

Abstract
The photoreactions of selected styrylpyridine derivatives to the corresponding benzo[c]quinolizinium ions are described. It is shown that these reactions are more efficient in aqueous solution (97–44%) than in organic solvents (78–20% in MeCN). The quinolizinium derivatives bind to DNA by intercalation with binding constants of 6–11 × 104 M−1, as shown by photometric and fluorimetric titrations as well as by CD- and LD-spectroscopic analyses. These ligand–DNA complexes can also be established in situ upon irradiation of the styrylpyridines and formation of the intercalator directly in the presence of DNA. In addition to the DNA-binding properties, the tested benzo[c]quinolizinium derivatives also operate as photosensitizers, which induce DNA damage at relative low concentrations and short irradiation times, even under anaerobic conditions. Investigations of the mechanism of the DNA damage revealed the involvement of intermediate hydroxyl radicals and C-centered radicals. Under aerobic conditions, singlet oxygen only contributes to marginal extent to the DNA damage.
Keywords: DNA intercalators, heterocycles, photocyclization, photosensitizer, styrylpyridines
Introduction
DNA intercalators – most often represented by small planar heteroaromatic compounds – play an important role as chemotherapeutic agents [1–4]. Specifically, upon intercalation into the DNA double helix such ligands can cause a change of the DNA structure or occupy binding sites of essential enzymes, which in turn may influence or even inhibit important biochemical processes, for example DNA replication or transcription [1–2]. As a result, the development of DNA-targeting drugs still involves the design of suitable DNA intercalators, and some currently applied anticancer drugs actually operate on the basis of intercalation [3]. Hence, several classes of compounds have been established, whose DNA-binding properties can be tailored and fine-tuned for that purpose, for example anthracyclines [5], indolocarbazoles [6], acridines [7], quinoxalines [8], naphthalimides [9], phenanthridines [10], cyanines [11], or indoles [12], as well as metal-organic complexes [13], and several others [1–2,4].
In this context, benzoquinolizinium derivatives and resembling polycyclic azoniahetarenes are an established class of DNA-binding compounds, which have been employed in biomedical imaging and as potential DNA-targeting anticancer agents [14–17]. More recently, a benzoquinolizinium-based fluorescent dye was reported to be used as imaging agent for inflammation and for the evaluation of the physiological response to anti-inflammatory drugs [18].
In this context, the benzo[c]quinolizinium structure provides some special features. First of all, it has the general requirements of a DNA intercalator, namely a planar, polycyclic heteroaromatic structure and a permanent positive charge [14]. Moreover, it has been shown that this DNA-binder and resembling intercalators can be directly generated upon irradiation of styrylazines under aerobic conditions, even in the presence of DNA, which provides local and temporal control of the DNA-binding event (Scheme 1) [14]. Specifically, the styrylpyridine, which does not bind to DNA, can be delivered without effect to the binding site, where the DNA-binding benzoquinolizinium ligand can then be generated as needed upon irradiation. Notably, the use of light for the activation of photo-controllable DNA ligands offers several advantages because it is easy to apply, traceless, and non-invasive [19]. As a result, several photoactive compounds have been developed, whose DNA-binding properties can be efficiently switched on and off by light [14,20–34].
Scheme 1.

Photoinduced formation of benzo[c]quinolizinium and its interaction with DNA upon intercalation.
In addition to their DNA-binding properties some annelated quinolizinium derivatives have also the ability to induce DNA damage upon irradiation [35–38], and may therefore be considered as promising basis for the development of new reagents for photodynamic (chemo)therapy (PDT). Notably, PDT has developed into an important therapeutic tool against several serious diseases, such as cancer [39], and bacterial, fungal, parasitic and viral infections [40–41]. In general, PDT operates on the basis of a photosensitizer, which generates reactive intermediates upon irradiation [42–45]. Hence, in the type-I mechanism the photosensitizer induces the formation of reactive oxygen species (ROS), such peroxyl, alkoxy and hydroxyl radicals, or carbon-centered radicals, which subsequently induce DNA strand cleavage. In the type-II mechanism, a triplet-excited photosensitizer reacts with molecular oxygen to give highly reactive singlet oxygen, 1O2, as reactive intermediate, which in turn oxidizes the DNA bases [46]. As a result, various classes of photosensitizers [47–49] have been established, for example, porphyrins [50], chlorins [51], phthalocyanines [52], porphycenes [53], metal-organic complexes [54–56], dye aggregates [57], as well as nano-drug carriers and metal-based nanoparticles [58–59]. But although these classes of compounds have been intensively studied and already contributed significantly to the field of PDT, there is still a demand for novel DNA-photodamaging ligands that could be applied for specific purposes, e.g., to improve efficacy or to limit side-effects. Therefore, the search for a class of photosensitizers is still a topical research area in photobiology [60]. To this end, benzo[c]quinolizinium derivatives may be considered as feasible photosensitizers because they can be formed readily in situ in the presence of DNA and because the structurally related alkaloids berberine [61–65] and coralyne [36,66–67] have been shown already to act as efficient photosensitizers for DNA damage. To the best of our knowledge, however, benzo[c]quinolizinium derivatives have not been investigated with respect to their DNA-photodamaging properties, so far. Therefore, we have synthesized selected benzo[c]quinolizinium derivatives and studied their DNA-binding and DNA-photodamaging properties.
Results and Discussion
Synthesis
The styrylpyridine derivatives 2a,c,d,f were synthesized by a piperidine- or Ca(OTf)2-catalyzed condensation reaction of 3,4-dimethoxybenzaldehyde with 5-substituted 2-picoline derivatives in low to moderate yields ranging from 13% (2f) to 65% (2a) (Scheme 2). The amino-substituted derivative 2b was synthesized by reduction of the nitrostyrylpyridine 2a with Pd/C and hydrazine in 83% yield. Subsequent acylation of the amine 2b gave the corresponding amide 2g in 28% yield. The chloro-substituted derivative 2e was synthesized in a Sandmeyer-reaction from 2b in 20% yield. The products 2a–g were identified and fully characterized by NMR spectroscopy (1H, 13C, COSY, HSQC, and HMBC), elemental analyses, and mass spectrometry. In all cases, the E-configuration of the alkene double bonds was indicated by characteristic coupling constants of the alkene protons (3JH–H = 16 Hz).
Scheme 2.

Synthesis of styrylpyridine derivatives 2a–g. Conditions: i: piperidine, MeOH, reflux (2a,c), ii: Ca(OTf)2, Bu4NPF6, 130 °C (2d,f), iii: N2H4·H2O, Pd/C, MeOH, reflux (2b), iv: NaNO2, CuCl, aq HCl (37%), room temp., 2 h, 60 °C, 30 min (2e), v: acetyl chloride, pyridine, THF, room temp. (2g).
Absorption properties and photoreactions of styrylpyridine derivatives
In acetonitrile solution, the styrylpyridines 2a–g exhibited long-wavelength absorption bands with maxima in a range from λmax = 333 nm for the ethyl-substituted compound 2f to λmax = 394 nm for the nitro-substituted derivative 2a (Figure 1A, Supporting Information File 1, Table S1). As compared with the absorption maximum of the parent compound at λmax = 330 nm [20], the derivatives 2b–g showed a slight bathochromic shift mostly in the range of λmax = 333–360 nm, whereas for the nitro-substituted compound 2a a stronger red shift of the absorption maximum was observed, presumably caused by the strong electron-withdrawing property of the nitro group resulting in a more pronounced intramolecular charge transfer [68]. In water, the absorption maxima showed only a small shift of the absorption maxima of 1–4 nm as compared with the ones in acetonitrile (Figure 1B, Supporting Information File 1, Table S1). The absorption spectra of compounds 2a and 2c could not be recorded because of their low water solubility.
Figure 1.

Absorption spectra of styrylpyridine derivatives 2a (black), 2b (red), 2c (blue), 2d (green), 2e (magenta), 2f (orange), and 2g (purple) in MeCN (A) and H2O (B) (c = 20 µM).
