

ABSTRACT
The isolation and selection of yeast strains to improve the quality of the cachaça—Brazilian Spirit—have been studied in our research group. Our strategy considers Saccharomyces cerevisiae as the predominant species involved in sugarcane juice fermentation and the presence of different stressors (osmolarity, temperature, ethanol content, and competition with other microorganisms). It also considers producing balanced concentrations of volatile compounds (higher alcohols and acetate and/or ethyl esters), flocculation capacity, and ethanol production. Since the genetic bases behind these traits of interest are not fully established, the whole genome sequencing of 11 different Saccharomyces cerevisiae strains isolated and selected from different places was analyzed to identify the presence of a specific genetic variation common to cachaça yeast strains. We have identified 20,128 single-nucleotide variants shared by all genomes. Of these shared variants, 37 were new variants (being six missenses), and 4,451 were identified as missenses. We performed a detailed functional annotation (using enrichment analysis, protein–protein interaction network analysis, and database and in-depth literature searches) of these new and missense variants. Many genes carrying these variations were involved in the phenotypes of flocculation, tolerance to fermentative stresses, and production of volatile compounds and ethanol. These results demonstrate the existence of a genetic profile shared by the 11 strains under study that could be associated with the applied selective strategy. Thus, this study points out genes and variants that may be used as molecular markers for selecting strains well suited to the fermentation process, including genetic improvement by genome editing, ultimately producing high-quality beverages and adding value.
IMPORTANCE
This work demonstrates the existence of new genetic markers related to different phenotypes used to select yeast strains and mutations in genes directly involved in producing flavoring compounds and ethanol, and others related to flocculation and stress resistance.
KEYWORDS: comparative genomics, biotechnology for the fermentation industry, Saccharomyces cerevisiae, SNVs
INTRODUCTION
Cachaça is the denomination of Brazilian spirit produced from distilling fermented sugarcane juice with 38%–48% (vol/vol) alcohol content at 20°C (1). Moreover, 1.3 billion liters are produced annually, making cachaça the third distilled alcoholic beverage worldwide after vodka and soju. Generally, sugarcane juice fermentation in cachaça distilleries is performed in open systems without temperature, pH, or microbial contamination controls, creating a unique environment with high inter-microorganism competition. However, it has been shown that Saccharomyces cerevisiae is the predominant species (2). Since cachaça production is a fed-batch open-fermentation process that takes place during long periods (up to 6 months, corresponding to the sugarcane harvest), fermentative yeast populations are constantly being changed by new strains from the sugarcane juice and the non-sterile production conditions (3–5). Accordingly, they must compete with many other types of yeast and bacteria, including high environmental temperature fluctuations, low aw (water activity) generated by sugarcane juice at 20% sucrose (18–20° Brix), and increasing ethanol levels during each fermentation cycle.
Considering the favorable traits described above, the first selective strategy consisted using inoculum samples from different distilleries, aiming to isolate Saccharomyces cerevisiae strains with the following characteristics: (i) higher resistance to different types of stress (temperatures up to 37°C; osmolarity up to 20% sucrose; and alcohol content up to 12–15%); (ii) resistance to drugs, such as to 5, 5′, 5″ trifluoro-D-leucine and cerulenin to test for the potential production of the flavoring compounds isoamyl acetate and ethyl caproate, respectively (6, 7); (iii) ability to flocculate since this trait facilitates separation of yeast cells from the fermented must at the end of fermentation, thereby facilitating the distillation process; and (iv) higher capacity to produce ethanol (8–11). The selected strains with a combination of traits were further examined, and their applications in different biotechnological fields, such as producing other beverages (beer), bread, and bioethanol, were evaluated. Our studies revealed that Saccharomyces cerevisiae isolated and selected from cachaça fermentation vats presented additional characteristics such as the capacity to ferment maltose or to be resistant against toxic compounds present in different hydrolyzed biomass that potentially allow their commercial utilization in such applications (12–14).
Meanwhile, through different molecular tools, previous studies demonstrated the possibility of using selected cachaça yeast strain polymorphisms to obtain a protected denomination of origin for different distilleries and/or regions (15, 16). Besides, other studies have demonstrated the coexistence of different types of strains in cachaça fermentations: (i) wine strains, exhibiting alleles related or identical to those present in European wine strains; (ii) native strains, containing alleles similar to those found in strains isolated from traditional fermentations from Latin America and other regions of the world; and (iii) intraspecific hybrids or “mestizo” strains, heterozygous for both types of alleles (17). Furthermore, it has also been suggested that cachaça yeast strains seem closer to wild yeast populations found in North America and Japan. Moreover, despite the wine genotype penetrating the wild Brazilian population (suggesting the impact of domesticated microbe lineages on the genetic structure of wild populations), hybridization events with an American population of Saccharomyces paradoxus led to gene enrichment in encoding secondary active transmembrane transporters. Such hybridization events facilitated the habitat transition accompanying the colonization of the tropical ecosystem (18).
