

Abstract
Cytochromes P450 (CYP450) are hemoproteins generally involved in the detoxification of the body of xenobiotic molecules. They participate in the metabolism of many drugs and genetic polymorphisms in humans have been found to impact drug responses and metabolic functions. In this study, we investigate the genetic diversity of CYP450 genes. We found that two clusters, CYP3A and CYP4F, are notably differentiated across human populations with evidence for selective pressures acting on both clusters: we found signals of recent positive selection in CYP3A and CYP4F genes and signals of balancing selection in CYP4F genes. Furthermore, an extensive amount of unusual linkage disequilibrium is detected in this latter cluster, indicating co-evolution signatures among CYP4F genes. Several of the selective signals uncovered co-localize with expression quantitative trait loci (eQTL), which could suggest epistasis acting on co-regulation in these gene families. In particular, we detected a potential co-regulation event between CYP3A5 and CYP3A43, a gene whose function remains poorly characterized. We further identified a causal relationship between CYP3A5 expression and reticulocyte count through Mendelian randomization analyses, potentially involving a regulatory region displaying a selective signal specific to African populations. Our findings linking natural selection and gene expression in CYP3A and CYP4F subfamilies are of importance in understanding population differences in metabolism of nutrients and drugs.
Keywords: Cytochromes P450, population genetics, linkage disequilibrium, co-evolution, gene expression
Significance.
Genetic diversity in Cytochromes P450 enzymes has been hypothesized to evolve subjected gene–gene interactions, potentially explaining their tendency of living in clusters within genomes. Here, we confirmed outstanding selective signatures in the CYP3A and CYP4F gene clusters, and identified a high level of unusual correlation between genetic markers far apart from each other, suggesting selection on combinations of genotypes in distinct regions, a signature of co-evolution due to epistasis.
Introduction
In the last decades, it has become clear that every individual has their own “fingerprint” of alleles encoding drug-metabolizing enzymes, playing central roles in the metabolism of endogenous and exogenous compounds. It was established that hydrophobic molecules are first modified by oxidation and subsequently excreted as water-soluble forms, two distinct steps now described as phases I and II. Phase I is performed mainly by Cytochromes P450 (CYP450) enzymes, able to catalyze a considerable variety of oxidation reactions for many structural classes of chemicals (including the majority of drugs) (Danielson 2002; Nebert and Dalton 2006). They metabolically activate parent compounds to electrophilic intermediates, while phase II enzymes conjugate these intermediates towards more easily excretable derivatives.
CYP450 genes are a super-family of genes which appeared more than 3.5 billion years ago (Wright et al. 2019), being present in fungi, plants, bacteria, and animals. Genes are grouped into families and subfamilies based on sequence similarity: genes from the same family have sequence similarity greater than 40% and, to be grouped into a subfamily, their sequence similarity must be greater than 55% (Nelson et al. 1996).
In humans, the CYP450 family includes 57 genes and 58 pseudogenes (Nelson et al. 2004) grouped in 18 families (Nebert et al. 2013). Several CYP450 genes are found in clusters in the human genome but some members of the subfamilies can be spread out across the genome. For example, the CYP4F subfamily has genes on chromosome 19 and pseudogenes on multiple chromosomes. The CYP2D6 gene is the most widely studied CYP450 gene in humans, due to its role in the metabolism of many drugs (Gaedigk 2013; Gaedigk et al. 2017) along with CYP3A4 and CYP3A5, members of the CYP3A subfamily (Wang et al. 2011; Elens et al. 2012; Lamba et al. 2012; Tavira et al. 2013; Rojas et al. 2015). However, not all CYP450 genes or families have been studied thoroughly, and details on the evolution and clinical significance are lacking for several families, such as the CYP4F subfamily.
Several CYP450 genes have been suggested to have undergone natural selection in humans (Carlson et al. 2005; Voight et al. 2006). Other studies of the genetic diversity for specific CYP450 subfamilies in human populations confirmed the presence of positive (Qiu et al. 2008; Bains et al. 2013), balancing (Janha et al. 2014), or purifying selection signatures (Yasukochi and Satta 2015). One example is CYP2C19, involved in the metabolism of clopidogrel (Scott et al. 2013; Brown and Pereira 2018), where signals of positive selection on its alleles conferring slow metabolism (CYP2C19*2 and CYP2C19*3) were detected using relative extended haplotype homozygosity (REHH) (Janha et al. 2014). CYP2C19*2 is detected worldwide, but CYP2C19*3 is only present in people of Asian descent. The selective advantages may have been caused by diet and environmental pollutants impacting humans over thousands of years and could differ between ethnic groups. Additionally, low values across CYP2C19 SNPs suggest balancing selection in CYP2C19 (Janha et al. 2014). The excess of alleles at intermediate frequencies could reflect the evolution of balanced polymorphisms, which is to be expected in evolutionarily old enzymes responsible for numerous critical life functions.
Moreover, the detection of natural selection signals in the CYP450 genes raises the possibility that the selective advantage acts on polymorphisms that modulate gene expression, widely known as expression quantitative trait loci (eQTL) (Kudaravalli et al. 2009). Detecting eQTLs linked to selection signals helps clarifying how gene expression is regulated and can lead to a better understanding of variants’ biological effects (Nica and Dermitzakis 2013). Furthermore, analyzing eQTLs helps in the detection of gene–gene interaction (Huang et al. 2013) and co-regulation between genes (Lehner 2011). Such gene–gene interactions can also be detected by looking at patterns of linkage disequilibrium (LD), as evolution will maintain co-evolving polymorphisms on the same haplotypes (Rohlfs et al. 2010), which can also be detected as balancing selection signatures.
Here, we investigated genetic diversity and selective pressures across human populations in CYP450 genes. Two subfamilies emerged from our analyses and were investigated in greater depth: the CYP3A and CYP4F families. Both subfamilies were generated by duplication events resulting in consecutive genes in the same genomic region, or gene cluster (supplementary fig. S1, Supplementary Material online). The CYP3A subfamily contains four genes and four pseudogenes located in a genomic region of about 220 KB on chromosome 7. They metabolize around 50% of common drugs. The CYP4F subfamily has six genes located in a genomic region of about 430 KB on chromosome 19 and have mostly been associated with metabolism of lipids. We found that both families exhibit selective pressures in human populations and that the SNPs under selection could impact gene expression levels in several tissues. Furthermore, our results suggest interactions between the genes in both CYP450 subfamilies, providing evidence of co-evolution and co-regulation within these gene clusters, that may vary between populations.
Results
We obtained genotypic data from the 1000 Genomes project phase 3 release (1000G) (Genomes Project Consortium et al. 2015). A total of 2,157 individuals were analyzed from 22 populations, which belongs to four of the five super-populations included in the project (i.e. Africa, Europe, East Asia, and South Asia).
Global Genetic Diversity Across Populations in CYP450 Genes
First, we aimed to identify global genetic patterns by calculating Tajima’s D values for each CYP450 genes in each population of the 1000G dataset to provide insights into the non-neutral forces that act on these genes. A total of 61,739 biallelic SNPs were analyzed in all of the 57 CYP450 genes, and for each gene, we computed the mean Tajima’s D per gene and also in 1 Kb windows. Significantly low Tajima’s D values indicate an excess of rare alleles, whereas significantly high values of Tajima’s D suggest an excess of intermediate frequency alleles, which can reflect the occurrence of balancing selection.
