

Abstract
Background:
Angiopoietin-like 3 (ANGPTL3) is a therapeutic target for reducing plasma levels of triglycerides and low-density lipoprotein cholesterol (LDL-C). A recent trial with vupanorsen, an antisense oligonucleotide targeting hepatic production of ANGPTL3, reported a dose-dependent increase in hepatic fat. It is unclear if this adverse effect is due to an on-target effect of inhibiting hepatic ANGPTL3.
Methods:
We recruited participants with ANGPTL3 deficiency due to ANGPTL3 loss-of-function (LoF) mutations along with wild-type participants from two previously characterized cohorts located in Campodimele, Italy and Saint Louis, Missouri. Magnetic resonance spectroscopy and magnetic resonance proton density fat fraction were performed to measure hepatic fat fraction (HFF) and the distribution of extrahepatic fat. To estimate the causal relationship between ANGPTL3 and hepatic fat, we generated a genetic instrument of plasma ANGPTL3 levels as a surrogate for hepatic protein synthesis and performed Mendelian randomization (MR) analyses with hepatic fat in the UK Biobank study.
Results:
We recruited participants with complete (N=6) or partial (N=32) ANGPTL3 deficiency due to ANGPTL3 LoF mutations along with wild-type participants (N=92) without LoF mutations. Participants with ANGPTL3 deficiency exhibited significantly lower total cholesterol (complete deficiency=78.5 mg/dL, partial deficiency=172 mg/dL, wild-type=188 mg/dL; P<0.05 for both deficiency groups as compared to wild-type) along with plasma triglycerides (complete deficiency=26 mg/dL, partial deficiency=79 mg/dL, wild-type=88mg/dL; P<0.05 for both deficiency groups compared to wild-type) without any significant difference in hepatic fat (complete deficiency=9.8%, partial deficiency=10.1%, wild-type=9.9%;; P>0.05 for both deficiency groups compared to wild-type) or severity of hepatic steatosis as assessed by magnetic resonance imaging. In addition, ANGPTL3 deficiency did not alter the distribution of extrahepatic fat. Results from MR analyses in 36,703 participants from the UK Biobank demonstrated that genetically determined plasma ANGPTL3 protein levels were causally associated with LDL cholesterol (P=1.7x10−17) and triglycerides (P=3.2x10−18) but not with hepatic fat (P=0.22).
Conclusions:
ANGPTL3 deficiency due to LoF mutations in ANGPTL3 as well as genetically determined reduction of plasma ANGPTL3 levels are not associated with hepatic steatosis. Therapeutic approaches to inhibit ANGPTL3 production in hepatocytes are not necessarily expected to result in the increased risk for hepatic steatosis that was observed with vupanorsen.
Keywords: Angiopoietin-like 3, ANGPTL3, drug-related adverse side effect, hepatic steatosis, lipids, Mendelian randomization, triglycerides
Introduction
Angiopoietin-like 3 (ANGPTL3) is a key factor in the regulation of lipoprotein transport, prompting increasing interest in its role on fat metabolism1,2. As a glycoprotein synthesized predominantly by hepatocytes, ANGPTL3 acts as circulating inhibitor of lipoprotein lipase (LPL), the rate-limiting enzyme for hydrolyzing triglycerides in TG-rich lipoproteins including chylomicrons and very-low-density lipoproteins (VLDL) 1. ANGPTL3 also acts as an inhibitor of endothelial lipase (EL), which raises high-density lipoprotein cholesterol (HDL-C). Consistent with this role, genetic and pharmacological inactivation of ANGPTL3 in mice are associated with de-repression of LPL and EL activity along with significant reductions in plasma total triglycerides (TG) and total cholesterol (TC)3.
In humans, complete ANGPTL3 deficiency due to bi-allelic inactivating mutations in ANGPTL3 causes familial combined hypolipidemia (FHBL2 [OMIM 605019]), a very rare lipid disorder characterized by a marked reduction of low-density lipoprotein cholesterol (LDL-C), HDL-C, and VLDL4–6. FHBL2 is associated with increased LPL mass and activity in post-heparin plasma7 and affected individuals have marked reduction of postprandial lipemia8 along with lower plasma free fatty acids (FFA) and improved insulin sensitivity5,7,9. In agreement with these favorable metabolic changes, we and others have reported that carriers of loss-of-function (LoF) mutations in ANGPTL3 displayed reductions in atherosclerotic burden and risk of coronary events10,11.
An important unanswered question related to the lipid-lowering mechanism of ANGPTL3 deficiency is if and how this condition influences the hepatic synthesis of VLDL. In the initial report of ANGPTL3 deficiency in humans, Musunuru et al. 6 showed that carriers of compound heterozygous ANGPTL3 LoF alleles exhibited reduced hepatic VLDL-apolipoproteinB (ApoB) production, thus raising the possibility of impaired TG export from the liver. It is well established that impaired VLDL secretion from hepatocytes is a major cause of fatty liver12,13. This condition is typically represented in abetalipoproteinemia (OMIM 200100) and familial hypobetalipoproteinemia (OMIM 615558), where hepatic secretion of VLDL is impaired due either to defects in the lipid chaperone protein microsomal triglyceride transfer protein (MTP) or to the reduced synthesis of the acceptor protein ApoB. In either setting, hepatic steatosis develops in the majority of patients14. However, Wang et al. 15 demonstrated in murine models that the inactivation of ANGPTL3 with a monoclonal antibody was not associated with an increase in hepatic TG content. In a previous study, we found that FHBL2 participants did not show increased prevalence of steatosis as evaluated by hepatic ultrasonography16. In contrast, it was recently reported that the inhibition of hepatic ANGPTL3 synthesis by vupanorsen, a specific antisense oligonucleotide against ANGPTL3, was associated with increases in liver steatosis in a dose dependent manner17. Therefore, the consequences of ANGPTL3 deficiency on hepatic fat accumulation remain to be firmly established. In addition, the interplay between ANGPTL3 and adipose tissue depots has been incompletely characterized in humans. Previous studies have shown that ANGPTL3 levels are increased in obesity18 and that body mass index (BMI) is an independent predictor of ANGPTL3 levels19. However, whether ANGPTL3 can influence the mass and distribution of adipose tissue through its action on FFA metabolism in humans remains unknown.
Studies of humans with naturally occurring LoF mutations in genes encoding drug targets can provide insight into the efficacy and potential adverse effects of inhibitory drugs directed at those targets20. Therefore, to estimate the effect of targeting ANGPTL3 production in hepatocytes, we first characterized hepatic fat content as well as extrahepatic fat distribution by using magnetic resonance spectroscopy (MR-S) and magnetic resonance proton density fat fraction (MR-PDFF) in participants with either partial or complete ANGPTL3 deficiency due to inactivating mutations in ANGPTL3. We then extended our study to a large population cohort to determine if reduced concentrations of ANGPTL3 were causally associated with changes in hepatic fat content using two-sample Mendelian randomization.
Methods
Study participants and protocols
The data that support the findings of this study are available from the corresponding authors upon reasonable request. Participants who were carriers of ANGPTL3 LoF mutations and wild-type controls were recruited from two previously characterized FHBL2 cohorts located in Campodimele, Italy and Saint Louis, Missouri. The procedures to characterize these individuals have been extensively described in previous reports7–9,16,21–25. Individuals from Campodimele received a community invitation to all inhabitants who have previously participated in a series of studies aimed at characterizing the metabolic consequences and natural history of humans with LoF mutations in ANGPTL37−9,16,21–25. All inhabitants of the Campodimele village were invited to participate in the current study without a priori selection. In addition to this population, participants were recruited from a large family carrying ANGPTL3 LoF mutations followed at Washington University in Saint Louis, MO. All LoF mutations in both cohorts were characterized in previous studies6–9,16,21–25, therefore repeat genotyping was not performed. In comparison to our previously published studies7–9,16,21–25, one participant with FHBL2 died, another woman with FHBL2 was unwilling to undergo the magnetic resonance due to limited mobility, and two additional FHBL2 subjects refused to participate in the imaging study due to claustrophobia. Ultimately, 130 participants (126 from Campodimele and 4 from Saint Louis) were included, comprised of 6 participants with homozygous or compound heterozygous LoF mutations, 32 with heterozygous LoF mutations, and 92 without LoF mutations. Among the Campodimele’s population, when considering second and first-degree relationships, 48 subjects were clustered in 15 families while 78 were the single representative of their family. Two of the four FHBL2 subjects from Campodimele are siblings. All four subjects from Saint Louis belong to the same family. All participants were of European ancestry.