The styrylpyridine derivatives 2a–g were irradiated in oxygen-saturated solutions in MeCN, H2O, MeOH, or MeCN/H2O with a high-pressure Hg lamp (λ > 220 nm), and the course of the photocyclization reaction was monitored by absorption spectroscopy (Figure 2). In general, the absorption maximum of the derivatives 2b–g decreased during the photoreaction with formation of new red-shifted absorption bands. Nevertheless, the new red-shifted absorption band of the amino-substituted styryl derivative 2b in MeCN was weak and very broad, indicating only negligible formation of the photocyclization product (Figure 2B). Likewise, in H2O solution with MeCN as co-solvent, no formation of a new absorption band was observed upon irradiation of 2b, either (Figure 2C). Moreover, the aminobenzoquinolizinium 3b could not be isolated after irradiation of 2b at larger scale. As an exception, the irradiation of the nitro-substituted styrylpyridine derivative 2a in MeCN or H2O led to disappearance of the long-wavelength absorption maximum with no formation of a distinct new band (Figure 2A), which usually indicates photoinduced decomposition. This observation is in agreement with reports on resembling aromatic p-nitro-substituted derivatives, which do not react in a photocyclization reaction [69].
Figure 2.

Changes of the absorption spectra during the irradiation of 2a in MeCN for 16 min (A), 2b in MeCN for 4 min (B), and 2b in H2O/MeCN 49:1 for 6 min (C) (c = 20 µM, λex > 220 nm, irradiated in a cuvette). Blue: spectrum of the starting material before irradiation; red: spectrum at the end of the irradiation.
Direct irradiation of the styrylpyridine derivatives 2c–g in MeCN led to the formation of new absorption bands in the range from λ = 389 nm (2f) to λ = 407 nm (2d) with bathochromic shifts of Δλ = 47–57 nm (Figure 3, Supporting Information File 1, Table S1), which indicated the formation of the benzo[c]quinolizinium ions 3c–g as photocyclization products [20]. The absence of isosbestic points during the photometric monitoring of the photoreactions indicated a stepwise formation of different intermediates in the reaction sequence starting with E–Z isomerization, followed by photocyclization and subsequent oxidation.
Figure 3.

Changes of the absorption spectra during the irradiation of 2c for 13 min (A), 2d for 12 min (B), 2e for 15 min (C), 2f for 10 min (D), and 2g for 7 min (E) (in MeCN, c = 20 µM, λex > 220 nm, irradiated in a cuvette). Blue: spectrum of the starting material before irradiation; red: spectrum at the end of the irradiation.
In general, the photoreaction was more efficient in polar, protic aqueous solvents (cf. Supporting Information File 1, Figures S2B–S6B) or in buffer solution (cf. Supporting Information File 1, Figures S3C–S6C) than in polar, aprotic MeCN solution. In MeOH the photoreactions were inefficient as indicated by the lack of red-shifted bands or by formation of broad, weak absorption bands (cf. Supporting Information File 1, Figures S1, S2A, S3A, S4A, S5A, and S6A). Because of the low water solubility of the styrylpyridine derivatives 2a–g, solvent mixtures of MeCN/H2O or pure MeCN were used for the preparative photocyclization reactions (Scheme 3, cf. Supporting Information File 1, Figure S2C,D) to provide sufficient solubility of the substrates, as well as optimal efficiencies for the formation of photocyclization products. But even under these optimized conditions, compounds 3c–g could be isolated only in low yields of <5–21%. Hence, to assess whether the products are generally formed to minor extent in the photoreaction or whether the low yields of isolated product result from significant losses during the purification process, the content and yield of the benzo[c]quinolizinium ions were determined directly after irradiation of 2c, 2e–g by photometric analysis of the reaction mixture (Figure 3, red spectra). The yield of the product 3d could not be determined because it was not available in pure form on preparative scale. With the absorption data, specifically the molar extinction coefficients, obtained from the isolated benzo[c]quinolizinium derivatives, the yields of the initially formed photoproducts 3c, 3e, 3f, and 3g in the reaction mixtures were determined to be 78%, 73%, 30%, and 20% in MeCN, >97%, 80%, 44% and 55% in H2O, and 32%, 76%, 53% and 38% in Na phosphate buffer. Overall, the initially formed amounts of photoproducts are significantly larger than the ones of the isolated compounds, in the case of 3c and 3e even with good yields, so that it may be concluded that the small amounts of isolated product result from losses during work-up and purification.
Scheme 3.

Photoinduced formation of styrylpyridine derivatives 2b–g to the benzo[c]quinolizinium ions 3b–g (yields in % refer to isolated products).
DNA-binding properties of benzoquinolizinium derivatives 3c,e–g
The styrylpyridines 2d–g and the benzo[c]quinolizinium derivatives 3c,e–g were investigated with respect to their DNA-binding properties with calf thymus (ct) DNA. The titrations of ct DNA to compounds 2d–g resulted in no or only negligible changes of the absorption and fluorescence spectra (cf. Supporting Information File 1, Figures S8–S11), indicating that these substrates do not interact significantly with DNA. In contrast, upon addition of DNA to compounds 3c,e–g, the absorption maxima at 398 nm, 393 nm, 386 nm and 394 nm were red-shifted with a hypochromic effect, and isosbestic points developed during all titrations (Figure 4, Table 1). Furthermore, the addition of DNA to substrates 3c,e–g led to efficient fluorescence quenching (Figure 5), which is commonly observed with this class of cationic ligands [3,70], mainly as a result of a photoinduced electron transfer from the excited, DNA-bound ligand with the DNA bases [71]. The binding isotherms obtained from the titration data were used to determine the binding constants, Kb, of the DNA ligands. Thus, the derivatives 3c, 3e, and 3g bind to ct DNA with Kb values of 6.0 × 104 M−1, 5.9 × 104 M–1, and 6.4 × 104 M−1, respectively, whereas the affinity of ligand 3f is slightly higher with Kb = 1.1 × 105 M−1, which is in the same range as the binding constant reported for the 8,9-dimethoxybenzo[c]quinolizinium (Kb = 1.2 × 105 M−1) [20]. The slightly lower binding constants of derivatives 3c, 3e, and 3g as compared with the one of compound 3f, may be explained by the larger substituents of the former ligands, which cause more steric repulsion within the binding site.
Figure 4.

Photometric titration of ct DNA to 3c (A) 3e (B) 3f (C) and 3g (D) (c = 20 µM) in Na phosphate buffer (pH 7.0, T = 20 °C, cNa+ = 16 mM). The arrows indicate the development of the absorption bands during titration. Inset: plot of absorption versus cDNA.
Table 1.
Absorption and emission maxima of 3c–g in the absence and presence of ct DNA and binding constants of their complexes with ct DNA.
| λabs / nma | Δλ / nm | Kb / 104 M−1b | ||
|
|
||||
| without DNA | with DNA | |||
|
| ||||
| 3c | 398 | 415 | 17 | 6.0 |
| 3d | – | – | – | – |
| 3e | 393 | 405 | 12 | 5.9 |
| 3f | 386 | 398 | 22 | 11 |
| 3g | 394 | 409 | 15 | 6.4 |
aIn Na phosphate buffer, cNa+ = 16 mM, pH 7.0, 20 °C. bDetermined from photometric titrations.
Figure 5.

Fluorimetric titration of ct DNA to 3c (A), 3e (B), 3f (C), and 3g (D) (c = 20 µM) in Na phosphate buffer (pH 7.0, T = 20 °C, cNa+ = 16 mM). The arrows indicate the development of the emission bands during titration. Inset: plot of relative emission intensity versus cDNA.
The binding mode of the benzo[c]quinolizinium derivatives 3c,e–g with DNA was further examined with circular dichroism (CD) and linear dichroism (LD) spectroscopy (Figure 6 and Supporting Information File 1, Figures S12–S14). Hence, with increasing ligand-DNA ratio (LDR) weak positive induced CD (ICD) signals developed in the long-wavelength absorption range of ligands 3e–g, that is, where the DNA bases do not absorb (Figure 6A and Supporting Information File 1, Figures S13A and S14A), which is a commonly observed indication of DNA binding as the ICD bands result from non-degenerative coupling of the dipole moments of the ligand with the DNA bases [72]. The association of the ligands 3e–g with DNA was further confirmed by the formation of negative LD bands with increasing LDR developing in the absorption range of the ligands (λ > 300 nm) (Figure 6B and Supporting Information File 1, Figures S13B and S14B), which is a characteristic indication of a coplanar alignment of the ligand relative to the base pairs in an intercalative binding mode [73]. In contrast, ligand 3c did not exhibit an ICD signal in the presence of DNA (Supporting Information File 1, Figure S12A) and gave rather weak and less structured LD bands (Figure S12B) as compared with the ones of ligands 3e–g. The lack of distinct ICD bands of DNA-bound 3c might be explained by the very weak signals, which are usually observed for this class of DNA binder because of unfavorable angles between transition moments of ligand and DNA bases [72].
