

Abstract
The population of cancer survivors is rapidly increasing due to improving healthcare. However, cancer therapies often have long-term side effects. One example is cancer therapy-related cardiac dysfunction (CTRCD) caused by doxorubicin: up to 9% of the cancer patients treated with this drug develop heart failure at a later stage. In recent years, doxorubicin-induced cardiotoxicity has been associated with an accelerated aging phenotype and cellular senescence in the heart. In this review we explain the evidence of an accelerated aging phenotype in the doxorubicin-treated heart by comparing it to healthy aged hearts, and shed light on treatment strategies that are proposed in pre-clinical settings. We will discuss the accelerated aging phenotype and the impact it could have in the clinic and future research.
Subject terms: Cardiovascular diseases, Senescence
Introduction
In recent years, the number of cancer survivors has increased. In Europe alone, 12 million patients were successfully treated for cancer1. Among this group, the long-term cardiac side-effects of cancer therapies are becoming evident. Between 1 and 5% of cancer survivors show signs of cancer therapy-related cardiac dysfunction (CTRCD), and 20% of these patients show asymptomatic LV function reduction2,3. In childhood cancer survivors who reached the age of 50, this percentage was even higher. The cumulative incidence of at least one symptom of cardiovascular disease was 45.5%, while this was only 15.7% in controls4,5. One of the chemotherapeutic drugs that is notorious for causing CTRCD, is doxorubicin3. Doxorubicin-induced cardiotoxicity (DCT) is defined by a decline in left ventricular ejection fraction of more than 10% to a value smaller than 53%2,6. This happens in up to 9% of the patient population treated with doxorubicin, depending on the received cumulative dose7,8.
The mechanisms underlying DCT are extensively investigated, but no conclusive evidence has been found. Proposed mechanisms include apoptosis, mitochondrial dysfunction, calcium dysregulation, inflammation, and oxidative stress8,9. One hypothesis that is supported by an increasing pool of evidence, is that doxorubicin causes a phenotype of accelerated aging, resulting to changes in the mentioned processes10–14. This is illustrated by studies in patients who were treated with doxorubicin during their childhood. These individuals show physical traits that are more similar to an older population, in terms of frailty, endurance, development of life-threatening diseases, and muscle strength10,13,15. Aging is also visible at the cellular level, where it is termed cellular senescence14,16–18.
Doxorubicin: molecular and biological characteristics of a potent cancer treatment
Doxorubicin was discovered in 1969 as a homolog of daunorubicin, and was isolated from a soil bacterium, Streptomyces peucetius8,19. It is a widely used chemotherapeutic drug to treat solid tumors, leukemia, and lymphoma both in adults and children6. The classic antitumor effect of doxorubicin is most often attributed to the inhibition of Topoisomerase IIb (TOP2B). TOP2B is an enzyme that prevents double stranded nuclear DNA breaks (DSBs) caused by stresses in the strands. These can build up due to transcription or replication20. Besides this, doxorubicin is involved in many other mechanisms directly and indirectly underlying DSBs, such as increased reactive oxygen species (ROS) production and mitochondrial dysfunction8.
Implementation of doxorubicin in cancer treatment regimens has been very beneficial, as reflected by the reduced the risk of death in ovarian cancer by 15% (HR = 0.85 [CI 0.76–0.95], p < 0.001)21. In metastatic breast cancer, doxorubicin led to a survival advantage of 13% (HR = 1.13 [CI 1.00–1.27])22. However, by this time it was also evident that doxorubicin could cause cardiotoxicity in a dose dependent manner. In a large meta-analysis, 22,815 doxorubicin-treated patients, 17.9% of these patients developed sub-clinical cardiac dysfunction, while 6.3% of them developed clinical heart failure. Cumulative dose was reported as the most important predictor of DCT23.
Many mechanisms for DCT have been proposed9. However, controversy remains due to varying doses of doxorubicin used in preclinical experiments. In human blood, the concentration after doxorubicin administration is typically between 0.025 and 0.250 µmol/L24–28, whereas in vitro experiments often apply doses of 1 µmol/L or higher. Additionally, mice can tolerate much higher doxorubicin concentrations, with tolerable blood concentrations reaching 0.7–2.1 µmol/L (10–30 mg/kg)29,30. Doxorubicin treatment has shown different pharmacological effects at different concentrations: At sub-micromolar concentrations, the primary mechanism of action is on DSB formation, while this shifts to free radical toxicity at 2–4 µmol/L19. Therefore, the accelerated aging phenotype might have been overlooked in experimental setups assessing cardiomyopathy31.
Phenotypic overlap between the aged and the doxorubicin-treated heart
As the heart ages, it undergoes many transformations, affecting morphology, electrophysiology gene expression, and metabolism, all of which have a substantial influence on cardiac output. Particularly, the heart of a patient with DCT exhibits more similarities with the heart of an aged individual than with that of healthy peers10,13,15. Both the aged and DCT heart show decreased atrial emptying rates, a measure of diastolic dysfunction31–36. This is linked to development of atrial fibrillation (AF)37–39 Incidence of AF increases with age: below the age of 55, 0.1–0.2% of the population has AF, while this number increases to 9.1–11.1% above 85 years40,41. In DCT patients (46 ± 15 years), the incidence of atrial fibrillation lies around 10%38,39. Additionally, both the aged and DCT treated heart show prolonged QT intervals, pointing at aberrant electrophysiology38,42,43.
On a morphological level, hypertrophy was observed in experimental models of both the aged44–50 and the DCT heart51–54, as well as in patients when measuring LV mass index32,55,56. When assessed microscopically, both the aged and DCT heart show accumulation of waste products, shown with lipofuscin staining and increased β-galactosidase activity18,44,57–59. Furthermore, increased fibrosis is observed (1.5x increase in the aged heart, 2.2x increase in the DCT heart18,60). In DCT mRNA expression of collagen fibers is not upregulated61, while protein levels are increased62. Besides, cardiac fibroblasts show increased expression of matrix metalloproteases (MMPs) and tissue inhibitors of MMPs (TIMPs), suggesting increased remodeling50,60,61. Inhibition of MMPs during doxorubicin treatment prevents DCT63.
Overall, there are marked similarities between aged and, often younger, DCT hearts, supporting the hypothesis that doxorubicin induces an accelerated aging phenotype.
Molecular consequences: cellular senescence as a hallmark of the aging process
The aging of the human body is accompanied by increasing numbers of senescent cells. Increased senescent cells are found in DCT models as well, for example in a transgenic model that permits the identification of cells expressing p16, a regulator of senescence64. Various cell types in the heart have been shown to become senescent upon doxorubicin treatment, including cardiomyocytes65, endothelial cells66, cardiac fibroblasts61, and cardiac progenitor cells67. While our primary focus is on cardiomyocytes, many of the processes described here are also applicable to the other cell types (Fig. 1)65.
Fig. 1. Overview of mechanisms in which both doxorubicin and healthy aging can influence heart function.

Doxorubicin induces DNA damage, but also affects epigenetics and telomere length. In the cytoplasm, doxorubicin inhibits reuptake of calcium, leading to an increased calcium concentration. In the mitochondria, doxorubicin can by itself cause increased ROS production. Besides, it inhibits the electron transport chain, which leads to additional ROS production. In the nucleus chromosomes are affected by time as well. DNA damage occurs, epigenetics are altered, and telomeres become shorter. In the cytoplasm, reuptake of calcium becomes less efficient, leading to increased calcium concentrations. Age also affects the mitochondria so that the electron transport chain becomes less efficient, and more ROS is produced. All these mechanisms lead to similar outputs in the form of hypertrophy, diastolic dysfunction, increased incidence of atrial fibrillation, prolonged QT interval, and fibrosis.
DNA damage and double-stranded break repair
DNA is a very stable molecule: intact DNA molecules of 700,000 years old have been recovered68. However, it is common knowledge that DNA damage arises on a day-to-day basis in several forms, including formation of oxidized nucleotides, single strand breaks, double strand breaks, and inter-strand crosslinks. The cell protects itself from the cytotoxic effects, by activating DNA damage response (DDR), which also consists of a range of mechanisms. These include base excision repair, non-homologous end-joining, and homologous recombination. Which of these is used depends on the type of damage and the speed and accuracy at which the DNA needs to be repaired. All these levels of protection cannot prevent accumulation of permanent mutations over time69. Doxorubicin also accelerates the DNA mutation rate. by interacting with DNA in two ways: by intercalating with the strands70–72, or by inhibiting TOP2b as mentioned above73–75. When doxorubicin intercalates with the DNA, it preferably binds to areas rich in GC basepairs76. The complex that is formed leads to supercoiling of the DNA strands, which results in increased stresses and the unwrapping of the DNA from the nucleosome77. The relevance of this mechanism in patients can be questioned, since only 4–5 adducts form every 107 base pairs at clinically relevant doses76. There is more evidence for the activity of the second mechanism within patients, inhibition of TOP2b. Doxorubicin can influence TOP2b in two ways: it can prevent the protein from binding to the DNA or it can inhibit the final ligation step74,78,79. Inhibition of binding is due to the intercalation of doxorubicin with the DNA, but this does not happen in relevant frequency at clinical doses74. Thus, the second mechanism is a more likely candidate. When TOP2b is knocked out both in vitro73,74 and in vivo75, DCT can be prevented74–76. Additional confirmation of role of TOP2b in DCT comes from the use of dexrazoxane. Dexrazoxane administration simultaneously with doxorubicin has shown reduction of DCT incidence. Initially, it was believed that dexrazoxane mitigated DCT through iron chelation, but recent research indicates that it depletes TOP2a and TOP2b from tissues, thereby protecting the heart from toxicity67,80.