In European populations, nine genes had Tajima’s D values consistently below 0 (Fig. 1a). We assessed significance based on the empirical (null) distribution, which allows to determine whether any genes have values that are higher or lower than expected while taking population-specific demographic factors into account (see Materials and Methods). The proportion of 1 Kb-windows of each gene lying outside the null distribution is shown in Fig. 1b. CYP26A1, CYP27B1, and CYP1A2 had the largest proportion of windows with significantly low D values; however, these genes are quite small (4.4, 4.9, and 7.8 Kb, respectively), meaning that the signal is driven by one or two windows only. Interestingly, the four CYP3A genes in our dataset were all included in this group of nine genes, suggesting that strong purifying selection pressures may be acting, however complete selective sweeps driven by positive selection can also create this lack of diversity (Kim and Stephan 2002). Notably, CYP3A5 has a low Tajima’s D average but no 1 Kb-window is significantly lower than expected, whereas other CYP3A genes have several windows showing significantly low Tajima’s D values. All CYP450 genes show negative Tajima’s D values, as expected in coding regions, but ten genes have a mean above 0, which suggests relaxation of purifying selection pressure. The presence of several 1 Kb-windows significantly enriched for high D values can also reflect the presence of localized balancing selection signatures within these genes. Of these ten genes, five are in the CYP4F subfamily: CYP4F3, CYP4F11, CYP4F12, CYP4F8, and CYP4F2. The strongest of these signals is seen on CYP4F12 (Fig. 1b). Interestingly, the only CYP4F gene that does not show this specific signature is CYP4F22, which is the ancestral gene of the CYP4F cluster (Kirischian and Wilson 2012). Notably, these analyses were also performed for each of the subpopulations, yielding similar results (supplementary fig. S2, Supplementary Material online).
Fig. 1.

Metrics of diversity and differentiation among CYP genes. a) Distribution of Tajima’s D values computed on windows of 1 Kb for each CYP450 genes in the European populations. The 2.5th percentile is marked by the orange vertical line on the left, and the 97.5th percentile is marked by the red vertical line on the right, representing the significance threshold. b) Proportion of Tajima’s D windows lying outside the null distribution for each CYP450 gene. For each gene, the total number of windows of Tajima’s D is shown beside the proportions, between brackets. The windows with Tajima’s D values below the 2.5th percentile is displayed in orange on the left side of the plot and over the 97.5th percentile is displayed in red on the right side of the plot. c) Distribution of values for each CYP450 gene calculated on 4 super-populations (AFR, EUR, EAS, and SAS). The mean of chromosome 22, the null distribution, is displayed with the gray horizontal line.
Because population differentiation can also help identifying natural selection signatures within genes, we calculated the mean fixation index () across CYP450 genes (Materials and Methods). measures the differentiation between populations using genotype frequencies, with high values indicating that the average pairwise heterozygosity is higher between than within populations. Figure 1c shows the distribution of values for each CYP450 gene calculated on 4 super-populations (AFR, EUR, EAS, and SAS). CYP4F genes are scattered across the CYP450 spectrum, with CYP4F12 having the second highest mean while CYP4F8 is in the bottom half of the distribution. Mean of genes of the CYP3A subfamily are in the highest values, meaning that these genes have a high divergence between population’s genotype frequencies. This could indicate that the low Tajima’s D in CYP3A reflects positive rather than extreme purifying selection.
Positive Selection in CYP3A and CYP4F Subfamilies
The global neutrality and differentiation analyses of CYP450 genes suggest that positive selection, either directional (CYP3A) or balancing (CYP4F), may be acting on subfamilies of CYP450 genes, possibly in a concerted fashion. To further validate positive selection signatures and identify specific putative sites, we used the integrated haplotype score (iHS), which leverages linkage disequilibrium (LD) patterns in a specific population (Voight et al. 2006). Typically, an absolute value of iHS greater than 2 at a SNP suggests that the region around the SNP is under selection (Voight et al. 2006).
In the CYP3A cluster, significant iHS values are detected (Fig. 2a), but signals of positive selection differ between populations. Many signals are detectable in Africans, in East Asians and in Europeans, while fewer signals are detectable in South Asians. Signals of positive selection are noticeable in CYP3A5, CYP3A51P, CYP3A4 and CYP3A43 among Africans. In particular, iHS values in CYP3A5 are consistently below 2, indicating that the derived alleles have quickly increased in frequency, a signature of positive selection. Interestingly, unlike populations of European descent where the CYP3A4 gene is typically the most expressed, the CYP3A5 gene is the most expressed in the African individuals (Kuehl et al. 2001; Burk and Wojnowski 2004). Among East Asians, the selective sweep is located from CYP3A51P to CYP3A4, and among South Asians, in CYP3A43. Lastly, for Europeans, signals of positive selection are detectable in the region between CYP3A7 and CYP3A4, a signal also present in the East Asian population. CYP3A43 is the only gene with signals in all super-populations. With these results, we establish robust evidence for positive selection acting on CYP3A genes, corroborating and extending observations from previous studies (Thompson et al. 2004; Voight et al. 2006; Qiu et al. 2008; Li et al. 2011).
Fig. 2.
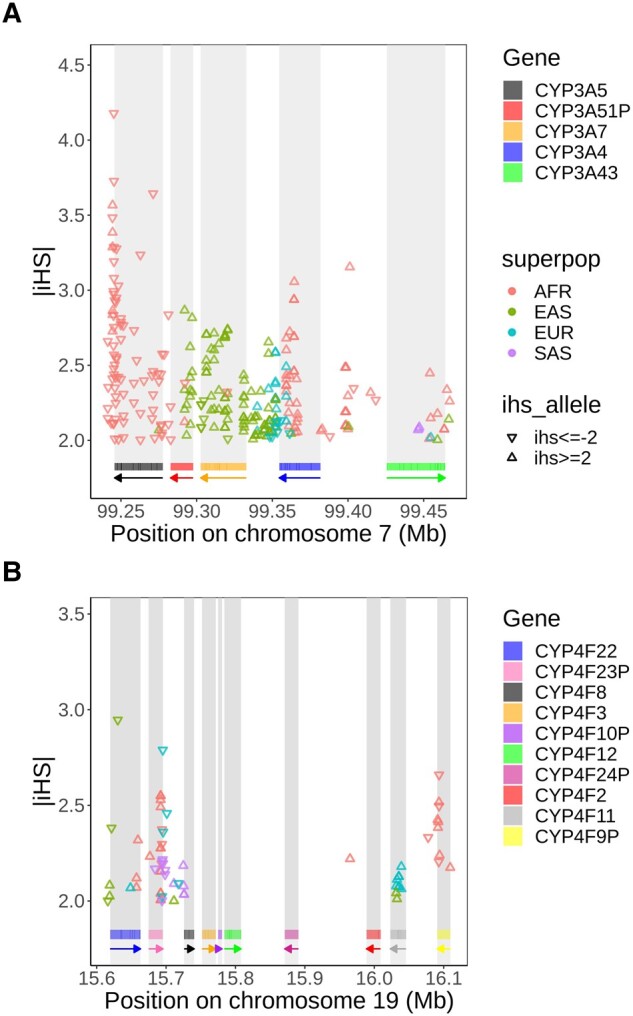
Distribution of SNPs with high values () in the a) CYP3A and b) CYP4F cluster. A triangle standing on its base means an value ≥2, indicating that the ancestral allele has increased in frequency, and a triangle standing on its point means an value , indicating that the positive selection is acting on the derived allele. SNPs located in repetitive elements and sequences are masked. Rectangles below the plot show the position of each gene, and arrows indicate on which strand the gene is located.
Positive selective pressure is also detected in the CYP4F cluster, but on a smaller scale. For the CYP4F cluster, signals of positive selection are visible in CYP4F22, CYP4F23P, CYP4F11, and CYP4F9P (Fig. 2b). The region between the pseudogene CYP4F23P and the gene CYP4F8 also shows high iHS values, indicating positive selection in every super-population. iHS values greater than 2 are present in CYP4F11 in Europeans and East Asians, indicating positive selection acting on ancestral alleles. CYP4F9P has significant iHS values in Africans. Again, most iHS values are greater than 2, indicating selective pressures on ancestral alleles, but the three strongest signals are seen for derived alleles (iHS below 2), suggesting these SNPs may be driving the signal.
