia di San Paolo) under the joint “Doctorate of Bioengineering and Medical-Surgical Sciences” of University of Turin and Politecnico di Torino and Fondazione “Franco e Marilisa Caligara. The laboratory of C.A. is supported by the Giovanni Armenise-Harvard Foundation.
Footnotes
Supplementary Materials
This PDF file includes:
Supplementary Materials and Methods
Other Supplementary Material for this manuscript includes the following:
MDAR Reproducibility Checklist
Competing interests: C.C.−M. works for Catapult Therapeutics that has developed the anti-CCR7 CAP100 antibody and is an employee and a shareholder of Immunological and Medical Products (IMMED S.L.).
Data and materials availability:
All data associated with this study are present in the paper or the Supplementary Materials. The RNA-seq data generated from ALCL cell lines sensitive and resistant to crizotinb are available at NCBI Gene Expression Omnibus (GEO) under accession number GSE173306 (token: ohqdkaawnfkpbon). scRNA-seq data generated from patients’ primary lymphoma samples and PDX are available at NCBI: www.ncbi.nlm.nih.gov/sra/PRJNA980417.
REFERENCES AND NOTES
- 1.Holla VR, Elamin YY, Bailey AM, Johnson AM, Litzenburger BC, Khotskaya YB, Sanchez NS, Zeng J, Shufean MA, Shaw KR, Mendelsohn J, Mills GB, Meric-Bernstam F, Simon GR, ALK: A tyrosine kinase target for cancer therapy. Cold Spring Harb. Mol. Case Stud 3, a001115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gambacorti-Passerini C, Orlov S, Zhang L, Braiteh F, Huang H, Esaki T, Horibe K, Ahn J-S, Beck JT, Edenfield WJ, Shi Y, Taylor M, Tamura K, Van Tine BA, Wu S-J, Paolini J, Selaru P, Kim TM, Long-term effects of crizotinib in ALK-positive tumors (excluding NSCLC): A phase 1b open-label study. Am. J. Hematol 93, 607–614 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mossé YP, Voss SD, Lim MS, Rolland D, Minard CG, Fox E, Adamson P, Wilner K, Blaney SM, Weigel BJ, Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: A children’s oncology group study. J. Clin. Oncol 35, 3215–3221 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rindone G, Aroldi A, Bossi E, Verga L, Zambrotta G, Tarantino S, Piazza R, Mussolin L, Chiarle R, Gambacorti-Passerini C, A monocentric analysis of the long-term safety and efficacy of crizotinib in relapsed/refractory ALK+ lymphomas. Blood Adv. 7, 314–316 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gambacorti Passerini C, Farina F, Stasia A, Redaelli S, Ceccon M, Mologni L, Messa C, Guerra L, Giudici G, Sala E, Mussolin L, Deeren D, King MH, Steurer M, Ordemann R, Cohen AM, Grube M, Bernard L, Chiriano G, Antolini L, Piazza R, Crizotinib in advanced, chemoresistant anaplastic lymphoma kinase-positive lymphoma patients. J. Natl. Cancer Inst 106, djt378 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Gambacorti-Passerini C, Mussolin L, Brugieres L, Abrupt relapse of ALK-positive lymphoma after discontinuation of crizotinib. N. Engl. J. Med 374, 95–96 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Graetz D, Crews KR, Azzato EM, Singh RK, Raimondi S, Mason J, Valentine M, Mullighan CG, Holland A, Inaba H, Leventaki V, Leukemic presentation of ALK-positive anaplastic large cell lymphoma with a novel partner, poly(A) binding protein cytoplasmic 1 (PABPC1), responding to single-agent crizotinib. Haematologica 104, e218–e221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bossi E, Aroldi A, Brioschi FA, Steidl C, Baretta S, Renso R, Verga L, Fontana D, Sharma GG, Mologni L, Mussolin L, Piazza R, Gambacorti-Passerini C, Phase two study of crizotinib in patients with anaplastic lymphoma kinase (ALK)-positive anaplastic large cell lymphoma relapsed/refractory to chemotherapy. Am. J. Hematol 95, E319–E321 (2020). [DOI] [PubMed] [Google Scholar]