Participants were invited to undergo clinical evaluation and blood drawings during a screening visit. Anthropometric and clinical characteristics of study participants were assessed as previously described5,8. Alcohol consumption was estimated both by a semi-quantitative scale as previously described5 and by using AUDIT score26 combined in a unique variable. At the time of screening, no participants were consuming special diets. To determine indices of insulin resistance, participants without T2DM were asked to undergo a 75 g oral glucose tolerance test (OGTT) 27.
Magnetic resonance imaging was performed using methods described below. After excluding participants in which a technical problem occurred in the execution of the exam (early termination due to participant request, for example), MR-S and MR-PDFF images were available in 96 and 123 participants, respectively.
The Sapienza University of Rome and Washington University School of Medicine Human Studies Committees approved the experimental protocols and procedures. All participants voluntarily participated in the study and written informed consent was obtained from all participants in accordance with the principles of the Helsinki Declaration.
Laboratory determinations
Blood samples for laboratory determinations were obtained in the morning after an overnight fast and collected in EDTA-containing tubes. Plasma was immediately obtained by centrifugation at 4° C. Aliquots were prepared with EDTA (0.04%), NaNO3 (0.05%), and phenylmethylsulphonyl fluoride (0.015%) to prevent lipid and lipoprotein modifications. Plasma lipid and blood glucose concentrations were measured from fresh aliquots within 2–4 h from acquisition. Other assays were performed on aliquots that were stored at −80° C after acquisition.
Plasma lipids, ApoB, ApoA1, fasting blood glucose, and liver transaminases were assayed after separation of lipoproteins by serial ultracentrifugation and precipitation methods as previously described5,8. Plasma glucose and insulin were measured using commercial assays on Roche Cobas 6000 automated analyzer (Roche Diagnostics, Mannheim, Germany) according with local procedures. Insulin sensitivity and secretion were estimated by HOMAIR28, HOMAβ28, QUICKI29, along with dynamic indices obtained during OGTT and insulin sensitivity index according to the Matsuda formula (ISIm) 27. Circulating plasma ANGPTL3 protein levels were measured using a human ANGPTL3 ELISA (BioVendor R&D).
Magnetic resonance imaging (MRI) protocol
Data were acquired on a 3T magnet (GE Discovery 750; General Electric Healthcare, Milwaukee, WI) with a peak gradient amplitude of 50 mT/m and a time to peak of 200 ms using a thirty-two-element body torso-array coil system. For the quantification of hepatic fat fraction (HFF) we adopted a 3D sequence (IDEAL IQ) to create T2* and triglyceride fat fraction maps from a single breath-hold (MR-PDFF). The sequence has the following parameters: TR, 12.9 ms; TE six different echos from 1.6 to 9.8 ms field of view, 42–38 cm; matrix, 128 x 128; bandwidth, 111 kHz; FA, 3°; section thickness, 10 mm with 26 sections; acquisition time, 25 s. The images were processed using the software provided by the manufacturer (IDEAL-IQ; GE Healthcare) to create water, fat, in-phase, out-of-phase, R2*, and fat fraction maps instantaneously30.
MR-S of the liver was performed using 20 mm × 20 mm × 20 mm voxels placed in the IVa, VII and VIII segments, avoiding major blood vessels and biliary ducts. All spectra were obtained in the stimulated echo acquisition mode (PRESS, TR 4000 ms, TE 30 ms), using a breath hold sequence with an acquisition time of approximately 24 s. Field homogeneity was automatically adjusted for each voxel, and saturation bands were not used31.
For the quantification of subcutaneous and visceral adipose tissue (SAT and VAT, respectively), a 3D spoiled GRE T1-weighted sequence in the axial plane (TR, 4.2; TE, 1.3; FA, 15; matrix, 320 192; section thickness, 5 mm, reconstructed 2.5 mm; intersection gap, 0) was acquired with the Dixon method, which enabled the separation between water and fat components. The sequence described above was also used for the estimation of the subcutaneous adipose tissue and intermuscular lipids of the thigh. Additional MRI methods are included in the Supplemental Material.
Mendelian randomization
Plasma levels of ANGPTL3 have previously been measured by aptamer-based technology (SomaScan) in the deCODE32 and INTERVAL33 studies of 35,559 Icelanders and 3,301 European participants, respectively. We performed an inverse-variance weighted meta-analysis of summary statistics from these two studies for plasma ANGPTL3 levels. We then selected independent (r2 ≤ 0.3 based on 1000Genomes European panel) markers within 150kb of ANGPTL3 that were associated with ANGPTL3 levels (as assessed by aptamer 10391-1) at a level of genome-wide statistical significance (P≤5x10−8) to generate an instrumental variable for plasma ANGPTL3 levels. We generated an instrumental variable for plasma ApoB levels using independent (r2 ≤ 0.3 based on 1000Genomes European panel) variants within 150kb of APOB that associated with protein levels (as assessed by aptamer 2797-56) at P≤1x10−4. Outcomes for hepatic fat were obtained from previously published summary statistics in a study of 36,703 participants from the UK Biobank study34. Outcomes for plasma TG and LDL-C were obtained from a previously published genome-wide association study of plasma lipid levels in the UK Biobank study35. Outcomes for alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were also obtained from a previously published genome-wide association study of laboratory biomarkers in the UK Biobank study36. Causal analysis was performed using the inverse-variant weighted method implemented in the R package TwoSampleMR37.
Statistical analysis
For descriptive statistics, continuous traits were presented as mean and standard deviation (SD) or as median and interquartile range (IQR) as appropriate. Categorical traits were shown as number and proportion. Comparisons were carried out by Mann-Whitney or Kruskal-Wallis test for non-normally distributed variables and by Student’s or ANOVA test for normally distributed variables. For differences between categorical traits, statistical significance was calculated by chi-square or by Fisher-exact test as appropriate. Multiple regression analysis was used to determine the best predictors of liver fat content and hepatic steatosis in all participants. The multiple models included age, male sex, BMI, HOMA IR index, and ANGPTL3 genotype as a co-dominant trait (using a variable with three categories). The analysis was conducted using a stepwise linear regression method. Non-normally distributed values were log-transformed before entering the model. Statistical analyses were performed using the IBM Statistical Package for Social Sciences (IBM SPSS, version 25.0, Inc. Chicago, IL). A P-value <0.05 was considered statistically significant.
Results
Subject characteristics
The demographic, anthropometric, and clinical characteristics of 130 study participants are shown in Table 1. Among all participants, 92 were wild-type (WT) and 38 were carriers of ANGPTL3 LoF mutations; 32 carried heterozygous mutations resulting in partial LoF, while 6 carried either homozygous or compound heterozygous mutations resulting in complete LoF. The only demographic differences observed between genotypes were older age in complete LoF participants and a higher prevalence of current/past heavy drinkers among ANGPTL3 LoF carriers as compared with WT controls (Ptrend=0.01). As expected, complete LoF ANGPTL3 participants had a pronounced reduction of all plasma lipoprotein fractions while partial LoF ANGPTL3 participants had an intermediate phenotype between WT and complete LoF. ANGPTL3 LOF carriers had a non-significant trend toward lower HOMA-IR and HOMAβ values as compared with WT (with an apparent gene-dose effect). No difference was found in the QUICKI index among genotypes. ISIm values did not differ by genotype although complete LoF participants had a slightly higher ISIm as compared to others (Table 1). Finally, biochemical laboratory assessment of liver function did not significantly differ between the groups although there was a trend toward lower gamma-glutamyl transferase levels in ANGPTL3 LoF carriers in an apparent gene dose-dependent fashion.
