

Abstract
Long-read sequencing platforms provide unparalleled access to the structure and composition of all classes of tandemly repeated DNA from STRs to satellite arrays. This review summarizes our current understanding of their organization within the human genome, their importance with respect to disease, as well as the advances and challenges in understanding their genetic diversity and functional effects. Novel computational methods are being developed to visualize and associate these complex patterns of human variation with disease, expression, and epigenetic differences. We predict accurate characterization of this repeat-rich form of human variation will become increasingly relevant to both basic and clinical human genetics.
Introduction.
Tandem repeat DNA are hotspots of genetic variation and have a longstanding association with human disease. At least five categories of tandem repeats are recognized, defined primarily based on the length of the underlying repeating sequence motif and the total size of the resulting repeat structure. The shortest, termed short tandem repeats (STRs; aka microsatellites, Table 1), typically have a repeat motif length ≤6 bp and were used during early human genetic mapping studies because they exhibit a high degree of heterozygosity among human haplotypes [1]. The overall length of STR arrays rarely exceeds 1 kbp for any given locus, although there are disease-associated exceptions such as the FMR1 CGG repeat that leads to Fragile X syndrome in a hyperexpanded disease state with over 200 triplet repeats[2]. The motif length of variable number tandem repeats (VNTRs; aka minisatellites) are larger (≥7 bp) with the majority of VNTRs harboring motif length repeats between 10–60 units [3]. Larger tandem repeats, typically >1 kbp with respect to the tandem repeat motif, are most often annotated as tandem segmental duplications (SDs; >1 kbp and >90% sequence identity) and are not annotated as other identified classes of repeats such as LINEs or endogenous retroviruses (ERVs), to name a few. Such tandem SDs are distinguished from interspersed SDs, which refer to non-retrotransposed repetitive segments of DNA [4] separated from the nearest identity paralog by >1 Mbp. The structure of these larger tandem SDs can be complex in their organization, often composites of different sequences, and particular subsets were recently dubbed as composite elements during the analysis of the finished human genome [5,6]. Finally, in humans the most abundant centromeric satellite repeats consist of a tandemly repeated alpha-satellite monomer repeat of 170–171 bp. Alpha-satellite repeats are often organized into higher-order repeats (HORs) of specific sets of monomers that expand to reach multiple megabase pairs in length. A subset of these alpha-satellite HORs correspond to the site of kinetochore attachment. While alpha-satellites have a well-established function for mitotic and meiotic segregation of chromosomes, the role of other satellite classes is less well understood. In humans, these are most often associated with shorter motifs (Table 1) and have expanded into megabase-pair structures mapping most frequently within heterochromatic pericentromeric and acrocentric portions of human chromosomes [6,7]. Satellite DNA (HSAT2 &3), for example, typically consists of thousands to tens of thousands of tandem repeats of the pentamer “CATTC” and, in contrast to STRs, are isolated as a distinct fraction from centrifugation gradients of human DNA representing multiple Mbp of contiguous sequence.
Table 1:
Classes of tandem repeats in the human genome
| Class | Motif length | Overall size* | |
|---|---|---|---|
| STR | Short tandem repeat | 1–6 bp | ~200 bp |
| VNTR | Variable number tandem repeat | ≥7 bp | ~500 bp |
| SD | Tandem segmental duplication | >1 kbp | 10–100 kbp |
| SAT | Alpha centromeric satellite DNA | ~171 bp | 500 kbp - multiple Mbp |
| Human satellite 1 (HSat1A) | 42 bp | 890 kbp - multiple Mbp | |
| Human satellite 1 (HSat1B) | ~2.4 kbp | ~14 Mbp | |
| Human satellite 2 (HSat2) | 5 bp | 620 kbp or ~12.6 Mbp | |
| Human satellite 3 (HSat3) | 5 bp | 890 kbp - multiple Mbp | |
| Human satellite 4 (HSat4) | 35 bp | 10kbp - 100kbp | |
| Beta satellite | 68 bp | Variable clusters | |
| Gamma satellite | 220 bp | 10–220 kbp clusters |
When plans to sequence and assemble the human genome were first laid out, there was considerable enthusiasm that the “anatomy” of these regions would be fully revealed [9]. Unfortunately, these regions were often the last to be sequence resolved [3,10–12]. The sequence identity and length of the tandem repeats, in particular, prevented both reliable assembly and cloning of the largest and most mutable regions of the human genome [12–15]. Other factors like skewed GC composition contributed to amplification and cloning biases, such that both the regions and their variation could not be properly assessed. For almost two decades, these regions remained gaps in all human genomic analyses—often regarded as sequence non grata [16]. With the advent of long-read sequencing (LRS) of native DNA (i.e., no bacterial cloning), the telomere-to-telomere (T2T) completion of the first human genome [11], and sequencing of more than 100 human genomes [17,18], a more complete view of the genomic architecture of these regions has emerged. In this review, we apprise our understanding of human genomic organization, emerging properties regarding their variation, and outstanding challenges for visualizing, classifying, and genotyping tandem repeats. While many dozens of diseases have now been described in association tandem repeats as summarized in this review, we anticipate many more as human genetic variation in these dynamic regions becomes more routinely assessed, often for the first time, in the context of controls and patients with disease.
Distribution of tandem repeats in the human genome.
Our ability to accurately resolve the sequence of tandem repeats has significantly improved through the combination of LRS from population genomes where parental data are available or where phasing information can be obtained from specialized next-generation sequencing (NGS) methods, such as Strand-seq and Hi-C [19,20]. As a result, the average full repeat length of VNTRs in the finished human genome (T2T) has more than doubled (585 bp) when compared to the average VNTR length reported for GRCh38 (215 bp). These larger tandem repeats have been placed into the context of chromosomal-level phased genome assemblies [21]. Concerted efforts from the Human Genome Structural Variation Consortium (HGSVC) [17], T2T Consortium [11], and Human Pangenome Reference Consortium (HPRC) [18] have produced valuable resources of long-read genome assemblies and structural variation from diverse human populations. These datasets have provided our first glimpse of the landscape of large tandem repeats in the human genome (Fig. 1).
Figure 1. Genome-wide distribution of tandem repeats in 47 HPRC human genomes.

The ideogram depicts the genome-wide distribution for VNTRs (n=166,918, purple) and STRs (n=131,679, green). We also show HOR regions of the genome enriched in satellite sequences (red) and tandem segmental duplications (SDs; blue) that map less than 1 Mbp apart. The average STR and VNTR lengths are 174 bp and 516 bp, while their average motif lengths are 3 bp and 49 bp, respectively. SDs and centromere satellite annotation are based only on the T2T reference genome.
Early studies of polymorphic tandem repeat markers identified an enrichment of variation in subtelomeric regions of the genome [22–24]. These studies were accurately quantified using long reads that found 55% of polymorphic VNTRs map to subtelomeric portions of human chromosome arms—the final 5 Mbp of sequence proximal to the telomere [25,26]. There is a modest yet significant association between the genome-wide distribution of VNTRs and sites of double-strand breaks (DSB, R2=0.23, P-value < 1E-22), suggesting a link between genomic regions prone to DSB and VNTR formation and we estimated that DSBs account for 23% of the variance in VNTR abundance across the genome. There are two distinct classes of VNTRs. The first is SINE-VNTR-Alu (SVA) retrotransposition-mediated events responsible for the expansion of VNTRs into GC-rich regions of the genome with a bias against genic regions. In contrast, non-SVA-associated VNTRs appear to accumulate preferentially near genes and are the most likely to be enriched in subtelomeric regions [3].
The recently published phased genome assemblies from 47 diverse humans confirm these original observations [18]. Using structural variants reported by the long-read base caller PBSV [27], we identify 145,966 polymorphic VNTRs with repeat motif ≥7 bp and an overall length <1 kbp. The subtelomeric bias is still observed though less pronounced as more genomes of greater diversity are included; 33.5% and 9.6% of the VNTRs and STRs are located in the subtelomere, respectively. The difference in subtelomeric enrichment is likely due to a combination of the increased population diversity in the HPRC genomes and any biases introduced by the different variant-calling methods employed in both studies. An additional 20,952 VNTRs are greater than 1 kbp in length and are potentially more likely to become unstable generationally. STR elements (n=131,679) are more uniformly distributed within the human genome and have an average size of 217 bp. Even larger tandem SDs and nearly complete satellite arrays have now been assembled, although these regions still pose challenges for the complete T2T assembly of diploid genomes [28]. SDs are shown to have higher mutation rates and interlocus gene conversion rates than unique DNA [14]. Figure 1 depicts our current understanding of the landscape of tandem repeats based on the analysis of 47 recently released HPRC genomes using Tandem Repeats Finder [29] and the GRCh38 UCSC table browser tracks for SDs and satellite sequences.
Advances in the discovery and alignment of tandem repeats.
Computational analysis of tandem repeat sequences is performed by de novo identification in reference genomes followed by targeted analysis of variation from sample sequence reads. While the sequence and assembly of large tandem repeat sequences have now been made possible by LRS, the characterization of their underlying variation has been challenging. Generalized algorithms for discovering variation from short reads, including single-nucleotide variants and small indels [30,31], as well as structural variation [32–35] have been shown to perform poorly in repetitive DNA [21,36–38]. Additionally, although variation in tandem repeats is accurately identified in current long-read data analyzed by context-agnostic LRS software [21,27], there are difficulties in comparing variant calls in repetitive DNA between methods and across individuals [27,39,40]. To address this, several sequence analysis tools have been developed to analyze specific classes of tandem repeat variation, with the computational approach governed by the relative length of repeat alleles to read lengths, prior knowledge of repeat domain structure, and read error profile.
Tools that discover variation in tandem repeats rely on de novo computational annotation of tandem repeat loci in reference genomes. RepeatMasker [41] performs a naive scan for mono, di, and trinucleotide repeats, while Tandem Repeats Finder (TRF) [29], TANTAN [42], and ULTRA [43] use more sophisticated algorithms based on k-mer repeats and hidden Markov models to find inexact repetitions of longer motif patterns. While multiple algorithms and theoretical studies have demonstrated more sensitive results for tandem repeat annotation than TRF [44–47], this method is used by RepeatMasker and in the University of California, Santa Cruz Genome Browser [48] annotations and, thus, forms the basis for most targeted repeat studies. Reliance on the human reference GRCh38 annotations misses some tandem repeat loci; an analysis of TRF annotation of 148 haplotype-resolved assemblies discovered an additional 5,294 annotations missing from the default annotations. Larger repeats including SD may be annotated using DupMasker [49], as well as SEDEF [50] and BISER [51] that are recently developed and are more computationally efficient. Finally, methods including SRF [52], RepeatNet [53], and Alpha-CENTAURI [54] have been written to annotate alpha-satellite repeats from sequencing reads because de novo assembly of centromeric DNA remains challenging.
The approaches to analyze tandem repeat variation using short reads fall into three groups: methods to genotype exact STR alleles fully contained by individual reads, targeted approaches to detect pathogenic repeat expansions, and methods that estimate VNTR length or composition by read depth. The RepeatSeq [55] and lobSTR [56] methods detect STR variation spanned by short reads (up to 100 bases) and provide exact counts of repeat motifs in each haplotype (Fig. 2a). The hipSTR method includes modeling of artifacts of polymerase chain reaction to improve genotype accuracy and enable identification of de novo repeat expansions. Furthermore, hipSTR identifies the non-reference repeat motifs common at highly variable loci.
Figure 2. Methods for discovery, genotyping and annotations.
