Abstract
Biotransformation is one of the main mechanisms used by the body to eliminate drugs. As drug molecules become more complicated, the involvement of drug metabolizing enzymes increases beyond those that are typically studied, such as the cytochrome P450 enzymes. In this review, we try to capture the many outstanding articles that were published in the past year in the field of biotransformation. We have divided the articles into two categories of (1) metabolites and drug metabolizing enzymes, and (2) bioactivation and safety.
This annual review is the fifth of its kind since 2016 (see references). This effort in itself also continues to evolve. We have followed the same format we used in previous years in terms of the selection of articles and the authoring of each section. I am pleased of the continued support of Rietjens, Miller, Zhang, Driscoll and Mitra to this review. We would like to welcome Klarissa D. Jackson as a new author for this year’s issue. We strive to maintain a balance of authors from academic and industry settings.
We would be pleased to hear your opinions of our commentary, and we extend an invitation to anyone who would like to contribute to a future edition of this review.
Cyrus Khojasteh, on behalf of the authors.
Figure 1.
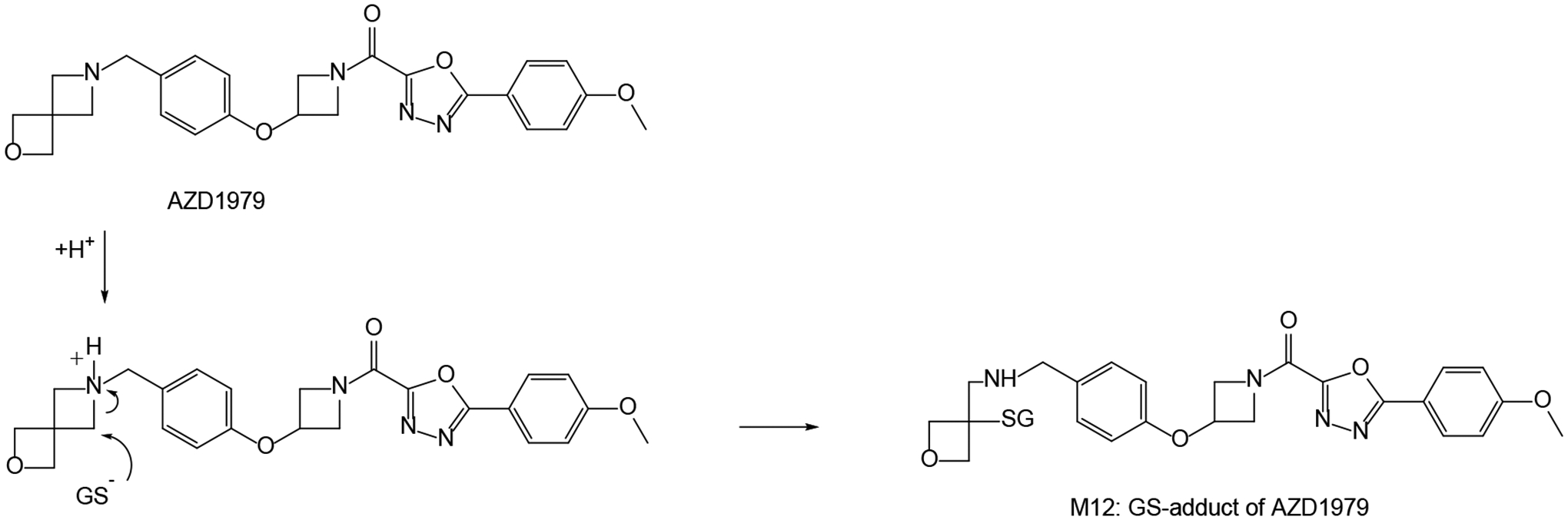
Glutathione conversion of AZD1979 in the active site of GST, proceeding via protonation of the cyclic aminyl nitrogen of the strained spiro-azetidinyl moiety and subsequent attack by deprotonated GSH (GS−).
Figure 4.
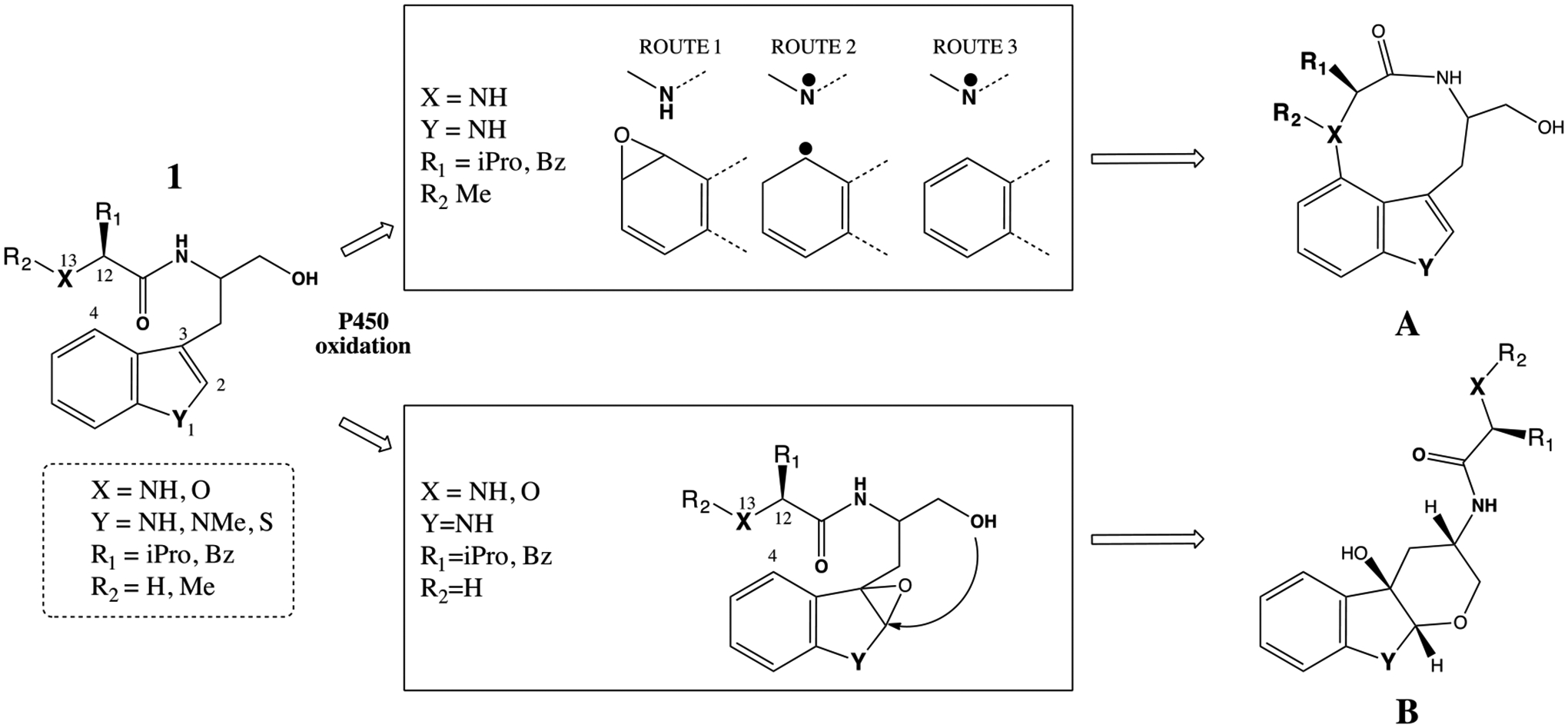
Bacterial P450-mediated oxidation for the formation of two cyclic products A and B. The specific requirement for the formation of these metabolites are captured in the middle boxes with the descriptions of X, Y, R1 and R2 substituents.
iPro=isopropyl and Bz= benzyl groups.