Figure 6.

CD (A) and LD (B) spectra of 3f and ct DNA (cDNA = 20 µM) in Na phosphate buffer (pH 7.0, T = 20 °C, cNa+ = 16 mM) at LDR = 0 (black), 0.2 (red), 0.5 (green), 1.0 (blue), 1.5 (orange), and 2.0 (magenta).
To gain additional information about the orientation of the benzo[c]quinolizinium ligand in the intercalation site, the reduced LD (LDr) spectrum was determined exemplarily for ligand 3f (Supporting Information File 1, Figure S15) [74–75]. The analysis of the data revealed a binding angle α = 59° between the ligand 3f and the DNA helix, thus indicating a tilted orientation of the ligand relative to the DNA base pairs within the binding site.
The DNA-binding ligands were also generated in situ in the presence of DNA. For that purpose, solutions of the styrylpyridines 2d–g and ct DNA were irradiated in phosphate buffer, and the formation of the dimethoxybenzo[c]quinolizinium ions and their subsequent binding to the DNA were shown photometrically by the development of the characteristic red-shifted absorption bands (Figure 7A, cf. Supporting Information File 1, Figure S7A), which matched the ones of the independently formed complexes of these ligands with DNA (see above). In the case of 3e–g, the binding event was also confirmed by CD spectroscopy, namely by the formation of weak ICD signals (Figure 7B, cf. Supporting Information File 1, Figure S7B). Because of the very low solubility of compound 2c in aqueous solutions, the former could not be irradiated in situ in the presence of DNA.
Figure 7.

Changes of the absorption (A) and CD (B) spectra during the irradiation of 2e (1) and 2f (2) (c = 20 μM) in the presence of ct DNA (cDNA = 0.1 mM) in Na phosphate buffer, cNa+ = 16 mM. Blue: spectrum of the starting material before irradiation; red: spectrum at the end of the irradiation.
Photoinduced DNA damage
In first orienting experiments, the photoinduced DNA damage by benzo[c]quinolizinium derivatives was examined exemplarily with ligand 3f and plasmid DNA pBR322 (Table 2). To assess the optimal parameters for the photoreaction, the ligand was irradiated in the presence of the DNA under anaerobic conditions at different irradiation times and concentrations of the ligand. The DNA-strand cleavage was analyzed by agarose-gel electrophoresis. In this assay, the DNA-strand cleavage in the supercoiled plasmid DNA pBR322 is indicated by the formation of the relaxed, open-circular form [76]. It has to be noted that under these conditions the DNA is already damaged in the absence of the photosensitizer. Therefore, each series of experiments is complemented for comparison with control experiments without photosensitizer. In general, a photoinduced DNA damage by the quinolizinium 3f was observed, whose extent increased with increasing irradiation time and with increasing concentration of 3f. Thus, after 2 min of irradiation with fixed concentration (c = 2.5 × 10−5 M) 38% of the supercoiled DNA were transformed to the open-circular form, whereas after 5 min, 73% of the DNA were damaged by strand cleavage (Table 2A). At the same time, experiments with varying ligand concentration revealed 81% of damaged DNA after 2 min of irradiation with c = 5.0 × 10−5 M, which was almost twice as much as the cleavage (41%) determined with c = 2.5 × 10−5 M Table 2B). In addition, a small series of benzo[c]quinolizinium ligands 3c,e–g was tested under conditions optimized for derivative 3f. Within this series, the amount of DNA cleavage ranged fom 39% (3c) to 51% (3e) after 2 min of irradiation (Table 2C).
Table 2.
Gel-electrophoretic analysis of photoinduced DNA-strand cleavage in the presence of 3f depending on the irradiation time (A), on the concentration (B), and in the presence of 3c,e–g (C). Lanes 1, 10 and 11 (A) and lane 11 (B): control experiment without 3f. Lanes 1 and 2 (B): control experiment without 3f, irradiated for 2 min. In all cases: c3 = 2.5 × 10−5 M, cDNA = 3.5 × 10−9 M, anaerobic conditions, irradiation time: 2 min, λmax = 366 nm.
| A | |||||||||||
| supercoiled open-circular |
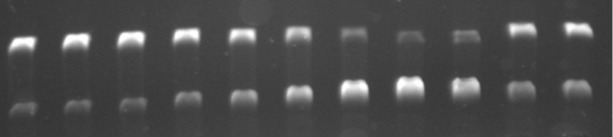
|
||||||||||
|
| |||||||||||
| lane | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|
| |||||||||||
| irradiation time / min | 0 | 0 | 0 | 0.5 | 1 | 2 | 5 | 10 | 15 | 15 | 15 |
| Strand cleavage / % | 11 | 9 | 9 | 16 | 25 | 38 | 73 | 83 | 73 | 24 | 26 |
|
| |||||||||||
| B | |||||||||||
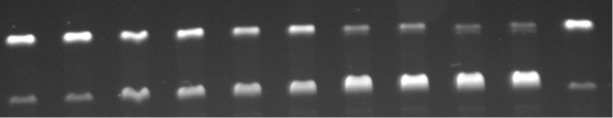
|
|||||||||||
|
| |||||||||||
| lane | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|
| |||||||||||
| c / 10−5 M | 0 | 0 | 1.3 | 1.3 | 2.5 | 2.5 | 3.8 | 3.8 | 5.0 | 5.0 | 0 |
| strand cleavage / % | 13 | 11 | 31 | 32 | 41 | 41 | 71 | 73 | 81 | 81 | 13 |
|
| |||||||||||
| C | |||||||||||
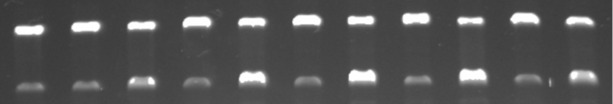
|
|||||||||||
|
| |||||||||||
| lane | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|
| |||||||||||
| irradiation time / min | 0 | 2 | 2 | 0 | 2 | 0 | 2 | 0 | 2 | 0 | 2 |
| substrate | − | − | ref | 3f | 3f | 3g | 3g | 3e | 3e | 3c | 3c |
| strand cleavage / % | 14 | 15 | 31 | 15 | 42 | 15 | 46 | 16 | 51 | 17 | 39 |
To investigate the mechanism of the DNA cleavage, commonly employed control experiments [46,77] were performed exemplarily with ligand 3f (Table 3 and Supporting Information File 1, Figure S16). To assess the influence of oxygen, the DNA–ligand mixture was irradiated for 2 min under ambient aerobic conditions as well as in argon- and oxygen-saturated medium (Supporting Information File 1, Figure S16A). Under anaerobic atmosphere, 50% of the supercoiled DNA were transformed to open-circular DNA after 2 min of irradiation, whereas under atmospheric conditions and oxygen-saturation only 36% and 30% of the DNA were cleaved, respectively. However, with prolonged irradiation time, even under oxygen atmosphere the photocleavage activity increased to give 65% strand cleavage after 5 min and 85% after 10 min of irradiation (Supporting Information File 1, Figure S16B).
Table 3.
Photoinduced DNA-strand cleavage by benzoquinolizinium 3f under different conditions.a
| Additive | c Additive | Atmosphere | Strand cleavage / %b |
|
| |||
| – | anaerobicc | 50 | |
| DMSO | 5 vol % | anaerobicc | 34 |
| t-BuOH | 5 vol % | anaerobicc | 28 |
| 2-PrOH | 5 vol % | anaerobicc | 23 |
| TEMPOd | 1.3 × 10−2 M | anaerobicc | 13 |
| 2-mercaptoethanol | 2.0 × 10−2 M | anaerobicc | 18 |
| 2-mercaptoethylamine·HCl | 2.0 × 10−2 M | anaerobicc | 16 |
| – | aerobic | 36 | |
| – | O2 saturated | 30 | |
| – | O2 saturatede | 65 | |
| – | O2 saturatedf | 85 | |
| NaN3 | 2.5 × 10−4 M | aerobic | 25 |
| D2O (solvent) | aerobic | 23 | |
aIn all cases: c3f = 2.5 × 10–5 M, cDNA = 3.5 × 10–9 M (pBr322), irradiation time = 2 min; λex = 366 nm. bDetermined by agarose gel electrophoresis and staining with ethidium bromide. cAr saturated. dTEMPO = 2,2,6,6-tetramethylpiperidine 1-oxyl. eIrradiation time = 5 min. fIrradiation time = 10 min.
