

Abstract
INTRODUCTION:
We aimed to describe baseline amyloid-beta (Aβ) and tau-positron emission tomograrphy (PET) from Longitudinal Early-onset Alzheimer’s Disease Study (LEADS), a prospective multi-site observational study of sporadic early-onset Alzheimer’s disease (EOAD).
METHODS:
We analyzed baseline [18F]Florbetaben (Aβ) and [18F]Flortaucipir (tau)-PET from cognitively impaired participants with a clinical diagnosis of mild cognitive impairment (MCI) or AD dementia aged < 65 years. Florbetaben scans were used to distinguish cognitively impaired participants with EOAD (Aβ+) from EOnonAD (Aβ−) based on the combination of visual read by expert reader and image quantification.
RESULTS:
243/321 (75.7%) of participants were assigned to the EOAD group based on amyloid-PET; 231 (95.1%) of them were tau-PET positive (A+T+). Tau-PET signal was elevated across cortical regions with a parietal-predominant pattern, and higher burden was observed in younger and female EOAD participants.
DISCUSSION:
LEADS data emphasizes the importance of biomarkers to enhance diagnostic accuracy in EOAD. The advanced tau-PET binding at baseline might have implications for therapeutic strategies in patients with EOAD.
Keywords: Alzheimer’s disease, amyloid-PET, atypical AD, centiloids, early-onset, EOAD, LEADS, sex differences, tau-PET
1 |. BACKGROUND
Approximately 5%–10% of all patients with Alzheimer’s disease (AD) develop symptoms before age 65, a cutoff that is used in the literature to define early-onset AD (EOAD).1 Over 90% of patients with EOAD do not carry a known disease-causing genetic mutation and have sporadic AD.2 Unlike late-onset AD (LOAD) and dominantly inherited AD, research in sporadic EOAD is limited to single-site studies with modest sample sizes. Patients with sporadic EOAD have typically been excluded from large-scale observational studies and clinical trials3 due to their young age or non-amnestic presentations.4 Furthermore, clinical diagnosis can also be challenging in EO patients due to non-amnestic phenotype that have substantial clinical overlap with non-AD conditions, particularly frontotemporal dementia (FTD).5 Positron emission tomography (PET) with radioligands that bind aggregated amyloid-beta (Aβ) or hyper-phosphorylated tau can aid diagnosis by establishing the presence of plaques and tangles in patients presenting clinically with EOAD. Furthermore, larger scale longitudinal studies on the evolution of amyloid and tau-PET changes in EOAD are needed, especially given the central role of PET in developing and implementing novel therapies such as anti-Aβ monoclonal antibodies now entering clinical practice.6,7
In patients with a clinical diagnosis of probable AD dementia, the prevalence of positive AD biomarkers (e.g., PET- or biofluid-based measures of amyloid) is very high in EOAD, approximating 85%–90%.8 However, most biomarker studies in EOAD have been conducted at academic centers with strong expertise in the diagnosis of young-onset dementia (AD and FTD), potentially over-estimating the prevalence of amyloid biomarker positivity in clinically diagnosed patients compared to more generalizable practice settings.
While AD is defined by the presence of amyloid and tau neuropathologies regardless of age of onset,9,10 both autopsy11,12 and tau-PET imaging13–15 studies have shown that EOAD is associated with higher burden of tau neuropathology (i.e., higher neurofibrillary tangle density) than LOAD. This greater tau burden seems to account for the more aggressive disease course observed in EOAD compared to LOAD, including greater atrophy rates and faster cognitive decline.11,16,17 Beyond the overall neuropathology burden, EOAD and LOAD also differ in terms of the regional distribution of tau. While LOAD is typically characterized by high tau burden in the temporal lobe, mirroring the typical amnestic presentation, patients with EOAD typically show more neocortical predominant tau patterns that parallel their frequent non-amnestic (e.g., visuo-spatial, language, executive) syndromes.11,15 Studies examining differences in the burden or distribution of amyloid between EOAD and LOAD have yielded mixed results, with some studies reporting subtle increases in cortical amyloid in EOAD and others reporting no association with age.18–21
Here we describe the baseline PET data from LEADS (Longitudinal Early-onset Alzheimer’s Disease Study), an ongoing multi-center, longitudinal investigation of patients with a clinical diagnosis of sporadic EOAD at the mild cognitive impairment (MCI) or mild dementia stage.4 The current paper presents an overview of the amyloid- and tau-PET data acquired at the baseline visit for clinically impaired and cognitively normal participants. We first describe our process for interpreting amyloid-PET in cognitively impaired participants, which is used to assign impaired participants to EOAD (positive/elevated amyloid) or early-onset non-AD (EOnonAD; negative/non-elevated amyloid) cohorts. We then report group differences in amyloid- and tau-PET measures, and correlations between amyloid- and tau-PET, demographic variables (age, sex) and disease severity (Mini-Mental State Examination, MMSE) within the EOAD group. Primary quantitative analyses of PET are focused on summary metrics (global amyloid burden measured in centiloids [CL22], tau-PET signal in the temporal cortex23,24), and exploratory analyses assessed PET patterns across all cortical regions.
2 |. METHODS
2.1 |. Participants
The present study analyzed baseline PET data from LEADS, which was launched in 2018, and has been described previously.4 LEADS recruits participants with clinical impairment who meet the 2011 criteria of the National Institute on Aging and the Alzheimer’s Association (NIA-AA) for MCI due to AD25 or mild dementia due to AD,26have a global Clinical Dementia Rating (CDR) ≤1, and are aged 40–64 years at the time of consent. Clinical diagnoses were made at each site through a multidisciplinary consensus process involving cognitive neurologists, neuropsychologists, geriatrician, psychiatrists, and/or nurse specialists during formal reviews.27 Expert clinicians carefully reviewed the standardized baseline clinical assessments, which included medical and family history, concurrent medication review, neurological, and neuropsychological examinations. Note that the 2011 NIA-AA criteria are not limited to amnestic-predominant clinical presentation and specifically mention language, visuo-spatial, and executive presentations of AD. As a consequence, individuals who meet consensus criteria for non-amnestic variants of AD, including dysexecutive AD,28,29 logopenic-variant of primary progressive aphasia,30 or posterior cortical atrophy,31 also fulfill criteria for MCI due to AD or dementia due to AD, depending on the clinical severity, and are eligible to enroll. When enrolling clinically impaired participants into LEADS, whether at the MCI or dementia stage, study investigators were asked to specify the clinical phenotype by selecting one of the four following options, based on their clinical assessment of the patient: “amnestic-predominant single or multi-domain cognitive impairment”, “posterior cortical atrophy syndrome”, “primary progressive aphasia syndrome”, or “non-amnestic single of multi-domain cognitive impairment, not above”.27 This classification was used for exploratory analyses in the current paper.
Participants with known pathogenic mutations in PSEN1, PSEN2, and APP are excluded. Cognitively normal (CN) participants were also recruited. CN participants were required to have MMSE ≥ 24, global CDR = 0, and normal neuropsychological testing compared to age-matched norms.
Written informed consent was obtained from all patients or their surrogates. The study was conducted under a central institutional review board based at Indiana University.
2.2 |. PET acquisition, quality control, and initial pre-processing
Detailed PET acquisition and processing protocols were described in a previous paper.4 Briefly, [18F]Florbetaben (FBB) PET (Aβ radiotracer) was acquired 90 to 110 min post-injection of ~8 mCi of FBB (four 5-min frames). [18F]Flortaucipir (FTP) PET (tau radiotracer) was acquired 75 to 105 min post-injection of ~10 mCi of FTP (six 5-min frames). Image acquisition and reconstruction parameters were aligned with the Alzheimer’s Disease Neuroimaging Initiative 3 (ADNI-3) PET procedures to enable comparisons. PET frames acquired at each site were uploaded to the Laboratory of Neuroimaging (LONI) at the University of Southern California.
Quality control and image standardization were performed at the University of Michigan following ADNI-3 procedures. Briefly, all images were downloaded from LONI, evaluated for statistical noise, motion across frames, full coverage of the brain, and common PET artifacts. Initial pre-processing steps were then performed: frames were realigned and averaged, set to a standard orientation, intensity-normalized and smoothed to standard 8 mm isotropic resolution using procedures developed for ADNI. Resulting images were then uploaded to LONI. Further analysis of PET data was performed at the University of California, San Francisco, based on these quality-controlled, pre-processed PET scans.
2.3 |. Amyloid-PET screening: centralized FBB-PET interpretation for cognitively impaired participants
In LEADS, amyloid-PET serves two main purposes, which are associated with two independent procedures.
First, amyloid-PET is used to determine amyloid positivity in cognitively impaired participants. This information is used to both (i) assign participants to the EOAD or EOnonAD group, which has consequences in subsequent study design (e.g. duration of follow-up, frequency of follow-up scans...4) and (ii) disclose amyloid status (positive /elevated or negative/non-elevated) to the site principal investigators and cognitively impaired participants. To ensure timely communication of amyloid-PET results and group assignment, this process is based on a PET-only processing pipeline, that is, not relying on getting a structural MRI, which may be acquired later depending on site logistics. This step, called “amyloid-PET screening” is detailed in subsequent paragraphs and in Figure 1, and involves both a visual read of the scan and a PET-only standardized uptake value ratio (SUVRPET-only) value.
FIGURE 1.
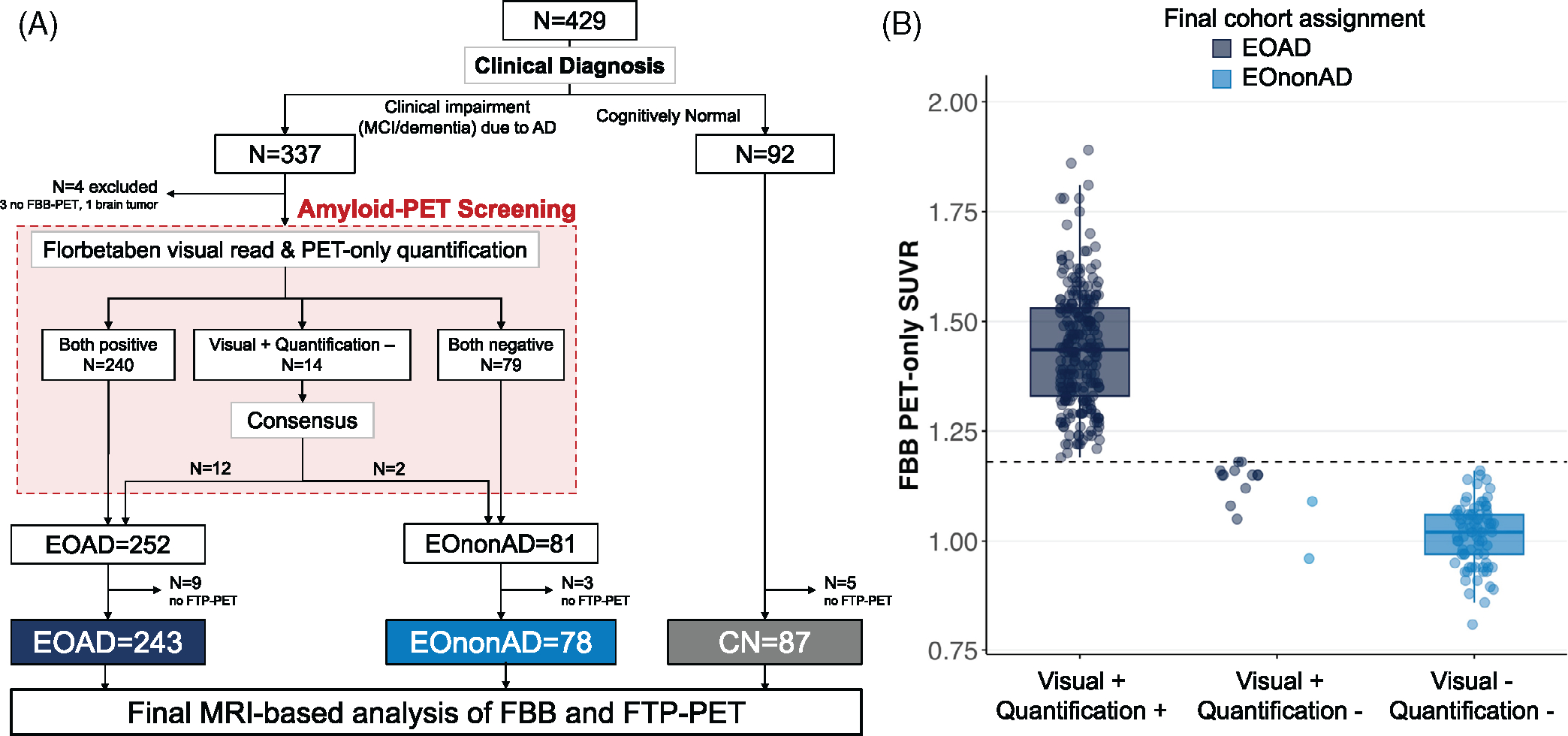
Study flow and amyloid-PET screening. (A) Flowchart of patient selection and cohort assignment. (B) PET-only quantification performed for amyloid-PET screening (dashed line indicate the SUVRPET-only threshold of 1.18). CI, cognitively impaired; CN, cognitively normal; EOAD, early-onset Alzheimer’s disease; EOnonAD, early-onset non-Alzheimer’s disease; FBB, 18F-Florbetaben; FTP, 18F-Flortaucipir; PET, positron emission tomography
Second, amyloid-PET is independently processed to quantify amyloid burden in all participants. This procedure is not as time sensitive as the previous one; it relies on a different pipeline that is identical to the ADNI procedure (to facilitate future comparisons) and requires structural MRI. This pipeline outputs MRI-based SUVR values that are then converted to centiloid values, which can then be used for any research project using amyloid-PET. Centiloid values can then be used as a continuous variable or binarized into negative and positive categories using predetermined thresholds.