Figure 5.
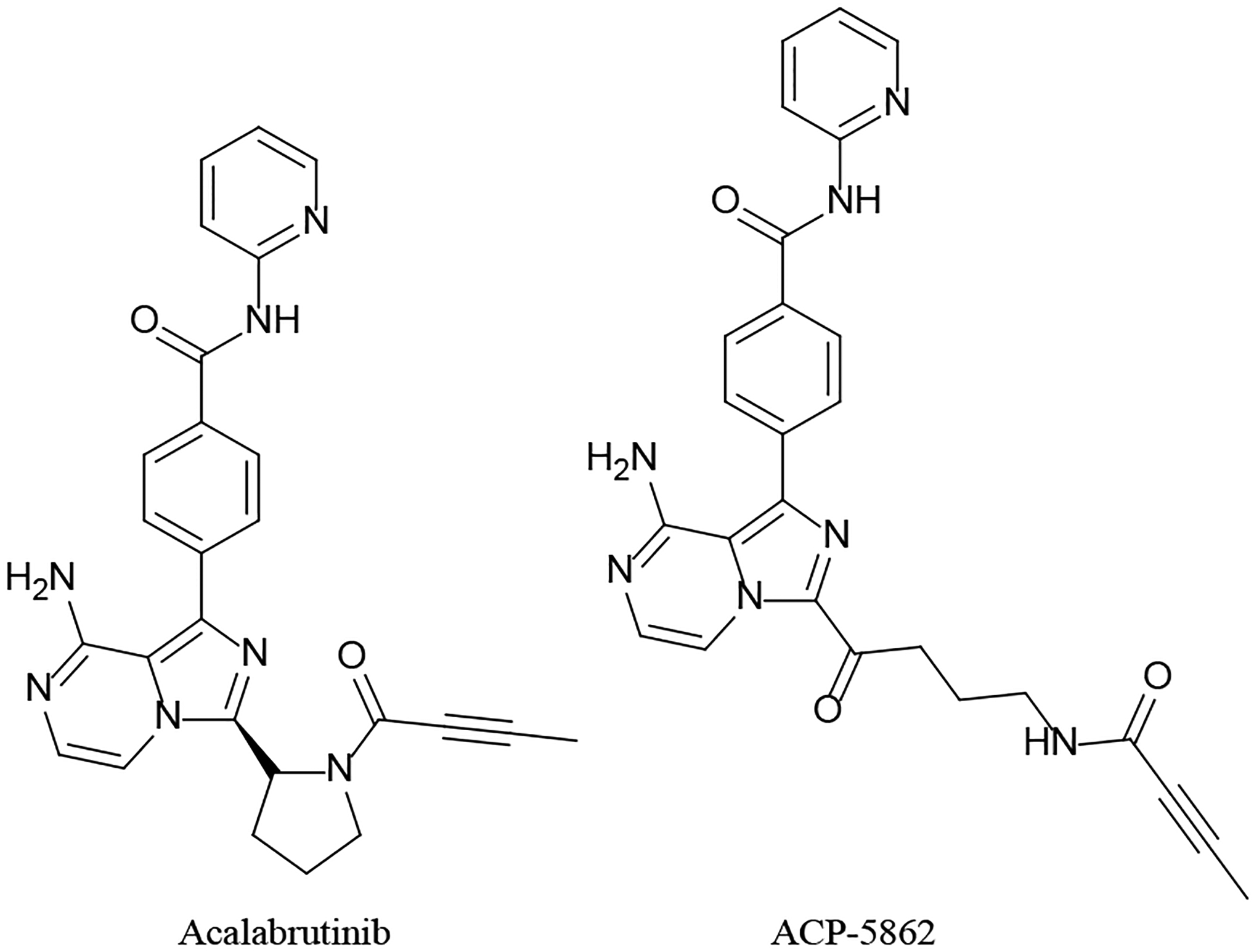
Acalabrutinib and its active metabolite
Figure 6:
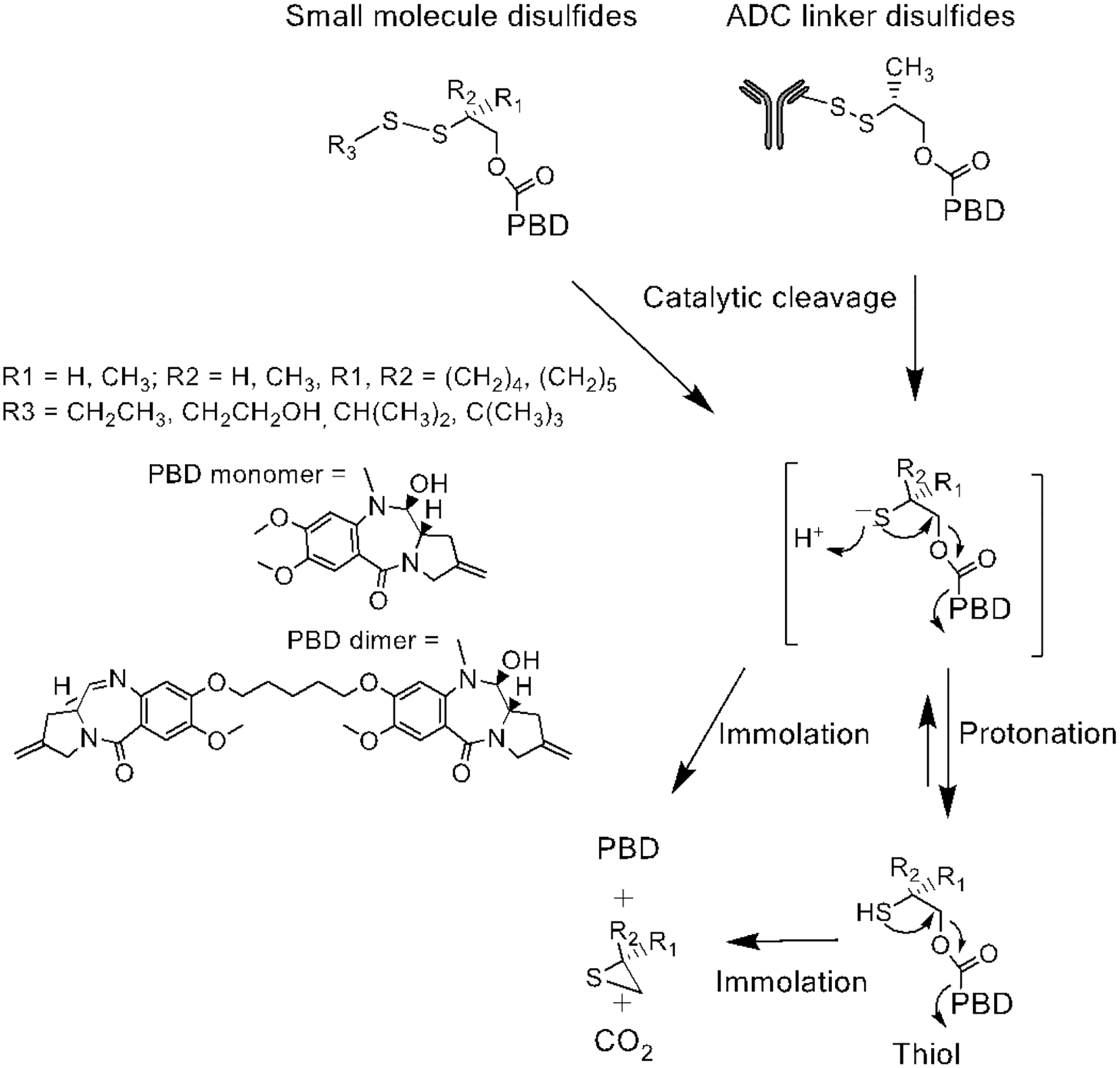
Catalytic cleavage of disulfide bonds in small molecules and ADC linkers.