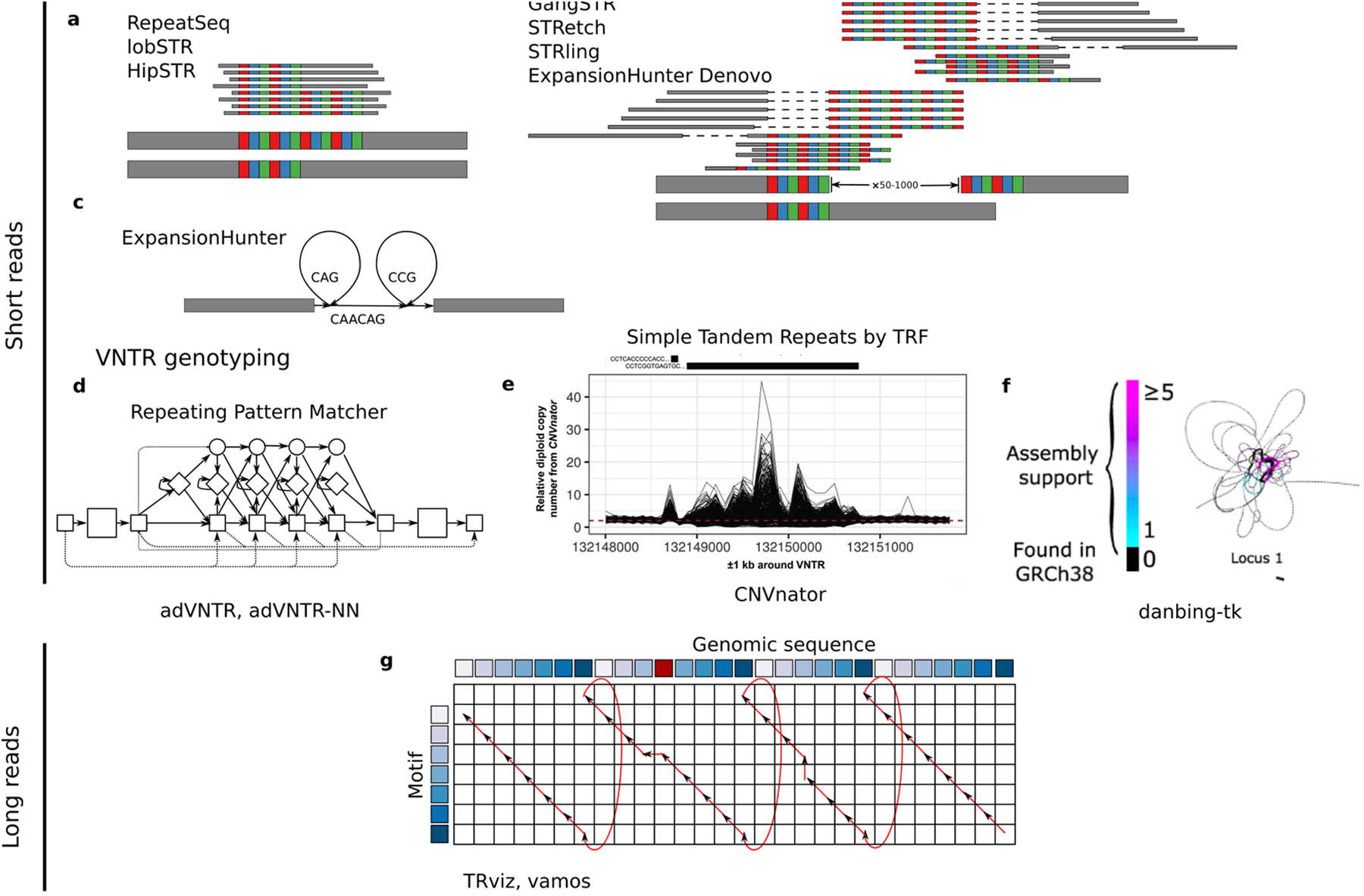
Top, methods to genotype STR expansions using short reads. a, STR genotyping methods that require reads to fully span alleles: RepeatSeq, lobSTR, and hipSTR. These may provide exact repeat counts as well as phased variants. b, STR genotyping methods that can genotype expansions larger than the length of a read or paired-end insert: TREDPARSE, GangSTR, STRetch, STRling, and ExpansionHunter Denovo. The resulting calls are an estimate of motif repeat counts. c, ExpansionHunter genotypes long expansions that fit predefined patterns. Middle, methods to genotype VNTR expansions using short reads. d, adVNTR and adVNTR-NN use a hidden Markov model to estimate repeat unit copy number. e, Copy number defined by CNVnator correlates with ground-truth copy number with sufficient accuracy for association analysis. f, Genotyping VNTR length using repeat pangenome graphs can detect changes in motif composition. Bottom, methods to genotype VNTR alleles with long reads. g, Schematic of algorithmic approach used by TRviz and vamos to annotate motif copies in LRS and genome assemblies using wrap-around dynamic programming that aligns an optimal number of copies of a motif sequence to a genomic sequence by copying alignment scores from the last row to the first and allowing the trace back path index (red) to wrap around from the beginning to the end of the motif.
Pathogenic repeat expansions such as those at the HTT and C9orf72 loci may be many times greater than the length of a short read and cannot be measured in short-read data using methods requiring reads to span the repeat allele. Nevertheless, they may be detected indirectly in short-read data by excess read alignments and paired-end discordance at known tandem repeat loci. TREDPARSE [57] and GangSTR [58] incorporate information from paired-end reads and likelihood models to genotype repeat expansions, including triplet repeat expansions with nearly 1,000 motif copies (FIg. 2b). exSTRa[59] uses similar read information but is designed to detect outliers rather than specific alleles. ExpansionHunter [60,61] genotypes pathogenic repeat expansions with known motif patterns, such as the HTT repeat expansion (CAG)*CAACAG(CCG)* that may have a variable number of CAG and CCG trinucleotide repeats (Fig. 2c). Because reads often misalign at tandem repeat loci, the GangSTR method and STRetch [62] include reads mapped to off-target or STR decoys to improve the estimated tandem repeat length. To allow the discovery of novel repeat expansions, STRling [63] and ExpansionHunter Denovo [64] can similarly estimate repeat expansions without specific tandem repeat intervals being specified but often fail to report both the true length or underlying sequence architecture.
The motif diversity and length of VNTR loci make them recalcitrant to variation analysis with software designed for STRs [65]. For relatively short VNTRs (up to 21 motif repeats), adVNTR and adVNTR-NN [65,66] use a hidden Markov model to estimate motif counts for targeted VNTR loci, with adVNTR-NN using a neural network to improve read recruitment (Fig. 2d). For relatively large VNTR loci, >100 bases with repeat motifs at least 10 bases, read depth measured by CNVnator [67] calibrated by flanking sequences provides a relatively accurate measurement of tandem repeat length [68] (Fig. 2e). Read alignment to a repeat pangenome graph constructed from VNTR sequences using danbing-tk can identify VNTR expansions [69] as well as expansions of individual motifs (Fig. 2f) [70]. Finally, there are bespoke methods for genotyping specific disease or biologically relevant loci, such as LPA [71] and telomeres [72].
Exact measurement of tandem repeat variation using short reads is thus limited to short loci, and remaining loci only have estimations of repeat variation. These challenges are solved using LRS because the long reads or their assemblies span the full length of the STR and VNTR loci enabling precise discovery of tandem repeat variation from alignments [21,73]. The initial methods for tandem repeat analysis using long reads focused on overcoming the high error rates of single-molecule sequencing to count repeat copy number from Pacific Biosciences (PacBio) continuous long reads [74], or from Oxford Nanopore Technologies (ONT) current signal data [75,76]. However, with the accuracy provided by PacBio HiFi (high-fidelity) sequencing [27] and pore improvements paired with consensus sequencing from ONT [77], low-level methods are not necessary. Instead, a greater challenge is in organizing highly polymorphic VNTR sequences at the population level. The SV-Merger [78] and Jasmine [40] methods use graph clustering to combine variant calls across individuals; however, they can underestimate diversity and do not distinguish tandem repeat motif composition. Two similar methods, TRviz [79] and vamos [80], annotate the motif composition of tandem repeats from long-read sequences. Both methods use an algorithmic approach called wrap-around dynamic programming (Fig. 2g) that in contrast to standard local alignment [81] aligns a single query sequence (motif) to a target (genomic VNTR sequence) and finds the optimal matching between repeated motifs to LRS or genome assemblies [82,83]. A summary of software and their targeted classes of variation is given in Table 2.
Table 2:
Methods for tandem repeat variant annotation and discovery
| Reference annotation | |||||
|---|---|---|---|---|---|
| Class of repeat | Method | Reference | |||
| STR, VNTR | Repeat Masker | [41] | |||
| STR, VNTR | Tandem Repeats Finder | [29] | |||
| STR, VNTR | TANTAN | [42] | |||
| STR, VNTR | ULTRA | [43] | |||
| Satellite | SRF | [52] | |||
| Satellite | Repeatnet | [53] | |||
| Satellite | Alpha-CENTAURI | [54] | |||
| Segmental Duplication | DupMasker | [49] | |||
| Segmental Duplication | SEDEF | [50] | |||
| Segmental Duplication | BISER | [51] | |||
| Variant discovery, short-reads | |||||
| Class of variant | Method | Size range* | Precision | Untargeted** | |
| STR | RepeatSeq | < RL | exact | [55] | |
| STR | LobSTR | < RL | exact | [56] | |
| STR | hipSTR | < RL | exact | [95] | |
| STR | TREDPARSE | < 500 bp | exact, estimate | [57] | |
| STR | ExpansionHunter | < 1kb | exact, estimate | [60] | |
| STR | exSTRa | Any | estimate | [59] | |
| STR | ExpansionHunter-denovo | Any | estimate | yes | [64] |
| STR | STRetch | Any | estimate | [62] | |
| STR | STRling | Any | exact, estimate | yes | [63] |
| STR,VNTR | GangSTR | Any | exact, estimate | [58] | |
| VNTR | adVNTR-NN | Any | estimate | [66] | |
| VNTR | CNVnator | Any | estimate | [67] | |
| VNTR | danbing-tk | Any | estimate | [69] | |
| Variant discovery, long-reads | |||||
| VNTR | TRViz | < RL | exact | [79] | |
| VNTR/STR | vamos | < RL | exact | [80] | |
Size range <RL are limited to the sample read length.
Untargeted methods do not require a list of input tandem repeat loci to analyze.
The PacBio and ONT technologies each currently offer distinct advantages in the analysis of tandem repeats. ONT supports an ultra-long sequencing protocol with N50 read lengths (minimum length contained by 50% of data) over 100 kbp [84]. In contrast, PacBio HiFi reads are consistently more accurate than ONT (r10.4) with error rates less than ~1 in 1000 base pairs. Furthermore, more computational support is available for phased genome assembly using PacBio HiFi reads [85]. This combination of HiFi and ONT reads can produce telomere-to-telomere assemblies that resolve the majority of centromeres [86]. This is particularly useful for fully resolving the longest tandem repeat structures and was critical for the contiguous sequence assembly of the first human centromeric satellites [7]. While both platforms can now be used to reliably detect CpG methylation [87], it is possible to determine longer range phasing of methylated sites using ONT reads. For practical considerations, ONT sequencing is capable of higher-throughput sequencing of up to 48 multiplex samples at once using the PromethION, offers portable sequencing using the MinION, and requires less up-front capital investment than the PacBio Revio. However, both technologies are rapidly advancing and future specifications may differ.
Expansions of tandem repeat sequences are often associated with changes in methylation [88–90], yet mapping methylated bases with the standard approach of bisulfite sequencing with short-read technologies is challenging because of the sequence divergence and alignment ambiguity. A promising application of LRS is the ability to detect methylated bases directly from native DNA. The Remora base caller for ONT sequences can detect 5-methylcytosine (5mC) bases from current (squiggle) data. These data have been used to characterize methylation patterns in satellite repeats of a complete human genome [91]. When combined with phasing, this can produce haplotype-resolved methylation maps that have a genome-wide correlation of 0.949 with whole-genome bisulfite sequencing[92]. Recently, PacBio released the Jasmine software to detect 5mC from HiFi sequencing data. Both approaches rely on detecting changes in kinetics unique to methylated bases from low-level sequencing data. These properties have been used to map protein-DNA interactions by treating DNA with a methyltransferase such that interacting bases are protected from methylation and interactions are read as the inverse of the methylation signal [93,94].
Advances in visualization methods.
Sequenced-based resolution of the tandem repeats has revealed complex higher-order structures even for alleles with the same overall repeat length necessitating the need to develop more sophisticated tools to investigate their underlying architecture. Several groups have begun to tackle the challenge of resolving tandem repeat architecture by creating programs to extract sequence information, often specifically for STRs, VNTRs, and centromere satellites. Some of the early strategies for demonstrating variability in VNTR sequence and structure involve color-coding internal repeats, as is the case for PRDM9 [96,97]. Dot plot graphs are often used to demonstrate commonality of repeat structure within a genomic region and variability of length; however, these matrices do little to demonstrate the diversity of sequence information (Fig. 3a). In the past, custom scripts were developed to align VNTRs and reveal their structure, such as in CACNA1C [98] and WDR7 [99,100]. Other programs are now emerging that specifically address the need to visualize VNTRs, producing, for example, “waterfall” plots [3] (Fig. 3b) that show changes in cassette architecture based on tracking of k-mers as a function of VNTR length. The TRviz program takes VNTR sequences that have been extracted, identifies the most common k-mers (repeat motifs), next assigns an ASCII character to the most common character, and then converts the character to a color to produce an alignment based on internal sequence homology, what we refer to as a “Seattle plot” (Fig. 3c) [79]. A similar program, vamos, adjusts parameters for generating efficient motif sets at VNTRs and has the benefit of collapsing repeats into fewer colors to help visualize patterns when merging rare or private motifs [80]. Other tools like TRVZ have been made available to visualize VNTRs from long-read sequences based on the knowledge of individual repeat motifs [101] in addition to the tandem repeat annotation library (TRAL), which has been used to generate what are termed “Mola plots” for several VNTRs in SLC6A3 [102].
Figure 3. Methods to visualize tandem repeats and their variation.
