

Abstract
Bacterial lipopolysaccharide induces tyrosine phosphorylation of paxillin, actin reorganization, and opening of the transendothelial paracellular pathway through which macromoles flux. In this study, lipid A was shown to be the bioactive portion of the lipopolysaccharide molecule responsible for changes in endothelial barrier function. We then studied whether endotoxin-neutralizing protein, a recombinant peptide that is derived from Limulus antilipopolysaccharide factor and targets lipid A, could block the effects of lipopolysaccharide on protein tyrosine phosphorylation, actin organization, and movement of 14C-bovine serum albumin across bovine pulmonary artery endothelial cell monolayers. In the presence of serum, a 6-h exposure to lipopolysaccharide (10 ng/ml) increased transendothelial 14C-albumin flux compared to the simultaneous media control. Coadministration of endotoxin-neutralizing protein (≥10 ng/ml) with lipopolysaccharide (10 ng/ml) protected against lipopolysaccharide-induced barrier dysfunction. This protection was dose dependent, conferring total protection at endotoxin-neutralizing protein/lipopolysaccharide ratios of ≥10:1. Similarly, endotoxin-neutralizing protein was capable of blocking the lipopolysaccharide-induced endothelial cell responses that are prerequisite to barrier dysfunction, including tyrosine phosphorylation of paxillin and actin depolymerization. Finally, endotoxin-neutralizing protein cross-protected against lipopolysaccharide derived from diverse gram-negative bacteria. Thus, endotoxin-neutralizing protein offers a novel therapeutic intervention for the vascular endothelial dysfunction of gram-negative sepsis and its attendant endotoxemia.
Gram-negative septic processes can be complicated by endothelial cell (EC) injury and/or dysfunction that contributes to systemic vascular collapse, disseminated intravascular coagulation, and vascular leak syndromes, including the adult respiratory distress syndrome (ARDS) (5, 26). Endotoxin or bacterial lipopolysaccharide (LPS) has been implicated as the bacterial component responsible for much of the EC injury associated with gram-negative bacterial infections. First, LPS bioactivity has been detected in the bloodstream of gram-negative septicemic patients, and in some studies, particularly those focusing on meningococcal sepsis, levels of circulating LPS predict development of multiple organ failure, including the adult respiratory distress syndrome (31). Second, administration of LPS alone to experimental animals reproduces the EC injury seen after gram-negative bacterial challenge (13, 26). Lastly, in some animal studies, interventions that specifically target the LPS molecule appear to protect against the EC complications associated with gram-negative sepsis or experimental LPS challenge (1, 33, 34). The efficacy of most of these interventions has yet to be evaluated in humans.
Most bactericidal antibiotics that target viable, replicating gram-negative bacteria do not diminish LPS activity and can actually liberate free LPS into the circulation (1). One notable exception, polymyxin B (PMB) derived from the bacteria Bacillus polymyxa (6, 23), can bind to the lipid A portion of LPS and neutralize it. In the past, however, PMB’s nephrotoxic properties have severely limited its therapeutic application. Other naturally occurring proteins which also bind to and neutralize LPS include bactericidal/permeability-increasing protein (BPI) and cationic antimicrobial protein 18 found in polymorphonuclear leukocytes (10, 17), high- and low-density lipoproteins (20, 25), and the Limulus anti-LPS factor (LALF) found in the horseshoe crab, Limulus polyphemus (22). LALF is a 11.8-kDa protein isolated from the amebocyte, the single blood cell type found in the horseshoe crab (22). The amebocyte-derived LALF as well as its recombinant form, endotoxin-neutralizing protein (ENP), each binds to and neutralizes LPS (22, 32). The LPS-binding site is 32 to 50 amino acids in length and forms an amphipathic loop which binds to the lipid A portion of LPS (18, 24, 32). ENP or LALF neutralizes LPS bioactivity in the Limulus amebocyte lysate assay (11, 32), prevents macrophage production of tumor necrosis factor in vitro (4), and protects against LPS challenge in vivo (11, 33).
LPS interaction with cells of monocytic lineage has been well characterized. These cells express membrane-bound CD14, a glycosylphosphatidylinositol-anchored protein which can recognize complexes of LPS and LPS-binding protein, resulting in cell activation (12, 30, 35). In EC, which lack membrane-bound CD14, a specific EC-binding site(s) or receptor(s), although implied, has not yet been demonstrated. Circulating LPS, in concert with the accessory molecules LPS-binding protein and soluble CD14, can be presented to the non-CD14-bearing EC, evoking EC responses through as yet unidentified mechanisms (12, 14). One such EC response involves a sequence of events comprised of protein tyrosine phosphorylation, actin depolymerization, intercellular gap formation, and loss of EC barrier function (3). The initial tyrosine phosphorylation events are clearly a prerequisite to LPS-induced actin changes and disruption of EC monolayer integrity (3). Further, prior F-actin stabilization of EC monolayers with phallicidin protects against LPS-induced increments in transendothelial albumin flux (15). We therefore studied whether a molecule such as ENP, which binds to lipid A and has been shown to confer protection against the deleterious effect of LPS in vivo, could block one or more of the sequential LPS-induced events leading to increased EC monolayer permeability. In this work, we have studied whether ENP protects against LPS-induced protein tyrosine phosphorylation, actin reorganization, and loss of endothelial barrier function.
MATERIALS AND METHODS
Reagents.
