

Abstract
Cerebral organoids are comprised of diverse cell types found in the developing human brain, and can be leveraged in the identification of critical cell types perturbed by genetic risk variants in common, neuropsychiatric disorders. There is great interest in developing high-throughput technologies to associate genetic variants with cell types. Here, we describe a high-throughput, quantitative approach (oFlowSeq) by utilizing CRISPR-Cas9, FACS sorting, and next-generation sequencing. Using oFlowSeq, we found that deleterious mutations in autism-associated gene KCTD13 resulted in increased proportions of Nestin+ cells and decreased proportions of TRA-1–60+ cells within mosaic cerebral organoids. We further identified that a locus-wide CRISPR-Cas9 survey of another 18 genes in the 16p11.2 locus resulted in most genes with > 2% maximum editing efficiencies for short and long indels, suggesting a high feasibility for an unbiased, locus-wide experiment using oFlowSeq. Our approach presents a novel method to identify genotype-to-cell type imbalances in an unbiased, high-throughput, quantitative manner.
Complex 3D human induced pluripotent stem cell (iPSC) derived systems such as brain organoids provide researchers with an in-vitro system comprised of diverse cell types (Lancaster and Knoblich 2014; Pasca et al. 2015; Qian et al. 2016; Quadrato et al. 2017), that can be used to identify the relationships between genes and cell types in common, complex human neurodevelopmental and neurodegenerative diseases such as autism spectrum disorders (ASD) and Alzheimer’s disease (AD) (Roth et al. 2020). Previously, we reported a transcriptomics-based approach using donorderived cerebral organoids (Orgo-Seq) (Lim et al. 2022), to identify critical cell types and candidate driver genes associated with an ASD-associated risk variant (16p11.2 deletion) (Kumar et al. 2008; Sanders et al. 2011; Sebat et al. 2007; Weiss et al. 2008). Deletions in the 16p11.2 locus were reported to be associated with increased head sizes in humans, while duplications were associated with decreased head sizes (Horev et al. 2011; Qureshi et al. 2014; Shinawi et al. 2010; Steinman et al. 2016). The reverse phenotypes indicate the importance of investigating cellular, morphological, and molecular differences.
Based on our prior research and research from other groups, we found a critical cell type (defined as a cell type whose proportions are affected by the mutation of interest, suggesting that the mutation might affect cell type specific molecular processes in the critical cell type) associated with 16p11.2 deletions (Lim et al. 2022; Urresti et al. 2021). The critical cell type mapped to intermediate progenitor cells (IPCs) and/or immature neurons (ImNs). This indicates that the 16p11.2 deletion mutation potentially affects proportions of IPCs and ImNs.
Using the co-expression of cell type specific genes, we further identified that there are three candidate driver genes within the 16p11.2 region (YPEL3, KCTD13 and INO80E) that are likely to be affecting the proportions of the critical cell type underlying 16p11.2 deletions. Of the three candidate driver genes, KCTD13 has been more well-studied compared to the other two genes. However, there were conflicting results from KCTD13-deficient animal studies for the role of KCTD13 in affecting head sizes in animal models (Arbogast et al. 2019; Escamilla et al. 2017; Golzio et al. 2012). At a cellular level, differences in head sizes (such as microcephaly) often affected the proportions of cell types such as neural progenitor cells in the brain (Chen et al. 2014; Doobin et al. 2016; Tang et al. 2016; Zhang et al. 2019).
To test the hypothesis if synthetic deleterious mutations in KCTD13 can affect the proportions of neural progenitor cells using human cerebral organoids, we developed a quantitative, sequencing- and fluorescence activated cell sorting (FACS)-based approach. Our method serves two primary purposes: first, to enable researchers to identify which synthetic mutations are likely to be benign mutations or deleterious mutations that are likely to impact mRNA transcripts or protein expression, by using functional readouts from FACS sorting; and second, to identify genotype-to-cell type associations (if any) using an unbiased screening of cells with deleterious or benign mutations, as well as unedited cells, within the same cerebral organoids that can control for experimental artifacts or batch effects.
We utilized an established approach named FlowSeq, that our group and other groups had previously described using bacteria and yeast (Kosuri et al. 2013; Raveh-Sadka et al. 2012; Sharon et al. 2012). We modified the concept used by FlowSeq to utilize CRISPR-Cas9 editing (Cong et al. 2013; Jinek et al. 2012; Mali et al. 2013; Yeo et al. 2018), combined with a mosaic cerebral organoid system targeting the KCTD13 gene, and termed our adapted approach as oFlowSeq for “organoid FlowSeq”. We found that deleterious mutations in KCTD13 resulted in increased proportions of Nestin+ neural progenitor cells, decreased proportions of TRA-1–60+ stem cells, but did not change the proportions of NeuN+ mature neurons. These results suggest that deleterious mutations in KCTD13 resulted in accelerated differentiation of TRA-1–60+ stem cells into Nestin+ cells.
The 16p11.2 locus encompasses 29 genes, out of which, 20 of these genes are expressed in bulk RNA sequence data from multiple donor-derived cerebral organoids (Lim et al. 2022). Editing in one of these expressed genes (ALDOA) resulted in inviable cells, and it was excluded from the analysis. KCTD13 was additionally excluded as it was tested in a single-gene experiment. We performed a locus-wide CRISPR-Cas9 editing of the remaining 18 genes in the 16p11.2 locus. We sequenced guide RNA (sgRNA) targeted regions for each gene, and calculated the editing efficiencies for each target region (defined as the ratio of mutant reads divided by reference reads). For each gene, we defined the highest editing efficiency across all target regions in the gene as the maximum editing efficiency. Our experiments achieved at least 2% maximum editing efficiencies, resulting in short or long indels in most of the 18 genes. Our results suggest that it is highly feasible to perform a targeted 16p11.2 locus-wide CRISPR-Cas9 survey of synthetic mutations to identify potential associations between deleterious mutations and cell type imbalances, utilizing wildtype reference reads in mosaic cerebral organoids to account for experimental biases.
Results
oFlowSeq method for unbiased discovery of gene-to-cell type relationships
We adapted a high-throughput concept from a previous method (FlowSeq) (Kosuri et al. 2013), to establish the oFlowSeq method to identify the relationships between genotypes to cell type proportions using cerebral organoids (Fig. 1A). We first performed CRISPR-Cas9 editing of the KCTD13 gene with ribonucleoprotein delivery of an sgRNA in induced pluripotent stem cells (iPSCs) from a single control donor. The pool of cells (wildtype and edited cells) were differentiated into mosaic cerebral organoids for three months. These mosaic organoids were comprised of several cell types with different KCTD13 genotypes. Subsequently, we dissociated these mosaic cerebral organoids into single cells for FACS, to obtain seven sorted pools of cells from four distinct markers (mouse IgG2A−, NeuN−, NeuN+, Nestin−, Nestin+, TRA-1–60−, and TRA-1–60+). The mouse IgG2A− cell population is equivalent to unsorted cells, and serves as an internal background control to ensure that any mutant reads observed in other sorted cell populations are similarly observed in the mouse I gG2A− cell population. We focused on three well-validated markers: NeuN as a marker for mature neurons, Nestin for neural progenitor cells and TRA-1–60 for stem cells (Supplementary Fig. 1). DNA from each sorted cell population was extracted for library preparation and high coverage targeted sequencing.