- 9.Hata AN, Niederst MJ, Archibald HL, Gomez-Caraballo M, Siddiqui FM, Mulvey H-EC, Maruvka YE, Ji F, Bhang HE, Krishnamurthy Radhakrishna V, Siravegna G, Hu H, Raoof S, Lockerman E, Kalsy A, Lee D, Keating CL, Ruddy DA, Damon LJ, Crystal AS, Costa C, Piotrowska Z, Bardelli A, Iafrate AJ, Sadreyev RI, Stegmeier F, Getz G, Sequist LV, Faber AC, Engelman JA, Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med 22, 262–269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin JJ, Riely GJ, Shaw AT, Targeting ALK: Precision medicine takes on drug resistance. Cancer Discov. 7, 137–155 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sharma GG, Mota I, Mologni L, Patrucco E, Gambacorti-Passerini C, Chiarle R, Tumor resistance against ALK targeted therapy-where it comes from and where it goes. Cancers (Basel) 10, 62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Prokoph N, Probst NA, Lee LC, Monahan JM, Matthews JD, Liang H-C, Bahnsen K, Montes-Mojarro IA, Karaca-Atabay E, Sharma GG, Malik V, Larose H, Forde SD, Ducray SP, Lobello C, Wang Q, Luan S-L, Pospíšilová Š, Gambacorti-Passerini C, Burke GAA, Pervez S, Attarbaschi A, Janíková A, Pacquement H, Landman-Parker J, Lambilliotte A, Schleiermacher G, Klapper W, Jauch R, Woessmann W, Vassal G, Kenner L, Merkel O, Mologni L, Chiarle R, Brugières L, Geoerger B, Barbieri I, Turner SD, IL10RA modulates crizotinib sensitivity in NPM1-ALK+ anaplastic large cell lymphoma. Blood 136, 1657–1669 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laimer D, Dolznig H, Kollmann K, Vesely PW, Schlederer M, Merkel O, Schiefer A-I, Hassler MR, Heider S, Amenitsch L, Thallinger C, Staber PB, Simonitsch-Klupp I, Artaker M, Lagger S, Turner SD, Pileri S, Piccaluga PP, Valent P, Messana K, Landra I, Weichhart T, Knapp S, Shehata M, Todaro M, Sexl V, Höfler G, Piva R, Medico E, Ruggeri BA, Cheng M, Eferl R, Egger G, Penninger JM, Jaeger U, Moriggl R, Inghirami G, Kenner L, PDGFR blockade is a rational and effective therapy for NPM-ALK–driven lymphomas. Nat. Med 18, 1699–1704 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G, The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat. Rev. Cancer 8, 11–23 (2008). [DOI] [PubMed] [Google Scholar]
- 15.Hallberg B, Palmer RH, Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat. Rev. Cancer 13, 685–700 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES, The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 127, 2375–2390 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karaca Atabay E, Mecca C, Wang Q, Ambrogio C, Mota I, Prokoph N, Mura G, Martinengo C, Patrucco E, Leonardi G, Hossa J, Pich A, Mologni L, Gambacorti-Passerini C, Brugieres L, Geoerger B, Turner SD, Voena C, Cheong T-C, Chiarle R, Tyrosine phosphatases regulate resistance to ALK inhibitors in ALK+ anaplastic large cell lymphoma. Blood 139, 717–731 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A, Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A, Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Redaelli S, Ceccon M, Antolini L, Rigolio R, Pirola A, Peronaci M, Gambacorti-Passerini C, Mologni L, Synergistic activity of ALK and mTOR inhibitors for the treatment of NPM-ALK positive lymphoma. Oncotarget 7, 72886–72897 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Redaelli S, Ceccon M, Zappa M, Sharma GG, Mastini C, Mauri M, Nigoghossian M, Massimino L, Cordani N, Farina F, Piazza R, Gambacorti-Passerini C, Mologni L, Lorlatinib treatment elicits multiple on- and off-target mechanisms of resistance in ALK-driven cancer. Cancer Res. 78, 6866–6880 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Boi M, Rinaldi A, Kwee I, Bonetti P, Todaro M, Tabbo F, Piva R, Rancoita PM, Matolcsy A, Timar B, Tousseyn T, Rodríguez-Pinilla SM, Piris MA, Beà S, Campo E, Bhagat G, Swerdlow SH, Rosenwald A, Ponzoni