Figure 7.
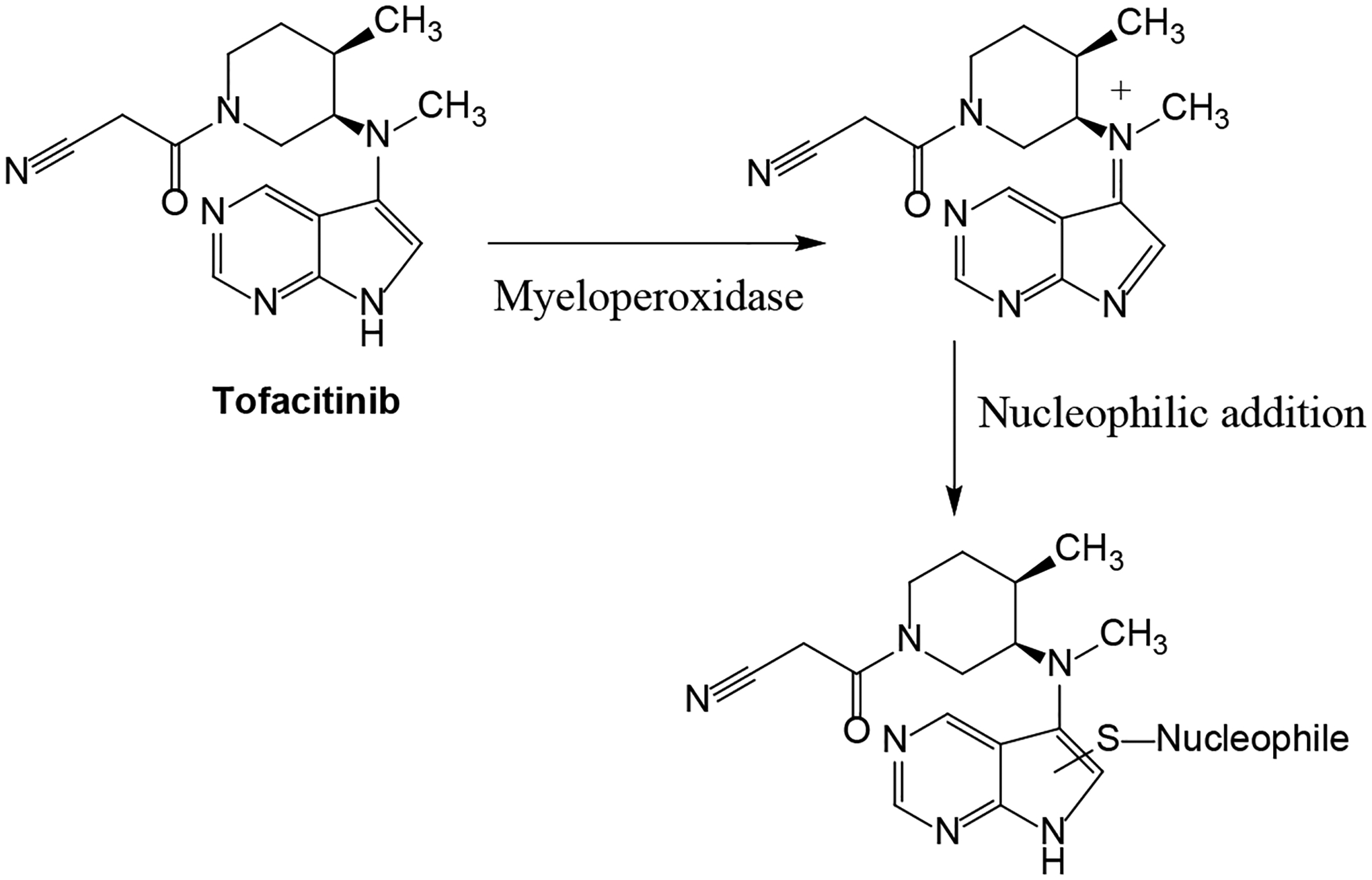
Proposed bioactivation mechanism of tofacitinib by myeloperoxidase.
Figure 10.