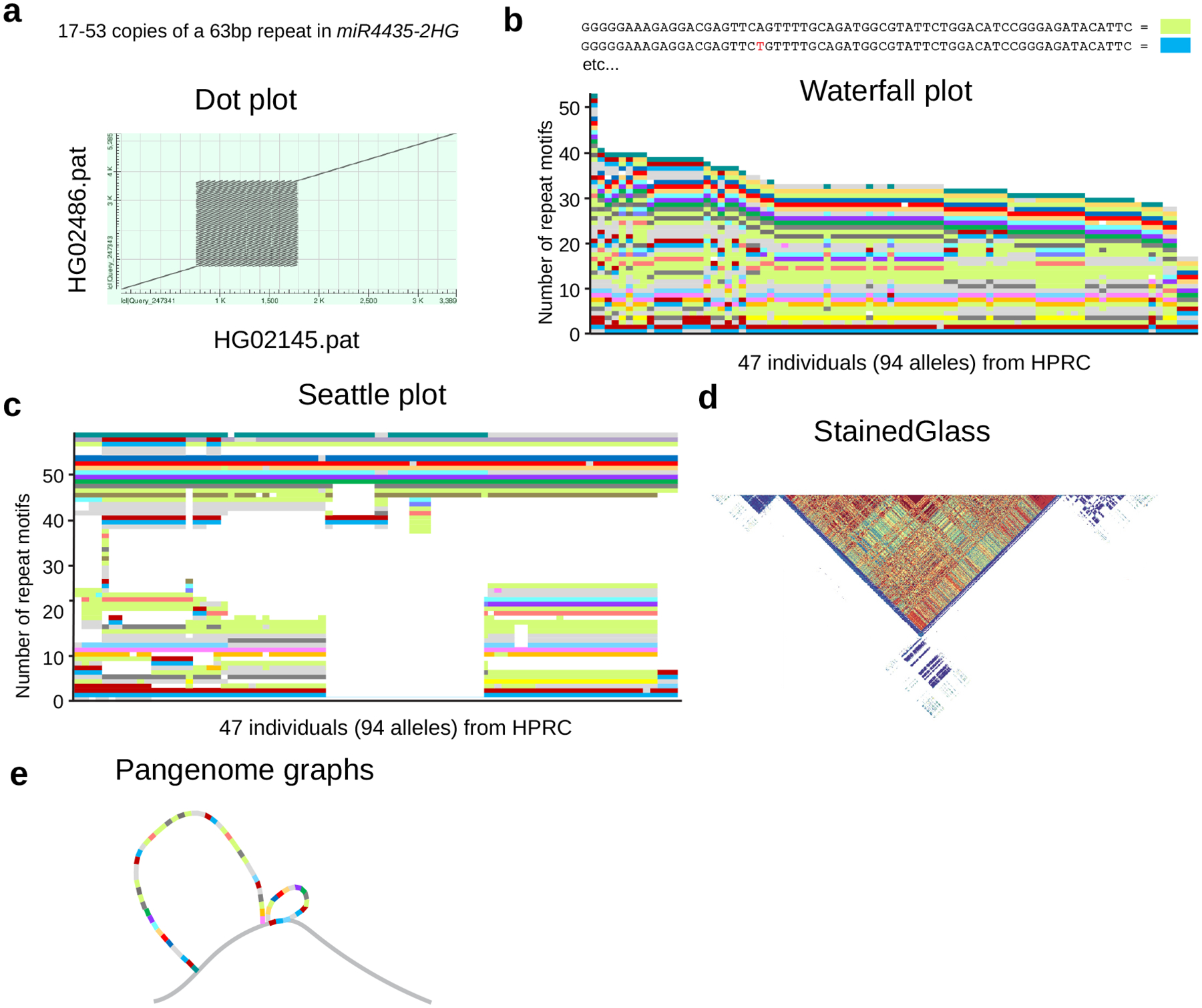
a, A 63 bp VNTR in MIR4435-2 host gene (MIR4435-2HG) is used as an example (top). A dot plot alignment of one allele each from two individuals is shown (bottom). b, A Waterfall plot where repeat motifs are assigned various colors based on sequence identity (top) and then sorted by total length (bottom). This strategy is also often used to demonstrate individual reads separated by allele for one individual. c, A Seattle plot, which takes color-coded alleles and organizes them by internal sequence similarity to highlight clusters of related alleles. Insertions and deletions (representing whitespace gaps) are observed in this context. d, A StainedGlass plot (reproduced from [104]) demonstrating sequence homology for a tandem repeat in heatmap form, often used for centromeric repeats. Heat map defines % identity of the higher-order repeat (red~99% identical versus blue ~70% identity) e, A Pangenome graph highlighting different common paths that a repeat can take in the context of the genome including other structural variants, like two that are shown here.
Some tools, such as StainedGlass and its derivative ModDotPlot, have been designed to interactively handle the massive amount of sequence associated with centromeric and pericentromeric satellite DNA [103]. StainedGlass, for example, generates colored heatmaps based on sequence identity across megabase pairs of DNA and can uncover long-range higher-order structures and regions of recent homogenization identifying distinct evolutionary layers in the formation of these regions (Fig. 3d) [104]. Given that higher sequence identity often reflects more recent evolutionary events, the tool provides a snapshot of evolutionary change and recent gene conversion across otherwise highly identical repeats. Other tools, such as DupMasker [49] and RepeatMasker [41], have proven very useful for defining the composition of tandem SDs by identifying the evolutionary structure of the underlying cassette that is tandemly repeated [5,11,103].
These programs notwithstanding, providing a robust and streamlined approach to visualize tandem repeats remains a challenge to the genetics community. All current programs, for example, prioritize stand-alone visualization of tandem repeats without consideration of how they vary among different individuals. Such realizations may be uncovered through integrating repeat structure and variation via pangenome graphs [69,105] (Fig. 3e) although these regions have proven particularly challenging. Most of the popular tools, unfortunately, do not adequately resolve these details at present, at least for the largest and most variable tandem repeats in our genome. Part of the difficulty in visualizing high-copy tandem repeats arises from the diversity and complexity of the underlying sequence that is present. VNTRs differ in repeat length but also internal sequence differences that often cannot be evenly divided into individual repeat units. In some VNTRs, the repeat motif length is highly variable, or harbor repeats-within-repeats (such as in a VNTR near PROP1 [106]), which presents obstacles to identifying the various internal repeat sequence motifs. Centromere satellites similarly contain new higher-order repeat units that are derivative from a larger higher-order repeat unit sequence already present in the surrounding sequence making alignments challenging. These issues are further compounded by assembly errors and sequence errors, especially at homopolymer runs creating artifacts of variation. Moreover, VNTR start and end positions are often not precisely recorded, so portions of a repeat motif will appear at the end of a VNTR. For example, the WDR7 repeat ends in 46 bp of the 69 bp motif [99]. Convention dictates that the repeat motif starts with the first nucleotide in the VNTR, but the true origination of the repeat may differ, and the repeat may originate and expand from a 3’ to 5’ (rather than 5’ to 3’) direction. Altogether, programs for visualizing VNTRs need to accommodate several pieces of information to produce an output that is accurate yet also clearly informative of the internal sequence.
Nevertheless, with appropriate methodology often geared to a particular class or even an individual locus, tandem repeat visualization methods have become powerful tools to quickly identify and demonstrate differences in repeat length and internal sequence composition. For instance, color-coding is effective at highlighting where a shift in repeat motif occurs in ATXN10 [107], or interruptions in the CAG repeat in HTT that correlate with age of onset of Huntington’s Disease. In the case of FMR1, the identification of alleles that have lost these interruptions has become a predictor of those that will ultimately become permutation alleles [108]. In a more complex example, a VNTR in ART1 ranges from 9 to 500 copies (0.5 to 31.5 kbp) in HGSVC samples. Only after breaking the repeat into color-coded repeat units do patterns emerge for samples, consisting of higher order ~3 kbp (47 repeat motif copies) duplication events[106]. Overall, a universal tool for handling each repeat is not currently available, but conceivably could consist of a combination of tools that provide a comprehensive overview of a repeat, beginning with a high level view of the repeat structure (e.g. via StainedGlass), breaking the repeat apart into individual repeat units and clustering individuals based on length or sequence similarity (e.g. via vamos or TRviz), and then providing a combined output via a pangenome graph. As increasing numbers of long-read genomes become available, having the appropriate programs to visualize tandem repeats will be critical in deducing patterns variation and mechanisms of expansion events as well as their evolutionary origin.
Tandem repeats and disease.
One of the primary motivations for systematic discovery and characterization of these loci has been their association with genomic instability and disease. Tandem repeats have been estimated to mutate at orders of magnitude higher than most other unique regions of the genome [109] and variation at these loci has been implicated in Mendelian, as well as seemingly non-Mendelian, and more complex genetic disease. Pathogenic mutations result typically when repeat lengths either go below or exceed some threshold often resulting in changes of expression of some nearby genes wherein the repeats are embedded. Such is the case with reductions of the D4Z4 repeat associated with facioscapulohumeral muscular dystrophy type I [110] where contraction below 11 copies leads to a permissive chromatin state and the expression of DUX4 in muscle tissue and disease. Similarly, reductions of the tandem SD encoding the two exons of the Kringle IV domain of lipoprotein A gene lead to higher levels of lipoprotein (a) in the circulating blood and is one of most significant genetic risk factors for coronary heart disease and stroke especially among individuals of African ancestry [111].
More often, however, an increase in repeat length is associated with pathogenicity. This is the case with many triplet repeat disorders where expanded repeats lead to disruption of the normal function of the protein or hypermethylation of the promoter and silencing [88–90]. In the case of Huntington’s disease, expansion of a protein-coding CAG repeat within HTT beyond 41 units results in the formation of an abnormal protein that accumulates in the brain, causing neurodegeneration [112]. In the case of myotonic dystrophy, expansion of the CTG nucleotide repeat beyond the normal range (5–37 repeats) in the 3′ untranslated region (UTR) of the dystrophia myotonica protein kinase (DMPK) gene leads to mRNA instability and decreased expression (Table 3) [113,114]. Several genes, such as XYLT1 (Baratela-Scott syndrome) and FMR1 (Fragile X syndrome), possess CGG repeats within the 5′ UTR of the gene. In both cases expansion of the CGG repeat beyond the premutation size range (100–200 repeats) results in hypermethylation of the nearby promoter region followed by silencing of gene expression and disease [88,115]. Importantly, the discovery of expanding triplet repeats and their transmission within families provided the molecular basis for genetic anticipation for several genetic disorders, including increased penetrance, severity, as well as earlier age of onset in subsequent generations [116].
Table 3:
Tandem repeats associated with disease.