M, Young KH, Piccaluga PP, Dummer R, Pileri S, Zucca E, Inghirami G, Bertoni F, PRDM1/BLIMP1 is commonly inactivated in anaplastic large T-cell lymphoma. Blood 122, 2683–2693 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Gambacorti-Passerini C, Messa C, Pogliani EM, Crizotinib in anaplastic large-cell lymphoma. N. Engl. J. Med 364, 775–776 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Slupianek A, Nieborowska-Skorska M, Hoser G, Morrione A, Majewski M, Xue L, Morris SW, Wasik MA, Skorski T, Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res. 61, 2194–2199 (2001). [PubMed] [Google Scholar]
- 25.Ambrogio C, Martinengo C, Voena C, Tondat F, Riera L, di Celle PF, Inghirami G, Chiarle R, NPM-ALK oncogenic tyrosine kinase controls T-cell identity by transcriptional regulation and epigenetic silencing in lymphoma cells. Cancer Res. 69, 8611–8619 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hassler MR, Pulverer W, Lakshminarasimhan R, Redl E, Hacker J, Garland GD, Merkel O, Schiefer A-I, Simonitsch-Klupp I, Kenner L, Weisenberger DJ, Weinhaeusel A, Turner SD, Egger G, Insights into the pathogenesis of anaplastic large-cell lymphoma through genome-wide DNA methylation profiling. Cell Rep. 17, 596–608 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Powell CE, Gao Y, Tan L, Donovan KA, Nowak RP, Loehr A, Bahcall M, Fischer ES, Janne PA, George RE, Gray NS, Chemically induced degradation of anaplastic lymphoma kinase (ALK). J. Med. Chem 61, 4249–4255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bai L, Zhou H, Xu R, Zhao Y, Chinnaswamy K, McEachern D, Chen J, Yang C-Y, Liu Z, Wang M, Liu L, Jiang H, Wen B, Kumar P, Meagher JL, Sun D, Stuckey JA, Wang S, A potent and selective small-molecule degrader of STAT3 achieves complete tumor regression in vivo. Cancer Cell 36, 498–511.e17 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chiarle R, Gong JZ, Guasparri I, Pesci A, Cai J, Liu J, Simmons WJ, Dhall G, Howes J, Piva R, Inghirami G, NPM-ALK transgenic mice spontaneously develop T-cell lymphomas and plasma cell tumors. Blood 101, 1919–1927 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Swat W, Montgrain V, Doggett TA, Douangpanya J, Puri K, Vermi W, Diacovo TG, Essential role of PI3Kdelta and PI3Kgamma in thymocyte survival. Blood 107, 2415–2422 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costa C, Barberis L, Ambrogio C, Manazza AD, Patrucco E, Azzolino O, Neilsen PO, Ciraolo E, Altruda F, Prestwich GD, Chiarle R, Wymann M, Ridley A, Hirsch E, Negative feedback regulation of Rac in leukocytes from mice expressing a constitutively active phosphatidylinositol 3-kinase gamma. Proc. Natl. Acad. Sci. U.S.A 104, 14354–14359 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horwitz SM, Koch R, Porcu P, Oki Y, Moskowitz A, Perez M, Myskowski P, Officer A, Jaffe JD, Morrow SN, Allen K, Douglas M, Stern H, Sweeney J, Kelly P, Kelly V, Aster JC, Weaver D, Foss FM, Weinstock DM, Activity of the PI3K-δ,γ inhibitor duvelisib in a phase 1 trial and preclinical models of T-cell lymphoma. Blood 131, 888–898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT, The PI3K pathway in human disease. Cell 170, 605–635 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fruman DA, Rommel C, PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov 13, 140–156 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamant L, de Reyniès A, Duplantier M-M, Rickman DS, Sabourdy F, Giuriato S, Brugières L, Gaulard P, Espinos E, Delsol G, Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK+ subtypes. Blood 109, 2156–2164 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Menotti M, Ambrogio C, Cheong T-C, Pighi C, Mota I, Cassel SH, Compagno M, Wang Q, Dall’Olio R, Minero VG, Poggio T, Sharma GG, Patrucco E, Mastini C, Choudhari