Fig. 1.
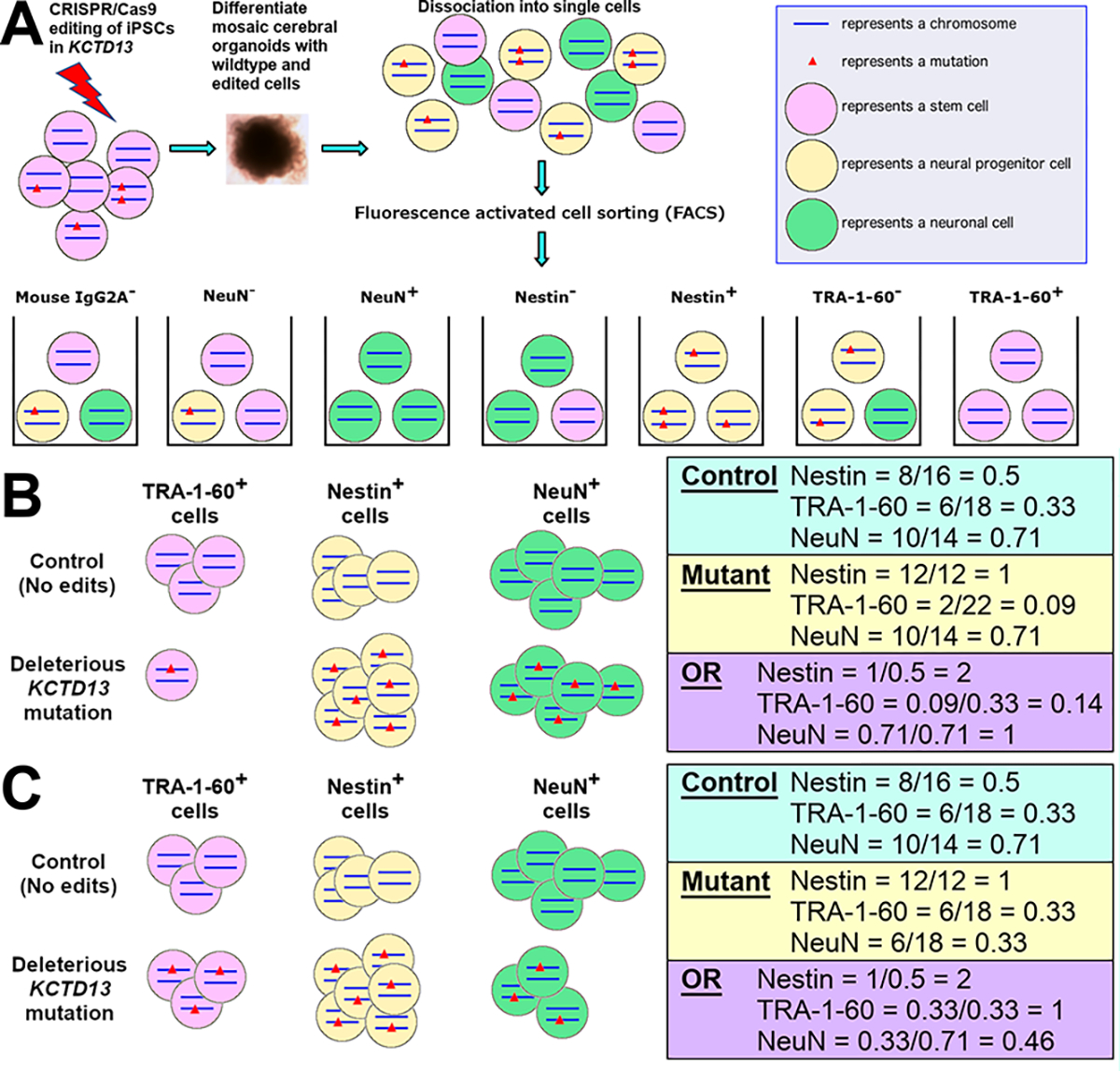
Hypothetical schematic of the oFlowSeq method to identify cell types affected by KCTD13 mutations in CRISPR-edited cerebral organoids. A Schematic of oFlowSeq method using CRISPR-Cas9 editing of the KCTD13 gene in iPSCs to differentiate mosaic cerebral organoids. These mosaic organoids, which comprised of different cell types with different KCTD13 genotypes, were dissociated into single cells for FACS into seven sorted pools of cells. B Hypothetical schematic of cell counts for a deleterious mutation in KCTD13 that enriches for Nestin+ cells while depleting TRA-1–60+ cells, with an OR > 1 representing an enrichment; OR < 1 representing a depletion; and OR = 1 representing no changes in cell proportions. C Hypothetical schematic of cell counts for a deleterious mutation in KCTD13 that enriches for Nestin+ cells while depleting NeuN+ cells
From the sequencing data, we identified reads that mapped to the reference (unedited) sequence, and reads that harbored mutations (insertions or deletions). For each mutation, we calculated a normalized ratio, which is defined as the number of reads for the mutation divided by the number of unedited reads. To identify if the normalized ratios of a mutation are different between two sorted cell populations, such as between Nestin− and Nestin+ cells, we calculated an odds ratio (OR) using the ratios from both sorted cell populations. An OR of greater than 1 indicates that the mutation is enriched in one sorted cell population compared to the other sorted cell population, and an OR of less than 1 indicates the reciprocal enrichment. An OR of 1 indicates that there is no enrichment of the mutation in either of the sorted cell populations.
Conceptually, we illustrate an example showing a control set of sorted cells that were not edited, and a set of cells that all contain a heterozygous deleterious mutation in the KCTD13 gene. If the deleterious mutation resulted in an increased proportion of Nestin+ cells and a decreased proportion of TRA-1–60+ cells, we expect to observe an OR = 2 for Nestin and OR = 0.33 for TRA-1–60 (Fig. 1B). These results would then suggest that there might be accelerated differentiation of TRA-1–60+ stem cells into Nestin+ cells, arising from the deleterious mutation.