| Gene | Repeat | Normal length | Pathogenic Length | Context | Cyto | Inheritance | Disease | Phenotype OMIM # | Clin testing for expansion? | Ref |
|---|---|---|---|---|---|---|---|---|---|---|
| ABCA7 | 25 bp | 12–427 | >200 | intron | 19p13.3 | AD | Susceptibility to Alzheimer’s disease | 608907 | n | [132] |
| AFF2 | CCG | 4–39 | >200–900 | 5’UTR | Xq28 | XLR | Intellectual developmental disorder, X-linked 109 | 309548 | n | [133] |
| AR | CAG | 9–36 | > 37–68 | exon | Xq12 | XLR | Spinal and bulbar muscular atrophy of Kennedy (SBMA) | 313200 | y | [134] |
| ARX | GCN | 12–16 | 20–23 | exon | Xp21.3 | XLR | Developmental and epileptic encephalopathy 1 (EIEE1) | 308350 | n | [135] |
| ATN1 | CAG | 3–35 | >47–93 | exon | 12p13.31 | AD | Dentatorubral-pallidoluysian atrophy (DRPLA) | 125370 | y | [136] |
| ATXN1 | CAG | 6–38 | >38–88 | exon | 6p22.3 | AD | Spinocerebellar ataxia 1 (SCA1) | 164400 | y | [137] |
| ATXN10 | ATTCT/ATTGT | 10–32 | >280–4500 | intron | 22q13 | AD | Spinocerebellar ataxia 10 (SCA10) | 611150 | n | [138] |
| ATXN2 | CAG | 13–31 | >31–500 | exon | 12q24.12 | AD | Spinocerebellar ataxia 2 (SCA2) | 183090 | y | [139] |
| ATXN3 | CAG | 12–44 | >54–87 | exon | 14q32.12 | AD | Spinocerebellar ataxia 3 (SCA3); Machado-Joseph disease | 109150 | y | [140] |
| ATXN7 | CAG | 4–33 | >36–460 | exon | 3p14.1 | AD | Spinocerebellar ataxia 7 (SCA7) | 164500 | y | [141] |
| ATXN8 / ATXN8OS | CAG/CTG | 15–50 | >74 | 3’UTR | 13q21 | AD | Spinocerebellar ataxia 8 (SCA8) | 608768 | y | [142] |
| BEAN1/TK2 | TAAAA* | variable | ≥ 110–760 | intron | 16q22 | AD | Spinocerebellar ataxia 31 (SCA31) | 117210 | n | [143] |
| C9orf72 | GGGGCC | 3–25 | >30 | 5’UTR/intron | 9p21.2 | AD | Frontotemporal dementia and/or amyotrophic lateral sclerosis 1 | 105550 | y | [144] |
| CACNA1A | CAG | 4–18 | 19–33 | exon | 19p13.13 | AD | Spinocerebellar ataxia 6 (SCA6) | 183086 | y | [145] |
| CACNA1C | 30 bp | variable | na**** | intron | 12p13.33 | AD | Schizophrenia and bipolar disorders | 620029 | n | [98] |
| CNBP | CCTG/CAGG | 11–30 | >50–11000 | intron | 3q21.3 | AD | Myotonic dystrophy type 2 (DM2) | 602668 | y | [146] |
| CSTB | CCCCGCCCCGCG | 2–3 | ≥ 30–75 | promoter/5’UTR | 21q22.3 | AR | Unverricht-Lundborg syndrome (EPM1) | 254800 | y | [147] |
| DAB1 | ATTTT** | 7–400 | >31–75 | intron | 1p32 | AD | Spinocerebellar ataxia 37 (SCA37) | 615945 | n | [148] |
| DMPK | CTG | 5 – 37 | >50 – 2,000 | 3’UTR | 19q13.32 | AD | Steinert myotonic dystrophy syndrome (DM1) | 160900 | y | [149] |
| FGF14 | GAA | 50 | >250 | intron | 13q33.1 | AD | Late-Onset Spinocerebellar Ataxia 27B | 620174 | y | [123] |
| FMR1 | CGG | 5–50 | >200 | 5’UTR | Xq27.3 | XLD | Fragile X syndrome (FXS) | 300624 | y | [150] |
| FMR1 | CGG | 5–50 | 55–200 | 5’UTR | Xq27.3 | XLR | Premature ovarian failure 1 (POF1) | 311360 | y | [151] |
| FMR1 | CGG | 5–50 | 55–200 | 5’UTR | Xq27.3 | XLR | Fragile X-associated tremor/ataxia syndrome (FXTAS) | 300623 | y | [152] |
| FOXL2 | GCN | 14 | 19–24 | exon | 3q22.3 | AD | Blepharophimosis, epicanthus inversus, and ptosis, type 1 (BPES) | 110100 | n | [153] |
| FXN | GAA | 5–34 | >66–1300 | intron | 9121.11 | AR | Friedreich ataxia 1 (FRDA) | 229300 | y | [154] |
| GIPC1 | CGG | 12–32 | ≥ 97–120 | 5’UTR | 19p13.12 | AD | Oculopharyngodistal myopathy 2 (OPDM2) | 618940 | n | [155] |
| GLS | GCA | 8–16 | ≥ 680 – 1400 | 5’UTR | 2q32.2 | AR | Global developmental delay, progressive ataxia, and elevated glutamine | 618412 | n | [156] |
| HOXA13 | GCG | 12–18 | 18–30 | exon | 7p15.2 | AD | HFGSHand-foot-uterus syndrome (HFU) | 140000 | n | [157] |
| HOXD13 | GCG | 15 | 24 | exon | 2q31.1 | AD | Synpolydactyly (SPD1) | 186000 | n | [158] |
| HTT | CAG | 6–35 | >36 | exon | 4p16.3 | AD | Huntington disease (HD) | 143100 | y | [159] |
| JPH3 | CAG | 6–28 | >40–58 | exon | 16q24.2 | AD | Huntington disease-like 2 (HDL2) | 606438 | n | [160] |
| LRP12 | CGG | 13–45 | 90–130 | 5’UTR | 8q22.3 | AD | Oculopharyngodistal myopathy 1 (OPDM1) | 164310 | n | [161] |
| MARCHF6 | ATTTT** | 10–30 | ≥ 660–2800 | intron | 5p15.2 | AD | Epilepsy, myoclonic, familial adult, 3 (FAME3) | 613608 | n | [120] |
| NOP56 | GGCCTG | 5–14 | ≥ 650–2500 | intron | 20p13 | AD | Spinocerebellar ataxia 36 (SCA36) | 614153 | n | [162] |
| NOTCH2NLC | CGG | 7–60 | ≥ 61–500 | 5’UTR/exon1 | 1q21.2 | AD | Neuronal intranuclear inclusion disease (NIID) | 603472 | n | [163] |
| NUTM2B-AS1 | CGG/CCG | 3–16 | 40–60 | noncoding RNA | 10q22.3 | AD | Oculopharyngeal myopathy with leukoencephalopathy 1 (OPML1) | 618637 | n | [121] |
| PABPN1 | GCG | 6–10 | >11–17 | exon | 14q11.2 | AD | Oculopharyngeal muscular dystrophy (OPMD) | 164300 | n | [164] |
| PHOX2B | GCN | 20 | 25–29 | exon | 4p13 | AD | Central hypoventilation syndrome 1 (CCHS) | 209880 | n | [165] |
| PLIN4 | 99 bp | 27–31 | 40 | exon | 19p13.3 | AD | Myopathy with rimmed ubiquitin-positive autophagic vacuolation (MPUPAV) | 613247+ | n | [166] |
| PPP2R2B | CAG | 4–32 | ≥ 43–78 | 5’UTR | 5q31 | AD | Spinocerebellar ataxia 12 (SCA12) | 604326 | y | [167] |
| PRDM12 | GCG | 12 | 18–19 | exon | 9q34.12 | AR | Neuropathy, hereditary sensory and autonomic, type VIII (HSAN8) | 616488 | n | [168] |
| PRNP | 24 bp | 1 | 6 | exon | 20p13 | AD | Creutzfeldt-Jakob Disease | 123400 | n | [169] |
| RAPGEF2 | ATTTT** | na | na | intron | 4q32.1 | AD | Epilepsy, myoclonic, familial adult, 7 (FAME7) | 618075 | n | [118] |
| RFC1 | AAAAG / AAAGG / AAGAG / AGAGG*** | variable | ≥ 400 – 2000 | intron | 4p14 | AR | Cerebellar ataxia with neuropathy and bilateral vestibular areflexia syndrome (CANVAS) | 614575 | y | [170] |
| RUNX2 | GCN | 17 | 27 | exon | 6p21.1 | AD | Cleidocranial dysplasia 1 | 119600 | n | [171] |
| SAMD12 | ATTTT** | 7-exp | ≥ 440–3680 | intron | 8q24 | AD | Epilepsy, myoclonic, familial adult, 1 (FAME1) | 601068 | n | [172] |
| SOX3 | GCN | 15 | 26 | exon | Xq27.1 | XLR | Intellectual developmental disorder, X-linked, with isolated growth hormone deficiency | 300123 | n | [173] |
| STARD7 | ATTTT** | 9–20 | ≥ 661–735 | intron | 2q11.2 | AD | Epilepsy, myoclonic, familial adult, 2 (FAME2) | 607876 | n | [119] |
| TAF1 | CCTCT | none | 30–55 | intron | Xq13.1 | XLR | X-linked Dystonia-Parkinsonism (XDP) | 314250 | n | [174] |
| TBP | CAG (or CAG/CAA) | 25–40 | >42–66 | exon | 6q27 | AD | Spinocerebellar ataxia 17 (SCA17) | 607136 | y | [175] |
| TCF4 | CTG | 5–31 | > 50 | intron | 18q21.2 | AD | Fuchs endothelial corneal dystrophy 3 (FECD3) | 613267 | y | [176] |
| TNRC6A | ATTTT** | na | na | intron | 16p12.1 | AD | Epilepsy, myoclonic, familial adult, 6 (FAME6) | 618074 | n | [118] |
| WDR7 | 69 bp | variable | na ***** | intron | 18q21.31 | AD | Amyotrophic lateral sclerosis (ALS) | 606640 | n | [99] |
| XYLT1 | GGC | 9–20 | > 100 | promoter | 16p12.3 | AR | Desbuquois dysplasia 2; Baratela-Scott Syndrome (BSS) | 615777 | n | [177] |
| YEATS2 | ATTTT** | 7–400 | na | intron | 3q27.1 | AD | Epilepsy, myoclonic, familial adult, 4 (FAME4) | 615127 | n | [178] |
| ZIC2 | GCG | 15 | 25 | exon | 13q32.3 | AD | Holoprosencephaly 5 (HPE5) | 609637 | n | [179] |
repeat motif is TGGAA/TAGAA when pathogenic
repeat motif is ATTTC when pathogenic
repeat motif is AAGGG when pathogenic
specific 30-mer sequences are associated with disease risk
longer repeat associated with ALS
MIM for PLIN4, as the phenotype is not in OMIM
Over the last decade other more heterogeneous complex disorders often neurological in nature with diverse genetic etiologies have been shown to result from the instability of tandem repeats. A hexanucleotide repeat expansion of the GGGGCC motif in C9orf72 to thousands of base pairs in length is the cause of chromosome 9p21-linked amyotrophic lateral sclerosis and frontotemporal dementia (ALS-FTD) [89,90]. Interestingly, even carriers of two intermediate-size alleles (<20 repeats) may be at risk for ALS and associated disorders [117]. Expansions of the TTTCA and TTTTA repeats in a variety of genes (e.g., SAMD12, TNRC6A, RAPGEF2) are now thought to underlie benign adult familial myoclonic epilepsy [118]. Later, the same pentanucleotide TTTCA expansion beyond 10 kbp in length was observed in association with familial autosomal myoclonic epilepsy, albeit mapping to the intron of different genes (MARCH6 and STARD7) [119,120]. The finding of the same pathogenic pentanucleotide expansion in genes that share no other property other than being highly expressed in the brain has led to speculation that it is the transcribed repeat itself instead of the specific function of the gene that underlies the epilepsy pathology. In a separate instance, a VNTR in an intron of CACNA1C was found to be associated with schizophrenia by motif composition rather than length [98]. In this light, understanding not only the repeat length but also the composition of the repeat appears critical to the pathogenic state [121] making both detection and deciphering the sequence of the expanded repeat structures critical to understanding disease risk (see above).
LRS has been used to discover novel repeat sequences in individuals with familial adult myoclonic epilepsy with negative clinical testing [122] and to identify new genes and expand the phenotype of genes associated with spinocerebellar ataxia [123,124]. For example, in a cohort with late-onset spinocerebellar ataxia 27B, Pellerin and colleagues used LRS to clarify a heterozygous trinucleotide GAA repeat expansion in the gene, FGF14, initially identified with short-read sequencing (SRS) [123]. LRS has also been used to identify disease-causing expansions within duplicated genes–so-called repeats within repeats, as in neuronal intranuclear inclusion disease (NIID, OMIM: 603472) and the expanding CGG repeat mapped to the 5′ UTR of NOTCH2NLC or NBPF19–a human-specific duplicated gene [118,125]. Expansion of this repeat beyond 100 repeat units has been associated not only with NIID but also with oculopharyngodistal myopathy with neurological manifestations (OMIM: 619473).
Until recently, clinical testing for tandem repeat disorders was limited to PCR-based approaches or Southern blot. Clinical testing labs have begun to evaluate limited loci using short-read-based approaches, likely due to the integration of ExpansionHunter [60] into the DRAGEN pipeline [126]. LRS has also been used to evaluate select repeats. In 2021, for example, Invitae began evaluating PCR products from FMR1 alleles with 55–90 triplet repeats on the PacBio platform [127]. While it is exciting to see both SRS and LRS approaches beginning to be used in the clinical space, there remains considerable need to broaden the number of tandem repeats that can be clinically evaluated (Table 3), which may be met by the introduction of LRS into the clinical environment [128]. Routine LRS of amplicons [129], CRISPR-CAS targets [122], and ultimately whole genomes will make routine clinical testing of these disorders possible [130] and perhaps drive novel therapeutic approaches [131].
Future prospects.
LRS has created a renaissance of interest in investigating the disease significance and biological mechanisms of expanding/contracting repeats and their epigenetic consequences. Tools are being developed to search for associations with gene expression [65,68,69,180] using GTEx genomes and trait association in the UK Biobank [181]. Intriguing candidate loci and genome-wide patterns of tandem repeat variation are being identified in families with autism and developmental delay[182,183] that will benefit from more extensive LRS characterization. Reports of enrichment of tandem repeat expansions in various cancer types [184] will justify LRS of tumor-matched patient samples. Still, the discovery of new disease or functional roles of tandem repeat variation is limited because almost all disease cohorts of sufficient sample size have been sequenced by short-read NGS platforms. New computational tools, such as PanGenie [73], are leveraging the sequence resolution and linkage disequilibrium provided by phased pangenomes to more accurately genotype and perform genome-wide associations in preexisting NGS datasets [17].