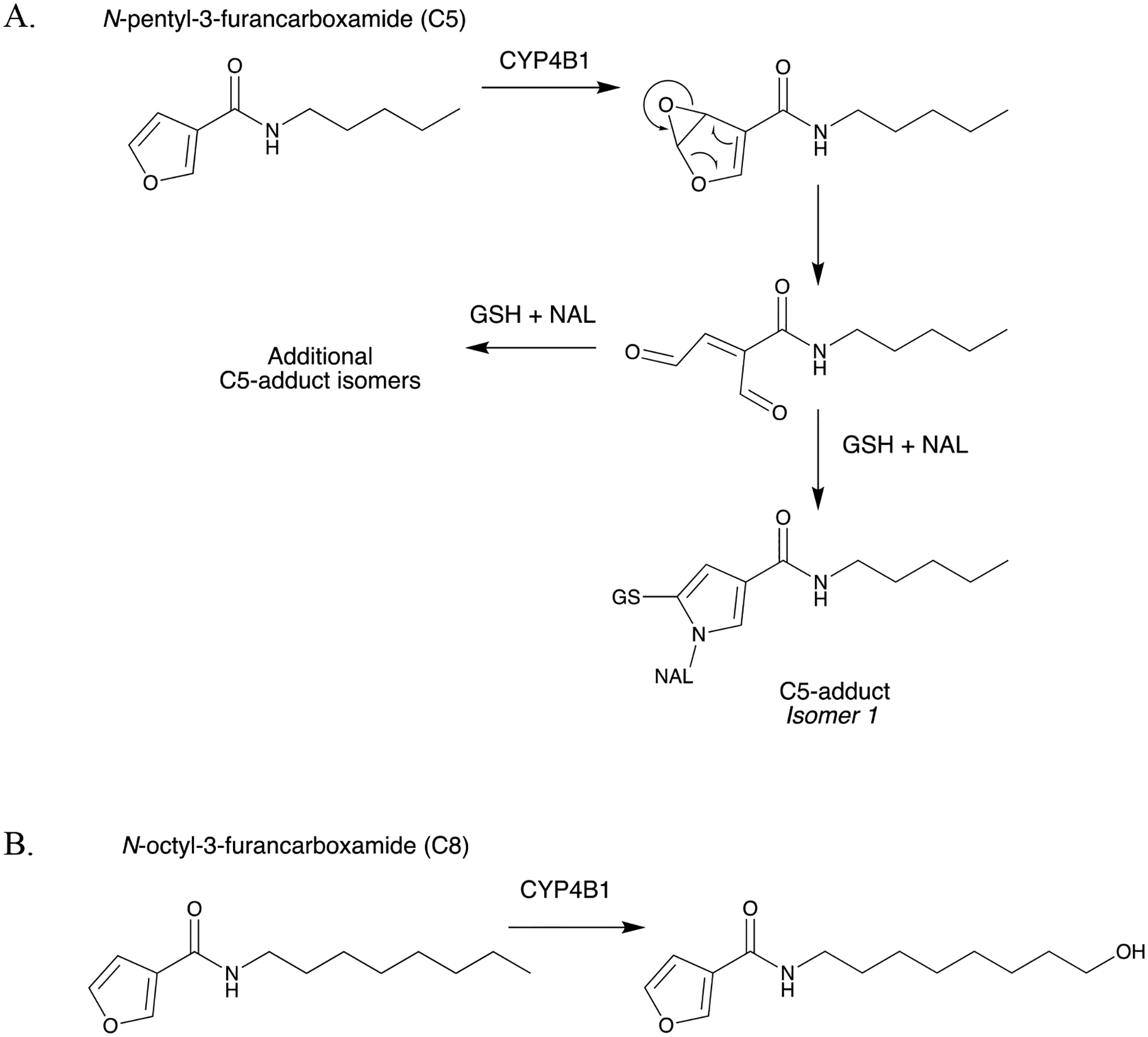
CYP4B1-mediated bioactivation vs. ω-hydroxylation of N-alkyl-3-furancarboxamides. (A) Bioactivation of N-pentyl-3-furancarboxamide (C5) to form GSH/NAL-trapped C5-adducts, and (B) ω-hydroxylation of N-octyl-3-furancarboxamide (C8) (Kowlaski et al., 2019).
Figure 13.
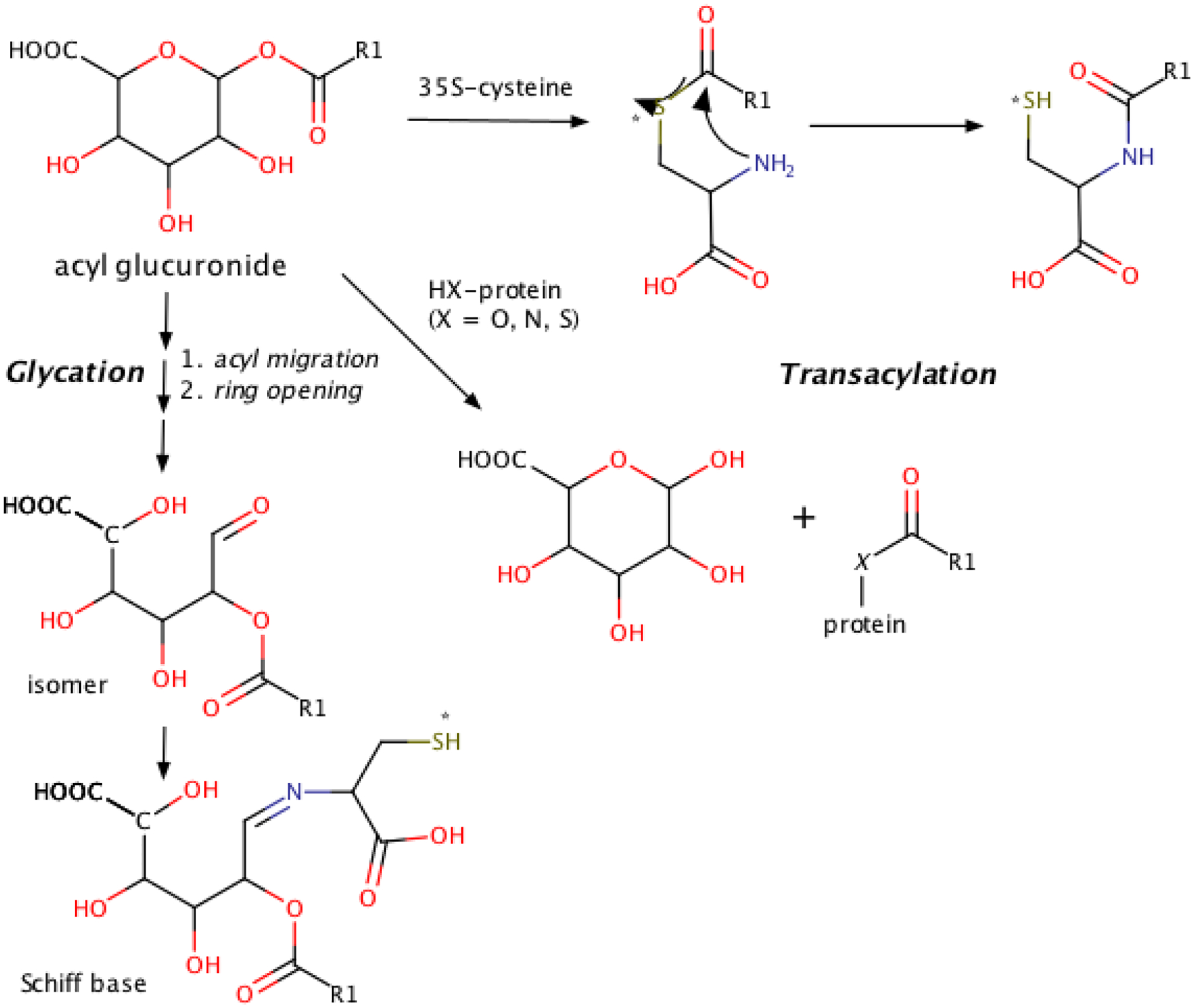
Proposed pathways leading to trapping of the glycation adduct as a Schiff base and the transacylation adduct ultimately as an amide. 35S-label indicated by asterisk (*).
Table 1.