R, Pich A, Zamo A, Piva R, Giliani S, Mologni L, Collings CK, Kadoch C, Gambacorti-Passerini C, Notarangelo LD, Anton IM, Voena C, Chiarle R, Wiskott-Aldrich syndrome protein (WASP) is a tumor suppressor in T cell lymphoma. Nat. Med 25, 130–140 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharma N, Benechet AP, Lefrançois L, Khanna KM, CD8 T cells enter the splenic T cell zones independently of CCR7, but the subsequent expansion and trafficking patterns of effector T cells after infection are dysregulated in the absence of CCR7 migratory cues. J. Immunol 195, 5227–5236 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buonamici S, Trimarchi T, Ruocco MG, Reavie L, Cathelin S, Mar BG, Klinakis A, Lukyanov Y, Tseng J-C, Sen F, Gehrie E, Li M, Newcomb E, Zavadil J, Meruelo D, Lipp M, Ibrahim S, Efstratiadis A, Zagzag D, Bromberg JS, Dustin ML, Aifantis I, CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature 459, 1000–1004 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Förster R, Davalos-Misslitz AC, Rot A, CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol 8, 362–371 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Reif K, Okkenhaug K, Sasaki T, Penninger JM, Vanhaesebroeck B, Cyster JG, Cutting edge: Differential roles for phosphoinositide 3-kinases, p110gamma and p110delta, in lymphocyte chemotaxis and homing. J. Immunol 173, 2236–2240 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Xu Y, Liu L, Qiu X, Liu Z, Li H, Li Z, Luo W, Wang E, CCL21/CCR7 prevents apoptosis via the ERK pathway in human non-small cell lung cancer cells. PLOS ONE 7, e33262 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bard JD, Gelebart P, Anand M, Amin HM, Lai R, Aberrant expression of IL-22 receptor 1 and autocrine IL-22 stimulation contribute to tumorigenicity in ALK+ anaplastic large cell lymphoma. Leu?kemia 22, 1595–1603 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Knörr F, Damm-Welk C, Ruf S, Singh VK, Zimmermann M, Reiter A, Woessmann W, Blood cytokine concentrations in pediatric patients with anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Haematologica 103, 477–485 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mellgren K, Hedegaard CJ, Schmiegelow K, Müller K, Plasma cytokine profiles at diagnosis in pediatric patients with non-hodgkin lymphoma. J. Pediatr. Hematol. Oncol 34, 271–275 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Matsuyama H, Suzuki HI, Nishimori H, Noguchi M, Yao T, Komatsu N, Mano H, Sugimoto K, Miyazono K, miR-135b mediates NPM-ALK-driven oncogenicity and renders IL-17-producing immunophenotype to anaplastic large cell lymphoma. Blood 118, 6881–6892 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Campisi M, Shin Y, Osaki T, Hajal C, Chiono V, Kamm RD, 3D self-organized micro-vascular model of the human blood-brain barrier with endothelial cells, pericytes and astrocytes. Biomaterials 180, 117–129 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang S, Wang H, Xu Z, Bai Y, Xu L, Lymphatic metastasis of NSCLC involves chemotaxis effects of lymphatic endothelial cells through the CCR7-CCL21 axis modulated by TNF-α. Genes (Basel) 11, 1309 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chiarle R, Simmons WJ, Cai H, Dhall G, Zamo A, Raz R, Karras JG, Levy DE, Inghirami G, Stat3 is required for ALK-mediated lymphomagenesis and provides a possible therapeutic target. Nat. Med 11, 623–629 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Okkenhaug K, Signaling by the phosphoinositide 3-kinase family in immune cells. Annu. Rev. Immunol 31, 675–704 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B, The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell Biol 11, 329–341 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, Pearce W, Meek SE, Salpekar A, Waterfield