Balancing Selection in CYP3A and CYP4F Subfamilies
The Tajima’s D analyses (Fig. 1) suggested balancing selection in the CYP4F cluster. To confirm this finding, we used the Beta score (Siewert and Voight 2017), a statistic which detects clusters of alleles with similar allele frequencies, developed to specifically test whether balancing selection is present at specific loci.
We considered β score in the top 1% of the whole chromosome as significant β scores (empirical ), which can vary between populations. In contrast to iHS, very few significant β score values are seen in the CYP3A cluster. Only one SNP in CYP3A43 meets this criteria in Africans, in the same region where balancing selection was also identified in a previous study (Aqil et al. 2023). The same signal can been seen in the other populations, but it is weaker and do not pass our 1% threshold (Fig. 3a). Overall, these results show no clear evidence of balancing selection acting on the CYP3A cluster. In line with Tajima’s D results, clearer signals are seen in the CYP4F cluster, which show larger β scores compared to the CYP3A cluster: the highest β score in the CYP4F cluster is almost twice as high as the highest CYP3A’s β score. SNPs in CYP4F12 show highly significant β scores, replicated among Africans, Europeans and South Asians, but not in the East Asians. Also, the region between CYP4F23P and CYP4F8 has the most extreme β score in the region, and the signal is visible in all super-populations (Fig. 3b). These consistent signals across populations provide convincing evidence of balancing selection acting around CYP4F8 and CYP4F12. Weaker signals, which do not pass our significance threshold but are seen consistently between populations, are seen in CYP4F23P and CYP4F11. Taken together, these results demonstrate evidence supporting the presence of balancing selective pressures in the CYP4F cluster but show a lack of evidence for balancing selection across the CYP3A cluster.
Fig. 3.

β score in the chromosomal region of the a) CYP3A and b) CYP4F cluster for the four super-populations analyzed. The β score was calculated on the 1000G dataset and the 99th percentile indicating the top 1% β score is displayed by the horizontal line in red. Rectangles below the plot show the position of each gene, and arrows indicate on which strand the gene is located.
Detection of Unusual Linkage Disequilibrium
Since CYP3A and CYP4F genes are in a gene cluster and selective pressures are acting on these genes, co-evolution could be occurring. Indeed, the different combinations of alleles which co-occurred during evolution can lead to concerted selective pressure, or co-evolution, depending on the resulting fitness of the individuals (Rohlfs et al. 2010). Such co-evolution signals can be revealed by analyzing patterns of linkage disequilibrium (LD) beyond local associations due to allelic proximity, in order to detect whether specific combinations of alleles (or genotypes) at two distinct loci are particularly overrepresented. To do so, we calculated the genotyped-based LD () between each pair of SNPs with minor allele frequency (MAF) above 0.05 in the two CYP450 clusters, across each 1000G subpopulation (Materials and Methods). Under neutrality, the LD association between SNPs is expected to decrease as genetic distance between the SNPs increases, allowing us to build an empirical distribution by considering clusters of genes of similar size genome-wide (Materials and Methods) to the clusters under investigation. Pairs of SNPs showing unusual LD (uLD) values, lying outside of this null distribution, are therefore likely transmitted together more often than expected, making it possible to identify candidate sites that are co-evolving.
In both clusters, strong signals of uLD are present (Fig. 4, supplementary fig. S3, Supplementary Material online) compared to matched gene clusters (Materials and Methods), with CYP4F showing much more extreme signals than CYP3A (8.1% vs 4.7% of pairs of SNPs in uLD), despite genetic distances in the CYP4F cluster being four times larger than in the CYP3A cluster (maximum distance of 0.60 cM vs 0.15 cM, respectively), whereas the physical size of the cluster is only twice (500 Kb vs 250 Kb, respectively). Significant uLD between CYP3A5 and CYP3A43 and between CYP3A7 and CYP3A43 can be seen in all European populations (supplementary fig. S3a, Supplementary Material online). CYP3A5 and CYP3A43 are the opposite to each other in term of physical location in the cluster while CYP3A7 and CYP3A43 are next to each other. Finland (FIN) and Toscani (TSI) populations have the most uLD signals across European populations, with FIN uniquely showing uLD between CYP3A5-CYP3A4, and TSI showing uLD between CYP3A4 and CYP3A43, a signal consistently seen in the East Asians. TSI also have the highest genetic distance interval in this region, likely due to a larger, more widespread, recombination rate in CYP3A4 compared to other populations (supplementary fig. S4, Supplementary Material online). Among East Asians, uLD signals are seen almost exclusively between SNPs in CYP3A4 and CYP3A43, two genes that are next to each other, with no clear recombination hotspot separating them, meaning that linkage disequilibrium can be expected (supplementary fig. S4, Supplementary Material online). SNPs in these genes are also in uLD in Gujarati Indian (GIH) population, but none of the other South Asian populations show any signal, which may be explained by the short genetic distances within this cluster in this super-population (SAS) (<0.05 cM). Finally, African populations show the most deviation from the null (Fig. 4a). SNPs in CYP3A4 are in uLD with all other genes and the signal also replicates the observations from the European populations, with SNPs in CYP3A43 in uLD with SNPs in all other genes (Fig. 4a, supplementary fig. S3a, Supplementary Material online).
Fig. 4.
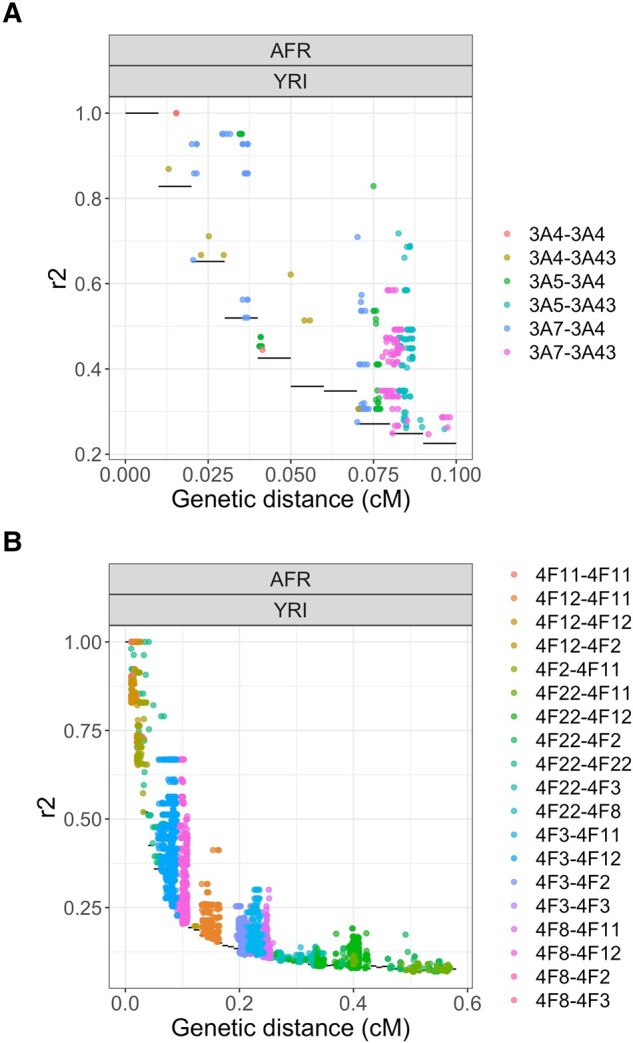
values between each pairs of SNPs in the a) CYP3A and b) CYP4F cluster in the Yoruba (YRI, AFR) population. The distance between the SNPs is in centimorgan (cM). Only values over the null distribution are shown. The null distribution is shown with black horizontal lines. Dots are colored according to which genes are involved in the pair.