Articles covered in this review.
| Title | Authors | Source | |
|---|---|---|---|
| Metabolites and drug metabolizing enzymes | |||
| 1 | Metabolism of Strained Rings: Glutathione S-transferase-Catalyzed Formation of a Glutathione-Conjugated Spiro-azetidine without Prior Bioactivation | Li XQ, Grönberg G, Bangur EH, Hayes MA, Catagnoli Jr. N, Weidolf L | Drug Metabolism Disposition 47:1247–1256, 2019 |
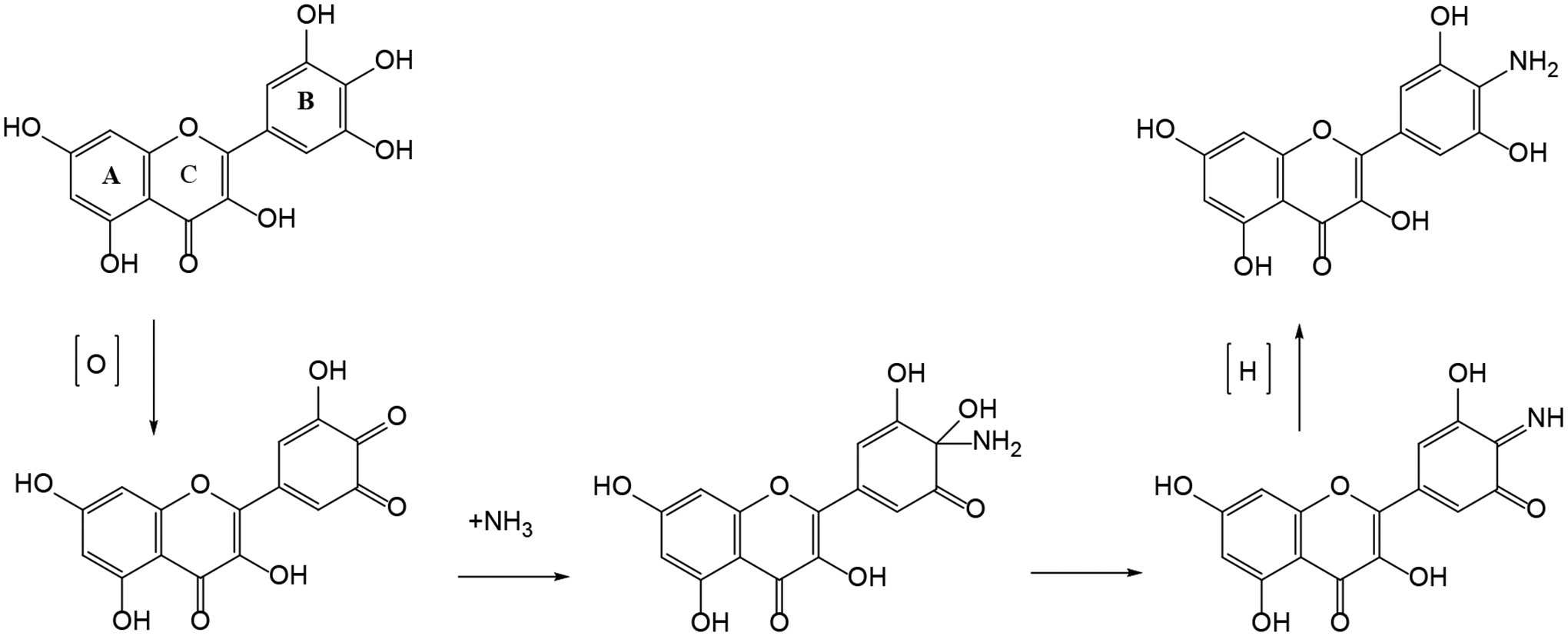
| 2 | Biotransformation of Myricetin: A Novel Metabolic Pathway to Produce Aminated Products in Mice | Zhang S, Wang R, Zhao Y, Tareq FS, Sang FS | Molecular Nutrition and Food Research 63(14):1900203, 2019 |
| 3 | The Impact of Carboxylesterases in Drug Metabolism and Pharmacokinetics | Di L | Current Drug Metabolism 20:91–102, 2019 |
| 4 | Biocatalytic Reversal of Advanced Glycation End Product Modification | Kim NY, Goddard TN, Sohn S, Spiegel DA, Crawford JM | ChemBioChem 20:2402–2410, 2019 |
| 5 | Molecular basis for the P450-catalyzed C–N bond formation in indolactam biosynthesis | He F, Mori T, Morita I, Nakamura H, Alblova M, Hoshino S, Awakawa T, Abe I | Nature Chemical Biology 15:1206–1213, 2019 |
| 6 | Bioavailability, Biotransformation, and Excretion of the Covalent Bruton Tyrosine Kinase Inhibitor Acalabrutinib in Rats, Dogs, and Humans | Podoll T, Pearson PG, Evarts J, Ingallinera T, Bibikova E, Sun H, Gohdes M, Cardinal K, Sanghvi M, Slatter JG | Drug Metabolism Disposition 47:145–154, 2019 |