In conclusion, two separate Florbetaben SUVR values are calculated from the same images: a SUVRPET-only, which is only used for amyloid-PET screening (Figure 1), and an MRI-based SUVR which is then converted to centiloids for research analyses (all other figures in the present manuscript). These two values are highly correlated (r = 0.956 in the 321 cognitively impaired patients; see supplementary eMethod) but slight differences in values and thresholds might lead to occasional differences in assigning amyloid-positivity status (see supplementary eMethod).
2.3.1 |. PET-only pipeline for amyloid-PET in cognitively impaired participants
A PET-only pipeline was used to quantify amyloid-PET scans for clinical interpretation as MRI scans were not always available at the time of clinical read, which had to be performed within 2 weeks of PET acquisition per study protocol. For this PET-only quantification, PET images undergo spatial normalization to an FBB PET template (see Supplementary eMethods). A SUVRPET-only was then calculated using a template space whole cerebellum region of interest (ROI) as a reference region and a composite ROI including frontal, parietal, and occipital cortices as the target region. A SUVRPET-only ≥1.18 was used as a quantitative threshold for amyloid-PET positivity; see Supplementary eMethods for more details on how this threshold was established based on 72 FBB scans from ADNI-3 in June 2018. Note that these SUVRPET-only values were only used for the cohort assignment in participants with cognitive impairment (“amyloid-PET screening”), not for any statistical analyses described in this paper.
2.3.2 |. Combining quantification and visual read
FBB-PET scans were visually read as amyloid-positive or amyloid-negative using validated criteria32 by one of the certified PET core physicians at UCSF (CLH, CCW, DSM, EZ, GDR, HC, JAT, or OLS). Each FBB scan was first read by a single physician, without access to participants’ clinical information or scan quantification. After reading the scan and saving their interpretation, each physician had access to the SUVRPET-only value. If both classifications were positive, the participant was assigned to the EOAD cohort; if both were negative, the participant was assigned to the EononAD cohort. If visual read and quantification-based classification was incongruent (e.g., visually read as positive but SUVRPET-only < 1.18, or vice-versa), a second visual read was performed by an additional reader who was blind to previous read and quantification. This second read was used as a tie breaker. This decision was motivated by our experience combining expert visual read and quantification of amyloid-PET, showing that extraction of PET signal from a large neocortical region of interest can sometimes miss early (i.e., focal) PET positivity.33,34 In addition, the amyloid-PET screening relied on a PET-only quantification, which might not be as accurate and precise as an MRI-based quantification; for instance, PET-only quantification might be particularly vulnerable to cortical atrophy, which is commonly severe in EOAD.
For two borderline positive amyloid scans (focal radiotracer uptake only), the participant’s clinical presentation was reviewed with site clinicians to determine the most appropriate cohort assignment as EOAD or EononAD.
2.4 |. MRI-based processing for amyloid and tau-PET analyses in all participants
MRI-based processing of amyloid- and tau-PET scans was used to extract quantification for all participants, regardless of clinical status and amyloid screening result, using data that were quality controlled at the University of Michigan (see Section 2.2).
2.4.1 |. SUVR extractions
FBB-PET and FTP-PET images were co-registered to the participants’ respective T1 structural MR images using Statistical Parametric Mapping 12 (Wellcome Department of Imaging Neuroscience, Institute of Neurology, London, UK). Reference regions were created via segmentation of the MRI using Freesurfer 7.1 to define the whole cerebellum for FBB-PET or Freesurfer 7.1 combined with the SUIT template to define the inferior cerebellar gray matter for FTP-PET.35,36 Mean SUVR values were extracted in native MRI space from ROIs defined in the Desikan-Killiany atlas37 labeled by Freesurfer 7.1. For FBB-PET, a composite neocortical SUVR was computed and converted to CL22 using the equation developed in ADNI with identical image pre-processing.38 For FTP-PET, average SUVR values were computed from a temporal meta-ROI which included the entorhinal, amygdala, parahippocampal, fusiform, inferior temporal, and middle temporal ROIs.23,24 These two measures (CL values derived from neocortical FBB-SUVR extraction and temporal meta-ROI FTP-SUVR) were used as primary summary metrics of amyloid- and tau-PET for the main analyses in this paper. For tau-PET, we repeated the main analyses using global cortical FTP-SUVR (weighted average of SUVR values extracted from all Freesurfer defined cortical regions) instead of the temporal FTP-SUVR to make less assumptions about the temporal predominance of the tau-PET signal in EOAD. Additional exploratory analyses were conducted to analyze brain patterns of amyloid and tau after extracting mean FBB- and FTP-SUVR values from all 68 cortical ROIs from the Desikan-Killiany atlas.
2.4.2 |. Amyloid- and Tau-PET positivity thresholds with MRI-based quantification
To determine amyloid- and tau-PET positivity based on quantification only, we employed a quantitative analysis method using MRI-based PET processing and previously published thresholds for our amyloid- and tau-PET summary metrics. For amyloid-PET positivity, scans were quantitatively classified as positive or negative based on a 25 CL threshold.39,40 FTP scans were quantitatively classified as tau-positive or tau-negative based on a temporal meta-ROI FTP-SUVR threshold of 1.27.24,41
2.5 |. Statistical analyses
Demographic and clinical data were analyzed via independent t-test or one-way Welsh analysis of variance (ANOVA) for continuous variables or chi-square test for categorical variables. We compared continuous variables between groups (EOAD, EOnonAD, and CN) using Welsh ANOVA and Games-Howell post-hoc tests, given large inhomogeneity of variance across groups (see Figures). Associations between continuous variables were assessed using bivariate correlations (Pearson’s r); ranked correlation coefficients were also used when there were outliers or non-linear associations. To better describe the amyloid-tau relationship in the whole group, we compared three nested models (linear, quadratic, and cubic regressions) using F tests. Each model’s fitness was also assessed using R2 and adjusted R2 (adjR2 higher is better), and Akaike Information Criterion (AIC, lower is better); the latter two metrics discourage overfitting by penalizing models with more independent variables. Analyses of covariance (ANCOVA) were also used to compare groups while controlling for age and sex.
Additional tests (bivariate correlations or t-tests) were used to characterize the association between PET summary metrics and demographic (age, sex) or clinical (MMSE) measures within the EOAD group. To assess whether demographic and clinical variables were more strongly associated with amyloid- and tau-PET metrics, we computed 95% confidence intervals of the difference for the corresponding effect sizes (e.g., rage-tau vs. rage-amyloid) using bootstrapped resampling with 1000 iterations.
Exploratory analyses were run on 68 ROIs covering the entire cerebral cortex. For these analyses, we report uncorrected (α = 0.05) and Bonferroni-corrected (α = 0.05/68 = 0.000735) results and regional effect size metrics (Cohen’s d for group comparisons, Pearson’s r for correlations).
Last, exploratory analyses were conducted to assess the association between clinical phenotypes (the four phenotypes captured in LEADS as described in section 2.1) and primary PET outcomes: (i) classification as EOAD or EOnonAD, (ii) amyloid and tau burden, measured with the PET summary metrics described in section 2.4.1, (iii) regional distribution of amyloid- and tau-PET. These results need to be considered exploratory due to the small sample size for the non-amnestic phenotypes (see result section). Phenotypes were compared using Chi-square tests for binary variable (e.g., amyloid positivity), and ANOVAs and ANCOVAs, controlling for age and sex for continuous variables (e.g., PET SUVR). Due to small sample size, no post hoc tests were conducted.
Statistical analyses were performed using R (version 4.1.1), and figures were created with ggplot (boxplots, scatter plots) and ggseg (regional brain analyses) packages.42,43
3 |. RESULTS
3.1 |. Participant selection and amyloid-PET screening
In total, 429 participants were enrolled at 19 sites in the United States between May 2018 and May 2022, including 337 with a clinical diagnosis of MCI or dementia due to AD and 92 CN participants (Figure 1a). One participant with clinical impairment was excluded from the study due to detection of a brain tumor during image quality control procedures, and three participants had not undergone FBB-PET by the time of analyses. The remaining 333 participants underwent amyloid-PET screening to classify them as EOAD or EOnonAD based on the approach described in Section 2.3 (Figure 1a). In this sample, visual read and SUVRPET-only-based classifications were congruent in 319/333 (95.8%) cases. A total of 240 participants were positive on both and, therefore, assigned to the EOAD cohort, while 79 participants were negative on both and assigned to the EOnonAD cohort (Figure 1b). Of 14/333 (4.2%) scans with a visual/quantification discrepancy, all had a positive visual read and a negative quantification (SUVRPET-only < 1.18). In all 14 cases, the second reader agreed with the original visual read, and all scans were eventually considered amyloid-positive. During the amyloid screening consensus process, 12 of these 14 cases were included in the EOAD group while 2 cases were assigned to the EOnonAD group due to very focal amyloid-PET signal and after consultation with the site clinician who then considered the clinical presentation most consistent with a non-AD etiology (Figure 1b). After excluding cases without FTP-PET, the final sample included 243 participants with EOAD and 78 participants with EOnonAD (Figure 1a).
This breakdown (243 EOAD out of 321 cognitively impaired participants, 75.7%) includes patients in the MCI and mild dementia stage. Among patients with MCI (n = 106), 65 (61.3%) were considered EOAD, while that proportion was higher in patients with dementia (178/213, 83.6%); Fisher’s exact test, p < 0.001.
The final sample also includes 87 CN controls, after excluding 5 cases without FTP-PET at the time of analysis. Controls were included regardless of their amyloid status and did not go through amyloid-PET clinical interpretation.