In the CYP4F cluster, several pairs of SNPs have patterns of LD that deviate significantly from the empirical distribution (supplementary fig. S3b, Supplementary Material online). There is uLD for CYP4F22-CYP4F11 and CYP4F22-CYP4F12 in almost every populations, even though these genes are far from each other (0.36 and 0.12 Mb, respectively). CYP4F22 and CYP4F2 are also in uLD in AFR, EUR, and EAS.
The African populations have more evidence of uLD than the other super-populations. One population in particular, the Yoruba (YRI) population, has even more extreme signals in comparison with other African populations and most uLD signal are driven by associations involving the CYP4F12 gene (Fig. 4b). Thus, we investigated whether a specific region in CYP4F12 is in strong LD with the other genes. Indeed, in the YRI population, there is evidence of uLD between a region in CYP4F12 (at 15.79–18.00 Mb on chromosome 19) and the CYP4F3 (supplementary fig. S5a, Supplementary Material online) and CYP4F8 genes (supplementary fig. S5b, Supplementary Material online). The extreme signals in this gene cluster are in line with the hypothesis that balancing selection acts via gene–gene interactions, or epistasis (Llaurens et al. 2017). As these patterns could be due to sequencing errors (Akey et al. 2001), we used the latest 1000G dataset which has high-coverage sequencing and is aligned on GRCh38 (Materials and Methods). These results were replicated in this second dataset, greatly reducing the possibility that the observed signal is due to sequencing errors or spurious mapping. Finally, in the Europeans, the FIN population has a specific pattern between CYP4F12 and CYP4F2, CYP4F8, and CYP4F3. Looking more closely, many SNPs in CYP4F12 are in uLD with one SNP in CYP4F3 (supplementary fig. S5a, Supplementary Material online) and two SNPs in CYP4F8 (supplementary fig. S5b, Supplementary Material online). No specific SNPs are in uLD with CYP4F2.
Detection of eQTLs
We next evaluated the effects of the SNPs identified as being under positive and balancing selection on the expression of the genes in each CYP450 cluster to test if these are eQTLs.
In the CYP3A cluster, three SNPs are under positive selection in the Punjabi population from South Asia (PJL): rs487813, rs679320, and rs568859. These SNPs are located in CYP3A43 and are significant eQTLs of CYP3A5 in lung (Fig. 5a). The SNP under balancing selection in the Luhya population (LWK) in CYP3A43, rs800667, is also an eQTL of CYP3A5 in lung (Fig. 5b). The effect size estimate for these significant eQTL is negative, indicating a reduction in CYP3A5 gene expression with each nonreference allele. This locus in CYP3A43 thus impact CYP3A5 expression in lung, even though CYP3A5 and CYP3A43 are at opposite ends of the cluster, 147.99 Kb apart. According to the ReMap density database (Hammal et al. 2022), this locus also displays regulatory signals, supporting the importance of this region at the transcriptional regulatory level. This result is in line with the LD analyses (Fig. 4a), which suggested uLD between SNPs in CYP3A5 and CYP3A43 in Europeans, Africans, and the Japanese. Those four SNPs were all in uLD with 11 SNPs in the Toscani population (TSI) and five other SNPs in Americans of African Ancestry (ASW).
Fig. 5.

P-values of the associations between SNPs under a) positive selection and b) balancing selection and CYP3A5’s gene expression in lung and P-values associated with SNPs c) under positive selection and d) balancing selection and tissue-specific gene expression of CYP4F12. CYP3A5 and CYP4F12 are shown in dark gray, as the expressions of these genes are tested. The triangle standing on its base indicates a positive effect size (), while a triangle standing on its point indicates a negative effect size (). The threshold, set to , is represented by the horizontal black line, meaning that a is a significant eQTL. Only tissues with significant eQTLS are displayed. As before, rectangles below each plot show the position of each gene, and arrows indicate on which strand the gene is located. Each gene has its own color to indicate its location.
In the CYP4F cluster, a SNP under positive selection, rs74459786 (supplementary table S1, Supplementary Material online), located in the intergenic region between CYP4F23P and CYP4F8, is an eQTL of CYP4F12 in adipose tissue (Fig. 5c), with a negative effect size. SNPs under balancing selection (supplementary table S2, Supplementary Material online) within CYP4F12 are eQTLs for CYP4F12 expression in the colon, esophagus, and skin, but interestingly, their effects in these tissues are in opposite directions, with positive effect sizes in the colon and skin, and negative ones for the esophagus. Furthermore, a SNP with a balancing selection signal is also an eQTL of CYP4F12 expression in adipose-subcutaneous tissue (Fig. 5d) with a negative effect size estimate. It lies in the intergenic region between CYP4F23P and CYP4F8, which is the same region as the SNP under positive selection (rs74459786) in Fig. 5c.
Another SNP under positive selection in this intergenic region, rs62115147 (supplementary table S1, Supplementary Material online), is also associated with CYP4F3 expression in one of the brain tissues (Brain-Spinalcord-cervicalc-1) and in nerve tissue (supplementary fig. S6a, Supplementary Material online). The CYP4F12 gene emerged repeatedly as a candidate in our balancing selection and uLD analyses, while the intergenic region between CYP4F23P and CYP4F8 is seen only in the balancing selection analysis.
Even if less positive selection is present in the CYP4F cluster compared to the CYP3A cluster, many of the SNPs showing high iHS values in the CYP4F cluster show up as eQTLs for different genes. SNPs under positive selection located in CYP4F11 (supplementary table S1, Supplementary Material online) are eQTLs of CYP4F2 in brain and skin tissues (supplementary fig. S6b, Supplementary Material online) with consistent, negative effect sizes. Additionally, the same SNPs under positive selection within CYP4F11 are associated with expression of CYP4F11 itself in multiple tissues (supplementary fig. S6c, Supplementary Material online). The direction of effect on gene expression is the same for all significant associations.
Phenotypic Associations
Using the UK Biobank cohort (UKb), we did a Phenome-Wide Association Study (PheWAS) to identify phenotypes potentially under selective pressure (Materials and Methods), using the available variants with selective signals in the CYP4F genes (166 variants from the 180 found under selection) and in the CYP3A genes (62 from the 125 variants found under selection). No significant associations were found for SNPs under selective pressure in CYP4F cluster.
In the CYP3A cluster, however, SNPs under positive selection in at least one studied populations were found associated with six phenotypes (Fig. 6a,b) in our PheWAS. Among the disease phenotypes, we found association with pelvic inflammatory disease (PID), which is female-specific, and for which the SNP with the strongest association () was under positive selection in European (CEU, GBR) and East Asian (CHB, CHS, CDX) populations. Among the continuous phenotypes investigated (Materials and Methods), we found association with pulse rate, for which the SNP with the strongest association () was also found under selective pressure in Europeans (CEU). Among the biomarker variables, the strongest associations with platelet count () and erythrocyte count () were both found with SNPs under selective pressure in the Japanese population.
Fig. 6.

Associations of CYP3A cluster with phenotypes in the UK biobank. Significant associations () for continuous traits a) and plasmatic biomarkers b) which are significant in at least one SNP under selection. SNPs under selection are represented as full dots, meanwhile other SNPs are represented as empty dots. As before, rectangles below each plot show the position of each gene, and arrows indicate on which strand the gene is located. Each gene has its own color to indicate its location. c) Causal relationship with CYP3A5 expression in lung for phenotypes showing significant association with its eQTLs. β represents the change of 1 standard deviation of CYP3A5 expressions on phenotypes, also in standard deviation units. P-value of three statistics (IVW, Egger, Weighted Median) are displayed with the β and the 95% confidence interval (CI) of the association for each phenotype in the gray box. For Egger, the P-value of the intercept is also displayed.
Lastly, for both high light scatter reticulocyte count and reticulocyte count, their strongest association (; respectively) were both found under selective pressure in African population (MSL and ACB, respectively). Using Mendelian randomization (Materials and Methods), we evaluated the causal relationship between CYP3A5 expression in lung, for which eQTLs were found under selective pressure above, and the phenotypes found to be associated with SNPs in the CYP3A cluster. We identified a significant causal association between CYP3A5 expression and both high light scatter reticulocyte count () and reticulocyte count (). We did not detect pleiotropy using MR-Egger and results were robust using the weighted median test (Materials and Methods).