Current population-level analyses of tandem repeats using LRS with digital readouts of motif counts and tandem repeat composition are from a relatively modest number of human diversity samples [17,18]. Improvement in accuracy from the ONT PromethION and increases in throughput and reduction in costs such as the PacBio recent release of the Revio system will make it possible to begin to more cost-effectively consider larger disease and population cohorts. The low-coverage LRS of 3,622 Icelandic individuals has already led to the association of two new loci with height and atrial fibrillation [78]. A smaller pilot study of familial LRS of simplex autism discovered de novo STR and VNTR variation missing from SRS [13]. As costs decrease and throughput increases, not only will more genomes be sequenced but so too will higher quality phased genomes of near T2T status providing access to the largest and most identical repeats. We anticipate the discovery of many new disease associations as LRS of unsolved Mendelian disorders and deeply phenotyped and diverse cohorts such as GTEx samples [185], autism families [186], tumor-normal cancer studies [187] and biobanks (e.g., All of Us) begin over the next few years.
Another important area of future development will be the application of new LRS platforms to investigate somatic variation and expression at the single-cell level. This will require the development of cost-effective methods to generate long-read sequences from low-input DNA and mRNA materials. New methods such as MAS-ISO-seq [188] are beginning to emerge, which allow quantitative sequencing of longer transcripts (~1 kbp) providing tissue-specific spliceform characterization. In light of the extensive mosaicism already associated with tandem repeat disorders, the characterization of somatic variability at the DNA will be critical and clinically relevant. For example, somatic instability has been observed in Huntington’s disease, ALS [90], and Fragile X syndrome [189]. In these cases, the severity and age of onset of the disease can depend on the number of repeat units, as well as the extent of somatic instability in affected tissues. Some tissues may have a higher propensity for somatic instability than others, as has been shown forFMR1 where the instability of the CGG repeat expansion can vary, with higher instability observed in brain tissues compared to blood [190]. Similarly, instability of the CAG repeat expansion in the HTT gene can depend on genetic modifiers in other genes, such as DNA repair enzymes [191]. A better understanding of repeat structure, expression of genes known to be associated with somatic instability, and response of individual cells to repeat expansions will expand our understanding of the pathogenesis, uncover previously unknown associations [192], and potentially guide novel treatment approaches [193].
Summary Points.
Tandem repeat sequences span multiple classes of DNA including short tandem repeats, variable-number tandem repeats, satellite DNA, and segmental duplications.
Long-read sequencing is rapidly improving our understanding of tandem repeat organization and variation in human genomes.
Current advancements in the scale and diversity of populations undergoing long-read sequencing have unveiled nearly 150,000 polymorphic VNTRs where 33.5% are located in the subtelomeric regions of the genome.
Many different computational approaches are required to discover tandem repeat variation with short reads, depending on the class and scale of the variation.
Standard approaches for variant analysis with long reads can detect variation in tandem repeats, however specific methods are required to organize and visualize tandem repeat variation.
Patterns within the complex nature of tandem repeat variation becomes more clear when length and motif variation are visualized.
Over 56 diseases linked to tandem repeat variation have been identified.
The pace that tandem repeat disease loci are being discovered is increasing due to long-read sequencing.
Detailed resolution of tandem repeats using new technologies may lead to novel therapeutic approaches
Acknowledgments
This work was supported, in part, by US National Institutes of Health (NIH) grants DP5OD033357 (DEM), R01HG010169 (E.E.E.), R01HG002385 (E.E.E.), U01HG10973 (M.J.P.C., E.E.E.), GR1056892 (M.J.P.C.), R01NS122766 (P.N.V.), and NSF CAREER 2046753 (M.J.P.C). E.E.E. is an investigator of the Howard Hughes Medical Institute.
Footnotes
COI (Conflicts of Interest) Statement
E.E.E. is a scientific advisory board (SAB) member of Variant Bio, Inc. DEM is on a scientific advisory board at Oxford Nanopore Technologies (ONT), is engaged in a research agreement with ONT, and ONT has paid for him to travel to speak on their behalf. A.S. is an employee of Cajal Neuroscience Inc.
Databases/Websites
DRAGEN-ExpansionHunter: https://www.illumina.com/science/genomics-research/articles/str-expansionhunter.html
Jasmine: https://github.com/PacificBiosciences/Jasmine
Remora: https://github.com/nanoporetech/remora
StainedGlass: https://mrvollger.github.io/StainedGlass/
RepeatMasker: https://repeatmasker.org/RepeatMasker/
DupMasker: https://www.repeatmasker.org/DupMaskerDownload.html
SafFire: https://mrvollger.github.io/SafFire/#dataset=default&ref=CHM13_v1.1&query=GRCh38
TRviz: https://github.com/Jong-hun-Park/trviz
TRVZ: https://github.com/PacificBiosciences/trgt/blob/main/docs/trvz-plots.md
vamos: https://github.com/ChaissonLab/vamos
ModDotPlot: https://github.com/marbl/ModDotPlot
HPRC assemblies: https://github.com/human-pangenomics/HPP_Year1_Assemblies
References
- 1.Dib C, Fauré S, Fizames C, Samson D, Drouot N, Vignal A, et al. (1996) A comprehensive genetic map of the human genome based on 5,264 microsatellites. Nature 380, 152–154 10.1038/380152a0 [DOI] [PubMed] [Google Scholar]
- 2.Fu YH, Kuhl DPA, Pizzutti A, Pieretti M and Richards S Fragile X site: A polymorphic and highly mutable CGG repeat in the FMR-1 gene. Cell 10.1111/j.1469-1809.2011.00694.x [DOI] [Google Scholar]
- 3.Sulovari A, Li R, Audano PA, Porubsky D, Vollger MR, Logsdon GA, et al. (2019) Human-specific tandem repeat expansion and differential gene expression during primate evolution. Proc. Natl. Acad. Sci. U. S. A 116, 23243–23253 10.1073/pnas.1912175116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bailey JA, Yavor AM, Massa HF, Trask BJ and Eichler EE (2001) Segmental duplications: organization and impact within the current human genome project assembly. Genome Res 11, 1005–1017 10.1101/gr.gr-1871r [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoyt SJ, Storer JM, Hartley GA, Grady PGS, Gershman A, de Lima LG, et al. (2022) From telomere to telomere: The transcriptional and epigenetic state of human repeat elements. Science 376, eabk3112 10.1126/science.abk3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vollger MR, Guitart X, Dishuck PC, Mercuri L, Harvey WT, Gershman A, et al. (2022) Segmental duplications and their variation in a complete human genome. Science 376, eabj6965 10.1126/science.abj6965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Altemose N, Logsdon GA, Bzikadze AV, Sidhwani P, Langley SA, Caldas GV, et al. (2022) Complete genomic and epigenetic maps of human centromeres. Science 376, eabl4178 10.1126/science.abl4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Altemose N (2022) A classical revival: Human satellite DNAs enter the genomics era. Semin. Cell Dev. Biol 128, 2–14 10.1016/j.semcdb.2022.04.012 [DOI] [PubMed] [Google Scholar]
- 9.Nakamura Y, Leppert M, O’Connell P, Wolff R, Holm T, Culver M, et al. (1987) Variable number of tandem repeat (VNTR) markers for human gene mapping. Science 235, 1616–1622 10.1126/science.3029872 [DOI] [PubMed] [Google Scholar]
- 10.Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, et al. (2001) The sequence of the human genome. Science 291, 1304–1351 10.1126/science.1058040 [DOI] [PubMed] [Google Scholar]
- 11.Nurk S, Koren S, Rhie A, Rautiainen M, Bzikadze AV, Mikheenko A, et al. (2022) The complete sequence of a human genome. Science 376, 44–53 10.1126/science.abj6987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, et al. (2002) Recent segmental duplications in the human genome. Science 297, 1003–1007 10.1126/science.1072047 [DOI] [PubMed] [Google Scholar]
- 13.Noyes MD, Harvey WT, Porubsky D, Sulovari A, Li R, Rose NR, et al. (2022) Familial long-read sequencing increases yield of de novo mutations. Am. J. Hum. Genet 109, 631–646 10.1016/j.ajhg.2022.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vollger MR, Dishuck PC, Harvey WT, DeWitt WS, Guitart X, Goldberg ME, et al. (2023) Increased mutation and gene conversion within human segmental duplications. Nature 617, 325–334 10.1038/s41586-023-05895-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Logsdon GA, Rozanski AN, Ryabov F, Potapova T, Shepelev VA, Mao Y, et al. (2023) The variation and evolution of complete human centromeres. bioRxiv 10.1101/2023.05.30.542849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eichler EE, Clark RA and She X (2004) An assessment of the sequence gaps: unfinished business in a finished human genome. Nat. Rev. Genet 5, 345–354 10.1038/nrg1322 [DOI] [PubMed] [Google Scholar]
- 17.Ebert P, Audano PA, Zhu Q, Rodriguez-Martin B, Porubsky D, Bonder MJ, et al. (2021) Haplotype-resolved diverse human genomes and integrated analysis of structural variation. Science 372 10.1126/science.abf7117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liao W-W, Asri M, Ebler J, Doerr D, Haukness M, Hickey G, et al. (2023) A draft human pangenome reference. Nature 617, 312–324 10.1038/s41586-023-05896-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Porubsky D, Human Genome Structural Variation Consortium, Ebert P, Audano PA, Vollger MR, Harvey WT, et al. (2020) Fully phased human genome assembly without parental data using single-cell strand sequencing and long reads. Nature Biotechnology 10.1038/s41587-020-0719-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jarvis ED, Formenti G, Rhie A, Guarracino A, Yang C, Wood J, et al. (2022) Semi-automated assembly of high-quality diploid human reference genomes. Nature 611, 519–531 10.1038/s41586-022-05325-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chaisson MJP, Sanders AD, Zhao X, Malhotra A, Porubsky D, Rausch T, et al. (2019) Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat. Commun 10, 1784 10.1038/s41467-018-08148-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Royle NJ, Clarkson RE, Wong Z and Jeffreys AJ (1988) Clustering of hypervariable minisatellites in the proterminal regions of human autosomes. Genomics 3, 352–360 10.1016/0888-7543(88)90127-9 [DOI] [PubMed] [Google Scholar]
- 23.Vergnaud G, Mariat D, Apiou F, Aurias A, Lathrop M and Lauthier V (1991) The use of synthetic tandem repeats to isolate new VNTR loci: cloning of a human hypermutable sequence. Genomics 11, 135–144 10.1016/0888-7543(91)90110-z [DOI] [PubMed] [Google Scholar]
- 24.Armour JAL, Wong Z, Wilson V, Royle NJ and Jeffreys AJ (1989) Sequences flanking the repeat arrays of human minlsatellites: association with tandem and dispersed repeat elements. Nucleic Acids Res 17, 4925–4936 10.1093/nar/17.13.4925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Audano PA, Sulovari A, Graves-Lindsay TA, Cantsilieris S, Sorensen M, Welch AE, et al. (2019) Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 176, 663–675.e19 10.1016/j.cell.2018.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Linthorst J, Meert W, Hestand MS, Korlach J, Vermeesch JR, Reinders MJT, et al. (2020) Extreme enrichment of VNTR-associated polymorphicity in human subtelomeres: genes with most VNTRs are predominantly expressed in the brain. Transl. Psychiatry 10, 369 10.1038/s41398-020-01060-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT, et al. (2019) Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol 37, 1155–1162 10.1038/s41587-019-0217-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porubsky D, Vollger MR, Harvey WT, Rozanski AN, Ebert P, Hickey G, et al. (2023) Gaps and complex structurally variant loci in phased genome assemblies. Genome Res 33, 496–510 10.1101/gr.277334.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27, 573–580 10.1093/nar/27.2.573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garrison E and Marth G (2012) Haplotype-based variant detection from short-read sequencing. arXiv [q-bio.GN] 10.48550/arXiv.1207.3907 [DOI] [Google Scholar]