To clarify whether the mechanism of the DNA photodamage proceeds through the formation of radicals, experiments with commmonly employed radical scavengers were conducted (Table 3, Supporting Information File 1, Figure S17). In the presence of hydroxyl-radical scavengers DMSO, t-BuOH, and 2-propanol [78] the cleavage of the DNA was reduced to 34%, 28%, and 23%, respectively, which may indicate that the cleavage of DNA involves hydroxyl radicals (Supporting Information File 1, Figure S17A). At the other hand, a clear decrease of DNA damage to ca. 13%, 18% and 16% in the presence of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO), 2-mercaptoethanol and 2-mercaptoethylamine hydrochloride, respectively, showed that C-centered radicals contribute even more to the DNA damage than hydroxyl radicals (Supporting Information File 1, Figure S17B). It should be noted, however, that these scavengers may also intercept hydroxyl radicals [79] or interfere with the DNA damage by alternative pathways [80]. In any case, the significant decrease of DNA damage in the presence of the radical scavengers indicated the formation and direct or indirect participation of carbon radicals and hydroxyl radicals in the photoinduced DNA damage.
In order to investigate the involvement of singlet oxygen in the DNA cleavage process, the samples were irradiated in the presence of NaN3 or in D2O (Table 3, Supporting Information File 1, Figure S18). The latter is known to extend the lifetime of singlet oxygen by a factor of ca. 10 as compared with H2O. Therefore, strand cleavage reactions induced by singlet oxygen are more efficient in D2O than in H2O [81]. However, irradiation of 3f in D2O resulted in essentially the same DNA cleavage (23%) as compared with the reaction in H2O (25%) under otherwise identical conditions (Supporting Information File 1, Figure S18A). In the presence of NaN3 (2.5 × 10−5 M), which is a known radical scavenger for singlet oxygen [82], a strand cleavage of 25% occurred, whereas 38% cleavage was observed in the absence of NaN3. Nevertheless, a larger access of the scavenger (2.5 × 10−4 M) resulted in a decrease of the cleavage to 17%. It has to be noted, however, that an inhibition of DNA cleavage in the presence of NaN3 may also be induced by direct deactivation of the excited photosensitizer by the azide and not only from quenching of singlet oxygen [83]. Similar, seemingly contradictory effects of D2O and NaN3 on the photoinduced DNA cleavage were observed with other photosensitizers [84–85]. In addition, it has been reported that a relative large excess of NaN3 is required to detect an efficient inhibition of DNA-photocleavage [86]. Overall, these results as well as the efficient photocleavage under oxygen-saturated conditions, at least with long irradiation times (Supporting Information File 1, Figure S18B), indicated the direct or indirect involvement of singlet oxygen in the overall mechanism; yet, only to a small extent.
Overall, the experiments on the photoinduced DNA damage by benzo[c]quinolizinium 3f revealed a more complex mechanistic scenario (Scheme 4). While it became clear that the irradiation of this substrate in the presence of DNA leads to efficient DNA-strand cleavage, the systematic assessment of parameters that influence this reaction revealed the formation of different reactive intermediates (Scheme 4). Under anaerobic conditions, the DNA damage is similar to the one observed with the isomeric benzo[c]quinolizinium ions [35]. In the latter case, it has been shown that frank DNA-strand breaks are induced by hydroxyl radicals, supposedly formed by photoinduced electron transfer (PET) reaction of the strongly oxidizing excited quinolizinium ion. Likewise, the results obtained with 3f point to the formation of hydoxyl radicals that are known to induce DNA-strand breaks. At the same time, the formation of C-centered radicals was indicated by the pronounced decrease of photocleavage in the presence of the corresponding radical scavengers. As there is no obvious reaction mechanism for the direct formation of C-radicals upon irradiation of 3f it is proposed that the reaction of the initially formed hydroxyl radicals with the benzoquinolizinium 3f leads to the formation of the C-centered radicals 4 and 5, namely by addition of the radical or by hydrogen abstraction at the methylene group of the ethyl substituent (Scheme 4). Subsequently, the intermediate radicals 4 and 5 induce DNA-strand breaks initiated by hydrogen abstraction reactions at the ribose residues [78,87].
Scheme 4.

Proposed mechanisms for the photoinduced DNA damage initiated by photoexcitation of benzoquinolizinium 3f.
Most notably, under aerobic conditions, a reduced DNA photocleavage was observed as compared with the reaction under anaerobic conditions. This result is somewhat surprising as the formation of singlet oxygen, 1O2, by the reaction of the triplet-excited 3f and oxygen was confirmed in control experiments. And the reactive intermediate 1O2 is known to induce DNA damage [78,88]. Nevertheless, these lesions, namely DNA base oxidations, often require alkaline treatment to lead to a strand cleavage, so that the DNA damage remained mainly unnoticed in the employed assay. At the same time, it has been reported that 1O2 can also efficiently induce single-strand breaks directly [89], so that the observed low DNA cleavage under aerobic conditions may be assigned to such a reaction. Moreover, it should be noted that both 3O2 and 1O2 [46] might react with other intermediates formed during the photoreaction, for example by the reaction with C-radicals 4 and 5 to give peroxides such as 6 (Scheme 4), by cycloaddition of 1O2 to alkene and diene units, or by deactivation of the excited state in a triplet-triplet annihilation [90], all of which leading to a reduced photocleavage efficiency. However, with much longer irradiation time under oxygen-saturated conditions, a more efficient photodamage was observed, which may be induced by those intermediates or secondary products formed from these 1O2 reactions.
In comparison with the already known quinolizinium-type photosensitizers [35,91], the photoinduced DNA damage by benzo[c]quinolizinium derivatives is more efficient under resembling conditions. Thus, the isomeric benzo[b]quinolizinium cations showed DNA damage to a lower extent after irradiation for 10 min with 15–20% and 20–25% DNA cleavage under anaerobic and aerobic conditions, respectively [35]. Likewise, naphthoquinolizinium salts exhibited DNA cleavage of about 20% and 50% after 5 min and 10 min, respectively, under anaerobic conditions, thus showing lower efficiencies as compared with photosensitizer 3f with DNA cleavage of 73% and 83%, respectively [91].
Conclusion
In summary, it was demonstrated that the photoinduced cyclization reaction of readily available styrylpyridine derivatives 2a–g gives the corresponding benzo[c]quinolizinium derivatives and that these reactions are more efficient in aqueous solutions than in organic solvents. The benzo[c]quinolizinium derivatives have the typical properties of DNA intercalators and bind to DNA with Kb values of 6.0–11 × 104 M–1. Importantly, the ligand–DNA complex may be accessed as needed in situ upon irradiation of the styrylpyridines 2c,e–g in the presence of DNA, which is a useful feature of DNA-targeting substrates, specifically for a spatio-temporal control of this biological activity.
Furthermore, we have discovered that this class of compounds has a large potential to operate as photosensitizer that induces DNA damage already at relatively low irradiation times and low concentrations. Most notably, the photoinduced DNA damage does not necessarily require oxygen, unlike type-II photosensitizers. In fact, the representative compound 3f is a more efficient DNA-damaging photosensitizer under anaerobic conditions, which may be an advantage for applications in hypoxic cancer cells. Preliminary investigations of the mechanism of the DNA damage revealed the involvement of intermediate hydroxyl radicals and C-centered radicals. Singlet oxygen, one of the most important reactive intermediates in conventional PDT, however, only contributes to marginal extent to the DNA damage. Therefore, these results are a promising starting point for the development of novel photosensitizers based on benzo[c]quinolizinium derivatives because their particular mode of activity may offer complementary applications in addition to the already established photosensitizers.
Overall, the benzo[c]quinolizinium scaffold offers some advantageous properties for its use as DNA-targeting agent, both as photo-controllable DNA binder and as DNA-damaging photosensitizer. Still, some key parameters have to be optimized by variation of the substitution pattern. For example, the water solubility of the styrylpyridine substrates has to be increased, and the excitation wavelength for the photocyclization reaction has to be red-shifted. But with a focus on the improvement of these properties the benzo[c]quinolizinium ion should be considered as a promising platform for further development of DNA-binding and DNA-photodamaging reagents.