Altogether, these results indicate that the selective pressure in the CYP3A cluster could be driven by the production of reticulocyte through the expression levels of CYP3A5 and also suggest that pulse rate could be impacted by genetic variation in CYP3A genes.
Among other associations identified in this cluster, three SNPs showed strong associations with anthropometric traits (Fig. 6a) and are under selective pressure in European population (CEU, IBS). Those SNPs were, however, found to be associated with expression of genes outside the CYP3A cluster (supplementary text 14.3, Supplementary Material online), prompting for further investigation of the relationship between this cluster and other neighboring genes to understand the different drivers at play.
Discussion
Drug metabolism is a rather complex system with the CYP450 genes metabolizing around 75% of common drugs. As shown by others (Thompson et al. 2004; Qiu et al. 2008; Chen et al. 2009; Li et al. 2011), we also found that selective pressure and genetic differentiation between populations were present in CYP450 genes. Here, we provide a deeper analysis of two CYP450 clusters, the widely studied CYP3A (Burk and Wojnowski 2004; Thompson et al. 2004, 2006; Chen et al. 2009) and the less well-known CYP4F clusters, identified thanks to their outlier patterns in neutrality and population differentiation analyses. These two CYP450 clusters exhibit multiple selective signatures (positive selection and balancing selection) and show population differentiation. We found that natural selection forces involved differ between the two clusters; the CYP3A cluster is evolving under positive selection, while the CYP4F cluster show signals of balancing selection. Furthermore, the CYP4F cluster shows strong evidence for co-evolution and co-regulation signals.
In the literature, the CYP450 genes are often studied independently. In our study, we considered the evolution of the entire family cluster, mostly CYP3A and CYP4F genes, and detected signatures of co-evolution between the paralogous genes, suggestive of potential epistatic interactions. As these clusters of genes are involved in drug metabolism (Danielson 2002; Nebert and Dalton 2006; Liang et al. 2012; Zhang et al. 2017), it is important to understand the impact of genetic variants on their gene expression, to help understand how these variants might impact drug response and refine disease treatments in a personalized way. Our results also show that the impact of specific variants may differ between populations, which could lead to a deeper understanding of differences in individual drug response (Singh et al. 2011; Guttman et al. 2019).
The CYP3A cluster contains four genes: CYP3A4, CYP3A5, CYP3A7, and CYP3A43. Signals of positive selection were detected in the CYP3A cluster, specifically in CYP3A4 and CYP3A7, which have been under recent positive selection in African, European and the Chinese populations, while CYP3A5 appears under positive selection in Europeans and CYP3A43 in non-Africans (Chen et al. 2009). Our analyses confirmed that CYP3A genes are evolving under positive selection as previously reported (Thompson et al. 2004; Voight et al. 2006; Qiu et al. 2008; Li et al. 2011).
We found that the locus known to cause nonexpression of CYP3A5 (Kuehl et al. 2001), rs10264272/CYP3A5*6, is under positive selection () in African populations (YRI, GWD, LWK). A second locus, known to cause low CYP3A5 expression, rs776746/CYP3A5*3, is under positive selection () in two African populations (YRI, GWD). These derived allele have thus swept up in frequency in several African populations. In the Toscani population, rs776746/CYP3A5*3 is found to be in uLD with the four SNPs under selective pressure in the CYP3A cluster, that are eQTLs of CYP3A5 in lung.
CYP3A43 is the ancestor gene of this cluster (McArthur et al. 2003; Qiu et al. 2008); however, its function is not well understood, unlike other CYP3A genes. Our analyses suggest that SNPs in CYP3A43 regulate CYP3A5 gene expression, at least in lung. Levels of expression of CYP3A5 in lung were causally associated to reticylocytes count and many of its eQTLs were under selection in Africans. Since Plasmodium vivax, a parasite causing malaria, affect mainly young reticulocytes (Clark et al. 2021) and that malaria is present in Africa, the selective pressure found in this population could be associated to this disease. Further studies need to be done to validate this hypothesis.
In the CYP4F cluster, we found both positive and balancing selection pressures acting. Furthermore, the SNPs evolving under selective pressures are associated with gene expression levels across the cluster in several tissues. For instance, a cis-eQTL of CYP4F12, rs74459786, is detected to be under positive selection in the Kinh population in East Asia (KHV). We also found that several SNPs in CYP4F11 are associated with CYP4F2 expression. Both genes are implicated in common metabolic function, such as the synthesis of 20-hydroxyeicosatetraenoic acid (20-HETE) from arachidonic acid (Yi et al. 2017). Thus, this could indicate a possible regulatory mechanism of common functions.
Finally, the region between CYP4F23P and CYP4F8 emerged multiple times in our analyses. This intergenic region shows strong signals for selection, with the same SNPs also being eQTLs of CYP4F3 (nerve) and CYP4F12 (adipose tissue). Given the implication of CYP4F12 in fatty acid metabolism (Stark et al. 2005), our results may point towards the identification of new regulatory elements involved in this process in adipose tissues.
A potential limitation in the current study is that population genetic statistics can be biased in the presence of fine-scale population structure. However, to mitigate this issue, we performed our analyses not only at the broader population level but also within individual subpopulations, ensuring that the values obtained from subpopulations were consistent with those from the overall superpopulation.
An important limitation to consider is the methodology used for calculating Tajima’s D using the vcftools software. This tool’s current implementation does not account for mappability and callability in whole genome sequencing data. This approach introduces a bias by implicitly considering uncalled positions as nonvariable, leading to an underestimation of diversity measures (Korunes and Samuk 2021). Although our strategy to exclude regions with high proportions of missing data likely minimizes this bias, we acknowledge that approaches that directly incorporate genomic accessibility considerations for a more precise estimation of genetic diversity should be used in future studies of CYP genes. While the impact on our results appears minimal, as evidenced by the high similarity of results after the removal of high-missing data regions, the potential for underestimation of diversity estimators should be considered when comparing these estimates across genomic regions.
In conclusion, our results demonstrate high heterogeneity across human populations, both in terms of selective signals and interaction between variants and expression levels, for the CYP3A and CYP4F genes. There could thus be important differences in metabolic regulation impacting drug response in individuals from different ethnicities. In particular, these variants could cause impaired efficacy, as well as side effects. As pharmacogenetic studies still typically focus on European populations, our results underline the importance of including individuals from several populations in order to capture all of the genetic diversity and its impact on disease treatment and metabolism.
Materials and Methods
1000 Genomes Genetic Data
The data analyzed are from the phase III of the 1000 Genomes project (1000G) (Genomes Project Consortium et al. 2015). The 1000 Genomes Project includes 2,504 individuals from 26 populations. These populations can be split into five distinct genetic ancestries, referred herein as super-populations, as defined by the 1000G consortium: African (AFR), European (EUR), South Asian (SAS), East Asian (EAS), and Admixed American (AMR). Data from the AMR population are not included in this study because the high degree of admixture may confound selection and linkage disequilibrium analyses. This left us with 22 subpopulations and four super-populations for study. The available variant call format (vcf) files of 1000G are under the GRCh37 genome build. VCFtools v0.1.14 (Danecek et al. 2011) was used to filter the 1000G dataset. Indels and nonbiallelic alleles were removed and only SNPs located in the 57 CYP450 genes were kept, extracted based on coordinates genomic coordinates obtained from the UCSC genes table using the UCSC Genome Browser (supplementary file 1, Supplementary Material online). After filtering, the CYP450 dataset included a total of 61,739 SNPs and 2,157 individuals. We refer to this as the “1000G CYP450 dataset.” A more recent dataset was also used as a validation dataset for the unusual linkage disequilibrium analysis, the resequencing dataset of 30X coverage, mapped on GRCh38 (Byrska-Bishop et al. 2021), which includes the 2157 individuals.