More recently, wine yeasts were demonstrated to constitute the main genetic source of cachaça yeast strains, and the multiple additional contributions originating from other domesticated populations, including native wild strains or the closer species, such as Saccharomyces paradoxus, contribute to shaping the unique genomes of cachaça yeast strains (19). Interestingly, in a study on the domestication and divergence of Saccharomyces cerevisiae beer yeasts (20), the sequencing of a set of 450 isolates of Saccharomyces cerevisiae allowed to construct a larger phylogeny based on nine genomic regions. It was observed that the evolutionary divergence of industrial yeasts is shaped by their industrial application and geographical origin. A clear separation between yeast strains used for industrial purposes and the wild-type or clinical strains previously sequenced became evident.
Moreover, it has been claimed that a great diversity of yeast cells can be applied in the beer industry to improve the quality of the products by taking advantage of the yeasts’ evolutionary history and biology (20). More recently, it has been shown that correlation maps between genotypes and relevant brewing phenotypes could further improve the search for novel craft beer starter yeasts (21). On the other hand, similar strategies have been used to select new yeast strains, considering phenotypic profiles and using data mining approaches to predict the potential of different yeast stains for winemaking (22). Continuing this line of work, it was demonstrated that genomic sequencing can be considered essential for understanding the individual variety of yeast strains as well as for studying mechanisms that explain relationships between genotype and genotype. However, for this purpose, only the sequencing of 11 polymorphic microsatellites was used (23).
Considering all these information, we decided to analyze the genome of 11 different cachaça yeast strains, selected by a unique protocol, to see the possibility of discovering formal signatures to confirm the origin of these strains and to validate the selective procedure following the expected genome constitution. Using different bioinformatics approaches, we demonstrated the existence of new variants occurring in genes related to different phenotypes used to select yeast strains, but also missense mutations in genes directly involved in producing flavoring compounds, flocculation, stress resistance, and ethanol.
MATERIALS AND METHODS
Yeast strains, culturing, and DNA extraction
S. cerevisiae strains belonging to the collection of the Laboratory of Cellular and Molecular Biology of the Pharmacy Department/School of Pharmacy of the Federal University of Ouro Preto were used. Such strains were isolated from different cachaça-producing units existing in the states of Minas Gerais, Bahia, Rio de Janeiro, and Espírito Santo and selected according to a previously established methodology (10, 13, 16), which includes producing volatile compounds since the presence of important flavors in fermented beverages must be considered (21, 24). Considering that the strains were selected using the same methodology, the choice of the 11 yeast strains in the collection that had their genome sequenced occurred following a proportionality criterion, considering the provenance of all existing strains in each of the Brazilian states mentioned.
Yeasts were cultivated for 24 h at 30°C in YP medium [yeast extract 1% (wt/vol) and peptone 2% (wt/vol)] plus 2% glucose, then subsequently collected by centrifugation at 4,000 rpm for 5 min. The pellet was resuspended in DNA extraction buffer (200 mM Tris HCl, pH 8, 250 mM NaCl, 25 mM EDTA, 0.5% SDS) and transferred to 2.0 mL microcentrifuge tubes, previously prepared with glass beads. The cells were vortexed for 10 min and transferred to new microcentrifuge tubes; then, an identical volume of phenol:chloroform solution (1:1) was added and vortexed again for 10 min. The mixture was centrifuged at 14,000 rpm for 20 min. The supernatants were transferred to 1.5 mL microcentrifuge tubes, 600 µL of isopropanol was added, and then centrifuged for another 5 min at 14,000 rpm. The supernatant was discarded, and the DNA was precipitated with 70% ethanol. Following another centrifugation at 14,000 rpm for 2 min, the supernatant was discarded, and each DNA pellet was resuspended in 50 µL of TE buffer with 1 µL RNase (10 mg/mL) and kept at 37°C for 1 h. The DNA was subsequently purified using the PowerClean DNA Clean-UP kit (MO Bio). For constructing the libraries, the Nextera DNA Library Preparation Kit (Illumina Inc., USA) was used. Paired-end sequencing (2 × 100 bp) was performed using the HiSeq2500 platform (Illumina Inc., USA). DNA extraction and genome sequencing were performed at the Brazilian Biorenewables National Laboratory (LNBR/CNPEM) in Campinas—São Paulo.