LPSs phenol extracted from Escherichia coli serotype O111:B4, E. coli O55:B5, Klebsiella pneumoniae, Pseudomonas aeruginosa, Salmonella minnesota, and Serratia marcescens (Sigma Chemical Co., St. Louis, Mo.) were suspended in phosphate-buffered saline (PBS) at 5 mg/ml, and these stock solutions were stored at 4°C. Lipid A from E. coli K-12 (List Biological Laboratories, Campbell, Calif.) was dissolved into chloroform (69%)–methanol (27%)–water (4%) and evaporated under nitrogen, and the dry residue was resuspended in water. To prepare the O-polysaccharide fraction, E. coli O111:B4 LPS was hydrolyzed at 100°C for 2 h with 1% acetic acid, neutralized with 1.0 M NaOH, and centrifuged. The supernatant containing the O polysaccharide was desalted on a Sephadex G-25 (Pharmacia Biotech, Piscataway, N.J.) column, using water as the elutant, and the fractions were tested for O polysaccharide by the phenol sulfuric acid method of Dubois et al. (9). The O-polysaccharide-positive fractions were pooled and lyophilized. ENP was a gift from the Associates of Cape Cod (Woods Hole, Mass.). ENP was reconstituted at 1 mg/ml in PBS, aliquoted, and stored at −20°C. PMB sulfate (Sigma) was reconstituted at 25.8 mg/ml and stored at 4°C.
EC culture.
Bovine pulmonary artery EC (American Type Culture Collection, Rockville, Md.) were cultured in Dulbecco’s modified Eagle’s medium enriched with 20% fetal bovine serum, 4 mM l-glutamine, nonessential amino acids, and vitamins in the presence of penicillin (50 U/ml) and streptomycin (50 μg/ml) as previously described (14, 15). Cell cultures were determined to be endothelial by uniform cobblestone morphology and quantitative determination of angiotensin-converting enzyme activity with commercially available 3H-benzoyl-Phe-Ala-Pro substrate (Ventex Laboratories, Portland, Maine).
Assay of transendothelial albumin flux.
Transendothelial 14C-bovine serum albumin (14C-BSA) flux was assayed as we have previously described (3, 14, 15). Briefly, polycarbonate filters (13-mm diameter; 0.4-μm pore size; Nucleopore Corp., Pleasanton, Calif.) were impregnated with pig skin gelatin (Fisher Scientific Co., Pittsburgh, Pa.), mounted in polystyrene chemotactic chambers (ADAPS Inc., Dedham, Mass.), inserted into wells of 24-well culture dishes, and sterilized with ethylene oxide. Each upper compartment was seeded with 2 × 105 EC and cultured for 72 h. 14C-BSA (Sigma) with a specific activity of 1.3 μCi/mg of protein was used as the tracer molecule. The baseline barrier function of each monolayer was determined by applying 14C-BSA to each upper compartment for 1 h, after which the lower compartment was counted in a liquid scintillation analyzer (Tri-Carb 1500; Packard Instruments Co.). Only EC monolayers retaining ≥97% of the 14C-BSA were studied. EC monolayers were exposed for 6 h to increasing concentrations of either the lipid A or the O-polysaccharide LPS fraction. In some experiments, native LPS was coadministered with increasing concentrations of either PMB or ENP. In other experiments, ENP was introduced either before or after the LPS challenge. Simultaneous controls with medium alone were performed. Transfer of 14C-BSA across EC monolayers was again assayed immediately following treatment.
The ability of ENP to cross-protect against endotoxins derived from diverse gram-negative bacterial strains was also studied. Due to the differences in the length of the O-specific polysaccharide and lipid A acyl chains, various endotoxins cannot be compared on a dry weight/weight basis. However, each LPS molecule, regardless of the bacterial origin, contains at least one 2-keto-3-deoxyoctonic acid (KDO) molecule. LPS preparations derived from E. coli O55:B5, K. pneumoniae, P. aeruginosa, S. minnesota, or S. marcescens each were standardized to 10 ng of LPS per ml derived from E. coli O111:B4 on the basis of KDO content (19). EC were exposed for 6 h to either medium alone, ENP (100 ng/ml), equivalent concentrations of LPS based on KDO content, or each LPS preparation coadministered with ENP.
Immunoblotting for phosphotyrosines.
EC were exposed for 1 h to either medium alone, ENP (1 μg/ml), LPS derived from E. coli O111:B4 (100 ng/ml), or LPS coadministered with ENP. EC were then processed for phosphotyrosine immunoblotting as previously described (3). Briefly, EC were lysed with ice-cold lysis buffer (50 mM Tris-HCl [pH 8.0], 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EGTA, 1 mM phenylmethylsulfonyl fluoride, 1 μg of leupeptin per ml, 1 μg of pepstatin per ml, 1 μg of aprotinin per ml, 1 μg of DNase I per ml, 1 mM sodium orthovanadate, 1 mM NaF, 1 mM disodium pyrophosphate, 1 mM phenylarsine oxide, 500 μM p-nitrophenyl phosphate), scraped, transferred to a tube, and centrifuged (16,000 × g, 10 min, 4°C). The supernatant from the EC lysate was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE; 25 μg of protein/lane) on an 8 to 16% Tris-glycine gradient gel (Novex Inc., San Diego, Calif.) and transferred for 3 h to a polyvinylidene fluoride membrane (PVDF). The blot was blocked with 3% dry milk, probed with biotinylated antiphosphotyrosine antibody 4G10 (0.7 μg/ml; Upstate Biotechnology Inc. [UBI], Lake Placid, N.Y.), washed, and incubated with horseradish peroxidase-conjugated streptavidin (0.5 μg/ml; UBI). The blot was developed with enhanced chemiluminescence (Amersham Life Sciences, Arlington Heights, Ill.) and exposed to Du Pont Reflection (NEF-406) film. The films were subsequently scanned for laser densitometric analysis (Molecular Dynamics, Sunnyvale, Calif.).
Immunolocalization of phosphotyrosine-containing proteins.