Genetic Diversity and Population Differentiation
Both Tajima’s D and statistics were obtained with VCFtools (Danecek et al. 2011) using the 1000G CYP450 dataset (Genomes Project Consortium et al. 2015). Tajima’s D values were calculated in the super-population (AFR, EUR, EAS, and SAS) separately on nonoverlapping windows of 1 Kb. We also performed these analyses excluding positions and windows with low mappability (see supplementary text 14.1 and fig. S2, Supplementary Material online). We computed the mean Tajima’s D value for each gene by averaging the window-based values, and sorted genes according to their mean. To create a null distribution, we computed Tajima’s D values for all SNPs associated with a gene name in the Combined Annotation Dependent Depletion (CADD) annotation file (Kircher et al. 2014) on chromosome 22, so that all SNPs used to compute the empirical distribution are located in genes. We computed the 2.5 and 97.5th percentile on the window-based values of chromosome 22. Values above the 97.5th percentile and below the 2.5th percentile were considered to be statistically significant (two sided empirical ). To ensure that our results were not biased by fine-scale population structure, we also perform the analyses in each of the subpopulations of EUR (see supplementary text 14.1 and fig. S2, Supplementary Material online). The values, from Weir and Cockerham derivation (Weir and Cockerham 1984), were calculated using four super-populations (AFR, EUR, EAS, and SAS) on a per-site basis. The per-gene mean was calculated on raw values and genes were sorted based on their mean . As in the previous analysis, chromosome 22 was used to create an empirical distribution. values were also computed on SNPs located in genes of the chromosome 22 (see above) and the per-gene mean was calculated.
Detecting Natural Selection
The method used to detect balancing selection is the β score (Siewert and Voight 2017). This score has already been calculated on the whole 1000 Genomes project data for each subpopulation. The approach used to detect signal of recent positive selection was iHS (integrated haplotype score) (Voight et al. 2006). The iHS computation was performed by us on the 1000G dataset, filtered to exclude INDELs and CNVs. Reference alleles from filtered 1000 Genomes vcf files were changed to the ancestral alleles retrieved from six primates EPO pipeline (version e59) using the fixref plugin of bcftools (Li 2011). The hapbin program v.1.3.0 (Maclean et al. 2015) was then used to compute iHS using per population-specific genetic maps computed by Adam Auton on the 1000 Genomes OMNI dataset (Genomes Project Consortium et al. 2015). When the genetic map was not available for a subpopulation, the genetic map from the closest subpopulation was selected according to their global value computed on the 1000G dataset. For all natural selection analyses, SNPs annotated to be in a repetitive region were identified using the RepeatMasker track available on the UCSC genome browser (Kent et al. 2002) and were removed.
Unusual Linkage Disequilibrium
Linkage disequilibrium between pairs of SNPs from the same cluster was assessed using the geno-r2 option from VCFTools on SNPs with minor allele frequencies (MAF) above 0.05. The genetic position of each SNP was calculated with PLINK v1.90 (Chang et al. 2015) using the population-specific genetic maps the same was as described in previous section.
To compute a null distribution to detect unusual linkage disequilibrium (uLD), the Human GRCh38 Gene transfer format (GTF) file from Ensembl v87 was screened per autosomal chromosome using an in-house python script to find windows matching the CYP4F cluster: windows of 430 Kb containing six genes were kept. In these windows, we excluded INDELs and SNPs with MAF <0.05. The for each pair of SNPs located within a selected window was computed using VCFTools with the geno-r2 option. We divided the genetic distance into bins of 0.01 cM and calculated the 99th percentile of values of each pair of SNPs lying in the bin. This process was done separately for each 1000G subpopulation, yielding a null distribution per subpopulation. values on pairs of SNPs in the extremes of the empirical distribution are considered to be significant for what we called unusual linkage disequilibrium (uLD).
To specifically confirm the signal seen between CYP4F12 and other CYP4F genes, we extracted only the SNPs showing significant uLD in the previous analysis and kept only those pairs where one SNP was located in CYP4F12. Using VCFTools, CYP4F genetic data were extracted from the 1000 Genomes 30X on GRCh38 dataset (Byrska-Bishop et al. 2021), and values were calculated as described above.
eQTLs Analysis of SNPs under Selection
The Genotype-Tissue Expression v8 (GTEx) (Lonsdale et al. 2013) was accessed through dbGaP (phs000424.v8.p2, dbgap project #19088) and contains gene expression across 54 tissues and 948 donors as well as genotyping information, compiled in a VCF file by GTEx on the GRCh38 genome build. The cohort comprises 67% males and 33% females, mainly of European descent (84.6%), aged between 20 and 79 years old. Analyses were done on 699 individuals of European descent, as described in supplementary text 14.2, Supplementary Material online. To take into account hidden factors, we calculated PEER factors on the normalized expressions. We removed tissues with less than 50 samples, leaving samples from 50 different tissues.
For eQTL analyses, we selected only SNPs that were identified to be under positive or balancing selection in CYP3A and CYP4F clusters in previous analyses and with a MAF above 5%. Since the positions of these SNPs were in the GRCh37 genome build, we converted these positions to the GRCh38 genome build to match GTEx v8 data, using the liftOver function of the rtracklayer R library (Lawrence et al. 2009). P-values of associations between each selected SNP and gene expression of every gene in the cluster were calculated with a linear model using the lm function in R. The linear model was calculated on each SNP individually. The covariates include the first five principal components (PCs) (see supplementary text 14.2, Supplementary Material online), age, sex, PEER factors, the collection site (SMCENTER), the sequencing platform (SMGEBTCHT), and total ischemic time (TRISCHD). To report genome-wide significant eQTL signals, we used a P-value threshold for significance at .
Lastly, we have searched for regulatory annotations of at eQTL signals using the UCSC Genome Browser, specifically looking at the data provided by the ReMap density database (Hammal et al. 2022).
Phenotypic Associations
The UK biobank (UKb) (Sudlow et al. 2015) was accessed through project 15357. We kept only individuals of European descent which were within 3 SD of the mean for the top 3 PCs, removed one individual for each pair of related individuals, and removed individuals whose genetic sex did not match self-identified sex. We extracted positions for CYP3A (chr7:99-244-812-99-470-881, GRCh38) and for CYP4F (chr19:15-618-335-16-110-830, GRCh38) families. We then removed positions with more than 10% of missing genotype and with a MAF under 1%, then removed individuals with more than 5% missing genotypes, leaving us with 399,149 individuals and 3,092 variants for the CYP4F genes, and 400,504 individuals and 374 variants for the CYP3A genes.
We used baseline values for continuous phenotypes. We selected phenotypes recommended by the UKb, as well as blood cells measurements, a total of 90 and 11 phenotypes respectively (supplementary table S3, Supplementary Material online). When many values were available at the baseline, we took the mean of those values. We also looked at diseases using phecode coding extracted from phewascatalog (Denny et al. 2013), which indicated ICD-10 to group and to exclude from controls. We kept only phecodes with more than 500 cases, leaving 603 for both sexes, 62 female-only and 11 male-only. Covariates used are the age at baseline, sex, top 10 PCs, deprivation index and the genotyping array. Analyses were done with plink2 (Chang et al. 2015) with linear transformation of the quantitative covariates. We used a P-value threshold for significance at , based on Bonferonni correction for the number of phenotypes evaluated ().