| 7 | Catalytic cleavage of disulfide bonds in small molecules and linkers of antibody- drug conjugates | Zhang D, Fourie-O’Donohue A, Dragovich PS, Pillow TH, Sadowsky JD, Kozak KR, Cass RT, Liu L, Deng Y, Liu Y, Hop CECA, Khojasteh SC | Drug Metabolism Disposition 47(10):1156–1163, 2019 |
| Bioactivation and Safety | |||
| 8 | Metabolic Activation of Tofacitinib Mediated by Myeloperoxidase in Vitro | Guo Y, Jia Y, Han L, Zhao Y, Li W, Zhang Z, Peng Y, Zheng J | Chemical Research in Toxicology 32:2459–2465, 2019 |
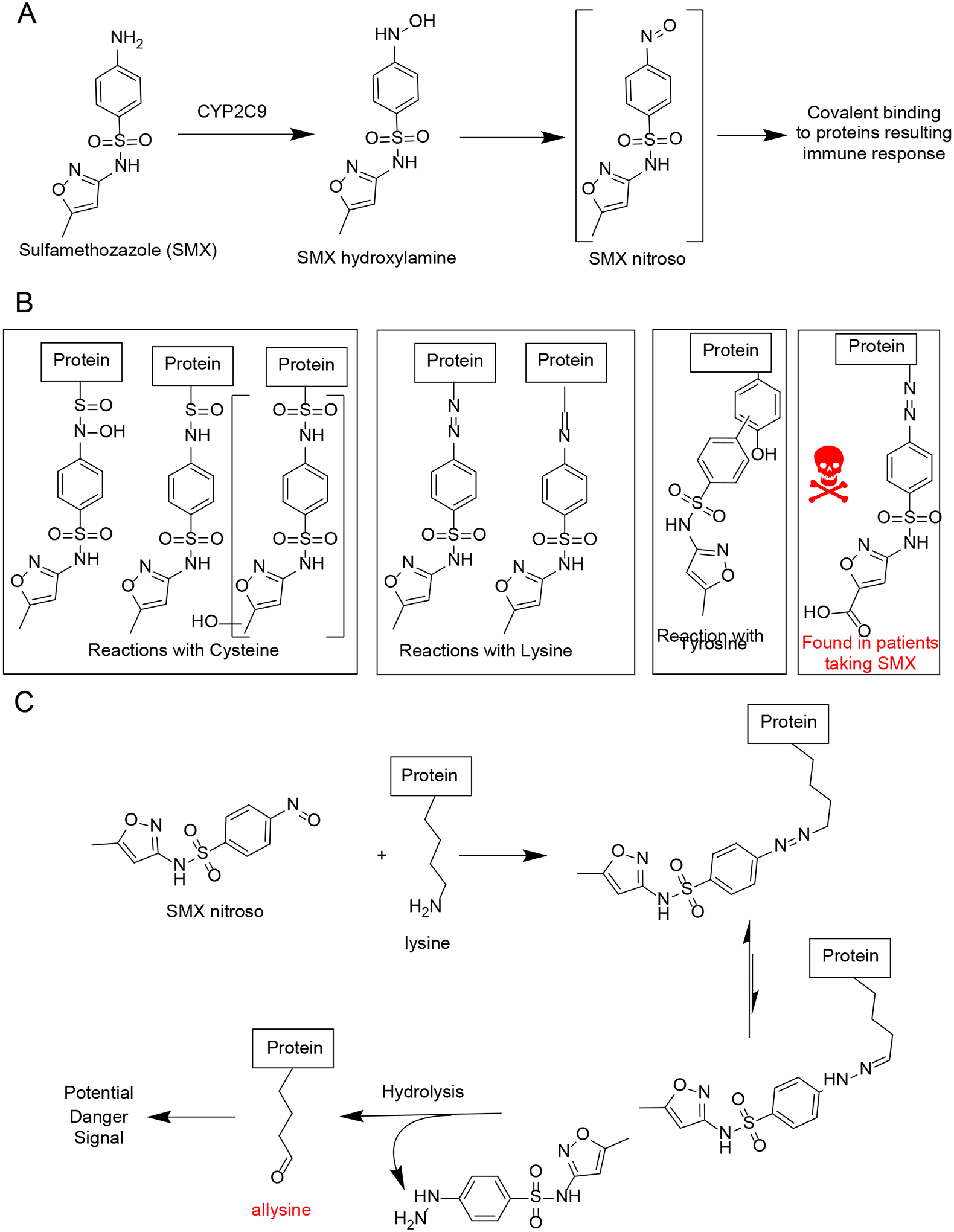
| 9 | Definition of Haptens Derived from Sulfamethoxazole: In Vitro and in Vivo | Tailor A, Waddington J, Hamlett J, Maggs J, Kafu L, Farrell J, Dear G, Whitaker P, Naisbitt D, Park K, Meng X | Chemical Research in Toxicology 32(10):2095–2106, 2019 |
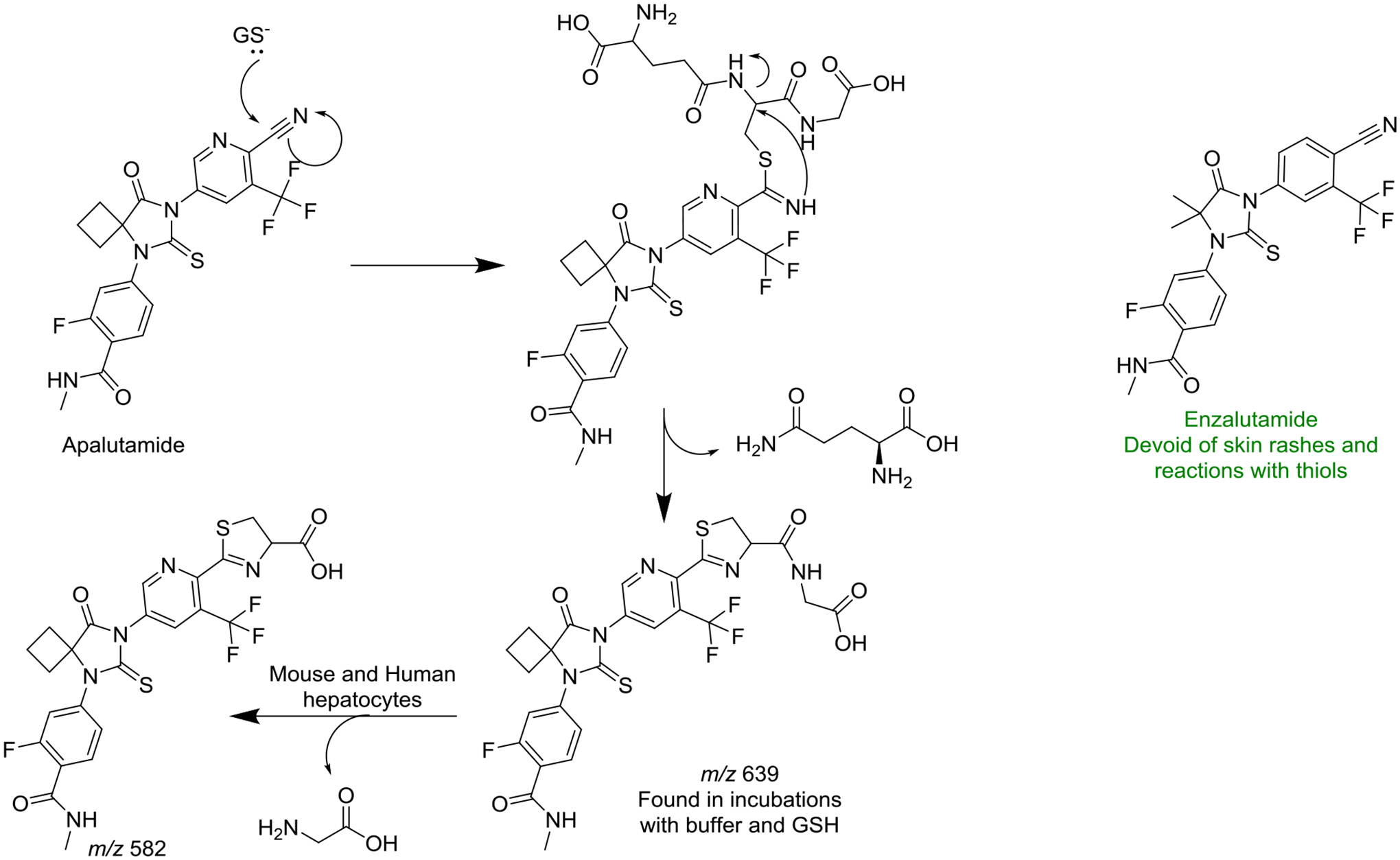
| 10 | Enzalutamide and Apalutamide: In Vitro Chemical Reactivity Studies and Activity in a Mouse Drug Allergy Model | Ji C, Guha M, Zhu X, Whritenour J, Hemkens M, Tse S, Walker G, Evans E, Khan N, Finkelstein M, Callegari E, Obach R | Chemical Research in Toxicology 32:2095–2106, 2019 |