DNA damage activates senescence-related pathways. The first proteins that react to DNA damage are Ataxia Telangiectasia Mutated (ATM), Ataxia Telangiectasia and Rad3-Related Protein (ATR), and Checkpoint Kinase 1 (Chk1) and 2 (Chk2). ATM-Chk2 is activated by double stranded breaks, while ATR-Chk1 reacts to single stranded breaks81. Inhibition of the cell cycle is accomplished through p53. In the healthy cell, p53 is a short-lived protein, which is rapidly ubiquitinated by Mouse Double Minute 2 homolog (MDM2) and MDM4 and degraded. ATM-Chk2 and ATR-Chk1 affect p53 in multiple ways. First, they stabilize it by inhibiting MDM2 and MDM4. Secondly, they also phosphorylate p5382. Once phosphorylated, p53 can either activate p53 upregulated modulator of apoptosis (PUMA) and NOXA, which are apoptosis regulators, or p21, which causes cellular senescence69. How it is determined which pathway is activated is not fully understood82, but Forkhead box protein O4 (FOXO4) is believed to be involved. Disruption of p53-FOXO4 complexes with a peptide causes senescent cells to go into apoptosis83. Another important senescence marker that activates upon DNA stress, independently from p53, is p16. Although postnatal cardiomyocytes are in cell cycle arrest at the G1/S restriction point, they show upregulation of these pathways regardless14,16,18 (Fig. 2).
Fig. 2. Pathways involved in DNA damage.
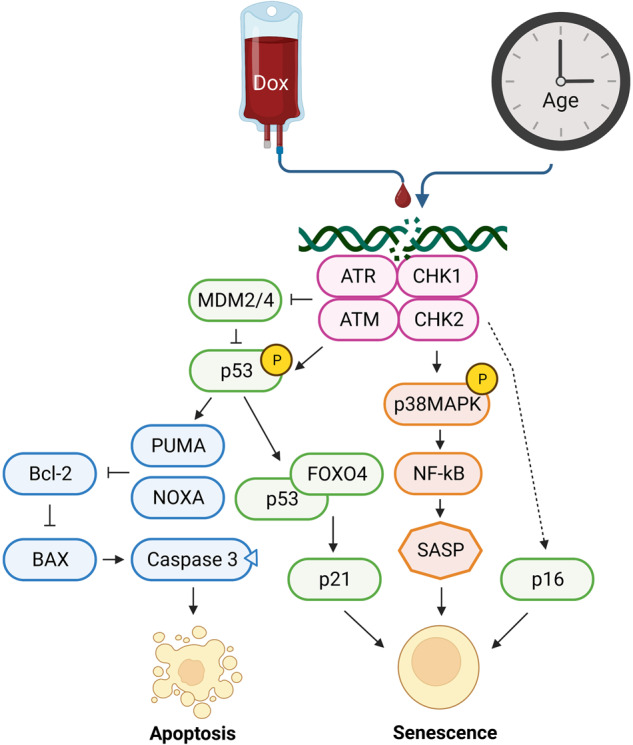
After induction of DNA damage, ATR-CHK1 and ATM-CHK2 are recruited to the damaged position. These lead to an activation and increased presence of p53, by inhibiting MDM2/4. Activated p53 in turn either binds to FOXO4, inducing increased expression of p21, or interacts with PUMA and NOXA, which activates the apoptosis cascade. Besides activation of p53, ATR-CHK1 and ATM-CHK2 are also responsible for phosphorylation of p38MAPK and transcription of p16. These two proteins activate senescence in the cell.
Another regulator of senescence is p38MAPK. It can be activated by DNA damage, oxidative stress, ER stress, metabolic stress, and inflammatory stress. When activated, it can drive many other pathways, including p53, p21 (independent of p53), p16, and NF-kB pathways84. Treatment with doxorubicin has been shown to activate p38MAPK14,85.
Telomeres
In addition to DNA damage, aging correlates with telomere shortening. To replicate DNA, DNA polymerase needs an RNA primer as a starting position. At the ends of the DNA strands, this results in the loss of basepairs with each replication cycle (the end replication problem). To protect the genetic material, the ends of chromosomes consist of telomeres. These are repeats of a random sequence, packaged in protein complexes known as shelterin. Shelterin prevents the recognition of chromosome ends as DNA damage45. With every replication, the telomeres become shorter, until they reach a critical length that disrupts the shelterin complex. This activates a sustained DDR and the cell stops proliferating. Telomerase can lengthen the telomeres again, but it is not expressed in all celltypes86. In a study of 530 autopsy subjects, it was shown that telomeres in the heart shorten at a rate of approximately 20 basepairs per year87. With age, an increase in DDR proteins was observed in the telomeres, independent from shortening45,88. In doxorubicin-treated human18 and mouse16 hearts, telomeres were shorter compared to controls. Telomere binding proteins89 and telomerase16 were also shown to be dysregulated in doxorubicin-treated hearts16.
Epigenetics
DNA in the nucleus is wrapped around proteins (histones) and organized into chromosomes. Modification of packaging can control gene expression: tightly packed DNA cannot be transcribed (heterochromatin), while loosely organized DNA is available for transcription (euchromatin). The tightness of packaging is adjusted by modifications of the histones and of the DNA itself. Acetylation promotes formation of euchromatin, while methylation promotes heterochromatin90. Aging reduces histone levels and when they are experimentally elevated, lifespan is increased91. Doxorubicin treatment causes nucleosome turnover around the promoters of active genes77 and histone eviction and degradation92, resulting in decreased cellular histone levels.
DNA bases can be methylated, mostly in CpG islands: stretches of DNA less than 200 basepairs long, which contain >50% C-G pairs. CpG islands are often located in proximity to promotors. More methylated bases results in decreased gene expression93. Aging leads to an overall decrease of DNA methylation93. Doxorubicin decreases expression of DNA methyltransferases94, and methylation levels95,96. Methylation levels can be used to determine biological age, which has been linked to cardiovascular health96–98. Although DNA methylation in doxorubicin-treated models has been measured, biological age has not been assessed.
Epigenetic regulation ties gene expression to metabolism. During aging and doxorubicin treatment availability of substrates such as ATP, NAD+, and acetyl co-enzyme declines, resulting in heterochromatin95,99. Sirtuins, NAD+ dependent histone deacetylases, are regulators of this process. When there is less energy, more NAD+ is available, and Sirtiuns deacetylate histones, leading to heterochromatin100. The involvement of these proteins in aging and DCT has been studied extensively. Overexpression of several Sirtuins increased lifespan, while knockout decreased lifespan100,101. Several studies showed increased activity of Sirtuins protects the heart from DCT51,85,102–104.
Mitochondrial dysfunction and mtDNA damage
Due to high energy demand, 40% of the cardiomyocyte’s cytoplasm is occupied by mitochondria105, Mitochondrial dysfunction, a primary characteristic of senescence, therefore plays a key role in cardiomyocytes. Improving mitochondrial function can suppress senescence65. Mitochondria have an inner (IMM) and an outer (OMM) mitochondrial membrane. In the IMM, the electron transport chain (ETC) is present consisting of four complexes (complex I-IV) that use NADH and oxygen to increase the proton content in the intermembrane compartment. ATP synthase (complex V) uses this gradient to phosphorylate ADP into ATP (oxidative phosphorylation). The IMM is folded into cristae, to increase the surface area available for oxidative phosphorylation106. During aging, organization of these cristae is disrupted107,108 and the activity of the ETC is decreased106,109. Both are a result of changing phospholipid content of the IMM and the resulting decreased membrane fluidity. This disrupts the organization of the ETC complexes, interrupting efficient function106. Cardiolipin, a phospholipid exclusive to the IMM, is a likely candidate. Its conical structure enables the membrane to form curves109. Cardiolipin content decreases with age and experimental addition of it to aged mitochondria increased function109,110. Doxorubicin accumulates in mitochondria, partially by binding to cardiolipin111,112. The phospholipid has an anionic charge, while doxorubicin has a cationic charge, resulting in the formation of acomplex9,113. Similar to aged-induced dysfunction, this bond disrupts the organization of the cristae104,114–116 and inhibits proper function of ETC117. Other processes that are affected in aged cells include a shift to glycolysis, increased proton leak, decreased membrane potential, and increased ROS production58,108,118,119. These changes also take place in doxorubicin-treated cells51,53,54,95,104,114,116,120–124. In senescent fibroblasts, mitochondrial function decreased, possibly due to impaired mitophagy125. Doxorubicin-treated fibroblasts show a similar phenotype, possibly mediated by p5361. Mitochondria have small circular mitochondrial DNA (mtDNA) strands. mtDNA encodes a few proteins, which all are involved in ATP-production. There are less repair mechanisms in place for mtDNA, therefore the mutation rate is 10–17 times higher than in nuclear DNA95. Over time, mutation in the mtDNA accumulate, resulting in mitochondrial dysfunction106. The role of mtDNA in aging was confirmed in a mouse-model expressing a defective mitochondrial DNA polymerase: it showed an accelerated aging phenotype119,126. Doxorubicin is a molecule that can interact with DNA. It increases mutation rate and dysfunction of mitochondria95,104.