On the other hand, if the heterozygous deleterious mutation in KCTD13 resulted in an increased proportion of Nestin+ cells and a decreased proportion of NeuN+ cells, we would expect to observe an OR = 2 for Nestin and OR = 0.46 for NeuN (Fig. 1C). These results would then suggest that there might be cell cycle arrest in Nestin+ cells due to the deleterious mutation, and that fewer mutant Nestin+ cells are differentiating into NeuN+ cells.
CRISPR-edited mosaic organoids show enrichment of KCTD13 mutants in Nestin+ cells
As a proof-of-concept experiment, as well as to follow up on our prior results (Lim et al. 2022), we created KCTD13 insertion and deletion mutations in iPSCs from a control individual using CRISPR-Cas9 and an sgRNA. Next, we differentiated mosaic cerebral organoids from a mixture of iPSCs harboring different KCTD13 mutations (edited cells) and iPSCs with reference sequences (unedited cells). After 84 days, we harvested the mosaic cerebral organoids and dissociated single cells from the organoids for FACS. DNA was extracted from the sorted cell populations, followed by targeted PCR, barcoding and MiSeq sequencing. We identified 19 different mutations in KCTD13 in these sorted cell populations, and calculated the ratios of reads for each mutation divided by the number of reference reads. The total percentage of obtaining one of the 19 mutant sequences versus reference sequences in KCTD13 was 8.3% in sorted mouse IgG2A− cells.
We divided the mutations into “short” mutations that resulted in ≤ 10-basepair (bp) insertions or deletions (indels), and “long” mutations that resulted in > 10-bp indels. For each of the sorted groups of cells, we calculated a mean ratio using the observed ratios from the long mutations, and normalized all ratios by the mean calculated ratio (Supplementary Table 1). This is to quantitatively account for experimental biases in the FACS sorting, such as differences in the concentrations of antibodies used, isotypes of the antibodies, or intensities of fluorophores. We calculated the mean ratio based on the long mutations, which showed a unimodal normal distribution compared to the distribution of ratios calculated based on the short mutations. We calculated the ORs for TRA-1–60, Nestin, and NeuN by dividing the normalized ratios between the TRA-1–60− versus TRA-1–60+ cells, Nestin− versus Nestin+ cells, and NeuN− versus NeuN + cells.
If all detected mutations in KCTD13 were benign mutations, or mutations that did not affect the proportions of a cell type, we would expect that the ORs for the mutations to be normally distributed with a mean OR of 1 (Fig. 2A). On the other hand, if a subset of the detected mutations in KCTD13 were deleterious mutations that affected the proportions of a cell type, we would expect to observe a bimodal distribution, where the deleterious mutations will have a mean OR of > 1 (Fig. 2B).
Fig. 2.
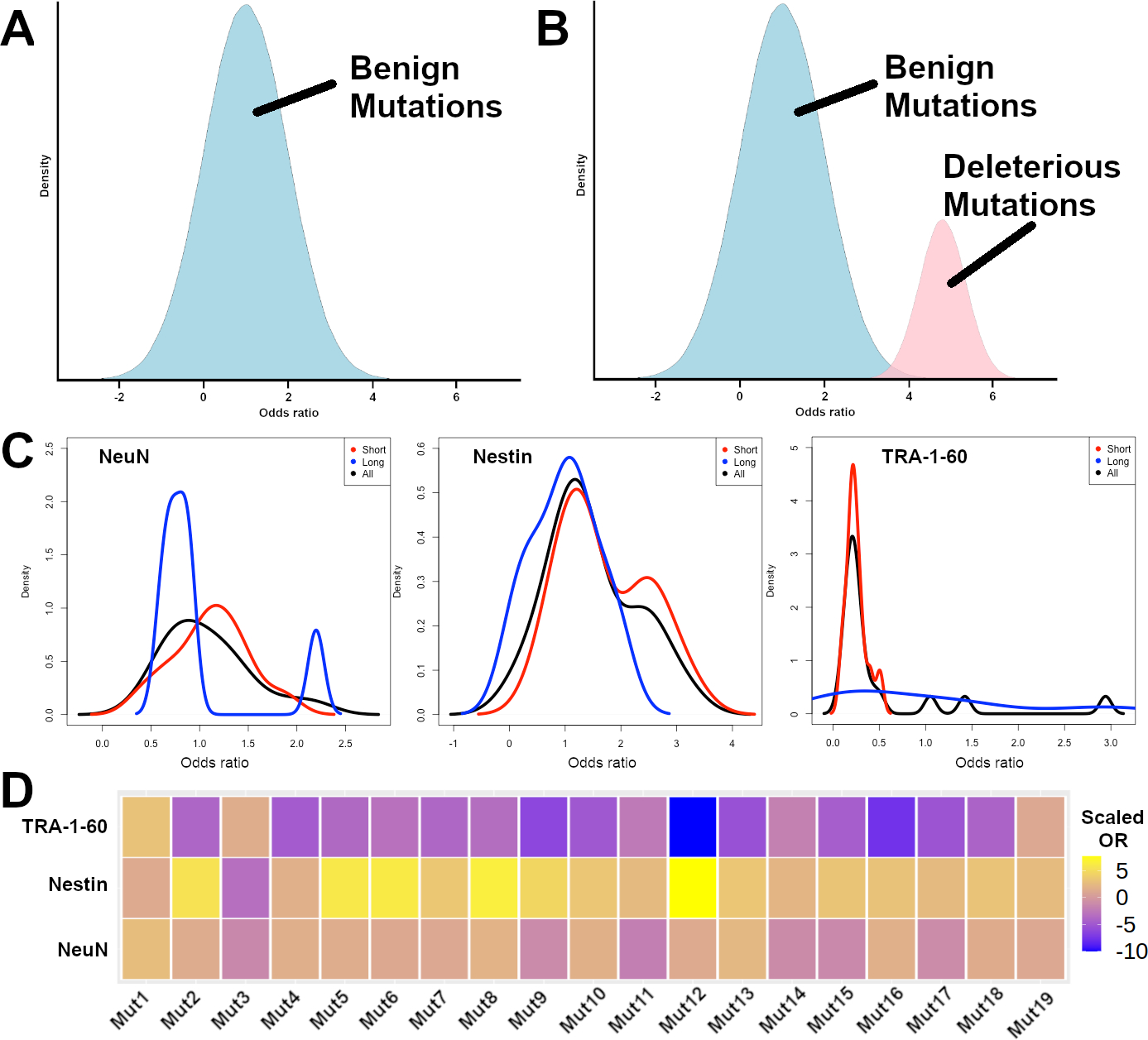
Odds ratios to correlate mutations with cell type proportions. A Hypothetical expected distribution of ORs for benign mutations that do not affect cell type proportions (in blue). B Hypothetical expected distribution of ORs for benign mutations that do not affect cell type proportions (in blue), and deleterious mutations that increase cell type proportions (in pink). C Observed density distributions of short mutations (red), long mutations (blue), and all mutations (black) for NeuN, Nestin, and TRA-1–60 positively stained cells. D Observed heatmap representation of the scaled odds ratios for each of the 19 synthetic mutations in KCTD13 for TRA-1–60, Nestin, and NeuN positively stained cells
We found that the density plot of Nestin ORs for short indels showed a bimodal distribution, but not for long indels (Fig. 2C), suggesting that a subset of the short indels in KCTD13 were likely to be deleterious mutations that affected the proportions of Nestin+ versus Nestin− cells. We observed that for the short indels, the mean ORs across all KCTD13 mutations for TRA-1–60, Nestin, and NeuN were 0.25, 1.7, and 1.1 respectively (Wilcoxon rank-sum test P = 0.0048, 0.014 and 0.61 respectively), suggesting that for the short indels, there may be differences in the proportions of TRA-1–60+ versus TRA-1–60− cells and Nestin+ versus Nestin− cells, but not in the proportions of NeuN+ versus NeuN− cells. As a control, we did not observe any differences in the normalized ratios for long indels across TRA-1–60, Nestin, and NeuN (Wilcoxon rank-sum test P = 0.82, 0.94 and 1 respectively).