Experimental
General methods
The commercially available chemicals (Alfa, Merck, Fluorochem or BLDpharm) were of reagent grade and used without further purification. 1H NMR spectra were recorded with a JEOL ECZ 500 (1H: 500 MHz and 13C: 125 MHz) and a Varian VNMR S600 (1H: 600 MHz and 13C: 150 MHz) at T = 25 °C. The 1H NMR and 13C{1H} NMR spectra were referenced to the residual proton signal of the solvent [CD3CN: δ(1H) = 1.94 ppm, δ(13C) = 118.36 ppm or DMSO-d5: δ(1H) = 2.50 ppm, δ(13C) = 39.52 ppm] or to an internal standard in CDCl3 [TMS: δ(1H) = 0.00 ppm, δ(13C) = 0.00 ppm]. Structural assignments were made with additional information from gCOSY, gHSQC, and gHMBC experiments. The spectra were processed with the MestreNova software. The mass spectra were recorded with a Finnigan LCQ Deca (driving current: 6 kV, collision gas: argon, capillary temperature: 200 °C, support gas: nitrogen) and an Orbitrap mass spectrometer Thermo Fisher Exactive (driving current: 3.5 kV, capillary temperature: 300 °C, capillary voltage: 45 V, injection rate: 5 μL/min, scanning range: 150−750 m/z, and resolution: ultra-high) and processed with the software Xcalibur. The CHNS analysis data were determined in-house with a HEKAtech EuroEA combustion analyzer. The melting points were measured with a melting point apparatus BÜCHI 545 (Büchi, Flawil, CH) and are uncorrected. The absorption spectra were recorded on a Varian Cary 100 Bio absorption spectrometer with Hellma quartz glass cuvettes 110-QS (layer thickness d = 10 mm). The emission spectra were recorded on a Varian Cary Eclipse fluorescence spectrometer with Hellma quartz glass cuvettes 115 FQS (layer thickness d = 10 mm). All measurements were recorded at T = 20 °C as adjusted with a thermostat, if not stated otherwise. The sample solutions in the DNA experiments were mixed with a reaction vessel shaker Top-Mix 11118 (Fisher Bioblock Scientific). E-Pure water was obtained with an ultrapure water system D 4632-33 (Wilhelm Werner GmbH, Leverkusen, Germany) with filters D 0835, D 0803, and D 5027 (2×). CD- and LD-spectroscopic measurements were performed on a Chirascan spectrometer (Applied Photophysics). For LD-spectroscopic experiments, the spectrometer was equipped with a High Shear Couette Cell Accessory. The samples were oriented in a rotating Couette with a shear gradient of 1200 s−1. Photochemical reactions were carried out with a high-pressure Hg lamp (Heraeus TQ 150) in a quartz glass photoreactor or in a cuvette. Photoreactions for the investigation of the DNA damage were performed with a Rayonet (RPR-100) photoreactor equipped with 12 ultraviolet lamps (8 W, λexc = 366 nm).
Synthesis
General procedure (GP)
A solution of the styrylpyridine derivatives (c = 0.24–0.95 mM) in MeCN or in a mixture of MeCN/H2O was saturated with oxygen gas for 5–15 min, and the solutions were irradiated in an immersion-well photoreactor with a high-pressure Hg lamp. The reaction was controlled by absorption spectroscopy. After completion of the reaction, the solvent was removed by distillation, and the residue was purified by washing with n-pentane, n-hexane or n-hexane and EtOAc and subsequently by column chromatography or recrystallization from MeOH with addition of HClO4.
2-Methoxycarbonyl-8,9-dimethoxybenzo[c]quinolizinium perchlorate (3c)
According to GP, a solution (c = 0.80 mM) of 2c (100 mg, 334 µmol) in MeCN/H2O (6:1, 400 mL) was irradiated for 49 min. The crude product was washed with n-hexane (2 × 3 mL) and EtOAc (3 × 5 mL) and recrystallized from MeOH with addition of HClO4 to give the product as brown amorphous solid (14 mg, 35 µmol, 10%); mp > 200 °C (decomp.); 1H NMR (600 MHz, CD3CN) δ 4.07 (s, 3H, OCH3), 4.09 (s, 3H, CO2CH3), 4.23 (s, 3H, OCH3), 7.71 (s, 1H, 7-H), 8.11 (s, 1H, 10-H), 8.15 (d, 3J = 9 Hz, 1H, 5-H), 8.49 (d, 3J = 9 Hz, 1H, 4-H), 8.61 (d, 3J = 9 Hz, 1H, 6-H), 8.67 (dd, 3J = 9 Hz, 4J = 1 Hz, 1H, 3-H), 10.15 (s, 1H, 1-H); 13C NMR (150 MHz, CD3CN) δ 54.3 (CO2CH3), 57.4 (OCH3), 58.2 (OCH3), 100.1 (C10), 110.1 (C7), 121.5 (C5), 124.4 (C6a), 127.3 (C2), 129.8 (C4), 131.9 (C10a), 135.4 (C1), 136.4 (C3), 139.5 (C6), 144.8 (C4a), 153.4 (C8), 156.4 (C9), 164.1 (CO2CH3); MS (ESI+) m/z: [M+] 298 (100); Anal. calcd for C17H16ClNO8·0.5HClO4: C, 45.58; H, 3.71; N, 3.13; found: C, 45.56; H, 3.89; N, 3.32.
2-Cyano-8,9-dimethoxybenzo[c]quinolizinium perchlorate (3d)
According to GP, a solution (c = 0.24 mM) of 2d (30.0 mg, 110 µmol) in H2O/MeCN (6:1, 420 mL) was irradiated for 35 min. The product was purified by recrystallization from MeOH with addition of HClO4 and by column chromatography (SiO2, CHCl3/MeOH 95:5 → 90:10). The crude product was filtered through celite, washed with n-pentane (3 × 10 mL), suspended in CHCl3 (1 mL) and filtered to give the product as brown amorphous solid, containing small amounts of impurities according to 1H NMR-spectroscopic analysis (4.0 mg, 11 µmol, 10%); 1H NMR (500 MHz, CD3CN) δ 4.06 (s, 3H, OCH3), 4.20 (s, 3H, OCH3), 7.73 (s, 1H, 10-H), 8.08 (s, 1H, 7-H), 8.15 (d, 3J = 9 Hz, 1H, 6-H), 8.41 (dd, 3J = 9 Hz, 4J = 1 Hz, 1H, 3-H), 8.50 (d, 3J = 9 Hz, 1H, 4-H), 8.67 (d, 3J = 9 Hz, 1H, 5-H), 10.22 (s, 1H, 1-H).
2-Chloro-8,9-dimethoxybenzo[c]quinolizinium perchlorate (3e)
According to GP, a solution (c = 0.70 mM) of 2e (44 mg, 0.16 mmol) in MeCN (300 mL) was irradiated for 35 min. The crude product was washed with n-hexane (3 mL) and recrystallized from MeOH with addition of HClO4. The remaining solid was dissolved in H2O (50 mL), filtered from a black precipitate and a saturated aq solution of NaBF4 (7 mL) was added to the solution. The aqueous layer was extracted with MeNO2 (2 × 30 mL) and the combined organic layers were washed with H2O (2 × 30 mL) and dried with Na2SO4. The solvent was evaporated to give a yellow oil. The residue was suspended in CHCl3 (1 mL) and the remaining solid was filtered off to give the product as yellow amorphous solid (2.5 mg, 6.9 µmol, <5%); mp 164–166 °C (decomp.); 1H NMR (500 MHz, CD3CN) δ 4.07 (s, 3H, OCH3), 4.20 (s, 1H, OCH3), 7.68 (s, 1H, 7-H), 8.04 (s, 1H, 10-H), 8.10 (d, 3J = 9 Hz, 1H, 5-H), 8.31 (dd, 3J = 9 Hz, 4J = 1 Hz, 1H, 3-H), 8.40 (d, 3J = 9 Hz, 1H, 4-H), 8.53 (d, 3J = 9 Hz, 1H, 6-H), 9.88 (d, 4J = 1 Hz, 1H, 1-H); 13C NMR (125 MHz, CD3CN) δ 57.4 (OCH3), 58.3 (OCH3), 100.0 (C10), 109.9 (C7), 121.5 (C5), 124.5 (C6a), 130.2 (C4), 131.0 (C10a), 131.8 (C1), 132.7 (C2), 137.8 (C6), 138.4 (C3), 142.0 (C4a), 153.5 (C8), 156.4 (C9); MS (ESI+) m/z: [M+] 274 (100); HRMS–ESI+ (m/z): [M]+ calcd. for C15H13NO2Cl, 274.0629; found, 274.0625.