Genomic sequencing, detection, and functional annotation of the variants
The quality of the reads was checked using the FastQC software (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were filtered using the Prinseq software (25) and used to discard the bases with a Phred quality index below Q30 and the sequences with less than 50 bp size using the sliding window of size 5. The Bowtie2 software (26) was used to align the filtered reads against the genome of the laboratory strain S. cerevisiae S288c (version S288c_reference_genome_R64-2-1_20150113) available in the SGD (Saccharomyces Genome Database, http://sgd-archive.yeastgenome.org/sequence/ S288C_reference/genome_releases/). The statistical analyses for each file obtained after mapping, including in-depth and in-extension coverage analyses, were performed using the SAMtools package (27). Genomic variants were identified using SAMtools mpileup (27) and validated using the GATK package (28). The identified single-nucleotide variants (SNVs) and InDels were filtered according to the criteria previously established (29). To identify the shared variants, the 11 files containing information about the variants (Variant Call Format, VCF) were compared to pairs using the VCFtools package (30). The file containing the genomic variants shared among the 11 LBCM strains was used for the functional annotation of variants using the web interface of the Ensembl Variant Effect Predictor software (VEP—https://www.ensembl.org/ Tools/VEP) (31). In fact, the VEP tool determines the effect of the variants (SNPs, insertions, deletions, CNVs, or structural variants) on genes, transcripts, and protein sequences, as well as regulatory regions. The VEP tool uses the coordinates of the variants under investigation and the nucleotide changes to find out the known variants. Therefore, the new variants are those not yet found in other sequenced yeast genomes already available in databank. In addition, we informed that we use the default parameters from the online version of the VEP.
Detailed functional annotation of genes with shared new and missense SNVs
To verify if the SNVs shared by the 11 strains occurred in genes related to the selection process, possibly representing the specialization of these genomes, a detailed annotation strategy for genes carrying new and missense variants was applied. This comprised the sequence of (i) the gene(s) carrying new variants to enrichment analysis, protein–protein interaction network analysis, and in-depth literature search; and (ii) the genes carrying missense variants to enrichment analysis and in-depth literature search. The enrichment analysis was performed in the online tool YeastEnrichr (32, 33) using the Gene Ontology databases (34) and KEGG (Kyoto Encyclopedia of Genes and Genomes) (35), considering the terms that presented a P-value < 0.05. Protein–protein interactions (PPI) network analysis was performed using the STRING database (33) to integrate all known and predicted associations between proteins, including physical interactions and functional associations. Two proteins contributing to a specific cellular process were considered functionally associated (36).
To perform the in-depth literature search, a specific reference list was built containing (i) genes described in the literature as involved in each of the phenotypes under study; (ii) genes involved with the phenotypes described in the GUILDify database, a tool for prioritizing candidate genes, scoring the relevance of genes in relation to keywords (36); and (iii) genes involved with the phenotypes described in the SGD (37). To create this list, we performed a bibliographic survey of articles describing genes related to flocculation, secondary compound synthesis, resistance to different types of stress, and ethanol production published until August 2021. The search terms used were thermotolerance, stress, ethanol stress, osmotic stress, flocculation, and ethanol production. To search for overlaps between the genes of the reference list containing the candidate genes and the genes carrying new and missense variants, in this study, the listcompare.py script (http://github.com/bioinfonupeb/sarcopenia) was used. This in-depth literature review strategy has been used in different approaches to understand gene functions in higher eukaryotic genome characterization as well as to understand the function of potentially functional variants (29, 38).
RESULTS AND DISCUSSION
Genomic sequencing of cachaça S. cerevisiae strains
Genomic DNAs from 11 S. cerevisiae strains were sequenced using the Illumina HiSeq2500 platform. The number of reads ranged from 10,256,698 (LBCM1037) to 34,929,508 (LBCM1076). Then, the reads were filtered and aligned against the genome of the laboratory strain S. cerevisiae S288c, achieving in-depth and in-extension coverages greater than 100 times and 98.5%, respectively (Supplementary Material, S1). However, in two cases (LBCM1050 and LBCM1106), although the amount of reads used for assembly was less than 90%, we were able to cover “in extension” 99.1% (LBCM1050) and 99.1% (LBCM1106), with confidence in depth coverage of 194.4 times (LBCM1050) and 182.5 times (LBCM1106). Particularly, all sequencing data are available at NCBI (NCBI Bioproject Accession ID: PRJNA906656).
For the 11 genomes under study, a total of 708,374 variants were identified, among which 677,259 are SNVs and 31,115 Insertions/Deletions (InDels). The identified variants were validated, resulting in a 0.03% reduction in the number of SNVs, including a 0.7% reduction in the number of InDels. After validation, the identified variants were filtered following the criteria previously established (29). After this step, the total number of variants was reduced to 699,014, with 669,878 SNVs and 29,136 InDels.
By comparing the variant files of each strain under study, identifying 20,128 variants shared by the LBCM strains was possible. Among these variants, 90% are in non-coding regions. Regarding variants in coding regions, about 66% are synonymous, and 33% are missense. Furthermore, among the shared variants, the annotation program classified 37 as “new variants” (variants that have not yet been identified in any S. cerevisiae SNVs database), of which 6 are missenses; 8 are synonymous; and 23 are localized in non-coding regions. All these variants were found in 37 different genes, and the average in-depth and in-extension coverage was greater than 52-fold and 92.9%, respectively. The chromosomal positions for each new variant, including information about the genes, are shown in Supplementary Material, S2.