The mutation with the highest normalized Nestin OR (Mut12) was a 2-bp deletion that resulted in amino acid changes starting at Phe93Trp, resulting in a truncated 129 amino acid sequence instead of the full-length 329 amino acid sequence (Fig. 2D, Supplementary Table 1). The mutation with the second highest normalized Nestin OR (Mut8) was a 1-bp insertion that resulted in changes starting at the same amino acid Phe93Leu, resulting in a truncated 130 amino acid sequence. Both mutations affected the BTB/POZ domain of the KCTD13 protein that is necessary for Cullin-3 interaction, similar to previously identified synthetic deleterious mutations in KCTD13 (Kizner et al. 2020).
Locus-wide CRISPR-editing in 16p11.2 reveal high rates of short and long indels
There are 29 genes in the 16p11.2 locus, and it is likely that multiple genes within or outside the locus can drive the cellular phenotypes in ASD associated with deletions in the 16p11.2 locus (Blumenthal et al. 2014; Lim et al. 2022; Pizzo et al. 2021; Roth et al. 2020; Tai et al. 2016; Urresti et al. 2021; Vysotskiy et al. 2021). Of the 29 genes, 20 of them (including KCTD13) are expressed in bulk RNA sequence data from cerebral organoids (Lim et al. 2022). As a feasibility study to evaluate if we can perform locus-wide CRISPR-editing for all the 20 genes with detectable rates of short indels, we surveyed the synthetic mutational landscape introduced by CRISPR-Cas9 editing for the other 19 genes, to identify the rates of mutations and types of mutations. For each of the 19 genes, we obtained a pool of 1–3 sgRNAs targeting the exons (Supplementary Table 2). We further designed multiple pairs of sequencing primers for each region targeted by each sgRNA, and selected the pair of sequencing primers that resulted in a single band for each region, indicating that these primer pairs are likely to be specific for amplifying the targeted region. In instances where several sets of sequencing primer pairs were specific, we used only one of the sets of primer pairs (Supplementary Table 3).
Of the 19 genes, one gene (ALDOA) has been shown to be associated with a rare autosomal recessive disease (Kara et al. 2021; Yao et al. 2004). However, we did not obtain any viable cells after nucleofection using the sgRNA pool for ALDOA initially. Using 1:10 and 1:100 dilutions of the sgRNA pool for ALDOA, we obtained viable cells that were then sequenced, but we did not detect any short indels that were 10-bp or shorter. It is possible that further optimization to reduce the concentrations of the sgRNA pool might yield viable cells with hemizygous short indels in ALDOA.
For the remaining 18 genes, we performed 3 sets of analyses using “ultra-short” indels that resulted in PCR products that were ≤ 2-bp shorter compared to the unedited PCR product, short indels that resulted in PCR products that were ≤ 10-bp shorter compared to the unedited PCR product, and long indels that resulted in PCR products that were > 10-bp longer compared to the unedited PCR product. We observed that for the long indels, there is a correlation between the total number of sequencing reads per sample with the editing efficiency for each sgRNA, which is calculated as the ratio of edited reads to the total number of reads (Pearson’s correlation r = 0.84, P = 4 × 1 0−14). However, for short and ultra-short indels, there were no correlations between the total number of sequencing reads per sample with the editing efficiency for each sgRNA (r = 0.076, P = 0.6 and r = 0.06, P = 0.67 respectively). These observations suggest that there are more long indels at lower frequencies than short or ultra-short indels with lower frequencies, so we will need to sequence each sample with higher coverage to accurately detect or quantify the rates of these long indels or that these long indel sequences might be enriched for sequencing artifacts. The full list of synthetic mutations and editing efficiencies for each pair of sequencing primers for each sgRNA is shown in Supplementary Table 4.
We calculated the editing efficiencies as the ratios of each set of mutant reads to reference reads, and identified the maximum editing efficiency for each gene, which was defined as the maximum editing efficiency across all sgRNAs for each gene (Supplementary Table 5). We found that 12 of the 18 genes had maximum editing efficiencies of > 5% for ultra-short indels, 14 genes had maximum editing efficiencies of > 5% for short indels, and 17 genes had maximum efficiencies of > 5% for long indels (Fig. 3A–D). For short and long indels, most genes had maximum editing efficiencies of more than 2% (Fig. 3A, B).
Fig. 3.