- 32.Layer RM, Chiang C, Quinlan AR and Hall IM (2014) LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15, R84 10.1186/gb-2014-15-6-r84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V and Korbel JO (2012) DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339 10.1093/bioinformatics/bts378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hormozdiari F, Hajirasouliha I, Dao P, Hach F, Yorukoglu D, Alkan C, et al. (2010) Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics 26, i350–7 10.1093/bioinformatics/btq216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kronenberg ZN, Osborne EJ, Cone KR, Kennedy BJ, Domyan ET, Shapiro MD, et al. (2015) Wham: Identifying Structural Variants of Biological Consequence. PLoS Comput. Biol 11, e1004572 10.1371/journal.pcbi.1004572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H, Bloom JM, Farjoun Y, Fleharty M, Gauthier L, Neale B, et al. (2018) A synthetic-diploid benchmark for accurate variant-calling evaluation. Nat. Methods 15, 595–597 10.1038/s41592-018-0054-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zook JM, Chapman B, Wang J, Mittelman D, Hofmann O, Hide W, et al. (2014) Integrating human sequence data sets provides a resource of benchmark SNP and indel genotype calls. Nat. Biotechnol 32, 246–251 10.1038/nbt.2835 [DOI] [PubMed] [Google Scholar]
- 38.Zhao X, Collins RL, Lee W-P, Weber AM, Jun Y, Zhu Q, et al. (2021) Expectations and blind spots for structural variation detection from long-read assemblies and short-read genome sequencing technologies. Am. J. Hum. Genet 108, 919–928 10.1016/j.ajhg.2021.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J and Chaisson MJP (2022) TT-Mars: structural variants assessment based on haplotype-resolved assemblies. Genome Biol 23, 110 10.1186/s13059-022-02666-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kirsche M, Prabhu G, Sherman R, Ni B, Battle A, Aganezov S, et al. (2023) Jasmine and Iris: population-scale structural variant comparison and analysis. Nat. Methods 20, 408–417 10.1038/s41592-022-01753-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smit AFA, Hubley R and Green P (1996) RepeatMasker https://repeatmasker.org
- 42.Frith MC (2011) A new repeat-masking method enables specific detection of homologous sequences. Nucleic Acids Res 39, e23 10.1093/nar/gkq1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olson D and Wheeler T (2018) ULTRA: A Model Based Tool to Detect Tandem Repeats. ACM BCB 2018, 37–46 10.1145/3233547.3233604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Boeva V, Regnier M, Papatsenko D and Makeev V (2006) Short fuzzy tandem repeats in genomic sequences, identification, and possible role in regulation of gene expression. Bioinformatics 22, 676–684 10.1093/bioinformatics/btk032 [DOI] [PubMed] [Google Scholar]
- 45.Wexler Y, Yakhini Z, Kashi Y and Geiger D (2005) Finding approximate tandem repeats in genomic sequences. J. Comput. Biol 12, 928–942 10.1089/cmb.2005.12.928 [DOI] [PubMed] [Google Scholar]
- 46.Pellegrini M, Renda ME and Vecchio A (2010) TRStalker: an efficient heuristic for finding fuzzy tandem repeats. Bioinformatics 26, i358–66 10.1093/bioinformatics/btq209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kolpakov R, Bana G and Kucherov G (2003) mreps: Efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res 31, 3672–3678 10.1093/nar/gkg617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuhn RM, Haussler D and Kent WJ (2013) The UCSC genome browser and associated tools. Brief. Bioinform 14, 144–161 10.1093/bib/bbs038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang Z, Hubley R, Smit A and Eichler EE (2008) DupMasker: a tool for annotating primate segmental duplications. Genome Res 18, 1362–1368 10.1101/gr.078477.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Numanagic I, Gökkaya AS, Zhang L, Berger B, Alkan C and Hach F (2018) Fast characterization of segmental duplications in genome assemblies. Bioinformatics 34, i706–i714 10.1093/bioinformatics/bty586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Išerić H, Alkan C, Hach F and Numanagić I (2022) Fast characterization of segmental duplication structure in multiple genome assemblies. Algorithms Mol. Biol 17, 4 10.1186/s13015-022-00210-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang Y, Chu J, Cheng H and Li H (2023) De novo reconstruction of satellite repeat units from sequence data. ArXiv 10.48550/arXiv.2304.09729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alkan C, Cardone MF, Catacchio CR, Antonacci F, O’Brien SJ, Ryder OA, et al. (2011) Genome-wide characterization of centromeric satellites from multiple mammalian genomes. Genome Res 21, 137–145 10.1101/gr.111278.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sevim V, Bashir A, Chin C-S and Miga KH (2016) Alpha-CENTAURI: assessing novel centromeric repeat sequence variation with long read sequencing. Bioinformatics 32, 1921–1924 10.1093/bioinformatics/btw101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Highnam G, Franck C, Martin A, Stephens C, Puthige A and Mittelman D (2013) Accurate human microsatellite genotypes from high-throughput resequencing data using informed error profiles. Nucleic Acids Res 41, e32 10.1093/nar/gks981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gymrek M, Golan D, Rosset S and Erlich Y (2012) lobSTR: A short tandem repeat profiler for personal genomes. Genome Res 22, 1154–1162 10.1101/gr.135780.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tang H, Kirkness EF, Lippert C, Biggs WH, Fabani M, Guzman E, et al. (2017) Profiling of Short-Tandem-Repeat Disease Alleles in 12,632 Human Whole Genomes. Am. J. Hum. Genet 101, 700–715 10.1016/j.ajhg.2017.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mousavi N, Shleizer-Burko S, Yanicky R and Gymrek M (2019) Profiling the genome-wide landscape of tandem repeat expansions. Nucleic Acids Res 47, e90 10.1093/nar/gkz501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tankard RM, Bennett MF, Degorski P, Delatycki MB, Lockhart PJ and Bahlo M (2018) Detecting Expansions of Tandem Repeats in Cohorts Sequenced with Short-Read Sequencing Data. Am. J. Hum. Genet 103, 858–873 10.1016/j.ajhg.2018.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dolzhenko E, Deshpande V, Schlesinger F, Krusche P, Petrovski R, Chen S, et al. (2019) ExpansionHunter: a sequence-graph-based tool to analyze variation in short tandem repeat regions. Bioinformatics 35, 4754–4756 10.1093/bioinformatics/btz431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dolzhenko E, van Vugt JJFA, Shaw RJ, Bekritsky MA, van Blitterswijk M, Narzisi G, et al. (2017) Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res 27, 1895–1903 10.1101/gr.225672.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dashnow H, Lek M, Phipson B, Halman A, Sadedin S, Lonsdale A, et al. (2018) STRetch: detecting and discovering pathogenic short tandem repeat expansions. Genome Biol 19, 121 10.1186/s13059-018-1505-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dashnow H, Pedersen BS, Hiatt L, Brown J, Beecroft SJ, Ravenscroft G, et al. (2022) STRling: a k-mer counting approach that detects short tandem repeat expansions at known and novel loci. Genome Biol 23, 257 10.1186/s13059-022-02826-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dolzhenko E, Bennett MF, Richmond PA, Trost B, Chen S, van Vugt JJFA, et al. (2020) ExpansionHunter Denovo: a computational method for locating known and novel repeat expansions in short-read sequencing data. Genome Biol 21, 102 10.1186/s13059-020-02017-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bakhtiari M, Shleizer-Burko S, Gymrek M, Bansal V and Bafna V (2018) Targeted genotyping of variable number tandem repeats with adVNTR. Genome Res 28, 1709–1719 10.1101/gr.235119.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bakhtiari M, Park J, Ding Y-C, Shleizer-Burko S, Neuhausen SL, Halldórsson BV, et al. (2021) Variable number tandem repeats mediate the expression of proximal genes. Nat. Commun 12, 2075 10.1038/s41467-021-22206-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Abyzov A, Urban AE, Snyder M and Gerstein M (2011) CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 21, 974–984 10.1101/gr.114876.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Garg P, Martin-Trujillo A, Rodriguez OL, Gies SJ, Hadelia E, Jadhav B, et al. (2021) Pervasive cis effects of variation in copy number of large tandem repeats on local DNA methylation and gene expression. Am. J. Hum. Genet 108, 809–824 10.1016/j.ajhg.2021.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lu T-Y, Human Genome Structural Variation Consortium and Chaisson, M.J.P. (2021) Profiling variable-number tandem repeat variation across populations using repeat-pangenome graphs. Nat. Commun 12, 4250 10.1038/s41467-021-24378-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lu T-Y, Smaruj PN, Fudenberg G, Mancuso N and Chaisson MJP (2023) The motif composition of variable-number tandem repeats impacts gene expression. Genome Res 10.1101/gr.276768.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Behera S, Belyeu JR, Chen X, Paulin LF, Nguyen NQH, Newman E, et al. (2023) Identification of allele-specific KIV-2 repeats and impact on Lp(a) measurements for cardiovascular disease risk. bioRxiv 2023.04.24.538128 10.1101/2023.04.24.538128 [DOI] [Google Scholar]
- 72.Feuerbach L, Sieverling LK, Deeg K, Hutter B, Buchhalter I, Mughal SS, et al. (2019) TelomereHunter: In Silico Estimation of Telomere Content and Composition from Cancer Genomes. BMC Bioinformatics 20 10.1186/s12859-019-2851-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ebler J, Ebert P, Clarke WE, Rausch T, Audano PA, Houwaart T, et al. (2022) Pangenome-based genome inference allows efficient and accurate genotyping across a wide spectrum of variant classes. Nat. Genet 54, 518–525 10.1038/s41588-022-01043-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ummat A and Bashir A (2014) Resolving complex tandem repeats with long reads. Bioinformatics 30, 3491–3498 10.1093/bioinformatics/btu437 [DOI] [PubMed] [Google Scholar]
- 75.De Roeck A, De Coster W, Bossaerts L, Cacace R, De Pooter T, Van Dongen J, et al. (2019) NanoSatellite: accurate characterization of expanded tandem repeat length and sequence through whole genome long-read sequencing on PromethION. Genome Biol 20, 239 10.1186/s13059-019-1856-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fang L, Liu Q, Monteys AM, Gonzalez-Alegre P, Davidson BL and Wang K (2022) DeepRepeat: direct quantification of short tandem repeats on signal data from nanopore sequencing. Genome Biol 23, 108 10.1186/s13059-022-02670-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Silvestre-Ryan J and Holmes I (2021) Pair consensus decoding improves accuracy of neural network basecallers for nanopore sequencing. Genome Biol 22, 38 10.1186/s13059-020-02255-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Beyter D, Ingimundardottir H, Oddsson A, Eggertsson HP, Bjornsson E, Jonsson H, et al. (2021) Long-read sequencing of 3,622 Icelanders provides insight into the role of structural variants in human diseases and other traits. Nat. Genet 53, 779–786 10.1038/s41588-021-00865-4 [DOI] [PubMed] [Google Scholar]
- 79.Park J, Kaufman E, Valdmanis PN and Bafna V (2023) TRviz: a Python library for decomposing and visualizing tandem repeat sequences. Bioinformatics Advances 3, vbad058 10.1093/bioadv/vbad058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ren J, Gu B and Chaisson MJP (2023) vamos: variable-number tandem repeats annotation using efficient motif sets. Genome Biol 24, 175 10.1186/s13059-023-03010-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smith TF and Waterman MS (1981) Identification of common molecular subsequences. J. Mol. Biol 147, 195–197 10.1016/0022-2836(81)90087-5 [DOI] [PubMed] [Google Scholar]
- 82.Sagot MF and Myers EW (1998) Identifying satellites and periodic repetitions in biological sequences. J. Comput. Biol 5, 539–553 10.1089/cmb.1998.5.539 [DOI] [PubMed] [Google Scholar]