We performed Mendelian randomization analyses. As instrument variables, we selected SNPs in CYP3A cluster showing strong associations with CYP3A5 expression in lung (exposure), with a F-statistic above 10 and a P-value under 0.001 (0.05/50 tissues), then removed SNPs in pair with a correlation above , estimated on Europeans from GTEx using the function ld_matrix from ieugwasr package in R (Lyon et al. 2021), leaving eight SNPs for analyses. Furthermore, we used the scale function on continuous traits and gene expression to estimate the change of 1 SD of the phenotypes for 1 SD of the gene expression. As outcomes, we used the six phenotypes for which SNPs under selection showed associations for both phenotype () and CYP3A5 expression (). Mendelian randomization analyses were performed using MendelianRandomization package in R (Yavorska and Burgess 2017) and correlation matrix generated using ld_matrix (Lyon et al. 2021) was given to the function to adjust for linkage disequilibrium. We performed Inverse Variance Weighted (IVW) as the main statistical test, and we performed MR-Egger to detect and correct for directional pleiotropy: we report MR-Egger results if the intercept was significant. Lastly, we performed weighted median test as a sensitivity test.
Supplementary Material
Acknowledgments
The authors thank the anonymous reviewers for their valuable suggestions. We also thank Claude Bherer and Marie-Pierre Dubé for their detailed review of this article. This work was completed thanks to computational resources provided by Digital Research Alliance of Canada clusters Graham, Narval and Beluga. This study was supported by funding from the Canada Foundation for Innovation (CFI) (#40157) and the Montreal Heart Institute Foundation. A.R.S.H. received an internship scholarship from the Canadian Institutes of Health Research (CIHR). I.G. is a Robert-Cedergren Bioinformatics Awardee at Université de Montréal. J.G.H. is a Fonds de Recherche du Québec en Santé (FRQS) Junior 2 research scholar.
Contributor Information
Alex Richard-St-Hilaire, Département de biochimie et médecine moléculaire, Université de Montréal, Montreal, QC, Canada; Sainte-Justine Hospital, Research Center, Montreal, QC, Canada.
Isabel Gamache, Département de biochimie et médecine moléculaire, Université de Montréal, Montreal, QC, Canada; Montreal Heart Institute, Research Center, Montreal, QC, Canada.
Justin Pelletier, Département de biochimie et médecine moléculaire, Université de Montréal, Montreal, QC, Canada; McGill CERC in Genomic Medicine, McGill University, Montreal, Canada.
Jean-Christophe Grenier, Montreal Heart Institute, Research Center, Montreal, QC, Canada.
Raphaël Poujol, Montreal Heart Institute, Research Center, Montreal, QC, Canada.
Julie G Hussin, Montreal Heart Institute, Research Center, Montreal, QC, Canada; Département de médecine, Université de Montréal, Montreal, QC, Canada; Mila-Quebec AI institute, Montreal, QC, Canada.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Author Contributions
A.R.S.H., I.G., and J.P. performed analyses. J.C.G., R.P., and I.G. pre-processed the data. A.R.S.H., I.G., and J.G.H. wrote the article, revised by J.C.G., J.P., and R.P. J.G.H. initiated and supervised the project.
Data Availability
The 1000 Genomes Project, GEUVADIS is freely available. The GTEx v8 dataset was accessed through dbGaP under project number #19088. The UK Biobank was accessed through data access approval under the project number #15357. Information to apply for data access can be found here: https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access.
Literature Cited
- Akey JM, Zhang K, Xiong M, Doris P, Jin L. The effect that genotyping errors have on the robustness of common linkage-disequilibrium measures. Am J Hum Genet. 2001:68(6):1447–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aqil A, Speidel L, Pavlidis P, Gokcumen O. Balancing selection on genomic deletion polymorphisms in humans. eLife. 2023:12:e79111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bains RK, Kovacevic M, Plaster CA, Tarekegn A, Bekele E, Bradman NN, Thomas MG. Molecular diversity and population structure at the Cytochrome P450 3A5 gene in Africa. BMC Genet. 2013:14:34–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown S-A, Pereira N. Pharmacogenomic impact of CYP2C19 variation on clopidogrel therapy in precision cardiovascular medicine. J Pers Med. 2018:8(1):8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burk O, Wojnowski L. Cytochrome P450 3A and their regulation. Naunyn Schmiedebergs Arch Pharmacol. 2004:369(1):105–124. [DOI] [PubMed] [Google Scholar]
- Byrska-Bishop M, Evani US, Zhao X, Basile AO, Abel HJ, Regier AA, Corvelo A, Clarke WE, Musunuri R, Nagulapalli K, et al. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios. bioRxiv 430068. 10.1101/2021.02.06.430068, 2021. [DOI] [PMC free article] [PubMed]
- Carlson CS, Thomas DJ, Eberle MA, Swanson JE, Livingston RJ, Rieder MJ, Nickerson DA. Genomic regions exhibiting positive selection identified from dense genotype data. Genome Res. 2005:15(11):1553–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation plink: rising to the challenge of larger and richer datasets. Gigascience. 2015:4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wang H, Zhou G, Zhang X, Dong X, Zhi L, Jin L, He F. Molecular population genetics of human CYP3A locus: signatures of positive selection and implications for evolutionary environmental medicine. Environ Health Perspect. 2009:117(10):1541–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MA, Kanjee U, Rangel GW, Chery L, Mascarenhas A, Gomes E, Rathod PK, Brugnara C, Ferreira MU, Duraisingh MT. Plasmodium vivax infection compromises reticulocyte stability. Nat Commun. 2021:12(1):1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. The variant call format and VCFtools. Bioinformatics. 2011:27(15):2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson PB. The cytochrome P450 superfamily: biochemistry, evolution and drug metabolism in humans. Curr Drug Metab. 2002:3(6):561–597. [DOI] [PubMed] [Google Scholar]
- Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, Mosley JD, Field JR, Pulley JM, Ramirez AH, Bowton E, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013:31(12):1102–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elens L, van Gelder T, Hesselink DA, Haufroid V, van Schaik RH. CYP3A4*22: promising newly identified CYP3A4 variant allele for personalizing pharmacotherapy. Pharmacogenomics. 2012:14(1):47–62. [DOI] [PubMed] [Google Scholar]
- Gaedigk A. Complexities of CYP2D6 gene analysis and interpretation. Int Rev Psychiatry. 2013:25(5):534–553. [DOI] [PubMed] [Google Scholar]
- Gaedigk A, Sangkuhl K, Whirl-Carrillo M, Klein T, Leeder J S. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017:19(1):69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genomes Project Consortium , et al. A global reference for human genetic variation. Nature. 2015:526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman Y, Nudel A, Kerem Z. Polymorphism in cytochrome P450 3A4 is ethnicity related. Front Genet. 2019:10:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammal F, de Langen P, Bergon A, Lopez F, Ballester B. Remap 2022: a database of human, mouse, drosophila and arabidopsis regulatory regions from an integrative analysis of DNA-binding sequencing experiments. Nucleic Acids Res. 2022:50(D1):D316–D325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Wuchty S, Przytycka TM. eQTL epistasis – challenges and computational approaches. Front Genet. 2013:4:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janha RE, Worwui A, Linton KJ, Shaheen SO, Sisay-Joof F, Walton RT. Inactive alleles of cytochrome P450 2C19 may be positively selected in human evolution. BMC Evol Biol. 2014:14:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002:12(6):996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Stephan W. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics. 2002:160(2):765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014:46(3):310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischian NL, Wilson JY. Phylogenetic and functional analyses of the cytochrome P450 family 4. Mol Phylogenet Evol. 2012:62(1):458–471. [DOI] [PubMed] [Google Scholar]