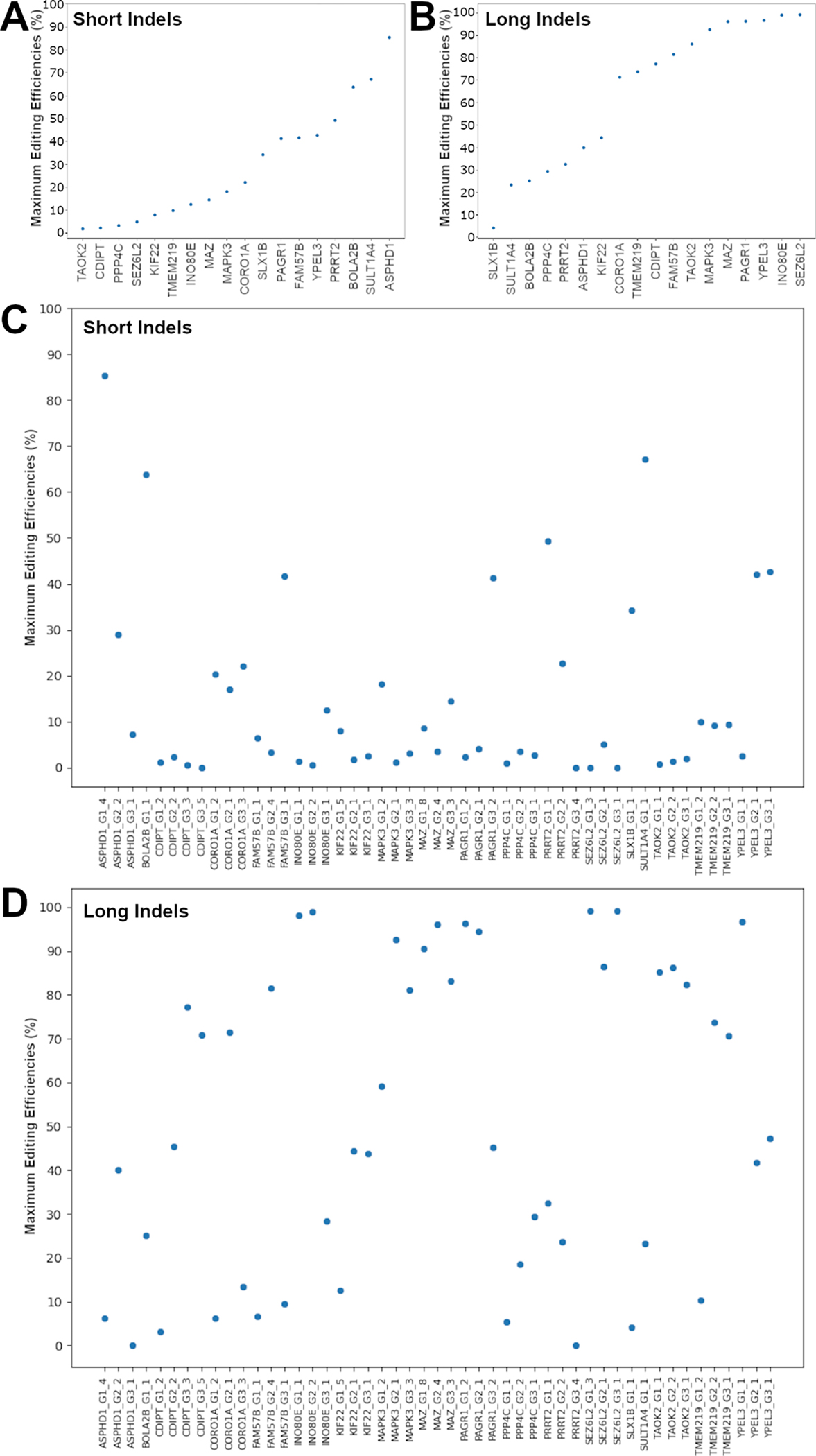
Maximum editing efficiencies for each gene. A Gene-level maximum editing efficiencies for short indels. B Gene-level maximum editing efficiencies for long indels. C Guide-level editing efficiencies for short indels. D Guide-level editing efficiencies for long indels.
Identifying the maximum editing efficiencies for gene each in our pilot study is important for future oFlowSeq experiments into the 16p11.2 locus. In our pilot study, we conducted 49 sets of PCR experiments on edited iPSCs and another 49 sets of PCR experiments on unedited iPSCs in parallel. However, it might not be feasible to perform the same number of PCR experiments in parallel for each FACS-sorted population of cells. If we use the same three markers (NeuN, Nestin, and TRA-1–60), we will need to perform 392 PCR experiments in total. However, given that we had identified the maximum editing efficiencies from multiple sgRNAs for each gene, we can now perform targeted PCR and sequencing for only a single region in each gene, resulting in a total of 144 PCR experiments. In addition, we can estimate the sequence coverage needed for each gene, as genes with higher maximum editing efficiencies that result in short indels will require less sequence coverage than genes with lower maximum editing efficiencies that result in short indels.
Given the high efficiencies of indels observed in our locus-wide CRISPR-Cas9 editing of the 18 genes in the 16p11.2 locus, our results show that it is highly feasible to perform locus-wide CRISPR-Cas9 editing, and utilize the oFlowSeq framework to identify deleterious mutations in each of the 18 genes that might affect the proportions of Nestin+ cells. An unbiased quantitative method such as oFlowSeq will be important for identifying deleterious from benign indels, and for utilizing the mosaic cerebral organoid system to identify potential associations between deleterious mutations to cell type imbalances by using reference unedited sequences as controls for batch effects or experimental artifacts.
Discussion
Cerebral organoids have been demonstrated to be complex systems comprised of diverse cell types that can be used to identify critical cell types that are important in common human neurological diseases. We describe the oFlowSeq system, where we harness the diversity of cell types found in cerebral organoids, combined with the diversity of synthetic mutations using CRISPR-Cas9, to identify genotype-to-cell type associations in an unbiased and quantitative way. A major strength of mosaic cerebral organoids differentiated from a mixture of edited and unedited iPSCs is that similar conditions are maintained for all the edited and unedited cells across different cell types that were differentiated from the same organoids. As such, we can leverage the heterogeneity of the cerebral organoids for creating a self-controlled mixture of cells for generating or validating hypotheses about cell types affected by disease-associated mutations.
Transcriptomics-based approaches using bulk or single-cell RNA sequencing to identify genotype-to-cell type associations, such as our previous Orgo-Seq framework (Lim et al. 2022), are becoming increasingly effective approaches for novel discoveries. Our oFlowSeq method provides an orthogonal approach to these transcriptomics-based approaches, and enables independent technological validation of discoveries to overcome biases that can exist in experimental technologies used. For instance, our previously described Orgo-Seq framework is a non-cell autonomous approach that uses bulk RNA sequence data from donor-derived cerebral organoids with naturally occurring variants (Lim et al. 2022). On the other hand, oFlowSeq is a cell autonomous, FACS-based approach that uses CRISPR-Cas9 induced synthetic mutations to discover or validate associations between genes and genotypes with cell type imbalances.
One limitation with the oFlowSeq framework is that we can only sort for cell types with well-validated cell type specific antibodies that are compatible for flow cytometry or FACS. There might be cellular subtypes of interest with unknown cell type markers, and it will be more suitable to first use a single-cell transcriptomics-based approach to identify novel cell type markers. It is also possible that a cell type specific antibody can sort for multiple cell types expressing the same marker. We can use multiple antibodies to perform oFlowSeq and obtain finer resolution into the identity of the relevant critical cell type.
