

Abstract
Inflammatory caspases are key enzymes in mammalian innate immunity, which control the processing and release of interleukin-1 (IL-1) family cytokines1,2. Despite the biological importance, the structural basis for inflammatory caspase-mediated cytokine processing remained unknown. To date, catalytic cleavage of IL-1 family members, including pro-IL-1β and pro-IL-18, has been attributed primarily to caspase-1 activities within canonical inflammasomes3. In this study, we demonstrate that the LPS receptor caspase-4 from humans and other mammalian species (except rodents) can cleave pro-IL-18 with an efficiency similar to pro-IL-1β and pro-IL-18 cleavage by the prototypical IL-1 converting enzyme caspase-1. This ability of caspase-4 to cleave pro-IL-18, combined with its previously defined ability to cleave and activate the lytic pore-forming protein gasdermin D (GSDMD)4,5, enables human cells to bypass the need for canonical inflammasomes and caspase-1 for IL-18 release. Cryogenic electron microscopy (cryo-EM) structure of the caspase-4/pro-IL-18 complex reveals that pro-lL-18 interacts with caspase-4 through two distinct interfaces: a protease exosite and an interface at the caspase-4 active site involving residues in the pro-domain of pro-IL-18, including the tetrapeptide caspase recognition sequence6. The mechanisms revealed for cytokine substrate capture and cleavage differ from those observed for the caspase substrate GSDMD7,8. These findings provide a structural framework for the discussion of caspase activities in health and disease.
Cytokines of the IL-1 family are central regulators of inflammation and immunity1. Unlike most cytokines, several members of the IL-1 family, including IL-1β and IL-18, are synthesized as inactive cytoplasmic pro-proteins whose N-termini must be cleaved to achieve bioactivity1. Upon release into the extracellular space from living cells or by a lytic form of cell death known as pyroptosis9–12, IL-1β and IL-18 can bind to inflammation-inducing receptors on neighbouring cells. Due to the potent inflammatory activities of the IL-1 family and their importance in immunity, attention has focused on defining regulators of cleavage and release of these cytokines3. Much progress in this area has come from the study of pro-IL-1β, whose principal mechanism of cleavage is via the actions of canonical inflammasomes13. Inflammasomes are cytoplasmic supramolecular organizing centres (SMOCs)14, whose assembly is seeded by proteins such as NLRP313. These SMOCs serve as the subcellular site of caspase-1 activation, which was originally identified as interleukin-1 converting enzyme (ICE)15. Inflammasome-associated caspase-1 cleaves pro-IL-1β and the latent pore forming protein gasdermin D (GSDMD) to mediate release of IL-1β4,10,11,15,16. Related caspases, such as human caspase-4, -5 and murine caspase-11, are not recruited into inflammasomes and cannot cleave IL-1β efficiently17–20. Rather, these latter enzymes mediate IL-1β cleavage indirectly, as they can act upstream of the NLRP3 inflammasome upon binding bacterial lipopolysaccharides (LPS)(known as non-canonical inflammasome activation)21–24. Caspase-4, but not caspase-5, has been implicated in cleavage of the IL-1 family cytokine pro-IL-18, but the cleavage preference for this cytokine is undefined18,25,26. Despite these insights into IL-1 biology, fundamental gaps in our knowledge of this cytokine family remain. In particular, no structural insight into how any caspase interacts with its cytokine substrate exists.
In this study, we report that pro-IL-18 is the preferred IL-1 family substrate of caspase-4, allowing for canonical inflammasome-independent cleavage and release of IL-18 from human myeloid cells. We also report a cryo-EM structure of the caspase-4/pro-IL-18 complex, revealing molecular insights into substrate recognition, cleavage and release by this inflammatory caspase.
Caspase-4 is an IL-18 converting enzyme
We began our study by examining the ability of caspase-4 to cleave multiple IL-1 family pro-cytokines in a quantitative in vitro cleavage assay using purified components. The catalytic domain of caspase-4 was expressed in E. coli, in which autoprocessing occurs to yield active caspases consisting of the large p20 and the small p10 catalytic subunits (Extended Data Fig. 1a). Serially diluted caspase-4 was incubated with a fixed concentration of substrate and kinetic parameters were derived by determining the enzyme concentration at which 50% of the substrate is consumed27. Most of the tested IL-1 family cytokines (pro-IL-1α, pro-IL-33, pro-IL-36α, pro-IL-36β, pro-IL-37) were not cleaved by caspase-4 (Fig. 1a and Extended Data Fig. 1b–i). Of the cytokines that were cleaved, pro-IL-18 was cleaved with 30- to >100-fold higher catalytic efficiency than others (pro-IL-1β and pro-IL-36γ) (Fig. 1a and Extended Data Fig. 1b, c, h). Notably, caspase-4 was as efficient at cleaving pro-IL-18 as the ICE caspase-1, in line with previous reports (Fig. 1b and Extended Data Fig. 1j)18. Contrary to the published literature18, pro-IL-18 was also cleaved efficiently by human caspase-5, an ortholog of caspase-4 (Fig. 1b and Extended Data Fig. 1k). Pro-IL-1β, which is cleaved with high efficiency by caspase-1, was poorly cleaved by caspase-4 or caspase-5 (Fig. 1c and Extended Data Fig. 1l, m). We noted that human caspase-4 and its murine homolog caspase-11 possessed low activity towards murine pro-IL-18, whereas human and murine caspase-1 cleaved murine pro-IL-18 efficiently (Fig. 1d and Extended Data Fig. 1n–q). We extended this analysis to caspase-4 homologs from other mammalian species. Analogous enzymes from dog, lemur, rabbit and sheep cleaved species-matched pro-IL-18 homologs with an efficiency comparable to human caspase-4 cleavage of human pro-IL18 (Fig. 1e and Extended Data Fig. 1r–u). By contrast, the caspase-4 homologs from mice and rats (known as caspase-11) displayed weak ability to cleave pro-IL-18 (Fig. 1e and Extended Data Fig. 1p, v). Murine caspase-11 also cleaved human pro-IL-18 with low efficiency (Extended Data Figure 1w, x). These results suggest that caspase-4 from multiple species, except rodents, represents an IL-18 converting enzyme.
Fig. 1 |. Human caspase-4 can efficiently cleave human pro-IL-18 but not other IL-1 family cytokines.
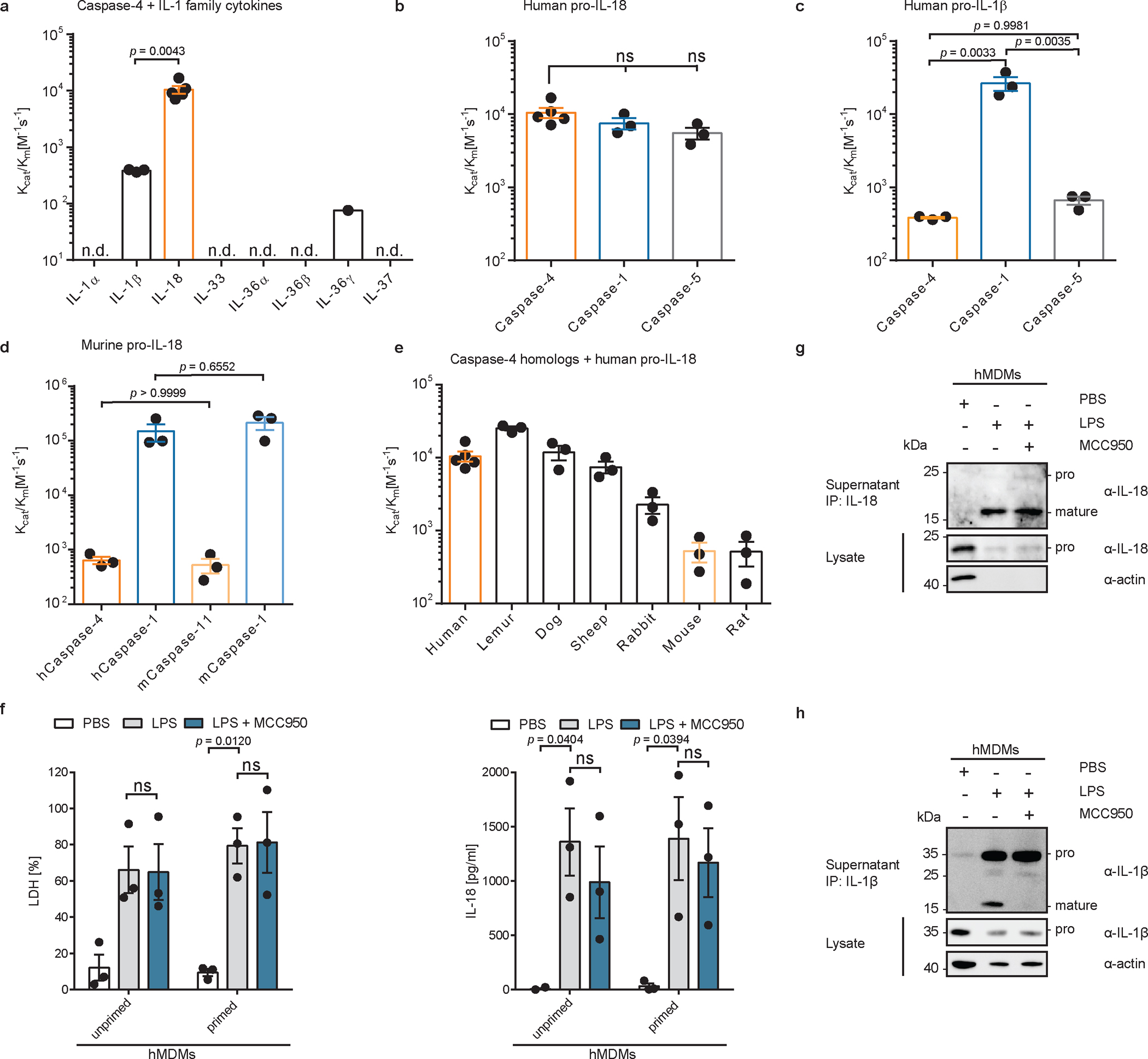
a, In vitro cleavage of human IL-1 family cytokines by human caspase-4. n.d. = cleavage not detected. b, c, d, In vitro cleavage of human or murine pro-IL-18, or human pro-IL-1β by indicated caspases. e, In vitro cleavage of different mammalian pro-IL-18 homologs by species-matched caspase-4 homologs. In a-e, n=3 biological replicates for all substrate/enzyme pairs, except for human caspase-4 + human pro-IL-18, which is n=4. f, g, h, Primary human monocyte-derived macrophages were primed with LPS, or left unprimed, and electroporated with LPS (or PBS) and LDH and IL-18 release into cell culture supernatant was quantified after 2 h. n=3 biological replicates with cells from different donors. IL-18 or IL-1β was immunoprecipitated from cell culture supernatants and analyzed by immunoblot. If indicated, NLRP3 was inhibited with 10 μM MCC950. Immunoblots are representative of three biological replicates. Each data point represents the result of one independent assay. Bars and error bars represent mean ± SEM. Statistical significance was determined by one-way ANOVA (a-d) or two-way ANOVA (f) with Tukey’s multiple comparisons test: ns = not significant (p > 0.05). For gel source data, see Supplementary Figure 1.
Caspase-4 links LPS detection to IL-18 release
The ability of human caspase-4 to bind LPS and cleave pro-IL-18 and GSDMD should, in principle, bypass the need for the NLRP3 inflammasome to mediate IL-18 release from cells. Conversely, the inability of murine caspase-11 to cleave pro-IL-18 should result in a dependence on inflammasome-associated caspase-1 to cleave and release IL-18 from cells. In human epithelial cells (e.g. Hela), which lack NLRP3, these predictions are supported by previous reports, where bacteria induced IL-18 release by a caspase-4-dependent but caspase-1-independent process25,26,28–30. This concept has yet to be tested in human cells competent for NLRP3 signalling, such as myeloid cells. We tested these predictions by delivering LPS into the cytosol of THP1 monocytes, where caspase-4 and pro-IL-18 are constitutively expressed21,31. We also examined THP1 monocytes in which NLRP3 signalling was disrupted by genetic deficiency or through the use of the small-molecule inhibitor MCC95032. Electroporation of wild type (WT) THP1 cells with LPS induced pyroptosis, as indicated by release of the cytosolic enzyme lactate dehydrogenase (LDH) into the extracellular space, while cells electroporated with PBS remained alive (Extended Data Fig. 2a). Pyroptosis was independent of NLRP3, as neither MCC950 nor NLRP3 knockout (KO) prevented LDH release. NLRP3 was not required for LPS-induced release of IL-18 into the cell culture supernatant as shown by ELISA (Extended Data Fig. 2b). Importantly, we confirmed that the IL-18 ELISA preferentially detects cleaved IL-18, as compared to pro-IL-18 (Extended Data Fig. 2c). We also confirmed by immunoblot that cleavage of pro-IL-18 still occurred when NLRP3 function was blocked (Extended Data Fig. 2d, e). By contrast, LPS-induced cleavage of pro-IL-1β was prevented in NLRP3-deficient or MCC950-treated THP1 cells following LPS electroporation (Extended Data Fig. 2f, g). Similar observations were made in primary human monocyte-derived macrophages (hMDMs), where LPS-induced processing and release IL-18, but not IL-1β, was unaffected by NLRP3 inhibition with MCC950 (Fig. 1f–h).
To test the caspase-dependency of IL-18 release from human cells, we generated clonal THP1 cell lines deficient for genes encoding caspase-1 or caspase-4 using CRISPR/Cas9 (Extended Data Figure 2h). Pyroptosis and IL-18 release in response to LPS electroporation was reduced in caspase-4-deficient, but not caspase-1-deficient cells, as compared to cells transfected with a non-targeting (NT) single guide RNA (sgRNA) (Extended Data Fig. 2i). By contrast, cleavage and release of IL-1β was diminished in either caspase-1-deficient or caspase-4-deficient cells (Extended Data Fig. 2j). We were unable to detect the caspase-5 protein in any of these cell lines (Extended Data Figure 2k). However, in line with its ability to efficiently cleave pro-IL-18 in vitro (Fig. 1b), expression of caspase-5 in caspase-4-deficient THP1 cells enabled cytosolic LPS-induced IL-18 release, which was insensitive to MCC950 (Extended Data Fig. 2l, m). Lastly, pyroptosis and IL-18 release from THP-1 macrophages was NLRP3-independent following infection with a flagellin-deficient strain of Salmonella enterica serovar Typhimurium33 (Extended Data Fig. 2n). No IL-18 release was observed when caspase-4-deficient THP1 macrophages were infected with these bacteria (Extended Data Fig. 2n). Collectively, these data demonstrate that caspase-4 and caspase-5 cleave pro-IL-18 in vitro and in cells, which allows these enzymes to intrinsically link LPS detection to IL-18 cleavage and release. Moreover, the requirement for NLRP3 and caspase-1 for IL-1β release under all conditions examined suggests that the weak in vitro cleavage activities of pro-IL-1β by caspase-4 are negligible in the context of cells.
In contrast to our findings in human cells, different observations were made in murine immortalized bone marrow-derived macrophages (iBMDMs). Consistent with previous reports24 and the poor cleavage of pro-IL-18 by caspase-11 in vitro (Fig. 1d), NLRP3 was required for IL-18 and IL-1β cleavage and release following LPS electroporation of iBMDMs (Extended Data Fig. 3a–e). To determine if we could convert human cells into cells that require NLRP3 for IL-18 release, akin to murine cells, we expressed caspase-11 in caspase-4-deficient THP1 cells (Extended Data Fig. 3f). We found that THP1 macrophages expressing caspase-11 underwent pyroptosis and released IL-18 in response to LPS electroporation but IL-18 release from caspase-11-expressing THP1 cells was reduced when NLRP3 activation was blocked by MCC950 (Extended Data Fig. 3g). These results reveal the ability of human caspase-4, but not murine caspase-11, to serve as a sensor and effector that mediates cleavage and release of IL-18, independent of canonical inflammasomes. This one-protein signalling pathway—whereby caspase-4 links LPS detection to pro-IL-18 cleavage and release via GSDMD—is akin to that we have described in several species of carnivora17. These animals, including felines, bears and seals, encode a caspase known as caspase-1/4, which intrinsically links LPS detection to IL-1β cleavage and release from cells17.
Structure of caspase-4/pro-IL-18 complex
To define the interaction between caspase-4 and pro-IL-18 at a molecular level, we determined the cryo-EM structure of caspase-4 in complex with pro-IL-18. We assembled a complex consisting of recombinant human caspase-4 (large p20 subunit carrying an inactivating C258A mutation + small p10 subunit) and human pro-IL-18 (Fig. 2a, Extended Data Fig. 4). Before making EM grids, the complex was stabilized with the chemical crosslinker BS3 and repurified by size-exclusion chromatography. Size and composition of the complex was validated by SDS-PAGE and blue native PAGE (Extended Data Fig. 4). Cryo-EM data were collected in 5,934 raw movies, and 2D classification revealed the existence of different orientations of the complex and molecular details (Fig. 2b). After multiple rounds of 2D and 3D classifications followed by refinement with imposed twofold symmetry of a best class consisting of 229,147 particles, we obtained a cryo-EM map of the caspase-4/pro-IL-18 complex at a resolution of 3.2 Å (Extended Data Fig. 5 and Extended Data Table 1). Overall, regions of the density comprising caspase-4 and the part of pro-IL-18 proximal to caspase-4 are best-defined, whereas the distal ends of pro-IL-18 are less ordered (Fig. 2c). We applied focused refinement on IL-18, which improved the overall resolution in this region of the map to 3.1 Å resolution (Fig. 2d), but the resulting map had similar quality at the caspase-4/pro-IL-18 interface as the complete map. The lower resolution in the distal part of the map is likely due to the dynamic nature of the complex shown by 3D variability analysis (Fig. 2c, d, Extended Data Fig. 5b, Supplementary Video 1).
Fig. 2 |. Overview of the caspase-4/pro-IL-18 complex.