To maintain EC under experimental conditions identical to those of our permeability assay, we directly stained and visualized EC monolayers cultured on polycarbonate filters as previously described (3, 15). Briefly, EC cultured to confluence on filters were exposed for 1 h to medium, LPS (100 ng/ml), ENP (1 μg/ml), or LPS coadministered with ENP. The monolayers were washed with 1 mM vanadate in PBS, fixed with 4% paraformaldehyde (30 min) followed by absolute methanol (8 min, −20°C), and stained with fluorescein isothiocyanate (FITC)-conjugated antiphosphotyrosine antibody (UBI) (5 μg/ml, 1 h) (3). The filters and their attached monolayers were mounted cell side up on microscope slides and photographed through a Zeiss Axioskop 20 microscope equipped for epifluorescence.
F-actin quantification.
EC F-actin was fluorimetrically measured as previously described (3, 15). EC were pretreated with cycloheximide (50 μg/ml) for 30 min prior to and throughout treatments with medium, ENP (100 ng/ml), LPS (10 ng/ml), or LPS coadministered with ENP. EC were washed with 75 mM KCl–1 mM EGTA–3 mM MgSO4–0.2 mM dithiothreitol–10 mM imidazole–10 μg of aprotinin per ml–0.1 mM phenylmethylsulfonyl fluoride (pH 7.2), fixed (3.7% formaldehyde, 15 min), permeabilized (0.2% Triton X-100, 5 min), incubated with NBD-phallicidin (Molecular Probes, Eugene, Oreg.) (1 U/well, 30 min), and extracted with methanol (overnight, −20°C). The extracts were assayed in a Perkin-Elmer LS30 luminescence spectrometer at 465-nm excitation (10-nm slit) and 535-nm emission (10-nm slit) and F-actin content expressed in arbitrary fluorescence units per milligram of total EC protein. Since methodologies for quantifying F- and G-actin pools preclude protein determinations on the same monolayers, EC were cultured under the same conditions as the monolayers assayed for F- and G-actin pools, lysed, and assayed for protein concentration with the standard Bio-Rad DC protein assay (Bio-Rad Chemical Division, Richmond, Calif.).
DNase I inhibition assay for G-actin.
EC G-actin was measured by using the DNase I inhibition assay as previously described (3, 15). This assay utilizes the ability of monomeric G-actin to inhibit DNase I hydrolysis of type 1 DNA into its component nucleotides. Briefly, DNase I obtained from bovine pancreas (Sigma) was mixed with calf thymus DNA type 1 (Sigma) in the cuvette of a spectrophotometer, and the slope of the linear portion of the ΔA260 recorded. Purified bovine skeletal muscle actin (Sigma) was used to calibrate the assay. DNase I inhibitory activity within a range of 30 to 70% inhibition is directly proportional to the concentration of monomeric G-actin. EC monolayers grown in six-well culture dishes were exposed for 6 h to medium, LPS (10 ng/ml), ENP (100 ng/ml), or LPS coadministered with ENP. The monolayers were permeabilized with lysing buffer containing 1% Triton X-100 for 5 min. The G-actin-containing supernatants were then tested in the DNase I inhibition assay to generate inhibitory activities that fell on the linear portion of the standard curve (i.e., 30 to 70% inhibition). The inhibitory activities were interpolated to G-actin concentrations, which were used to calculate G-actin expressed in micrograms per milligram of total EC protein.
Statistical analysis.
One-way analysis of variance was used to compare the mean responses among experimental and control groups for all experiments. The Bonferroni post hoc comparison test was used to determine between which groups significant differences existed. A P value of ≤0.05 was considered significant.
RESULTS
Structure-function studies of LPS-induced endothelial barrier dysfunction.
The mean ± standard error (SE) pretreatment baselines reflecting functional integrity for monolayers to be exposed to either lipid A or the O-specific polysaccharide fraction were 0.016 ± 0.002 and 0.013 ± 0.002 pmol/h, respectively (Fig. 1A). The mean ± SE 14C-BSA flux across naked filters without endothelial monolayers was 0.215 ± 0.015 pmol/h. Increasing concentrations of lipid A caused dose-dependent increases in 14C-BSA flux across endothelial monolayers, whereas identical concentrations of the O-specific polysaccharide fraction did not (Fig. 1A). The lowest lipid A concentration that after a 6-h exposure increased 14C-BSA flux compared to the simultaneous medium control was 15 ng/ml. Further dose-dependent increments were evident at concentrations up to 15 μg/ml. The polysaccharide fraction at concentrations up to 15 μg/ml failed to increase 14C-BSA compared to the simultaneous medium control.
FIG. 1.

Effects of LPS fractions on transendothelial 14C-BSA flux. (A) Transendothelial 14C-BSA flux across monolayers was assayed after exposure for 6 h to increasing concentrations of either the lipid A fraction or the O-specific polysaccharide fraction derived from E. coli O111:B4 LPS. Each bar represents mean (± SE) transendothelial 14C-BSA flux. Pretreatment baseline 14C-BSA flux across monolayers exposed to either lipid A or O-specific polysaccharide fractions as well as 14C-BSA flux across naked filters are also shown. ∗, significantly increased compared with simultaneous medium control. n, number of monolayers studied. (B) EC monolayers were assayed for transendothelial 14C-BSA flux immediately after 6-h exposures to medium alone, PMB, LPS, or LPS (10 ng/ml) coadministered with increasing concentrations of PMB. Mean (± SE) pretreatment baseline 14C-BSA flux is also shown ∗, significantly increased compared to medium control; ∗∗, significantly decreased compared to LPS. n, number of monolayers studied.