Another limitation with the oFlowSeq framework is that it can only be used to evaluate genes that are dosage sensitive, i.e. if heterozygous deleterious mutations and homozygous deleterious mutations similarly result in loss-of-function on gene function. If heterozygous deleterious mutations in a gene result in loss-of-function effect on the gene and increase the proportions of a particular critical cell type, but homozygous deleterious mutations in the gene result in gain-of-function effect on the gene and decrease the proportions of the same critical cell type, the differences in proportion of the critical cell type will be negated.
Currently, we performed the experiments for CRISPR-Cas9 editing and PCR in parallel. To improve the throughput for surveying many mutations and genes, such as across hundreds of genes, it will be important to develop a truly multiplexed system. Additional optimization will be needed to evaluate the conditions for performing CRISPR-Cas9 editing on multiple genes and performing multiplexed PCR for sequencing.
As a proof-of-concept experiment to demonstrate the oFlowSeq framework using KCTD13, we had performed the experiments once. Additional replication and validation experiments will be needed to fully interpret the biological effects of KCTD13 in the 16p11.2 locus. In addition to the KCTD13 gene, we also performed locus-wide CRISPR-Cas9 editing for 18 genes in the 16p11.2 locus, and demonstrated high maximum editing efficiencies for detecting short or long indels in all genes. In the future, it will be exciting to explore the genotype to cell type associations for all genes in the 16p11.2 locus using the oFlowSeq framework, and we can evaluate the contributions of all genes locus-wide using an unbiased quantitative approach. In addition, it will be exciting to perform a mutational scan to identify novel or known domains within each gene that can modulate the proportions of cell types. The oFlowSeq method can also be applied to identify locus-wide genotype-to-cell type associations for other copy number variants associated with neuropsychiatric disorders.
Materials and methods
Standard protocol approval
Research performed on samples and data of human origin was conducted according to protocols approved by the institutional review board of UMass Chan Medical School.
Source of iPSC
The control donor iPSC line used in our study (PGP1 iPSC) was a kind gift from Professor George Church’s lab, and we had previously characterized and sequenced the line (Lim et al. 2022). We performed flow cytometry (MACSquant VYB) to confirm that > 90% of the PGP1 iPSCs are positive for TRA-1–60 (Novus Biologicals NB100–730F488).
CRISPR-Cas9 editing of KCTD13 in iPSCs
iPSCs were cultured with mTeSR1 media (Stemcell Technologies 85850) on 6-well plates coated with Matrigel Basement Membrane Matrix (Corning 354234), until they were ~ 75% confluent. Accutase (BioLegend 423201) was used to dissociate the cells, and 800,000 cells were counted using an automated cell counter (NanoEnTek EVE). We used an sgRNA targeting the second exon of KCTD13 (chr16:29923312–29923331; sequence 5′-UGAG GAUUGU AC CAA AGU GA-3′) and Cas9 2NLS nuclease from Synthego, and incubated 3 μl of 100 μM sgRNA with 2 μl of 20 μM Cas9 nuclease at room temperature for 10 min prior to nucleofection. Nucleofection was performed using the HSC-1 kit and B-016 protocol on an Amaxa nucleofector (Lonza Bioscience). After nucleofection, the iPSCs were passaged onto a single well of a 24-well plate coated with Matrigel in 1 ml of mTeSR1 media with 10 μM Y-27632 (Santa Cruz sc-216067A) and 1:100 SMC4 cocktail (BioVision S231) for 24 h, and expanded for another 6 days on 6-well plates coated with Matrigel.
Differentiation of mosaic cerebral organoids
As previously described (Lim et al. 2022), we adapted our cerebral organoid differentiation protocol according to a previously described protocol (Lancaster et al. 2013). Briefly, 900,000 iPSCs were re-suspended in 15 ml of mTeSR1 medium with 50 μM Y-27632, and 150 μl was seeded into individual wells of a 96-well ultra-low attachment Corning plate (ThermoFisher CLS7007). On Day 6, 50 μl of mTeSR medium with a single embryoid body was transferred to individual wells of 24-well ultra-low attachment Corning plates (ThermoFisher CLS3473) with 500 μl of neural induction media per well. On Day 8, another 500 μl of neural induction media was added to each well of the 24-well plates. On Day 10, a droplet comprised of 10 μl of neural induction media with an organoid was placed onto a single dimple on Parafilm substrate, and 40 μl of Matrigel (Corning 354234) was added to each organoid to encapsulate it. The Matrigel droplets were incubated at 37 °C for 15 min before they were scraped into single wells of the 24-well plates using a cell scraper. 1 ml of differentiation media with 10% penicillin streptomycin (ThermoFisher 15140122) per well was used to passage the organoids every 2–4 days, and the plates of organoids were placed on an orbital shaker at 90 rpm in the incubator.
Preparation of mosaic cerebral organoids for antibody staining and FACS
Mosaic cerebral organoids were collected after 84 days, and washed twice using 1 × DPBS at 4 °C for 10 min for each wash. 0.25% Trypsin–EDTA (ThermoFisher 25200056) was added to dissociate the cells for 30 min at 37 °C on a shaking heat block at 300 rpm, before inactivating the trypsin using mTeSR1 medium and washed twice using 1 × DPBS (ThermoFisher 14190144). To remove residual Matrigel, dissociated cells were filtered through 30 μm cell strainers (Miltenyi Biotec 130–041-407). Cells were counted using an automated cell counter and 12 million cells were fixed and permeabilized using equal volumes of 4% paraformaldehyde and permeabilization buffer (DPBS, 0.02% sodium azide, 2% FBS and 0.1% saponin) for 45 min. 3 million cells were used for each antibody and staining was performed for an hour, followed by FACS.
The antibodies used were commercially available and Alexa Fluor 488-conjugated: mouse IgG2A control (R&D Systems IC003G), NeuN (Novus Biologicals NBP-92693AF488), Nestin (R&D Systems IC1259G), and TRA-1–60 (Novus Biologicals NB100–730F488). For each antibody, 1 million cells were sorted using a Sony SH800 FACS sorter using a 100 μm sorting chip and sorting speed at 4000 events per second. Live positive cells were gated using the mouse IgG2A− cells.
DNA extraction, PCR and MiSeq sequencing
Cells were washed twice with 1 × DPBS and DNA extraction was performed using the standard protocol for the DNA FFPE Tissue Kit (Qiagen 56,404). PCR was performed using the standard protocol for Q5 Hot Start Master Mix (New England BioLabs M094S). We also performed PCR using unedited DNA extracted from PGP1 iPSCs as a control for background.