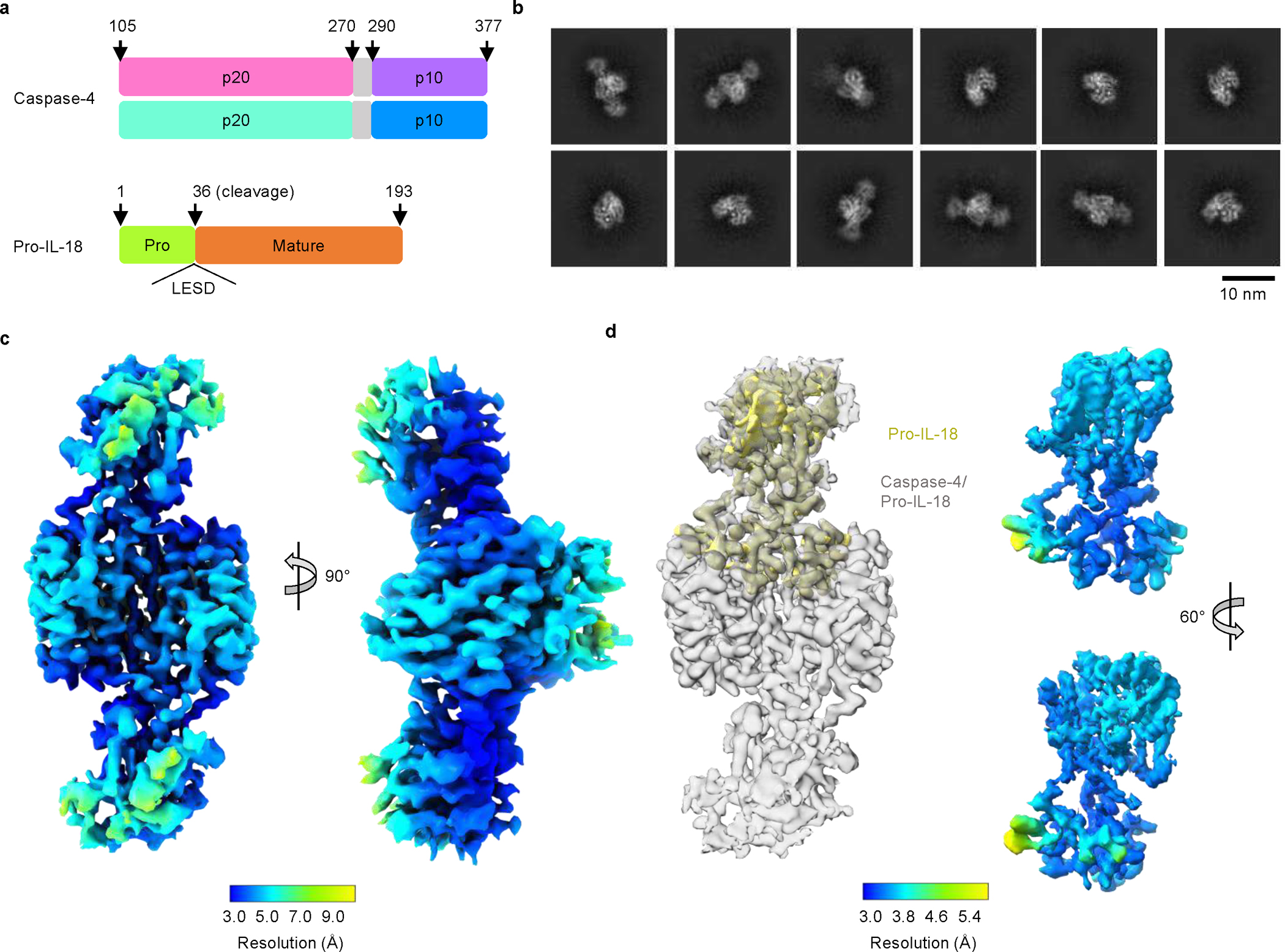
a, Domain organization of caspase-4 and pro-IL-18. Caspase-4 large subunits (p20) are in cyan or pink; small subunits (p10) are in purple or blue; linker regions between p20 and p10 are in grey, which is absent in the model; the pro-domain and mature domain of pro-IL-18 are in green and orange, respectively. b, Representative 2D classes of the caspase-4/pro-IL-18 complex. c, The complete map of the caspase-4/pro-IL-18 complex at 3.2 Å overall resolution (contour level: 3.0 σ), coloured by local resolution. d, Focus-refined map of pro-IL-18 (yellow) at 3.1 Å overall resolution (contour level: 3.4 σ), superimposed with the complete map (grey) (left) or coloured by local resolution in two views (right).
The cryo-EM structure of the caspase-4/pro-IL-18 complex has a symmetric candy-shaped architecture, in which two pro-IL-18 molecules are bound to the caspase-4 dimer-of-heterodimers (heterotetramer) consisting of two copies of p20/p10 (Fig. 3a). The density fitting was initiated with the crystal structure of caspase-47 and mature IL-1834. While the caspase-4 structure matched the density well, the IL-18 structure exhibited substantial differences from the density of pro-IL-18. We traced these differences to construct an atomic model of pro-IL-18. The final model includes the caspase-4 heterotetramer and pro-IL-18 with the near complete pro-domain (residues 6–36). Two regions in pro-IL-18 (residues 1–5, 53–80) are absent, likely due to disorder. The model was independently validated by crosslinking mass spectrometry, with all intermolecular crosslinks consistent with the structure (Extended Data Fig. 6a, b).
Fig. 3 |. Cryo-EM map and model of the caspase-4/pro-IL-18 complex.
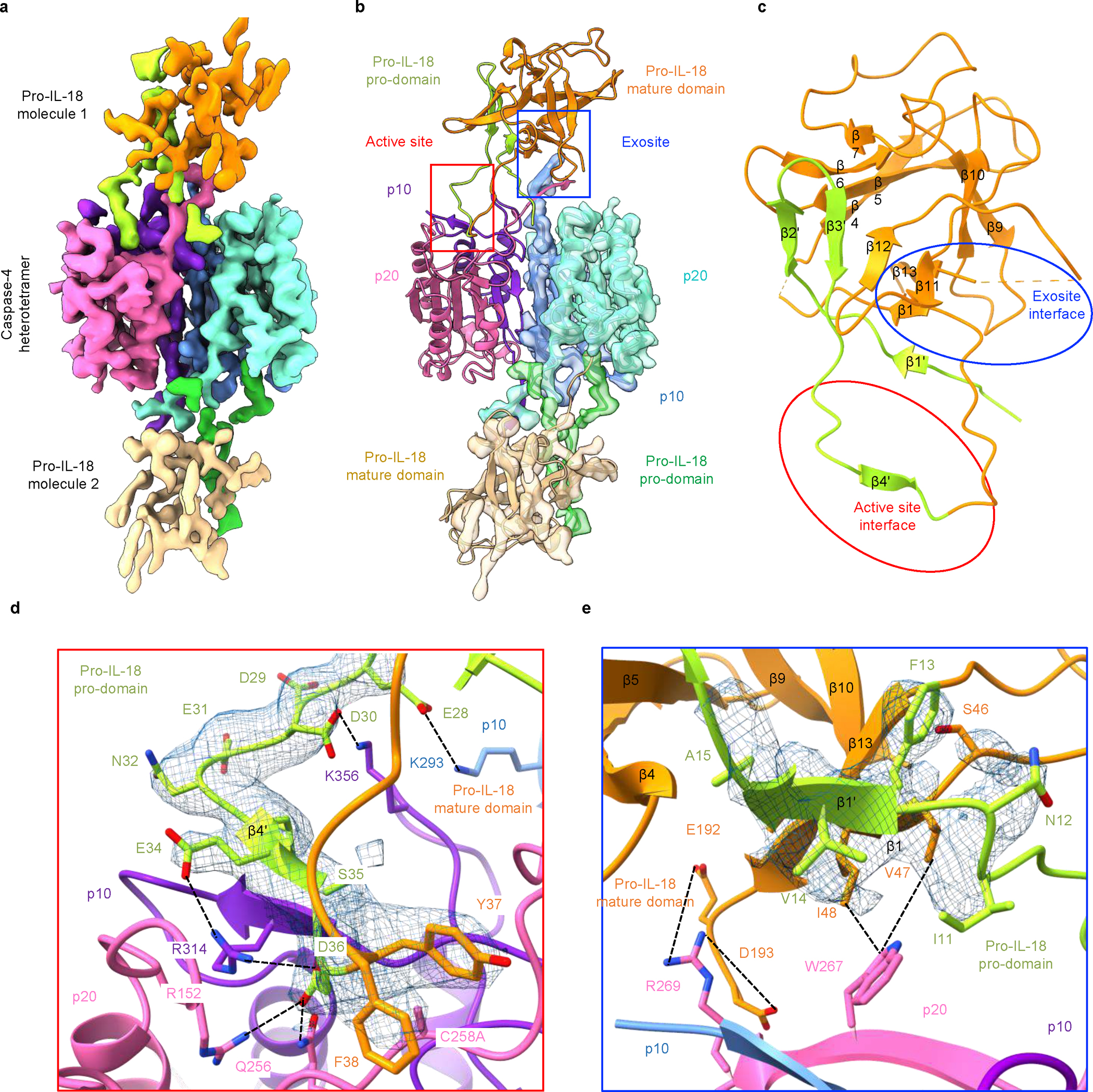
a, Cryo-EM map of the caspase-4/pro-IL-18 complex coloured by domains (contour level: 3.0 σ). b, The caspase-4/pro-IL-18 complex model shown in the colour code as in (a), alone for one half, and superimposed with the cryo-EM map for the other half. Active site and exosite are enclosed in squares of red and blue, respectively. c, Pro-IL-18 structure with secondary structures labelled and the two interfaces enclosed in red and blue ovals. d, Detailed interaction interface at the active site. e, Detailed interaction interface at the exosite. The cryo-EM density for the relevant pro-IL-18 region is shown in blue mesh in (d) and (e).
The cryo-EM structure of caspase-4/pro-IL-18 revealed two binding interfaces between caspase-4 and pro-IL-18, defined as the active site interface and the exosite interface (Fig. 3b, c). Pro-IL-18 buries extensive surface areas of approximately 800 Å2 and 650 Å2 at the active site and exosite, respectively35. Caspase substrates are often defined by the so-called tetrapeptide sequences, meaning the four amino acid residues directly upstream of the scissile peptide bond (33-LESD-36 in pro-IL-18)6. In our structure, ordered density exists for the region preceding and after D36 in pro-IL-18 (β4’ and the adjacent linkers), among which residues E28-F38 interact with caspase-4 to form the active site interface (Fig. 3d). The active site interface is dominated by electrostatic interactions, which position this tetrapeptide in the substrate-binding cleft of caspase-4. We observed paired charge-charge interactions between E28 and K293, D30 and K356, and E34 and R314 in pro-IL-18 and caspase-4, respectively. The cleavage site residue D36 interacts with R152, Q256 and R314 of caspase-4. The alanine mutation on the catalytic C258 residue of caspase-4 allows the binding of the pro-IL-18 substrate at the active site without being cleaved. However, in an active caspase-4, the scissile peptide bond at the C-terminus of D36 and the catalytic centre of caspase-4 (C258) would be in proximity, enabling cleavage (Extended Data Fig. 6c).
In contrast to the electrostatic active site interface, the exosite interface is defined by hydrophobic and electrostatic interactions (Fig. 3e). W267 of caspase-4 is inserted into a hydrophobic pocket formed by V47 and I48 of pro-IL-18 (Fig. 3e). The interaction is enhanced by the interactions of E192 and D193 of pro-IL-18 with R269 in the p10 subunit of caspase-4 (Fig. 3e). Similar interactions have been observed at the exosite of inflammatory caspase/GSDMD complexes, suggesting a common mechanism for inflammatory caspases to capture different substrates to enhance their cleavage activity7,8. Of note, due to limited resolution of the cryo-EM density in this region we suspect that additional conformations may exist in which R49 of pro-IL-18 interacts with W267 via cation-π-interactions.
The conformational difference between pro-IL-18 in complex with caspase-4 and mature IL-18 can be visualized as the insertion of the pro-domain as β-strands into the β-trefoil fold in the mature domain (Extended Data Fig. 6d, e). The N-terminal β2 and β3 strands of the mature domain partly unfold and change conformation in the bound conformation of pro-IL-18, to form the hydrophobic pocket at the exosite interface. These observations indicate that newly cleaved IL-18 undergoes conformational changes to arrive at the structure of the mature IL-18. Crosslinking mass spectrometry of pro-IL-18 alone and in the caspase-4/pro-IL-18 complex suggest conformational differences in pro-IL-18 before and after caspase-4 interaction (Extended Data Fig. 6f, g). AlphaFold36 predicted a structure of pro-IL-18, in which the pro-domain is localized at the opposite end of where the pro-domain is localized in our caspase-bound structure. Nevertheless, the predicted pro-domain contains certain β-strand elements consistent with the caspase-4/pro-IL-18 complex (Extended Data Fig. 6h). Thus, it appears that pro-IL-18 is conformationally adaptable and may have at least three conformations: apo state, upon caspase binding, and post cleavage.
Active site interactions define cleavage
Upon engagement of a hydrophobic exosite, inflammatory caspases cleave GSDMD in a tetrapeptide-independent manner7. This is not the case for pro-IL-18, as mutating the tetrapeptide sequence in pro-IL-18 from LESD to AAAD decreased cleavage by caspase-4 (Fig. 4a and Extended Data Fig. 7a). Thus, the tetrapeptide of pro-IL-18 promotes cleavage by caspase-4. However, the tetrapeptide is not sufficient for efficient cleavage by caspase-4, as we found that murine pro-IL-18, which shares the same tetrapeptide sequence with human pro-IL-18, is poorly cleaved (Fig. 1d and Extended Data Fig. 7b). Consistent with this idea, a chimeric pro-IL-18 variant consisting of the pro-domain of murine pro-IL-18 and mature domain of human IL-18 was cleaved less efficiently by caspase-4 than WT human pro-IL-18 and failed to form a stable complex with caspase-4 (Fig. 4a, b and Extended Data Fig. 7c). These results indicate that the higher catalytic efficiency of caspase-4 for human versus murine pro-IL-18 can at least partially be explained by amino acid residues in the pro-domain of human pro-IL-18. To test this idea, we determined the impact of amino acid swaps between human and murine pro-IL-18 on cleavage by caspase-4. Our cryo-EM structure suggests that K356 in caspase-4 forms electrostatic interactions with D30 in pro-IL-18. We found that swapping a stretch of amino acids surrounding this site in murine pro-IL-18 to the respective human-specific residues (ENGD28DDEN) led to an increase in the catalytic efficiency of caspase-4 (Fig. 4c). By combining this mutation with additional human-specific mutations (N36Y and H41E), we generated a ‘humanized’ murine pro-IL-18 triple mutant, which is cleaved by caspase-4 almost as efficiently as human pro-IL-18 in vitro (Fig. 4c and Extended Data Fig. 7d–h). We detected higher amounts of IL-18 in the supernatants of IL-18-deficient THP1 cells expressing humanized murine pro-IL-18, as compared to those expressing WT murine pro-IL-18 following LPS electroporation in the presence of MCC950 (Fig. 4d, e). These data indicate that residues in the pro-domain, including but not limited to the tetrapeptide, promote binding and cleavage of pro-IL-18 by caspase-4.
Fig. 4 |. Electrostatic interactions between pro-IL-18 pro-domain and caspase-4 promote cytokine processing.
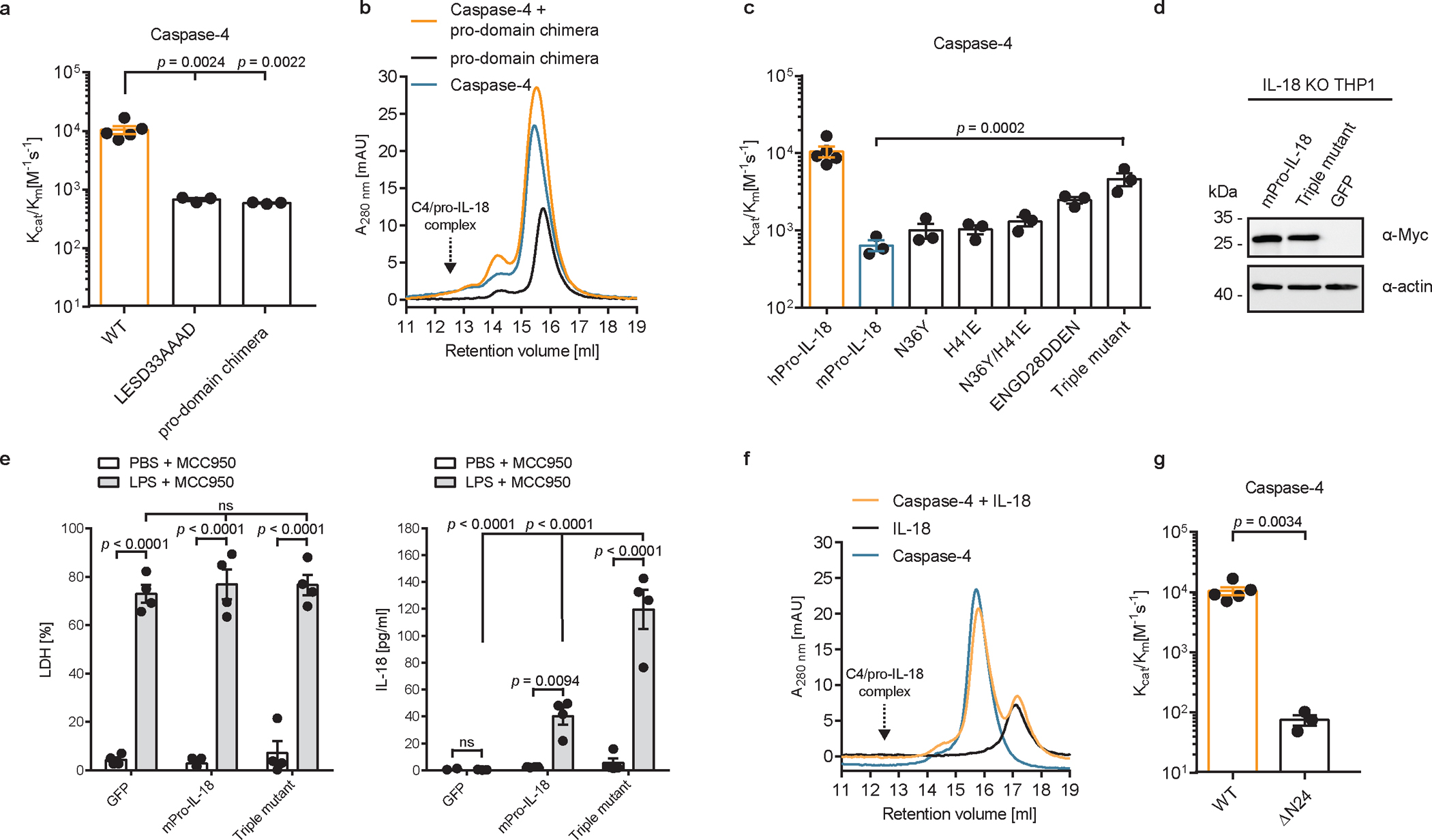
a, In vitro cleavage of human pro-IL-18 mutants by human caspase-4. Pro-domain chimera consists of murine pro-domain fused to human mature domain. b, Analytical SEC showing no interaction between caspase-4 and pro-domain chimera. Retention volume of caspase-4/pro-IL-18 complex is marked. c, In vitro cleavage of murine pro-IL-18 variants by human caspase-4. Triple mutant combines all human-specific mutations (N36Y, H41E, ENGD28DDEN). d, Expression of Myc-tagged murine pro-IL-18 variants in IL-18-deficient THP1 cells. e, IL-18-deficient THP1 macrophages expressing murine pro-IL-18 variants were primed with LPS and electroporated with LPS (or PBS) in the presence of MCC950 and LDH and IL-18 release into cell culture supernatant was quantified after 2 h. f, Analytical SEC showing no interaction between mature IL-18 and caspase-4. g, In vitro cleavage of human pro-IL-18 ΔN24, which lacks the first 24 amino acids, by human caspase-4. SEC profiles and immunoblots are representative of three biological replicates. In a, c, and g, n=3 biological replicates for all substrate/enzyme pairs, except for WT caspase-4 + WT pro-IL-18, which is n=4. a, c and g. In e, n=3 biological replicates. Each data point represents the result of one independent assay. Bars and error bars represent mean ± SEM. Statistical significance was determined by unpaired, two-sided student’s t-test (g), one-way ANOVA (a, c) or two-way ANOVA (e) with Tukey’s multiple comparisons test: ns = not significant. For gel source data, see Supplementary Figure 1.