- Korunes KL, Samuk K. pixy: unbiased estimation of nucleotide diversity and divergence in the presence of missing data. Mol Ecol Resour. 2021:21(4):1359–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudaravalli S, Veyrieras J-B, Stranger BE, Dermitzakis ET, Pritchard JK. Gene expression levels are a target of recent natural selection in the human genome. Mol Biol Evol. 2009:26(3):649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehl P, Zhang J, Lin Y, Lamba J, Assem M, Schuetz J, Watkins PB, Daly A, Wrighton SA, Hall SD, et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat Genet. 2001:27(4):383–391. [DOI] [PubMed] [Google Scholar]
- Lamba J, Hebert JM, Schuetz EG, Klein TE, Altman RB. PharmGKB summary: very important pharmacogene information for CYP3A5. Pharmacogenet Genomics. 2012:22(7):555–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Gentleman R, Carey V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics. 2009:25(14):1841–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner B. Molecular mechanisms of epistasis within and between genes. Trends Genet. 2011:27(8):323–331. [DOI] [PubMed] [Google Scholar]
- Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011:27(21):2987–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhang L, Zhou H, Stoneking M, Tang K. Global patterns of genetic diversity and signals of natural selection for human ADME genes. Hum Mol Genet. 2011:20(3):528–540. [DOI] [PubMed] [Google Scholar]
- Liang R, Wang C, Zhao H, Huang J, Hu D, Sun Y. Influence of CYP4F2 genotype on warfarin dose requirement—a systematic review and meta-analysis. Thromb Res. 2012:130(1):38–44. [DOI] [PubMed] [Google Scholar]
- Llaurens V, Whibley A, Joron M. Genetic architecture and balancing selection: the life and death of differentiated variants. Mol Ecol. 2017:26(9):2430–2448. [DOI] [PubMed] [Google Scholar]
- Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, et al. The genotype-tissue expression (GTEx) project. Nat Genet. 2013:45(6):580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon MS, Andrews SJ, Elsworth B, Gaunt TR, Hemani G, Marcora E. The variant call format provides efficient and robust storage of GWAS summary statistics. Genome Biol. 2021:22(1):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean CA, Chue Hong NP, Prendergast JG. hapbin: an efficient program for performing haplotype-based scans for positive selection in large genomic datasets. Mol Biol Evol. 2015:32(11):3027–3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur AG, Hegelund T, Cox RL, Stegeman JJ, Liljenberg M, Olsson U, Sundberg P, Celander MC. Phylogenetic analysis of the cytochrome P450 3 (CYP3) gene family. J Mol Evol. 2003:57(2):200–211. [DOI] [PubMed] [Google Scholar]
- Nebert D, Dalton T. The role of cytochrome P450 enzymes in endogenous signalling pathways and environmental carcinogenesis. Nat Rev Cancer. 2006:6(12):947–960. [DOI] [PubMed] [Google Scholar]
- Nebert DW, Wikvall K, Miller WL. Human cytochromes P450 in health and disease. Philos Trans R Soc B Biol Sci. 2013:368(1612):20120431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson DR, Koymans L, Kamataki T, Stegeman JJ, Feyereisen R, Waxman DJ, Waterman MR, Gotoh O, Coon MJ, Estabrook RW, et al. P450 superfamily: update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996:6(1):1–42. [DOI] [PubMed] [Google Scholar]
- Nelson DR, Zeldin DC, Hoffman SMG, Maltais LJ, Wain HM, Nebert DW. Comparison of cytochrome P450 (CYP) genes from the mouse and human genomes, including nomenclature recommendations for genes, pseudogenes and alternative-splice variants. Pharmacogenet Genomics. 2004:14(1):1–18. [DOI] [PubMed] [Google Scholar]
- Nica AC, Dermitzakis ET. Expression quantitative trait loci: present and future. Philos Trans R Soc B Biol Sci. 2013:368(1620):20120362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu H, Taudien S, Herlyn H, Schmitz J, Zhou Y, Chen G, Roberto R, Rocchi M, Platzer M, Wojnowski L. CYP3 phylogenomics: evidence for positive selection of CYP3A4 and CYP3A7. Pharmacogenet Genomics. 2008:18(1):53–66. [DOI] [PubMed] [Google Scholar]
- Rohlfs RV, Swanson WJ, Weir BS. Detecting coevolution through allelic association between physically unlinked loci. Am J Hum Genet. 2010:86(5):674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas L, Neumann I, Herrero MJ, Bosó V, Reig J, Poveda JL, Megías J, Bea S, Aliño SF. Effect of CYP3A5*3 on kidney transplant recipients treated with tacrolimus: a systematic review and meta-analysis of observational studies. Pharmacogenomics J. 2015:15(1):38–48. [DOI] [PubMed] [Google Scholar]
- Scott SA, Sangkuhl K, Stein CM, Hulot J-S, Mega JL, Roden DM, Klein TE, Sabatine MS, Johnson JA, Shuldiner AR. Clinical pharmacogenetics implementation consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther. 2013:94(3):317–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siewert KM, Voight BF. Detecting long-term balancing selection using allele frequency correlation. Mol Biol Evol. 2017:34(11):2996–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh O, Sandanaraj E, Subramanian K, Lee LH, Chowbay B. Influence of CYP4F rs2108622 (V433M) on warfarin dose requirement in Asian patients. Drug Metab Pharmacokinet. 2011:26(2):130–136. [DOI] [PubMed] [Google Scholar]
- Stark K, Wongsud B, Burman R, Oliw EH. Oxygenation of polyunsaturated long chain fatty acids by recombinant CYP4F8 and CYP4F12 and catalytic importance of Tyr-125 and Gly-328 of CYP4F8. Arch Biochem Biophys. 2005:441(2):174–181. [DOI] [PubMed] [Google Scholar]
- Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M, et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015:12(3):e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavira B, Coto E, Diaz-Corte C, Alvarez V, López-Larrea C, Ortega F. A search for new CYP3A4 variants as determinants of tacrolimus dose requirements in renal-transplanted patients. Pharmacogenet Genomics. 2013:23(8):445–448. [DOI] [PubMed] [Google Scholar]
- Thompson EE, Kuttab-Boulos H, Witonsky D, Yang L, Roe BA, Di Rienzo A. CYP3A variation and the evolution of salt-sensitivity variants. Am J Hum Genet. 2004:75(6):1059–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson EE, Kuttab-Boulos H, Yang L, Roe BA, Di Rienzo A. Sequence diversity and haplotype structure at the human CYP3A cluster. Pharmacogenomics J. 2006:6(2):105–114. [DOI] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK, Hurst L. A map of recent positive selection in the human genome. PLoS Biol. 2006:4(3):e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Guo Y, Wrighton SA, Cooke GE, Sadee W. Intronic polymorphism in CYP3A4 affects hepatic expression and response to statin drugs. Pharmacogenomics J. 2011:11(4):274–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984:38(6):1358–1370. [DOI] [PubMed] [Google Scholar]
- Wright WC, Chenge J, Chen T. Structural perspectives of the CYP3A family and their small molecule modulators in drug metabolism. Liver Res. 2019:3(3–4):132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukochi Y, Satta Y. Molecular evolution of the CYP2D subfamily in primates: purifying selection on substrate recognition sites without the frequent or long-tract gene conversion. Genome Biol Evol. 2015:7(4):1053–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavorska OO, Burgess S. Mendelianrandomization: an R package for performing mendelian randomization analyses using summarized data. Int J Epidemiol. 2017:46(6):1734–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Cho S-A, Min J, Kim DH, Shin J-G, Lee S-J. Functional characterization of a common CYP4F11 genetic variant and identification of functionally defective CYP4F11 variants in erythromycin metabolism and 20-HETE synthesis. Arch Biochem Biophys. 2017:620:43–51. [DOI] [PubMed] [Google Scholar]
- Zhang JE, Klein K, Jorgensen AL, Francis B, Alfirevic A, Bourgeois S, Deloukas P, Zanger UM, Pirmohamed M. Effect of genetic variability in the CYP4F2, CYP4F11, and CYP4F12 genes on liver mRNA levels and warfarin response. Front Pharmacol. 2017:8:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The 1000 Genomes Project, GEUVADIS is freely available. The GTEx v8 dataset was accessed through dbGaP under project number #19088. The UK Biobank was accessed through data access approval under the project number #15357. Information to apply for data access can be found here: https://www.ukbiobank.ac.uk/enable-your-research/apply-for-access.