The following primers were used for sequencing the KCTD13 locus:
Forward primer: 5′-CAC CAG GTA GTA GCG TGC TT-3′
Reverse primer: 5′-GCA GCA AAG CCA TCT TTC CC-3′
Barcoding was performed using Nextera indexes, followed by DNA clean-up using the Monarch PCR & DNA Cleanup Kit (New England BioLabs T1030S). Library preparations were quantified using the using KAPA library quantification kit (Kapa Biosystems KK4824) and pooled in equal concentrations prior to MiSeq v3 sequencing with 15% phiX control spike-in (Illumina FC-110–3001).
Analyses on MiSeq data from CRISPR-edited organoids
Unique sequences detected from the MiSeq sequencing data were counted for each of the 8 samples (Mouse I gG2A−, NeuN−, NeuN+, Nestin−, Nestin+, TRA-1–60− and TRA-1–60+ sorted cells, as well as PGP1 unedited cells). To identify sequences that were specific to the edited cells but were not present in unedited cells, sequences with less than 25 reads in unedited cells and had at least 25 reads in each of the seven edited samples, were identified for further analyses. The number of reads for each mutant sequence was divided by the number of reads for the reference sequence in each sample to obtain ratios. For each sorted pool of cells, the ratios were divided by the mean ratios from the long mutations to obtain normalized ratios. Two-sided Wilcoxon ranked sum test was performed to test the normalized ratios between the NeuN− and NeuN+, Nestin− and Nestin+, TRA-1–60− and TRA-1–60+ samples. ORs were further calculated for each mutant sequence and each antibody marker by dividing the normalized ratios for the positive bins ( NeuN+, Nestin+ or TRA-1–60+) by the normalized ratios for the negative bins (NeuN−, Nestin− or TRA-1–60−) respectively.
| (1) |
| (2) |
| (3) |
| (4) |
CRISPR-Cas9 editing of 19 genes in the 16p11.2 locus in iPSCs
iPSCs were cultured with StemFlex media (Thermo Scientific A3349401) on 6-well plates coated with Matrigel, until they were ~ 75–90% confluent. Accutase was used to dissociate the cells, and 500,000 cells were counted. For each gene, we used a pool of sgRNAs from Synthego comprised of 1–3 sgRNAs for each gene, and TrueCut Cas9 protein v2 (Thermo Scientific A36496). We incubated 3 μl of 100 μM sgRNA with 2 μl of 20 μM Cas9 nuclease at room temperature for 10 min prior to nucleofection. Nucleofection was performed using the P3 L kit and CA-137 protocol on a Lonza 4D nucleofector. After nucleofection, the iPSCs were passaged onto a single well of a 24-well plate coated with Matrigel in 1 ml of StemFlex media with 10 μM Y-27632 and 1:100 SMC4 cocktail for 24 h, and expanded for another 6 days on 6-well plates coated with Matrigel.
Locus-wide DNA extraction, PCR, MiSeq sequencing and analyses
iPSCs for each gene were washed twice with 1 × DPBS and DNA extraction was performed using the standard protocol for the AccuPrep Genomic DNA Extraction Kit (Bioneer K-3032G). PCR was performed using the standard protocol for Q5 Hot Start Master Mix. Primers were designed for each sgRNA within each gene knockout pool (Supplementary Table 3). Barcoding was performed using Nextera indexes, followed by DNA clean-up using the Monarch PCR & DNA Cleanup Kit. Library quantification was performed at the UMass Chan Medical School Deep Sequencing Core Facility and all samples were pooled in equal concentrations with a whole-genome human sample from our laboratory, prior to HiSeq sequencing. In parallel, we performed the same sets of sequencing experiments using DNA from unedited iPSCs to identify potential sequencing artifacts.
Sequences with ≤ 0.2% reads compared to the total number of reads for each sample, as well as sequences that did not match the primers used for the samples, were filtered prior to downstream analyses. All sequences that were found in the edited iPSCs but were also present in unedited iPSCs (read count ≥ 1), were further filtered as these sequences might be potential sequencing artifacts. We can estimate the fraction of cells with low-frequency mutations as 2 × the fraction of mutant reads divided by the total number of reads, given that most low-frequency mutations are more likely to be heterozygous mutations than homozygous mutations.
Supplementary Material
Funding
This study was supported by the National Institutes of Health grants (NHGRI RM1HG008525 to GMC; NIMH R01MH113279 to GMC), and Robert Wood Johnson Foundation Grant (74178 to GMC), and startup funds by UMass Chan Medical School (ETL, YC).
Footnotes
Conflict of interest The authors declare no competing interests.
Supplementary Information The online version contains supplementary material available at https://doi.org/10.1007/s00439-023-02534-4.
Data availability
The sequencing data generated from our work will be deposited into SRA.
References
- Arbogast T, Razaz P, Ellegood J, McKinstry S, Erdin S, Currall B, Aneichyk T, Lerch JP, Qiu LR, Rodriguiz RM et al. (2019) Kctd13-deficient mice display short-term memory impairment and sex-dependent genetic interactions. Hum Mol Genet [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal I, Ragavendran A, Erdin S, Klei L, Sugathan A, Guide JR, Manavalan P, Zhou JQ, Wheeler VC, Levin JZ et al. (2014) Transcriptional consequences of 16p11.2 deletion and duplication in mouse cortex and multiplex autism families. Am J Hum Genet 94:870–883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Zhang Y, Wilde J, Hansen KC, Lai F, Niswander L (2014) Microcephaly disease gene Wdr62 regulates mitotic progression of embryonic neural stem cells and brain size. Nat Commun 5:3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doobin DJ, Kemal S, Dantas TJ, Vallee RB (2016) Severe NDE1-mediated microcephaly results from neural progenitor cell cycle arrests at multiple specific stages. Nat Commun 7:12551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escamilla CO, Filonova I, Walker AK, Xuan ZX, Holehonnur R, Espinosa F, Liu S, Thyme SB, Lopez-Garcia IA, Mendoza DB et al. (2017) Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature 551:227–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golzio C, Willer J, Talkowski ME, Oh EC, Taniguchi Y, Jacquemont S, Reymond A, Sun M, Sawa A, Gusella JF et al. (2012) KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature 485:363–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horev G, Ellegood J, Lerch JP, Son YE, Muthuswamy L, Vogel H, Krieger AM, Buja A, Henkelman RM, Wigler M et al. (2011) Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc Natl Acad Sci USA 108:17076–17081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara E, Kor D, Bulut FD, Herguner O, Ceylaner S, Koseci B, Burgac E, Mungan NO (2021) Glycogen storage disease type XII; an ultra rare cause of hemolytic anemia and rhabdomyolysis: one new case report. J Pediatr Endocrinol Metab 34:1335–1339 [DOI] [PubMed] [Google Scholar]