It is important to emphasize that the chromosomal positions showing allele alterations relative to the reference, the laboratory S. cerevisiae S288c strain, were considered shared variants present in the 11 genomes under study. For the new shared variants, the analysis of the alleles identified in each of these positions for the 11 strains was also performed (Supplementary Material, S3). In 26 of the 37 positions analyzed, the alleles presented by the 11 strains were the same, differing only in the S. cerevisiae S288c strain. This is the case of missense variants identified in the YIL058W, TPK1, and SLD2 genes. Furthermore, for the other 11 positions, at least one different allele was identified among the 11 genomes under study, including missense variants identified in the NUP116, MSS11, and AGA1 genes. However, even in these cases, all the identified alleles differed from those of the S. cerevisiae S288c strain (Supplementary Material, S3). These results show that these strains share a unique genetic profile that the selection strategy could favor.
Moreover, many variants have already been identified throughout the genome of 1011 S. cerevisiae strains (39). These variants have been deposited in the ENSEMBL databases. The VEP tool, used here to annotate the variants shared by the 11 strains in this study, carried out a search in this database. Thus, the variants described as new in our article are those that have not yet been identified in any S. cerevisiae SNVs database already deposited. Furthermore, in the work of Peter and coworkers (39), a search for genes or variants associated with 35 stress conditions was also performed. Then, we compared the genes described in that work with the genes carrying new variants shown in our paper, and no overlap was observed. These results highlight the originality of our approach.
Although the genome sequences of other cachaça yeast strains are available in databases (18), the detected SNVs were not deposited in public databases, which makes the comparison with the SNVs described in this work difficult. In addition, it is noteworthy that the aim of our work was to verify whether the variants shared by the 11 genomes that present the same phenotypic profile may be associated with these phenotypes. All 11 strains in our study underwent the same selection process. On the other hand, Barbosa and coworkers published cachaça yeast strains’ genomes and the SNVs of cachaça yeast strains of the 28 strains, but in such work, the strains did not undergo the same screening (18). We do not know if they all share the same phenotype as those in our study, and, therefore, if we did a comparison with such strains, we could be mistakenly excluding variants that are associated with the phenotypes of interest.
Functional characterization of the new genetic variants of cachaça S. cerevisiae strains
To verify if the genetic profile shared by the 11 strains would be related to the selection process, the new variants shared by the 11 strains and identified for the first time in the selected cachaça yeast strains were annotated using a detailed strategy developed in this study (see more details about the pipeline in the subtitle: Detailed functional annotation of genes with shared new and missense SNVs). The detailed search in the literature allowed the construction of a reference list with 526 genes already described as involved with the phenotypes of interest, of which 35 are related to flocculation, 74 are involved in the production of volatile compounds, 420 are associated with tolerance to different types of stress, and 18 genes are involved in ethanol production (10, 40–51). Using the Python script called “listcompare.py,” a comparison was made between the created reference list (Supplementary Material S4) and the 37 genes carrying new genetic variants. PLB2, FLO5, AGA1, MSS11, HSP26, MTL1, TPK1, and KIN3 genes were identified in both lists, therefore suggesting their relationship with the phenotypes of interest. The new variants identified in the HSP26 and KIN3 genes are upstream variants. The new variants identified in the PLB2, FLO5, and MTL1 genes are synonymous, while those identified in the AGA1, MSS11, and TPK1 genes are missense.
The AGA1 gene composes a second group of genes of the FLO family and is induced by sexual interactions between yeasts from the expression of complementary cell surface glycoproteins, a- and α-agglutinin, which promote aggregation between cells (52). The MSS11 gene plays a central role in regulating flocculation and controlling the expression of FLO1 and FLO11 (53, 54). Finally, the TPK1 gene encodes for the catalytic subunit of cAMP-dependent protein kinase (PKA), which is very important under stress conditions since PKA is down-regulated, resulting in growth arrest, trehalose accumulation, and activation of the protective mechanism (40).
Therefore, in addition to the existence of a genetic profile shared by the cachaça yeast strains, as evidenced by the presence of new genetic variants in these genomes, the identification of variants in genes associated with flocculation and tolerance to fermentative stresses could indicate the relationship between the shared genetic profile and the phenotypes under study that have been used as selection criteria (8), supporting the hypothesis that the strategy used would favor the selection of strains with this genetic profile.
Enrichment analysis of gene groups with new variants
Gene Set Enrichment Analysis (GSEA), also called functional enrichment, is a tool used to identify classes of over-represented genes in a large set of genes associated with biological signatures, particular processes, and phenotypes (55). Therefore, the functions performed by the 37 genes related to the new variants, including the metabolic pathways or processes in which they act, were analyzed using the GSEA through the Enrichr program (32, 33).
Regarding the molecular functions corresponding to the 37 genes, six GO terms were enriched, considering the adjusted P-value lower than 0.05 (Table 1). The most significantly enriched term was DNA binding (adjusted P = 0.0045), which included five (SLD2, TOG1, GAT1, ECM23, and ACA1) of the 37 genes with new variants, followed by the sequence-specific DNA binding term, which included four genes (TOG1, GAT1, ECM23, and ACA1). GAT1, MSS11, and ACA1 genes were associated with three different GO terms enriched, all related to the transcriptional activity of RNA polymerase II. In addition, the protein kinase activity term was enriched, which included three genes with new variants (KIN3, TPK1, and PSK2).