3.2 |. Demographic characteristics
All analyses presented in the subsequent sections of this manuscript only include participants with both amyloid and tau-PET available at the time of analyses (Figure 1a). Main demographic and clinical information are described in Table 1. The EOAD and EOnonAD groups did not significantly differ in terms of age or years of education. Groups did not differ in terms of race or ethnicity and included predominantly non-Hispanic White participants. Compared to EOnonAD, the EOAD group included a higher proportion of females (54% vs. 35%, p = 0.003) and Apolipoprotein ε4 (APOE ε4) carriers (54.7% vs. 41.7%, p = 0.056). Across clinical variables, participants with EOAD had more severe impairment than EOnonAD participants: lower MMSE or Montreal Cognitive Assessment (MOCA) scores, higher CDR sum of boxes (CDR-SB), and higher frequency of dementia diagnosis; see Table 1 for more information.
TABLE 1.
Demographic and clinical characteristics of the participants included in the final sample
| N | EOAD 243 | EOnonAD 78 | CN 87 |
|---|---|---|---|
| Age, y | 59.2 ± 4.1a | 58.7 ± 5.9 | 56.9 ± 5.9 |
| Sex, male/female (%female) | 112/131 (54%)b | 51/27 (35%)a,b | 33/54 (62%) |
| Education, y | 15.4 ± 2.4a | 15.5 ± 2.5a | 16.7 ± 2.1 |
| APOE ε4 alleles, 0/1/2 (% ε4 carriers) | 97/85/32 (55%)c | 42/28/2 (42%) | 49/28/8 (42%) |
| Ethnicity, Hispanic (%) | 8 (3%) | 3 (4%) | 7 (8%) |
| Race, White/Black/Asian/more than one/unknown (% White) | 226/8/4/4/1 (93%)a | 68/4/1/3/2 (90%)a | 63/15/5/3/1 (73%) |
| Clinical characteristics | |||
| CDR sum of boxes | 3.7 ± 1.8a,b | 3.0 ± 2.a,b | 0.0 ± 0.1 |
| MMSE | 21.6 ± 5.1a,b | 25.5 ± 4.2a,b | 29.2 ± 0.9 |
| MOCA | 16.1 ± 6.0a,b | 21.6 ± 4.7a,b | 27.0 ± 2.5 |
| MCI/dementia, (% dementia) | 64/117 (73%)b | 42/36 (46%) | n.a. |
Note: For continuous variables,data is shown as mean ± SD and Welch’s ANOVA were conducted withGames-Howell post-hoctests. Forcategoricalvariables, data is presented as n and percentages. For group comparisons, categorical variables with more than two levels were binned to avoid cells with small values (ApoE coded as ε4 carriers vs. non carriers; race coded as White vs. other races), and chi-squared tests were conducted.
Abbreviations: APOE, apolipoprotein E; CDR, Clinical Dementia Rating; CN, cognitively normal; EOAD, early-onset Alzheimer’s disease; EOnonAD, early- onset non-Alzheimer’s disease; MCI, mild cognitive impairment; MMSE, Mini-Mental State Examination.; MOCA, Montreal Cognitive Assessment; n.a., not applicable.
p < 0.05 group is different from CN.
p < 0.05 between EOAD and EOnonAD group.
p = 0.055 for both EOAD versus EOnonAD and EOAD versus CN.
3.3 |. Summary metrics of amyloid- and tau-PET across groups
All the results presented below are based on amyloid and tau-PET measures derived from the MRI-based processing pipeline.
3.3.1 |. Amyloid-PET: CL
Amyloid burden was, by study design, elevated in the EOAD group (mean ± SD = 96.1 ± 26.2 CL) compared to EOnonAD (5.9 ± 10.2) and controls (9.8 ± 13.6); see Figure 2a. One-way ANOVA was significant (F(2226.9)) = 1075.4, p < 0.001) but post-hoc Games-Howell tests only showed significant differences between EOAD and the other two groups (both comparisons p < 0.001), while the comparison between EOnonAD and CN groups did not reach significance (p = 0.097).
FIGURE 2.
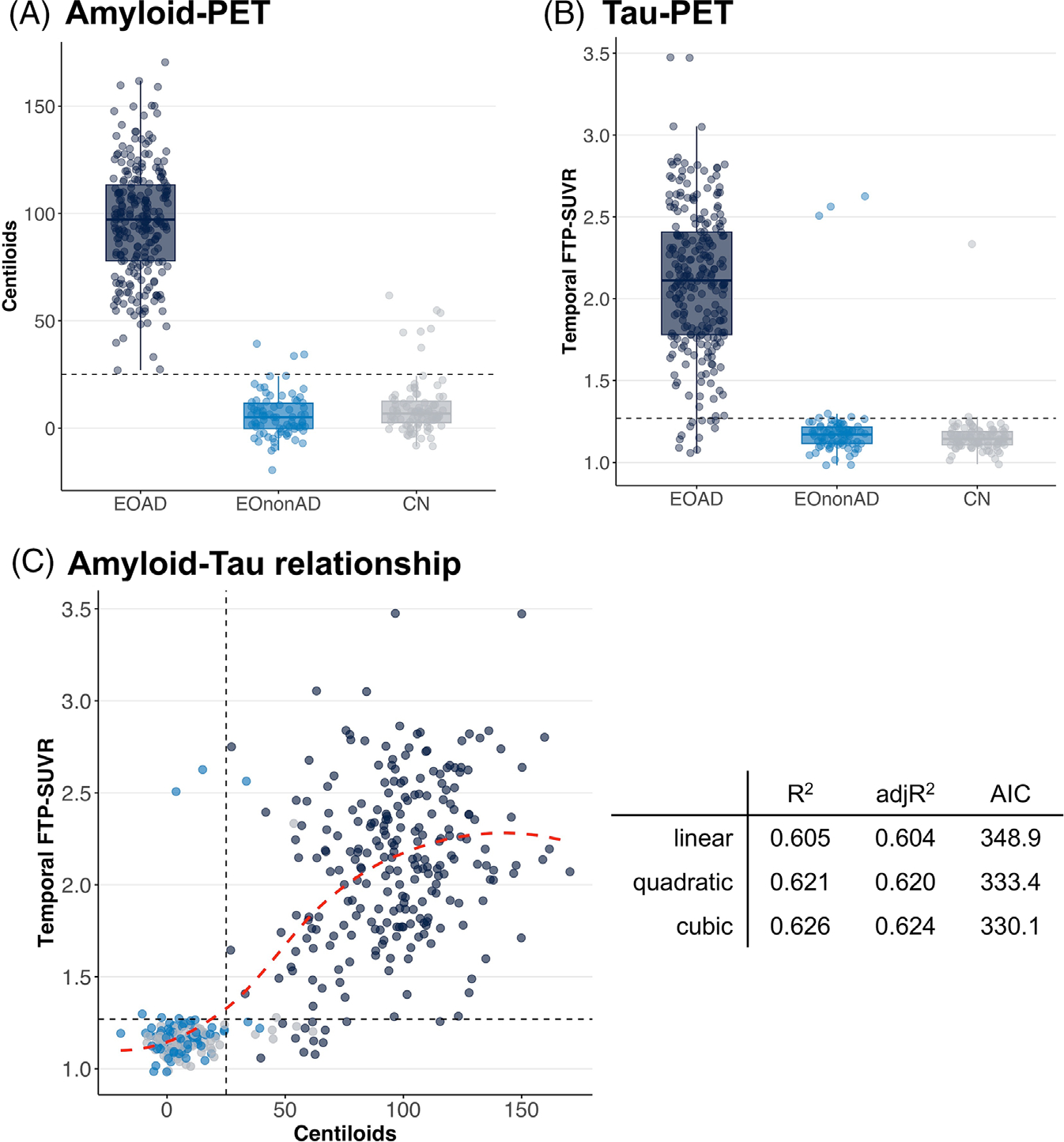
Summary metrics of amyloid- and tau-PET across groups. Distribution of Centiloid (A) and temporal FTP-SUVR (B) values across the three groups. (C) The association between Centiloid and temporal FTP-SUVR, with summary metrics for linear, quadratic, and cubic models. The red dotted line shows a data-driven smooth regression line using the locally estimated scatterplot smoothing (LOESS) method. Dotted lines indicate pre-determined positivity thresholds of 25 Centiloid for amyloid-PET (A and C) and 1.27 temporal meta-ROI FTP-SUVR (B and C). See Figure S2 for tau-PET analyses using global cortical SUVR instead of temporal SUVR. FTP, 18F-Flortaucipir; PET, positron emission tomography; ROI, region of interest; SUVR, standardized uptake value ratio
All 243 EOAD participants had amyloid-PET quantification above the pre-determined 25 CL threshold (min = 26.9, max = 170.4). The EOnonAD group included three (3.8%) cases above the 25 CL threshold (min = 33.6, max = 39.2). Note that these three cases were different from the two EOnonAD cases that were read as amyloid positive during the amyloid screening step (section 3.1). Finally, seven (8.0%) controls were above the 25 CL threshold (min = 37.4, max = 61.7).
3.3.2 |. Tau-PET: temporal meta-ROI SUVR
The tau-PET signal in the temporal lobe showed a similar pattern as amyloid-PET. Group differences were significant (Welsh’s ANOVA: F(2198.5) = 430.5, p < 0.001) and driven by the EOAD group (p < 0.001 compared to the other two groups), without significant differences between EOnonAD and CN (Games-Howell p = 0.26); see Figure 2b.
The EOAD group showed high variability in temporal SUVR (mean ± SD = 2.11 ± 0.45); 12 participants (4.9%) had values below the 1.27 threshold. The other two groups had low temporal FTP-SUVR with limited intra-group variability, apart from a few notable outliers. The EOnonAD group had 6 cases (7.7%) with FTP SUVR > 1.27 but three of them had mildly elevated values (< 1.30). In contrast, the other three were unambiguously elevated with SUVR values above the third quartile of the EOAD distribution (Figure 2b). Among these three EOnonAD cases, only one had amyloid-PET quantification above the 25 CL threshold (33.6), and none were read as amyloid-positive on amyloid-PET visual interpretation (Section 3.1). Visual inspection of FTP-PET scans from these cases showed typical AD binding patterns, with temporo-parietal cortex binding and additional frontal tracer uptake (Figure S1). In the CN group, two individuals (2.3%) had temporal SUVR values above the 1.27 threshold (1.28 and 2.33), and both were amyloid positive (46.2 and 53.6 CL, respectively).
Patterns of group differences were unchanged when using total cortical FTP-SUVR instead of temporal SUVR (Figure S2b).
3.3.3 |. Amyloid-tau relationships in the whole sample
When including all participants in the analyses, we observed a significant correlation between CL and temporal FTP-SUVR (Spearman’s rho = 0.762, p < 0.001, Figure 2c). This relationship was unchanged when controlling for age and sex (Table S1). Based on scatterplot inspection, we tested the non-linearity of the amyloid/tau association by comparing a series of polynomial models with up to 3 degrees. With increasing model complexity, we observed a mild but significant increase in R2 (from 0.605 to 0.626) and adjR2 (0.604 to 0.624) and a decrease in AIC (348.9 to 330.0). Comparison of nested models showed that each model was significantly better than the previous one (quadratic vs. simple, p < 0.001; cubic vs. quadratic, p = 0.022). Adding higher degree polynomials did not further improve model fit.
Amyloid-tau association was similar when using total cortical FTP-SUVR instead of temporal SUVR (Figure S2b).
3.3.4 |. Amyloid- and tau-PET in the EOAD group
Correlation between CL and temporal FTP-SUVR was still significant when restricted to the EOAD group (r = 0.316, Spearman’s rho = 0.285, both p < 0.001). This relationship was unchanged when controlling for age and sex (standardized estimate = 0.300, p < 0.001; Table S1), or when using total cortical FTP-SUVR instead of temporal SUVR (r = 0.341, p < 0.001; Figure S2b).