- 83.Dvorkina T, Bzikadze AV and Pevzner PA (2020) The string decomposition problem and its applications to centromere analysis and assembly. Bioinformatics 36, i93–i101 10.1093/bioinformatics/btaa454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Logsdon GA, Vollger MR and Eichler EE (2020) Long-read human genome sequencing and its applications. Nat. Rev. Genet 21, 597–614 10.1038/s41576-020-0236-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cheng H, Concepcion GT, Feng X, Zhang H and Li H (2021) Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 18, 170–175 10.1038/s41592-020-01056-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rautiainen M, Nurk S, Walenz BP, Logsdon GA, Porubsky D, Rhie A, et al. (2023) Telomere-to-telomere assembly of diploid chromosomes with Verkko. Nat. Biotechnol 10.1038/s41587-023-01662-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ni P, Nie F, Zhong Z, Xu J, Huang N, Zhang J, et al. (2023) DNA 5-methylcytosine detection and methylation phasing using PacBio circular consensus sequencing. Nat. Commun 14, 4054 10.1038/s41467-023-39784-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.LaCroix AJ, Stabley D, Sahraoui R, Adam MP, Mehaffey M, Kernan K, et al. (2019) GGC Repeat Expansion and Exon 1 Methylation of XYLT1 Is a Common Pathogenic Variant in Baratela-Scott Syndrome. Am. J. Hum. Genet 104, 35–44 10.1016/j.ajhg.2018.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 10.1016/j.neuron.2011.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 10.1016/j.neuron.2011.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gershman A, Sauria MEG, Guitart X, Vollger MR, Hook PW, Hoyt SJ, et al. (2022) Epigenetic patterns in a complete human genome. Science 376, eabj5089 10.1126/science.abj5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kolmogorov M, Billingsley KJ, Mastoras M, Meredith M, Monlong J, Lorig-Roach R, et al. (2023) Scalable Nanopore sequencing of human genomes provides a comprehensive view of haplotype-resolved variation and methylation. Nat. Methods 20, 1483–1492 10.1038/s41592-023-01993-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stergachis AB, Debo BM, Haugen E, Churchman LS and Stamatoyannopoulos JA (2020) Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science 368, 1449–1454 10.1126/science.aaz1646 [DOI] [PubMed] [Google Scholar]
- 94.Altemose N, Maslan A, Smith OK, Sundararajan K, Brown RR, Mishra R, et al. (2022) DiMeLo-seq: a long-read, single-molecule method for mapping protein-DNA interactions genome wide. Nat. Methods 19, 711–723 10.1038/s41592-022-01475-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Willems T, Zielinski D, Yuan J, Gordon A, Gymrek M and Erlich Y (2017) Genome-wide profiling of heritable and de novo STR variations. Nat. Methods 14, 590–592 10.1038/nmeth.4267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Berg IL, Neumann R, Sarbajna S, Odenthal-Hesse L, Butler NJ and Jeffreys AJ (2011) Variants of the protein PRDM9 differentially regulate a set of human meiotic recombination hotspots highly active in African populations. Proc. Natl. Acad. Sci. U. S. A 108, 12378–12383 10.1073/pnas.1109531108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, et al. (2010) PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840 10.1126/science.1183439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Song JHT, Lowe CB and Kingsley DM (2018) Characterization of a Human-Specific Tandem Repeat Associated with Bipolar Disorder and Schizophrenia. Am. J. Hum. Genet 103, 421–430 10.1016/j.ajhg.2018.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Course MM, Gudsnuk K, Smukowski SN, Winston K, Desai N, Ross JP, et al. (2020) Evolution of a Human-Specific Tandem Repeat Associated with ALS. Am. J. Hum. Genet 107, 445–460 10.1016/j.ajhg.2020.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Course MM, Gudsnuk K and Valdmanis PN (2022) Long-Read Sequencing and Analysis of Variable Number Tandem Repeats. In: Proukakis C, editor Genomic Structural Variants in Nervous System Disorders New York, NY: Springer US; 2022.. p. 79–94. 10.1007/978-1-0716-2357-2_5 [DOI] [Google Scholar]
- 101.Dolzhenko E, English A, Dashnow H, De Sena Brandine G, Mokveld T, Rowell WJ, et al. (2023) Resolving the unsolved: Comprehensive assessment of tandem repeats at scale. bioRxiv 2023.05.12.540470 10.1101/2023.05.12.540470 [DOI] [Google Scholar]
- 102.Apsley AT, Domico ER, Verbiest MA, Brogan CA, Buck ER, Burich AJ, et al. (2023) A novel hypervariable variable number tandem repeat in the dopamine transporter gene (SLC6A3). Life Sci Alliance 6 10.26508/lsa.202201677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vollger MR, Kerpedjiev P, Phillippy AM and Eichler EE (2022) StainedGlass: interactive visualization of massive tandem repeat structures with identity heatmaps. Bioinformatics 38, 2049–2051 10.1093/bioinformatics/btac018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Logsdon GA, Vollger MR, Hsieh P, Mao Y, Liskovykh MA, Koren S, et al. (2021) The structure, function and evolution of a complete human chromosome 8. Nature 593, 101–107 10.1038/s41586-021-03420-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Li H, Feng X and Chu C (2020) The design and construction of reference pangenome graphs with minigraph. Genome Biol 21, 265 10.1186/s13059-020-02168-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Course MM, Sulovari A, Gudsnuk K, Eichler EE and Valdmanis PN (2021) Characterizing nucleotide variation and expansion dynamics in human-specific variable number tandem repeats. Genome Res 31, 1313–1324 10.1101/gr.275560.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schüle B, McFarland KN, Lee K, Tsai Y-C, Nguyen K-D, Sun C, et al. (2017) Parkinson’s disease associated with pure ATXN10 repeat expansion. NPJ Parkinsons Dis 3, 27 10.1038/s41531-017-0029-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Eichler EE, Holden JJ, Popovich BW, Reiss AL, Snow K, Thibodeau SN, et al. (1994) Length of uninterrupted CGG repeats determines instability in the FMR1 gene. Nat. Genet 8, 88–94 10.1038/ng0994-88 [DOI] [PubMed] [Google Scholar]
- 109.Steely CJ, Watkins WS, Baird L and Jorde LB (2022) The mutational dynamics of short tandem repeats in large, multigenerational families. Genome Biol 23, 253 10.1186/s13059-022-02818-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.van der Maarel SM, Tawil R and Tapscott SJ (2011) Facioscapulohumeral muscular dystrophy and DUX4: breaking the silence. Trends Mol. Med 17, 252–258 10.1016/j.molmed.2011.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Utermann G, Kraft HG, Menzel HJ, Hopferwieser T and Seitz C (1988) Genetics of the quantitative Lp(a) lipoprotein trait. Hum. Genet 78, 41–46 10.1007/bf00291232 [DOI] [PubMed] [Google Scholar]
- 112.MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 72, 971–983 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- 113.Fu YH, Pizzuti A, Fenwick RG Jr, King J, Rajnarayan S, Dunne PW, et al. (1992) An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 255, 1256–1258 10.1126/science.1546326 [DOI] [PubMed] [Google Scholar]
- 114.Musova Z, Mazanec R, Krepelova A, Ehler E, Vales J, Jaklova R, et al. (2009) Highly unstable sequence interruptions of the CTG repeat in the myotonic dystrophy gene. Am. J. Med. Genet. A 149A, 1365–1374 10.1002/ajmg.a.32987 [DOI] [PubMed] [Google Scholar]
- 115.Pieretti M, Zhang FP, Fu YH, Warren ST, Oostra BA, Caskey CT, et al. (1991) Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66, 817–822 10.1016/0092-8674(91)90125-i [DOI] [PubMed] [Google Scholar]
- 116.Zoghbi HY and Warren ST (2010) Neurogenetics: advancing the “next-generation” of brain research. Neuron 68, 165–173 10.1016/j.neuron.2010.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kaivola K, Salmi SJ, Jansson L, Launes J, Hokkanen L, Niemi A-K, et al. (2020) Carriership of two copies of C9orf72 hexanucleotide repeat intermediate-length alleles is a risk factor for ALS in the Finnish population. Acta Neuropathol Commun 8, 187 10.1186/s40478-020-01059-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ishiura H, Doi K, Mitsui J, Yoshimura J, Matsukawa MK, Fujiyama A, et al. (2018) Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet 50, 581–590 10.1038/s41588-018-0067-2 [DOI] [PubMed] [Google Scholar]
- 119.Corbett MA, Kroes T, Veneziano L, Bennett MF, Florian R, Schneider AL, et al. (2019) Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat. Commun 10, 4920 10.1038/s41467-019-12671-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Florian RT, Kraft F, Leitão E, Kaya S, Klebe S, Magnin E, et al. (2019) Unstable TTTTA/TTTCA expansions in MARCH6 are associated with Familial Adult Myoclonic Epilepsy type 3. Nat. Commun 10, 4919 10.1038/s41467-019-12763-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ishiura H, Shibata S, Yoshimura J, Suzuki Y, Qu W, Doi K, et al. (2019) Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet 51, 1222–1232 10.1038/s41588-019-0458-z [DOI] [PubMed] [Google Scholar]
- 122.Maroilley T, Tsai M-H, Mascarenhas R, Diao C, Khanbabaei M, Kaya S, et al. (2023) A novel FAME1 repeat configuration in a European family identified using a combined genomics approach. Epilepsia Open 8, 659–665 10.1002/epi4.12702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rafehi H, Read J, Szmulewicz DJ, Davies KC, Snell P, Fearnley LG, et al. (2023) An intronic GAA repeat expansion in FGF14 causes the autosomal-dominant adult-onset ataxia SCA50/ATX-FGF14. Am. J. Hum. Genet 110, 105–119 10.1016/j.ajhg.2022.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tan D, Wei C, Chen Z, Huang Y, Deng J, Li J, et al. (2023) CAG Repeat Expansion in THAP11 Is Associated with a Novel Spinocerebellar Ataxia. Mov. Disord 10.1002/mds.29412 [DOI] [PubMed] [Google Scholar]
- 125.Fiddes IT, Pollen AA, Davis JM and Sikela JM (2019) Paired involvement of human-specific Olduvai domains and NOTCH2NL genes in human brain evolution. Hum. Genet 138, 715–721 10.1007/s00439-019-02018-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.DRAGEN-STR. STR-ExpansionHunter https://www.illumina.com/science/genomics-research/articles/str-expansionhunter.html
- 127.Invitae test. Invitae-test https://www.invitae.com/en/providers/test-catalog/test-56022 [Google Scholar]
- 128.Kaplun L, Krautz-Peterson G, Neerman N, Stanley C, Hussey S, Folwick M, et al. (2023) ONT long-read WGS for variant discovery and orthogonal confirmation of short read WGS derived genetic variants in clinical genetic testing. Front. Genet 14, 1145285 10.3389/fgene.2023.1145285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ebbert MTW, Farrugia SL, Sens JP, Jansen-West K, Gendron TF, Prudencio M, et al. (2018) Long-read sequencing across the C9orf72 “GGGGCC” repeat expansion: implications for clinical use and genetic discovery efforts in human disease. Mol. Neurodegener 13, 46 10.1186/s13024-018-0274-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Erdmann H, Schöberl F, Giurgiu M, Leal Silva RM, Scholz V, Scharf F, et al. (2023) Parallel in-depth analysis of repeat expansions in ataxia patients by long-read sequencing. Brain 146, 1831–1843 10.1093/brain/awac377 [DOI] [PubMed] [Google Scholar]
- 131.Fang L, Monteys AM, Dürr A, Keiser M, Cheng C, Harapanahalli A, et al. (2023) Haplotyping SNPs for allele-specific gene editing of the expanded huntingtin allele using long-read sequencing. HGG Adv 4, 100146 10.1016/j.xhgg.2022.100146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Van den Bossche T, Sleegers K, Cuyvers E, Engelborghs S, Sieben A, De Roeck A, et al. (2016) Phenotypic characteristics of Alzheimer patients carrying an ABCA7 mutation. Neurology 86, 2126–2133 10.1212/WNL.0000000000002628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Bensaid M, Melko M, Bechara EG, Davidovic L, Berretta A, Catania MV, et al. (2009) FRAXE-associated mental retardation protein (FMR2) is an RNA-binding protein with high affinity for G-quartet RNA forming structure. Nucleic Acids Res 37, 1269–1279 10.1093/nar/gkn1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Harding AE, Thomas PK, Baraitser M, Bradbury PG, Morgan-Hughes JA and Ponsford JR (1982) X-linked recessive bulbospinal neuronopathy: a report of ten cases. J. Neurol. Neurosurg. Psychiatry 45, 1012–1019 10.1136/jnnp.45.11.1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kato M, Saitoh S, Kamei A, Shiraishi H, Ueda Y, Akasaka M, et al. (2007) A longer polyalanine expansion mutation in the ARX gene causes early infantile epileptic encephalopathy with suppression-burst pattern (Ohtahara syndrome). Am. J. Hum. Genet 81, 361–366 10.1086/518903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vinton A, Fahey MC, O’Brien TJ, Shaw J, Storey E, Gardner RJM, et al. (2005) Dentatorubral-pallidoluysian atrophy in three generations, with clinical courses from nearly asymptomatic elderly to severe juvenile, in an Australian family of Macedonian descent. Am. J. Med. Genet. A 136, 201–204 10.1002/ajmg.a.30355 [DOI] [PubMed] [Google Scholar]
- 137.Bergeron D, Lapointe C, Bissonnette C, Tremblay G, Motard J and Roucou X (2013) An out-of-frame overlapping reading frame in the ataxin-1 coding sequence encodes a novel ataxin-1 interacting protein. J. Biol. Chem 288, 21824–21835 10.1074/jbc.M113.472654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Matsuura T, Yamagata T, Burgess DL, Rasmussen A, Grewal RP, Watase K, et al. (2000) Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat. Genet 26, 191–194 10.1038/79911 [DOI] [PubMed] [Google Scholar]
- 139.Pulst SM, Nechiporuk A, Nechiporuk T, Gispert S, Chen XN, Lopes-Cendes I, et al. (1996) Moderate expansion of a normally biallelic trinucleotide repeat in spinocerebellar ataxia type 2. Nat. Genet 14, 269–276 10.1038/ng1196-269 [DOI] [PubMed] [Google Scholar]