Table 1.
Baseline Characteristics of study participants
| Variables | Wild type carriers (N=92) | ANGPTL3 deficiency due to LoF mutations (N=38) | |
|---|---|---|---|
|
| |||
| Partial (N=32) | Complete (N=6) | ||
|
| |||
| Demographic | |||
|
| |||
| European Ancestry, n (%) | 92 (100) | 32 (100) | 6 (100) |
|
| |||
| Median age (IQR), years | 54.5 (44.5-63.0) | 50.0 (40.0-61.7) | 64.0 (59.7-70.5) ^° |
| Male, n (%) | 47 (51.1) | 18 (56.3) | 4 (66.7) |
|
| |||
| Life-style habit and past medical history | |||
|
| |||
| Mean BMI (IQR), kg/m2 | 28.5 (25.9-30.2) | 27.7 (24.7-30.8) | 30.8 (28.4-32.3) |
| Current/past heavy alcohol use*, n (%) | 3 (3.7) | 4 (13.3) | 2 (33.3) ^° |
| T2DM, n (%) | 12 (13.3) | 1 (3.1) | 1 (16.7) |
| HTN, n (%) | 36 (40.0) | 10 (31.2) | 3 (50.0) |
| CHD, n (%) | 5 (5.6) | 0 | 0 |
| Cancer, n (%) | 6 (6.7) | 1 (3.1) | 0 |
|
| |||
| ANGPLT3, ng/ml (Mean ± SD) | |||
|
| |||
| 177.2±63.1 | 98.6 ±47.1 | 0 | |
|
| |||
| Plasma Lipids (Mean and IQR) | |||
|
| |||
| TC, mg/dl | 188.0 (159.5-210.2) | 172.0 (147.2-190.5) § | 78.5 (66.7-86.7) ^° |
| HDL-C, mg/dl | 58.5 (50.0-68.0) | 51.5 (45.0-60.7) § | 26.0 (17.2-32.0) ^° |
| TG, mg/dl | 88.0 (65.0-129.2) | 79.0 (54.2-98.7) § | 26.0 (26.7-48.7) ^° |
| LDL-C, mg/dl | 104.0 (85.0-126.2) | 101.3 (76.2-113.0) | 42.0 (34.5-50.5) ^° |
| ApoA1, mg/dl | 145.0 (121.0-178.0) | 139.0 (119.0-173.0) | 86.5 (63.5-129.0) ^° |
| ApoB, mg/dl | 92.5 (72.0-109.2) | 83.0 (70.0-102.7) | 38.0 (34.5-88.7) |
|
| |||
| Glucose Metabolism (Mean and IQR) | |||
|
| |||
| FPG, mg/dl | 94.0 (88.0-104.0) | 94.0 (86.0-101.0) | 91.0 (88.5-109.2) |
| Fasting Insulin, U/L | 9.9 (6.5-11.9) | 7.7 (5.9-9.9) | 6.6 (4.2-9.9) |
| HOMA IR | 2.1 (1.5-2.8) | 1.7 (1.3-2.8) | 1.5 (1.0-2.2) |
| HOMAβ | 106.5 (63.9-140.5) | 93.6 (70.4-138.4) | 90.4 (37.5-124.9) |
| Mean QUICKI ± SD | 0.3±0.03 | 0.3±0.03 | 0.4±0.02 |
| Mean ISIm* ± SD | 5.0±2.7 | 4.9±2.5 | 6.7±5.2 |
|
| |||
| Liver Function Test (Mean ± SD) | |||
|
| |||
| AST, U/L | 21.9±7.4 | 23.4±9.7 | 22.8±6.7 |
| ALT, U/L | 24.4±12.3 | 24.0±10.1 | 31.3±14.2 |
| GGT, U/L | 27.7±27.1 | 21.5±14.6 | 16.5±4.4 |
Alcohol use as defined in methods. Data on alcohol consumption were missing for 9 wildtype, 2 heterozygous, and 3 homozygous ANGPTL3 LoF carriers.
P<0.05 for comparison between wild-type and heterozygous ANGPTL3 LoF carriers;
P<0.05 for comparison between wild-type and homozygous ANGPTL3 LoF carriers;
P<0.05 for comparison between heterozygous and homozygous ANGPTL3 LoF carriers.
LoF, loss of function; BMI, body mass index; T2DM, type 2 diabetes; HTN, hypertension; CHD, coronary heart disease; TC, total cholesterol, HDL-C, high density lipoprotein cholesterol; TG, triglyceride; LDL-C, low density cholesterol; FPG, fasting plasma glucose; Quicki, Quantitative Insulin Sensitivity Check Index; HOMA IR, Homeostasis Model Assessment insulin resistance; HOMAβ Homeostasis Model Assessment β; ISI Matsuda, insulin sensitivity index; AST, aspartate aminotransferase; ALT, alanine aminotransferase; GGT, gamma-glutamyl transferase
Complete or partial ANGPTL3 deficiency is not associated with increase in hepatic fat fraction or liver steatosis
As shown in Figure 1, the HFF as estimated by either MR-S (n=96) or MR-PDFF (n=123) was not significantly different between ANGPTL3 LoF carriers and WT participants. Mean HFF assessed by MR-S was 9.9% (standard deviation 15.2%) in WT, 10.1% (standard deviation 15.9%) in partial LoF carriers, and 9.8% (standard deviation 8.6%) in complete LoF carriers (P=NS). Similarly, mean HFF assessed by MR-PDFF was 6.9% (standard deviation 5.9%) in WT, 6.4% (standard deviation 5.4%) in partial LoF carriers, and 9.7% (standard deviation 8.9%) in complete LoF carriers (P=NS). Furthermore, we did not observe a significant difference between genotype groups when sub-categorizing the severity of hepatic steatosis (Figure 2). Among WT participants, we found that 40 (59.7%) had no liver steatosis, while 21 (31.3%) had mild steatosis, five (7.5%) had moderate steatosis, and one had severe (1.5%) steatosis. Similarly, among ANGPTL3 LoF carriers, 16 (55.2%) had no liver steatosis, 10 (34.5%) had mild steatosis, and 3 (10.3%) had moderate steatosis. Comparable results were observed when HFF was assessed by MR-PDFF (Figure 2, Panel B). As assessed by both MRI techniques, ANGPTL3 LoF carriers did not have severe hepatic steatosis and the prevalence of hepatic steatosis among partial and complete LoF carriers was comparable to WT controls (Figure 2).
Figure 1. ANGPTL3 deficiency and quantity of hepatic fat.