| 11 | Structure-activity Relationships of CYP4B1 Bioactivation of 4-ipomeanol Congeners: Direct Correlation Between Cytotoxicity and Trapped Reactive Intermediates. | Kowalski JP, McDonald MG, Whittington D, Guttman M, Scian M, Girhard M, Hanenberg H, Wiek C, Rettie AE | Chemical Research in Toxicology 32:2488–2498, 2019 |
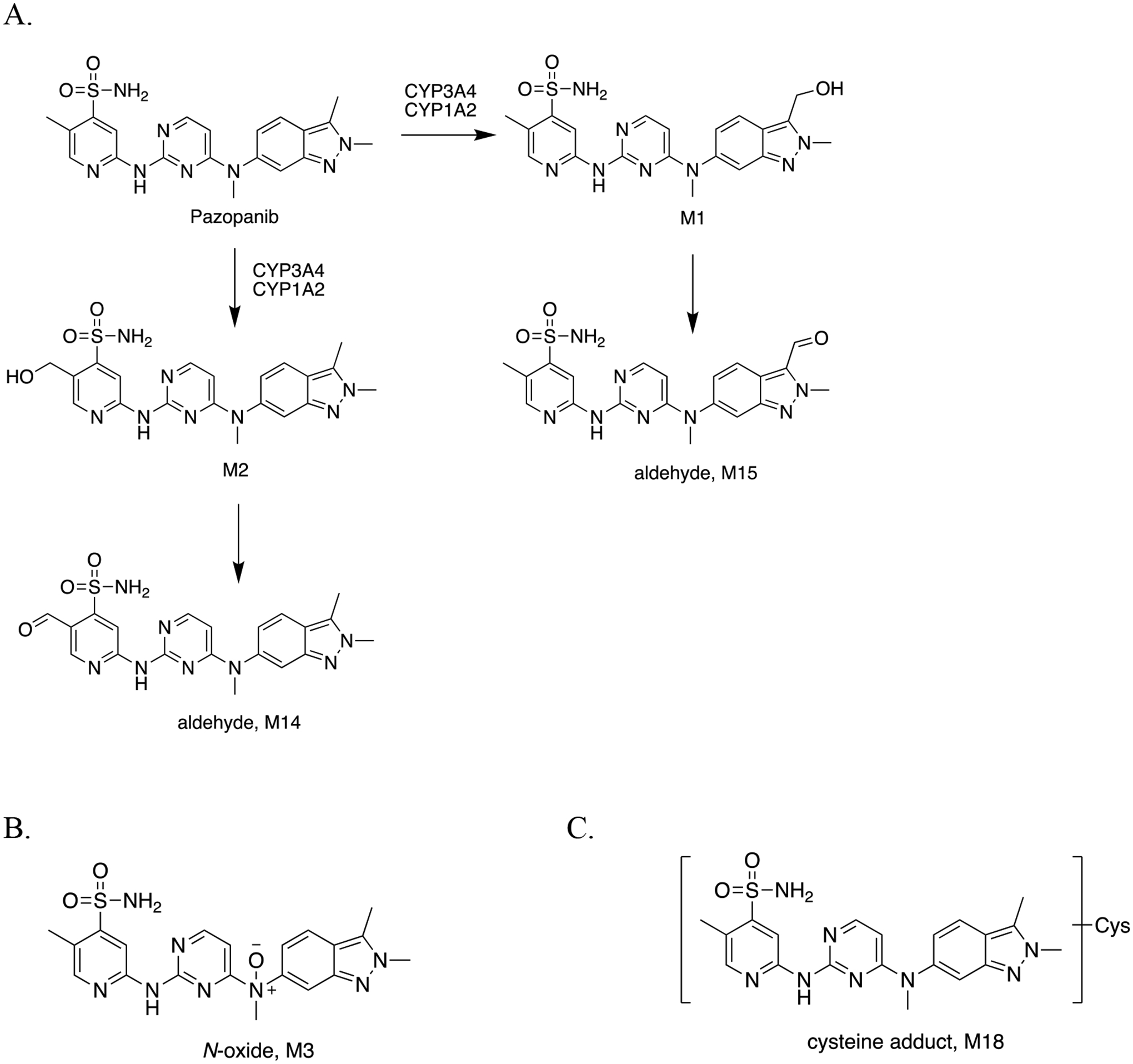
| 12 | A Metabolomic Perspective of Pazopanib-induced Acute Hepatotoxicity in Mice. | Wang YK, Yang XN, Liang WQ, Xiao Y, Zhao Q, Xiao, Gonzalez FJ, Li F | Xenobiotica 49(6):655–670, 2019 |
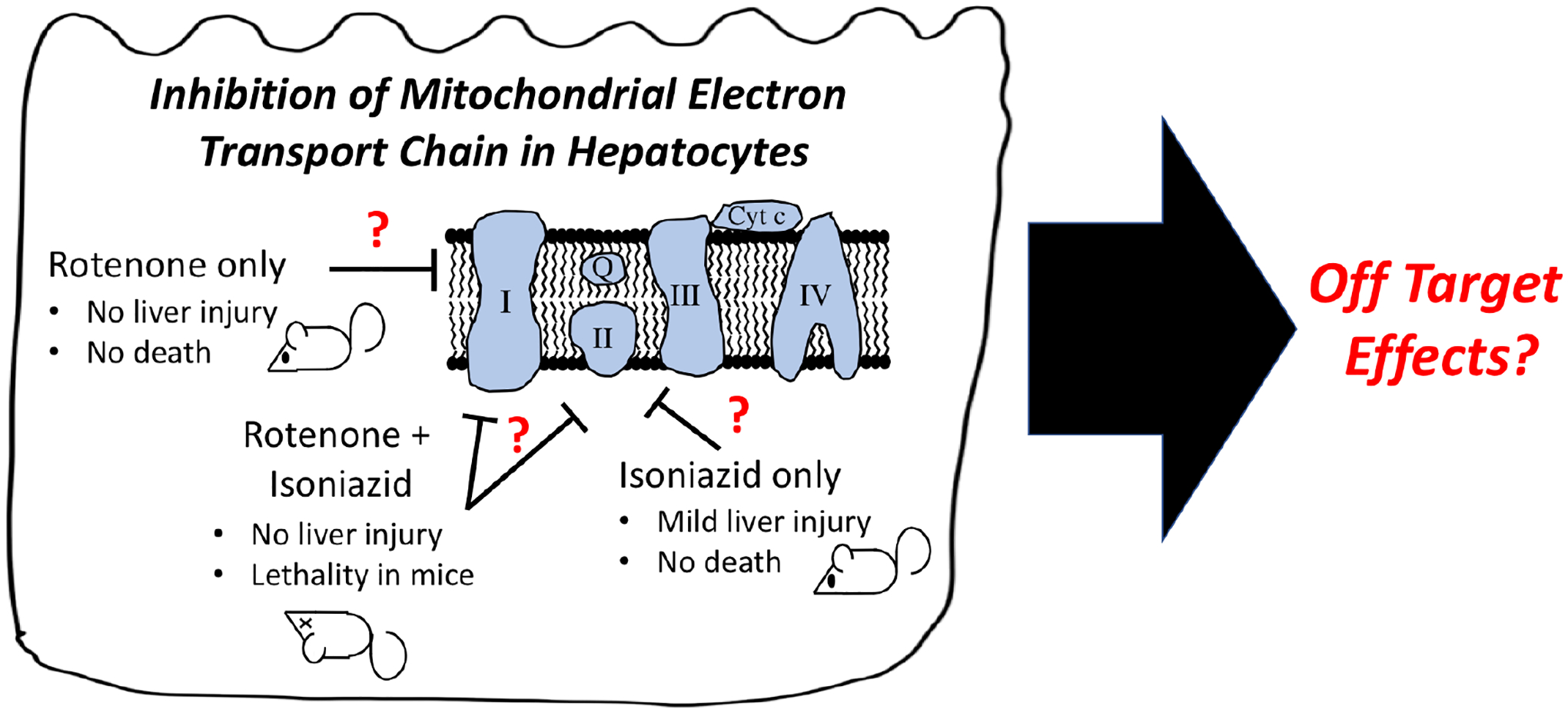
| 13 | Rotenone Increases Isoniazid Toxicity but Does Not Cause Significant Liver Injury: Implications for the Hypothesis that Inhibition of the Mitochondrial Electron Transport Chain Is a Common Mechanism of Idiosyncratic Drug-Induced Liver Injury | Cho T, Wang X, Uetrecht J | Chemical Research in Toxicology 32:1423–1431, 2019 |
| 14 | Mechanism of Idiosyncratic Drug-Induced Liver Injury Quantitative Evaluation of Reactivity and Toxicity of Acyl Glucuronides by [35S]-Cysteine Trapping | Harada H, Toyoda Y, Abe Y, Endo T, Takeda H | Chemical Research in Toxicology 32:1955–1964, 2019 |
Funding:
This work was supported by the US National Institutes of Health (NIH) grants No. R01LM012482 and R01LM012222, and the National Library of Medicine (LM).