Reactive oxygen species and oxidative stress
Mitochondria are a source of free radicals such as ROS. In a healthy cell, there is a balance between the production of free radicals and the activity of antioxidant enzymes and molecules. However, when this balance is disrupted, the cell will enter a state of oxidative stress65. The heart is particularly vulnerable to oxidative stress, due to high metabolic activity, production of radicals for signaling purposes, and low amounts of antioxidants and antioxidant enzymes19,117,127. The presence of free radicals causes sulfhydryl oxidation, lipid peroxidation, cardiolipin reduction, and mtDNA damage in the mitochondria. This leads to a further increase in free radical production. Free radicals can cross over to organelles that are in close proximity, such as the endoplasmatic reticulum. The cell will respond by uncoupling the oxidative phosphorylation from ATP production (proton leak), increasing activity of the ETC complexes106,117. It has been shown that both aging processes and doxorubicin treatment increases ROS production in cardiomyocytes19,128 and cardiac fibroblasts61,129.
During aging, the activity of the ETC complexes decreases. This increases ROS molecule production106. In doxorubicin-treated cells, activity of the ETC also decreases with similar results. Besides that, doxorubicin is a quinone molecule which can be reduced by several enzymes, including cytosolic xanthine oxidase, NADH dehydrogenase (complex I), and NADPH-dependent cytochrome P450 reductases. After reduction, it auto-oxidizes to its neutral state. During this second step, a superoxide anion is generated130,131. Doxorubicin also reduces expression of antioxidant enzymes65,132.
Overall, it can be concluded that ROS production increases in both aging cells and in doxorubicin-treated cells. In recent years, several studies suggest that increasing ROS may not be the causative factor of both aging19 and DCT133. While suppression of ROS production is beneficial in aged animal models134, in the healthy aged population suppression of ROS production decreased lifespan in some cases, possibly due to the signaling function some of these molecules have135–137. With doxorubicin treatment, similar objections have been suggested. Firstly, free radicals do not form immediately after treatment19. Antioxidant enzymes could not prevent doxorubicin-induced DNA damage, indicating another mechanism74. Secondly, clinical trials showed that antioxidants could not prevent the accelerated aging phenotype in DCT patients19,138,139, while in animals upregulation of antioxidant enzymes has a beneficial effect48,127. Lastly, it remains unclear whether antioxidant therapy influences the cancer efficacy of doxorubicin, rendering it impractical for clinical use140.
Calcium flux
Calcium regulates contraction of cardiomyocytes. During an action potential, Ryanodine Receptor 2 (RyR2) releases calcium from the sarcoplasmic reticulum (SR). This calcium spike activates the sarcomeres, and the cell shortens. Subsequently, SR Ca2+-stimulated ATPase (SERCA) pumps calcium back into the SR and the cell relaxes. In both aged and doxorubicin-treated mice the amount of phosphorylated troponin I increased, indicating that relaxation of the cardiomyocyte is impaired16. In human aged cells, calcium concentration remains high for longer and relaxation is slower44,141. RyR2 and SERCA are responsible for these changes. The likelihood of RyR2 being in its open conformation increases with age (leaking), while it is activated by doxorubicin treatment142–144. SERCA activity has been shown to be decreased both with age and doxorubicin treatment123,144. This is partially due to decreased phosphorylation of phospholamban, an inhibitor of SERCA, and decreased ATP content123.
Involvement of the immune system in aging and DCT
The immune system’s response to aging and DCT illustrates a complex interplay, characterized by the secretion of chemokines, cytokines, and growth factors, collectively forming the senescence-associated secretory phenotype (SASP). During aging, the upregulation of pro-inflammatory cytokines such as IL-6 and TNF-α, alongside chemokines like CCL2 and CXCL12, establishes a chronic inflammatory state within the heart145,146. This inflammatory environment induces cellular stress and fosters cardiac cell senescence, contributing to the decline in cardiac function.
In the context of DCT, a similar pro-inflammatory response is demonstrated, marked by the elevation of SASP factors. Studies have demonstrated that expression of cytokines such as IFN-γ, CCL27, and MIF is increased in patients undergoing doxorubicin treatment, linking them to the DCT process64,147–149. This response is part of a broader systemic reaction, where the immune system attempts to mitigate the damage caused by the chemotherapeutic agent but instead contributes to the deterioration of cardiac tissue.
The role of growth factors within the SASP, such as TGF-β and PDGF, is critically involved in cardiac remodeling and fibrosis, processes that are prominent in both aging and DCT64,150. The presence of increased SASP production both in DCT and aging underscores the concept of an accelerated aging phenotype in DCT. This similarity in cytokine, chemokine, and growth factor expression and activity between aging and DCT suggests shared mechanisms, offering potential therapeutic targets or biomarkers151,152. Modulating the inflammatory response could present new strategies for mitigating the effects of both aging and DCT. For example, Zymosan A has been shown to improve cardiac healing and ameliorate doxorubicin-induced ventricular remodeling. This was achieved by a heightened cardiac inflammatory response, leading to enhanced repair in DCT mice153–155.
Cardiac senescence in clinical practice
Senescence as a biomarker
Senescence could be used as a biomarker for predicting outcomes of DCT. Senescent cells secrete SASP factors, which can be detected in the blood. Several SASP factors have already been linked to worse prognosis in cardiovascular disease. Examples are IGFBP7156,157, interleukin-6 (IL-6)158,159, and GDF-1565,160. Patients with chronic heart failure (BIOSTAT-CHF) with elevated levels of IGFBP7 showed a 44% (HR = 1.44 [CI 1.23–1.70], p < 0.001) increase in combined adverse endpoints156. Increased IL-6 levels have also been reported in women who experienced cardiovascular events versus healthy women (1.65 versus 1.30 pg/ml, p = 0.003)159. GDF-15 is upregulated upon doxorubicin treatment14 and has been proposed as a cardiomyocyte-specific SASP factor65. Higher levels of GDF-15 in the blood predict worse outcome: A significant correlation between GDF-15 and death (HR = 1.66 [SD 1.07–1.26] p < 0.001), heart failure (HR = 1.52 [SD 1.29–1.78], p < 0.001), and major cardiovascular events (HR = 1.26 [SD 1.14–1.41], p < 0.001) was found160. However, large trials investigating SASP levels in the blood of patients treated with doxorubicin have not been conducted.
Senotherapeutics
Research has indicated that a promising approach to protect the heart against DCT involves the removal of senescent cells. By employing a transgenic model that enables the targeted elimination of p16-expressing cells, cardiac function was successfully restored64. Consequently, two classes of drugs, known as senolytics and senomorphics, hold potential for both DCT treatment and the broader context of combating aging. Senolytics cause cell death of senescent cells specifically, while senomorphics prevent cells from becoming senescent by modulating SASP expression.
Senescent cells can resist apoptosis by upregulating anti-apoptotic genes such as Bcl-2129. Senolytics target this phenotype to remove senescent cells from a tissue. This has beneficial effects on health-span in general161. The first senolytic compounds to be discovered, were dasatinib and quercetin. When administered together, heart function in 24-month-old mice was improved162, but they were never tested in DCT. Another compound that was designed to inhibit anti-apoptotic proteins is navitoclax163. Navitoclax is being investigated in clinical trials as an anti-tumor drug for multiple cancer types and the results are promising164. It has also been shown to improve cardiac function in doxorubicin-treated mice165. FOXO4-DRI is a senolytic that was designed to disrupt p53-FOXO4 complexes, causing p53 to initiate apoptosis instead of senescence83. This peptide can protect against doxorubicin-induced liver damage, but its effect on DCT has not been investigated83. Fisetin is a bioflavonoid that can be found in many types of plants and fruits, including strawberries and apples. It has senolytic properties and increases healthspan166. In doxorubicin-treated rats, it has a beneficial effect on heart function167.
Senomorphics include a wide range of compounds, many of them natural products. They affect the cells in multiple ways, making their exact mechanism of action difficult to establish161,168. Several have been shown to be effective against DCT in animal models: resveratrol169, metformin170, PJ34171, and 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR)172. For example, rats that were co-treated with doxorubicin and AICAR had a preserved ejection fraction after six weeks172. Human data on the efficacy of these compounds in DCT is scarce and mostly based on in vitro research173–175. The effect of resveratrol in combination with other compounds on cardiovascular disease has been investigated. The results of these trials were marginally positive, but the effect of resveratrol alone was not assessed173. Besides, conventional heart failure therapies have limited efficacy in DCT patients176–178. Therefore, the effect of resveratrol on DCT could differ from what is observed in other cardiovascular etiologies. This was supported by a recent study from our lab. iPSC-CM-based dynamic engineered heart tissues were subjected to four clinically relevant doses of doxorubicin over the course of four weeks and co-treated either with resveratrol or AICAR. Neither resveratrol nor AICAR was able to improve the phenotype induced by doxorubicin. It was suggested that prevention of senescence resulted in apoptosis and ultimately in worsening of the function in the case of the AICAR treated tissues14. This indicates that treatment with senomorphics in DCT could be detrimental and that preclinical experiments using animals might not be accurate predictors of the outcome in humans.