We found that caspase-4 is unable to bind to mature IL-18, which lacks the pro-domain, as assessed by analytical SEC or isothermal titration calorimetry (ITC) (Fig. 4f and Extended Data Fig. 7i). Similarly, a truncated version of pro-IL-18, which lacks the first 24 amino acid residues of the pro-domain (named ΔN24) does not bind caspase-4 and is cleaved less efficiently by caspase-4 (Fig. 4g and Extended Data Fig. 7j, k). Removal of the pro-domain of pro-IL-18 upon cleavage thus may facilitate release of the bioactive cytokine from the enzyme.
Exosite mutations abolish IL-18 cleavage
Introduction of charge-reversing mutations in either of the interaction surfaces (K356D or R269D) or mutation of the hydrophobic residue at the core of the exosite to a polar residue (W267N) disrupted caspase-4 binding to pro-IL-18 in vitro (Fig. 5a and Extended Data Fig. 8a, b). As a result, in vitro cleavage of pro-IL-18 by caspase-4 variants carrying these mutations was reduced, with the W267N mutation having the greatest effect (Fig. 5b and Extended Data Fig. 8c–e). These mutations in caspase-4 did not interfere with intrinsic enzymatic activities, as the caspase-4 mutants retained the ability to cleave the chromogenic peptide substrate Ac-WEHD-pNA (Fig. 5c). Caspase-4 W267N showed a slight reduction in Ac-WEHD-pNA processing (as described before7), but this effect is unlikely to explain the more than 100-fold reduction in its ability to cleave pro-IL-18 (Fig. 5b). Reciprocal mutations in pro-IL-18 yielded symmetrical results to the mutagenesis of caspase-4. Perturbing the hydrophobic pocket or charge-charge interactions at the exosite interface by the introduction of V47N/I48N or E192K/D193K mutations decreased binding of pro-IL-18 to caspase-4 (Fig. 5d and Extended Data 8f, g). Consequently, in vitro cleavage of these pro-IL-18 mutants by caspase-4 was decreased (Fig. 5e and Extended Data Fig. 8h–k).
Fig. 5 |. A hydrophobic exosite is required for recognition and cleavage of pro-IL-18 by caspase-4.
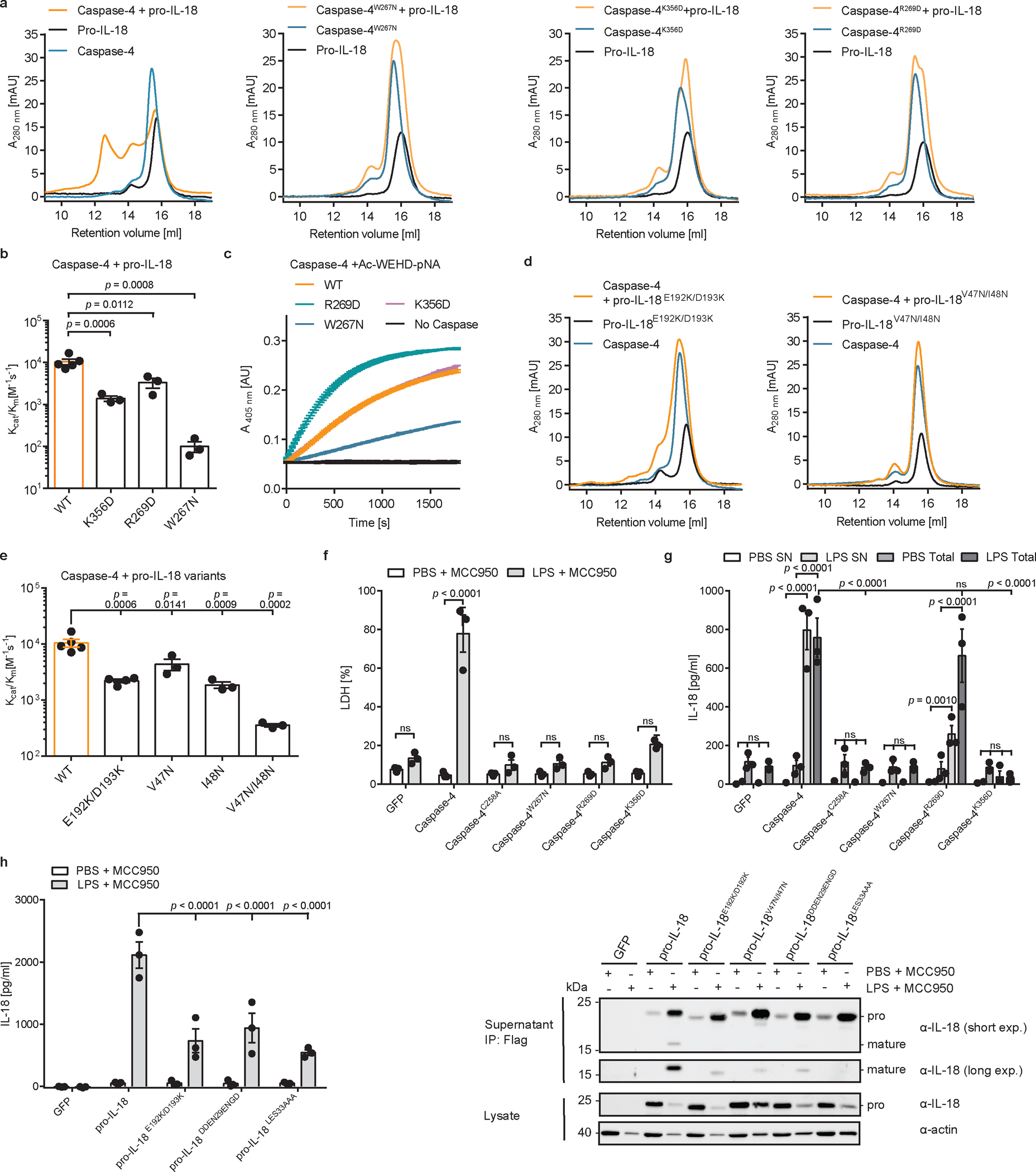
a, Analytical SEC investigating complex formation between pro-IL-18 (produced in insect cells) and indicated caspase-4 p20/p10 mutants. b, In vitro cleavage of human pro-IL-18 by caspase-4 mutants. c, Kinetic analysis of cleavage of chromogenic peptide substrate Ac-WEHD-pNA by caspase-4 mutants. Curves show mean ± SD of two technical replicates and are representative of three biological replicates. d, Analytical SEC investigating complex formation between caspase-4 p20/p10 and indicated pro-IL-18 mutants. Pro-IL-18 mutants were produced in E. coli. e, In vitro cleavage of human pro-IL-18 mutants by human caspase-4. f, g, Caspase-4-deficient THP1 macrophages expressing caspase-4 variants were primed with LPS and electroporated with LPS (or PBS) in the presence of MCC950 and LDH release was quantified after 2 h. IL-18 levels in the supernatants or total IL-18 (combined supernatants + cell lysates) were quantified by ELISA. h, I, IL-18-deficient THP1 macrophages expressing pro-IL-18 variants were primed with LPS and electroporated with LPS (or PBS) in the presence of MCC950 and IL-18 levels in supernatants were quantified after 2 h. IL-18 was immunoprecipitated from supernatants using Flag-tag and analyzed by immunoblot. Immunoblots and SEC profiles are representative of three biological replicates. In b, and e, n=3 biological replicates for all substrate/enzyme pairs, except for WT caspase-4 + WT pro-IL-18 and pro-IL-18E192K/D193K, which are n=4. In f-h, n=3 biological replicates. Each data point represents the result of one independent assay. Bars and error bars represent mean ± SEM. Statistical significance was determined by one-way ANOVA (b, e) or two-way ANOVA (f, g, h) with Tukey’s multiple comparisons test: ns = not significant (p > 0.05). For gel source data, see Supplementary Figure 1.
To correlate these in vitro results with activities in cells, we reconstituted caspase-4-deficient THP1 cells with WT caspase-4 or caspase-4 mutants deficient in pro-IL-18 interaction (C258A, K356D, R269D, W267N) (Extended Data Fig. 9a). These cells were electroporated with LPS in the presence of MCC950 to assess caspase-4-dependent pyroptosis and IL-18 release. Cells expressing WT caspase-4 underwent pyroptosis and released IL-18 in response to LPS electroporation, while cells expressing the catalytically inactive caspase-4 C258A did not (Fig. 5f, g). Cytosolic LPS-induced LDH and IL-18 release from cells expressing caspase-4 carrying W267N, R269D or K356D mutations was reduced compared to cells expressing WT caspase-4 (Fig. 5f, g). This finding can be explained by the role of the same interfaces for GSDMD cleavage7,8, which we confirmed experimentally in vitro and in cells (Extended Data Figure 9b–g). To assess pro-IL-18 cleavage independently of GSDMD activities, we took advantage of the fact that our IL-18 ELISA is specific for cleaved IL-18 (Extended Data Fig. 3c). We assessed IL-18 abundance in combined cell lysates and supernatants by in-well lysis of cells, followed by ELISA and immunoblot. These assays revealed that caspase-4 W267N or K356D mutations were as defective as the catalytic site mutant C258A in IL-18 generation (Fig. 5g and Extended Data Fig. 9h). Caspase-4 R269D displayed the least severe defect in IL-18 generation after LPS electroporation (Fig. 5g and Extended Data Figure 9h). We also reconstituted IL-18-deficient THP1 cells with WT pro-IL-18 or pro-IL-18 variants carrying mutations in the exosite or active site interfaces (E192K/D193K, V47N/I48N, DDEN29ENGD, LESD33AAAD) (Extended Data Fig. 9i). Upon LPS electroporation in the presence of MCC950, cells expressing all pro-IL-18 variants underwent pyroptosis to similar extent (Extended Data Fig. 9j). In contrast, we detected reduced levels of IL-18 in the supernatants of cells expressing pro-IL-18 E192K/D193K, pro-IL-18 DDEN29ENGD or pro-IL-18 LESD33AAAD compared to cells expressing WT pro-IL18 (Fig. 5h). We excluded cells expressing pro-IL-18 V47N/I48N from this analysis, as this mutant displayed severely reduced reactivity with the ELISA reagent (Extended Data Fig. 9k). However, we confirmed reduced levels of LPS-induced cleavage of all tested mutant proteins (including pro-IL-18 V47N/I48N) compared to WT pro-IL-18 by immunoblot (Fig. 5i). These collective data indicate that hydrophobic and charge-charge interactions determine the ability of caspase-4 to operate as an IL-18 converting enzyme.
Structure-based engineering of caspase-11
Lastly, we determined if our structural insights may explain the differential pro-IL-18 cleavage abilities of human caspase-4 and murine caspase-11. We fitted a crystal structure of caspase-117 into our caspase-4/pro-IL-18 map to generate a model of a hypothetical caspase-11/pro-IL-18 complex (Fig. 6a). In this complex, critical exosite residues W263 and R265 (corresponding to W267 and R269 in caspase-4) fit into the hydrophobic pocket in pro-IL-18, similar to what we observed in the caspase-4/pro-IL-18 complex (Fig. 6b). However, several critical interactions in the active site interface are absent in the caspase-11/pro-IL-18 complex (Fig. 6b). Specifically, we found that the regions surrounding K356 and K293 in caspase-4, which interact with E28 and D30 in pro-IL-18, are not conserved in caspase-11 (Extended data Fig. 10a). Instead, caspase-11 displays non-charged residues (H352, L289) at these sites. Introducing caspase-4-specific mutations at these sites in caspase-11 increased its ability to cleave pro-IL-18 in vitro to a level similar to caspase-4 (Fig. 6c and Extended Data Fig. 10b–d). We expressed this caspase-11 double mutant in caspase-4-deficient THP1 cells (Fig. 6d). When stimulated with cytosolic LPS in the presence of MCC950, we detected more mature IL-18 in the supernatants of cells expressing the caspase-11 double mutant compared to cells expressing WT caspase-11 (Fig. 6e). These data demonstrate that the differential efficiencies of pro-IL-18 cleavage between caspase-11 and caspase-4 can be explained by residues in the active site interface.
Fig. 6 |. Engineering of caspase-11 into an efficient IL-18 converting enzyme.
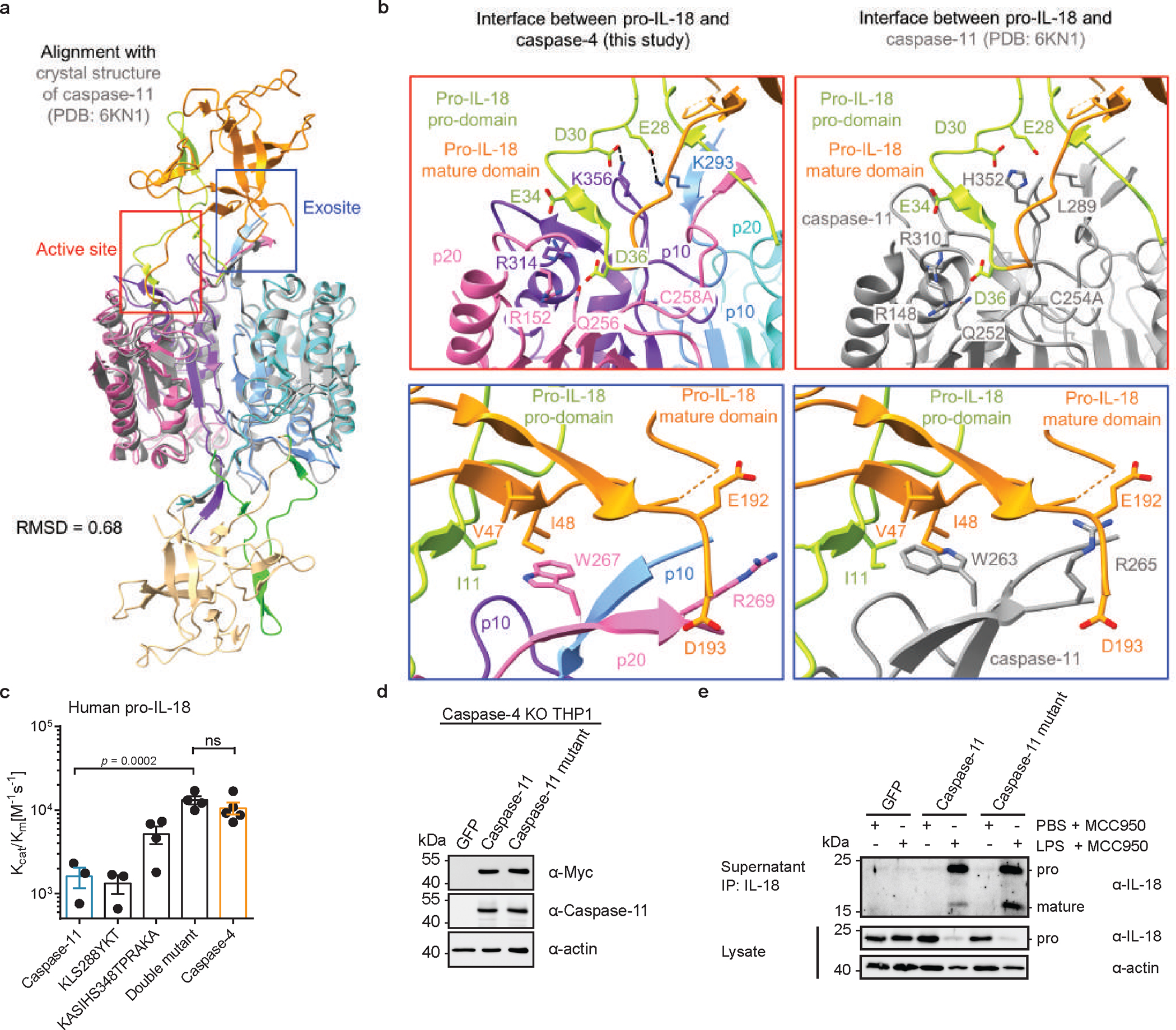
a, Structural model of a caspase-11/pro-IL-18 complex aligned with experimentally determined caspase-4/pro-IL-18 cryo-EM structure. b, Active site and exosite interfaces in caspase-11/pro-IL-18 model compared to caspase-4/pro-IL-18 structure. c, In vitro cleavage of human pro-IL-18 by caspase-11 mutants (n=4 biological replicates for caspase-4 and KASIHS348, n=3 for all other proteins). d, Expression of caspase-11 variants in caspase-4-deficient THP1 cells. e, Caspase-4-deficient THP1 macrophages expressing caspase-11 variants were primed with LPS and electroporated with LPS (or PBS) in the presence of MCC950. After 2h, IL-18 was immunoprecipitated from cell culture supernatants and analyzed by immunoblot. Immunoblots are representative of two (d) or three (e) biological replicates. Each data point represents the result of one independent assay. Bars and error bars represent mean ± SEM of at least three biological replicates. Statistical significance was determined by one-way ANOVA with Tukey’s multiple comparisons test: ns = not significant (p > 0.05). For gel source data, see Supplementary Figure 1.
Discussion
Based on the data presented, we propose a model for cytokine substrate capture, cleavage and release by caspase-4. Pro-IL-18 uses its pro-domain to interact with both neighbouring p10 subunits within the caspase-4 heterotetramer, which may stimulate dimerization of caspase-4 to promote its catalytic activity. We propose that pro-IL-18 binding and cleavage by caspase-4 relies on a unique bivalent mode of recognition. In addition to binding of a hydrophobic pocket in pro-IL-18 via an exosite, caspase-4 interacts with charged residues in and near the tetrapeptide in the pro-domain of pro-IL-18. This mode of interaction is different from how caspases recognize GSDMD and pro-IL-1β. Conversely, pro-IL-1β cleavage by caspase-1 appears to be largely independent of exosite residues in the caspase17. We observe different conformations of caspase-4-bound pro-IL-18 versus free cleaved IL-18, and caspase-4 is unable to bind mature IL-18, suggesting that cleavage may trigger a conformational change in IL-18 and its release from the caspase. Our studies therefore reveal molecular insights into the catalytic mechanism of inflammatory caspases and may provide a framework for the design of inhibitors of caspase-4 and other inflammatory caspases as tools for research and drug candidates.