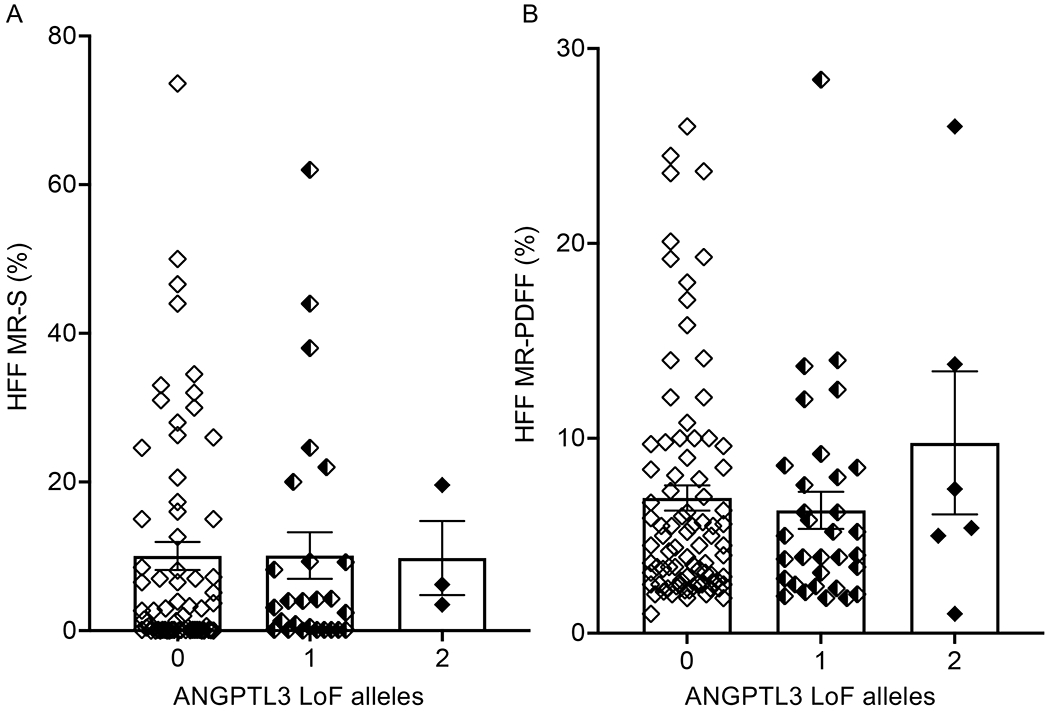
Hepatic fat fraction as estimated by MR-S (A) and MR-PDFF (B) is shown according to degree of ANGPTL3 deficiency. Boxes and error bars represent mean and SEM, respectively. P>0.05 between genotype groups by Kruskal-Wallis test. ANGPTL3: angiopoietin-like 3 protein; LoF: loss of function; MR-S: magnetic resonance spectroscopy; MR-PDFF: magnetic resonance proton density fat fraction.
Figure 2. ANGPTL3 deficiency and severity of hepatic steatosis.
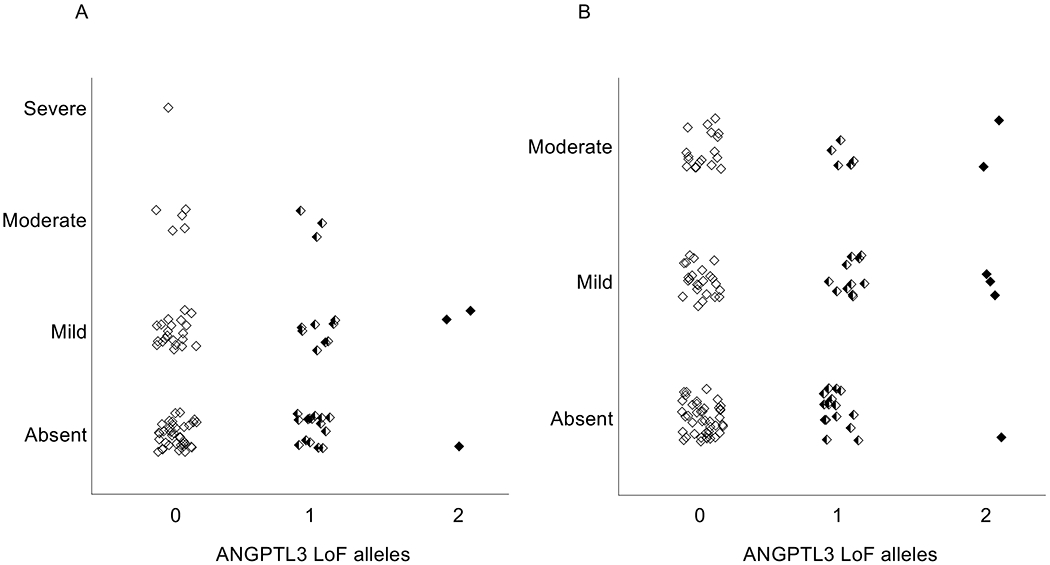
Hepatic steatosis severity as estimated by MR-S (A) and MR-PDFF (B) is shown according to degree of ANGPTL3 deficiency. Severity of hepatic steatosis was calculated as reported in the methods. ANGPTL3: angiopoietin-like 3 protein; LoF: loss of function; MR-S: magnetic resonance spectroscopy; MR-PDFF: magnetic resonance proton density fat fraction.
In multiple regression analyses, we found that older age (P=0.016), male sex (P=0.018), higher BMI (P<0.001), and higher HOMAIR (P< 0.001) were the best predictors of HFF content as estimated by MR-PDFF. Age (P=0.009) was the only significant predictor of HFF as estimated by MR-S.
Using circulating plasma levels of ANGPTL3 measured by ELISA, we performed additional analyses aimed at evaluating if a larger reduction in ANGPTL3 was associated with hepatic steatosis. Across all participants, the mean level of ANGPTL3 was 148.4 μg/L (standard deviation, 75.2 μg/L) with a progressive increase from FHBL2 to wild type (Table 1). Mean ANGPTL3 levels were significantly different among genotypes: wild type = 177.2 μg/L (standard deviation, 63.1 μg/L), heterozygous deficiency = 98.62 μg/L (standard deviation, 47.1 μg/L), complete deficiency = 0 μg/L (P<0.0001). In a regression analysis including all participants, plasma levels of ANGPTL3 had no association with HFF assessed by MR-S (P=0.95) or MR-PDFF (P=0.45). In contrast, in this sub-analysis we found age to be a significant positive predictor of HFF as estimated by MR-S (P=0.015) while age (P=0.026), BMI (P=0.002), male gender (P=0.044) and HOMA IR (P<0.001) were significant positive predictors of HFF as estimated by MR-PDFF. We also grouped participants with ANGPTL3 levels of either ≤ 48 μg/L or ≤ 59 μg/L, corresponding to the circulating ANGPTL3 levels achieved by vupanorsen dosing that was associated with significant increases in hepatic fat. We compared these groups to those without ANGPTL3 LoF mutations (to mimic individuals on placebo) and did not find that lower levels of ANGPTL3 associated with increased HFF (Table S1). A post hoc power calculation based on these data determined there was >80% power at an α of 0.05 to detect an increase in HFF of the same magnitude reported for both vupanorsen dosing in both groups17.
Complete or partial ANGPTL3 deficiency does not alter extrahepatic fat accumulation
We also determined the distribution of extrahepatic fat according to ANGPTL3 genotype groups. No significant differences were found between genotype carriers in any of the examined tissues (Figure 3). Results from multivariable analyses showed that male sex (P<0.001), increased HOMAIR (P=0.01), increased BMI (P=0.001), and older age (P=0.006) were significantly and positively associated with VAT expansion while increased BMI (P<0.001) and female sex (P=0.007) were significantly and positively associated with increased SAT.
Figure 3. ANGPTL3 deficiency and extrahepatic fat distribution.