Future perspectives
Further research into the underlying mechanisms linking cardiovascular disease and cancer is one of the Gaps in Evidence highlighted by the ESC 2022 Guidelines on Cardio-Oncology. In this review, we have discussed the evidence of an accelerated aging phenotype in the hearts of DCT patients. A comparison was made between the healthy aged heart and the doxorubicin-treated heart. On a macroscopic level, there were some similarities, such as hypertrophy and fibrosis. Most resemblances were found at cell level, where we described an increased number of mutations, upregulation of senescence markers, shorter telomeres, declining histone levels, decreased DNA methylation, decreased Sirtuin activity, less availability of substrates for epigenetic modifications, decreased energy production, increased mtDNA damage, increased ROS production, and dysfunctional calcium handling. Doxorubicin was in all instances able to initiate mechanisms that would otherwise happen gradually with time, suggesting doxorubicin treatment leads to an accelerated aging phenotype. Although this review discusses most well-known processes involved in both aging and doxorubicin treatment, not all are elaborately explained. Mechanisms for which less evidence exists, or for which evidence is controversial were omitted, for example autophagy49,179,180, and inflammation45,59.
More research on the involvement of senescence in DCT is merited. However, special care should be taken in selecting the experimental model. Rats and mice are much more resilient to doxorubicin than humans, meaning that higher dosages of doxorubicin have to be administered. It is known that different concentrations of doxorubicin activate separate mechanisms, suggesting that DCT in mice and rats might have a different origin than in humans. Besides that, we have seen that the effect of senolytics is positive in mice and rats with DCT, while it worsens function in human 3D models. Parallel to this, it has been observed that antioxidant molecules have a beneficial effect on experimental animal models of DCT and not in clinical trials. This discrepancy could be due a difference in underlying mechanism, or to higher regeneration rates of cardiomyocytes in mice (1.3–4%181) compared to humans (0.3–1%182).
All this knowledge is instrumental in developing cardioprotective strategies. Numerous avenues are being explored to protect the heart from DCT, including the use of dexrazoxane80, honokiol104, and curcumin183, many of which target either mitochondrial function or inflammation. Another cardioprotective strategy involves co-treating with doxorubicin and SGLT-2 inhibitors. A retrospective study including 32 patients who had received doxorubicin and SGLT-2 inhibitors showed no development of DCT184. In a pre-clinical model of DCT, empagliflozin showed decreased fibrosis, decreased inflammation, decreased oxidative stress, decreased lipid peroxidation, and increased mitochondrial biogenesis and left ventricular function185.
All this evidence taken together suggests that the heart undergoes an accelerated aging phenotype after doxorubicin exposure. Since many differences between the human heart and the heart of animals are observed, this should be taken into account when designing new experiments.
Acknowledgements
A.N.L. and P.v.d.M. are supported by the European Research Council (StG 715732 and CoG. 101045236; DISSECT-HF to P.v.d.M.). C.G.T. is supported by two grants from the Italian Ministry of Health (PNRR-MAD-2022-12376632 and RF-2016-02362988). Both figures were created in BioRender.com.
Author contributions
A.N.L., I.B.D. and N.B.: Conceptualization, collecting references, writing. P.v.d.M., T.L.F. and C.G.T.: Conceptualization.
Competing interests
The University Medical Center Groningen, which employs several of the authors, has received research grants and/or fees from AstraZeneca, Vifor Pharma, Pharmacosmos, Pharma Nord, Ionis, Abbott, Bristol Myers Squibb, Novartis, Novo Nordisk, and Roche. P.v.d.M. has received consultancy fees and/or grants from Novartis, Pharmacosmos, Vifor Pharma, AstraZeneca, Pfizer, Pharma Nord, BridgeBio, Novo Nordisk, and Ionis, all paid to the institution. C.G.T. reports honoraria or consultation fees from VivaLyfe, Univers Formazione, Solaris, Summeet, Astra Zeneca, Myocardial Solutions; funding from Amgen and MSD; listed as an inventor of two patents related to HF, outside the submitted work. All other authors have reported that they have no relationships relevant to the contents of this paper to disclose.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ahrenkilde Hansen, P. A cancer plan for Europe. https://ec.europa.eu/info/strategy/priorities-2019-2024/promoting-our-european-way-life/european-health-union/cancer-plan-europe_en#relatedlinks (2022).
- 2.Spetz J, Moslehi J, Sarosiek K. Radiation-induced cardiovascular toxicity: mechanisms, prevention, and treatment. Curr. Treat. Options Cardiovasc. Med. 2018;20:1–1. doi: 10.1007/s11936-018-0627-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lyon AR, et al. ESC guidelines on cardio-oncology developed in collaboration with the European Hematology Association (EHA), the European Society for Therapeutic Radiology and Oncology (ESTRO) and the International Cardio-Oncology Society (IC-OS) Eur. Heart J. 2022;43:4229–4361. doi: 10.1093/eurheartj/ehac244. [DOI] [PubMed] [Google Scholar]
- 4.Bhakta N, et al. Cumulative burden of cardiovascular morbidity in paediatric, adolescent, and young adult survivors of Hodgkin’s lymphoma: an analysis from the St Jude Lifetime Cohort Study. Lancet Oncol. 2016;17:1325–1334. doi: 10.1016/S1470-2045(16)30215-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ehrhardt MJ, et al. Systematic review and updated recommendations for cardiomyopathy surveillance for survivors of childhood, adolescent, and young adult cancer from the International Late Effects of Childhood Cancer Guideline Harmonization Group. Lancet Oncol. 2023;24:e108–e120. doi: 10.1016/S1470-2045(23)00012-8. [DOI] [PubMed] [Google Scholar]
- 6.Godishala A, Yang S, Asnani A. Cardioprotection in the modern era of cancer chemotherapy. Cardiol. Rev. 2018;26:113–121. doi: 10.1097/CRD.0000000000000194. [DOI] [PubMed] [Google Scholar]
- 7.Cardinale D, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. 2015;131:1981–1988. doi: 10.1161/CIRCULATIONAHA.114.013777. [DOI] [PubMed] [Google Scholar]
- 8.van der Zanden SY, Qiao X, Neefjes J. New insights into the activities and toxicities of the old anticancer drug doxorubicin. FEBS J. 2021;288:6095–6111. doi: 10.1111/febs.15583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rawat PS, Jaiswal A, Khurana A, Bhatti JS, Navik U. Doxorubicin-induced cardiotoxicity: an update on the molecular mechanism and novel therapeutic strategies for effective management. Biomed. Pharmacother. 2021;139:1–14. doi: 10.1016/j.biopha.2021.111708. [DOI] [PubMed] [Google Scholar]
- 10.Cupit-Link MC, et al. Biology of premature ageing in survivors of cancer. ESMO Open. 2017;2:1–8. doi: 10.1136/esmoopen-2017-000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serrano M, Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nat. Rev. Mol. Cell Biol. 2007;8:715–722. doi: 10.1038/nrm2242. [DOI] [PubMed] [Google Scholar]
- 12.Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448:767–774. doi: 10.1038/nature05985. [DOI] [PubMed] [Google Scholar]
- 13.Henderson TO, Ness KK, Cohen HJ. Accelerated aging among cancer survivors: from pediatrics to geriatrics. Am. Soc. Clin. Oncol. Educ. Book. 2014;34:e423–e430. doi: 10.14694/EdBook_AM.2014.34.e423. [DOI] [PubMed] [Google Scholar]
- 14.Linders, A. N. et al. Evaluation of Senescence and its prevention in doxorubicin-induced cardiotoxicity using dynamic engineered heart tissues. JACC Cardio Oncol.10.1016/j.jaccao.2023.03.012 (2023). [DOI] [PMC free article] [PubMed]
- 15.Lipshultz SE, et al. Late cardiac effects of doxorubicin therapy for acute lymphoblastic leukemia in childhood. N. Eng. J. Med. 1991;324:808–815. doi: 10.1056/NEJM199103213241205. [DOI] [PubMed] [Google Scholar]
- 16.Maejima Y, Adachi S, Ito H, Hirao K, Isobe M. Induction of premature senescence in cardiomyocytes by doxorubicin as a novel mechanism of myocardial damage. Aging Cell. 2008;7:125–136. doi: 10.1111/j.1474-9726.2007.00358.x. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu I, Minamino T. Cellular senescence in cardiac diseases. J. Cardiol. 2019;74:313–319. doi: 10.1016/j.jjcc.2019.05.002. [DOI] [PubMed] [Google Scholar]
- 18.Piegari, E. et al Doxorubicin induces senescence and impairs function of human cardiac progenitor cells. 10.1007/s00395-013-0334-4 (2013). [DOI] [PubMed]