Our study also highlights a disconnect in the regulation of IL-18 between human and mice. Caspase-4 homologs in rodents are unable to cleave pro-IL-18 efficiently, despite sharing almost 70% sequence identity with human caspase-4. Using our caspase-4/pro-IL18 structure as a blueprint, we mapped the regions responsible for these differential cleavage specificities to regions near the active site, which allowed us to engineer a caspase-11 variant with increased IL-18 converting enzyme activity. Thus, while caspase-4 proteins from other mammalian species can intrinsically link LPS detection to IL-1 family cytokine cleavage and release, murine cells require inflammasomes to operate downstream from their caspase-4 homologs to execute this process. The implications of these biochemical and pathway differences for immunity on an organismal level remain to be uncovered. The organism-specific pathway differences uncovered in this study provide a mandate to consider the impact of species-specific innate immunity when designing pre-clinical animal models to test therapeutics that involve IL-18.
Methods
No statistical methods were used to predetermine sample size. Experiments were not randomized, and investigators were not blinded during experiments and during outcome assessment. Statistical analyses were performed using GraphPad Prism (v6.01). Statistical tests are indicated in the figure legends.
Ligand and chemical reconstitution.
E. coli LPS (serotype O:111 B4) was purchased from Enzo Biosciences as a ready-to-use stock solution of 1 mg/ml or purchased from Invivogen and reconstituted in ultrapure water at a stock concentration of 1 mg/ml and used at a working concentration of 1 μg/ml. Pam3CSK4 was purchased from Invivogen and reconstituted in ultrapure water at a stock concentration of 1 mg/ml and used at a working concentration of 1 μg/ml. MCC950 (from Invivogen) were resuspended in sterile DMSO to a concentration of 20 mM and used at a final concentration of 10 μM. Recombinant human M-CSF (CHO expressed, carrier-free) was bought from R&D Systems and dissolved in sterile PBS pH 7.4 (stock concentration 100 μg/ml). Phorbol 12-myristate 13-acetate (PMA; from SigmaAldrich) was reconstituted at 50 μg/ml in DMSO. Ac-WEHD-pNA was from Enzo Biosciences and the stock solution was prepared at 20 mM in DMSO. BS3 was purchased from ThermoFisher in a No-Weigh format (2 mg per aliquot) and prepared freshly before each use in ultrapure water at 25 mM and used at 1 mM.
Antibodies for immunoblotting.
Primary antibodies for immunoblotting were purchased from the following vendors: monoclonal rabbit anti-human caspase-1, polyclonal rabbit anti-human caspase-4, monoclonal rabbit anti-human caspase-5, monoclonal rabbit anti-Myc tag, monoclonal mouse anti-Myc-tag, monoclonal rabbit anti-murine IL-18 (all from Cell Signaling), monoclonal mouse-anti caspase-4 (Enzo Biosciences), monoclonal mouse anti-mouse and human NLRP3 (Adipogen), polyclonal rabbit anti-human IL-18 (MBL), monoclonal rabbit anti-human GSDMD, monoclonal rabbit anti-cleaved GSDMD-NT, monoclonal rabbit-anti murine IL-18 (all from Abcam), monoclonal rat anti-actin, monoclonal rat-anti caspase-11 (both from Biolegend), monoclonal mouse anti-canine IL-18 (R&D Systems), polyclonal rabbit anti-human IL-1β, polyclonal rabbit anti-murine IL-1β (both from Genetex), monoclonal mouse-anti actin (SigmaAldrich). All primary antibodies were used at a dilution of 1:1000 in PBS pH7.4 + 0.02% Tween and 5% BSA, except for mouse anti-actin, which was used at 1:5000 in the same buffer.
Constructs.
Constructs encoding N-terminally Myc-tagged full-length human caspase-1, caspase-4 and murine caspase-11 in pMSCV-IRES-EGFP were described previously17 and are available from Addgene (#183359, 183358, 183356). Sequence encoding full-length human caspase-5 with an N-terminal Myc-tag were ordered Integrated DNA technologies and cloned into pMSCV-IRES-EGFP vector using NotI and SalI restriction sites. Expression plasmid for caspase-1 p20 and p10 were kindly provided by Sam Xiao (Case Western Reserve University). Constructs for bacterial expression of the catalytic domains of human caspase-4 and murine caspase-11, human and murine pro-IL-18 were described before17 and deposited on Addgene (Addgene #183381, 183382, 183389, 183390). For expression in insect cells, sequence for human pro-IL-18 was cloned into pFastBac HTb using BamHI and XhoI restriction sites. For retroviral transduction, N-terminal Myc-tag and C-terminal Flag-tag were added to human pro-IL-18 by PCR and sequence was cloned into pMSCV-IRES-EGFP vector using NotI and SalI restriction sites. Sequences encoding catalytic domains of human caspase-5 (AA133–434) as well as catalytic domains of caspase-4 homologs from lemur M. murinus, sheep O. aries, rabbit O. cuniculus, dog C. lupus familiaris were subcloned by PCR from full-length sequences and cloned into pET28a with an N-terminal His6-tag and TEV cleavage site using BamHI and EcoRI cleavage sites. Full-length sequences for these caspase-4 homologs are available on Addgene (Addgene # 183365, 183374, 183375, 183376). cDNAs encoding the catalytic domain of rat caspase-4, pro-IL-18 homologs from all mammalian species listed above as well as human pro-IL-1β, pro-IL-1α, pro-IL-33, pro-IL-36α, pro-IL-36β, pro-IL-36γ and pro-IL-37 were synthesized by Integrated DNA Technologies and cloned into pET28a with an N-terminal His-tag and TEV cleavage site. C-terminal Myc-tags were added to cytokine sequences by PCR. All point mutations and deletions were introduced using the Q5 site-directed mutagenesis kit (NEB) according to manufacturer’s instructions. Sequence encoding p20 subunit of caspase-4 (with C258A mutation) or p10 subunit of caspase-4 were subcloned from full-length sequence by PCR and cloned into a pET28a vector (+ N-terminal His-tag and TEV cleavage site) or a modified pET16b vector (without tags), respectively. The modified pET16b vector was a kind gift by Philip Kranzusch (Harvard Medical School). All constructs generated in this paper were sequence confirmed by Sanger sequencing.
Cell lines.
All cells were cultured in humidified incubators at 37 °C and 5% CO2. Immortalized bone marrow-derived macrophages (iBMDMs) were generated in the Jonathan Kagan Laboratory from the bone-marrow of female WT C57BL/6J or NLRP3 KO mice (B6.129S6-Nlrp3tm1Bhk/J)37. iBMDMs and HEK293T cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS), Penicillin + Streptomycin, L-Glutamine and Sodium pyruvate, herafter referred to as complete DMEM (cDMEM) and cultured in tissue-culture treated 10 cm dishes or T175 tissue culture flasks (Corning). iBMDMs and HEK293T cells were passaged using PBS + 4 mM EDTA or 0.25% trypsin + EDTA (Gibco), respectively. WT THP1 cells were obtained from ATCC and NLRP3 KO THP1 cells were obtained from Invivogen. THP1 cells were cultured in RPMI supplemented with 10% FBS, Penicillin + Streptomycin, L-Glutamine and Sodium pyruvate, herafter referred to as complete RPMI (cRPMI) and cultured in suspension culture in T75 or T175 tissue culture flasks (Corning). For differentiation into macrophages, THP1 cells treated with 100 ng/ml of PMA for 18 – 24h. Sf9 insect cells were purchased from ThermoFisher Scientific and grown in suspension culture in sterile glass flask in Hyclone SFX insect cell media at 28 °C and ambient CO2 while shaking at 120 rpm. All cell lines were verified by manufacturer’s website and identity were checked by their morphological features. THP1 cells tested negative for mycoplasma contamination using MycoStrip kit (Invivogen).
Primary cell culture.
Buffy coats from healthy human donors were purchased from BioIVT. Primary cells were cultured in cRPMI. Blood was diluted at a 1:1 ratio with sterile PBS pH 7.4 + 2.5 mM EDTA before layering 30 ml of diluted blood over 15 ml of FiColl Paque PLUS density gradient media (GE Healthcare). Density gradient centrifugation was performed at 800 × g for 35 min at 20 °C. Total peripheral blood mononuclear cells were havested from the interphase and washed twice with MACS buffer (PBS pH 7.4, 2.5 mM EDTA, 1% FBS). Red blood cells were lysed by resuspending the pellet in 10 ml ACK lysis buffer and incubation for 5 min at RT. After a final washing step in MACS buffer, CD14-positive PBMCs were isolated by magnetic-activated cell sorting (MACS). Cells were resupended in 800 μl of MACS buffer mixed with 200 ul of human CD14 microbeads (Miltenyi). After incubation on ice in the dark for 15 mins, cells were washed one more time in 20 ml of MACS buffer, resuspended in 2.5 ml and poured over an LS column (Miltenyi) placed in a magnetic holder and pre-equilibrated with 2.5 ml of MACS buffer. Column was washed three times with 2.5 ml of MACS buffer. Column was then removed from the magnetic holder and CD14-positive PBMCs were eluted in 5 ml of MACS buffer using the provided plunger. Cells were then seeded in T75 cell culture flasks (20×106 CD14-positive PBMCs per flask) in 15 ml complete RPMI supplemented with 30 ng/ml of recombinant human M-CSF (R&D Systems) for 6 days. Media was replenished with fresh complete RPMI containing M-CSF every 2 – 3 days.
Generation of KO cell lines using CRISPR/Cas9 technology.
CRISPR KO cell lines were generated by electroporation of in vitro assembled ribonucleoproteins (RNPs) consisting of synthetic sgRNAs and recombinant Cas9 protein (Alt-R S.p. HiFi Cas9 nuclease from IDT) using the Neon transfection system (Thermo Fisher). RNPs were assembled by mixing 0.3 μl of Cas9 protein (62 μM) with 0.5 μl of sgRNA (ordered from IDT and resuspended in nuclease-free water at a concentration of 100 μM) and incubated at RT for 15–20 min. 1.2×106 THP1 cellswere resuspended in 12 μl of T buffer, mixed with the assembled RNPs and electroporated using a 10 μl electroporation pipette tip with two 10 ms pulses at a voltage of 1400 V. Cells were then dispensed directly into a 6-well plate containing 3 ml of cRPMI and cultured for 3–5 days before assessing bulk KO efficiency by immunoblot. These cell lines were further single cell cloned by limited serial dilution in single cell cloning media (40% THP1 conditioned media, 40% RPMI, 20% FBS) to obtain clonal KO cell populations with complete ablation of the target protein. sgRNA sequences were pre-designed by IDT (Non-target sgRNA: AAAUGUGAGAUCAGAGUAAU; Caspase-1 sgRNA: CGGCTTGACTTGTCCATTAT; Caspase-4 sgRNA: AGGGATTCCAACACCTTAAG; IL-18 sgRNA: CAAATAGAGGCCGATTTCCT).
Retroviral transduction.
HEK293T cells were used as packaging cells for retroviral vectors. For the production of retroviral particles, 2.5×106 HEK293T cells were seeded in a 10 cm cell culture dish. After overnight incubation at 37 °C, cells were transfected with 10 μg of pMSCV IRES EGFP encoding the protein of interest, 6 μg of pCL-ECO and 3 μg of pCMV-VSVG using Lipofectamine 2000 (ThermoFisher) according to the manufacturer’s instructions. After 18–24 h at 37 °C, media was changed to 6 ml of fresh cRPMI (for transduction of THP1 cells) and virus containing supernatant was collected 24 h post media change. Supernatants were clarified from cellular debris by centrifugation (400 × g, 5 min) and filtered through a 0.45 μm PVDF syringe filter. 1 × 106 THP1 cells were resuspended in 4.5 ml of viral supernatant supplemented with Polybrene (1:2000; EMD Millipore) and plated in a 6-well plate followed by centrifugation for 1 h at 1250 × g and 30 °C. This procedure was performed twice on two consecutive days to maximize transduction efficiency. GFP+ cells were sorted twice on a FACSAria or FACSMelody cell sorter (BD Biosciences) to obtain cell lines with stable and homogenous expression of the target protein. Transgene expression was confirmed by immunobloting using a rabbit anti-Myc-tag or mouse anti-Myc-tag primary antibody (both from Cell Signaling Technologies).
Recombinant protein expression and purification.
Expression and purification of caspase catalytic domains and IL-1 family cytokine substrates for in vitro cleavage assays were expressed in bacteria and purified as described before17,27. Chemically competent Rosetta (DE3) pLysS cells (EMD Millipore) were transformed with the pET28 plasmid encoding the protein of interested and plated on LB agar plates with kanamycin (25 μg/ml). Overnight precultures were inoculated with a single colony and grown at 30°C and 250 rpm in 2× YT media containing kanamycin (25 μg/ml) and chloramphenicol (50 μg/ml). Individual expression cultures of 50 to 500 ml were inoculated with overnight cultures at a ratio of 1:100 and incubated at 37°C and 250 rpm until the OD600 reached a value between 0.7 and 0.8. After a cooling step on ice for 15 mins, protein expression was induced by adding isopropyl-β-d-thiogalactopyranoside (IPTG) to a final concentration of 0.25 mM, and expression was allowed to proceed overnight at 18°C. Bacterial pellets were harvested by centrifugation (5000g for 20 to 30 min at 4°C) and stored at −20°C if not immediately used for protein purification. To purify recombinant proteins, bacterial pellets were resuspended in resuspension buffer (25 mM HEPES-NaOH (pH 7.4), 150 mM NaCl, and 10 mM imidazole) and lysed by ultrasonication. Cell lysates were clarified by centrifugation (30 – 45 min, 20,000g, 4°C) and filtered through a 0.22-μm syringe filter before pouring them into a gravity flow column containing a bed of nickel–nitrilotriacetic acid (Ni-NTA) agarose beads (Qiagen). Beads were washed with at least 10 bed volumes of wash buffer (25 mM HEPES (pH 7.4), 400 mM NaCl, and 25 mM imidazole) and bound protein was eluted stepwise in resuspension buffer supplemented with 40 to 250 mM imidazole. Elution fractions were analyzed by SDS-PAGE followed by InstantBlue staining (Expedeon) to identify fractions containing proteins of interest and confirm autocatalytic processing of caspase catalytic domains into active p20 and p10 subunits. Fractions containing the protein of interest were pooled and buffer-exchanged into SEC buffer (25 mM HEPES (pH 7.4), 150 mM NaCl) using a PD-10 desalting column (GE Healthcare). Pro-IL-18 variants used for binding assays were further purified by SEC using a BioRad NGC Quest10 Chromatography system equipped with a Superdex 200 Increase 10/300 column in SEC buffer. Peak fractions containing the protein of interest were identified by SDS-PAGE and InstantBlue staining and combined. Lastly, protein was concentrated by centrifugal ultrafiltration using an Amicon Ultra-15 centrifugal filter unit with 10 kDa cut-off (EMD Millipore). Glycerol was added to a total concentration of 10% and aliquots were snap-frozen in liquid nitrogen and stored at −80°C.
For the expression of catalytically inactive caspase-4 p20/p10 complexes for binding assays and structural studies, chemically competent Rosetta (DE3) cells (EMD Millipore) were co-transformed with a pET28a plasmid encoding the N-terminally His-tagged large p20 subunit (with a C258A mutation) and a pET16b plasmid encoding the untagged p10 subunit and plated on LB agar plates with 25 μg/ml kanamycin and 100 μg/ml ampicillin. Overnight precultures were inoculated with a single colony and grown at 30°C and 250 rpm in 2× YT media containing kanamycin (25 μg/ml), ampicillin (100 μg/ml) and chloramphenicol (50 μg/ml). 500 ml of 2× YT were inoculated with overnight cultures at a ratio of 1:100 and incubated at 37°C and 250 rpm until the OD600 reached a value between 0.7 and 0.8. After cooling on ice for 15 mins, protein expression was induced by IPTG to a final concentration of 0.25 mM, and expression was allowed to proceed overnight at 18°C. Bacterial pellets were harvested by centrifugation (5000 × g for 15 min at 4°C), washed once with PBS, pH 7.4 and stored at −20°C if not immediately used for protein purification. For purification, bacterial pellets were resuspended in resuspension buffer and lysed by ultrasonication. Cell lysates were clarified by centrifugation (30 – 45 min, 20,000g, 4°C) and filtered through a 0.22-μm syringe filter before pouring them into a gravity flow column containing a bed of Ni-NTA agarose beads. Beads were washed with at least 10 bed volumes of wash buffer and bound protein was eluted in resuspension buffer supplemented with 250 mM imidazole. Proteins were further purified by SEC using a BioRad NGC Quest10 Chromatography system equipped with a Superdex 200 Increase 10/300 column in SEC buffer. Peak fractions containing the protein of interest were identified by SDS-PAGE and InstantBlue staining (Expedeon), pooled and protein was concentrated by centrifugal ultrafiltration using Amicon Ultra-15 centrifugal filter unit with 10 kDa cut-off (EMD Millipore). Glycerol was added to a total concentration of 10% and aliquots were snap-frozen in liquid nitrogen and stored at −80°C.
For expression of human pro-IL-18 and mature IL-18 in insect cells, chemically competent DH10Bac cells (ThermoFisher) were transformed with a pFastBac vector encoding the protein of interest and cells were plated on LB agar plates supplemented with 25 μg/ml kanamycin, 10 μg/ml tetracycline, 7 μg/ml gentamycin, 50 μg/ml X-Gal and and 40 μg/ml IPTG and incubated overnight at 37 °C for blue/white screening. 20 ml of LB medium with 25 μg/ml kanamycin, 10 μg/ml tetracycline, 7 μg/ml gentamycin were inoculated with one positive (white) colony and incubated overnight at 37 °C and 250 rpm. Bacmid DNA was isolated using buffer components from the GeneJET Plasmid Miniprep kit (ThermoFisher) and precipitated using isopropanol. The DNA pellet was washed once with 70 % ethanol, air-dried and resuspended in 40 μl of sterile, ultrapure water. 10 μl of bacmid DNA was diluted with 100 μl of Hyclone SFX insect cell media. 10 μl of CellFectin II (ThermoFisher) transfection reagent was mixed with 100 μl of media and added to the DNA mixture. After incubating at RT for 30 min, 100 μl of the transfection mix was added dropwise to 0.8 × 106 Sf9 cells seeded in the wells of a 6-well plate in 3 ml of media. Routinely, two wells were used for each construct. After 2 – 3 days of incubation at 28 °C, the baculovirus containing supernatant was collected, filtered using a 0.45 μm syringe filter and stored at 4 °C. To amplify the initial P1 virus, Sf9 cells were grown at a density of 1.0 × 106 cells/ml in a total volume of 25 ml, infected with 2 ml of the initial virus and incubated at 28 °C and 120 rpm. After 3 days, virus was collected by centrifuging the cells at 1000 g for 10 min and taking off the baculovirus-containing supernatant, which was then passed through a 0.45 μm syringe filter and stored at 4 °C. For protein expression, Sf9 cells were seeded at a density of 1.5 × 106 cells/ml in a suspension culture with a volume of up to 1 l, infected with 1% (v/v) of the amplified virus and incubated at 28 °C and 120 rpm. Cells were harvested via centrifugation (1000 g, 15 min), 48–72 h after a proliferation arrest occurred. Cell pellets were frozen in liquid nitrogen and stored at −20 °C until protein purification. For purification, insect cell pellets were resuspended in resuspension buffer and lysed by ultrasonication. Cell lysates were clarified by centrifugation (30–45 min, 20,000g, 4°C) and filtered through a 0.22-μm syringe filter before pouring them into a gravity flow column containing a bed of Ni-NTA agarose beads. Beads were washed with at least 10 bed volumes of wash buffer and bound protein was eluted in resuspension buffer supplemented 250 mM imidazole. Proteins were further purified by SEC using a BioRad NGC Quest10 Chromatography system equipped with a Superdex 200 Increase 10/300 column in SEC buffer. Peak fractions containing the protein of interest were identified by SDS-PAGE and InstantBlue staining (Expedeon), pooled and protein was concentrated by centrifugal ultrafiltration using Amicon Ultra-15 centrifugal filter unit with 10 kDa cut-off (EMD Millipore). Glycerol was added to a total concentration of 10% and aliquots were snap-frozen in liquid nitrogen and stored at −80°C.