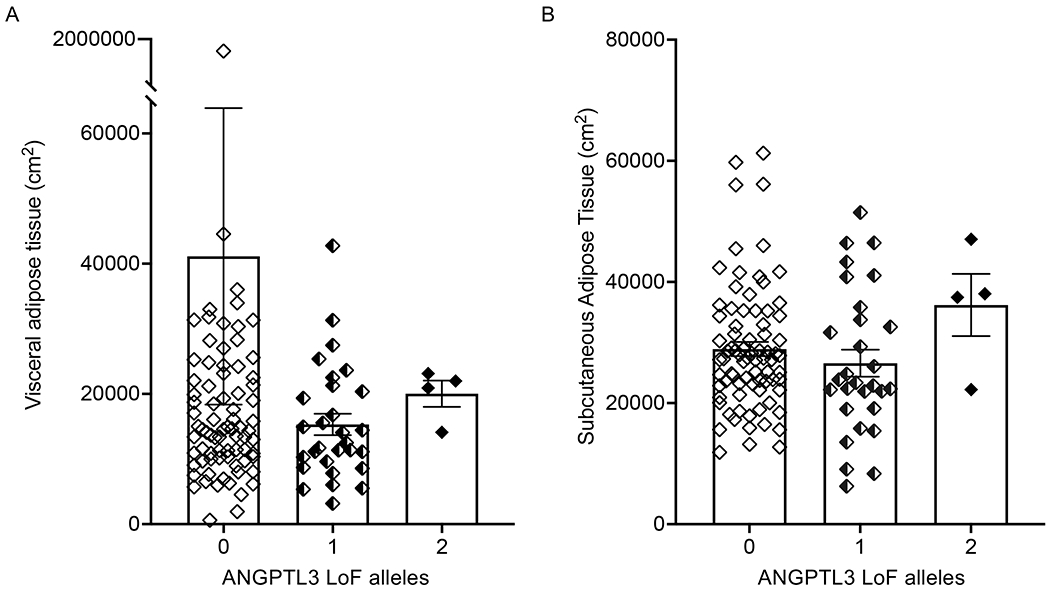
Distribution of extrahepatic fat in (A) visceral adipose tissue, (B) subcutaneous adipose tissue, (C) skeletal intramuscular adipose tissue, and (D) skeletal subcutaneous adipose tissue is shown according to degree of ANGPTL3 deficiency. Boxes and error bars represent means and SEM, respectively. P>0.05 between genotype groups by Kruskal-Wallis test. ANGPTL3: angiopoietin-like 3 protein; LoF: loss of function.
ANGPTL3 is not causally associated with hepatic fat by Mendelian randomization
To validate our finding that ANGPTL3 deficiency does not increase risk for hepatic steatosis, we performed Mendelian randomization to determine if there was a causal association between ANGPTL3 and hepatic fat in 36,703 participants from the UK Biobank with available abdominal MRI34. First, we generated an instrumental variable of genetically determined plasma ANGPTL3 levels as described in the methods. To validate our approach, we then determined if this instrument was causally associated with plasma TG and LDL-C in data from 441,016 and 440,546 participants of the UK Biobank study, respectively. As expected, genetically determined plasma ANGPTL3 levels were significantly positively associated with plasma TG (P=3.2x10−18; Figure 4A) and LDL-C (P=1.7x10−17; Figure 4B). In contrast, we did not find a significant association between genetically determined plasma ANGPTL3 levels and hepatic fat in UK Biobank (P=0.22; Figure 4C). We found a significant positive association between genetically determined plasma ANGPTL3 with both ALT (P=4.7x10−7; Figure S1A) and AST (P=2.6x10−4; Figure S1B). As a positive control, we performed the same analysis using an instrument to reflect genetically determined ApoB levels in UK Biobank. Genetically determined plasma ApoB levels were positively associated with plasma LDL-C (P=0.009; Figure S2A), but negatively associated with hepatic fat (P=0.005; Figure S2B), ALT (P=3.7x10−4; Figure S3C), and AST (P=4.2x10−4; Figure S3D). These findings are consistent with the observation that individuals with reduced hepatic ApoB production exhibit decreased levels of plasma LDL-C but increased hepatic steatosis.
Figure 4. Mendelian Randomization of genetically determined plasma ANGPTL3 with plasma lipids and hepatic fat in the UK Biobank study.
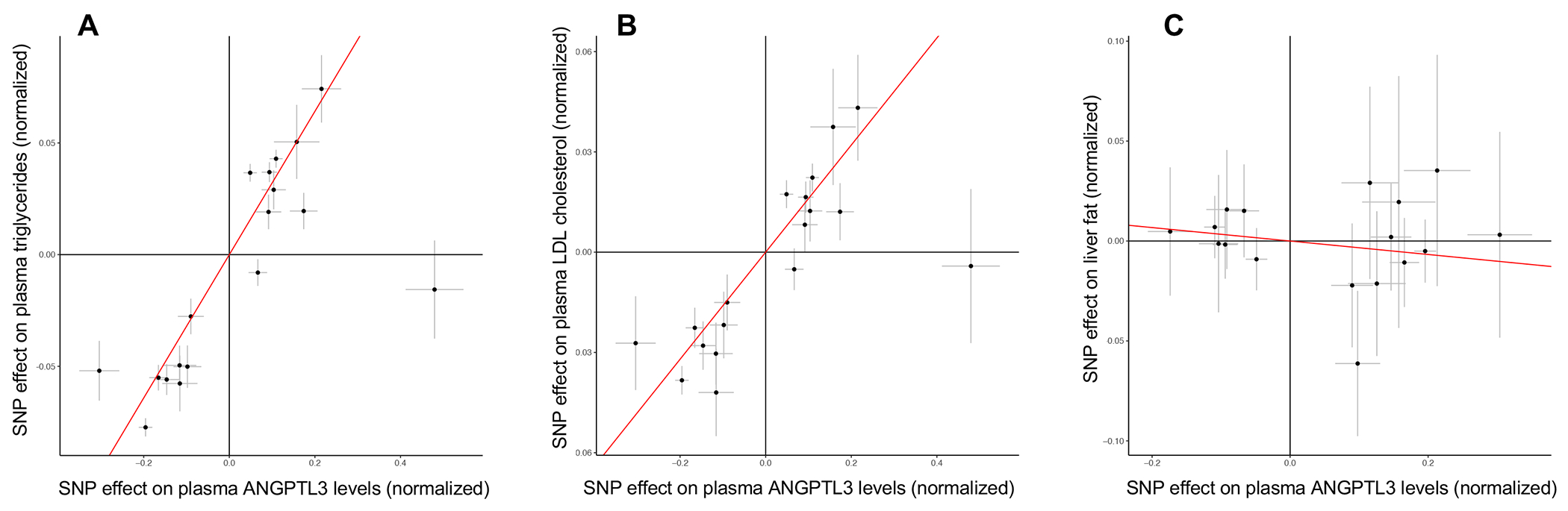
(A) Estimated effect (with 95% confidence intervals) of each variant included in the Mendelian Randomization analysis on ANGPTL3 levels for (A) plasma triglycerides (P=3.2x10−18), (B) plasma LDL cholesterol (P=1.7x10−17), and (C) hepatic fat (P=0.22). Red lines indicate the causal effect estimates.
Discussion
To our knowledge, this is the first study to fully investigate the consequences of ANGPTL3 deficiency on body fat distribution in humans. Leveraging our cohorts of participants with either partial or total ANGPTL3 deficiency, we found no evidence that either reduced concentration or total absence of ANGPTL3 is associated with altered liver fat content, risk of hepatic steatosis, or differences in the extrahepatic distribution of fat.
Although previous studies suggested that FHBL2 was not associated with increased hepatic steatosis, those studies mainly relayed on the use of abdominal ultrasound examination or laboratory assessment of liver function16. Ultrasound is a suboptimal technique for measuring hepatic fat, reliably detecting fat accumulation only in the presence of greater than 15% steatotic hepatocytes. Likewise, biochemical liver function tests reflect hepatocyte damage or inflammation and are only indirect markers of increased fat content16. To overcome this shortcoming, we employed MR-S and MR-PDFF, which are the methods of choice for the non-invasive evaluation of liver fat content with a sensitivity comparable to liver biopsy38.