Biographies
S Cyrus Khojasteh heads the Biotransformation Function at Genentech (South San Francisco). His research focuses on the mechanisms of biotransformation in drug discovery and development, from small molecules, antibody drug conjugates, and macrocyclic peptides. Cyrus received his Ph.D. in Medicinal Chemistry from the University of Washington under the direction of Professor Sidney D. Nelson.
James P. Driscoll is an accomplished Scientist and Project Leader in the Drug Metabolism and Pharmacokinetics department at MyoKardia, Inc, a clinical stage biopharmaceutical company focused on genetic heart disease. He has over 17 years of industry experience with Pfizer, Genentech, Theravance and MyoKardia. His expertise includes reactive metabolite identification, drug metabolism, and bioanalysis of small molecules. His responsibilities include driving early stage projects forward by identifying structure activity relationships, avoiding off-target toxicity, interrogating potential new targets in drug discovery, and leading a team focused on in vitro compound optimization. James received his B.S. in Human Biology from the State University of New York at Albany.
Klarissa D. Jackson is an assistant professor in the Division of Pharmacotherapy and Experimental Therapeutics at the University of North Carolina at Chapel Hill, UNC Eshelman School of Pharmacy. Jackson obtained her B.S. degree in Chemistry from Jackson State University and her Ph.D. in Pharmacology from Vanderbilt University in the laboratory of Drs. Jason Morrow and L. Jackson Roberts, II. She completed her postdoctoral training at the University of Washington School of Pharmacy in the Department of Medicinal Chemistry under the mentorship of Drs. Allan Rettie and Sidney Nelson. Jackson’s research interests focus on drug metabolism and toxicology to better understand the mechanisms and risk factors of adverse drug reactions.
Grover P. Miller is a full Professor in the Department of Biochemistry and Molecular Biology with a joint appointment in Biomedical Informatics at the University of Arkansas for Medical Sciences. He received BS degrees in Biochemistry and Chemistry with minors in English and French from the Louisiana State University. He obtained his PhD in Chemistry mentored by Stephen J. Benkovic at the Pennsylvania State University and subsequent postdoctoral training as an NIH NRSA fellow mentored by F. Peter Guengerich at Vanderbilt University. His research spans experimental and computational approaches to assess metabolic activation and detoxification of drugs, pollutants, and dietary compounds from the perspective of a chemist.
Kaushik Mitra heads the biotransformation group in Springhouse, PA at Janssen Pharmaceuticals. He is also the DMPK therapeutic area lead for the cardiovascular and metabolic disease portfolio. Previous to Janssen, Kaushik held successive positions at Merck as the head of biotransformation/ADME groups and the Molecular and Investigative Toxicology group. His research interests focus on mechanisms of drug-induced adverse effects such as liver injury and genetic toxicity. Kaushik received his undergraduate degree, with Honors in Chemistry, from St. Xavier’s College in Kolkata, India, Ph.D. in Organic Chemistry from the University of Missouri, Columbia under the mentorship of Prof. Kent S. Gates, and post-doctoral training in Biological Engineering with Prof. John M. Essigmann from the Massachusetts Institute of Technology.
Ivonne M.C.M. Rietjens is full professor and head of the division of Toxicology at Wageningen University, The Netherlands. She obtained her BSc and MSc degrees (cum laude) in Molecular Sciences with a major in Toxicology and a PhD in Toxicology under supervision of Prof. dr. Jan Koeman at Wageningen University. She completed post-doctoral trainings at the National Institute of Public Health and the Environment Health in The Netherlands and at the National Institutes of Health (Bethesda, USA). She is a member of the Royal Netherlands Academy of Arts and Sciences (KNAW), and chair or vice chair of many national and international advisory committees in the field of risk assessment. Her research focuses on the risk evaluation of food borne natural toxins with an emphasis on the role of reactive intermediates and DNA adduct formation, physiologically based kinetic (PBK) models for bioactivation and detoxification, genetic polymorphisms and consequences of life style factors for individual sensitivity and risk assessment and alternatives for animal testing.
Donglu Zhang a Principal scientist at Genentech. His research interests include applying drug metabolism studies in drug design and development of both small molecule drugs and antibody-drug conjugates (ADCs). He received the Sir James Black Award for discovery of and original research on Eliquis from British Pharmacological Society (2018) and the Ondetti and Cushman Award for invention of mass defect filtering method (MDF) from Bristol-Myers Squibb (2007). He received a Ph.D. in Organic Chemistry from University of Utah under direction of Professor C Dale Poulter.
Footnotes
This article is dedicated to the late Professor Judy L. Bolton from the University of Illinois at Chicago for her contribution to advancing the mechanistic understanding of estrogens and antiestrogens, and investigating natural alternatives to hormone replacement therapy.
References:
- Khojasteh SC, Bumpus NN, Driscoll JP, Miller GP, Mitra K, Rietjens IMCM, Zhang D. (2019) Biotransformation and bioactivation reactions – 2018 literature highlights. Drug Metab Rev. 51(2): 121–161. [DOI] [PubMed] [Google Scholar]
- Khojasteh SC, Miller GP, Mitra K, Rietjens IMCM. (2018) Biotransformation and bioactivation reactions – 2017 literature highlights. Drug Metab Rev. 50(3): 221–255. [DOI] [PubMed] [Google Scholar]
- Khojasteh SC, Rietjens IMCM, Dalvie D, Miller G. (2017) Biotransformation and bioactivation reactions - 2016 literature highlights. Drug Metab Rev. 49(3): 285–317. [DOI] [PubMed] [Google Scholar]
- Baillie TA, Dalvie D, Rietjens IMCM, Khojasteh SC. (2016) Biotransformation and bioactivation reactions - 2015 literature highlights. Drug Metab Rev. 48(2): 113–138. [DOI] [PubMed] [Google Scholar]