PMB binds to and neutralizes the lipid A moiety of LPS (6, 23). To determine whether lipid A was essential for LPS presentation to the non-CD14-bearing EC, the ability of PMB to block native LPS-induced barrier dysfunction was also studied (Fig. 1B). 14C-BSA flux was assayed immediately after 6-h exposures to medium, LPS (10 ng/ml), PMB (10 μg/ml), or LPS coadministered with increasing concentrations of PMB (10 to 10,000 ng/ml). LPS increased 14C-BSA flux compared to the simultaneous medium control, whereas PMB alone did not. PMB at a PMB/LPS dry weight-to-weight ratio of 1:1 did not significantly diminish the LPS effect. At PMB concentrations of ≥1,000 ng/ml (PMB/LPS dry weight-to-weight ratio of 100:1), PMB completely protected against the LPS effect, returning barrier function to medium control levels.
Effect of ENP on LPS-induced changes in transendothelial 14C-BSA flux.
ENP protected against LPS-induced barrier dysfunction (Fig. 2). 14C-BSA flux was assayed immediately after 6-h exposures to medium, LPS (10 ng/ml), ENP (10 μg/ml), or LPS coadministered with increasing concentrations of ENP (10 to 10,000 ng/ml). Again, LPS alone (10 ng/ml) increased 14C-BSA flux compared to the simultaneous medium control. Protection against LPS-induced increases in 14C-BSA flux by coadministration of ENP was dose dependent. ENP at ≥10 ng/ml significantly decreased LPS-induced barrier dysfunction. Partial protection was seen at an ENP concentration of 10 ng/ml (ENP/LPS dry weight-to-weight ratio of 1:1). Total protection was seen with ENP concentrations of ≥100 ng/ml (ENP/LPS ratio of ≥10:1). When ENP was introduced prior to a 5-min LPS challenge for either 0.5 or 1.0 h and subsequently removed by thorough washing, no protection was observed (Table 1). Similarly, if ENP was introduced immediately following the LPS, again no protection could be demonstrated.
FIG. 2.

Dose-dependent effects of ENP on LPS-induced barrier dysfunction. Baseline barrier function was determined for all monolayers prior to treatment. Transendothelial 14C-BSA flux across monolayers was assayed after exposure for 6 h to medium, ENP, LPS, or LPS coadministered with increasing concentrations of ENP. Each bar represents mean (± SE) transendothelial 14C-BSA flux. ∗, significantly increased compared to medium control; ∗∗, significantly decreased compared to LPS alone. n, number of monolayers studied.
TABLE 1.
ENP treatment either before or after LPS challenge does not protect against LPS-induced loss of endothelial monolayer barrier function
| Pretreatment | Treatment (5 min) | Posttreatment (6 h) | n | Mean 14C-BSA flux (pmol/h)a ± SE |
|---|---|---|---|---|
| —b | — | — | 70 | 0.010 ± 0.001 |
| Medium | Medium | Medium | 15 | 0.021 ± 0.001 |
| Medium | LPS | Medium | 25 | 0.087 ± 0.004c |
| ENP (0.5 h) | LPS | Medium | 8 | 0.070 ± 0.005c,d |
| ENP (1.0 h) | LPS | Medium | 8 | 0.078 ± 0.005c,d |
| Medium | LPS | ENP | 14 | 0.082 ± 0.008c,d |
14C-BSA flux across LPS (10 ng/ml)- or medium control-exposed monolayers that were either pre- or posttreated with ENP (1.0 μg/ml) or medium alone.
—, pretreatment baseline. The baseline barrier function for each monolayer was established prior to study; only monolayers which retained ≥97% of the 14C-BSA tracer were studied.
Significantly increased (P < 0.001) compared to medium alone.
Not significantly different (P > 0.05) compared to LPS alone.
A comparison of the protective effects of PMB (molecular size = 1,450 g/mol) and ENP (molecular size = 12,189 g/mol) suggests that ENP is more effective at blocking LPS-induced loss of EC barrier function. Complete protection against LPS (10 ng/ml)-induced transendothelial 14C-albumin flux was observed with either ≥6.90 × 10−7 M PMB (PMB/LPS dry weight-to-weight ratio of ≥100:1) or ≥8.2 × 10−9 M ENP (ENP/LPS dry weight-to-weight ratio of ≥10:1). Partial protection was observed with ≥6.90 × 10−8 M PMB (PMB/LPS dry weight-to-weight ratio of ≥10:1) or 8.20 × 10−10 M ENP (ENP/LPS dry weight-to-weight ratio of ≥1:1).
Effect of ENP on LPS-induced tyrosine phosphorylation of EC proteins.
Lysates obtained from EC exposed for 1 h to medium alone, ENP (1.0 μg/ml), LPS (100 ng/ml), or LPS coadministered with ENP were resolved by SDS-PAGE and transferred to PVDF, and the blot was probed for phosphotyrosines (Fig. 3). Monolayers exposed to LPS demonstrated a 2.2-fold increase in tyrosine phosphorylation of a 66-kDa band compared to medium alone; those monolayers exposed to LPS in the presence of ENP demonstrated only a 1.3-fold increase in tyrosine phosphorylation of this same 66-kDa protein. The increased tyrosine phosphorylation of this band in LPS-exposed EC relative to those exposed to medium alone was inhibited by 75% when LPS was coadministered with ENP.
FIG. 3.
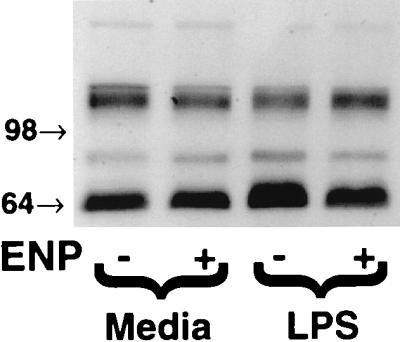
Effect of ENP on LPS-induced tyrosine phosphorylation of a 66-kDa EC protein. For Western blot analysis of protein tyrosine phosphorylation, EC monolayers were exposed to medium, ENP (1.0 μg/ml), LPS (100 ng/ml), or LPS coadministered with ENP for 1 h. The EC lysates were resolved by SDS-PAGE, transferred to PVDF, and probed for phosphotyrosines. Molecular weights (in thousands) are indicated by arrows on the left. The blot is representative of three separate experiments.