- 19.Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 20.Pommier Y, Leo E, Zhang H, Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.A’hern, R. P. & Gore, M. E. Impact of doxorubicin on survival in advanced ovarian cancer. J. Clin. Oncol. 13, 726–732 (1995). [DOI] [PubMed]
- 22.Fossati, R. et al. Cytotoxic and hormonal treatment for metastatic breast cancer: a systematic review of published randomized trials involving 31,510 women. Natl. Health Serv. Executive.16, 3439–3460 (1998). [DOI] [PubMed]
- 23.Lotrionte, M. et al. Review and meta-analysis of incidence and clinical predictors of anthracycline cardiotoxicity. Am. J. Cardiol. 112, 1980–1984 Preprint at 10.1016/j.amjcard.2013.08.026 (2013). [DOI] [PubMed]
- 24.Speth P, Linssen P, Boezeman J, Wessels H, Haanen C. Cellular and plasma adriamycin concentrations in long-term infusion therapy of leukemia patients. Cancer Chemother. Pharmacol. 1987;20:305–310. doi: 10.1007/BF00262581. [DOI] [PubMed] [Google Scholar]
- 25.Muller C, et al. Cellular pharmacokinetics of doxorubicin in patients with chronic lymphocytic leukemia: comparison of bolus administration and continuous infusion. Cancer Chemother. Pharmacol. 1993;32:379–384. doi: 10.1007/BF00735923. [DOI] [PubMed] [Google Scholar]
- 26.Greene RF, Collins JM, Jenkins JF, Speyer JL, Myers CE. Plasma pharmacokinetics of adriamycin and adriamycinol: implications for the design of in vitro experiments and treatment protocols. Cancer Res. 1983;43:3417–3421. [PubMed] [Google Scholar]
- 27.Gunvén P, Theve NO, Peterson C. Serum and tissue concentrations of doxorubicin after IV administration of doxorubicin or doxorubicin-DNA complex to patients with gastrointestinal cancer. Cancer Chemother. Pharmacol. 1986;17:153–156. doi: 10.1007/BF00306745. [DOI] [PubMed] [Google Scholar]
- 28.Rodvold KA, Rushing DA, Tewksbury DA. Doxorubicin clearance in the obese. J. Clin. Oncol. 1988;6:1321–1327. doi: 10.1200/JCO.1988.6.8.1321. [DOI] [PubMed] [Google Scholar]
- 29.Sturgeon K, et al. Concomitant low-dose doxorubicin treatment and exercise. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014;307:R685–R692. doi: 10.1152/ajpregu.00082.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Choi WG, et al. Liquid chromatography-tandem mass spectrometry for the simultaneous determination of doxorubicin and its metabolites doxorubicinol, doxorubicinone, doxorubicinolone, and 7-deoxydoxorubicinone in mouse plasma. Molecules. 2020;25:1–13. doi: 10.3390/molecules25051254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi J, Guo Y, Cheng L, Song F, Shu X. Early change in left atrial function in patients treated with anthracyclines assessed by real-time three-dimensional echocardiography. Sci. Rep. 2016;6:25512. doi: 10.1038/srep25512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerstenblith G, et al. Echocardiographic assessment of a normal adult aging population. Circulation. 1977;56:273–278. doi: 10.1161/01.CIR.56.2.273. [DOI] [PubMed] [Google Scholar]
- 33.Cheng S, et al. Age-related left ventricular remodeling and associated risk for cardiovascular outcomes the multi-ethnic study of atherosclerosis. Circ. Cardiovasc. Imaging. 2009;2:191–198. doi: 10.1161/CIRCIMAGING.108.819938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Grootel RWJ, et al. In-depth echocardiographic analysis of left atrial function in healthy adults using speckle tracking echocardiography and volumetric analysis. Echocardiography. 2018;35:1956–1965. doi: 10.1111/echo.14174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hausdorf G, et al. Long term doxorubicin cardiotoxicity in childhood: non-invasive evaluation of the contractile state and diastolic filling. Heart. 1988;60:309–315. doi: 10.1136/hrt.60.4.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yaylali, Y. T., Saricopur, A., Yurtdas, M., Senol, H. & Gokoz-Dogu, G. Atrial function in patients with breast cancer after treatment with Anthracyclines. Arq. Bras. Cardiol.10.5935/abc.20160146 (2016). [DOI] [PMC free article] [PubMed]
- 37.Roberts JD, et al. Epigenetic age and the risk of incident atrial fibrillation. Circulation. 2021;144:1899–1911. doi: 10.1161/CIRCULATIONAHA.121.056456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lefrak EA, Pitha J, Rosenheim S, Gottlieb JA. A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer. 1973;32:302–314. doi: 10.1002/1097-0142(197308)32:2<302::AID-CNCR2820320205>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 39.Kilickap S, et al. Early and late arrhythmogenic effects of doxorubicin. South Med. J. 2007;100:262–265. doi: 10.1097/01.smj.0000257382.89910.fe. [DOI] [PubMed] [Google Scholar]
- 40.Go AS, et al. Prevalence of diagnosed atrial fibrillation in adults. JAMA. 2001;285:2370. doi: 10.1001/jama.285.18.2370. [DOI] [PubMed] [Google Scholar]
- 41.Nattel S. Aging and protein kinase activation is it the missing link between age and atrial fibrillation? Circ. Res. 2018;122:799–801. doi: 10.1161/CIRCRESAHA.118.312786. [DOI] [PubMed] [Google Scholar]
- 42.Rabkin SW. Aging effects on QT interval: implications for cardiac safety of antipsychotic drugs. J. Geriatr. Cardiol. 2014;11:20–25. doi: 10.3969/j.issn.1671-5411.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rabkin SW, Cheng XBJ, Thompson DJS. Detailed analysis of the impact of age on the QT interval. J. Geriatr. Cardiol. 2016;13:740–748. doi: 10.11909/j.issn.1671-5411.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Acun A, Nguyen TD, Zorlutuna P. In vitro aged, hiPSC-origin engineered heart tissue models with age-dependent functional deterioration to study myocardial infarction. Acta Biomater. 2019;94:372–391. doi: 10.1016/j.actbio.2019.05.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson R, et al. Length‐independent telomere damage drives post‐mitotic cardiomyocyte senescence. EMBO J. 2019;38:e100492. doi: 10.15252/embj.2018100492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lieber SC, et al. Aging increases stiffness of cardiac myocytes measured by atomic force microscopy nanoindentation. Am. J. Physiol.-Heart Circ. Physiol. 2004;287:H645–H651. doi: 10.1152/ajpheart.00564.2003. [DOI] [PubMed] [Google Scholar]
- 47.Hua Y, et al. Chronic akt activation accentuates aging-induced cardiac hypertrophy and myocardial contractile dysfunction: role of autophagy. Basic Res. Cardiol. 2011;106:1173–1191. doi: 10.1007/s00395-011-0222-8. [DOI] [PubMed] [Google Scholar]
- 48.Dai DF, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Häseli S, Deubel S, Jung T, Grune T, Ott C. Cardiomyocyte contractility and autophagy in a premature senescence model of cardiac aging. Oxid. Med. Cell. Longev. 2020;2020:1–14. doi: 10.1155/2020/8141307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Horn MA, Trafford AW. Aging and the cardiac collagen matrix: novel mediators of fibrotic remodelling. J. Mol. Cell. Cardiol. 2016;93:175–185. doi: 10.1016/j.yjmcc.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Du Q, Zhu B, Zhai Q, Yu B. Sirt3 attenuates doxorubicin-induced cardiac hypertrophy and mitochondrial dysfunction via suppression of Bnip3. Am. J. Transl. Res. 2017;9:3360–3373. [PMC free article] [PubMed] [Google Scholar]
- 52.Shi J, Surma M, Wei L. Disruption of ROCK1 gene restores autophagic flux and mitigates doxorubicin-induced cardiotoxicity. Oncotarget. 2018;9:12995–13008. doi: 10.18632/oncotarget.24457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burridge PW, et al. Human induced pluripotent stem cell–derived cardiomyocytes recapitulate the predilection of breast cancer patients to doxorubicin-induced cardiotoxicity. Nat. Med. 2016;22:547–556. doi: 10.1038/nm.4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adamcova M, Skarkova V, Seifertova J, Rudolf E. Cardiac troponins are among targets of doxorubicin-induced cardiotoxicity in hiPCS-CMs. Int. J. Mol. Sci. 2019;20:2638. doi: 10.3390/ijms20112638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hees PS, Fleg JL, Lakatta EG, Shapiro EP. Left ventricular remodeling with age in normal men versus women: novel insights using three-dimensional magnetic resonance imaging. Am. J. Cardiol. 2002;90:1231–1236. doi: 10.1016/S0002-9149(02)02840-0. [DOI] [PubMed] [Google Scholar]
- 56.Tan TC, et al. Time trends of left ventricular ejection fraction and myocardial deformation indices in a cohort of women with breast cancer treated with Anthracyclines, Taxanes, and Trastuzumab. J. Am. Soc. Echocardiogr. 2015;28:509–514. doi: 10.1016/j.echo.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 57.Kakimoto Y, et al. Myocardial lipofuscin accumulation in ageing and sudden cardiac death. Sci. Rep. 2019;9:3304. doi: 10.1038/s41598-019-40250-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kim YY, et al. Anti-aging effects of vitamin C on human pluripotent stem cell-derived cardiomyocytes. Age. 2012;35:1545–1557. doi: 10.1007/s11357-012-9457-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lewis-McDougall FC, et al. Aged‐senescent cells contribute to impaired heart regeneration. Aging Cell. 2019;18:e12931. doi: 10.1111/acel.12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Debessa, G., Beatriz, L. & Maifrino, M. Age related changes of the collagen network of the human heart. 122, 1049–1058 (2001). [DOI] [PubMed]