TABLE 1.
The Gene Ontology (GO) enrichment analysis of the 37 genes carrying new variants
| GO term | P-value | Adjusted P-valuea | Genes |
|---|---|---|---|
| Molecular function | |||
| DNA-binding transcription activator activity, RNA polymerase II-specific (GO: 0001228) | 0.0001855 | 0.004531 | GAT1, MSS11, ACA1 |
| Proximal promoter DNA-binding transcription activator activity, RNA polymerase II-specific (GO: 0001077) | 0.0001764 | 0.004531 | GAT1, MSS11, ACA1 |
| Sequence-specific DNA binding (GO: 0043565) | 0.0002614 | 0.004531 | TOG1, GAT1, ECM23, ACA1 |
| DNA binding (GO: 0003677) | 0.0003691 | 0.004798 | SLD2, TOG1, GAT1, ECM23, ACA1 |
| DNA-binding transcription factor activity, RNA polymerase II-specific (GO: 0000982) | 0.0006744 | 0.007014 | GAT1, MSS11, ACA1 |
| Protein kinase activity (GO: 0004672) | 0.00206 | 0.01786 | KIN3, TPK1, PSK2 |
P-value adjusted by Fisher’s exact test for multiple tests.
Using the KEGG database (Kyoto Encyclopedia of Genes and Genomes), 18 metabolic pathways were associated with the analyzed genes. However, none of them had an adjusted P-value lower than 0.05, the considered limit for the existence of statistical correlation (Table 2). Although the result was not statistically significant, highlighting the enrichment of the mitogen-activated protein kinase (MAPK) signaling pathway is important since this pathway is associated with stress tolerance, and the “protein kinase activity” GO term was significantly enriched (56–58). The MTL1 and MSS11 genes are involved in two different processes controlled by the MAPK pathway. Mtl1p is involved in stress response mechanisms due to its role in maintaining cell wall integrity, while Mss11p acts as a central element in regulating invasive growth due to its role as a positive regulator of FLO11 gene transcription (53, 59).
TABLE 2.
The KEGG pathway enrichment analysis of the 37 genes carrying new variants
| Pathway | P-value | Adjusted P-valuea | Gene |
|---|---|---|---|
| Biosynthesis of ubiquinone and another terpenoid-quinone | 0.01288 | 0.1141 | COQ6 |
| Meiosis | 0.02407 | 0.1141 | TPK1, APC11 |
| MAPK signaling pathway | 0.01884 | 0.1141 | MTL1 e MSS11 |
| Fructose and mannose metabolism | 0.03816 | 0.1141 | FBP1 |
| Methane metabolism | 0.04526 | 0.1141 | FBP1 |
| Ribosome | 0.03939 | 0.1141 | RPS25A e RPP1A |
| Cysteine and methionine metabolism | 0.06801 | 0.1141 | UTR4 |
| Pentose phosphate pathway | 0.0488 | 0.1141 | FBP1 |
| Glycerophospholipid metabolism | 0.06973 | 0.1141 | PLB2 |
| Longevity regulation pathway | 0.06454 | 0.1141 | TPK1 |
| Ubiquitin-mediated proteolysis | 0.08344 | 0.1252 | APC11 |
| Peroxisome | 0.06973 | 0.1141 | PEX1 |
| Glycolysis/Gluconeogenesis | 0.09528 | 0.1319 | FBP1 |
| Autophagy | 0.1459 | 0.1595 | TPK1 |
| RNA transport | 0.1459 | 0.1595 | NUP116 |
| Processing of proteins in the endoplasmic reticulum | 0.1507 | 0.1595 | HSP26 |
| Cell cycle | 0.2087 | 0.2087 | APC11 |
| Aminoacyl-tRNA biosynthesis | 0.1279 | 0.1595 | SLM5, tQ(UUG)E1 |
P-value adjusted by Fisher's exact test for multiple tests.
Analysis of PPI network between genes with new variants
Among the analyses of interactions between genes, also called biomolecular networks, one of the most useful and comprehensive types is the protein–protein association network (60), which encompasses all protein-coding genes in each genome and highlights their functional associations. Since proteins can interact in various ways, any two proteins that jointly contribute to a specific cellular process are functionally associated (36). Thus, the PPI network analysis allows the functional annotation of certain genes, considering their interaction with other genes of known function. However, in one of the genes where new variants were identified, the tQ(UUG)E1 gene is identified as a non-protein-coding transfer RNA gene. Thus, it was impossible to identify interactions between this gene and the others. Figure 1 shows the initial result of the analysis of the interaction between the proteins encoded by the 36 genes carrying new SNVs. According to this type of analysis, and considering only the genes presenting missense variation (AGA1, MSS11, and TPK1), it was possible to observe interactions between the genes MSS11, AGA1, and FLO5 (genes associated with flocculation and cell adhesion phenotypes) and of the gene TPK1 (associated with tolerance to some of the different types of stress to which yeasts are exposed during the fermentation process) with the genes FBP1, ACA1, GAT1, and ECM23, which are involved in ethanol production in S. cerevisiae.