The human caspase-1 catalytic domain was expressed and purified as described before38. In brief, two non-tagged p20 and p10 subunits were expressed as inclusion bodies in E. coli BL21 (DE3), respectively. The two subunits were assembled by denaturing, refolding, and further purified by HiTrap SP cation exchange chromatography (GE Healthcare Life Sciences).
Human GSDMD was purified as described before39.
Assembly of caspase-4/pro-IL-18 complex for structural studies.
Purified caspase-4 p20/p10 with an inactivating C258A mutation was mixed with insect cell-purified human pro-IL-18 at a molar ratio of ~1:2 in SEC buffer and incubated at 37 °C for 20 mins. The mixture was then centrifuged at 12,000g for 5 min at 4 °C to spin down any precipitate that may have formed and fractionated by SEC using a BioRad NGC Quest10 Chromatography system equipped with a Superdex 200 Increase 10/300 column in SEC buffer. Fractions containing the assembled complex of caspase-4 and pro-IL-18 (indicated by a shift in retention volume and confirmed by SDS-PAGE and InstantBlue staining) were combined and the chemical crosslinker BS3 (ThermoFisher) was added at a final concentration of 1 mM. After incubation on ice for 1h, BS3 was quenched by adding 50 mM Tris (pH 7.4). For crosslinking mass-spectrometry, BS3 was quenched using 100 mM hydroxylamine. The protein complex was concentrated by centrifugal ultrafiltration using Amicon Ultra-15 centrifugal filter unit with 10 kDa cut-off (EMD Millipore) and again purified by SEC (Superdex 200 Increase 10/300) in SEC buffer.
Negative staining EM.
The peak fractions of SEC containing crosslinked caspase-4/pro-IL-18 were diluted to a final concentration of ~0.015 mg/ml (as determined by Nanodrop) for negative staining EM. 6 μl sample was added to each copper grid with carbon support film (Electron Microscopy Sciences) that had been glow-discharged for 30 seconds using a Pelco EasyGlow (Ted Pella) instrument. After 1 min, the sample was stained by 6 μl 2% uranyl acetate solution (Electron Microscopy Sciences) twice for 30 seconds each, with removal of excess buffer using filter papers (Whatman) after each round of staining. The negatively stained grids were imaged on Transmission Electron Microscope (Joel JEM1400) at 120 keV.
Cryo-EM data collection.
The peak fractions of crosslinked caspase-4/pro-IL-18 from SEC were concentrated to ~0.22 mg/ml (as determined by Nanodrop) for cryo-EM grid preparation. 3.3 μl sample was placed onto each glow-discharged cryo-EM grid (Quantifoil R1.2/1.3 gold grid with 400 mesh, Electron Microscopy Sciences) before being blotted for 3–5 s under 100% humidity at 4 °C and plunged into liquid ethane using a Mark IV Vitrobot (ThermoFisher). Before data collection, all the grids were pre-screened and optimized at the Harvard Cryo-EM Center for Structural Biology or at cryo-EM facility of University of Massachusetts (UMASS) to check ice thickness and particle distribution.
The final datasets were collected at UMASS on a Titan Krios electron microscope (Thermo Fisher Scientific) equipped with a K3 Summit direct electron detector (Gatan) and a post-column energy filter (Gatan). 5,934 movies were collected under the super resolution mode at 105,000× magnification (0.4125 Å per pixel) with a defocus range between −1.0 and −2.5 μm, and three shots were recorded for each hole per stage movement. For each movie stack with 40 frames, the total dose was 55.4 electrons per Å2. SerialEM was used for automated data collection40.
Cryo-EM data processing.
The computer support and software for data processing was provided by SBGrid consortium41. Raw movies were corrected by gain reference and beam-induced motion, and binned by two fold with or without dose weighting using the Relion 3.08 implementation of the MotionCor2 algorithm42. The motion-corrected micrographs were imported into CryoSPARC43. Patch CTF-estimation was performed to determine the local defocus values in each micrograph. Blob picking on a random subset of 500 micrographs was used to generate a template that was used to pick a total of 10,802,676 particles. Multiple rounds of 2D classification were performed, resulting in 1,234,038 good particles that were used for ab initio 3D reconstruction. Heterogeneous refinement was first carried out to classify the particles into five classes. The dominant class with 229,147 particles was further processed by homogeneous refinement with C2 symmetry to achieve a final map at a corrected resolution of 3.2 Å. The reported resolutions under different processing conditions were estimated based on the gold-standard Fourier shell correlation (FSC) = 0.143 criterion. The cryo-EM map was further sharpened by applying a negative B factor using automated procedures in Phenix44. Local resolution estimation of all the cryo-EM maps were performed in Phenix45.
Model fitting and building.
The crystal structure of caspase-4 dimer (ProteinDataBank (PDB) code 6KMZ7) and mature IL-18 (PDB code 3WO234) were used as initial templates for map fitting in the UCSF ChimeraX software46. The caspase-4/pro-IL-18 complex structure was refined by real space refinement and validated by model validation in Phenix. Protein-protein interaction analysis was conducted using the PISA webserver35. Per residue Cα–Cα distances in the caspase-4/pro-IL-18 complex were calculated with the Distances function in UCSF ChimeraX, and used to correlate with crosslinking mass spectrometry data. To generate model of a caspase-11/pro-IL-18 complex, the crystal structure of caspase-11 (PDB code 6KN17) was fitted into the electron density and aligned with our caspase-4 model in UCSF ChimeraX.
AlphaFold implementation in the ColabFold notebooks running on Google Colaboratory36,47 was used to predict the structure for pro-IL-18 alone. The top five models predicted by AlphaFold were aligned and checked for consistency. pLDDT (predicted local distance difference test, 0–100 with 100 being the best) score computed by AlphaFold was used to indicate the reliability of prediction.
Crosslinking mass-spectrometry.
BS3-crosslinked caspase-4/pro-IL-18 complex was purified as described above. For analysis of pro-IL-18 before binding to caspase-4, human pro-IL-18 purified from insect cells was diluted to 0.34 mg/ml in SEC buffer and crosslinked with 1 mM of BS3 on ice for 1 h. BS3 was then quenched with 100 mM hydroxylamine for 10 min on ice. Lyophilized samples were resuspended in a buffer containing 8M Urea and 50mM EPPS, pH 8.3 and reduced for 0.5 h with 10 mM TCEP. Proteins were alkylated with 30 mM iodoacetamide in the dark for 1 h, before quenching with 50 mM β-mercaptoethanol. Samples were then diluted 8-fold with 50 mM EPPS, pH 8.3 to dilute out Urea and digested with trypsin overnight at 37 °C (Promega; 1:25 ratio of enzyme:substrate). 10% formic acid was added to a final pH of 2. Samples were desalted using stage tips with Empore C18 SPE extraction discs (3M) and dried under vacuum. Peptides were then reconstituted in 5% formic acid, 5% acetonitrile, and analysed in the Orbitrap Eclipse mass spectrometer (ThermoFisher) coupled to an EASY-nLC 1200 (ThermoFisher) ultra-high pressure liquid chromatography pump, as well as a high-field asymmetric waveform ion mobility spectrometry (FAIMS) FAIMSpro interface. Peptides were separated on an in-house column with 100 μM inner diameter and packed with 30 cm of Accucore C18 resin (2.6 μm, 150 Å, ThermoFisher), in a gradient of 5–35% (acetonitrile, 0.125% formic acid) over 120 min at about 500 nl min−1. Instrument was operated in data-dependent mode. FTMS1 spectra were collected at a resolution of 120,000, with an automatic gain control target of 4 × 105, and a maximum injection time of 50 ms. The most intense ions were selected for tandem mass spectrometry for 1.5 s in top-speed mode, while switching among three FAIMS compensation voltages (−40, −60, and −80 V). Previously investigated precursors were excluded using a dynamic exclusion window of 90 s. MS2 precursors were isolated with a quadrupole mass filter set to a width of 0.7 Th and analysed by FTMS2, with the Orbitrap operating at 30,000 resolution, an automatic gain control target of 125,000 and a maximum injection time of 150 ms. Precursors were then fragmented by high-energy collision dissociation at a 30% normalized collision energy.
Mass spectra were processed and searched using the PIXL search engine48 against a sequence database containing most abundant contaminants from S.frugiperda and E.coli in addition to caspase and IL-18 construct sequences with a precursor tolerance of 15 ppm and fragment ion tolerance of 10 ppm. Methionine oxidation was set as a variable modification. Mass of BS3 was set as +156.0786 or +138.0681 for mono-linked or crosslink, respectively. All crosslinked searches included 50 most abundant protein sequences to ensure sufficient statistics for estimation of the false-discovery rate. Matches were filtered to 1% false-discovery rate on the unique peptide level using linear discriminant features48.
In vitro protein cleavage assay.
Proteolytic cleavage of purified full-length protein substrates by caspases was assessed as described before27. Two-fold dilution series of the indicated recombinant caspase was incubated with substrate protein at a final concentration of 50 nM in 40 μl of caspase assay buffer (10 mM PIPES pH 7.2, 10% sucrose, 10 mM DTT, 100 mM NaCl, 1 mM EDTA, 0.1% CHAPS) for 30 min at 37 °C. Reactions were stopped by adding 10 μl of 5x SDS loading dye with reducing agent and boiling at 65 °C for 10 min. Cleavage products were separated by SDS-PAGE and analyzed via immunoblotting using rabbit anti-human IL-18 (MBL), rabbit anti-murine IL-18 (Abcam), rabbit anti-Myc (Cell Signaling), rabbit anti-human IL-1β (Genetex) or rabbit anti-GSDMD (Cell Signaling) primary antibodies. Band intensities were quantified using ImageJ to determine EC50 values and catalytic efficiencies were calculated using the following equation:
In vitro peptide cleavage assay.
For peptide cleavage assays, recombinant caspase-4 carrying indicated mutations were first diluted to a concentration of 1 μM in caspase assay buffer. To start the reaction, 50 μl of the diluted caspase was then mixed with 50 μl of the chromogenic tetrapeptide substrate Ac-WEHD-pNA (100 μM final concentration) in the same buffer (final concentration of caspase was 500 nM in a total volume of 100 μl) in a clear 96-well plate. Absorbance at a wavelength of 405 nm was measured every 20 s for 30 min using a Tecan Spark plate reader with temperature control set to 37°C. Substrate solution was prewarmed to 37°C before adding to the caspase to ensure homogeneous assay conditions.
LPS electroporation.
The Neon transfection system (ThermoFisher) was used to deliver LPS into the cytoplasm of cells. For priming, iBMDMs, THP1 macrophages or primary human macrophages were treated with 1 μg/ml of LPS for 4 h. Monocytic THP1s were primed with 1 μg/ml of Pam3CSK4 for 4 h. Murine iBMDMs or human primary macrophages were resuspended in R buffer at a density of 10 × 106 cell/ml in a volume of 120 μl per sample. THP1 cells (both monocytes and macrophages) were resuspended in T buffer at a density between 250 × 106 cell/ml in a volume of 12 μl per sample. LPS or sterile PBS pH 7.4 (as negative control) was mixed with the cell supension (1 μg of LPS per 1 × 106 iBMDM or primary macrophage cells, 0.5 μg of LPS per 1 × 106 THP1 cells) before aspirating the cell suspension into the Neon electroporation pipette equipped with a 100 μl tip (for iBMDMs and primary macrophages) or 10 μl tip (for THP1s) and performing electroporation with two pulses with a pulse width of 10 ms each and a voltage of 1400 V. Cells were then dispensed into an appropriate cell culture media (5 × 105 cell/ml in cDMEM for iBMDMs and 3.33 × 105 cell/ml in cRPMI for human primary macrophages or 1.25 × 106 cell/ml in cRPMI for THP1 cells). THP1 cells or iBMDMs were then plated in 96-well plates (200 μl per well) for LDH assay and ELISA or 12-well plates (2 ml per well) for IL-1 cytokine immunoprecipitation experiments and incubated for 2 h. Primary human macrophages were seeded in 12-well and 6-well plates (in 1.5 ml or 3 ml of media, respectively) and LDH assay, ELISA and cytokine pulldowns were performed from the supernatants after 2 h.
Bacterial infections.
Salmonella strain deficient for flagellin (SL1344 fliC/fljB) was a kind gift from Igor Brodsky (University of Pennsylvania) and infections were performed as described before with minor modifications49. Bacteria were streaked on LB agar plates containing 25 μg/ml kanamycin, grown overnight at 37 °C and plates were stored at 4 °C for up to 2 weeks. Overnight cultures (3 ml LB + 25 μg/ml kanamycin and 25 μg/ml chloramphenicol) were inoculated with a single bacterial colony and grown at 37 °C while shaking at 250 rpm. On the next morning, bacterial culture was diluted into high salt LB (3 ml LB + 100 μl of overnight culture + 78 μl of sterile 5 M NaCl) and incubated for another 3 h at 37 °C without shaking. 1×105 THP1 cells were seeded in 96-well plates cRPMI without Pen/Strep and differentiated overnight with 100 ng/ml PMA. Bacteria were washed once cRPMI without Pen/Strep and added to cells at an MOI of 100 in 200 μl cRPMI without Pen/Strep (based on an assumption of 5 ×108 CFU/ml at OD=1). If indicated MCC950 was present in the media at a concentration of 10 μM. Synchronized infection was facilitated by spinning the plates at 500 × g for 5 min right after addition of bacteria-containing media and cells were incubated at 37 °C and 5 % CO2. After 1 h, gentamicin was added to a final concentration of 100 μg/ml to kill extracellular bacteria. Cell culture supernatants for downstream analyses (LDH assay and ELISA) were harvested at 24 h post-infection.
LDH assay.
Lytic cell death was quantified employing the lactate dehydrogenase (LDH) release assay using the CyQuant LDH cytotoxicity assay kit (Thermo Fisher). In a 96-well plate, 50 μl of cell-free supernatants were mixed with 50 μl of LDH assay buffer and incubated for 15–20 min at 37 °C in the dark. Absorbance at 490 nm and 680 nm was meaured on a Tecan Spark plate reader and signal was normalized to equally treated control samples in which cells were lysed using a detergent-containing lysis buffer.
Cytokine measurements by ELISA.
Cytokine release into the supernatants of cell was assessed by ELISA using the IL1 beta mouse uncoated ELISA kit (Thermo Fisher), the Total human IL-18 DuoSet kit (R&D Systems) and the Mouse IL-18 ELISA kit (MBL) respectively, according to manufacturer’s protocols. To quantify IL-18 levels in combined supernatant and lysate samples, cells were first lysed by adding the detergent-containing lysis buffer provided as part of the CyQuant LDH cytotoxicity assay kit (Thermo Fisher) directly to the wells (at 1:10 ratio) before performing ELISA analysis. We confirmed that all ELISA kits used in this study show a strong preference for the cleaved over the uncleaved form of the cytokine to be meausured.
Immunoprecipitation of IL-1 cytokines from cell culture supernatants.
Supernatants from 0.5 – 5.0 × 106 cells (cell number consistent within each individual experiment; generally murine IL-1β and IL-18 were immunoprecipitated from the supernatants of 1 × 106 iBMDMs; human IL-1β and IL-18 were immunoprecipitated from the supernatants of at least 2.5 × 106 THP1 cells or 0.5 – 1.0 × 106 primary macrophages) stimulated as indicated were transferred into microcentrifuge tubes and depleted of cells and debris by spinning at 400 × g for 5 min. Cell-free supernatants were transferred into new tubes and rotated overnight at 4 °C in the presence of 0.5 μg of biotinylated goat anti-murine IL-1β or goat anti-human IL-1β antibodies or 0.75 μg of detection antibodies from human and murine IL-18 ELISA kits (all from R&D Systems) and 20 μl neutravidin agarose beads (Thermo Fisher). IL-18 mutant proteins carrying C-terminal Flag-tags, were immunoprecipitated using 15 μl of Anti-Flag matrix (Thermo Fisher). The remaining cells in the well were lysed in 1x SDS loading dye and served as lysate control. Beads were washed three times with PBS pH 7.4 before eluting bound proteins in 50 μl of 1x SDS loading dye. Immunoprecipitated and cell-associated cytokines were detected by immunoblotting using a rabbit anti-murine IL-1β antibody, rabbit anti-human IL-1β antibody (both from Genetex), rabbit anti-human IL-18 antibody (MBL) or rabbit anti-murine IL-18 (Cell Signaling). Cell-associated actin was detected as a loading control using a mouse anti-actin antibody from Sigma.
Assessment of GSDMD and pro-IL-18 cleavage in cells.
Processing of pro-IL-18 and GSDMD in cells was analyzed by immunoblotting using a rabbit anti-IL-18 antibody (MBL) or rabbit anti-GSDMD or rabbit anti-cleaved NT-GSDMD monoclonal antibodies (both from Abcam), respectively. THP1 macrophages were electroporated with LPS as described above. In order to capture proteins present in both the cell lysate and the cell culture supernatant, cells were resuspended in 1 ml of serum-free Opti-MEM instead of cRPMI. After incubation for 2 h, samples for immunoblotting were prepared by adding 250 μl of 5x SDS loading buffer directly to the well and heated to 65 °C for 10 min to fully denature proteins.
Binding assays.
To test whether two proteins form a stable complex, analytical SEC was performed. Catalytically inactive caspase-4 p20/p10 (with indicated additional mutations) was mixed with pro-IL-18 (with indicated mutations) at an equimolar concentration of 25 μM in SEC buffer. After incubation at 37 °C for 20 min, SEC was performed on a BioRad NGC Quest10 Chromatography system equipped with a Superdex 200 Increase 10/300 column in SEC buffer.