Figure 3 shows the correlations between amyloid/tau binding and age, sex, and MMSE. Older age at PET was associated with lower temporal FTP-SUVR (r = −0.357, p < 0.001) but not associated with CL (r = 0.034, p = 0.60, Figure 3a); the difference between these two correlation coefficients was significant (Δr = 0.391, bootstrap 95%CI = [0.219, 0.554], p < 0.001). In contrast, sex was associated with both amyloid and tau measures: female participants had higher CL (d = 0.429, p < 0.001) and higher temporal FTP-SUVR (d = 0.378, p = 0.004) than males (Figure 3B). The magnitude of these sex differences was not significantly different for amyloid versus tau (Δd = 0.051, bootstrap 95%CI = [−0.244, 0.368], p = 0.77), and sex differences in temporal FTP-SUVR were still significant when controlling for CL values (p = 0.045). Note that female and male patients with EOAD did not differ in terms of age (mean ± SD = 59.3 ± 4.1 vs. 59.1 ± 4.1, two-sample t-test p = 0.70) or MMSE (21.6 ± 4.8 vs. 21.7 ± 5.4, p = 0.82).
FIGURE 3.
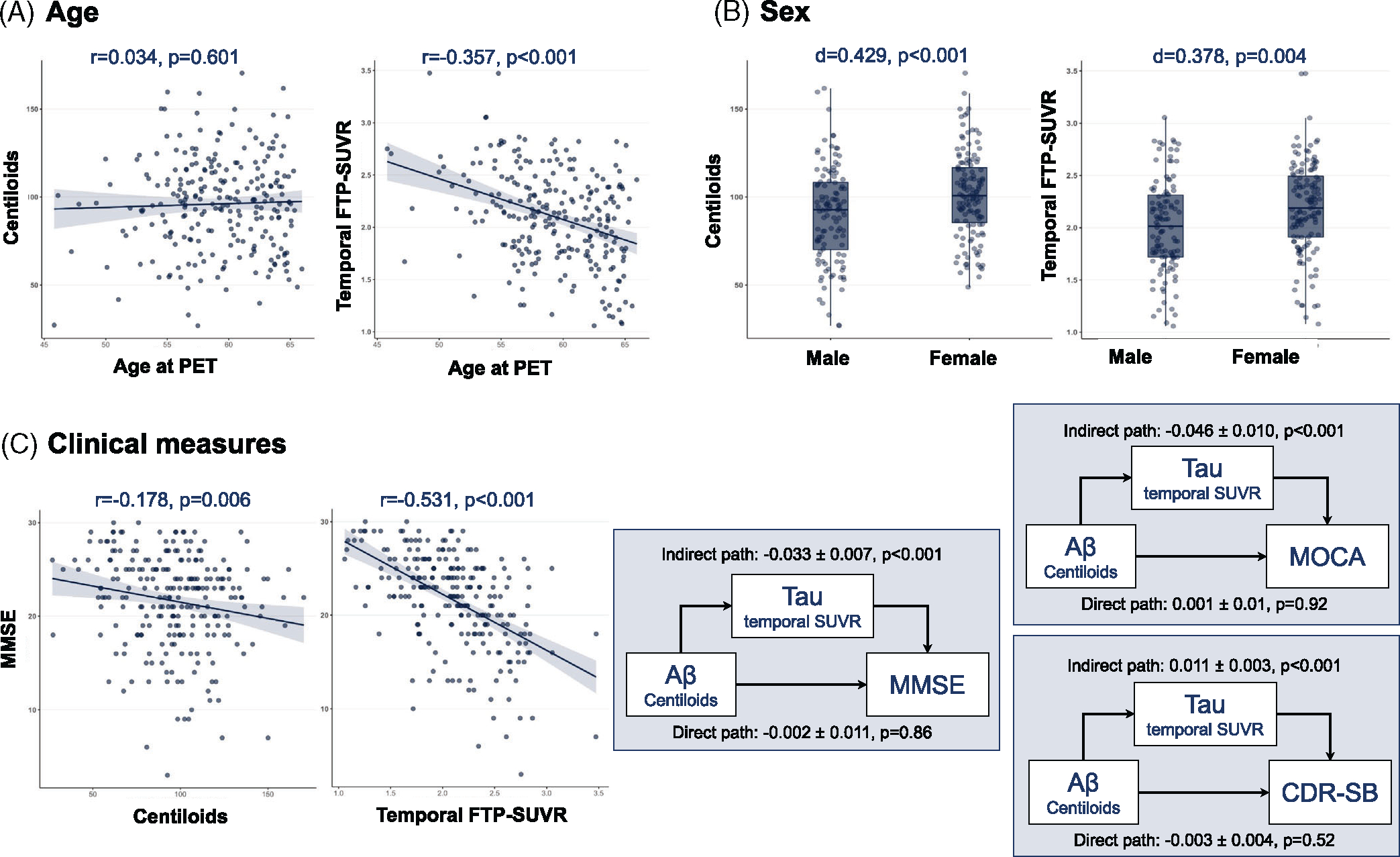
Summary metrics of amyloid- and tau-PET in the EOAD group, Association between amyloid- and tau-PET and age (A), sex (B), and MMSE (C) in the EOAD group. The rightmost part of panel C shows the result of a mediation analysis; numbers indicate point estimates ± standard error. Mediation model was repeated using the MOCA or CDR-SB scores as alternative dependent variables. Full path values are −0.035 ± 0.012 for MMSE (p = 0.005, n = 240), −0.044 ± 0.016 for MOCA (p = 0.006, n = 182) and 0.009 ± 0.004 for CDR-SB (p = 0.057, n = 225). See Figure S2 for tau-PET analyses using global cortical SUVR instead of temporal SUVR. CDR-SB, Clinical Dementia Rating sum of boxes; EOAD, early-onset Alzheimer’s disease; MOCA, Montreal Cognitive Assessment; MMSE, Mini-Mental State Examination; PET, positron emission tomography
Both PET measures were negatively associated with MMSE (Figure 3C), but the association was stronger for temporal FTP-SUVR (r = −0.531, p < 0.001) than CL (r = −0.178, p = 0.006), and the difference between these correlations was significant (Δr = 0.353, bootstrap 95%CI = [0.247,0.465], p < 0.001). Mediation analyses showed that the effect of amyloid on MMSE was fully mediated by tau (indirect effect: p < 0.001, direct effect p = 0.84, 94.6% mediation). Similar results were observed for MOCA or CDR-SB as the measure of clinical severity (Figure 3C).
Associations with age, sex, and MMSE were unchanged when using total cortical FTP-SUVR instead of temporal SUVR (Figure S2C).
3.4 |. Region-wise analysis of amyloid and tau-PET
3.4.1 |. Regional patterns of group differences
Figure 4 shows mean FBB- and FTP-SUVR values extracted from all 68 ROIs in all three groups. Compared to CN, participants with EOAD had significantly elevated FBB-SUVR in all 68 ROIs using a stringent Bonferroni-corrected threshold (Table S2 for details). Regional variations were noted (Figure 4a), with maximal differences in the bilateral precuneus (Cohen’s d = 3.76 on the left, 3.74 on the right). In contrast, bilateral occipital (cuneus, lingual, pericalcarine) and entorhinal cortices had the mildest differences (Cohen’s d < 2). Group differences between EOAD and CN were also marked in FTP-SUVR, with all 68 ROIs being significant at the Bonferroni-corrected threshold (Table S2). Maximal differences (Cohen’s d > 2.2) were seen in bilateral parietal and temporal cortices (Figure 4B), while differences were milder, but still significant (Cohen’s d < 1), in the bilateral pericalcarine, paracentral, and anterior cingulate cortices.
FIGURE 4.
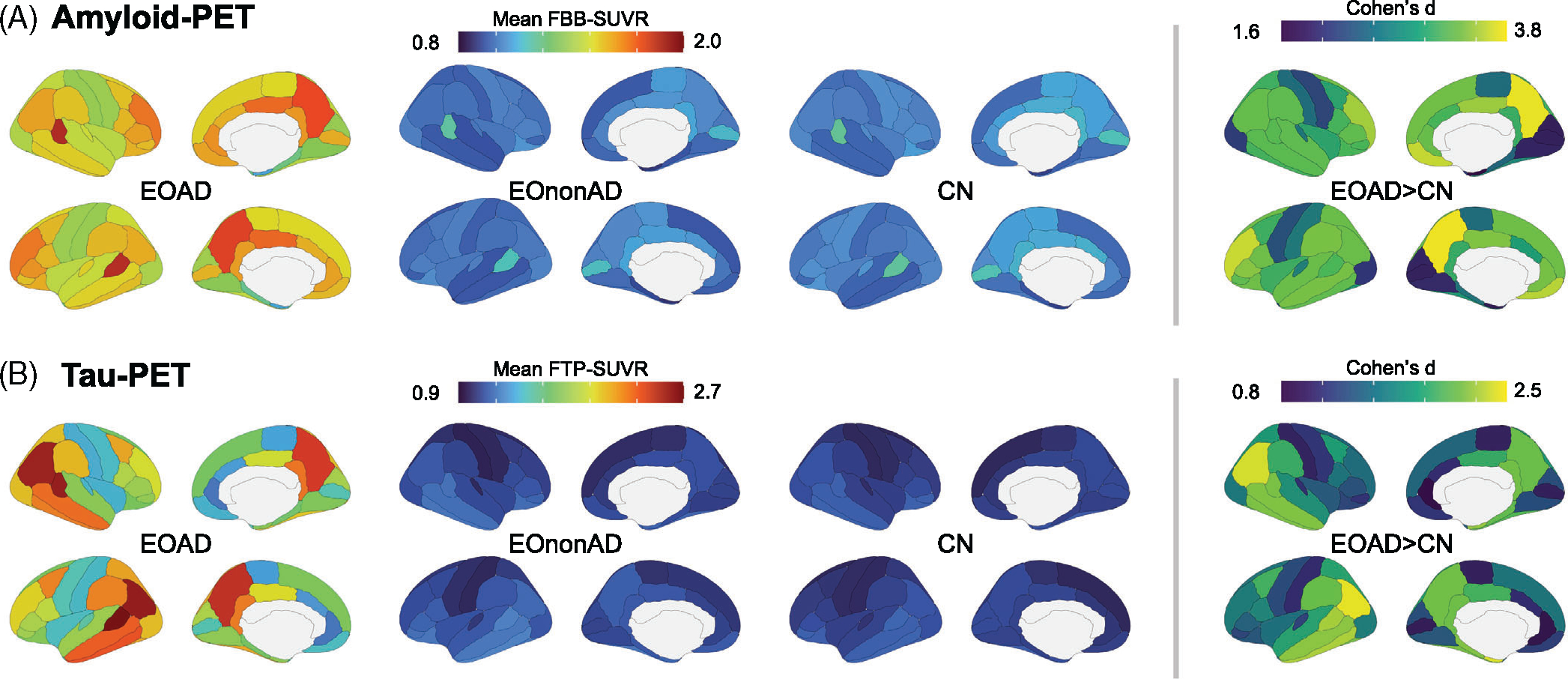
Regional distribution of amyloid- and tau-PET signal in the EOAD group. Regional distribution of amyloid- (A) and tau (B)-PET signal in all groups. Mean SUVR values were extracted from 68 cortical ROIs from the Desikan-Killiany atlas. The right panel shows group differences between EOAD and CN groups, quantified as Cohen’s d effect sizes (all 68 ROIs are different with Bonferroni-corrected p < 0.05). All pairwise group comparisons are described in detail in Table S1. CN, cognitively normal; EOAD, early-onset Alzheimer’s disease; PET, positron emission tomography; ROI, region of interest; SUVR, standardized uptake value ratio
The comparison of regional patterns of amyloid- and tau-PET between EOAD and EOnonAD resulted in very similar results as the comparison of EOAD and CN (Table S2). The comparison of EOnonAD to CN showed weak differences (Table S2). At the group level, FBB-SUVR were higher in CN than in EOnonAD, with 34 regions being significant at an uncorrected α = 0.05 threshold but only the left temporal pole and the right transverse temporal cortex survived Bonferroni correction. It is worth noting that, while participants were assigned to the EOnonAD group based on a negative amyloid-PET, controls were included regardless of amyloid status (Figure 1). These group differences were likely driven by the presence of a few amyloid-positive controls (Figure 2, Section 3.3.1). Last, the group comparison between EOnonAD and CN revealed little differences in FTP-PET, with only three ROIs showing uncorrected p < 0.05, all in the EOnonAD > CN direction: left entorhinal, temporal pole, and lateral orbitofrontal cortex. None of these regions survived Bonferroni correction (Table S2).