FIG 1.
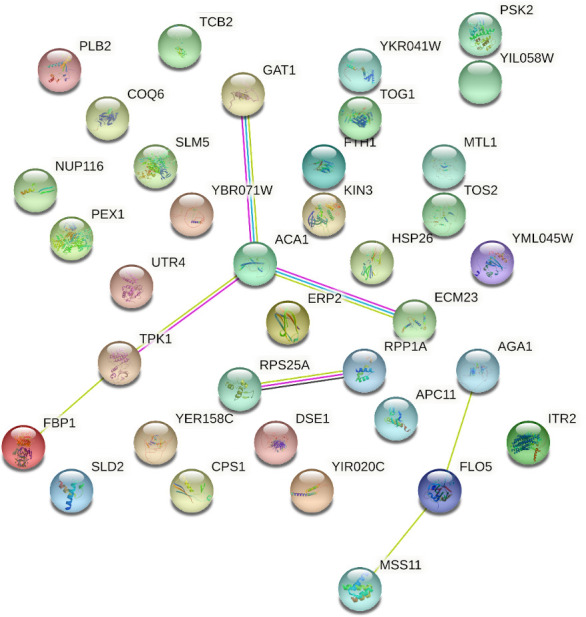
Interactome of the 36 genes carrying new SNVs shared among the 11 LBCM strains. The network shows, in each node, the prediction of functional linkage between the genes. The yellow, pink, blue, and black lines correspond to text mining, experiments supporting, databases, and co-expression of the relationship among the genes, respectively.
To make the interaction analysis more specific, the genes involved in flocculation, volatile compounds, and ethanol production, included in the reference list (see Supplementary Material, S4), were added to the list of the 36 genes carrying new SNVs. In addition to the FLO5, AGA1, and MSS11 genes involved in flocculation, among the 36 genes analyzed, another 13 showed interactions with genes related to flocculation: ACA1, DSE1, YER158C, GAT1, MTL1, COQ6, TPK1, FBP1, PLB2, TCB2, PSK2, ECM23, and TOG1 (Fig. 2). Regarding volatile compounds related to the aroma in fermented beverages, we evaluated the interaction between the 36 genes carrying new SNVs and the 74 genes from the reference list (Supplementary Material, S4), described in the literature to produce higher alcohols, acetate esters and medium-chain fatty acids, terpenoids, vicinal diketones, sulfur compounds, and phenolic compounds. Thirteen genes carrying new SNVs (HSP26, FTH1, APC11, UTR4, GAT1, MTL1, CPS1, TPK1, FBP1, ITR2, PSK2, ACA1, and PLB2) interact with 42 genes from the reference list related to the production of higher alcohols and esters (Fig. 3). In addition, eight genes carrying new SNVs (GAT1, HSP26, ACA1, COQ6, TPK1, SLD2, FBP1, and AGA1) interact with 32 genes from the reference list related to the synthesis of terpenoids, vicinal diketones, sulfur compounds, and phenolic compounds (Fig. 4). Regarding ethanol production, we observed that 11 genes with new SNVs (ECM23, GAT1, ACA1, TPK1, FBP1, YER158C, HSP26, FTH1, PEX1, ITR2, and SLD2) interact with genes already described to be involved in ethanol production in S. cerevisiae (Fig. 5).
FIG 2.

Interactome of the 36 genes carrying new SNVs shared between the 11 LBCM strains and the 35 reference list genes involved in flocculation in S. cerevisiae. The network shows, in each node, a gene predicted to have functional links with other genes involved in flocculation traits. In the figure, yellow, pink, light blue, black, red, blue, and green lines correspond, respectively, to text mining, experiments supporting, co-expression of the relationship among the genes, gene fusion, co-occurrence, databases, and neighboring genes.
FIG 3.

Interactome of the 16 genes carrying new SNVs shared between the 11 LBCM strains and the 42 reference list genes involved in producing higher alcohols and esters in S. cerevisiae. The network shows, in each node, a gene predicted to have functional links with other genes involved in producing higher alcohol and ester traits. In the figure, yellow, pink, light blue, black, red, blue, and green lines correspond, respectively, to text mining, experiments supporting, databases, co-expression of the relationship among the genes, gene fusion, co-occurrence, and neighboring genes.
FIG 4.

Interactome of the 16 genes carrying new SNVs shared between the 11 LBCM strains and the 32 reference list genes involved in producing terpenoids, vicinal diketones, sulfur compounds, and phenolic compounds in S. cerevisiae. The network shows, in each node, a gene predicted to have functional links with other genes involved in the production of terpenoids, vicinal diketones, sulfur compounds, and phenolic compounds traits. In the figure, yellow, pink, light blue, black, red, blue, and green lines correspond, respectively, to text mining, experiments supporting, databases, co-expression of the relationship among the genes, gene fusion, co-occurrence, and neighboring genes.