Thermodynamic parameters of binding between pro-IL-18 and caspase-4 p20/p10 were determined by isothermal titration calorimetry (ITC) on a MicroCal ITC200 (Malvern Pananalytical). Proteins were buffer exchanged into SEC buffer using PD-10 Desalting columns (Cytiva) right before the measurements to minimize buffer mismatch. Sample cell was filled with 25 μM of indicated caspase-4 p20/p10 complex and syringe was filled with pro-IL-18 at a concentration of 150 – 200 μM. After an initial delay period of 60 s, a total of 19 injections (first injection of 0.4 μl in 0.8 s, 2 μl in 4 s for residual injections) was performed while stirring at 750 rpm with a twisted paddle syringe at a constant temperature of 37 °C. Spacing between individual injections was 180 s with a filter period of 5 s. Reference power was set to 6 μcal/s and feedback mode/gain was set to high. Reference titration of pro-IL-18 into SEC buffer was performed on each experimental day. Peaks were fitted and integrated using Origin 7.0 software. If appropriate integrations and baseline were adjusted manually and data from first injection and extreme outliers was removed. Data from reference experiments was subtracted to correct for heat of dilutions. Binding heat was plotted in dependence of the molar ratio and thermodynamic parameters were obtained by fitting with a ‘One Set of Sites’ binding curve with at least 200 iterations.
Extended Data
Extended Data Fig. 1 |. Comparative analysis of IL-1 cytokine cleavage across mammals.

a, 10 μg of indicated proteins were separated by SDS-PAGE and stained with InstantBlue. b-k, In vitro cleavage of pro-forms of indicated human IL-1 family cytokines by caspase-4, caspase-1 and caspase-5. Immunoblots are representative of three independent repeats. Asterisk marks signal due to cross-reaction of primary antibody with His-tagged p20 subunit of the caspase. l,m, Immunoblots showing in vitro cleavage of human pro-IL-1β by human caspase-1 and caspase-5. Asterisk marks signal due to cross-reaction of primary antibody with His-tagged p20 subunit of the caspase. n-q, Immunoblots showing in vitro cleavage of murine pro-IL-18 by human caspase-1 and caspase-4, and murine caspase-1 and caspase-11. r–v, Immunoblots showing in vitro cleavage of pro-IL-18 from indicated mammalian species by caspase-4 homolog from the same species. w, x, Immunoblot and quantification of in vitro cleavage of human pro-IL-18 mutants by murine caspase-11. n=4 biological replicates for caspase-4 and n=3 biological replicates for caspase-11. All immunoblots are representative of at least three biological replicates. Bars and error bars represent mean ± SEM. Statistical significance was determined by unpaired, two-sided student’s t-test. For gel source data, see Supplementary Figure 1.
Extended Data Fig. 2 |. Cytosolic LPS induces NLRP3-independent IL-18 release from human cells.
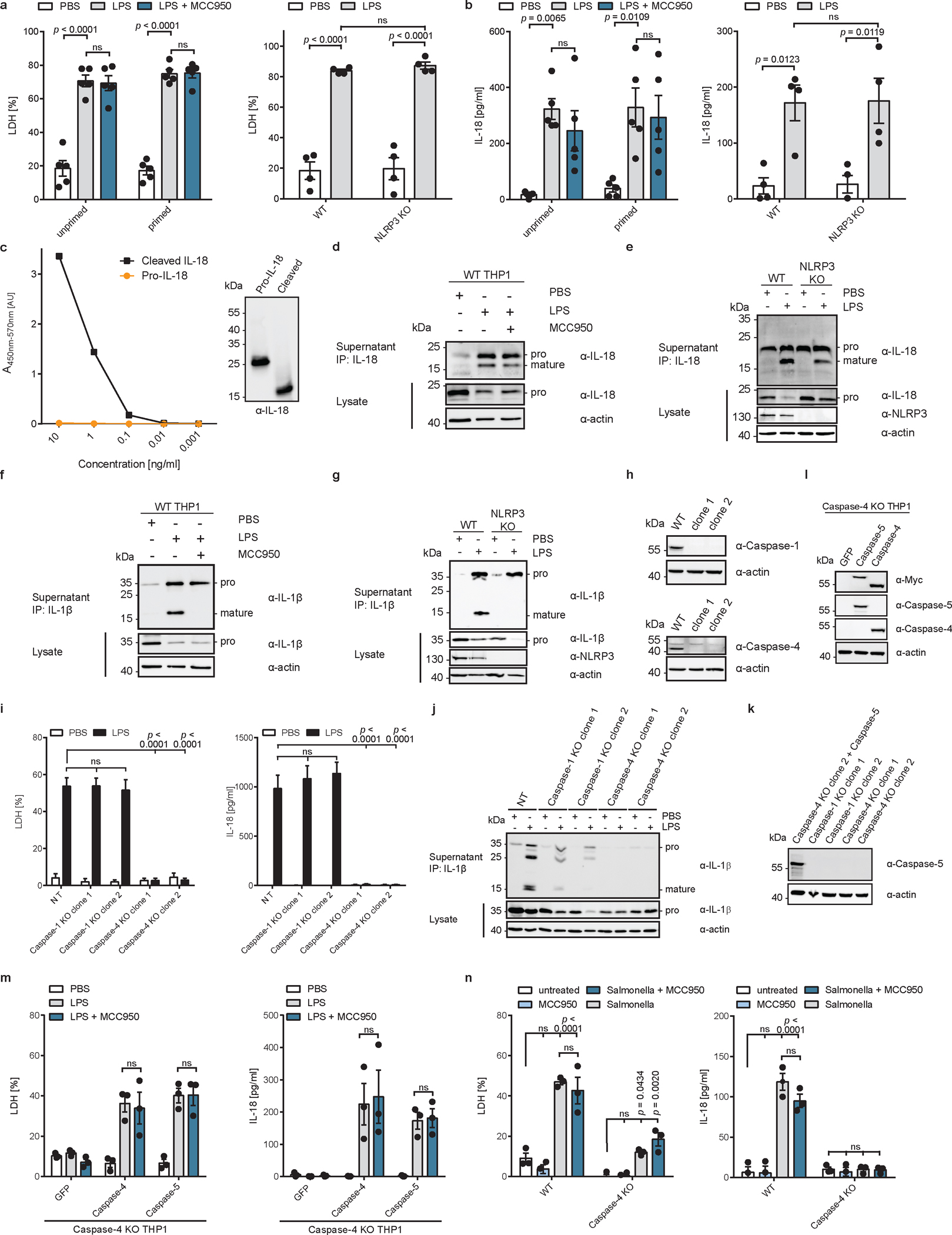
a, b, WT or NLRP3-deficient THP1 monocytes were primed with Pam3CSK4, or left unprimed, and electroporated with LPS (or PBS) in presence or absence of MCC950 and LDH and IL-18 release into supernatant was quantified after 2 h. c, ELISA analysis of purified pro-IL-18 and mature IL-18. Mature IL-18 was generated by cleavage of pro-IL-18 with recombinant caspase and complete cleavage was confirmed by immunoblot. Immunoblot and ELISA results are representative of two experiments. d, e, WT or NLRP3-deficient THP1 monocytes electroporated with LPS (or PBS) in the presence of absence of MCC950. IL-18 from cell culture supernatants was immunoprecipitated and analyzed by immunoblot. f, g, Pam3CSK4-primed WT or NLRP3-deficient THP1 monocytes were electroporated with LPS (or PBS) in the presence of absence of MCC950. IL-1β was immunoprecipitated from supernatants and analyzed by immunoblot. h, Immunoblot analysis of THP1 cells in which expression of caspase-1 or caspase-4 was disrupted by CRISPR/Cas9. i, j, LPS-primed THP1 macrophages deficient for caspase-1, caspase-4 or treated with a non-target sgRNA (NT) were electroporated with LPS (or PBS) and LDH release and IL-18 levels in supernatants were quantified after 2 h. IL-1β from supernatants was immunoprecipitated and analyzed by immunoblot. Immunoblot is representative of three biological replicates. k, Immunoblot analysis of LPS-primed caspase-1 or caspase-4-deficient THP1 macrophages compared to caspase-4-deficient THP1 cells reconstituted with caspase-5 by retroviral transduction. Cells were differentiated into macrophages and stimulated with LPS for 4 h. l, Immunoblot analysis of caspase-4-deficient THP1 cells reconstituted with caspase-4 or caspase-5 by retroviral transduction. m, Caspase-4-deficient THP1 macrophages expressing GFP only, caspase-4 or caspase-5 were primed with LPS before delivery of LPS into the cytosol by electroporation. LDH and IL-18 release into supernatants was quantified after 2 h. n, WT or caspase-4-deficient THP1 macrophages were infected with a flagellin-deficient strain of Salmonella and LDH and IL-18 release was quantified after 24 h. Immunoblots are representative of three (d, e, f, g, h, j) or two (h, l, k, c) biological replicates. Bars and error bars represent mean ± SEM of three biological replicates. Statistical significance was determined by two-way ANOVA with Tukey’s multiple comparisons test: ns = not significant (p > 0.05). For gel source data, see Supplementary Figure 1.
Extended Data Fig. 3 |. Cytosolic LPS-induced IL-18 release from murine cells is NLRP3-dependent.
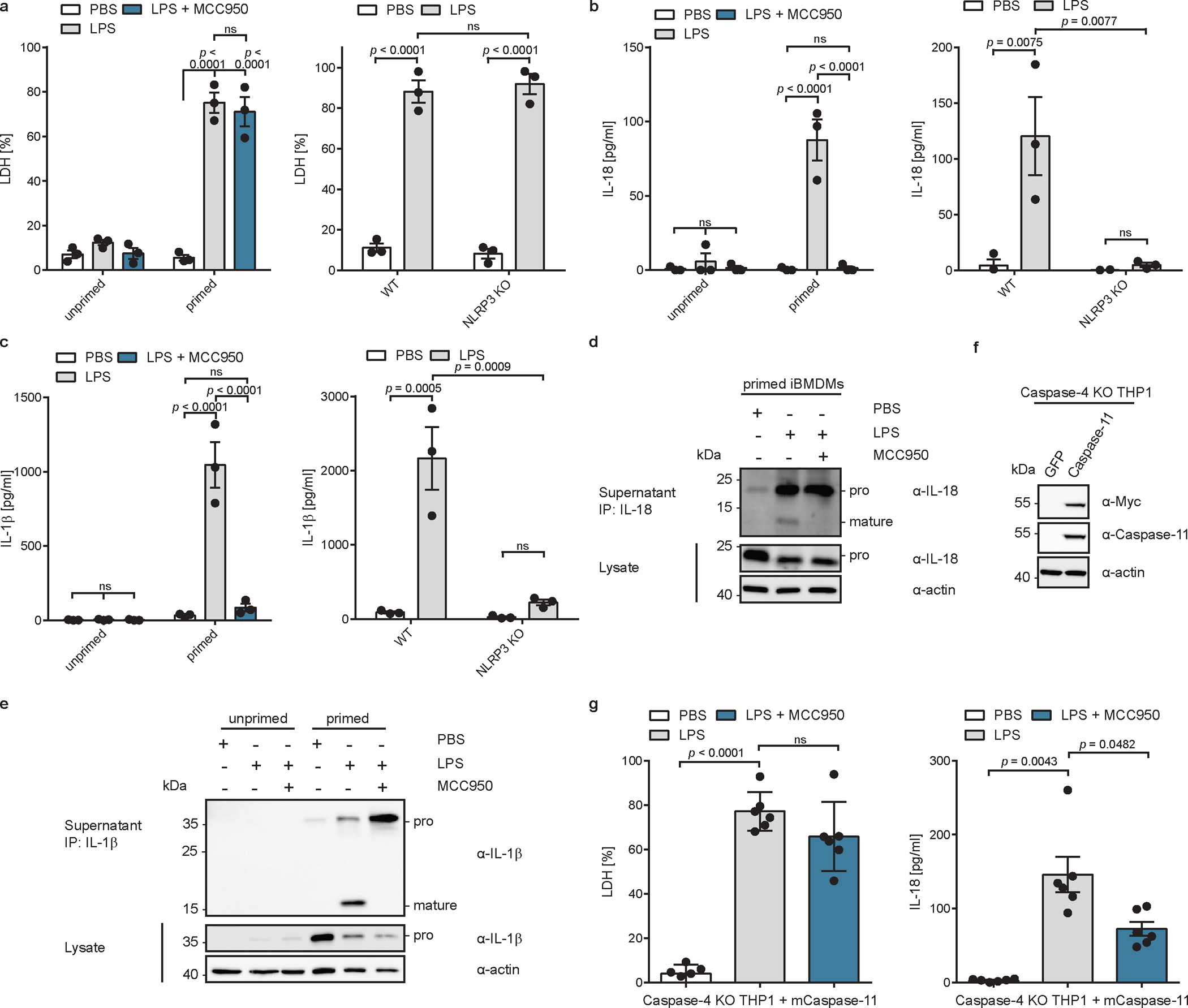
a, b, c, WT or NLRP3-deficient iBMDMs were primed with extracellular LPS, or left unprimed, and electroporated with LPS (or PBS) in the presence of absence of MCC950. LDH, IL-18 and IL-1β release into supernatant was quantified after 2 h. n=3 biological replicates. d, e, LPS-primed WT iBMDMs were electroporated with LPS (or PBS) in the presence or absence of MCC950. IL-18 and IL-1β were immunoprecipitated from supernatants and analyzed by immunoblot (representative of three biological replicates). f, Immunoblot analysis caspase-4-deficient THP1 cells reconstituted with caspase-11 by retroviral transduction (representative of two independent repeats). g, Caspase-4-deficient THP1 macrophages expressing caspase-11 were primed with extracellular LPS, or left unprimed, and electroporated with LPS (or PBS) in the presence of absence of MCC950. LDH, and IL-18 release into supernatant was quantified. n=6 biological replicates. Bars and error bars represent mean ± SEM. Statistical significance was determined by two-way ANOVA (a,b,c) or one-way ANOVA (g) with Tukey’s multiple comparisons test: ns = not significant (p > 0.05). For gel source data, see Supplementary Figure 1.
Extended Data Fig. 4 |. Assembly and purification of a recombinant caspase-4/pro-IL-18 complex.
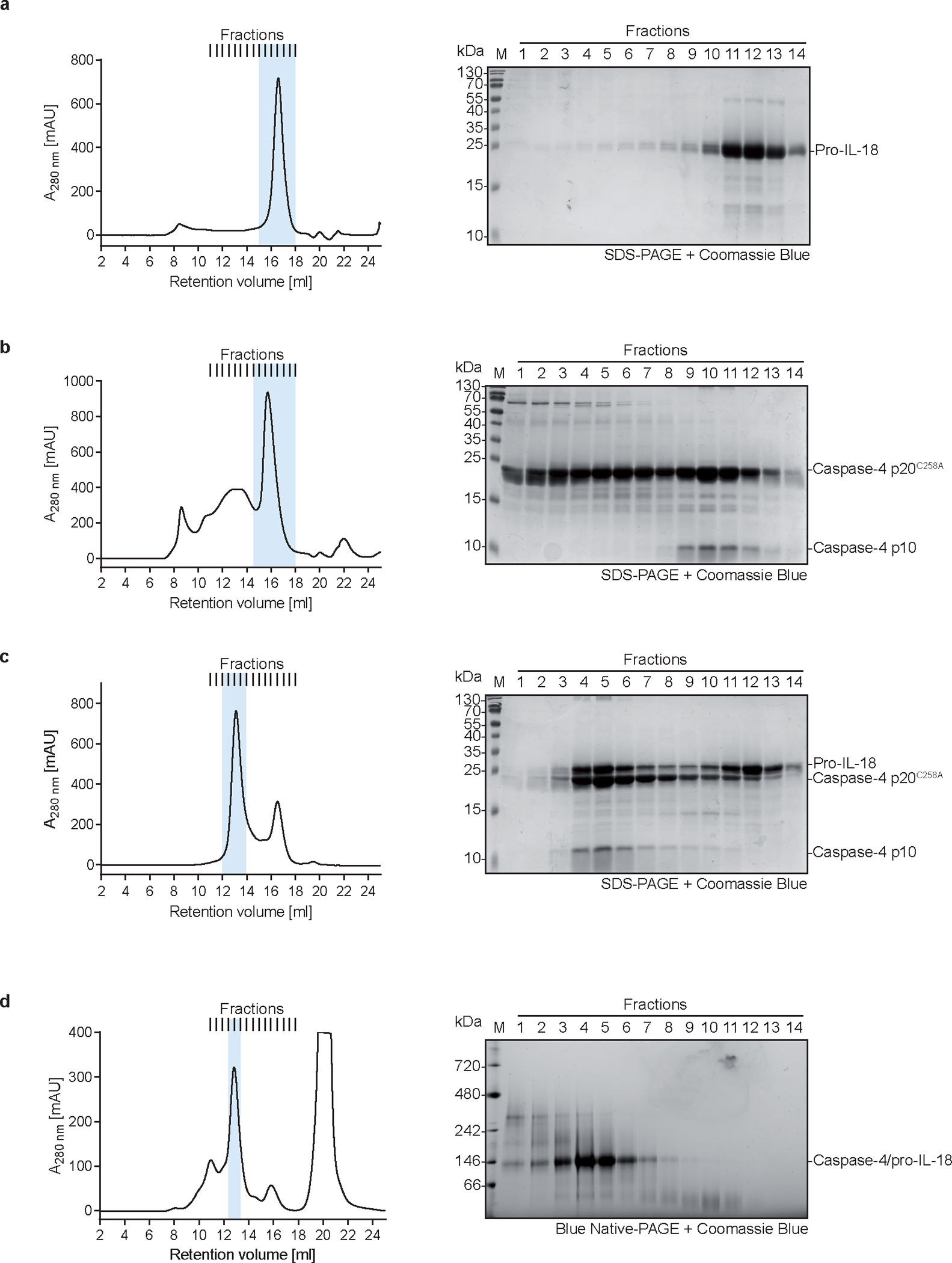
a, SEC profile and corresponding coomassie-stained SDS-PAGE gel of purified pro-IL-18 expressed in insect cells.. b, SEC profile and corresponding coomassie-stained SDS-PAGE gel of purified catalytically inactive caspase-4 p20/p10 expressed in E. coli. c, SEC profile and corresponding coomassie-stained SDS-PAGE gel of a complex consisting of caspase-4 p20/p10 and pro-IL-18. Complex was assembled by co-incubation at 37 °C for 20 min. d, SEC profile and corresponding coomassie-stained Blue Native-PAGE gel of a complex consisting of caspase-4 p20/p10 and pro-IL-18 crosslinked with BS3. Gels and SEC profiles are representative of at least two independent purifications. Peak fractions that were combined for downstream applications are highlighted in blue. For gel source data, see Supplementary Figure 1.
Extended Data Fig. 5 |. Cryo-EM data processing for the caspase-4/pro-IL-18 complex.