EC monolayers exposed for 1 h to medium, to ENP (1.0 μg/ml), to LPS (100 ng/ml), or to LPS coadministered with ENP were probed with an FITC-conjugated antiphosphotyrosine antibody, processed for epifluorescence microscopy, and photographed (Fig. 4). At 1 h, LPS-exposed EC (Fig. 4C) displayed increased tyrosine phosphorylation of proteins immunolocalized to the intercellular boundaries compared to both medium and ENP controls (Fig. 4A and B, respectively). Monolayers treated with both ENP and LPS (Fig. 4D) could not be distinguished from either the medium or ENP controls.
FIG. 4.

Effect of ENP on phosphotyrosine-containing proteins in LPS-exposed EC. EC monolayers grown on filters were exposed for 1 h to medium (A), ENP (1.0 μg/ml; B), LPS (100 ng/ml; C), or LPS coadministered with ENP (D). The monolayers were fixed, probed with FITC-conjugated antiphosphotyrosine antibody, and photographed through an epifluorescence microscope. Arrows indicate phosphotyrosine signal at intercellular boundaries. Magnification, ×600.
Effects of ENP on the LPS-induced changes in the F- and G-actin pools.
The effect of ENP on the LPS-induced decrement in EC F-actin, expressed as fluorescence units/milligram of total EC protein, was studied (Fig. 5A). There were no significant differences in F-actin content between the medium and ENP controls. A 6-h exposure to LPS (10 ng/ml) decreased F-actin compared to either simultaneous medium or ENP (100 ng/ml) controls. ENP coadministered with LPS diminished the LPS-induced F-actin decrement compared to LPS alone. In fact, F-actin content in EC treated with both LPS and ENP did not significantly differ from that for the simultaneous medium control.
FIG. 5.

Effects of ENP on LPS-induced changes in the F- and G-actin pools. For G- and F-actin measurements, monolayers were exposed for 6 h to medium, ENP, LPS, or LPS coadministered with ENP. (A) For the F-actin studies, monolayers were fixed, permeabilized, incubated with NBD-phallicidin, and extracted with methanol. The extracts were spectrofluorimetrically assayed, and F-actin concentrations were expressed as mean (± SE) fluorescent units per milligram of total EC protein. ∗, significantly decreased compared to medium control; ∗∗, significantly increased compared to LPS alone but not significantly decreased compared to medium alone. (B) For quantitation of the G-actin pool, EC were permeabilized and the G-actin-containing supernatants were tested in the DNase I inhibition assay standardized to pure G-actin. Each bar represents mean (± SE) G-actin expressed. ∗, significantly increased compared to medium control; ∗∗, significantly decreased compared to LPS alone but not significantly increased compared to medium alone. n for each experimental group is indicated in each bar.
The effect of ENP on the LPS-induced increment in EC G-actin, expressed in micrograms/milligram of total EC protein, was also studied (Fig. 5B). A 6-h exposure to LPS (10 ng/ml) increased G-actin compared to either the simultaneous medium or ENP (100 ng/ml) controls. ENP coadministered with LPS reduced the LPS-induced G-actin increment compared to that observed after exposure to LPS alone. This reduction was complete, reducing G-actin to the basal levels seen in the medium control. Therefore, LPS provokes reciprocal shifts between the F- and G-actin pools indicative of EC actin depolymerization, and these changes are completely blocked by ENP.
ENP cross-protects against LPS derived from diverse gram-negative bacterial strains.
ENP offered protection against loss of barrier function in response to a variety of endotoxins normalized on the basis of KDO content to 10 ng of LPS per ml derived from E. coli O111:B4 (Fig. 6). Monolayers were assayed for 14C-BSA flux immediately after 6-h exposures to the following: medium, LPS derived from E. coli O111:B4, E. coli O55:B5, K. pneumoniae, P. aeruginosa, S. minnesota, or S. marcescens or an equivalent concentration of each LPS preparation coadministered with ENP (100 ng/ml). All LPS preparations except that derived from S. marcescens induced comparable increments in 14C-BSA flux. ENP completely protected against LPS-induced increments in 14C-BSA transendothelial flux for all endotoxins tested.
FIG. 6.

ENP cross-protects against a wide variety of endotoxins. Transendothelial 14C-BSA flux was assayed immediately following 6-h exposures to medium (open bar), equivalent concentrations based on KDO content of LPS derived from E. coli O111:B4 (10 ng/ml), E. coli 055:B5, P. aeruginosa, K. pneumoniae, S. marcescens, or S. minnesota (cross-hatched bars), or these same LPS preparations coadministered with ENP (100 ng/ml) (gray bars). Each bar represents mean (± SE) transendothelial 14C-BSA flux. Baseline barrier function for all monolayers studied is also indicated. ∗, significantly decreased compared to LPS alone at P < 0.05 but not significantly increased compared to medium control.
DISCUSSION
In this report, we first confirmed that lipid A was the portion of the LPS molecule presented to the non-CD14-bearing EC. Neither the LPS polysaccharide fraction nor native LPS coadministered with either PMB or ENP simulated the LPS-induced loss of endothelial barrier function. Lipid A has previously been shown to be the active LPS fraction that stimulates CD14-bearing cells (27). Because EC do not express CD14, it was unclear whether EC barrier dysfunction was due to the lipid A portion of LPS. The lipid A fraction in concentrations up to 15 μg/ml induced dose-dependent increases in 14C-BSA flux, whereas the O-polysaccharide fraction did not. This lipid A-induced dose-dependent increase in EC monolayer permeability was similar to the native LPS effect previously described (14). PMB binds with 1:1 stoichiometry to the lipid A portion of LPS and has been shown to neutralize LPS in a number of in vitro and in vivo models (6, 23). Coadministration of LPS with PMB blocked LPS-induced EC barrier dysfunction, demonstrating that in native endotoxin, lipid A is the portion responsible for inducing increments in permeability.