- 61.Mancilla TR, Davis LR, Aune GJ. Doxorubicin-induced p53 interferes with mitophagy in cardiac fibroblasts. PLoS One. 2020;15:e0238856. doi: 10.1371/journal.pone.0238856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tanaka R, et al. Reactive fibrosis precedes doxorubicin‐induced heart failure through sterile inflammation. ESC Heart Fail. 2020;7:588–603. doi: 10.1002/ehf2.12616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chan BYH, et al. MMP inhibitors attenuate doxorubicin cardiotoxicity by preventing intracellular and extracellular matrix remodelling. Cardiovasc. Res. 2021;117:188–200. doi: 10.1093/cvr/cvaa017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Demaria M, et al. Cellular senescence promotes adverse effects of chemotherapy and cancer relapse. Cancer Discov. 2017;7:165–176. doi: 10.1158/2159-8290.CD-16-0241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tang X, Li PH, Chen HZ. Cardiomyocyte senescence and cellular communications within myocardial microenvironments. Front. Endocrinol. 2020;11:1–13. doi: 10.3389/fendo.2020.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hwang HJ, et al. Endothelial cells under therapy-induced senescence secrete CXCL11, which increases aggressiveness of breast cancer cells. Cancer Lett. 2020;490:100–110. doi: 10.1016/j.canlet.2020.06.019. [DOI] [PubMed] [Google Scholar]
- 67.De Angelis A, et al. Doxorubicin cardiotoxicity and target cells: a broader perspective. Cardio-Oncol. 2016;2:2. doi: 10.1186/s40959-016-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bailleul, A. M. et al. Evidence of proteins, chromosomes and chemical markers of DNA in exceptionally preserved dinosaur cartilage. Natl. Sci. Rev.10.1093/nsr/nwz206 (2020). [DOI] [PMC free article] [PubMed]
- 69.Ou H-L, Schumacher B. DNA damage responses and p53 in the aging process. Blood. 2018;131:488–495. doi: 10.1182/blood-2017-07-746396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Agudelo D, Bourassa P, Bérubé G, Tajmir-Riahi HA. Intercalation of antitumor drug doxorubicin and its analogue by DNA duplex: structural features and biological implications. Int. J. Biol. Macromol. 2014;66:144–150. doi: 10.1016/j.ijbiomac.2014.02.028. [DOI] [PubMed] [Google Scholar]
- 71.Chen NT, et al. Probing the dynamics of Doxorubicin-DNA intercalation during the initial activation of apoptosis by Fluorescence Lifetime Imaging Microscopy (FLIM) PLoS One. 2012;7:3–10. doi: 10.1371/journal.pone.0044947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Skladanowski A, Konopa J. Interstrand DNA crosslinking induced by anthracyclines in tumour cells. Biochem. Pharmacol. 1994;47:2269–2278. doi: 10.1016/0006-2952(94)90265-8. [DOI] [PubMed] [Google Scholar]
- 73.Vavrova A, et al. Catalytic inhibitors of Topoisomerase II differently modulate the toxicity of anthracyclines in cardiac and cancer cells. PLoS One. 2013;8:e76676. doi: 10.1371/journal.pone.0076676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yi LL, et al. Topoisomerase IIβ-mediated DNA double-strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res. 2007;67:8839–8846. doi: 10.1158/0008-5472.CAN-07-1649. [DOI] [PubMed] [Google Scholar]
- 75.Zhang S, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012;18:1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 76.Yang F, Teves SS, Kemp CJ, Henikoff S. Doxorubicin, DNA torsion, and chromatin dynamics. Biochim. Biophys. Acta. 2014;1845:84–89. doi: 10.1016/j.bbcan.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang F, Kemp CJ, Henikoff S. Doxorubicin enhances nucleosome turnover around promoters. Curr. Biol. 2013;23:782–787. doi: 10.1016/j.cub.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Congras A, et al. Doxorubicin-induced loss of DNA topoisomerase II and DNMT1- dependent suppression of MiR-125b induces chemoresistance in ALK-positive cells. Oncotarget. 2018;9:14539–14551. doi: 10.18632/oncotarget.24465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Capranico G, Kohn KW, Pommier Y. Local sequence requirements for DNA cleavage by mammalian topoisomerase II in the presence of doxorubicin. Nucleic Acids Res. 1990;18:6611–6619. doi: 10.1093/nar/18.22.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Deng S, et al. Dexrazoxane may prevent doxorubicin-induced DNA damage via depleting both Topoisomerase II isoforms. BMC Cancer. 2014;14:842. doi: 10.1186/1471-2407-14-842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smith, J., Tho, L. M., Xu, N. & Gillespie, D. A. The ATM–Chk2 and ATR–Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 108, 73–112 (Elsevier Inc., 2010). [DOI] [PubMed]
- 82.Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25:104–113. doi: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Baar MP, et al. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017;169:132–147.e16. doi: 10.1016/j.cell.2017.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Thornton TM, Rincon M. Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int. J. Biol. Sci. 2009;5:44–52. doi: 10.7150/ijbs.5.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ruan Y, et al. SIRT1 suppresses Doxorubicin-induced cardiotoxicity by regulating the oxidative stress and p38MAPK pathways. Cell. Physiol. Biochem. 2015;35:1116–1124. doi: 10.1159/000373937. [DOI] [PubMed] [Google Scholar]
- 86.Kajstura J, et al. The Telomere–Telomerase axis and the heart. Antioxid. Redox Signal. 2006;8:2125–2141. doi: 10.1089/ars.2006.8.2125. [DOI] [PubMed] [Google Scholar]
- 87.Terai M, et al. Association of telomere shortening in myocardium with heart weight gain and cause of death. Sci. Rep. 2013;3:2401. doi: 10.1038/srep02401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hewitt G, et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat. Commun. 2012;3:1–9. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Spallarossa, P. et al. Doxorubicin induces senescence or apoptosis in rat neonatal cardiomyocytes by regulating the expression levels of the telomere binding factors 1 and 2. Am. J. Physiol. Heart Circ. Physiol. 2169–2181 10.1152/ajpheart.00068.2009 (2009). [DOI] [PubMed]
- 90.Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016;17:487–500. doi: 10.1038/nrg.2016.59. [DOI] [PubMed] [Google Scholar]
- 91.Cakouros D, Gronthos S. Epigenetic regulation of bone marrow stem cell aging: revealing epigenetic signatures associated with hematopoietic and mesenchymal stem cell aging. Aging Dis. 2019;10:174. doi: 10.14336/AD.2017.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pang B, et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat. Commun. 2013;4:1908. doi: 10.1038/ncomms2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jones MJ, Goodman SJ, Kobor MS. DNA methylation and healthy human aging. Aging Cell. 2015;14:924–932. doi: 10.1111/acel.12349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sakai K, et al. Effects of doxorubicin on sperm DNA methylation in mouse models of testicular toxicity. Biochem. Biophys. Res. Commun. 2018;498:674–679. doi: 10.1016/j.bbrc.2018.03.044. [DOI] [PubMed] [Google Scholar]
- 95.Ferreira A, et al. Altered mitochondrial epigenetics associated with subchronic doxorubicin cardiotoxicity. Toxicology. 2017;390:63–73. doi: 10.1016/j.tox.2017.08.011. [DOI] [PubMed] [Google Scholar]
- 96.Nordgren KKS, Hampton M, Wallace KB. Editor’s highlight: the altered DNA Methylome of chronic doxorubicin exposure in sprague dawley rats. Toxico. Sci. 2017;159:470–479. doi: 10.1093/toxsci/kfx150. [DOI] [PubMed] [Google Scholar]
- 97.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14:R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Levine ME, et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging. 2018;10:573–591. doi: 10.18632/aging.101414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chaudhari U, et al. Metabolite signatures of doxorubicin induced toxicity in human induced pluripotent stem cell-derived cardiomyocytes. Amino Acids. 2017;49:1955–1963. doi: 10.1007/s00726-017-2419-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Booth LN, Brunet A. The aging epigenome. Mol. Cell. 2016;62:728–744. doi: 10.1016/j.molcel.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shimazu T, Hirschey MD, Huang J-Y, Ho LTY, Verdin E. Acetate metabolism and aging: an emerging connection. Mech. Ageing Dev. 2010;131:511–516. doi: 10.1016/j.mad.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 102.De Angelis A, et al. SIRT1 activation rescues doxorubicin-induced loss of functional competence of human cardiac progenitor cells. Int. J. Cardiol. 2015;189:30–44. doi: 10.1016/j.ijcard.2015.03.438. [DOI] [PubMed] [Google Scholar]