2-Ethyl-8,9-dimethoxybenzo[c]quinolizinium perchlorate (3f)
According to GP, a solution (c = 0.95 mM) of 2f (102 mg, 379 µmol) in MeCN (400 mL) was irradiated for 45 min and the crude product was washed with n-hexane (3 mL) and recrystallized from MeOH with addition of HClO4 to give a yellow amorphous solid (29 mg, 79 µmol, 21%); mp 215–217 °C (decomp.); 1H NMR (500 MHz, CD3CN) δ 1.42 (t, 3J = 8 Hz, 3H, CH3), 3.08 (q, 3J = 8 Hz, 2H, CH2), 4.05 (s, 3H OCH3), 4.20 (s, 1H, OCH3), 7.63 (s, 1H, 7-H), 8.01 (d, 3J = 9 Hz, 1H, 5-H), 8.05 (s, 1H, 10-H), 8.26 (dd, 3J = 9 Hz, 4J = 2 Hz, 1H, 3-H), 8.33 (d, 3J = 9 Hz, 1H, 4-H), 8.40 (d, 3J = 9 Hz, 1H, 6-H), 9.56 (s, 1H, 1-H); 13C NMR (125 MHz, CD3CN) δ 15.6 (CH3), 27.0 (CH2), 57.3 (OCH3), 58.1 (OCH3), 99.9 (C10), 110.0 (C7), 121.5 (C5), 124.0 (C6a), 129.0 (C4), 130.9 (C10a), 131.4 (C1), 136.2 (C6), 139.3 (C3), 141.9 (C4a), 142.1 (C2), 152.9 (C9), 155.7 (C8); MS (ESI+) m/z: [M+] 268 (100); Anal. calcd for C17H18ClNO6: C, 55.52; H, 4.93; N, 3.81; found: C, 55.71; H, 4.89; N, 3.83.
2-Acetylamino-8,9-dimethoxybenzo[c]quinolizinium tetrafluoroborate (3g)
According to GP, a solution (c = 0.25 mM) of (E)-5-acetylamino-2-(3,4-dimethoxystyryl)pyridine (30.0 mg, 101 µmol) in a mixture of MeCN/H2O (7:3, 400 mL) was irradiated for 33 min and the crude product was washed with n-hexane (3 mL) and redissolved in dest. H2O (5 mL). A saturated aq solution of NaBF4 (3 mL) was added to the solution. A brown precipitate was removed by filtration. The filtrate was extracted with MeNO2 (3 × 10 mL) and the solvent was evaporated to give a brown solid, which was purified by column chromatography (SiO2, CHCl3/MeOH, 95:5, Rf = 0.1) and washed with pentane (3 × 1 mL) to give the product as yellow amorphous solid (5.0 mg, 13 µmol, 13%); mp > 260 °C (decomp.); 1H NMR (500 MHz, CD3CN) δ 2.27 (s, 3H, COCH3), 4.05 (s, 3H, OCH3), 4.17 (s, 3H, OCH3), 7.61 (s, 1H, 7-H), 7.85 (s, 1H, 10-H), 7.97 (d, 3J = 9 Hz, 1H, 5-H), 8.30 (d, 3J = 9 Hz, 1H, 4-H), 8.34 (d, 3J = 9 Hz, 1H, 6-H), 8.36 (dd, 3J = 9 Hz, 4J = 1 Hz, 1H, 3-H), 9.48 (brs, 1H, NH), 10.45 (d, 4J = 1 Hz, 1H, 1-H); 13C NMR (125 MHz, CD3CN) δ 24.4 (COCH3), 57.3 (OCH3), 57.5 (OCH3), 99.3 (C10), 110.0 (C7), 121.4 (C5), 121.9 (C1), 124.2 (C6a), 129.5 (C4), 130.6 (C3), 135.2 (C6), 137.3 (C4a), 139.5 (C10a), 153.1 (C9), 155.6 (C8), 171.5 (COCH3); HRMS–ESI+ (m/z): [M]+ calcd. for C17H17N2O3+, 297.1234; found, 297.1235.
Supporting Information
Detailed experimental procedures, additional spectroscopic data and 1H NMR spectra.
Acknowledgments
We thank Christina Böhringer and Sandra Uebach for technical assistance and Manuel Heinelt and Cristian Camilo Escobar Carranza for HRMS measurement (Department of Chemistry and Biology, University of Siegen).
Funding Statement
Financial support by the Deutsche Forschungsgemeinschaft (Ih24/15-1) and the University of Siegen is gratefully acknowledged.
Data Availability
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.
References
- 1.Bhaduri S, Ranjan N, Arya D P. Beilstein J Org Chem. 2018;14:1051–1086. doi: 10.3762/bjoc.14.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rescifina A, Zagni C, Varrica M G, Pistarà V, Corsaro A. Eur J Med Chem. 2014;74:95–115. doi: 10.1016/j.ejmech.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 3.Venugopal S, Sharma V, Mehra A, Singh I, Singh G. Chem Biol Drug Des. 2022;100:580–598. doi: 10.1111/cbdd.14116. [DOI] [PubMed] [Google Scholar]
- 4.Sharma V, Gupta M, Kumar P, Sharma A. Curr Pharm Des. 2021;27:15–42. doi: 10.2174/1381612826666201118113311. [DOI] [PubMed] [Google Scholar]
- 5.Martins‐Teixeira M B, Carvalho I. ChemMedChem. 2020;15(11):933–948. doi: 10.1002/cmdc.202000131. [DOI] [PubMed] [Google Scholar]
- 6.Zenkov R G, Ektova L V, Vlasova O A, Belitskiy G A, Yakubovskaya M G, Kirsanov K I. Chem Heterocycl Compd (N Y, NY, U S) 2020;56:644–658. doi: 10.1007/s10593-020-02714-4. [DOI] [Google Scholar]
- 7.Prasher P, Sharma M. Med Chem Commun. 2018;9:1589–1618. doi: 10.1039/c8md00384j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park Y-S, Shin W-S, Kim C-S, Ahn C M, Qi X-F, Kim S-K. Mol Cell Toxicol. 2018;14:9–18. doi: 10.1007/s13273-018-0002-8. [DOI] [Google Scholar]
- 9.Tomczyk M D, Walczak K Z. Eur J Med Chem. 2018;159:393–422. doi: 10.1016/j.ejmech.2018.09.055. [DOI] [PubMed] [Google Scholar]
- 10.Tumir L-M, Radić Stojković M, Piantanida I. Beilstein J Org Chem. 2014;10:2930–2954. doi: 10.3762/bjoc.10.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ma X, Shi L, Zhang B, Liu L, Fu Y, Zhang X. Anal Bioanal Chem. 2022;414:4551–4573. doi: 10.1007/s00216-022-03995-8. [DOI] [PubMed] [Google Scholar]
- 12.Kadagathur M, Sujat Shaikh A, Panda B, George J, Phanindranath R, Kumar Sigalapalli D, Bhale N A, Godugu C, Nagesh N, Shankaraiah N, et al. Bioorg Chem. 2022;122:105706. doi: 10.1016/j.bioorg.2022.105706. [DOI] [PubMed] [Google Scholar]
- 13.Schatzschneider U. Metallo-Drugs: Development and Action of Anticancer Agents. Vol. 18. Berlin, Germany: De Gruyter; 2018. Metallointercalators and Metalloinsertors: Structural Requirements for DNA Recognition and Anticancer Activity; pp. 387–436. [DOI] [PubMed] [Google Scholar]
- 14.Aliyeu T M, Berdnikova D V, Fedorova O A, Gulakova E N, Stremmel C, Ihmels H. J Org Chem. 2016;81:9075–9085. doi: 10.1021/acs.joc.6b01695. [DOI] [PubMed] [Google Scholar]
- 15.Benner K, Ihmels H, Kölsch S, Pithan P M. Org Biomol Chem. 2014;12:1725–1734. doi: 10.1039/c3ob42140f. [DOI] [PubMed] [Google Scholar]
- 16.Bosch P, Sucunza D, Mendicuti F, Domingo A, Vaquero J J. Org Chem Front. 2018;5:1916–1927. doi: 10.1039/c8qo00236c. [DOI] [Google Scholar]
- 17.Bosch P, Marcelo G, Matamoros-Recio A, Sucunza D, Mendicuti F, Domingo A, Vaquero J J. Dyes Pigm. 2021;192:109443. doi: 10.1016/j.dyepig.2021.109443. [DOI] [Google Scholar]
- 18.Lee S B, Han Y R, Jeon H-J, Jun C-H, Kim S-K, Chin J, Lee S-J, Jeong M, Lee J-E, Lee C-H, et al. J Mater Chem B. 2019;7:7326–7331. doi: 10.1039/c9tb01266d. [DOI] [PubMed] [Google Scholar]