FIG 5.

Interactome of the 36 genes carrying new SNVs shared between the 11 LBCM strains and the 17 reference list genes involved in ethanol production in S. cerevisiae. The network shows, in each node, a gene predicted to have functional links with other genes involved in ethanol production traits. In the figure, yellow, pink, light blue, black, red, blue, and green lines correspond, respectively, to text mining, experiments supporting, databases, co-expression of the relationship among the genes, gene fusion, co-occurrence, and neighboring genes.
In this context, 25 of the 36 genes with new variants interact with genes involved in at least one of the phenotypes associated with the selection strategy, suggesting a possible relationship between these variants and the selection methodology used.
Functional characterization of the missense variants in genes of cachaça S. cerevisiae strains
Among the 20,128 SNVs shared by the genomes of the 11 LBCM strains, 4,451 are missenses located in 2,165 genes. To verify whether these mutations were occurring in genes involved in the phenotypes of flocculation, tolerance to fermentative stresses and volatile compounds, and ethanol production, a comparison was made between the reference list of genes related to the phenotypes of interest (see Supplementary Material, S4) and the list of 2,165 genes carrying missense variants. In fact, 181 of the 2,165 genes carrying missense variants had already been described as being involved with the phenotypes of interest, of which 13 genes were associated with the flocculation process, 151 were associated with tolerance to fermentative stresses, 23 participated in the production pathways of volatile compounds, and one gene related to ethanol production (Supplementary Material, S5). In addition, to know the molecular role of genes carrying missense variants and the metabolic pathways in which they are involved, an enrichment analysis was performed. Tables 3 and 4 present the statistically significant results for the molecular functions (Gene Ontology) and metabolic pathways (KEGG) related to these genes.
TABLE 3.
The Gene Ontology (GO) enrichment analysis of the genes carrying missense SNVs
| GO term | P-value | Adjusted P-valuea | Number of genes |
|---|---|---|---|
| Molecular function | |||
| DNA binding (GO: 0003677) | 4.902e-64 | 2.471e-61 | 158 |
| Sequence-specific DNA binding (GO: 0043565) | 1.381e-42 | 3.479e-40 | 90 |
| Kinase activity (GO: 0016301) | 2.225e-25 | 3.739e-23 | 68 |
| Phosphotransferase activity, alcohol group as acceptor (GO: 0016773) | 3.953e-24 | 4.981e-22 | 65 |
| Protein kinase activity (GO: 0004672) | 7.383e-24 | 7.442e-22 | 61 |
| Protein serine/threonine kinase activity (GO: 0004674) | 2.153e-22 | 1.808e-20 | 55 |
| RNA binding (GO: 0003723) | 1.316e-21 | 8.717e-20 | 125 |
| DNA-binding transcription factor activity, RNA polymerase II-specific (GO: 0000982) | 1.384e-21 | 8.717e-20 | 47 |
| mRNA binding (GO: 0003729) | 4.954e-21 | 2.774e-19 | 71 |
| RNA polymerase II transcription regulatory region sequence-specific DNA binding (GO: 0000977) | 4.228e-20 | 2.131e-18 | 48 |
P-value adjusted by Fisher’s exact test for multiple tests.
TABLE 4.
The KEGG pathway enrichment analysis of the genes carrying missense SNVs
| Pathway | P-value | Adjusted P-valuea | Number of genes |
|---|---|---|---|
| Cell cycle | 5.51e-15 | 5.234e-16 | 51 |
| MAPK signaling pathway | 1.51e-12 | 7.160e-14 | 45 |
| Meiosis | 8.82e-14 | 2.793e-12 | 46 |
| Endocytosis | 9.39e-13 | 2.229e-11 | 32 |
| Protein processing in the reticulum endoplasmic | 1.60e-12 | 3.039e-11 | 35 |
| RNA transport | 7.93e-11 | 1.255e-9 | 32 |
| Spliceosome | 7.22e-10 | 9.798e-9 | 28 |
| Autophagy | 1.88e-9 | 2.237e-8 | 30 |
| Ribosome biogenesis in eukaryotes | 3.41e-8 | 3.604e-7 | 26 |
| RNA degradation | 4.92e-8 | 4.676e-7 | 23 |
P-value adjusted by Fisher's exact test for multiple tests.
One of the enriched metabolic pathways was the MAPK signaling pathway (adjusted P-value = 7.160e – 14). Remarkably, 45 genes carrying missense SNVs identified in the present study play a role in this metabolic pathway (Table 4). Among these genes, SHO1, HKR1, STE20, STE50, SSK22, PTP2, and MSN2 can be highlighted as being involved in the osmotic stress response; MSS11 and FLO11 are associated with flocculation and invasive growth in the absence of glucose or other fermentable sugar; and RLM1 is involved in the cellular integrity pathway and flocculation in S. cerevisiae. It was already demonstrated that deleting the RLM1 gene caused a reduction in the flocculation of a naturally flocculating strain (50). Other pathways, such as fatty acid biosynthesis, amino acid biosynthesis, and glycolysis, were also enriched by genes in which missense variants were identified.