Group comparisons were repeated after controlling for age and sex, and results were virtually unchanged (Table S2).
3.4.2 |. Regional analyses in the EOAD group
We assessed association between regional PET SUVR and age, sex, and MMSE within EOAD participants (n = 243 for age and sex; n = 240 for MMSE, due to missing values). Results are presented in Figure 5 and detailed statistics are provided in Table S3.
FIGURE 5.
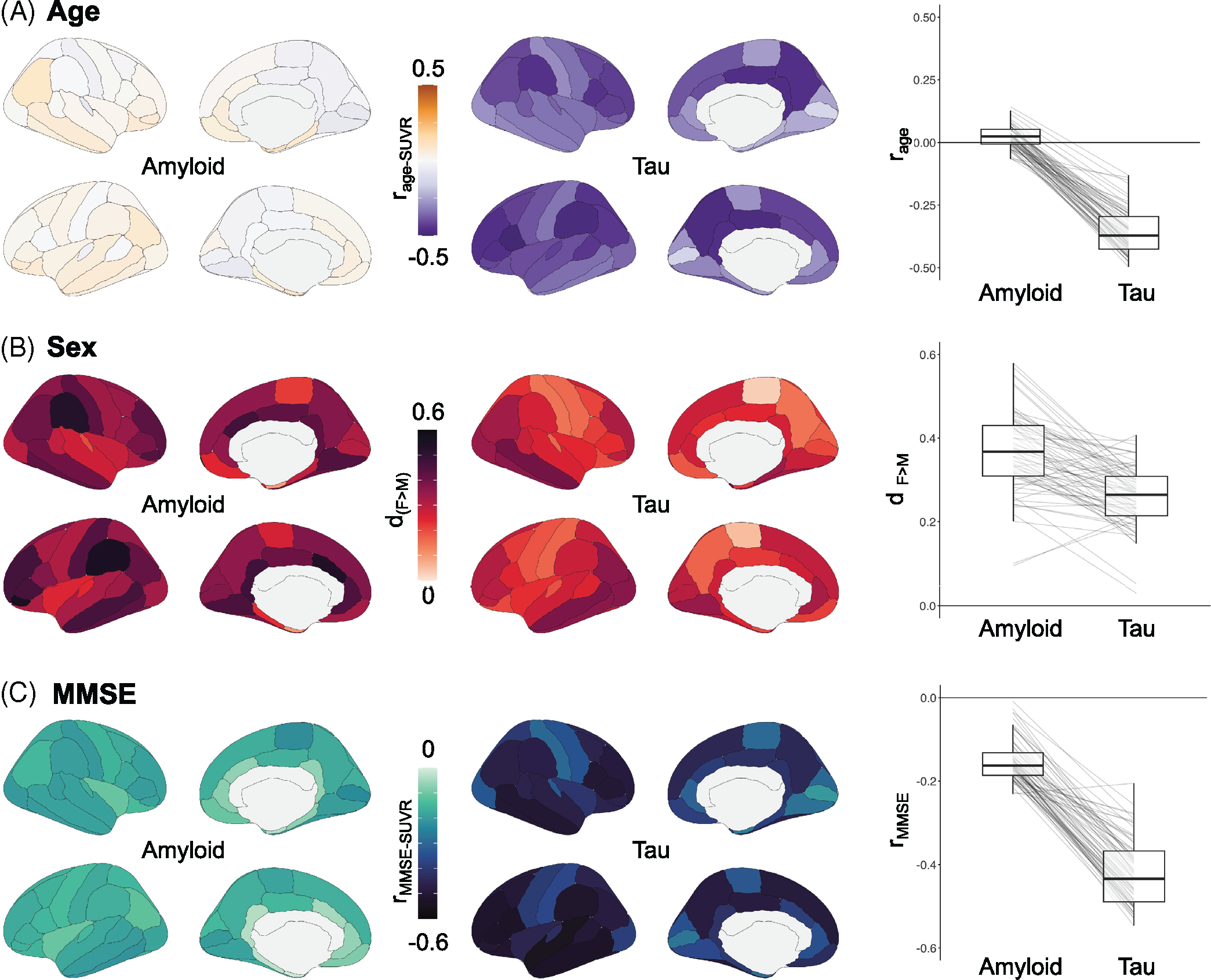
Association between regional PET SUVR and age, sex, and MMSE in the EOAD group. Regional analyses between PET SUVR, extracted from 68 cortical ROIs from the Desikan-Killiany atlas (left column: amyloid-PET with Florbetaben; right column: tau-PET with Flortaucipir), and age (A), sex (B), and MMSE (C). The right most panel shows the distribution of effect sizes (correlation coefficients for A and C, Cohen’s d for B) in all 68 ROIs. Details on the statistical measures are available in Table S2. EOAD, early-onset Alzheimer’s disease; MMSE, Mini-Mental State Examination; PET, positron emission tomography; ROI, region of interest; SUVR, standardized uptake value ratio
Older age at scan was associated with lower FTP-SUVR across all cortical ROIs (Figure 5A). All 68 regions had negative r values with uncorrected p < 0.05, and 63/68 regions remained significant after applying stringent Bonferroni correction. Highest association between tau and age was seen in the parietal lobe (both laterally and medially) and in the frontal cortex (r < −0.45), while temporal and occipital regions had less negative r values (e.g., r = −0.21 and −0.25 for right and left entorhinal cortices; Table S3). In contrast, regional FBB-SUVR were unrelated to participant age, with r values distributed around 0 (Figure 5A). Only 2 regions (left and right temporal poles) reached uncorrected p < 0.05 and showed weak positive association between FBB-SUVR and age (r = 0.143, p = 0.026; r = 0.128, p = 0.045). The relationship between age and SUVR was stronger for FTP than for FBB in all 68 ROIs (Figure 5A, right panel).
Female participants tended to have higher FBB- and FTP-SUVR values than males across regions (Figure 5B). For amyloid-PET, differences were significant at uncorrected p < 0.05 in 59/68 ROIs, and 16 survived Bonferroni correction. Highest differences were seen in the bilateral supramarginal gyri and frontal poles (Cohen’s d > 0.50). Sex differences were milder with tau-PET, with only 37 ROIs significant at uncorrected p < 0.05, and none surviving Bonferroni correction (Table S3). Highest sex difference was observed in the bilateral temporal and occipital cortex, with Cohen’s d exceeding 0.35.
Finally, higher FBB- and FTP-SUVR was associated with lower MMSE across regions (Figure 5C). Associations were relatively weak for FBB-PET, with significant correlations in 51 ROIs using an uncorrected threshold, and only 2 regions surviving Bonferroni correction, the right paracentral (r = −0.226, p = 0.0004) and right pericalcarine (r = −0.230, p = 0.0003) cortices. In contrast, all 68 ROIs showed significant association between FTP-SUVR and MMSE, with 67 surviving Bonferroni correction (Table S3). Highest associations were seen in temporal and frontal areas, with correlation coefficients below −0.50.
3.5 |. Exploratory analyses: clinical phenotypes in cognitively impaired participants
At the current stage of LEADS data collection, the majority of cognitively impaired patients were described by as having “amnestic-predominant single or multi-domain cognitive impairment” (257 out of 321, 80%) by the study clinician. Other syndromes included posterior cortical atrophy (PCA, 20 patients 6%), primary progressive aphasia (PPA, 19 patients, 6%), or “non-amnestic single of multi-domain cognitive impairment, not above” (“non-amnestic, other”, 25 patients, 8%). Due to the small number of patients in these non-amnestic syndromes, the following analyses need to be considered exploratory.
Each syndrome included patients who were assigned to both EOAD and EOnonAD cohorts based on amyloid-PET screening results (Figure 1), and no significant association was found between clinical phenotype and amyloid-PET positivity (Figure 6a).
FIGURE 6.
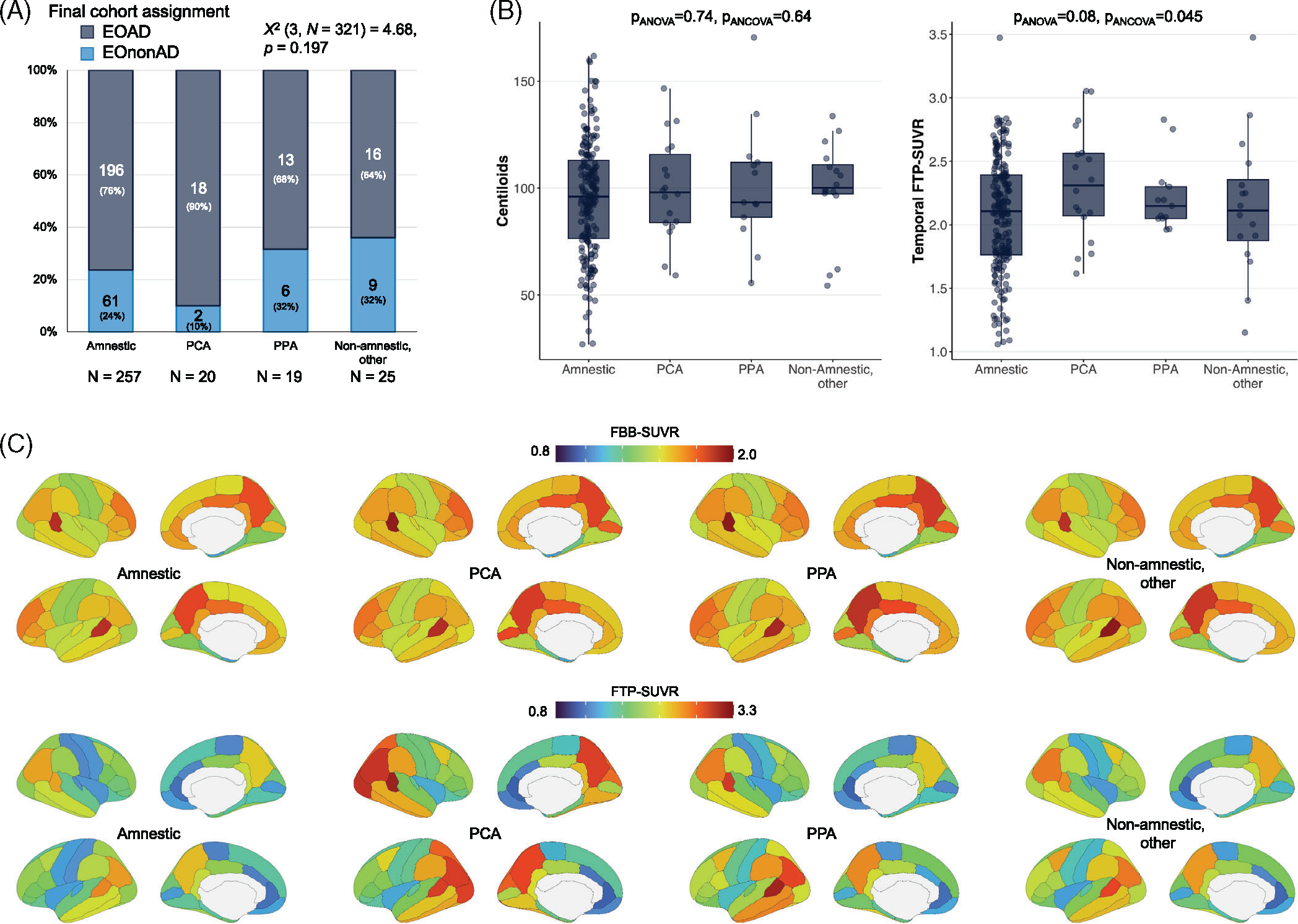
Clinical phenotypes in clinically impaired participants: exploratory analyses. (A) Proportion of cases that were assigned to the EOAD versus EOnonAD groups based on the amyloid-PET screening (see Figure 1a). (B) Summary metrics of amyloid and tau PET for each clinical phenotype in the EOAD group (n = 196 amnestic, 18 PCA, 13 PPA, 16 other non-amnestic cases); see Figure S3 for demographic and clinical information, and Figure S3 for tau-PET analyses using global cortical SUVR instead of temporal SUVR. Groups were compared using Welch’s analysis of variance (“pANOVA”) and using an analysis of covariance controlling for age and sex (“pANCOVA”). (C) Regional distribution of amyloid (Florbetaben, top) and tau (Flortaucipir, bottom) PET in each clinical phenotype in the EOAD group (same Ns as in panel B). See Table S4 and Figure S3 for statistical comparisons. EOAD, early-onset Alzheimer’s disease; EOnonAD, early-onset non-Alzheimer’s disease; PCA, posterior cortical atrophy; PET, positron emission tomography; PPA, primary progressive aphasia; SUVR, standardized uptake value ratio
Within patients with EOAD (see Figure S3a for demographic and clinical metrics), amyloid burden measured using centiloids was not related to clinical phenotypes (Figure 6b). The tau-PET measured in the temporal lobe (Figure 6b) or the whole cortex (Figure S2d) showed significant variability across phenotypes (p values between 0.002 and 0.08, depending on the tau-PET metrics and statistical model). While post hoc tests were limited due to sample size, it is to note that for both tau-PET metrics (temporal or whole cortical region), the amnestic EOAD subgroup has the lowest average SUVR values.