On the basis of these structure-function studies, we tested ENP, which directly targets the lipid A portion of LPS. ENP at concentrations of ≥100 ng/ml totally blocked LPS (10 ng/ml)-induced increases in transendothelial albumin flux. These studies were performed in the presence of serum at concentrations which support the LPS effect. This finding suggests that ENP successfully operates in the presence of serum proteins known to bind to the lipid A portion of LPS, notably LBP and soluble CD14 (14). BPI is a protein found in the azurophilic granules of human neutrophils that is homologous to LBP (4, 10, 28). However, the differences between the two molecules are sufficient that LBP facilitates and enhances LPS bioactivity (28) whereas BPI is inhibitory (10). This homology between LBP and BPI promotes competition between the two proteins for binding to LPS (7, 16). In the circulation, BPI is present in lower concentrations than LBP. In addition, LBP is an acute-phase protein that can increase from basal levels of 2 to 6 μg/ml to 50 μg/ml within 24 h after induction of the acute-phase response (28). Therefore, any effective clinical intervention for endotoxemia must be capable of performing in the presence of LBP as ENP apparently has in these experiments.
ENP and PMB have each been previously reported to bind to lipid A with a 1:1 stoichiometry (6, 23, 32). The LPS-neutralizing activities of these two proteins were compared in the barrier function assay in the presence of serum (Fig. 1B and 2). On a molar basis, ENP (molecular size = 12,189 g/mol) was ∼80-fold more effective at blocking LPS-induced loss of endothelial barrier function than was PMB (molecular size = 1,450 g/mol).
The signal transduction pathway for LPS-induced EC barrier dysfunction is still poorly understood. Protein tyrosine phosphorylation has been shown to be operative in the activation of CD14-bearing monocytes and macrophages (29, 35). More recently, the response of the non-CD14-bearing EC to LPS has been shown to also involve protein tyrosine phosphorylation (2, 3). LPS stimulates tyrosine phosphorylation of several mitogen-activated protein kinases, and protein tyrosine kinase inhibition blocks LPS-induced interleukin-6 biosynthesis and lactic dehydrogenase release. Recently, we have demonstrated that protein tyrosine kinase inhibition protects against LPS-induced barrier dysfunction (3). In addition, we have shown that LPS induces tyrosine phosphorylation of EC proteins localized to the intercellular boundaries as well as of paxillin, a 66-kDa protein which links the actin cytoskeleton to areas of the plasma membrane involved in cell-matrix adhesion (3).
On the basis of the ability of ENP to neutralize LPS bioactivity in the permeability assay, we attempted to show whether ENP blocks the previously described EC responses that are prerequisites to LPS-induced barrier dysfunction. ENP prevented LPS-induced tyrosine phosphorylation of paxillin and EC proteins localized to the intercellular boundaries as well as the G-actin increase and the reciprocal F-actin decrease that is indicative of F-actin depolymerization. Thus, inhibition of LPS-induced protein tyrosine phosphorylation by ENP at ≤1 h prevents the subsequent actin reorganization and loss of barrier function seen at ≥2 h, suggesting that ENP acts at an early step in the pathway proximal to tyrosine phosphorylation.
To our knowledge, only one previous report has studied the ability of a Limulus-derived factor to neutralize LPS bioactivity in an EC system (8). That work, however, differed in several aspects. First, the authors used EC derived from a different species and anatomical source. Second, they used anti-LPS factor derived from Tachypleus tridentatus, the Japanese horseshoe crab. We used a recombinant form of anti-LPS factor, ENP, which was derived from anti-LPS factor isolated from the American horseshoe crab. ENP differs from anti-LPS factor in its mannose content (11). Further, the LPS-induced EC response they attempted to block was EC adhesiveness for neutrophils. Unlike LPS-induced increments in transendothelial albumin flux (15), the EC response they report requires de novo protein synthesis. Finally, these and other investigators who have studied the ability of anti-LPS factor or ENP to neutralize endotoxins derived from various gram-negative bacteria have standardized their LPS preparations on a dry weight/weight basis (8, 32). The variability in the length of the lipid A acyl and O-specific polysaccharide chains precludes such comparison. We, therefore, standardized the endotoxins studied to 10 ng of E. coli O111:B4 per ml on the basis of KDO content (19).
In this study, ENP was able to protect against a wide variety of endotoxins. Clinical intervention for gram-negative septic shock and its complications must offer protection against the enormous diversity of endotoxins found in nature. ENP has been previously shown to protect against LPS-induced tissue injury indirectly mediated through host-derived factors (4, 11, 34). In animal models, ENP protects against LPS-induced pulmonary hypertension, systemic hypotension, metabolic acidosis, neutropenia, and pyrogenicity (1, 34). ENP also reduces circulating levels of free LPS and tumor necrosis factor (1). Pretreatment, coadministration, or even administration of ENP following the LPS challenge results in increased survival rates in sheep, mice, and rats (1, 34). In addition to the ability of LPS to indirectly cause injury through the hosts’ mediator systems, LPS is able to directly provoke increases in vascular permeability (3, 14, 15, 21). In this study, we have demonstrated that ENP is capable of preventing the direct effects of LPS on the endothelial barrier. The ability of ENP to protect against the effects of LPS on the host mediator systems and against the direct effects of LPS on the endothelium, offers a potential therapeutic intervention to protect against gram-negative sepsis and its vascular complications.