Our findings raise the question of why participants with ANGPTL3 LoF mutations, despite reduced VLDL secretion, did not show variation in hepatic fat content. Previous studies demonstrated alterations in VLDL particle size and neutral lipid content in participants receiving evinacumab39. Those findings suggest as one possibility that FHBL2 participants do not accumulate excess liver fat through compensatory reductions in the synthesis of triglycerides by hepatocytes40. Indeed, Wang et al. 15 demonstrated that inactivating ANGPTL3 with a monoclonal antibody in mice reduced hepatic production of VLDL-TG but not VLDL-ApoB, apparently due to a decreased supply of FFA from the circulation into the liver for TG synthesis. Interestingly, the impairment in hepatic VLDL secretion in this mouse model was not associated with an increase in liver triglyceride content. The importance of altered FFA delivery is in line with the observation that plasma concentration of non-esterified fatty acid is reduced in participants with FHBL27. In addition, we have previously reported that participants with complete ANGPTL3 deficiency exhibit increased concentrations of beta-hydroxybutyrate9 which is a major oxidative product of hepatic fatty acid metabolism41. Taken together, these findings may suggest that hepatic ANGPTL3 deficiency directs fatty acids towards oxidation rather than to the synthesis of triglycerides. Alternatively, we have demonstrated that ANGPTL3 deficiency in humans causes markedly reduced postprandial lipemia, probably due to faster catabolism of intestinally derived TG-rich lipoproteins8. Such an effect could be explained by enhanced LPL activity in oxidative tissues and possibly reduced adipose tissue lipolysis, resulting in a reduced flux of FFA into the liver. For example, we have recently found that the fibrinogen-like domain of ANGPTL3 enhances lipolysis in cultured adipocytes derived from 3T3-L1 cells42, raising the possibility that FHBL2 may result in a lower rate of in vivo adipose tissue lipolysis. It is possible that all of these mechanisms in concert might prevent an imbalance between the synthesis of triglycerides and ApoB within hepatocytes, thus preventing the development of excessive hepatic lipid accumulation. Further mechanistic studies are needed to establish which of these pathways is the most relevant in humans.
The data provided here are informative in predicting the potential adverse long-term hepatic effects of drugs inhibiting ANGPLT3. The development of vupanorsen, an antisense oligonucleotide targeting the hepatic production of ANGPTL3, was recently discontinued after hepatic steatosis and increases in AST or ALT were observed in a Phase 2b trial17. Our imaging and Mendelian randomization results suggest all of these adverse effects are unlikely to be due to the on-target effect of inhibiting ANGPTL3. Alternatively, it is possible that compensatory mechanisms could be induced by lifelong deficiency which might not be present with acute, relatively short-term inhibition of ANGPTL3. Regardless, therapeutically inhibiting ANGPTL3 by other approaches is not necessarily expected to result in the adverse effects observed with high doses of vupanorsen.
We also found that there were no differences across ANGPTL3 genotype groups in the distribution of fat in the abdominal cavity and skeletal muscle. This finding, which now extends to humans what was previously observed in mice43, provides definitive support to the notion that inactivating ANGPTL3 does not adversely affect body fat distribution.
Several limitations of our study deserve consideration, beginning with the small number of participants with complete ANGPTL3 deficiency due to bi-allelic LoF mutations. FHBL2 is very rare and the current study represents the largest available investigation of the direct impact of this condition on hepatic steatosis worldwide. We studied all available participants who had complete LoF ANGPTL3 mutations; although we would have liked to include more data from this population, we were unable to identify any additional complete LoF carriers for inclusion into our study. Nevertheless, the lack of association between an instrument reflecting a genetically determined lifelong reduction in plasma ANGPTL3 – a reasonable proxy of reduced hepatic protein expression and secretion of the protein since ANGPTL3 is nearly exclusively expressed in hepatocytes – with increased liver fat in a very large cohort from the UK Biobank significantly strengthens our conclusions. Finally, our study was performed while participants were fasting and ANGPTL3 appears to have important regulatory actions during the prandial state. Although this might have hypothetically limited the possibility to appreciate some effects of ANGPTL3 inactivation, we evaluated participants with lifelong inactivation of ANGPTL3 and their fat distribution is unlikely to change in response to feeding. Future kinetic studies will be needed to better clarify the interplay between FFA trafficking and lipid metabolism in the liver as well as in storage and oxidative tissues.
In summary, we have provided evidence that lifelong genetic ANGPTL3 deficiency in humans is not associated with appreciable changes in hepatic and extrahepatic fat distribution. Long term genetic ANGPTL3 inhibition appears to be safe and well tolerated, suggesting a favorable safety profile of pharmacological interventions aimed at inhibiting the activity of ANGPTL3.
Supplementary Material
Clinical Perspective.
What is New?
Partial or complete genetic deficiency of ANGPTL3 is not associated with increased hepatic fat as assessed by magnetic resonance imaging.
Mendelian randomization supports that ANGPTL3 is causally related to triglycerides and low-density lipoprotein cholesterol but not causally related to levels of hepatic fat.
What are the Clinical Implications?
Increased hepatic fat associated with vupanorsen does not appear to be the result of an on-target effect of inhibiting the hepatic production of ANGPTL3.
Therapeutic approaches to inhibit hepatic ANGPTL3 production should not generally be expected to result in increased hepatic steatosis.
Acknowledgments
We thank all research participants for their continuous support and help in our studies focused on the understanding of ANGPTL3 functioning and the consequences of its inactivation. We thank Teresa Roediger for assistance with study coordination and participant recruitment.
Funding Sources
This study was funded in part by grants from: the National Institutes of Health to NOS (R01HL159171, R01HL131961, UM1HG008853, and P01HL151328), KHB (T32GM007200 and T32HL134635), and NOD (R01DK119437 and P30DK052574); the Foundation for Barnes-Jewish Hospital to NOS; and the American Heart Association to KHB (899589). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Conflicts of Interest
NOS has received investigator-initiated research funding from Regeneron Pharmaceuticals related to ANGPTL3. MA has received research grant support from Amryth Pharmaceutical, Amgen, IONIS, Akcea Therapeutics, Daichi-Sankio, Novartis, Pfizer, and Sanofi, has served as a consultant for Amgen, Akcea Therapeutics, Daichi-Sankio, Novartis, Pfizer, Sanofi, and Alfasigma, and has received lecturing fees from Amgen, Amryth Pharmaceutical, Daichi-Sankio, Regeneron, Sanofi, and AlfaSigma. LD has received personal fees for public speaking, consultancy, or grant support from Amryt Pharmaceuticals, Akcea Therapeutics, Pfizer, SOBI, Amgen, and Sanofi.