Average patterns of amyloid- and tau-PET binding in each phenotype are presented in Figure 6c; results of statistical comparison are available in Figure S3B and Table S4. Briefly, amyloid-PET patterns were mostly identical in all four phenotypes, with 9 regions showing group differences at uncorrected p < 0.05, but none surviving Bonferroni correction. Tau-PET patterns varied across phenotypes, both visually (Figure 6c), and as evidenced by formal comparison with 32 regions showing p < 0.05, 14 of which survived Bonferroni correction, mostly in the occipital,temporal, and parietal cortices (Figure S3B and Table S4).
4 |. DISCUSSION
We described the baseline PET characteristics of the first 408 participants enrolled in LEADS. Among participants who met clinical criteria for MCI or dementia due to AD, 76% were amyloid-PET-positive, of whom 95% were also tau-PET-positive. Overall, 72% of cognitively impaired participants meeting clinical criteria for an EOAD clinical syndrome fulfilled the biomarker-based definition of AD (A+T+) according to the NIA-AA Research Framework.10
A recent meta-analysis of 10,139 participants from 50 cohorts confirmed that amyloid-PET positivity rates vary with age and clinical diagnosis.8 This dataset indicated that around age 60 (our group’s average age), 39% of patients with MCI and 85% of patients with a clinical diagnosis of AD dementia were amyloid-PET-positive. While our cohort showed comparable results for patients with dementia (83.6%), amyloid-positivity rate in LEADS participants with MCI (61.3%) was higher than reported in the meta-analysis. This discrepancy might be due to most existing studies focusing on amnestic MCI or all MCI subtypes, regardless of whether AD was the suspected etiology, while our study included MCI due to suspected AD, including non-amnestic variants. Previous research has shown that non-amnestic phenotypes like PCA or logopenic variant PPA are very highly predictive of amyloid-positivity.29 In LEADS, amyloid positivity rates did not significantly vary across clinical phenotypes (Figure 6a), but this result should be interpreted with caution given the small sample size. Overall, the prevalence of amyloid-negativity in LEADS is consistent with the appropriate use criteria for amyloid-PET,44 which highlight the need for biomarkers in patients with suspected EOAD.
The tau-PET positivity was highly related to amyloid-PET results (Figure 2): 95% of amyloid-PET-positive (EOAD) patients were tau-PET positive, and the 12 A+T− EOAD participants had lower amyloid burden than A+T+ cases, consistent with previous publications.45 In contrast, only six (7.7%) of the EOnonAD patients were tau-PET positive (A-T+), three of whom were unambiguously positive, with higher tau-PET values than the average EOAD patient (Figure S1). While other studies have documented this rare (“A-T++”) biomarker pattern,46,47 interpretation remains ambiguous. Additional genetic and biofluid analyses, and ultimately autopsy data, will be helpful to better interpret these PET results and clarify the underlying cause of cognitive impairment in these patients. A likely hypothesis, supported by Krishnadas et al.,47 is that these patients could have underlying AD with a false negative amyloid-PET due to a relative low burden of neuritic plaques39 or rare Aβ conformation that are not detected by the radiotracer.48–51 Alternatively, PET results could reflect a false positive tau-PET or a true tau tangle-only disease, such as primary age-related tauopathy52 or MAPT variants associated with tau-PET-positive Frontotemporal lobar degeneration (FTLD)-tauopathy.53
Apart from these few tau-PET-positive cases, most EOnonAD participants had clearly negative amyloid- and tau-PET scans. However, it cannot be excluded that some of these participants have underlying AD, with neuropathology burden below PET detection levels. This could be explained by the imperfect sensitivity of amyloid- and tau-PET to detect intermediate levels of AD neuropathology,39,54 and consistent with the milder clinical severity observed in this group (Table 1). Overall, the EOnonAD group likely includes a mix of various etiologies, as suggested by previous findings in older patients.55,56 Yet, the non-AD etiologies mimicking a clinical presentation of EOAD are expected to be different from LOAD-like diseases. While the latter includes limbic-predominant age-related TDP-43 encephalopathy,57 argyrophilic grain disease,58 and/or cerebrovascular lesions,59 these are less likely to play a role in patients in their 50s or early 60s. Instead, these non-AD etiologies are more likely to include FTLD subtypes and non-neurodegenerative conditions, including psychiatric disorders. We plan to analyze [ź8F]fluorodeoxyglucose-PET scans acquired in the EOnonAD participants to identify patterns of hypometabolism suggestive of non-AD diseases. Last, the longitudinal follow-up of patients with EOnonAD with clinical, neuropsychological, imaging, genetic, and biomarker data will help better understand which conditions mimic EOAD.
Quantitative measures of amyloid- and tau-PET were correlated (Figure 2c). This association was not linear, showing maximal association in the midrange of CL, as previously noted.60,61 The amyloid-tau relationship was still significant in the EOAD group only, and the association between amyloid-PET and clinical measures was mediated by tau-PET. These findings are crucial in the context of anti-Aβ therapies. Clinical trials with several anti-Aβ monoclonal antibodies have shown that significant reduction in Aβ plaques (as measured by PET) is associated with lowering of soluble concentrations of phosphorylated tau in plasma and CSF, slowing of accumulation of tangles (as measured by tau-PET), and less progression of cognitive and functional decline.62,63 The correlation between amyloid- and tau-PET, particularly in intermediate ranges of pathology supports the premise that lowering amyloid in this range may slow the tau accumulation. In the phase 2 study of donanemab,64 patients in the lowest baseline tau-PET tertile showed the greatest clinical benefit from amyloid removal. Few data have been published on the efficacy of anti-amyloid drugs in EOAD, but exploratory analyses in the lecanemab trial suggested that younger patients might show less clinical benefit than older patients.65 Based on the high baseline FTP-SUVR values seen in EOAD, one can hypothesize that lower efficacy of amyloid removal may be due to the higher tau burden in younger patients. The relationship between pretreatment tau-PET and clinical response to anti-Aβ therapies should be examined in future clinical trials and longitudinal registries of novel therapies such as the Alzheimer’s Network for Treatment and Diagnostics (ALZNET). If future research confirms the link between tau burden and efficacy of anti-amyloid treatments, it will be crucial to study earlier (potentially preclinical) stages of EOAD, when tau burden is lower and interventions are more likely to benefit patients. Yet, the preclinical phase of sporadic EOAD has not been studied, and the timing of pathological events needs to be established in EOAD. In addition, the present data suggest that, even in early clinical stages of the disease, most patients with EOAD may benefit less from amyloid-lowering as a sole therapy. Targeting inflammation or tau early on (e.g., preventing aggregation, spread, or expression using antisense oligonucleotides66) may be particularly important for this population.
In EOAD patients, the magnitude of cortical tau-PET, but not amyloid-PET was higher in younger patients, consistent with previous studies showing higher cortical tau-PET signal in EOAD compared to LOAD14,15,21 and studies showing a negative correlation between frontal and parietal tau-PET and age of onset in cohorts including both EOAD and LOAD.13 This finding of an age effect within an EOAD group emphasizes that age of onset is a continuum rather than a binary distribution split around an arbitrary 65-year-old threshold.67 Moreover, we replicated previous findings of female patients harboring more advanced amyloid- and tau-PET burden than males, in spite of similar cognitive testing scores.68 As discussed in previous publications, these observations could be explained by younger and female patients having higher resilience to pathology or by differences in the dynamic of disease progression. Longitudinal clinical and imaging data will help test these hypotheses.
CN participants in LEADS had very low frequency of amyloid-positivity (7/87, 8%) and only two cases (2%) were also positive on tau-PET (A+T+), consistent with the published low prevalence of preclinical AD in this age range.8,69–71 We found a low prevalence of amyloid-PET positivity in young CN in LEADS despite apparent enrichment of APOE ε4 carriers in the CN group (42.4%, double the expected ε4 prevalence in cognitively unimpaired adults). These findings suggest that detectable preclinical amyloid accumulation typically occurs after age 65, even in APOE-ε4 carriers. It should be noted that the CN group did not undergo the same amyloid-PET screening process as patients with clinical impairment (Figure 1); amyloid positivity was purely based on MRI-based FBB-PET quantification, and a 25 CL threshold. While this cut-off was based on a PET-to-autopsy study39 showing optimal discrimination of intermediate-to-high from absent-to-low AD neuropathological changes,9 thresholds in the 15–25 range, could improve sensitivity for the earliest amyloid stages to detect preclinical AD.33,38–40,72–75
This study has limited generalizability because participants were recruited from tertiary memory research centers in the United States and were mostly non-Hispanic Whites. Our interpretation was also limited by the cross-sectional nature of the analyses, but we are following-up participants over time with longitudinal PET imaging. We plan to combine amyloid- and tau-PET data with other measures acquired in LEADS, including clinical, neuropsychological, genetic, biofluid, and MRI data to answer more integrated questions about EOAD pathophysiology. Last, we plan to perform a direct comparison of PET data acquired in LEADS with ADNI to formally compare EOAD and LOAD.
In conclusion, the analysis of baseline PET data from LEADS showed that clinical diagnosis of EOAD is predictive of A+T+ PET biomarker profile in 70%–75% of patients. Biomarker-positive EOAD patients already show advanced stage of tau accumulation in the neocortex. Younger patients and female patients show greater tau burden. This information should be considered when selecting treatments and planning clinical trials for EOAD to identify patients who are most likely to benefit from targeted interventions.
Supplementary Material
HIGHLIGHTS.
72% of patients with clinical EOAD were positive on both amyloid- and tau-PET.
Amyloid-positive patients with EOAD had high tau-PET signal across cortical regions.
In EOAD, tau-PET mediated the relationship between amyloid-PET and MMSE.
Among EOAD patients, younger onset and female sex were associated with higher tau-PET.
RESEARCH IN CONTEXT.
1. Systematic review:
Authors reviewed existing literature on positron emission tomography (PET) imaging in early-onset alzheimer’s disease (EOAD) (PubMed, Google Scholar). Published studies, especially those combining amyloid- and tau-PET, were typically based on single site cohorts with relatively small samples.