- 140.Todd PK and Paulson HL (2010) RNA-mediated neurodegeneration in repeat expansion disorders. Ann. Neurol 67, 291–300 10.1002/ana.21948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.David G, Abbas N, Stevanin G, Dürr A, Yvert G, Cancel G, et al. (1997) Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet 17, 65–70 10.1038/ng0997-65 [DOI] [PubMed] [Google Scholar]
- 142.Ikeda Y, Daughters RS and Ranum LPW (2008) Bidirectional expression of the SCA8 expansion mutation: one mutation, two genes. Cerebellum 7, 150–158 10.1007/s12311-008-0010-7 [DOI] [PubMed] [Google Scholar]
- 143.Amino T, Ishikawa K, Toru S, Ishiguro T, Sato N, Tsunemi T, et al. (2007) Redefining the disease locus of 16q22.1-linked autosomal dominant cerebellar ataxia. J. Hum. Genet 52, 643–649 10.1007/s10038-007-0154-1 [DOI] [PubMed] [Google Scholar]
- 144.Reddy K, Zamiri B, Stanley SYR, Macgregor RB, Jr CE (2013) The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J. Biol. Chem 288, 9860–9866 10.1074/jbc.C113.452532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Li L, Saegusa H and Tanabe T (2009) Deficit of heat shock transcription factor 1-heat shock 70 kDa protein 1A axis determines the cell death vulnerability in a model of spinocerebellar ataxia type 6. Genes Cells 14, 1253–1269 10.1111/j.1365-2443.2009.01348.x [DOI] [PubMed] [Google Scholar]
- 146.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. (2001) Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 293, 864–867 10.1126/science.1062125 [DOI] [PubMed] [Google Scholar]
- 147.Lafrenière RG, Rochefort DL, Chrétien N, Rommens JM, Cochius JI, Kälviäinen R, et al. (1997) Unstable insertion in the 5’ flanking region of the cystatin B gene is the most common mutation in progressive myoclonus epilepsy type 1, EPM1. Nat. Genet 15, 298–302 10.1038/ng0397-298 [DOI] [PubMed] [Google Scholar]
- 148.Seixas AI, Loureiro JR, Costa C, Ordóñez-Ugalde A, Marcelino H, Oliveira CL, et al. (2017) A Pentanucleotide ATTTC Repeat Insertion in the Non-coding Region of DAB1, Mapping to SCA37, Causes Spinocerebellar Ataxia. Am. J. Hum. Genet 101, 87–103 10.1016/j.ajhg.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Jansen G, Mahadevan M, Amemiya C, Wormskamp N, Segers B, Hendriks W, et al. (1992) Characterization of the myotonic dystrophy region predicts multiple protein isoform-encoding mRNAs. Nat. Genet 1, 261–266 10.1038/ng0792-261 [DOI] [PubMed] [Google Scholar]
- 150.Devys D, Lutz Y, Rouyer N, Bellocq JP and Mandel JL (1993) The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet 4, 335–340 10.1038/ng0893-335 [DOI] [PubMed] [Google Scholar]
- 151.Murray A, Webb J, Grimley S, Conway G and Jacobs P (1998) Studies of FRAXA and FRAXE in women with premature ovarian failure. J. Med. Genet 35, 637–640 10.1136/jmg.35.8.637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jacquemont S, Hagerman RJ, Leehey M, Grigsby J, Zhang L, Brunberg JA, et al. (2003) Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet 72, 869–878 10.1086/374321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Crisponi L, Deiana M, Loi A, Chiappe F, Uda M, Amati P, et al. (2001) The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat. Genet 27, 159–166 10.1038/84781 [DOI] [PubMed] [Google Scholar]
- 154.Al-Mahdawi S, Pinto RM, Varshney D, Lawrence L, Lowrie MB, Hughes S, et al. (2006) GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 88, 580–590 10.1016/j.ygeno.2006.06.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Deng J, Yu J, Li P, Luan X, Cao L, Zhao J, et al. (2020) Expansion of GGC Repeat in GIPC1 Is Associated with Oculopharyngodistal Myopathy. Am. J. Hum. Genet 106, 793–804 10.1016/j.ajhg.2020.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.van Kuilenburg ABP, Tarailo-Graovac M, Richmond PA, Drögemöller BI, Pouladi MA, Leen R, et al. (2019) Glutaminase Deficiency Caused by Short Tandem Repeat Expansion in. N. Engl. J. Med 380, 1433–1441 10.1056/NEJMoa1806627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Goodman FR, Bacchelli C, Brady AF, Brueton LA, Fryns JP, Mortlock DP, et al. (2000) Novel HOXA13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am. J. Hum. Genet 67, 197–202 10.1086/302961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Goodman F, Giovannucci-Uzielli ML, Hall C, Reardon W, Winter R and Scambler P (1998) Deletions in HOXD13 segregate with an identical, novel foot malformation in two unrelated families. Am. J. Hum. Genet 63, 992–1000 10.1086/302070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, et al. (1993) A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 72, 971–983 10.1016/0092-8674(93)90585-e [DOI] [PubMed] [Google Scholar]
- 160.Margolis RL, O’Hearn E, Rosenblatt A, Willour V, Holmes SE, Franz ML, et al. (2001) A disorder similar to Huntington’s disease is associated with a novel CAG repeat expansion. Ann. Neurol 50, 373–380 10.1002/ana.1312 [DOI] [PubMed] [Google Scholar]
- 161.Qing J, Wei D, Maher VM and McCormick JJ (1999) Cloning and characterization of a novel gene encoding a putative transmembrane protein with altered expression in some human transformed and tumor-derived cell lines. Oncogene 18, 335–342 10.1038/sj.onc.1202290 [DOI] [PubMed] [Google Scholar]
- 162.Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, et al. (2011) Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am. J. Hum. Genet 89, 121–130 10.1016/j.ajhg.2011.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Sone J, Mitsuhashi S, Fujita A, Mizuguchi T, Hamanaka K, Mori K, et al. (2019) Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet 51, 1215–1221 10.1038/s41588-019-0459-y [DOI] [PubMed] [Google Scholar]
- 164.Brais B, Bouchard JP, Xie YG, Rochefort DL, Chrétien N, Tomé FM, et al. (1998) Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet 18, 164–167 10.1038/ng0298-164 [DOI] [PubMed] [Google Scholar]
- 165.Amiel J, Laudier B, Attié-Bitach T, Trang H, de Pontual L, Gener B, et al. (2003) Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat. Genet 33, 459–461 10.1038/ng1130 [DOI] [PubMed] [Google Scholar]
- 166.Ruggieri A, Naumenko S, Smith MA, Iannibelli E, Blasevich F, Bragato C, et al. (2020) Multiomic elucidation of a coding 99-mer repeat-expansion skeletal muscle disease. Acta Neuropathol 140, 231–235 10.1007/s00401-020-02164-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Holmes SE, O’Hearn EE, McInnis MG, Gorelick-Feldman DA, Kleiderlein JJ, Callahan C, et al. (1999) Expansion of a novel CAG trinucleotide repeat in the 5’ region of PPP2R2B is associated with SCA12. Nat. Genet 23, 391–392 10.1038/70493 [DOI] [PubMed] [Google Scholar]
- 168.Chen Y-C, Auer-Grumbach M, Matsukawa S, Zitzelsberger M, Themistocleous AC, Strom TM, et al. (2015) Transcriptional regulator PRDM12 is essential for human pain perception. Nat. Genet 47, 803–808 10.1038/ng.3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Owen F, Poulter M, Lofthouse R, Collinge J, Crow TJ, Risby D, et al. (1989) Insertion in prion protein gene in familial Creutzfeldt-Jakob disease. Lancet 1, 51–52 10.1016/s0140-6736(89)91713-3 [DOI] [PubMed] [Google Scholar]
- 170.Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yau WY, et al. (2019) Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet 51, 649–658 10.1038/s41588-019-0372-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, et al. (1997) Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 89, 773–779 10.1016/s0092-8674(00)80260-3 [DOI] [PubMed] [Google Scholar]
- 172.Bennett MF, Oliver KL, Regan BM, Bellows ST, Schneider AL, Rafehi H, et al. (2020) Familial adult myoclonic epilepsy type 1 SAMD12 TTTCA repeat expansion arose 17,000 years ago and is present in Sri Lankan and Indian families. Eur. J. Hum. Genet 28, 973–978 10.1038/s41431-020-0606-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Laumonnier F, Ronce N, Hamel BCJ, Thomas P, Lespinasse J, Raynaud M, et al. (2002) Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am. J. Hum. Genet 71, 1450–1455 10.1086/344661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Makino S, Kaji R, Ando S, Tomizawa M, Yasuno K, Goto S, et al. (2007) Reduced neuron-specific expression of the TAF1 gene is associated with X-linked dystonia-parkinsonism. Am. J. Hum. Genet 80, 393–406 10.1086/512129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Saito F, Yamamoto T, Horikoshi M and Ikeuchi T (1994) Direct mapping of the human TATA box-binding protein (TBP) gene to 6q27 by fluorescence in situ hybridization. Jpn. J. Hum. Genet 39, 421–425 10.1007/BF01892387 [DOI] [PubMed] [Google Scholar]
- 176.Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, et al. (2012) A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2–2) gene predicts Fuchs corneal dystrophy. PLoS One 7, e49083 10.1371/journal.pone.0049083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Bui C, Huber C, Tuysuz B, Alanay Y, Bole-Feysot C, Leroy JG, et al. (2014) XYLT1 mutations in Desbuquois dysplasia type 2. Am. J. Hum. Genet 94, 405–414 10.1016/j.ajhg.2014.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yeetong P, Ausavarat S, Bhidayasiri R, Piravej K, Pasutharnchat N, Desudchit T, et al. (2013) A newly identified locus for benign adult familial myoclonic epilepsy on chromosome 3q26.32–3q28. Eur. J. Hum. Genet 21, 225–228 10.1038/ejhg.2012.133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Brown SA, Warburton D, Brown LY, Yu CY, Roeder ER, Stengel-Rutkowski S, et al. (1998) Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat. Genet 20, 180–183 10.1038/2484 [DOI] [PubMed] [Google Scholar]
- 180.Gymrek M, Willems T, Guilmatre A, Zeng H, Markus B, Georgiev S, et al. (2016) Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet 48, 22–29 10.1038/ng.3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Mukamel RE, Handsaker RE, Sherman MA, Barton AR, Zheng Y, McCarroll SA, et al. (2021) Protein-coding repeat polymorphisms strongly shape diverse human phenotypes. Science 373, 1499–1505 10.1126/science.abg8289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Mitra I, Huang B, Mousavi N, Ma N, Lamkin M, Yanicky R, et al. (2021) Patterns of de novo tandem repeat mutations and their role in autism. Nature 589, 246–250 10.1038/s41586-020-03078-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Annear DJ, Vandeweyer G, Sanchis-Juan A, Raymond FL and Kooy RF (2022) Non-Mendelian inheritance patterns and extreme deviation rates of CGG repeats in autism. Genome Res 32, 1967–1980 10.1101/gr.277011.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Erwin GS, Gürsoy G, Al-Abri R, Suriyaprakash A, Dolzhenko E, Zhu K, et al. (2023) Recurrent repeat expansions in human cancer genomes. Nature 613, 96–102 10.1038/s41586-022-05515-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.GTEx Consortium (2020) The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 369, 1318–1330 10.1126/science.aaz1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Antaki D, Guevara J, Maihofer AX, Klein M, Gujral M, Grove J, et al. (2022) A phenotypic spectrum of autism is attributable to the combined effects of rare variants, polygenic risk and sex. Nat. Genet 54, 1284–1292 10.1038/s41588-022-01145-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Li Y, Roberts ND, Wala JA, Shapira O, Schumacher SE, Kumar K, et al. (2020) Patterns of somatic structural variation in human cancer genomes. Nature 578, 112–121 10.1038/s41586-019-1913-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Al’Khafaji AM, Smith JT, Garimella KV, Babadi M, Sade-Feldman M, Gatzen M, et al. (2021) High-throughput RNA isoform sequencing using programmable cDNA concatenation. bioRxiv 2021.10.01.462818 10.1101/2021.10.01.462818 [DOI] [PubMed] [Google Scholar]
- 189.Kacher R, Lejeune F-X, Noël S, Cazeneuve C, Brice A, Humbert S, et al. (2021) Propensity for somatic expansion increases over the course of life in Huntington disease. Elife 10 10.7554/eLife.64674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Pretto DI, Mendoza-Morales G, Lo J, Cao R, Hadd A, Latham GJ, et al. (2014) CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J. Med. Genet 51, 309–318 10.1136/jmedgenet-2013-102021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Lee J-M, Huang Y, Orth M, Gillis T, Siciliano J, Hong E, et al. (2022) Genetic modifiers of Huntington disease differentially influence motor and cognitive domains. Am. J. Hum. Genet 109, 885–899 10.1016/j.ajhg.2022.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Aishworiya R, Hwang YH, Santos E, Hayward B, Usdin K, Durbin-Johnson B, et al. (2023) Clinical implications of somatic allele expansion in female FMR1 premutation carriers. Sci. Rep 13, 7050 10.1038/s41598-023-33528-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Hwang YH, Hayward BE, Zafarullah M, Kumar J, Durbin Johnson B, Holmans P, et al. (2022) Both cis and trans-acting genetic factors drive somatic instability in female carriers of the FMR1 premutation. Sci. Rep 12, 10419 10.1038/s41598-022-14183-0 [DOI] [PMC free article] [PubMed] [Google Scholar]