ACKNOWLEDGMENTS
This work was supported in part by the Office of Research and Development, Department of Veterans Affairs and the U.S. Army Medical Research and Development Command (grant DAMD17-94-J-4117). Douglas D. Bannerman and Michael J. Fitzpatrick are each a recipient of a Department of Defense Augmentation Award for Science and Engineering Research Training.
REFERENCES
- 1.Alpert G, Baldwin G, Thompson C, Wainwright N, Novitsky T J, Gillis Z, Parsonnet J, Fleisher G R, Siber G R. Limulus antilipopolysaccharide factor protects rabbits from meningococcal endotoxin shock. J Infect Dis. 1992;165:494–500. doi: 10.1093/infdis/165.3.494. [DOI] [PubMed] [Google Scholar]
- 2.Arditi M, Zhou J, Torres M, Durden D L, Stins M, Kim K S. Lipopolysaccharide stimulates the tyrosine phosphorylation of mitogen-activated protein kinase p44, p42, and p41 in vascular endothelial cells in a soluble CD14-dependent manner. Role of protein tyrosine phosphorylation in lipopolysaccharide-induced stimulation of endothelial cells. J Immunol. 1995;155:3994–4003. [PubMed] [Google Scholar]
- 3.Bannerman D D, Goldblum S E. Endotoxin induces endothelial barrier dysfunction through protein tyrosine phosphorylation. Am J Physiol. 1997;273:L217–L226. doi: 10.1152/ajplung.1997.273.1.L217. [DOI] [PubMed] [Google Scholar]
- 4.Battafaraono R J, Dahlberg P S, Ratz C A, Johnston J W, Gray B H, Haseman J R, Mayo K H, Dunn D L. Peptide derivatives of three distinct lipopolysaccharide binding proteins inhibit lipopolysaccharide-induced tumor necrosis factor-alpha secretion in vitro. Surgery. 1995;118:318–324. doi: 10.1016/s0039-6060(05)80340-x. [DOI] [PubMed] [Google Scholar]
- 5.Brigham K L, Meyrick B. Endotoxin and lung injury. Am Rev Respir Dis. 1986;133:913–927. [PubMed] [Google Scholar]
- 6.Cooperstock M S. Inactivation of endotoxin by polymyxin B. Antimicrob Agents Chemother. 1974;6:422–425. doi: 10.1128/aac.6.4.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dentener M A, Von Asmuth E J, Francot G J, Marra M N, Buurman W A. Antagonistic effects of lipopolysaccharide binding protein and bacterial/permeability-increasing protein on lipopolysaccharide-induced cytokine release by mononuclear phagocytes. Competition for binding to lipopolysaccharide. J Immunol. 1993;151:4285–4265. [PubMed] [Google Scholar]
- 8.Desch CE, O’Hara P, Harlan J M. Antilipopolysaccharide factor from horseshoe crab, Tachypleus tridentatus, inhibits lipopolysaccharide activation of cultured human endothelial cells. Infect Immun. 1989;57:1612–1614. doi: 10.1128/iai.57.5.1612-1614.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubois M, Giles K, Hamilton J, Rebers P, Smith F. Colorimetric method for determination of sugars and related substances. Anal Chem. 1956;28:350–356. [Google Scholar]
- 10.Evans T J, Carpenter A, Moyes D, Martin R, Cohen J. Protective effects of a recombinant amino-terminal fragment of human bactericidal/permeability-increasing protein in an animal model of gram negative sepsis. J Infect Dis. 1995;171:153–160. doi: 10.1093/infdis/171.1.153. [DOI] [PubMed] [Google Scholar]
- 11.Fletcher M A, Mckena T M, Quance J L, Wainwright N R, Williams T J. Lipopolysaccharide detoxification by endotoxin neutralizing protein. J Surg Res. 1993;55:147–154. doi: 10.1006/jsre.1993.1122. [DOI] [PubMed] [Google Scholar]
- 12.Frey E A, Miller D S, Jahr T G, Sundan A, Bazil V, Espevik T, Finlay B B, Wright S D. Soluble CD14 participates in the response of cells to lipopolysaccharide. J Exp Med. 1992;176:1665–1671. doi: 10.1084/jem.176.6.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Glauser M P, Zanetti G, Baumgartner J D, Cohen J. Septic shock: pathogenesis. Lancet. 1991;338:732–736. doi: 10.1016/0140-6736(91)91452-z. [DOI] [PubMed] [Google Scholar]
- 14.Goldblum S E, Brann T W, Ding X, Pugin J, Tobias P S. Lipopolysaccharide (LPS)-binding protein and soluble CD14 function as accessory molecules for LPS-induced changes in endothelial barrier function, in vitro. J Clin Invest. 1994;93:692–702. doi: 10.1172/JCI117022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldblum S E, Ding X, Brann T W, Campbell-Washington J. Bacterial lipopolysaccharide induces actin reorganization, intercellular gap formation, and endothelial barrier dysfunction in pulmonary vascular endothelial cells: concurrent F-actin depolymerization and new actin synthesis. J Cell Physiol. 1993;157:13–23. doi: 10.1002/jcp.1041570103. [DOI] [PubMed] [Google Scholar]
- 16.Heumann D, Galley P, Betz-Corradin S, Barras C, Baumgartner J D, Glauser M P. Competition between bactericidal/permeability-increasing protein and lipopolysaccharide-binding protein for lipopolysaccharide binding to monocytes. J Infect Dis. 1993;167:1351–1357. doi: 10.1093/infdis/167.6.1351. [DOI] [PubMed] [Google Scholar]