MD, Smith AJ, Vanhaesebroeck B, Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science 297, 1031–1034 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Alcázar I, Marqués M, Kumar A, Hirsch E, Wymann M, Carrera AC, Barber DF, Phosphoinositide 3-kinase γ participates in T cell receptor-induced T cell activation. J. Exp. Med 204, 2977–2987 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Malcolm TIM, Villarese P, Fairbairn CJ, Lamant L, Trinquand A, Hook CE, Burke GA, Brugières L, Hughes K, Payet D, Merkel O, Schiefer A-I, Ashankyty I, Mian S, Wasik M, Turner M, Kenner L, Asnafi V, Macintyre E, Turner SD, Anaplastic large cell lymphoma arises in thymocytes and requires transient TCR expression for thymic egress. Nat. Commun 7, 10087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ceccon M, Merlo ME, Mologni L, Poggio T, Varesio LM, Menotti M, Bombelli S, Rigolio R, Manazza AD, Di Giacomo F, Ambrogio C, Giudici G, Casati C, Mastini C, Compagno M, Turner SD, Gambacorti-Passerini C, Chiarle R, Voena C, Excess of NPM-ALK oncogenic signaling promotes cellular apoptosis and drug dependency. Oncogene 35, 3854–3865 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rajan SS, Amin AD, Li L, Rolland DC, Li H, Kwon D, Kweh MF, Arumov A, Roberts ER, Yan A, Basrur V, Elenitoba-Johnson KSJ, Chen XS, Puvvada SD, Lussier YA, Bilbao D, Lim MS, Schatz JH, The mechanism of cancer drug addiction in ALK-positive T-cell lymphoma. Oncogene 39, 2103–2117 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dil N, Marshall AJ, Role of phosphoinositide 3-kinase p110 δ in TLR4- and TLR9-mediated B cell cytokine production and differentiation. Mol. Immunol 46, 1970–1978 (2009). [DOI] [PubMed] [Google Scholar]
- 57.Saxena V, Li L, Paluskievicz C, Kasinath V, Bean A, Abdi R, Jewell CM, Bromberg JS, Role of lymph node stroma and microenvironment in T cell tolerance. Immunol. Rev 292, 9–23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hauser MA, Legler DF, Common and biased signaling pathways of the chemokine receptor CCR7 elicited by its ligands CCL19 and CCL21 in leukocytes. J. Leukoc. Biol 99, 869–882 (2016). [DOI] [PubMed] [Google Scholar]
- 59.Till KJ, Lin K, Zuzel M, Cawley JC, The chemokine receptor CCR7 and alpha4 integrin are important for migration of chronic lymphocytic leukemia cells into lymph nodes. Blood 99, 2977–2984 (2002). [DOI] [PubMed] [Google Scholar]
- 60.Cuesta-Mateos C, Lopez-Giral S, Alfonso-Perez M, de Soria VG, Loscertales J, Guasch-Vidal S, Beltran AE, Zapata JM, Munoz-Calleja C, Analysis of migratory and prosurvival pathways induced by the homeostatic chemokines CCL19 and CCL21 in B-cell chronic lymphocytic leukemia. Exp. Hematol 38, 756–764 (2010). [DOI] [PubMed] [Google Scholar]
- 61.Pro B, Casulo C, Jacobsen E, Mead M, Mehta-Shah N, Zain JM, Zinzani PL, Lustgarten S, Youssoufian H, Horwitz SM, Duvelisib in patients with relapsed/refractory peripheral T-cell lymphoma from the phase 2 Primo trial: Dose optimization efficacy update and expansion phase initial results, paper presented at the 62nd ASH Annual Meeting, 5 to 8 December 2020. [Google Scholar]
- 62.Flinn IW, O’Brien S, Kahl B, Patel M, Oki Y, Foss FF, Porcu P, Jones J, Burger JA, Jain N, Kelly VM, Allen K, Douglas M, Sweeney J, Kelly P, Horwitz S, Duvelisib, a novel oral dual inhibitor of PI3K- δ,γ, is clinically active in advanced hematologic malignancies. Blood 131, 877–887 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Alfonso-Pérez M, López-Giral S, Quintana NE, Loscertales J, Martín-Jiménez P, Muñoz C, Anti-CCR7 monoclonal antibodies as a novel tool for the treatment of chronic lymphocyte leukemia. J. Leukoc. Biol 79, 1157–1165 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Mateu-Albero T, Juárez-Sánchez R, Loscertales J, Mol W, Terrón F, Muñoz-Calleja