- 103.Dolinsky VW. The role of sirtuins in mitochondrial function and doxorubicin-induced cardiac dysfunction. Biol. Chem. 2017;398:955–974. doi: 10.1515/hsz-2016-0316. [DOI] [PubMed] [Google Scholar]
- 104.Pillai VB, et al. Honokiol, an activator of Sirtuin-3 (SIRT3) preserves mitochondria and protects the heart from doxorubicin-induced cardiomyopathy in mice. Oncotarget. 2017;8:34082–34098. doi: 10.18632/oncotarget.16133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tang H, et al. Doxorubicin-induced cardiomyocyte apoptosis: Role of mitofusin 2. Int. J. Biochem. Cell Biol. 2017;88:55–59. doi: 10.1016/j.biocel.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 106.Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ. Res. 2016;118:1593–1611. doi: 10.1161/CIRCRESAHA.116.307505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.El’darov ChM, Vays VB, Vangeli IM, Kolosova NG, Bakeeva LE. Morphometric examination of mitochondrial ultrastructure in aging cardiomyocytes. Biochemistry. 2015;80:604–609. doi: 10.1134/S0006297915050132. [DOI] [PubMed] [Google Scholar]
- 108.Brandt T, et al. Changes of mitochondrial ultrastructure and function during ageing in mice and Drosophila. Elife. 2017;6:e24662. doi: 10.7554/eLife.24662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br. J. Pharmacol. 2014;171:2029–2050. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Paradies G, Ruggiero FM, Petrosillo G, Quagliariello E. Age-dependent decline in the cytochrome c oxidase activity in rat heart mitochondria: role of cardiolipin. FEBS Lett. 1997;406:136–138. doi: 10.1016/S0014-5793(97)00264-0. [DOI] [PubMed] [Google Scholar]
- 111.Ichikawa Y, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014;124:617–630. doi: 10.1172/JCI72931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Anderson AB, Arriaga EA. Subcellular metabolite profiles of the parent CCRF-CEM and the derived CEM/C2 cell lines after treatment with doxorubicin. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2004;808:295–302. doi: 10.1016/j.jchromb.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 113.Goormaghtigh E, Chatelain P, Caspers J, Ruysschaert JM. Evidence of a complex between adriamycin derivatives and cardiolipin: possible role in cardiotoxicity. Biochem. Pharmacol. 1980;29:3003–3010. doi: 10.1016/0006-2952(80)90050-7. [DOI] [PubMed] [Google Scholar]
- 114.Liu L, et al. Over-expression of heat shock protein 27 attenuates doxorubicin-induced cardiac dysfunction in mice. Eur. J. Heart Fail. 2007;9:762–769. doi: 10.1016/j.ejheart.2007.03.007. [DOI] [PubMed] [Google Scholar]
- 115.Wan Q, et al. miR-499-5p attenuates mitochondrial fission and cell Apoptosis via p21 in Doxorubicin Cardiotoxicity. Front. Genet. 2019;9:734. doi: 10.3389/fgene.2018.00734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Carvalho RA, et al. Metabolic remodeling associated with subchronic doxorubicin cardiomyopathy. Toxicology. 2010;270:92–98. doi: 10.1016/j.tox.2010.01.019. [DOI] [PubMed] [Google Scholar]
- 117.Carvalho FS, et al. Doxorubicin-induced cardiotoxicity: from bioenergetic failure and cell death to cardiomyopathy. Med. Res. Rev. 2013;34:106–135. doi: 10.1002/med.21280. [DOI] [PubMed] [Google Scholar]
- 118.Zhang H, et al. Reduction of elevated proton leak rejuvenates mitochondria in the aged cardiomyocyte. eLife. 2020;9:1–18. doi: 10.7554/eLife.60827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Martín-Fernández B, Gredilla R. Mitochondria and oxidative stress in heart aging. Age. 2016;38:225–238. doi: 10.1007/s11357-016-9933-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Maillet A, et al. Modeling Doxorubicin-induced cardiotoxicity in human pluripotent stem cell derived-cardiomyocytes. Sci. Rep. 2016;6:25333. doi: 10.1038/srep25333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jeyaseelan R, Poizat C, Wu H, Kedes L. Molecular mechanisms of Doxorubicin-induced cardiomyopathy. J. Biol. Chem. 1997;272:5828–5832. doi: 10.1074/jbc.272.9.5828. [DOI] [PubMed] [Google Scholar]
- 122.Pointon AV, et al. Doxorubicin in vivo rapidly alters expression and translation of myocardial electron transport chain genes, leads to ATP loss and caspase 3 activation. PLoS One. 2010;5:1–17. doi: 10.1371/journal.pone.0012733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhang Y, et al. Doxorubicin induces sarcoplasmic reticulum calcium regulation dysfunction via the decrease of SERCA2 and phospholamban expressions in rats. Cell Biochem. Biophys. 2014;70:1791–1798. doi: 10.1007/s12013-014-0130-2. [DOI] [PubMed] [Google Scholar]
- 124.Bordoni A, Biagi P, Hrelia S. The impairment of essential fatty acid metabolism as a key factor in doxorubicin-induced damage in cultured rat cardiomyocytes. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 1999;1440:100–106. doi: 10.1016/S1388-1981(99)00113-4. [DOI] [PubMed] [Google Scholar]
- 125.Guan R, et al. Bone morphogenetic protein 4 inhibits pulmonary fibrosis by modulating cellular senescence and mitophagy in lung fibroblasts. Eur. Respir. J. 2022;60:2102307. doi: 10.1183/13993003.02307-2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Trifunovic A, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 127.Sangomla S, Saifi MA, Khurana A, Godugu C. Nanoceria ameliorates doxorubicin induced cardiotoxicity: possible mitigation via reduction of oxidative stress and inflammation. J. Trace Elements Med. Biol. 2018;47:53–62. doi: 10.1016/j.jtemb.2018.01.016. [DOI] [PubMed] [Google Scholar]
- 128.Tang WHW, Mullens W. Cardiorenal syndrome in decompensated heart failure. Heart. 2010;96:255–260. doi: 10.1136/hrt.2009.166256. [DOI] [PubMed] [Google Scholar]
- 129.Hernandez-Segura A, Nehme J, Demaria M. Hallmarks of cellular senescence. Trends Cell Biol. 2018;28:436–453. doi: 10.1016/j.tcb.2018.02.001. [DOI] [PubMed] [Google Scholar]
- 130.Berthiaume JM, Wallace KB. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol. Toxicol. 2007;23:15–25. doi: 10.1007/s10565-006-0140-y. [DOI] [PubMed] [Google Scholar]
- 131.Bachur NR, Gee MV, Friedman RD. Nuclear catalyzed antibiotic free radical formation. Cancer Res. 1982;42:1078–1081. [PubMed] [Google Scholar]
- 132.Zhu H, et al. Doxorubicin redox biology: redox cycling, topoisomerase inhibition, and oxidative stress. React. Oxyg. Species. 2016;1:189–198. doi: 10.20455/ros.2016.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kalyanaraman B. Teaching the basics of the mechanism of doxorubicin-induced cardiotoxicity: have we been barking up the wrong tree? Redox. Biol. 2020;29:1–9. doi: 10.1016/j.redox.2019.101394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Treuting PM, et al. Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J. Gerontol. A Biol. Sci. Med. Sci. 2008;63:813–822. doi: 10.1093/gerona/63.8.813. [DOI] [PubMed] [Google Scholar]
- 135.Wenz T. Mitochondria and PGC-1α in aging and age-associated diseases. J. Aging Res. 2011;2011:1–12. doi: 10.4061/2011/810619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Cesselli D, et al. Cardiac cell senescence and redox signaling. Front. Cardiovasc. Med. 2017;4:38. doi: 10.3389/fcvm.2017.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention. JAMA. 2007;297:842. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 138.Dresdale AR, et al. Prospective randomized study of the role of N-acetyl cysteine in reversing doxorubicin-induced cardiomyopathy. Am. J. Clin. Oncol. 1982;5:657–664. doi: 10.1097/00000421-198212000-00015. [DOI] [PubMed] [Google Scholar]