- 17.Hirata M, Zhong J, Wright S C, Larrick J W. Structure and functions of endotoxin-binding peptides derived from CAP18. Prog Clin Biol Res. 1995;392:317–326. [PubMed] [Google Scholar]
- 18.Hoess A, Watson S, Siber G R, Liddington R. Crystal structure of endotoxin-neutralizing protein from the horseshoe crab, Limulus anti-LPS factor, at 1.5 A resolution. EMBO J. 1993;12:3351–3356. doi: 10.1002/j.1460-2075.1993.tb06008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karkhanis Y D, Zeltner J Y, Jackson J J, Carlo D J. A new and improved microassay to determine 2-keto-3-deoxyoctonate in lipopolysaccharide of Gram-negative bacteria. Anal Biochem. 1978;85:595–601. doi: 10.1016/0003-2697(78)90260-9. [DOI] [PubMed] [Google Scholar]
- 20.Levine D M, Parker T S, Donelly T M, Walsh A, Rubin A L. In vivo protection against endotoxin by plasma high density lipoprotein. Proc Natl Acad Sci USA. 1993;90:12040–12044. doi: 10.1073/pnas.90.24.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyrick B O, Ryan U S, Brigham K L. Direct effect of E. coli endotoxin on structure and permeability of pulmonary endothelial monolayers and the endothelial layer of intimal explants. Am J Pathol. 1986;122:140–151. [PMC free article] [PubMed] [Google Scholar]
- 22.Morita T, Ohtsubo S, Nakamura T, Tanaka S, Iwanaga S, Ohashi K, Niwa M. Isolation and biological activities of limulus anticoagulant (anti-LPS factor) which interacts with lipopolysaccharide (LPS) J Biochem (Tokyo) 1985;97:1611–1620. doi: 10.1093/oxfordjournals.jbchem.a135218. [DOI] [PubMed] [Google Scholar]
- 23.Morrison D C, Jacobs D M. Binding of polymyxin B to the lipid A portion of bacterial lipopolysaccharides. Immunochemistry. 1976;13:813–818. doi: 10.1016/0019-2791(76)90181-6. [DOI] [PubMed] [Google Scholar]
- 24.Muta T, Miyata T, Tokunaga F, Nakamura T, Iwanaga S. Primary structure of anti-lipopolysaccharide factor from American horseshoe crab, Limulus polyphemus. J Biochem (Tokyo) 1987;101:1321–1330. doi: 10.1093/oxfordjournals.jbchem.a121999. [DOI] [PubMed] [Google Scholar]
- 25.Netea M G, Demacker P N M, Kullberg B J, Boerman O C, Verschueren I, Stalenhoef A F H, van der Meer J W M. Low-density lipoprotein receptor-deficient mice are protected against lethal endotoxemia and severe Gram-negative infections. J Clin Invest. 1996;97:1366–1372. doi: 10.1172/JCI118556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Parrillo J E, Parker M M, Natanson C, Suffredini A F, Danner R L, Cunnion R E, Ognibene F P. Septic shock in humans. Advances in the understanding of pathogenesis, cardiovascular dysfunction, and therapy. Ann Intern Med. 1990;113:227–242. doi: 10.7326/0003-4819-113-3-227. [DOI] [PubMed] [Google Scholar]
- 27.Rietschel E T, Kirikae T, Schade F U, Mamat U, Schmidt G, Loppnow H, Ulmer A J, Zahringer U, Seydel U, Di Padova F, Schreier M, Brade H. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 28.Schumann R R, Leong S R, Flaggs G W, Gray P W, Wright S D, Mathison J C, Tobias P S, Ulevitch R J. Structure and function of lipopolysaccharide binding protein. Science. 1990;249:1429–1433. doi: 10.1126/science.2402637. [DOI] [PubMed] [Google Scholar]
- 29.Štefanová I, Corcoran M L, Horak E M, Wahl L M, Bolen J B, Horak I D. Lipopolysaccharide induces activation of CD14-associated protein tyrosine kinase p53/56lyn. J Biol Chem. 1993;268:20725–20728. [PubMed] [Google Scholar]
- 30.Ulevitch R J, Tobias P S. Recognition of endotoxin by cells leading to transmembrane signaling. Curr Opin Immunol. 1994;6:125–130. doi: 10.1016/0952-7915(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 31.Waage A, Brandtzaeg P, Espevik T, Halstensen A. Current understanding of the pathogenesis of gram-negative shock. Infect Dis Clin North Am. 1991;5:781–791. [PubMed] [Google Scholar]
- 32.Wainwright N R, Miller R J, Paus E, Novitsky T J, Fletcher M A, McKenna T M, Williams T. Endotoxin binding and neutralizing activity by a protein from Limulus polyphemus. In: Levin J, Alving C R, Munford R S, Stutz P L, editors. Cellular and molecular aspects of endotoxin reactions. New York, N.Y: Elsevier Science Publishers; 1990. pp. 315–325. [Google Scholar]
- 33.Warren H S, Novitsky T J, Bucklin A, Kania S A, Siber G R. Endotoxin neutralization with rabbit antisera to Escherichia coli J5 and other gram-negative bacteria. Infect Immun. 1987;55:1668–1673. doi: 10.1128/iai.55.7.1668-1673.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Warren H S, Glennon M L, Wainwright N, Amato S F, Black K M, Kirsch S J, Riveau G R, Whyte R I, Zapol W M, Novitsky T J. Binding and neutralization of endotoxin by Limulus antilipopolysaccharide factor. Infect Immun. 1992;60:2506–2513. doi: 10.1128/iai.60.6.2506-2513.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weinstein S L, June C H, DeFranco A L. Lipopolysaccharide-induced protein tyrosine phosphorylation in human macrophages is mediated by CD14. J Immunol. 1993;151:3829–3838. [PubMed] [Google Scholar]