C, Cuesta-Mateos C, Effect of ibrutinib on CCR7 expression and functionality in chronic lymphocytic leukemia and its implication for the activity of CAP-100, a novel therapeutic anti-CCR7 antibody. Cancer Immunol. Immunother 71, 627–636 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jaeger K, Bruenle S, Weinert T, Guba W, Muehle J, Miyazaki T, Weber M, Furrer A, Haenggi N, Tetaz T, Huang C-Y, Mattle D, Vonach J-M, Gast A, Kuglstatter A, Rudolph MG, Nogly P, Benz J, Dawson RJP, Standfuss J, Structural basis for allosteric ligand recognition in the human CC chemokine receptor 7. Cell 178, 1222–1230.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cuesta-Mateos C, Juárez-Sánchez R, Mateu-Albero T, Loscertales J, Mol W, Terrón F, Muñoz-Calleja C, Targeting cancer homing into the lymph node with a novel anti-CCR7 therapeutic antibody: The paradigm of CLL. MAbs 13, 1917484 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Del Baldo G, Abbas R, Woessmann W, Horibe K, Pillon M, Burke A, Beishuizen A, Rigaud C, Le Deley M-C, Lamant L, Brugières L, Neuro-meningeal relapse in anaplastic large-cell lymphoma: Incidence, risk factors and prognosis—A report from the European intergroup for childhood non-Hodgkin lymphoma. Br. J. Haematol 192, 1039–1048 (2021). [DOI] [PubMed] [Google Scholar]
- 68.Pitt LA, Tikhonova AN, Hu H, Trimarchi T, King B, Gong Y, Sanchez-Martin M, Tsirigos A, Littman DR, Ferrando AA, Morrison SJ, Fooksman DR, Aifantis I, Schwab SR, CXCL12-producing vascular endothelial niches control acute T cell leukemia maintenance. Cancer Cell 27, 755–768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crescenzo R, Abate F, Lasorsa E, Tabbo’ F, Gaudiano M, Chiesa N, Giacomo FD, Spaccarotella E, Barbarossa L, Ercole E, Todaro M, Boi M, Acquaviva A, Ficarra E, Novero D, Rinaldi A, Tousseyn T, Rosenwald A, Kenner L, Cerroni L, Tzankov A, Ponzoni M, Paulli M, Weisenburger D, Chan WC, Iqbal J, Piris MA, Zamo’ A, Ciardullo C, Rossi D, Gaidano G, Pileri S, Tiacci E, Falini B, Shultz LD, Mevellec L, Vialard JE, Piva R, Bertoni F, Rabadan R, Inghirami G; European T-Cell Lymphoma Study Group; T-Cell Project: Prospective Collection of Data in Patients with Peripheral T-Cell Lymphoma and the AIRC 5xMille Consortium “Genetics-Driven Targeted Management of Lymphoid Malignancies, Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell 27, 516–532 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen J, Zhang Y, Petrus MN, Xiao W, Nicolae A, Raffeld M, Pittaluga S, Bamford RN, Nakagawa M, Ouyang ST, Epstein AL, Kadin ME, Del Mistro A, Woessner R, Jaffe ES, Waldmann TA, Cytokine receptor signaling is required for the survival of ALK−anaplastic large cell lymphoma, even in the presence of JAK1/STAT3 mutations. Proc. Natl. Acad. Sci. U.S.A 114, 3975–3980 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prutsch N, Gurnhofer E, Suske T, Liang HC, Schlederer M, Roos S, Wu LC, Simonitsch-Klupp I, Alvarez-Hernandez A, Kornauth C, Leone DA, Svinka J, Eferl R, Limberger T, Aufinger A, Shirsath N, Wolf P, Hielscher T, Sternberg C, Aberger F, Schmoellerl J, Stoiber D, Strobl B, Jäger U, Staber PB, Grebien F, Moriggl R, Müller M, Inghirami GG, Sanda T, Look AT, Turner SD, Kenner L, Merkel O, Dependency on the TYK2/STAT1/MCL1 axis in anaplastic large cell lymphoma. Leukemia 33, 696–709 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Choudhari R, Minero VG, Menotti M, Pulito R, Brakebusch C, Compagno M, Voena C, Ambrogio C, Chiarle R, Redundant and nonredundant roles for Cdc42 and Rac1 in lymphomas developed in NPM-ALK transgenic mice. Blood 127, 1297–1306 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fiore D, Cappelli LV, Zumbo P, Phillips JM, Liu Z, Cheng S, Yoffe L, Ghione P, Di Maggio F, Dogan A, Khodos I, de Stanchina E, Casano J, Kayembe C, Tam