- Kizner V, Naujock M, Fischer S, Jager S, Reich S, Schlotthauer I, Zuckschwerdt K, Geiger T, Hildebrandt T, Lawless N et al. (2020) CRISPR/Cas9-mediated knockout of the neuropsychiatric risk gene KCTD13 causes developmental deficits in human cortical neurons derived from induced pluripotent stem cells. Mol Neurobiol [DOI] [PubMed] [Google Scholar]
- Kosuri S, Goodman DB, Cambray G, Mutalik VK, Gao Y, Arkin AP, Endy D, Church GM (2013) Composability of regulatory sequences controlling transcription and translation in Escherichia coli. Proc Natl Acad Sci USA 110:14024–14029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, Gilliam TC, Nowak NJ, Cook EH Jr, Dobyns WB et al. (2008) Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet 17:628–638 [DOI] [PubMed] [Google Scholar]
- Lancaster MA, Knoblich JA (2014) Generation of cerebral organoids from human pluripotent stem cells. Nat Protoc 9:2329–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, Knoblich JA (2013) Cerebral organoids model human brain development and microcephaly. Nature 501:373–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ET, Chan Y, Dawes P, Guo X, Erdin S, Tai DJC, Liu S, Reichert JM, Burns MJ, Chan YK et al. (2022) Orgo-Seq integrates single-cell and bulk transcriptomic data to identify cell type specific-driver genes associated with autism spectrum disorder. Nat Commun 13:3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM (2013) RNA-guided human genome engineering via Cas9. Science 339:823–826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasca AM, Sloan SA, Clarke LE, Tian Y, Makinson CD, Huber N, Kim CH, Park JY, O’Rourke NA, Nguyen KD et al. (2015) Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods 12:671–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzo L, Lasser M, Yusuff T, Jensen M, Ingraham P, Huber E, Singh MD, Monahan C, Iyer J, Desai I et al. (2021) Functional assessment of the “two-hit” model for neurodevelopmental defects in Drosophila and X. laevis. PLoS Genet 17:e1009112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian X, Nguyen HN, Song MM, Hadiono C, Ogden SC, Hammack C, Yao B, Hamersky GR, Jacob F, Zhong C et al. (2016) Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165:1238–1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrato G, Nguyen T, Macosko EZ, Sherwood JL, Min Yang S, Berger DR, Maria N, Scholvin J, Goldman M, Kinney JP et al. (2017) Cell diversity and network dynamics in photosensitive human brain organoids. Nature 545:48–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi AY, Mueller S, Snyder AZ, Mukherjee P, Berman JI, Roberts TP, Nagarajan SS, Spiro JE, Chung WK, Sherr EH et al. (2014) Opposing brain differences in 16p11.2 deletion and duplication carriers. J Neurosci 34:11199–11211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raveh-Sadka T, Levo M, Shabi U, Shany B, Keren L, Lotan-Pompan M, Zeevi D, Sharon E, Weinberger A, Segal E (2012) Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat Genet 44:743–750 [DOI] [PubMed] [Google Scholar]
- Roth JG, Muench KL, Asokan A, Mallett VM, Gai H, Verma Y, Weber S, Charlton C, Fowler JL, Loh KM et al. (2020) 16p11.2 microdeletion imparts transcriptional alterations in human iPSC-derived models of early neural development. Elife 9:e58178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SJ, Ercan-Sencicek AG, Hus V, Luo R, Murtha MT, MorenoDe-Luca D, Chu SH, Moreau MP, Gupta AR, Thomson SA et al. (2011) Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron 70:863–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, Yamrom B, Yoon S, Krasnitz A, Kendall J et al. (2007) Strong association of de novo copy number mutations with autism. Science 316:445–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharon E, Kalma Y, Sharp A, Raveh-Sadka T, Levo M, Zeevi D, Keren L, Yakhini Z, Weinberger A, Segal E (2012) Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat Biotechnol 30:521–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinawi M, Liu P, Kang SH, Shen J, Belmont JW, Scott DA, Probst FJ, Craigen WJ, Graham BH, Pursley A et al. (2010) Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet 47:332–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman KJ, Spence SJ, Ramocki MB, Proud MB, Kessler SK, Marco EJ, Green Snyder L, D’Angelo D, Chen Q, Chung WK et al. (2016) 16p11.2 deletion and duplication: characterizing neurologic phenotypes in a large clinically ascertained cohort. Am J Med Genet A 170:2943–2955 [DOI] [PubMed] [Google Scholar]
- Tai DJ, Ragavendran A, Manavalan P, Stortchevoi A, Seabra CM, Erdin S, Collins RL, Blumenthal I, Chen X, Shen Y et al. (2016) Engineering microdeletions and microduplications by targeting segmental duplications with CRISPR. Nat Neurosci 19:517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H, Hammack C, Ogden SC, Wen Z, Qian X, Li Y, Yao B, Shin J, Zhang F, Lee EM et al. (2016) Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 18:587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urresti J, Zhang P, Moran-Losada P, Yu NK, Negraes PD, Trujillo CA, Antaki D, Amar M, Chau K, Pramod AB et al. (2021) Cortical organoids model early brain development disrupted by 16p11.2 copy number variants in autism. Mol Psychiatry 26:7560–7580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vysotskiy M, Zhong X, Miller-Fleming TW, Zhou D, Autism Working Group of the Psychiatric Genomics C, Bipolar Disorder Working Group of the Psychiatric Genomics C, Schizophrenia Working Group of the Psychiatric Genomics C, Cox NJ, Weiss LA (2021) Integration of genetic, transcriptomic, and clinical data provides insight into 16p112 and 22q112 CNV genes. Genome Med 13:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, Saemundsen E, Stefansson H, Ferreira MA, Green T et al. (2008) Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med 358:667–675 [DOI] [PubMed] [Google Scholar]
- Yao DC, Tolan DR, Murray MF, Harris DJ, Darras BT, Geva A, Neufeld EJ (2004) Hemolytic anemia and severe rhabdomyolysis caused by compound heterozygous mutations of the gene for erythrocyte/muscle isozyme of aldolase, ALDOA(Arg303X/Cys-338Tyr). Blood 103:2401–2403 [DOI] [PubMed] [Google Scholar]
- Yeo NC, Chavez A, Lance-Byrne A, Chan Y, Menn D, Milanova D, Kuo CC, Guo X, Sharma S, Tung A et al. (2018) An enhanced CRISPR repressor for targeted mammalian gene regulation. Nat Methods 15:611–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Yang SL, Yang M, Herrlinger S, Shao Q, Collar JL, Fierro E, Shi Y, Liu A, Lu H et al. (2019) Modeling microcephaly with cerebral organoids reveals a WDR62-CEP170-KIF2A pathway promoting cilium disassembly in neural progenitors. Nat Commun 10:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data generated from our work will be deposited into SRA.