- 19.Protti S, Fagnoni M, Albini A. Photochemical Synthesis. In: Zhang W, Cue B W, editors. Green Techniques for Organic Synthesis and Medicinal Chemistry. 2nd ed. Hoboken, NJ, USA: John Wiley & Sons; 2018. pp. 373–406. [DOI] [Google Scholar]
- 20.Berdnikova D V, Aliyeu T M, Paululat T, Fedorov Y V, Fedorova O A, Ihmels H. Chem Commun. 2015;51:4906–4909. doi: 10.1039/c5cc01025j. [DOI] [PubMed] [Google Scholar]
- 21.Berdnikova D V, Heider J, Ihmels H, Sewald N, Pithan P M. ChemPhotoChem. 2020;4:520–525. doi: 10.1002/cptc.202000015. [DOI] [Google Scholar]
- 22.Simeth N A, Kobayashi S, Kobauri P, Crespi S, Szymanski W, Nakatani K, Dohno C, Feringa B L. Chem Sci. 2021;12:9207–9220. doi: 10.1039/d1sc02194j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Linares M, Sun H, Biler M, Andréasson J, Norman P. Phys Chem Chem Phys. 2019;21:3637–3643. doi: 10.1039/c8cp05326j. [DOI] [PubMed] [Google Scholar]
- 24.Nakagawa Y, Hishida T, Sumaru K, Morishita K, Kirito K, Yokojima S, Sakamoto Y, Nakamura S, Uchida K. J Med Chem. 2023;66:5937–5949. doi: 10.1021/acs.jmedchem.3c00164. [DOI] [PubMed] [Google Scholar]
- 25.Chen H, Li R, Li S, Andréasson J, Choi J H. J Am Chem Soc. 2017;139:1380–1383. doi: 10.1021/jacs.6b10821. [DOI] [PubMed] [Google Scholar]
- 26.Dohmen C, Ihmels H. Org Biomol Chem. 2023;21:1958–1966. doi: 10.1039/d3ob00013c. [DOI] [PubMed] [Google Scholar]
- 27.Cheng H-B, Zhang S, Bai E, Cao X, Wang J, Qi J, Liu J, Zhao J, Zhang L, Yoon J. Adv Mater (Weinheim, Ger) 2022;34:2108289. doi: 10.1002/adma.202108289. [DOI] [PubMed] [Google Scholar]
- 28.Kobauri P, Dekker F J, Szymanski W, Feringa B L. Angew Chem, Int Ed. 2023;62:e202300681. doi: 10.1002/anie.202300681. [DOI] [PubMed] [Google Scholar]
- 29.Leistner A-L, Pianowski Z L. Eur J Org Chem. 2022:e202101271. doi: 10.1002/ejoc.202101271. [DOI] [Google Scholar]
- 30.Heinrich B, Bouazoune K, Wojcik M, Bakowsky U, Vázquez O. Org Biomol Chem. 2019;17:1827–1833. doi: 10.1039/c8ob02343c. [DOI] [PubMed] [Google Scholar]
- 31.Gao M, Kwaria D, Norikane Y, Yue Y. Nat Sci. 2022;3:e20220020. doi: 10.1002/ntls.20220020. [DOI] [Google Scholar]
- 32.Sharma M, Friedman S H. ChemPhotoChem. 2021;5(7):611–618. doi: 10.1002/cptc.202100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez J, Mosquera J, Learte-Aymamí S, Vázquez M E, Mascareñas J L. Acc Chem Res. 2020;53(10):2286–2298. doi: 10.1021/acs.accounts.0c00415. [DOI] [PubMed] [Google Scholar]
- 34.Ihmels H, Mattay J, May F, Thomas L. Org Biomol Chem. 2013;11:5184–5188. doi: 10.1039/c3ob40930a. [DOI] [PubMed] [Google Scholar]
- 35.Bohne C, Faulhaber K, Giese B, Häfner A, Hofmann A, Ihmels H, Köhler A-K, Perä S, Schneider F, Sheepwash M A L. J Am Chem Soc. 2005;127:76–85. doi: 10.1021/ja046189m. [DOI] [PubMed] [Google Scholar]
- 36.Ihmels H, Salbach A. Photochem Photobiol. 2006;82(6):1572–1576. doi: 10.1111/j.1751-1097.2006.tb09813.x. [DOI] [PubMed] [Google Scholar]
- 37.Barbafina A, Amelia M, Latterini L, Aloisi G G, Elisei F. J Phys Chem A. 2009;113:14514–14520. doi: 10.1021/jp9040315. [DOI] [PubMed] [Google Scholar]
- 38.Schäfer K, Ihmels H, Porcù E, Viola G. Chem – Eur J. 2017;23:370–379. doi: 10.1002/chem.201603807. [DOI] [PubMed] [Google Scholar]
- 39.Triesscheijn M, Baas P, Schellens J H M, Stewart F A. Oncologist. 2006;11:1034–1044. doi: 10.1634/theoncologist.11-9-1034. [DOI] [PubMed] [Google Scholar]
- 40.Donnelly R F, McCarron P A, Tunney M M. Microbiol Res. 2008;163:1–12. doi: 10.1016/j.micres.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Wiehe A, O’Brien J M, Senge M O. Photochem Photobiol Sci. 2019;18(11):2565–2612. doi: 10.1039/c9pp00211a. [DOI] [PubMed] [Google Scholar]
- 42.Kessel D. Photochem Photobiol. 2023;99:199–203. doi: 10.1111/php.13616. [DOI] [PubMed] [Google Scholar]
- 43.Piksa M, Lian C, Samuel I C, Pawlik K J, Samuel I D W, Matczyszyn K. Chem Soc Rev. 2023;52:1697–1722. doi: 10.1039/d0cs01051k. [DOI] [PubMed] [Google Scholar]
- 44.Gunaydin G, Gedik M E, Ayan S. Front Chem (Lausanne, Switz) 2021;9:691697. doi: 10.3389/fchem.2021.691697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim M M, Darafsheh A. Photochem Photobiol. 2020;96:280–294. doi: 10.1111/php.13219. [DOI] [PubMed] [Google Scholar]
- 46.Baptista M S, Cadet J, Greer A, Thomas A H. Photochem Photobiol. 2023;99:313–334. doi: 10.1111/php.13774. [DOI] [PubMed] [Google Scholar]
- 47.Aires-Fernandes M, Botelho Costa R, Rochetti do Amaral S, Mussagy C U, Santos-Ebinuma V C, Primo F L. Molecules. 2022;27:6848. doi: 10.3390/molecules27206848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao X, Liu J, Fan J, Chao H, Peng X. Chem Soc Rev. 2021;50:4185–4219. doi: 10.1039/d0cs00173b. [DOI] [PubMed] [Google Scholar]
- 49.Mariño‐Ocampo N, Dibona‐Villanueva L, Escobar‐Álvarez E, Guerra‐Díaz D, Zúñiga‐Núñez D, Fuentealba D, Robinson‐Duggon J. Photochem Photobiol. 2023;99(2):469–497. doi: 10.1111/php.13749. [DOI] [PubMed] [Google Scholar]
- 50.Melissari Z, Williams R M, Senge M O. Porphyrinoids for Photodynamic Therapy. In: Lang H, Rueffer T, editors. Applications of Porphyrinoids as Functional Materials. London, UK: The Royal Society of Chemistry; 2021. pp. 252–291. [DOI] [Google Scholar]
- 51.Korolchuk A M, Zolottsev V A, Misharin A Y. Top Curr Chem. 2023;381:10. doi: 10.1007/s41061-023-00421-0. [DOI] [PubMed] [Google Scholar]
- 52.Rennie C C, Edkins R M. Dalton Trans. 2022;51:13157–13175. doi: 10.1039/d2dt02040h. [DOI] [PubMed] [Google Scholar]
- 53.Sánchez-García D. J Porphyrins Phthalocyanines. 2023;27:1009–1014. doi: 10.1142/s1088424623300124. [DOI] [Google Scholar]
- 54.Smith C B, Days L C, Alajroush D R, Faye K, Khodour Y, Beebe S J, Holder A A. Photochem Photobiol. 2022;98:17–41. doi: 10.1111/php.13467. [DOI] [PubMed] [Google Scholar]
- 55.Teeuwen P C P, Melissari Z, Senge M O, Williams R M. Molecules. 2022;27:6967. doi: 10.3390/molecules27206967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karges J. Angew Chem, Int Ed. 2022;61:e202112236. doi: 10.1002/anie.202112236. [DOI] [PubMed] [Google Scholar]