2. Interpretation:
In this large, multi-site study of participants with a clinical diagnosis of EOAD, ~25% individuals did not have biomarker evidence for underlying AD. Among amyloid-positive participants, high cortical tau-PET burden indicated that patients are already at an advanced stage of tau accumulation at the time of diagnosis.
3. Future directions:
PET data acquired in Longitudinal Early-onset Alzheimer’s Disease Study (LEADS) will be combined with other clinical, genetic, and biomarker (magnetic resonance imaging, cerebrospinal fluid, plasma) data for cross-modal correlations. Longitudinal PET data will soon be available to assess disease progression. Future comparison of LEADS and ADNI cohorts will allow direct comparison between EOAD and more typical late-onset AD.
ACKNOWLEDGMENTS
We thank participants and families for their trust and commitment to research. Avid Radiopharmaceuticals enabled the use of Flortaucipir but did not provide direct funding and were not involved in data analysis or interpretation. Life Molecular Imaging enabled the use of Florbetaben but did not provide direct funding and were not involved in data analysis or interpretation. LEADS is funded by the National Institute on Aging (NIA) U01-AG057195 and NIA R56-AG057195. GDR receives additional funding from NIA P30-AG062422, NIA R35-AG072362, NINDS R21-NS120629, NINDS U19-NS110456, Alzheimer’s Association ZEN-21-848216, American College of Radiology, Rainwater Charitable Foundation, Alliance for Therapies in Neuroscience.
Footnotes
CONFLICT OF INTEREST STATEMENT
Dr. Cho has nothing to disclose. Ms. Mundada has nothing to disclose. Dr. Apostolova served as a paid consultant for Biogen and Two Labs, serves on a DSMB for IQVIA, and receives research support from the NIH, the Alzheimer’s Association, Roche, Life Molecular Imaging, and Eli Lilly. Dr. Carrillo has nothing to disclose. Ms. Shankar has nothing to disclose. Ms. Amuiri has nothing to disclose. Dr. Zeltzer has nothing to disclose. Dr. Windon has nothing to disclose. Dr. Soleimani-Meigooni has nothing to disclose. Dr. Tanner has nothing to disclose. Dr. Lawhn Heath has nothing to disclose. Dr. Lesman-Segev has nothing to disclose. Dr. Aisen reports grants from NIA, FNIH, the Alzheimer’s Association, Janssen, Lilly, and Eisai, and personal fees from Merck, Roche, Biogen, Lundbeck, ImmunoBrain Checkpoint, and Samus. Dr. Eloyan has nothing to disclose. Dr. Lee has nothing to disclose. Dr. Hammers has nothing to disclose. Ms. Kirby has nothing to disclose. Dr. Dage has nothing to disclose. Dr. Fagan has received research support from Biogen, Fujirebio, and Roche Diagnostics. She is a member of the scientific advisory boards for Roche Diagnostics, Genentech, and AbbVie and consults for Araclon/Griffols, DiademRes, and Otsuka Pharmaceuticals. Dr. Foroud has nothing to disclose. Dr. Grinberg has nothing to disclose. Dr. Jack serves on an independent data monitoring board for Roche and has consulted for and served as a speaker for Eisai, but he receives no personal compensation from any commercial entity. He receives research support from the NIH and the Alexander Family Alzheimer’s Disease Research Professorship of the Mayo Clinic. Dr. Kramer has nothing to disclose. Dr. Kukull is supported by the NIA. Dr. Murray served as a paid consultant for AVID Radiopharmaceuticals and receives research support from the NIH, the Alzheimer’s Association, and the state of Florida. Dr. Nudelman has nothing to disclose. Dr. Toga has nothing to disclose. Dr. Vemuri has nothing to disclose. Dr. Atri has nothing to disclose. Dr. Day is supported by the NIA; he serves as a topic editor on dementia for DynaMed Plus (EBSCO Industries, Inc), is the clinical director for the Anti-NMDA Receptor Encephalitis Foundation and holds stocks in ANI Pharmaceuticals. Dr. Duara has nothing to disclose. Dr. Graff-Radford receives research support from Lilly, AbbVie, and Biogen. Dr. Honig has nothing to disclose. Dr. Jones has nothing to disclose. Dr. Masdeu served on the speaker’s bureau and consulted for Biogen. He received research support from the NIH, Avanir, Abbvie, Biogen, Eli Lilly, Esai, and Novartis. Dr. Mendez receives grant support from NIA and has received support from Biogen. Dr. Musiek received research funding from Eisai Pharmaceuticals Inc. Dr. Onyike has nothing to disclose. Dr. Rogalski has nothing to disclose. Dr. Salloway received consultation fees and research support from Biogen, Eisai, Lilly, Genentech, and Roche. Dr. Sha has nothing to disclose. Dr. Turner has nothing to disclose. Dr. Wingo has nothing to disclose. Dr. Wolk received grants from Eli Lilly/Avid Radiopharmaceuticals, Merck, and Biogen, and consulted for Merck, Janssen, and GE Healthcare. Dr. Koeppe has nothing to disclose. Dr. Iaccarino is currently a full-time employee of Eli Lilly and Company / Avid Radiopharmaceuticals and a minor shareholder of Eli Lilly and Company. His contribution to the work presented in this manuscript was performed while he was affiliated with the University of California San Francisco. Dr. Dickerson receives research support from the NIH and the Alzheimer’s Drug Discovery Foundation. He consulted for Arkuda, Axovant, Lilly, Biogen, Merck, Novartis, and Wave LifeSciences. He is editor for Neuroimage: Clinical and Cortex. He receives royalties from Oxford University Press and Cambridge University Press. Dr. La Joie is an Associate Editor for Alzheimer’s Research & Therapy. Dr. Rabinovici receives research funding from Avid Radiopharmaceuticals, GE Healthcare, Life Molecular Imaging, Genentech. He has received consulting fees from Alector, Eli Lilly, Genentech, Roche, Merck. He receives fees for serving on a DSMB for Johnson & Johnson. He is an Associate Editor for JAMA Neurology The authors have no conflicts of interest to declare supporting information.
CONSENT STATEMENT
Written informed consent was obtained from all patients or their surrogates. The study was conducted under a central institutional review board based at Indiana University.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
REFERENCES
- 1.Zhu XC, Tan L, Wang HF, et al. Rate of early onset Alzheimer’s disease: a systematic review and meta-analysis. Ann Transl Med. 2015;3:38. doi: 10.3978/j.issn.2305-5839.2015.01.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sirkis DW, Bonham LW, Johnson TP, La Joie R, Yokoyama JS. Dissecting the clinical heterogeneity of early-onset Alzheimer’s disease. Mol Psychiatry. 2022;27:2674–2688. doi: 10.1038/s41380-022-01531-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Szigeti K, Doody RS. Should EOAD patients be included in clinical trials? Alzheimers Res Ther. 2011;3:4. doi: 10.1186/alzrt63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Apostolova LG, Aisen P, Eloyan A, et al. The Longitudinal Early-onset Alzheimer’s Disease Study (LEADS): framework and methodology. Alzheimers Dement. 2021;17:2043–2055. doi: 10.1002/alz.12350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Swartz JR, Miller BL, Lesser IM, et al. Behavioral phenomenology in Alzheimer’s disease, frontotemporal dementia, and late-life depression: a retrospective analysis. J Geriatr Psychiatry Neurol. 1997;10:67–74. doi: 10.1177/089198879701000206 [DOI] [PubMed] [Google Scholar]
- 6.Lecanema T US Veterans Health Administration will cover cost of new Alzheimer’s drug. BMJ. 2023;380:628. doi: 10.1136/bmj.p628 [DOI] [PubMed] [Google Scholar]
- 7.Cummings J, Apostolova L, Rabinovici GD, et al. Lecanemab: appropriate use recommendations. J Prev Alzheimers Dis. 2023;10(3):362–377. doi: 10.14283/jpad.2023.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jansen WJ, Janssen O, Tijms BM, et al. Prevalence estimates of amyloid abnormality across the Alzheimer disease clinical spectrum. JAMA Neurology. 2022;79:228–243. doi: 10.1001/jamaneurol.2021.5216 [DOI] [PubMed] [Google Scholar]
- 9.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: a practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jack CR, Bennett DA, Blennow K, et al. NIA-AA Research Framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018;14:535–562. doi: 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spina S, La Joie R, Petersen C, et al. Comorbid neuropathological diagnoses in early versus late-onset Alzheimer’s disease. Brain. 2021;144:2186–2198. doi: 10.1093/brain/awab099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marshall GA, Fairbanks LA, Tekin S, Vinters HV, Cummings JL. Early-onset Alzheimer’s disease is associated with greater pathologic burden. J Geriatr Psychiatry Neurol. 2007;20:29–33. doi: 10.1177/0891988706297086 [DOI] [PubMed] [Google Scholar]
- 13.La Joie R, Visani AV, Lesman-Segev OH, Baker SL, et al. Association of APOE4 and clinical variability in Alzheimer disease with the pattern of Tau- and Amyloid-PET. Neurology. 2021;96:e650–e661. doi: 10.1212/WNL.0000000000011270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pontecorvo MJ, Devous MD, Navitsky M, et al. Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain. 2017;140:748–763. doi: 10.1093/brain/aww334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stage EC, Svaldi D, Phillips M, et al. Neurodegenerative changes in early- and late-onset cognitive impairment with and without brain amyloidosis. Alzheimers Res Ther. 2020;12:93. doi: 10.1186/s13195-020-00647-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med. 2020;12. doi: 10.1126/scitranslmed.aau5732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho H, Jeon S, Kang SJ, et al. Longitudinal changes of cortical thickness in early- versus late-onset Alzheimer’s disease. Neurobiology of Aging. 2013;34(1921):e9–1921.e15. doi: 10.1016/j.neurobiolaging.2013.01.004 [DOI] [PubMed] [Google Scholar]
- 18.Cho H, Seo SW, Kim JH, et al. Amyloid deposition in early onset versus late onset Alzheimer’s disease. J Alzheimers Dis. 2013;35:813–821. doi: 10.3233/JAD-121927 [DOI] [PubMed] [Google Scholar]
- 19.Rabinovici GD, Furst AJ, Alkalay A, et al. Increased metabolic vulnerability in early-onset Alzheimer’s disease is not related to amyloid burden. Brain. 2010;133:512–528. doi: 10.1093/brain/awp326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ossenkoppele R, Zwan MD, Tolboom N, et al. Amyloid burden and metabolic function in early-onset Alzheimer’s disease: parietal lobe involvement. Brain. 2012;135:2115–2125. doi: 10.1093/brain/aws113 [DOI] [PubMed] [Google Scholar]
- 21.Tanner JA, Iaccarino L, Edwards L, et al. Amyloid, tau and metabolic PET correlates of cognition in early and late-onset Alzheimer’s disease. Brain. 2022;145:4489–4505. doi: 10.1093/brain/awac229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klunk WE, Koeppe RA, Price JC, et al. The Centiloid Project: standardizing quantitative amyloid plaque estimation by PET. Alzheimers Dement. 2015;11:1–15.e1–4. doi: 10.1016/j.jalz.2014.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jack CR, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement. 2017;13:205–216. doi: 10.1016/j.jalz.2016.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ossenkoppele R, Rabinovici GD, Smith R, et al. Discriminative accuracy of [18F]flortaucipir positron emission tomography for Alzheimer disease vs other neurodegenerative disorders. JAMA. 2018;320:1151–1162. doi: 10.1001/jama.2018.12917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–279. doi: 10.1016/j.jalz.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKhann GM, Knopman DS, Chertkow, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–269. doi: 10.1016/j.jalz.2011.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hammers DB, Eloyan A, Taurone A, et al. Profiling baseline performance on the Longitudinal Early-Onset Alzheimer’s Disease Study (LEADS) cohort near the midpoint of data collection. Alzheimer’s & Dementia nd;n/a. 2023. doi: 10.1002/alz.13160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Townley RA, Graff-Radford J, Mantyh WG, et al. Progressive dysexecutive syndrome due to Alzheimer’s disease: a description of 55 cases and comparison to other phenotypes. Brain Commun. 2020;2:fcaa068. doi: 10.1093/braincomms/fcaa068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ossenkoppele R, Pijnenburg YAL, Perry DC, et al. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain. 2015;138:2732–2749. doi: 10.1093/brain/awv191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorno-Tempini ML, Dronkers NF, Rankin KP, et al. Cognition and anatomy in three variants of primary progressive aphasia. Ann Neurol. 2004;55:335–346. doi: 10.1002/ana.10825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Crutch SJ, Schott JM, Rabinovici GD, et al. Consensus classification of posterior cortical atrophy. Alzheimers Dement. 2017;13:870–884. doi: 10.1016/j.jalz.2017.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabri O, Sabbagh MN, Seibyl J, et al. Florbetaben PET imaging to detect amyloid beta plaques in Alzheimer’s disease: phase 3 study. Alzheimers Dement. 2015;11:964–974. doi: 10.1016/j.jalz.2015.02.004 [DOI] [PubMed] [Google Scholar]