- 139.Singh K, Bhori M, Kasu YA, Bhat G, Marar T. Antioxidants as precision weapons in war against cancer chemotherapy induced toxicity – Exploring the armoury of obscurity. Saudi Pharm. J. 2018;26:177–190. doi: 10.1016/j.jsps.2017.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Vincent DT, Ibrahim YF, Espey MG, Suzuki YJ. The role of antioxidants in the era of cardio-oncology. Cancer Chemother. Pharmacol. 2013;72:1157–1168. doi: 10.1007/s00280-013-2260-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Herraiz-Martínez A, et al. Ageing is associated with deterioration of calcium homeostasis in isolated human right atrial myocytes. Cardiovasc. Res. 2015;106:76–86. doi: 10.1093/cvr/cvv046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Janczewski AM, Lakatta EG. Modulation of sarcoplasmic reticulum Ca2+ cycling in systolic and diastolic heart failure associated with aging. Heart Fail. Rev. 2010;15:431–445. doi: 10.1007/s10741-010-9167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hamilton S, Terentyev D. Altered intracellular calcium homeostasis and arrhythmogenesis in the aged heart. Int. J. Mol. Sci. 2019;20:2386. doi: 10.3390/ijms20102386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hanna AD, Lam A, Tham S, Dulhunty AF, Beard NA. Adverse effects of Doxorubicin and its metabolic product on cardiac RyR2 and SERCA2A. Mol. Pharmacol. 2014;86:438–449. doi: 10.1124/mol.114.093849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Franceschi C, et al. Inflamm‐aging: an evolutionary perspective on immunosenescence. Ann. N Y Acad. Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 146.Chung HY, et al. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res. Rev. 2009;8:18–30. doi: 10.1016/j.arr.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Childs BG, et al. Senescent cells: an emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017;16:718–735. doi: 10.1038/nrd.2017.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lu X, et al. The role of CXC chemokines in cardiovascular diseases. Front. Pharmacol. 2022;12:765768. doi: 10.3389/fphar.2021.765768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Aukrust P, et al. Chemokines and cardiovascular risk. Arterioscler. Thromb. Vasc. Biol. 2008;28:1909–1919. doi: 10.1161/ATVBAHA.107.161240. [DOI] [PubMed] [Google Scholar]
- 150.Tchkonia T, et al. Fat tissue, aging, and cellular senescence. Aging Cell. 2010;9:667–684. doi: 10.1111/j.1474-9726.2010.00608.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Baker DJ, et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.van Deursen JM. The role of senescent cells in ageing. Nature. 2014;509:439–446. doi: 10.1038/nature13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Xu G, et al. Zymosan a improved doxorubicin‐induced ventricular remodeling by evoking heightened cardiac inflammatory responses and healing in mice. J. Am. Heart Assoc. 2023;12:e030200. doi: 10.1161/JAHA.123.030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Campisi JA. Cellular senescence, and cancer. Annu. Rev. Physiol. 2013;75:685–705. doi: 10.1146/annurev-physiol-030212-183653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Palmer AK, Kirkland JL. Aging and adipose tissue: potential interventions for diabetes and regenerative medicine. Exp. Gerontol. 2016;86:97–105. doi: 10.1016/j.exger.2016.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Bracun V, et al. Insulin‐like growth factor binding protein 7 (IGFBP7), a link between heart failure and senescence. ESC Heart Fail. 2022;9:4167–4176. doi: 10.1002/ehf2.14120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhang L, et al. Insulin-like growth factor-binding protein-7 (IGFBP7) links senescence to heart failure. Nat. Cardiovasc. Res. 2022;1:1195–1214. doi: 10.1038/s44161-022-00181-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ridker PM, Rane M. Interleukin-6 Signaling and Anti-Interleukin-6 therapeutics in cardiovascular disease. Circ. Res. 2021;128:1728–1746. doi: 10.1161/CIRCRESAHA.121.319077. [DOI] [PubMed] [Google Scholar]
- 159.Ridker, P. M., Rifai, N., Stampfer, M. J. & Hennekens, C. H. Plasma Concentration of Interleukin-6 and the Risk of Future Myocardial Infarction Among Apparently Healthy Men. 101 http://www.circulationaha.org (2000). [DOI] [PubMed]
- 160.Wang TJ, et al. Prognostic utility of novel biomarkers of cardiovascular stress: the framingham heart study. Circulation. 2012;126:1596–1604. doi: 10.1161/CIRCULATIONAHA.112.129437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zhang, L., Pitcher, L. E., Prahalad, V., Niedernhofer, L. J. & Robbins, P. D. Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. FEBS J.10.1111/febs.16350 (2022). [DOI] [PubMed]
- 162.Zhu Y, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015;14:644–658. doi: 10.1111/acel.12344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhu Y, et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl‐2 family of anti‐apoptotic factors. Aging Cell. 2016;15:428–435. doi: 10.1111/acel.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mohamad Anuar NN, Nor Hisam NS, Liew SL, Ugusman A. Clinical review: navitoclax as a pro-apoptotic and anti-fibrotic agent. Front. Pharmacol. 2020;11:1–16. doi: 10.3389/fphar.2020.564108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Lérida-Viso A, et al. Pharmacological senolysis reduces doxorubicin-induced cardiotoxicity and improves cardiac function in mice. Pharmacol. Res. 2022;183:106356. doi: 10.1016/j.phrs.2022.106356. [DOI] [PubMed] [Google Scholar]
- 166.Yousefzadeh MJ, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. 2018;36:18–28. doi: 10.1016/j.ebiom.2018.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ma T, Kandhare AD, Mukherjee-Kandhare AA, Bodhankar SL. Fisetin, a plant flavonoid ameliorates doxorubicin-induced cardiotoxicity in experimental rats: the decisive role of caspase-3, COX-II, cTn-I, iNOs and TNF-α. Mol. Biol. Rep. 2019;46:105–118. doi: 10.1007/s11033-018-4450-y. [DOI] [PubMed] [Google Scholar]
- 168.Kirkland JL, Tchkonia T. Cellular senescence: a translational perspective. EBioMedicine. 2017;21:21–28. doi: 10.1016/j.ebiom.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gu J, Hu W, Zhang D. Resveratrol, a polyphenol phytoalexin, protects against doxorubicin‐induced cardiotoxicity. J. Cell. Mol. Med. 2015;19:2324–2328. doi: 10.1111/jcmm.12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Zilinyi R, et al. The cardioprotective effect of metformin in Doxorubicin-induced cardiotoxicity: the role of autophagy. Molecules. 2018;23:1184. doi: 10.3390/molecules23051184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Szenczi O, et al. Poly(ADP-ribose) polymerase regulates myocardial calcium handling in doxorubicin-induced heart failure. Biochem. Pharmacol. 2005;69:725–732. doi: 10.1016/j.bcp.2004.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Choksey, A. et al. AICAR prevents doxorubicin-induced heart failure in rats by ameliorating cardiac atrophy and improving fatty acid oxidation. BioRxiv 10.1101/2022.08.17.504253 (2022).
- 173.Singh AP, et al. Health benefits of resveratrol: evidence from clinical studies. Med. Res. Rev. 2019;39:1851–1891. doi: 10.1002/med.21565. [DOI] [PubMed] [Google Scholar]
- 174.Justice JN, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. 2019;40:554–563. doi: 10.1016/j.ebiom.2018.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hickson LTJ, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. 2019;47:446–456. doi: 10.1016/j.ebiom.2019.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Vuong JT, Stein-Merlob AF, Cheng RK, Yang EH. Novel therapeutics for anthracycline induced cardiotoxicity. Front. Cardiovasc. Med. 2022;9:1–25. doi: 10.3389/fcvm.2022.863314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Huang S, et al. Protective role of beta-blockers in chemotherapy-induced cardiotoxicity—a systematic review and meta-analysis of carvedilol. Heart Fail. Rev. 2019;24:325–333. doi: 10.1007/s10741-018-9755-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Avila MS, et al. Carvedilol for prevention of chemotherapy-related cardiotoxicity: the CECCY Trial. J. Am. Coll. Cardiol. 2018;71:2281–2290. doi: 10.1016/j.jacc.2018.02.049. [DOI] [PubMed] [Google Scholar]
- 179.Kawaguchi T, et al. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc. Res. 2012;96:456–465. doi: 10.1093/cvr/cvs282. [DOI] [PubMed] [Google Scholar]
- 180.Corsetti G, et al. Autophagy and Oncosis/Necroptosis are enhanced in cardiomyocytes from heart failure patients. Med. Sci. Monit. Basic Res. 2019;25:33–44. doi: 10.12659/MSMBR.913436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Malliaras K, et al. Cardiomyocyte proliferation and progenitor cell recruitment underlie therapeutic regeneration after myocardial infarction in the adult mouse heart. EMBO Mol. Med. 2013;5:191–209. doi: 10.1002/emmm.201201737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bergmann O, et al. Evidence for cardiomyocyte renewal in humans. Science. 2009;324:98–102. doi: 10.1126/science.1164680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Wu R, et al. Zn(ii)-Curcumin supplementation alleviates gut dysbiosis and zinc dyshomeostasis during doxorubicin-induced cardiotoxicity in rats. Food Funct. 2019;10:5587–5604. doi: 10.1039/C9FO01034C. [DOI] [PubMed] [Google Scholar]
- 184.Gongora CA, et al. Sodium-Glucose Co-Transporter-2 inhibitors and cardiac outcomes among patients treated with anthracyclines. JACC Heart Fail. 2022;10:559–567. doi: 10.1016/j.jchf.2022.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Quagliariello V, et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc. Diabetol. 2021;20:150. doi: 10.1186/s12933-021-01346-y. [DOI] [PMC free article] [PubMed] [Google Scholar]