Non-standard Abbreviations and Acronyms
- ALT
alanine aminotransferase
- ANGPTL3
Angiopoietin-like 3
- ApoB
apolipoproteinB
- AST
aspartate aminotransferase
- BMI
body mass index
- EL
endothelial lipase
- FFA
free fatty acids
- FHBL2
familial combined hypolipidemia
- HDL-C
high-density lipoprotein cholesterol
- HFF
hepatic fat fraction
- LDL-C
low-density lipoprotein cholesterol
- LoF
loss-of-function
- LPL
lipoprotein lipase
- MRI
magnetic resonance imaging
- MR-PDFF
magnetic resonance proton density fat fraction
- MR-S
magnetic resonance spectroscopy
- MTP
microsomal triglyceride transfer protein
- OGTT
oral glucose tolerance test
- SAT
subcutaneous adipose tissue
- TC
total cholesterol
- TG
triglycerides
- VAT
visceral adipose tissue
- VLDL
very-low-density lipoproteins
- WT
wild-type
References
- 1.Sylvers-Davie KL, Davies BSJ. Regulation of lipoprotein metabolism by ANGPTL3, ANGPTL4, and ANGPTL8. Am J Physiol Endocrinol Metab. 2021;321:E493–E508. doi: 10.1152/ajpendo.00195.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bini S, D’Erasmo L, Di Costanzo A, Minicocci I, Pecce V, Arca M. The Interplay between Angiopoietin-Like Proteins and Adipose Tissue: Another Piece of the Relationship between Adiposopathy and Cardiometabolic Diseases? Int J Mol Sci. 2021;22. doi: 10.3390/ijms22020742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kersten S ANGPTL3 as therapeutic target. Curr Opin Lipidol. 2021;32:335–341. doi: 10.1097/MOL.0000000000000789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arca M, D’Erasmo L, Minicocci I. Familial combined hypolipidemia: angiopoietin-like protein-3 deficiency. Curr Opin Lipidol. 2020;31:41–48. doi: 10.1097/MOL.0000000000000668 [DOI] [PubMed] [Google Scholar]
- 5.Minicocci I, Montali A, Robciuc MR, Quagliarini F, Censi V, Labbadia G, Gabiati C, Pigna G, Sepe ML, Pannozzo F, et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. Journal of Clinical Endocrinology and Metabolism. 2012;97:E1266–1275. doi: 10.1210/jc.2012-1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Musunuru K, Pirruccello JP, Do R, Peloso GM, Guiducci C, Sougnez C, Garimella KV, Fisher S, Abreu J, Barry AJ, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363:2220–2227. doi: 10.1056/NEJMoa1002926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Robciuc MR, Maranghi M, Lahikainen A, Rader D, Bensadoun A, Oorni K, Metso J, Minicocci I, Ciociola E, Ceci F, et al. Angptl3 deficiency is associated with increased insulin sensitivity, lipoprotein lipase activity, and decreased serum free fatty acids. Arterioscler Thromb Vasc Biol. 2013;33:1706–1713. doi: 10.1161/ATVBAHA.113.301397 [DOI] [PubMed] [Google Scholar]
- 8.Minicocci I, Tikka A, Poggiogalle E, Metso J, Montali A, Ceci F, Labbadia G, Fontana M, Di Costanzo A, Maranghi M, et al. Effects of angiopoietin-like protein 3 deficiency on postprandial lipid and lipoprotein metabolism. J Lipid Res. 2016;57:1097–1107. doi: 10.1194/jlr.P066183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tikkanen E, Minicocci I, Hallfors J, Di Costanzo A, D’Erasmo L, Poggiogalle E, Donini LM, Wurtz P, Jauhiainen M, Olkkonen VM, et al. Metabolomic Signature of Angiopoietin-Like Protein 3 Deficiency in Fasting and Postprandial State. Arterioscler Thromb Vasc Biol. 2019;39:665–674. doi: 10.1161/ATVBAHA.118.312021 [DOI] [PubMed] [Google Scholar]
- 10.Stitziel NO, Khera AV, Wang X, Bierhals AJ, Vourakis AC, Sperry AE, Natarajan P, Klarin D, Emdin CA, Zekavat SM, et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J Am Coll Cardiol. 2017;69:2054–2063. doi: 10.1016/j.jacc.2017.02.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dewey FE, Gusarova V, Dunbar RL, O’Dushlaine C, Schurmann C, Gottesman O, McCarthy S, Van Hout CV, Bruse S, Dansky HM, et al. Genetic and Pharmacologic Inactivation of ANGPTL3 and Cardiovascular Disease. N Engl J Med. 2017;377:211–221. doi: 10.1056/NEJMoa1612790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908–922. doi: 10.1038/s41591-018-0104-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sookoian S, Pirola CJ, Valenti L, Davidson NO. Genetic Pathways in Nonalcoholic Fatty Liver Disease: Insights From Systems Biology. Hepatology. 2020;72:330–346. doi: 10.1002/hep.31229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schonfeld G Familial hypobetalipoproteinemia: a review. J Lipid Res. 2003;44:878–883. doi: 10.1194/jlr.R300002-JLR200 [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Gusarova V, Banfi S, Gromada J, Cohen JC, Hobbs HH. Inactivation of ANGPTL3 Reduces Hepatic VLDL-triglyceride Secretion. J Lipid Res. 2015. doi: 10.1194/jlr.M054882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Di Costanzo A, Di Leo E, Noto D, Cefalu AB, Minicocci I, Polito L, D’Erasmo L, Cantisani V, Spina R, Tarugi P, et al. Clinical and biochemical characteristics of individuals with low cholesterol syndromes: A comparison between familial hypobetalipoproteinemia and familial combined hypolipidemia. J Clin Lipidol. 2017;11:1234–1242. doi: 10.1016/j.jacl.2017.06.013 [DOI] [PubMed] [Google Scholar]
- 17.Bergmark BA, Marston NA, Bramson CR, Curto M, Ramos V, Jevne A, Kuder JF, Park JG, Murphy SA, Verma S, et al. Effect of Vupanorsen on Non-High-Density Lipoprotein Cholesterol Levels in Statin-Treated Patients With Elevated Cholesterol: TRANSLATE-TIMI 70. Circulation. 2022. doi: 10.1161/CIRCULATIONAHA.122.059266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abu-Farha M, Al-Khairi I, Cherian P, Chandy B, Sriraman D, Alhubail A, Al-Refaei F, AlTerki A, Abubaker J. Increased ANGPTL3, 4 and ANGPTL8/betatrophin expression levels in obesity and T2D. Lipids Health Dis. 2016;15:181. doi: 10.1186/s12944-016-0337-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cinkajzlova A, Mraz M, Lacinova Z, Klouckova J, Kavalkova P, Kratochvilova H, Trachta P, Krizova J, Haluzikova D, Skrha J, et al. Angiopoietin-like protein 3 and 4 in obesity, type 2 diabetes mellitus, and malnutrition: the effect of weight reduction and realimentation. Nutr Diabetes. 2018;8:21. doi: 10.1038/s41387-018-0032-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Young EP, Stitziel NO. Capitalizing on Insights from Human Genetics to Identify Novel Therapeutic Targets for Coronary Artery Disease. Annu Rev Med. 2019;70:19–32. doi: 10.1146/annurev-med-041717-085853 [DOI] [PubMed] [Google Scholar]
- 21.Fazio S, Minnier J, Shapiro MD, Tsimikas S, Tarugi P, Averna MR, Arca M, Tavori H. Threshold Effects of Circulating Angiopoietin-Like 3 Levels on Plasma Lipoproteins. J Clin Endocrinol Metab. 2017;102:3340–3348. doi: 10.1210/jc.2016-4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minicocci I, Cantisani V, Poggiogalle E, Favari E, Zimetti F, Montali A, Labbadia G, Pigna G, Pannozzo F, Zannella A, et al. Functional and morphological vascular changes in subjects with familial combined hypolipidemia: an exploratory analysis. Int J Cardiol. 2013;168:4375–4378. doi: 10.1016/j.ijcard.2013.05.053 [DOI] [PubMed] [Google Scholar]