- 33.Ozlen H, Pichet Binette A, Köbe T, et al. Spatial extent of Amyloid-β levels and associations with Tau-PET and cognition. JAMA Neurol. 2022;79:1025–1035. doi: 10.1001/jamaneurol.2022.2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villeneuve S, Rabinovici GD, Cohn-Sheehy BI, et al. Existing Pittsburgh Compound-B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain. 2015;138:2020–2033. doi: 10.1093/brain/awv112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maass A, Landau S, Baker SL, et al. Comparison of multiple tau-PET measures as biomarkers in aging and Alzheimer’s disease. Neuroimage. 2017;157:448–463. doi: 10.1016/j.neuroimage.2017.05.058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diedrichsen J, Balsters JH, Flavell J, Cussans E, Ramnani N. A probabilistic MR atlas of the human cerebellum. Neuroimage. 2009;46:39–46. doi: 10.1016/j.neuroimage.2009.01.045 [DOI] [PubMed] [Google Scholar]
- 37.Desikan RS, Ségonne F, Fischl B, et al. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. Neuroimage. 2006;31:968–980. doi: 10.1016/j.neuroimage.2006.01.021 [DOI] [PubMed] [Google Scholar]
- 38.Royse SK, Minhas DS, Lopresti BJ, et al. Validation of amyloid PET positivity thresholds in centiloids: a multisite PET study approach. Alzheimers Res Ther. 2021;13:99. doi: 10.1186/s13195-021-00836-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.La Joie R, Ayakta N, Seeley WW, et al. Multisite study of the relationships between antemortem [11C]PIB-PET Centiloid values and postmortem measures of Alzheimer’s disease neuropathology. Alzheimers Dement. 2019;15:205–216. doi: 10.1016/j.jalz.2018.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van der Kall LM, Truong T, Burnham SC, et al. Association of β-Amyloid level, clinical progression, and longitudinal cognitive change in normal older individuals. Neurology. 2021;96:e662–e670. doi: 10.1212/WNL.0000000000011222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sonni I, Lesman Segev OH, Baker SL, et al. Evaluation of a visual interpretation method for tau-PET with 18F-flortaucipir. Alzheimers Dement (Amst). 2020;12:e12133. doi: 10.1002/dad2.12133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wickham H ggplot2. Springer International Publishing; 2016. doi: 10.1007/978-3-319-24277-4 [DOI] [Google Scholar]
- 43.Mowinckel AM, Vidal-Piñeiro D. Visualisation of Brain Statistics with R-packages ggseg and ggseg3d. 2019. doi: 10.48550/arXiv.1912.08200 [DOI] [Google Scholar]
- 44.Johnson KA, Minoshima S, Bohnen NI, et al. Appropriate use criteria for amyloid PET: a report of the Amyloid imaging task force, the society of nuclear medicine and molecular imaging, and the Alzheimer’s association. Alzheimers Dement. 2013;9:e-1–16. doi: 10.1016/j.jalz.2013.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ossenkoppele R, Leuzy A, Cho H, et al. The impact of demographic, clinical, genetic, and imaging variables on tau PET status. Eur J Nucl Med Mol Imaging. 2021;48:2245–2258. doi: 10.1007/s00259-020-05099-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoon B, Guo T, Provost K, et al. Abnormal tau in amyloid PET negative individuals. Neurobiol Aging. 2022;109:125–134. doi: 10.1016/j.neurobiolaging.2021.09.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krishnadas N, Doré V, Laws SM, et al. Exploring discordant low amyloid beta and high neocortical tau positron emission tomography cases. Assessment & Disease Monitoring. 2022;14:e12326. doi: 10.1002/dad2.12326. Alzheimer’s & Dementia: Diagnosis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chhatwal JP, Schultz SA, McDade E, et al. Variant-dependent heterogeneity in amyloid β burden in autosomal dominant Alzheimer’s disease: cross-sectional and longitudinal analyses of an observational study. Lancet Neurol. 2022;21:140–152. doi: 10.1016/S1474-4422(21)00375-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rosen RF, Ciliax BJ, Wingo TS, et al. Deficient high-affinity binding of Pittsburgh compound B in a case of Alzheimer’s disease. Acta Neuropathol. 2010;119:221–233. doi: 10.1007/s00401-009-0583-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tomiyama T, Nagata T, Shimada H, et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann Neurol. 2008;63:377–387. doi: 10.1002/ana.21321 [DOI] [PubMed] [Google Scholar]
- 51.Schöll M, Wall A, Thordardottir S, et al. Low PiB PET retention in presence of pathologic CSF biomarkers in Arctic APP mutation carriers. Neurology. 2012;79:229–236. doi: 10.1212/WNL.0b013e31825fdf18 [DOI] [PubMed] [Google Scholar]
- 52.Crary JF, Trojanowski JQ, Schneider JA, et al. Primary age-related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol. 2014;128:755–766. doi: 10.1007/s00401-014-1349-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsai RM, Bejanin A, Lesman-Segev O, et al. 18F-flortaucipir (AV-1451) tau PET in frontotemporal dementia syndromes. Alzheimers Res Ther. 2019;11(1):13. doi: 10.1186/s13195-019-0470-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Soleimani-Meigooni DN, Iaccarino L, La Joie R, et al. 18F-flortaucipir PET to autopsy comparisons in Alzheimer’s disease and other neurodegenerative diseases. Brain. 2020;143:3477–3494. doi: 10.1093/brain/awaa276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chételat G, Ossenkoppele R, Villemagne VL, et al. Atrophy, hypometabolism and clinical trajectories in patients with amyloid-negative Alzheimer’s disease. Brain. 2016;139:2528–2539. doi: 10.1093/brain/aww159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Landau SM, Horng A, Fero A, Jagust WJ. Amyloid negativity in patients with clinically diagnosed Alzheimer disease and MCI. Neurology. 2016;86:1377–1385. doi: 10.1212/WNL.0000000000002576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nelson PT, Dickson DW, Trojanowski JQ, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019;142:1503–1527. doi: 10.1093/brain/awz099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grinberg LT, Heinsen H. Argyrophilic grain disease: an update about a frequent cause of dementia. Dement Neuropsychol. 2009;3:2–7. doi: 10.1590/S1980-57642009DN30100002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gladman JT, Corriveau RA, Debette S, et al. Vascular contributions to cognitive impairment and dementia: research consortia that focus on etiology and treatable targets to lessen the burden of dementia worldwide. Alzheimers Dement (N Y). 2019;5:789–796. doi: 10.1016/j.trci.2019.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Doré V, Krishnadas N, Bourgeat P, et al. Relationship between amyloid and tau levels and its impact on tau spreading. Eur J Nucl Med Mol Imaging. 2021;48:2225–2232. doi: 10.1007/s00259-021-05191-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rowe CC, Doré V, Krishnadas N, et al. Tau Imaging with 18F-MK6240 across the Alzheimer’s Disease spectrum. 2022:2022.02.13.22270894. doi: 10.1101/2022.02.13.22270894 [DOI]
- 62.Pontecorvo MJ, Lu M, Burnham SC, et al. Association of donanemab treatment with exploratory plasma biomarkers in early symptomatic Alzheimer disease: a secondary analysis of the TRAILBLAZER-ALZ randomized clinical trial. JAMA Neurology. 2022;79:1250–1259. doi: 10.1001/jamaneurol.2022.3392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shcherbinin S, Evans CD, Lu M, et al. Association of Amyloid reduction after donanemab treatment with tau pathology and clinical outcomes: the TRAILBLAZER-ALZ randomized clinical trial. JAMA Neurology. 2022;79:1015–1024. doi: 10.1001/jamaneurol.2022.2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in Early Alzheimer’s disease. N Engl J Med. 2021;384:1691–1704. doi: 10.1056/NEJMoa2100708 [DOI] [PubMed] [Google Scholar]
- 65.van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in Early Alzheimer’s disease. N Engl J Med. 2023;388:9–21. doi: 10.1056/NEJMoa2212948 [DOI] [PubMed] [Google Scholar]
- 66.Mummery CJ, Börjesson-Hanson A, Blackburn DJ, et al. Tau-targeting antisense oligonucleotide MAPTRx in mild Alzheimer’s disease: a phase 1b, randomized, placebo-controlled trial. Nat Med. 2023;29:1437–1447. doi: 10.1038/s41591-023-02326-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Reitz C, Rogaeva E, Beecham GW. Late-onset vs nonmendelian early-onset Alzheimer disease. Neurol Genet. 2020;6:e512. doi: 10.1212/NXG.0000000000000512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edwards L, La Joie R, Iaccarino L, et al. Multimodal neuroimaging of sex differences in cognitively impaired patients on the Alzheimer’s continuum: greater tau-PET retention in females. Neurobiol Aging. 2021;105:86–98. doi: 10.1016/j.neurobiolaging.2021.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jack CR Jr, Therneau TM, Weigand SD, et al. Prevalence of biologically vs clinically defined Alzheimer spectrum entities using the National Institute on Aging–Alzheimer’s Association Research Framework. JAMA Neurology. 2019;76:1174–1183. doi: 10.1001/jamaneurol.2019.1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ossenkoppele R, Pichet Binette A, Groot C, et al. Amyloid and tau PET-positive cognitively unimpaired individuals are at high risk for future cognitive decline. Nat Med. 2022;28:2381–2387. doi: 10.1038/s41591-022-02049-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Strikwerda-Brown C, Hobbs DA, Gonneaud J, et al. Association of elevated amyloid and tau positron emission tomography signal with near-term development of Alzheimer disease symptoms in older adults without cognitive impairment. JAMA Neurology. 2022;79:975–985. doi: 10.1001/jamaneurol.2022.2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Farrell ME, Jiang S, Schultz AP, et al. Defining the lowest threshold for Amyloid-PET to Predict future cognitive decline and Amyloid accumulation. Neurology. 2021;96:e619–e631. doi: 10.1212/WNL.0000000000011214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Salvadó G, Molinuevo JL, Brugulat-Serrat A, et al. Centiloid cut-off values for optimal agreement between PET and CSF core AD biomarkers. Alzheimers Res Ther. 2019;11:27. doi: 10.1186/s13195-019-0478-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Amadoru S, Doré V, McLean CA, et al. Comparison of amyloid PET measured in Centiloid units with neuropathological findings in Alzheimer’s disease. Alzheimers Res Ther. 2020;12:22. doi: 10.1186/s13195-020-00587-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bullich S, Roé-Vellvé N, Marquié M, et al. Early detection of amyloid load using 18F-florbetaben PET. Alzheimers Res Ther. 2021;13:67. doi: 10.1186/s13195-021-00807-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.