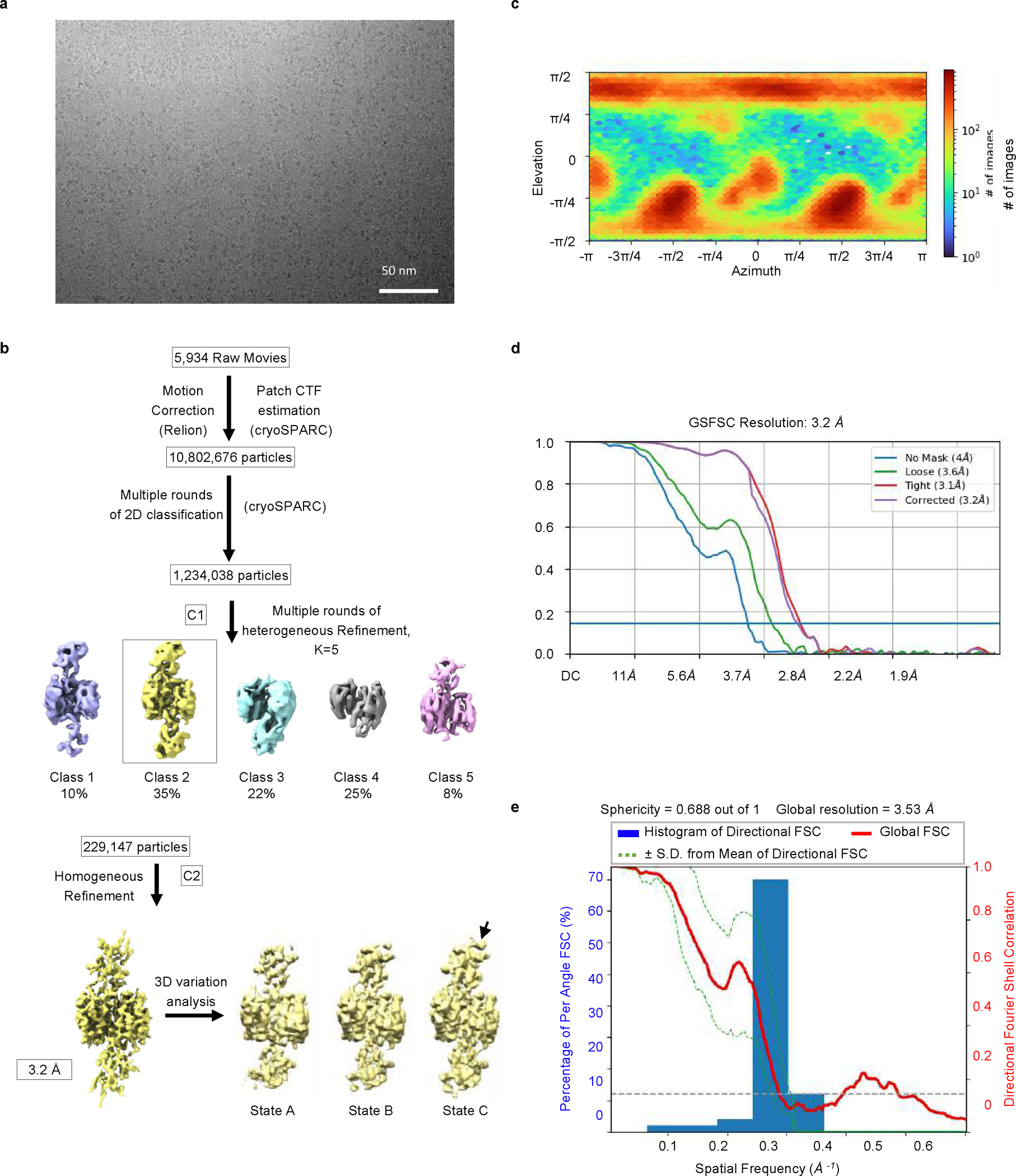
a, Cryo-EM raw image (representative of two data collections). b, Cryo-EM data processing flow chart. The black arrow pointing to “State C” shows domain flexibility indicated by 3D variability analysis. c, Heat map for the orientation of particles used for the final reconstruction. d, Fourier shell correlation (FSC) plots. e, 3D FSC plot.
Extended Data Fig. 6 |. Crosslinking mass spectrometry analysis of caspase-4/pro-IL-18 complex and structural comparison between caspase-4-bound pro-IL-18 and free IL-18.
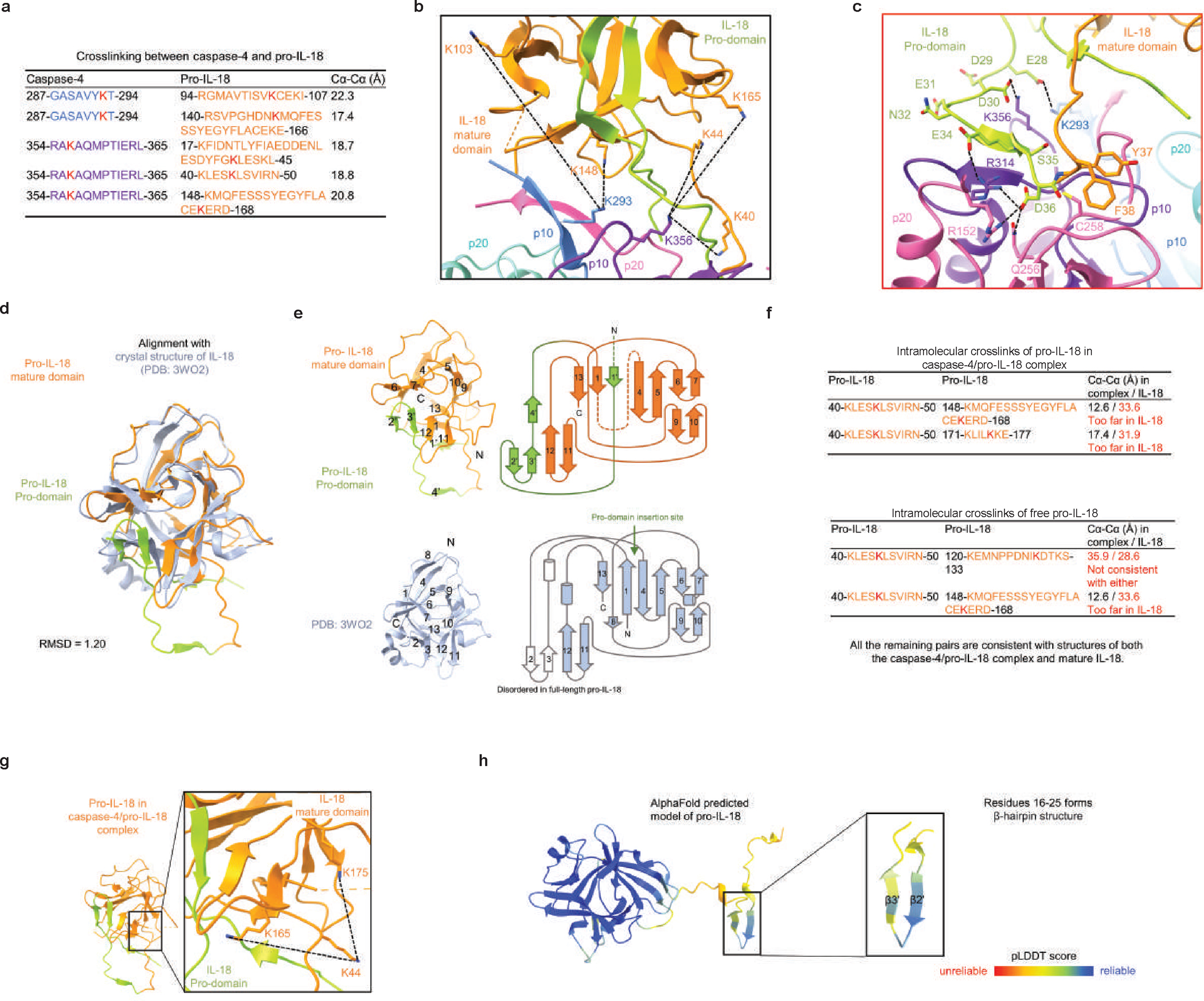
a, Summary of BS3 crosslinking between caspase-4 and pro-IL-18. The crosslinked peptides with high confidence are shown with residue ranges, and colour labelled by their domain colour except for the crosslinked Lys residues which are in red. b, Crosslinked lysine pairs between caspase-4 and pro-IL-18 mapped onto the structure of the caspase-4/pro-IL-18 complex and indicated by black dash lines. c, The molecular interaction of the active form of caspase-4 interacting with pro-IL-18. Model was derived by replacing A258 with Cys. d, e, Structural alignment between pro-IL-18 bound to caspase-4 and the crystal structure of mature IL-18 (d) and in topology diagrams (e). β-strands are labelled sequentially with those in the pro-domain denoted by a prime (‘). f, Crosslinked lysine pairs within pro-IL-18 in the caspase-4/pro-IL-18 complex (upper table), and those within pro-IL-18 alone before incubating with caspase-4 (lower table). The former pairs are consistent with the caspase-4/pro-IL-18 complex structure, but not the mature IL-18 structure, and the latter pairs are not consistent with any IL-18 structures, suggesting that the pro-IL-18 conformation may be different before caspase-4 binding. g, Crosslinked lysine pairs within pro-IL-18 mapped onto the structure of the caspase-4/pro-IL-18 complex and indicated by black dash lines. h, AlphaFold predicted model of pro-IL-18 coloured by per-residue pLDDT score, ranging from unreliable in red to reliable in blue. Residues 16–25 of the pro-domain form a β-hairpin structure, consistent with our cryo-EM structure pro-IL-18 in complex with caspase-4.
Extended Data Fig. 7 |. The pro-domain of pro-IL-18 mediates the interaction with caspase-4.
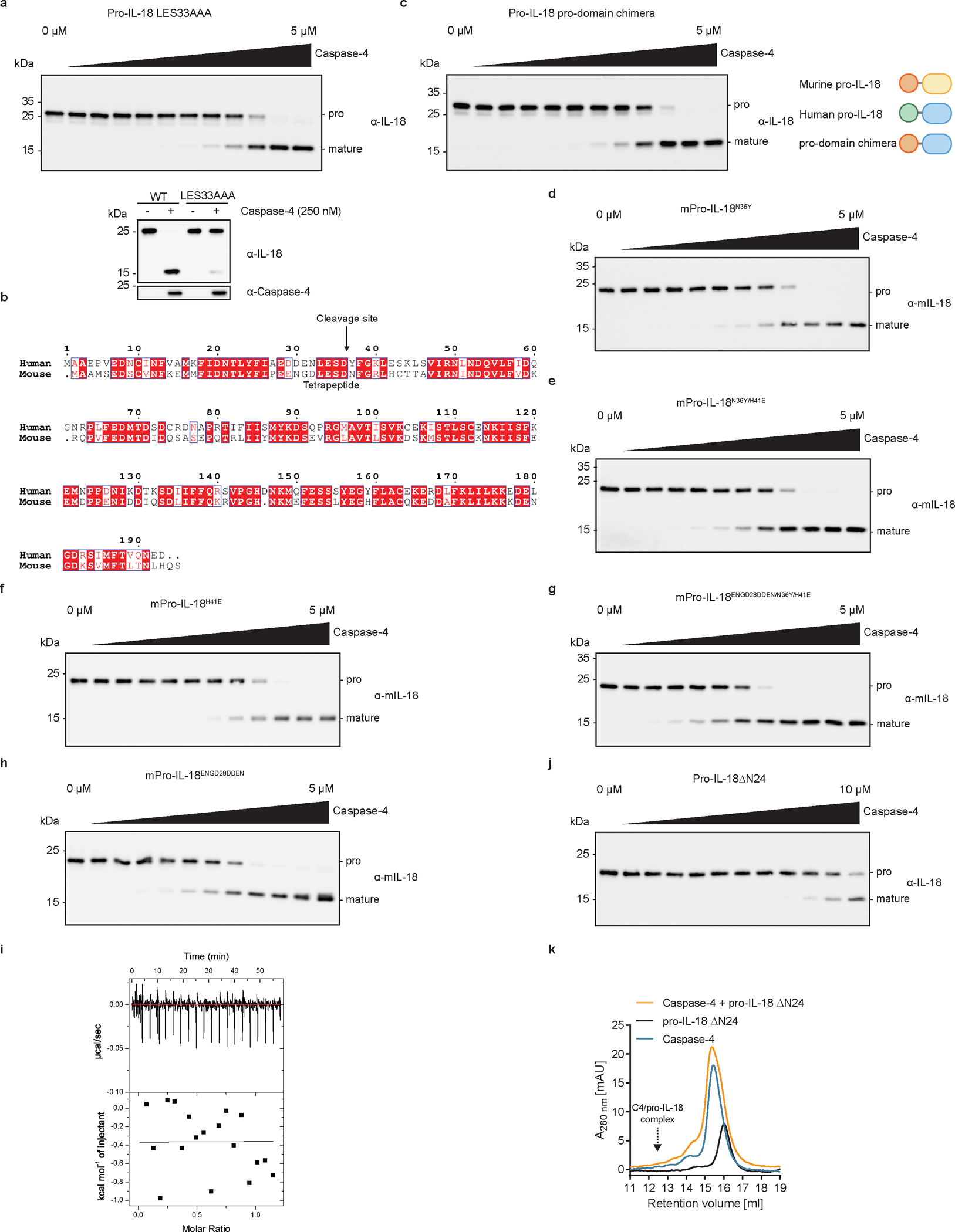
a, c, Immunoblots showing in vitro cleavage of pro-IL-18 mutants by caspase-4. The pro-domain chimera consists of the pro-domain of murine pro-IL-18 fused to the mature domain of human pro-IL-18. Immunoblots are representative of three biological replicates. b, Sequence alignment of human and murine pro-IL-18. Identical and similar amino acids are highlighted in red or white boxes, respectively. Sequences were aligned using ClustalOmega online tool and plotted in ESPript 3.0. d-h, Immunoblots showing in vitro cleavage of murine pro-IL-18 mutants by caspase-4. i, ITC analysis of binding of mature IL-18 to WT caspase-4. j, Immunoblots showing in vitro cleavage of human pro-IL-18 ΔN24 by caspase-4. Pro-IL-18 ΔN24 lacks the first 24 amino acids at the N-terminus. k, Analytical SEC demonstrating no binding between pro-IL-18 ΔN24 and caspase-4. All data are representative of three independent repeats. For gel source data, see Supplementary Figure 1.
Extended Data Fig. 8 |. Biochemical characterization of caspase-4 and pro-IL-18 mutants that disrupt interaction interfaces with pro-IL-18.
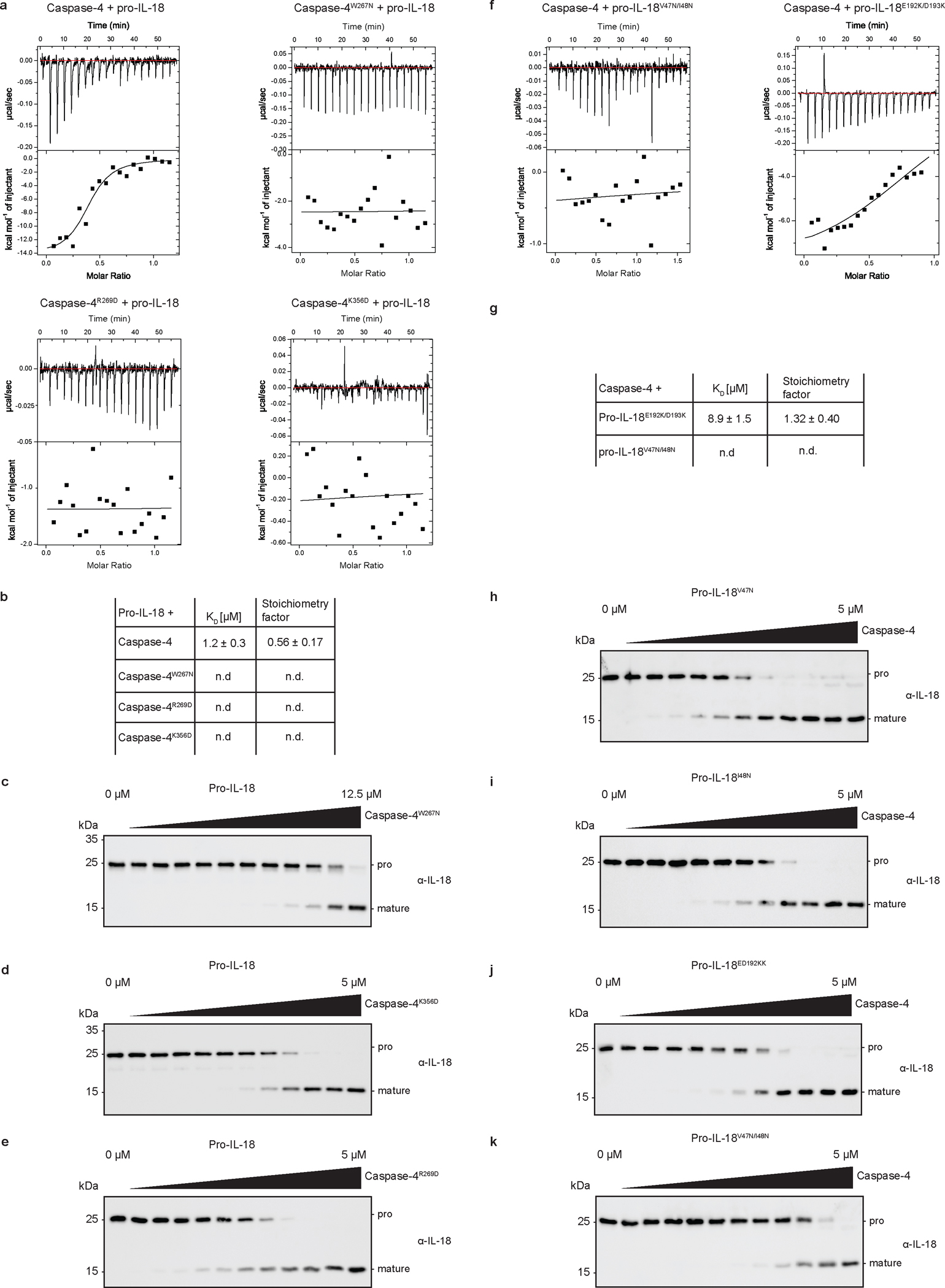
a, ITC analysis of binding of pro-IL-18 (purified from insect cells) to caspase-4 mutants. b, Thermodynamic parameters of binding of indicated caspase-4 mutants to pro-IL-18 as determined by ITC. c, d, e, Immunoblots showing in vitro cleavage of human pro-IL-18 by indicated caspase-4 mutants. f, ITC analysis of binding of indicated pro-IL-18 mutants (purified from E. coli) to WT caspase-4. Graphs are representative of at least three biological replicates. g, Thermodynamic parameters of binding of caspase-4 mutants to pro-IL-18 as determined by ITC. h-k, Immunoblots showing in vitro cleavage of indicated human pro-IL-18 variants by caspase-4. Immunoblots are representative of three biological replicates. ITC graphs are representative of three independent repeats. ITC results represent mean ± SD of three independent measurements. n.d. = no binding detected. For gel source data, see Supplementary Figure 1.