- 23.Minicocci I, Montali A, Robciuc MR, Quagliarini F, Censi V, Labbadia G, Gabiati C, Pigna G, Sepe ML, Pannozzo F, et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J Clin Endocrinol Metab. 2012;97:E1266–1275. doi: 10.1210/jc.2012-1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minicocci I, Santini S, Cantisani V, Stitziel N, Kathiresan S, Arroyo JA, Marti G, Pisciotta L, Noto D, Cefalu AB, et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: a pooled analysis. J Lipid Res. 2013;54:3481–3490. doi: 10.1194/jlr.P039875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pinzon Grimaldos A, Pacella I, Bini S, Tucci G, Cammarata I, Di Costanzo A, Minicocci I, D’Erasmo L, Arca M, Piconese S. ANGPTL3 deficiency associates with the expansion of regulatory T cells with reduced lipid content. Atherosclerosis. 2022;362:38–46. doi: 10.1016/j.atherosclerosis.2022.09.014 [DOI] [PubMed] [Google Scholar]
- 26.Saunders JB, Aasland OG, Babor TF, de la Fuente JR, Grant M. Development of the Alcohol Use Disorders Identification Test (AUDIT): WHO Collaborative Project on Early Detection of Persons with Harmful Alcohol Consumption--II. Addiction. 1993;88:791–804. doi: 10.1111/j.1360-0443.1993.tb02093.x [DOI] [PubMed] [Google Scholar]
- 27.Matsuda M, DeFronzo RA. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462 [DOI] [PubMed] [Google Scholar]
- 28.Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and beta-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. doi: 10.1007/BF00280883 [DOI] [PubMed] [Google Scholar]
- 29.Katz A, Nambi SS, Mather K, Baron AD, Follmann DA, Sullivan G, Quon MJ. Quantitative insulin sensitivity check index: a simple, accurate method for assessing insulin sensitivity in humans. J Clin Endocrinol Metab. 2000;85:2402–2410. doi: 10.1210/jcem.85.7.6661 [DOI] [PubMed] [Google Scholar]
- 30.Idilman IS, Aniktar H, Idilman R, Kabacam G, Savas B, Elhan A, Celik A, Bahar K, Karcaaltincaba M. Hepatic steatosis: quantification by proton density fat fraction with MR imaging versus liver biopsy. Radiology. 2013;267:767–775. doi: 10.1148/radiol.13121360 [DOI] [PubMed] [Google Scholar]
- 31.McPherson S, Jonsson JR, Cowin GJ, O’Rourke P, Clouston AD, Volp A, Horsfall L, Jothimani D, Fawcett J, Galloway GJ, et al. Magnetic resonance imaging and spectroscopy accurately estimate the severity of steatosis provided the stage of fibrosis is considered. J Hepatol. 2009;51:389–397. doi: 10.1016/j.jhep.2009.04.012 [DOI] [PubMed] [Google Scholar]
- 32.Ferkingstad E, Sulem P, Atlason BA, Sveinbjornsson G, Magnusson MI, Styrmisdottir EL, Gunnarsdottir K, Helgason A, Oddsson A, Halldorsson BV, et al. Large-scale integration of the plasma proteome with genetics and disease. Nat Genet. 2021;53:1712–1721. doi: 10.1038/s41588-021-00978-w [DOI] [PubMed] [Google Scholar]
- 33.Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, Burgess S, Jiang T, Paige E, Surendran P, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–79. doi: 10.1038/s41586-018-0175-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haas ME, Pirruccello JP, Friedman SN, Wang M, Emdin CA, Ajmera VH, Simon TG, Homburger JR, Guo X, Budoff M, et al. Machine learning enables new insights into genetic contributions to liver fat accumulation. Cell Genom. 2021;1. doi: 10.1016/j.xgen.2021.100066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Richardson TG, Sanderson E, Palmer TM, Ala-Korpela M, Ference BA, Davey Smith G, Holmes MV. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: A multivariable Mendelian randomisation analysis. PLoS Med. 2020;17:e1003062. doi: 10.1371/journal.pmed.1003062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sinnott-Armstrong N, Tanigawa Y, Amar D, Mars N, Benner C, Aguirre M, Venkataraman GR, Wainberg M, Ollila HM, Kiiskinen T, et al. Genetics of 35 blood and urine biomarkers in the UK Biobank. Nat Genet. 2021;53:185–194. doi: 10.1038/s41588-020-00757-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hemani G, Zheng J, Elsworth B, Wade KH, Haberland V, Baird D, Laurin C, Burgess S, Bowden J, Langdon R, et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife. 2018;7. doi: 10.7554/eLife.34408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di Martino M, Pacifico L, Bezzi M, Di Miscio R, Sacconi B, Chiesa C, Catalano C. Comparison of magnetic resonance spectroscopy, proton density fat fraction and histological analysis in the quantification of liver steatosis in children and adolescents. World J Gastroenterol. 2016;22:8812–8819. doi: 10.3748/wjg.v22.i39.8812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adam RC, Mintah IJ, Alexa-Braun CA, Shihanian LM, Lee JS, Banerjee P, Hamon SC, Kim HI, Cohen JC, Hobbs HH, et al. Angiopoietin-like protein 3 governs LDL-cholesterol levels through endothelial lipase-dependent VLDL clearance. J Lipid Res. 2020;61:1271–1286. doi: 10.1194/jlr.RA120000888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adiels M, Olofsson SO, Taskinen MR, Boren J. Overproduction of very low-density lipoproteins is the hallmark of the dyslipidemia in the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:1225–1236. doi: 10.1161/ATVBAHA.107.160192 [DOI] [PubMed] [Google Scholar]
- 41.Moller N Ketone Body, 3-Hydroxybutyrate: Minor Metabolite - Major Medical Manifestations. J Clin Endocrinol Metab. 2020;105. doi: 10.1210/clinem/dgaa370 [DOI] [PubMed] [Google Scholar]
- 42.Bini S, Pecce V, Di Costanzo A, Polito L, Ghadiri A, Minicocci I, Tambaro F, Covino S, Arca M, D’Erasmo L. The Fibrinogen-like Domain of ANGPTL3 Facilitates Lipolysis in 3T3-L1 Cells by Activating the Intracellular Erk Pathway. Biomolecules. 2022;12. doi: 10.3390/biom12040585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, McNutt MC, Banfi S, Levin MG, Holland WL, Gusarova V, Gromada J, Cohen JC, Hobbs HH. Hepatic ANGPTL3 regulates adipose tissue energy homeostasis. Proc Natl Acad Sci U S A. 2015;112:11630–11635. doi: 10.1073/pnas.1515374112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Middleton MS, Heba ER, Hooker CA, Bashir MR, Fowler KJ, Sandrasegaran K, Brunt EM, Kleiner DE, Doo E, Van Natta ML, et al. Agreement Between Magnetic Resonance Imaging Proton Density Fat Fraction Measurements and Pathologist-Assigned Steatosis Grades of Liver Biopsies From Adults With Nonalcoholic Steatohepatitis. Gastroenterology. 2017;153:753–761. doi: 10.1053/j.gastro.2017.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qu Y, Li M, Hamilton G, Zhang YN, Song B. Diagnostic accuracy of hepatic proton density fat fraction measured by magnetic resonance imaging for the evaluation of liver steatosis with histology as reference standard: a meta-analysis. Eur Radiol. 2019;29:5180–5189. doi: 10.1007/s00330-019-06071-5 [DOI] [PubMed] [Google Scholar]
- 46.Di Costanzo A, Pacifico L, D’Erasmo L, Polito L, Martino MD, Perla FM, Iezzi L, Chiesa C, Arca M. Nonalcoholic Fatty Liver Disease (NAFLD), But not Its Susceptibility Gene Variants, Influences the Decrease of Kidney Function in Overweight/Obese Children. Int J Mol Sci. 2019;20. doi: 10.3390/ijms20184444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yokoo T, Shiehmorteza M, Hamilton G, Wolfson T, Schroeder ME, Middleton MS, Bydder M, Gamst AC, Kono Y, Kuo A, et al. Estimation of hepatic proton-density fat fraction by using MR imaging at 3.0 T. Radiology. 2011;258:749–759. doi: 10.1148/radiol.10100659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boettcher M, Machann J, Stefan N, Thamer C, Haring HU, Claussen CD, Fritsche A, Schick F. Intermuscular adipose tissue (IMAT): association with other adipose tissue compartments and insulin sensitivity. J Magn Reson Imaging. 2009;29:1340–1345. doi: 10.1002/jmri.21754 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.