W, Betel D, Foa R, Cerchietti L, Rabadan R, Horwitz S, Weinstock DM, Inghirami G, A novel JAK1 mutant breast implant-associated anaplastic large cell lymphoma patient-derived xenograft fostering pre-clinical discoveries. Cancers (Basel) 12, 1603 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.George RE, Sanda T, Hanna M, Fröhling S, Luther II W, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, Webb TR, Gray NS, Gilliland DG, Diller L, Greulich H, Morris SW, Meyerson M, Look AT, Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455, 975–978 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Frankish A, Diekhans M, Ferreira A-M, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, Barnes I, Berry A, Bignell A, Sala SC, Chrast J, Cunningham F, Domenico TD, Donaldson S, Fiddes IT, Girón CG, Gonzalez JM, Grego T, Hardy M, Hourlier T, Hunt T, Izuogu OG, Lagarde J, Martin FJ, Martínez L, Mohanan S, Muir P, Navarro FCP, Parker A, Pei B, Pozo F, Ruffier M, Schmitt BM, Stapleton E, Suner M-M, Sycheva I, Uszczynska-Ratajczak B, Xu J, Yates A, Zerbino D, Zhang Y, Aken B, Choudhary JS, Gerstein M, Guigó R, Hubbard TJP, Kellis M, Paten B, Reymond A, Tress ML, Flicek P, GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 47, D766–D773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C, Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Soneson C, Love MI, Patro R, Hussain S, Malhotra D, Robinson MD, A junction coverage compatibility score to quantify the reliability of transcript abundance estimates and annotation catalogs. Life Sci Alliance 2, e201800175 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McCarthy DJ, Chen Y, Smyth GK, Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kanehisa M, Goto S, KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aref AR, Campisi M, Ivanova E, Portell A, Larios D, Piel BP, Mathur N, Zhou C, Coakley RV, Bartels A, Bowden M, Herbert Z, Hill S, Gilhooley S, Carter J, Cañadas I, Thai TC, Kitajima S, Chiono V, Paweletz CP, Barbie DA, Kamm RD, Jenkins RW, 3D microfluidic ex vivo culture of organotypic tumor spheroids to model immune checkpoint blockade. Lab. Chip 18, 3129–3143 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Voena C, Conte C, Ambrogio C, Boeri Erba E, Boccalatte F, Mohammed S, Jensen ON, Palestro G, Inghirami G, Chiarle R, The tyrosine phosphatase Shp2 interacts with NPM-ALK and regulates anaplastic lymphoma cell growth and migration. Cancer Res. 67, 4278–4286 (2007). [DOI] [PubMed] [Google Scholar]
- 82.Hirsch FR, Varella-Garcia M, Bunn PA Jr., M. V. Di Maria, R. Veve, R. M. Bremmes, A. E. Baron, C. Zeng, W. A. Franklin, Epidermal growth factor receptor in non-small-cell lung carcinomas: Correlation between gene copy number and protein expression and impact on prognosis. J. Clin. Oncol 21, 3798–3807 (2003). [DOI] [PubMed] [Google Scholar]
- 83.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Li B, Dewey CN, RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Anders S, Huber W, Differential expression analysis for sequence count data. Genome Biol. 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hao Y, Hao S, Andersen-Nissen E, Mauck WM 3rd, Zheng S, Butler A, Lee MJ, Wilk AJ, Darby C, Zager M, Hoffman P, Stoeckius M, Papalexi E, Mimitou EP, Jain J, Srivastava A, Stuart T, Fleming LM, Yeung B, Rogers AJ, McElrath JM, Blish CA, Gottardo R, Smibert P, Satija R, Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or the Supplementary Materials. The RNA-seq data generated from ALCL cell lines sensitive and resistant to crizotinb are available at NCBI Gene Expression Omnibus (GEO) under accession number GSE173306 (token: ohqdkaawnfkpbon). scRNA-seq data generated from patients’ primary lymphoma samples and PDX are available at NCBI: www.ncbi.nlm.nih.gov/sra/PRJNA980417.