Extended Data Fig. 9 |. Effect of structure-based mutations in caspase-4 on GSDMD and pro-IL-18 cleavage in cells.
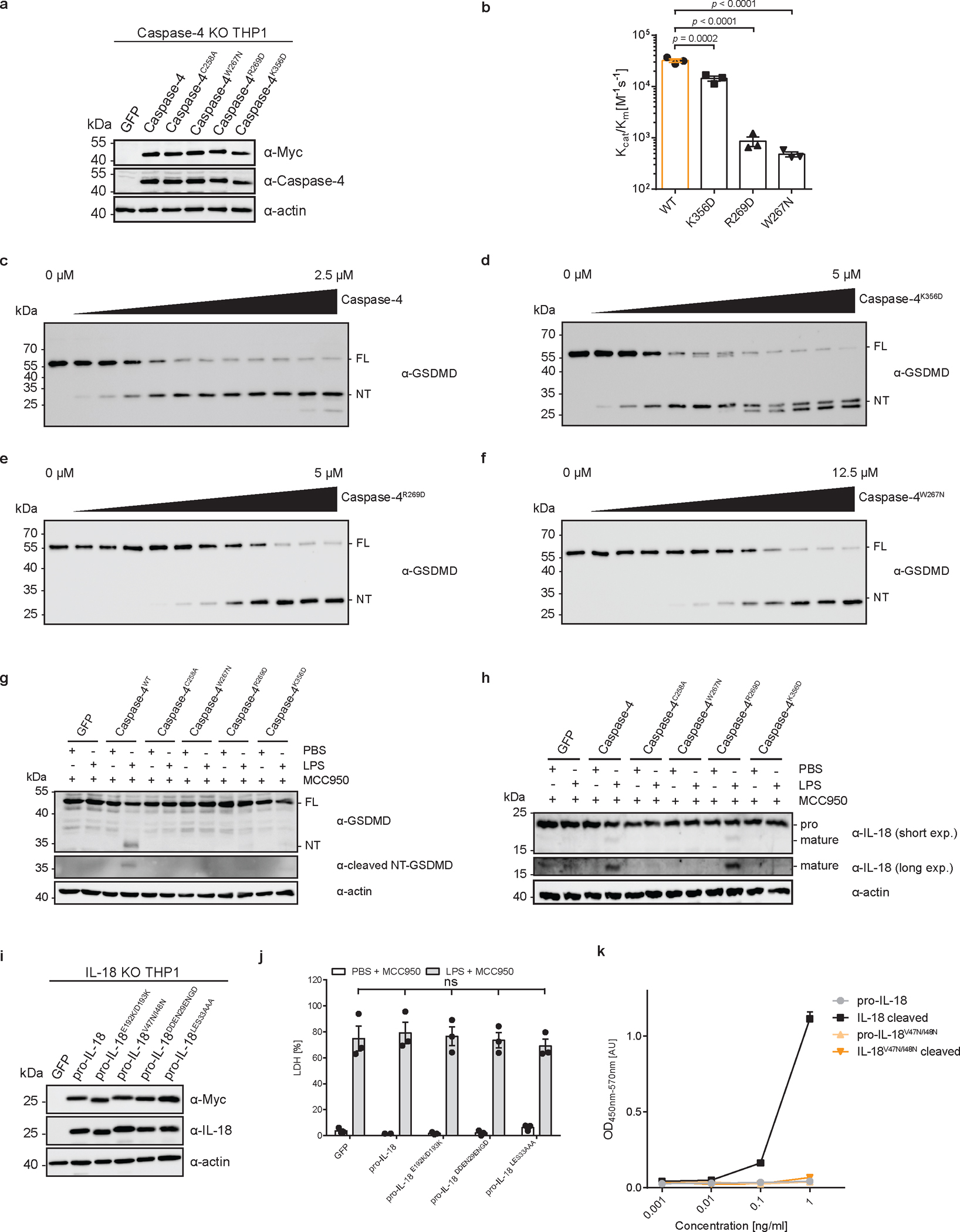
a, Expression of indicated caspase-4 mutants in caspase-4-deficient THP1 cells (representative of two independent repeats). b, In vitro cleavage of human GSDMD by caspase-4 mutants. c–f, Immunoblots showing in vitro cleavage of pro-IL-18 by caspase-4 mutants. Immunoblots are representative of three independent repeats. g, h, Caspase-4-deficient THP1 macrophages expressing caspase-4 mutants were primed with LPS and electroporated with LPS (or PBS). Processing of GSDMD and pro-IL-18 was analyzed by immunoblot. Immunoblots are representative of three biological replicates. i, Immunoblot showing expression of indicated pro-IL-18 mutants in IL-18-deficient THP1 cells (representative of two independent repeats). j, IL-18-deficient THP1 cells expressing pro-IL-18 mutants were electroporated with LPS (or PBS) in the presence of MCC950 and release of LDH into cell culture supernatant was quantified after 2 h. k, ELISA analysis demonstrating that pro-IL-18V47N/I48N is not recognized by the used ELISA reagent. ELISA results are displayed as mean ± SD of two technical replicates and are representative of two independent biological replicates. Bars and error bars in b, j represent mean ± SEM of three biological replicates. Each data point represents result of one independent experiment. Statistical significance was determined by two-way ANOVA with Tukey’s multiple comparisons test: ns = not significant (p > 0.05). For gel source data, see Supplementary Figure 1.
Extended Data Fig. 10 |. Mutagenesis of murine caspase-11 based on structural and sequence alignments.
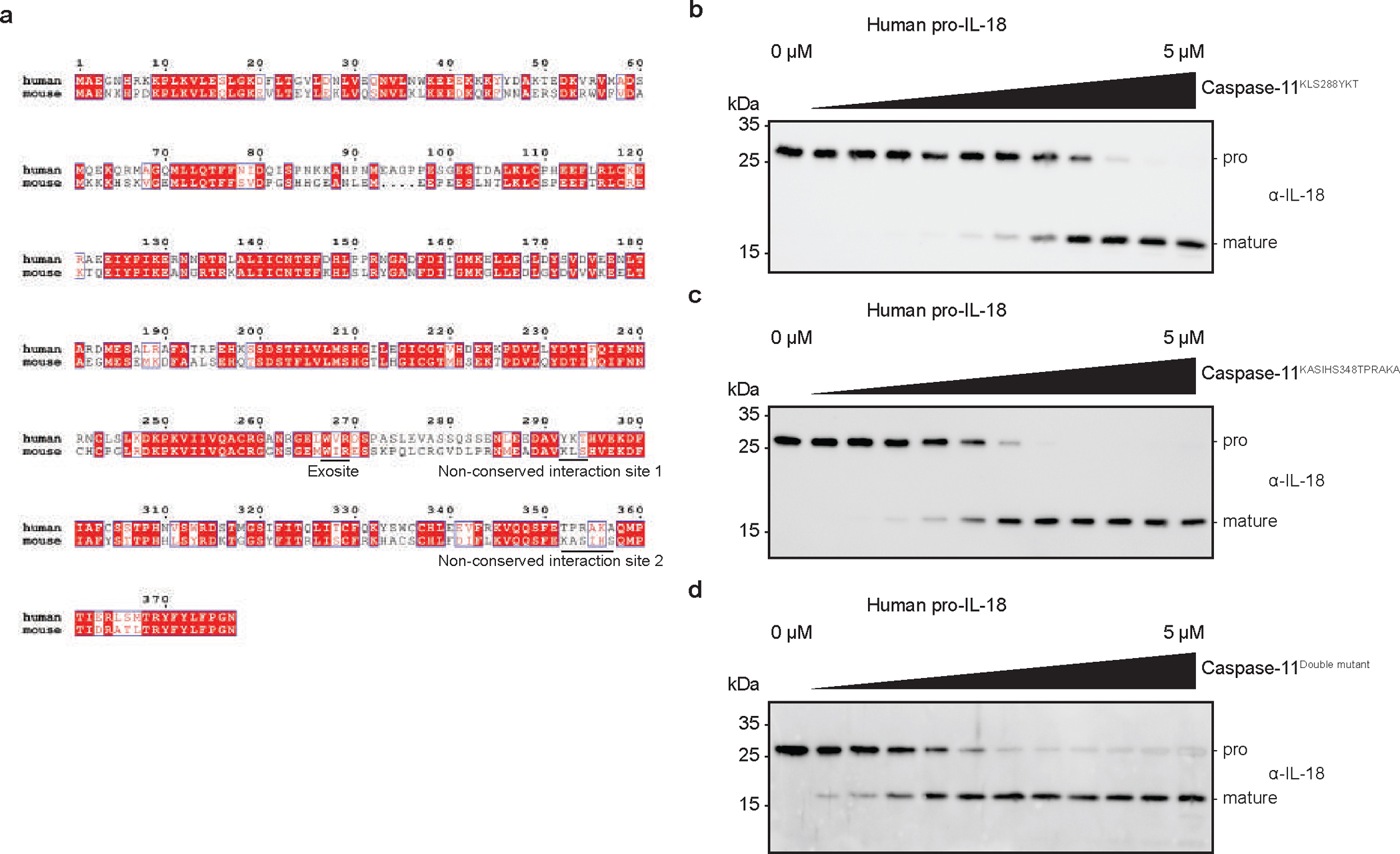
a, Sequence alignment of human caspase-4 and murine caspase-11. Identical and similar amino acids are highlighted in red or white boxes, respectively. Sequences were aligned using ClustalOmega and plotted in ESPript 3.0. b, c, d, Immunoblots showing in vitro cleavage of human pro-IL-18 by caspase-11 mutants. Immunoblots are representative of three biological replicates. For gel source data, see Supplementary Figure 1.
Extended Data Table 1.
Data Collection, data processing and validation statistics
| Data Collection and Processing | |
|---|---|
|
| |
| Microscope | Titan Krios |
| Voltage (keV) | 300 |
| Camera | K3 |
| Magnification | 105,000 |
| Pixel size at detector (Å/pixel) | 0.83 |
| Total electron exposure (e−/Å2) | 60.8 |
| Exposure rate (e−/pixel/sec) | 28.6 |
| Number of frames collected during exposure | 50 |
| Defocus range (μm) | −1.0 to −2.5 |
| Automation software | SerialEM 3.8 |
| Energy filter slit width (eV) | 20 |
| Micrographs collected (no.) | 5,934 |
| Micrographs used (no.) | 28,309 |
| Total extracted particles (no.) | 10,802,676 |
| Refinement | |
| Refined particles (no.) / Final particles (no.) | 1,234,038/229,147 |
| Symmetry parameters | C2 |
| Map resolution (Å) | 3.2 |
| FSC 0.143 (unmasked / masked) | 4.0/3.2 |
| Resolution range (Å) | 2.9 to 11.1 |
| Resolution range due to anisotropy (Å) | 3.3 to 3.7 |
| Map sharpening B factor range (Å2) | −144.2 |
| Map sharpening methods | LocalDeblur |
| Model composition | |
| Chains | 6 |
| Protein residues | 816 |
| Validation | |
| Model-Map scores | |
| CC (correlation coefficients) | 0.77 |
| Average FSC (0 / 0.143 / 0.5) | 2.2/3.3/3.5 |
| R.m.s. deviations from ideal values | |
| Bond lengths (Å) | 0.004 |
| Bond angles (°) | 0.787 |
| MolProbity score | 1.85 |
| CaBLAM outliers | 3.02 |
| Clashscore | 6.81 |
| Poor rotamers (%) | 0.26 |
| C-beta outliers (%) | 0.00 |
| Ramachandran plot | |
| Favoured (%) | 92.36 |
| Allowed (%) | 7.64 |
| Outliers (%) | 0.00 |
Supplementary Material
Acknowledgments
We thank Daniel Okin for providing primary cells and other members of the Kagan and Wu labs for helpful discussions. We thank T. Sam Xiao for the caspase-1 expression plasmid and Igor Brodsky for the Salmonella strain. We thank R. Walsh, S. Sterling. M. Mayer and S. Rawson at the Harvard Cryo-EM Center for Structural Biology for cryo-EM grid screening and data collection and Dr. K. Song at the University of Massachusetts cryo-EM Core for cryo-EM grids screening and dataset collection. We thank SBGrid for software and computing support. This work was supported by NIH grants AI67993, AI116550 and P30DK34854 (to J.C.K), AI124491 and AI139914 (to H.W.), a PhD fellowship by the Boehringer Ingelheim Fonds (to P.D.), and a postdoctoral fellowship from the Charles A. King Trust (to Y.D.). This paper is dedicated to the International Union of Crystallography on the occasion of its 75th anniversary.
Footnotes
Competing interests: J.C.K consults and holds equity in Corner Therapeutics, Larkspur Biosciences and Neumora Therapeutics. H.W. is a co-founder and chair of the Scientific Advisory Board of Ventus Therapeutics. None of these relationships influenced this study. The authors declare no other competing interests.
Data and materials availability. All data and materials reported in the main and supplementary data are available upon request. Raw data needed to recreate plots and uncropped gel images are accessible in the supplementary material of this paper. The electron density maps of caspase-4/pro-IL-18 at 3.2 Å have been deposited in the Electron Microscopy Data Bank (EMDB) with accession codes EMD-40678. The atomic coordinates for caspase-4/pro-IL-18 have been deposited in the Protein Data Bank with the accession code of 8SPB. Atomic coordinates for mature IL-18 and caspase-11 were downloaded from the Protein Data Bank under the accession code 3WO2 and 6KN1, respectively.
References
- 1.Dinarello CA Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 281, 8–27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bateman G, Hill B, Knight R & Boucher D Great balls of fire: activation and signalling of inflammatory caspases. Biochem. Soc. Trans. 49, 1311–1324 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chan AH & Schroder K Inflammasome signaling and regulation of interleukin-1 family cytokines. J. Exp. Med. 217, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 6.Julien O & Wells JA Caspases and their substrates. Cell Death Differ. 24, 1380–1389 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang K et al. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 180, 941–955.e20 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Liu Z et al. Caspase-1 Engages Full-Length Gasdermin D through Two Distinct Interfaces That Mediate Caspase Recruitment and Substrate Cleavage. Immunity 53, 106–114.e5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi J, Gao W & Shao F Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 42, 245–254 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Evavold CL et al. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heilig R et al. The Gasdermin-D pore acts as a conduit for IL-1β secretion in mice. Eur. J. Immunol. 48, 584–592 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Xia S et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593, 607–611 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barnett KC, Li S, Liang K & Ting JP-YA 360° view of the inflammasome: Mechanisms of activation, cell death, and diseases. Cell 186, 2288–2312 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kagan JC, Magupalli VG & Wu H SMOCs: supramolecular organizing centres that control innate immunity. Nat. Rev. Immunol. 14, 821–826 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thornberry NA et al. A novel heterodimeric cysteine protease is required for interleukin-1βprocessing in monocytes. Nature 356, 768–774 (1992). [DOI] [PubMed] [Google Scholar]
- 16.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Devant P, Cao A & Kagan JC Evolution-inspired redesign of the LPS receptor caspase-4 into an interleukin-1β–converting enzyme. Sci. Immunol. 6, eabh3567 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bibo-Verdugo B, Snipas SJ, Kolt S, Poreba M & Salvesen GS Extended subsite profiling of the pyroptosis effector protein gasdermin D reveals a region recognized by inflammatory caspase-11. J. Biol. Chem. jbc.RA120.014259 (2020) doi: 10.1074/jbc.RA120.014259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faucheu C et al. A novel human protease similar to the interleukin-1 beta converting enzyme induces apoptosis in transfected cells. EMBO J. 14, 1914–22 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kamens J et al. Identification and Characterization of ICH-2, a Novel Member of the Interleukin-1β-converting Enzyme Family of Cysteine Proteases. J. Biol. Chem. 270, 15250–15256 (1995). [DOI] [PubMed] [Google Scholar]
- 21.Shi J et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Rühl S & Broz P Caspase-11 activates a canonical NLRP3 inflammasome by promoting K + efflux. Eur. J. Immunol. 45, 2927–2936 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Baker PJ et al. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur. J. Immunol. 45, 2918–2926 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Kayagaki N et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Wandel MP et al. Guanylate-binding proteins convert cytosolic bacteria into caspase-4 signaling platforms. Nat. Immunol. (2020) doi: 10.1038/s41590-020-0697-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knodler LA et al. Noncanonical Inflammasome Activation of Caspase-4/Caspase-11 Mediates Epithelial Defenses against Enteric Bacterial Pathogens. Cell Host Microbe 16, 249–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devant P & Kagan JC Protocol to purify recombinant inflammatory caspases and assess their catalytic activity in vitro. STAR Protoc. 3, 101848 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holly MK et al. Salmonella enterica Infection of Murine and Human Enteroid-Derived Monolayers Elicits Differential Activation of Epithelium-Intrinsic Inflammasomes. Infect. Immun. 88, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Naseer N et al. Salmonella enterica Serovar Typhimurium Induces NAIP/NLRC4- and NLRP3/ASC-Independent, Caspase-4-Dependent Inflammasome Activation in Human Intestinal Epithelial Cells. Infect. Immun. 90, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobayashi T et al. The Shigella OspC3 Effector Inhibits Caspase-4, Antagonizes Inflammatory Cell Death, and Promotes Epithelial Infection. Cell Host Microbe 13, 570–583 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Gritsenko A et al. Priming Is Dispensable for NLRP3 Inflammasome Activation in Human Monocytes In Vitro. Front. Immunol. 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Coll RC et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 21, 248–55 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reyes Ruiz VM et al. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc. Natl. Acad. Sci. 114, 13242–13247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsutsumi N et al. The structural basis for receptor recognition of human interleukin-18. Nat. Commun. 5, 5340 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krissinel E & Henrick K Inference of Macromolecular Assemblies from Crystalline State. J. Mol. Biol. 372, 774–797 (2007). [DOI] [PubMed] [Google Scholar]
- 36.Jumper J et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods references
- 37.Evavold CL et al. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 184, 4495–4511.e19 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roschitzki-Voser H et al. Human caspases in vitro: Expression, purification and kinetic characterization. Protein Expr. Purif. 84, 236–246 (2012). [DOI] [PubMed] [Google Scholar]
- 39.Hu JJ et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat. Immunol. 21, 736–745 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mastronarde DN Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 41.Morin A et al. Collaboration gets the most out of software. Elife 2, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zheng SQ et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Punjani A, Rubinstein JL, Fleet DJ & Brubaker MA cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Klaholz BP Deriving and refining atomic models in crystallography and cryo-EM: the latest Phenix tools to facilitate structure analysis. Acta Crystallogr. Sect. D Struct. Biol. 75, 878–881 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 66, 213–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goddard TD et al. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mirdita M et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mintseris J & Gygi SP High-density chemical cross-linking for modeling protein interactions. Proc. Natl. Acad. Sci. 117, 93–102 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wynosky-Dolfi MA et al. Oxidative metabolism enables Salmonella evasion of the NLRP3 inflammasome. J. Exp. Med. 211, 653–668 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.