- 57.Meng Z, Xue H, Wang T, Chen B, Dong X, Yang L, Dai J, Lou X, Xia F. J Nanobiotechnol. 2022;20(1):344. doi: 10.1186/s12951-022-01553-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aghajanzadeh M, Zamani M, Rajabi Kouchi F, Eixenberger J, Shirini D, Estrada D, Shirini F. Pharmaceutics. 2022;14(2):322. doi: 10.3390/pharmaceutics14020322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gierlich P, Mata A I, Donohoe C, Brito R M M, Senge M O, Gomes-da-Silva L C. Molecules. 2020;25:5317. doi: 10.3390/molecules25225317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pham T C, Nguyen V-N, Choi Y, Lee S, Yoon J. Chem Rev. 2021;121:13454–13619. doi: 10.1021/acs.chemrev.1c00381. [DOI] [PubMed] [Google Scholar]
- 61.An Y-W, Jin H-T, Yuan B, Wang J-C, Wang C, Liu H-Q. Oncol Lett. 2021;21(5):359. doi: 10.3892/ol.2021.12620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fang J, Huang X, Yang Y, Wang X, Liang X, Liu J. Biochem Biophys Res Commun. 2021;552:183–190. doi: 10.1016/j.bbrc.2021.02.147. [DOI] [PubMed] [Google Scholar]
- 63.Oliveira P M, Lopes T Z, Tedesco A C, Rahal P, Calmon M F. Photodiagn Photodyn Ther. 2020;32:102045. doi: 10.1016/j.pdpdt.2020.102045. [DOI] [PubMed] [Google Scholar]
- 64.Lopes T Z, de Moraes F R, Tedesco A C, Arni R K, Rahal P, Calmon M F. Biomed Pharmacother. 2020;123:109794. doi: 10.1016/j.biopha.2019.109794. [DOI] [PubMed] [Google Scholar]
- 65.Liu H-Q, An Y-W, Hu A-Z, Li M-H, Cui G-H. Open Chem. 2019;17:413–421. doi: 10.1515/chem-2019-0048. [DOI] [Google Scholar]
- 66.Bhattacharyya R, Gupta P, Bandyopadhyay S K, Patro B S, Chattopadhyay S. Chem-Biol Interact. 2018;285:27–39. doi: 10.1016/j.cbi.2018.02.032. [DOI] [PubMed] [Google Scholar]
- 67.Patro B S, Bhattacharyya R, Gupta P, Bandyopadhyay S, Chattopadhyay S. J Photochem Photobiol, B. 2019;194:140–148. doi: 10.1016/j.jphotobiol.2019.03.011. [DOI] [PubMed] [Google Scholar]
- 68.Bolduc A, Dong Y, Guérin A, Skene W G. Phys Chem Chem Phys. 2012;14:6946–6956. doi: 10.1039/c2cp40293a. [DOI] [PubMed] [Google Scholar]
- 69.Kölsch S, Ihmels H, Mattay J, Sewald N, Patrick B O. Beilstein J Org Chem. 2020;16:111–124. doi: 10.3762/bjoc.16.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dsouza R N, Pischel U, Nau W M. Chem Rev. 2011;111:7941–7980. doi: 10.1021/cr200213s. [DOI] [PubMed] [Google Scholar]
- 71.Fukui K, Tanaka K, Fujitsuka M, Watanabe A, Ito O. J Photochem Photobiol, B. 1999;50:18–27. doi: 10.1016/s1011-1344(99)00063-9. [DOI] [Google Scholar]
- 72.Šmidlehner T, Piantanida I, Pescitelli G. Beilstein J Org Chem. 2018;14:84–105. doi: 10.3762/bjoc.14.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nordén B, Rodger A, Dafforn T. Linear Dichroism and Circular Dichroism. Cambridge, UK: The Royal Society of Chemistry; 2010. [DOI] [Google Scholar]
- 74.Berdnikova D V, Fedorova O A, Tulyakova E V, Li H, Kölsch S, Ihmels H. Photochem Photobiol. 2015;91:723–731. doi: 10.1111/php.12405. [DOI] [PubMed] [Google Scholar]
- 75.Matsuoka Y, Nordén B. Biopolymers. 1982;21:2433–2452. doi: 10.1002/bip.360211208. [DOI] [PubMed] [Google Scholar]
- 76.Michov B. Electrophoresis: Theory and Practice. Berlin, Germany: De Gruyter; 2020. [DOI] [Google Scholar]
- 77.Dąbrowski J M. Reactive Oxygen Species in Photodynamic Therapy: Mechanisms of Their Generation and Potentiation. In: van Eldik R, Hubbard C D, editors. Advances in Inorganic Chemistry: Inorganic Reaction Mechanisms. Cambridge, MA, USA: Academic Press; 2017. pp. 343–394. [DOI] [Google Scholar]
- 78.von Sonntag C. Free-Radical-Induced DNA Damage and Its Repair. A Chemical Perspective. Berlin, Heidelberg: Springer; 2006. [DOI] [Google Scholar]
- 79.Zhang B, Xu L, Zhou Y, Zhang W, Wang Y, Zhu Y. Luminescence. 2020;35:305–311. doi: 10.1002/bio.3728. [DOI] [PubMed] [Google Scholar]
- 80.Lin Y, Chen J, Xie P, Wang J, Hou Y, Zhang L, Zhang B, Cao Y. Res Chem Intermed. 2000;26:793–803. doi: 10.1163/156856700x00705. [DOI] [Google Scholar]
- 81.Merkel P B, Kearns D R. J Am Chem Soc. 1972;94:7244–7253. doi: 10.1021/ja00776a003. [DOI] [Google Scholar]
- 82.Simic M G, Taylor K A, Ward J F, et al., editors. Oxygen Radicals in Biology and Medicine. New York, NY, USA: Plenum Press; 1988. [Google Scholar]
- 83.Armitage B. Chem Rev. 1998;98:1171–1200. doi: 10.1021/cr960428+. [DOI] [PubMed] [Google Scholar]
- 84.Nicotera T M, Munson B R, Fiel R J. Photochem Photobiol. 1994;60:295–300. doi: 10.1111/j.1751-1097.1994.tb05107.x. [DOI] [PubMed] [Google Scholar]
- 85.OhUigin C, McConnell D J, Kelly J M, van der Putten W J M. Nucleic Acids Res. 1987;15:7411–7427. doi: 10.1093/nar/15.18.7411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhu X-J, Wang P, Leung H W C, Wong W-K, Wong W-Y, Kwong D W J. Chem – Eur J. 2011;17:7041–7052. doi: 10.1002/chem.201003342. [DOI] [PubMed] [Google Scholar]
- 87.Cadet J, Wagner J R. Arch Biochem Biophys. 2014;557:47–54. doi: 10.1016/j.abb.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 88.Fujii J, Soma Y, Matsuda Y. Molecules. 2023;28:4085. doi: 10.3390/molecules28104085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang C, Ebel K, Heinze K, Resch-Genger U, Bald I. Chem – Eur J. 2023;29:e202203719. doi: 10.1002/chem.202203719. [DOI] [PubMed] [Google Scholar]
- 90.Baptista M S, Cadet J, Di Mascio P, Ghogare A A, Greer A, Hamblin M R, Lorente C, Nunez S C, Ribeiro M S, Thomas A H, et al. Photochem Photobiol. 2017;93:912–919. doi: 10.1111/php.12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Viola G, Bressanini M, Gabellini N, Vedaldi D, Dall‘Acqua F, Ihmels H. Photochem Photobiol Sci. 2002;1(11):882–889. doi: 10.1039/b204275d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Detailed experimental procedures, additional spectroscopic data and 1H NMR spectra.
Data Availability Statement
All data that supports the findings of this study is available in the published article and/or the supporting information to this article.