It is notable that among the genes that presented new and missense mutations, TPK1, AGA1, and MSS11 genes show a profile of interaction with other genes also involved in the production of ethanol and flavoring compounds, and others also related to flocculation or invasive growth. Therefore, from these data, it is possible to speculate on the possibility that the new missense variants found in these genes could contribute to the display of a broader set of phenotypes considered important in yeasts, making them more appropriate for cachaça production. These results point to the existence of a genetic profile shared by the strains of the LBCM collection and demonstrate that this profile may be associated with the methodology used for the selection of these strains, since the shared variants are new.
Conclusions
In the present study, the whole genomes of 11 different strains of Saccharomyces cerevisiae found in the fermentative process of cachaça, the Brazilian Spirit, were analyzed to verify if there is a genetic profile shared by strains subjected to rigorous selection processes for producing industrial fermented beverages.
More than 20,000 SNVs were found to be shared by the 11 strains. Of these, 37 were new variants, and 4,451 were missense variants. After detailed functional annotation, many of the new and missense shared variants were found in genes involved with the response to different types of stress, the production of volatile compounds, flocculation ability, and ethanol production. Results show a genetic profile shared by these 11 strains from the fermentative process of cachaça, the Brazilian Spirit, comprising new and missense variants in genes involved with those phenotypes. Therefore, the results point to the possibility of using this shared profile to develop molecular markers for selecting strains well suited for the fermentation process, including for genetic improvement by genome editing in pursuing higher quality beverages and added value.
ACKNOWLEDGMENTS
This work was supported by grants from FAPEMIG (Process APQ-01625–16) and CNPq (Process 305183/2021–4 – Research fellowship to RLB; Process 315302/2018–6).
We thank CNPEM and LNBR for helpful assistance with yeast genome sequencing. We are grateful to the Bioinformatics Platform (RPT04B) of the Instituto René Rachou FIOCRUZ MINAS, Brazil, and the Multi-user Bioinformatics Laboratory at the Federal University of Ouro Preto, Brazil, for providing the servers and support in the bioinformatics analyses.
Contributor Information
Rogelio Lopes Brandão, Email: rlbrand@ufop.edu.br.
Yvonne Nygård, Chalmers University of Technology, Gothenburg, Sweden.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/aem.01759-23.
Summary of the results of the sequencing and mapping of the genomes of the 11 strains.
Sequencing coverage.
Alleles observed in the 11 LBCM strains related to the new variants in relation to S. cerevisiae strain S288c.
Reference list of genes involved in the production of flavoring compounds; flocculation; stress resistance and ethanol production.
Genes carrying missense variants shared by the 11 strains involved with the phenotypes of interest.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Brazil . 2004. Lei 10958/04. Republic Pot, Brasilia. [Google Scholar]
- 2. Freitas Schwan R, Mendonça AT, da Silva Jr. JJ, Rodrigues V, Wheals AE. 2001. Microbiology and physiology of Cachaca (Aguardente) fermentations. Antonie Van Leeuwenhoek 79:89–96. doi: 10.1023/A:1010225117654 [DOI] [PubMed] [Google Scholar]
- 3. Guerra JB, Araújo RA, Pataro C, Franco GR, Moreira ES, Mendonça-Hagler LC, Rosa CA. 2001. Genetic diversity of Saccharomyces cerevisiae strains during the 24 h fermentative cycle for the production of the artisanal Brazilian cachaça. Lett Appl Microbiol 33:106–111. doi: 10.1046/j.1472-765x.2001.00959.x [DOI] [PubMed] [Google Scholar]
- 4. Pataro C, Guerra JB, Petrillo-Peixoto ML, Mendonça-Hagler LC, Linardi VR, Rosa CA. 2000. Yeast communities and genetic polymorphism of Saccharomyces cerevisiae strains associated with artisanal fermentation in Brazil. J Appl Microbiol 89:24–31. doi: 10.1046/j.1365-2672.2000.01092.x [DOI] [PubMed] [Google Scholar]
- 5. Morais PB. 1997. Characterization and succession of yeast populations associated with spontaneous fermentations during the production of Brazilian sugar-cane aguardente, p 241–243. Spring-Verlag, London. [Google Scholar]
- 6. Bussey H, Umbarger HE. 1970. Biosynthesis of the branched-chain amino acids in yeast: a leucine-binding component and regulation of leucine uptake. J Bacteriol 103:277–285. doi: 10.1128/jb.103.2.277-285.1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ichikawa E, Hosokawa N, Hata Y, Abe Y, Suginami K, Imayasu S. 1991. Breeding of a sake yeast with improved ethyl caproate productivity. Agric Biol Chem 55:2153–2154. doi: 10.1080/00021369.1991.10870932 [DOI] [Google Scholar]
- 8. Vicente M de A, Fietto LG, Castro I de M, dos Santos ANG, Coutrim MX, Brandão RL. 2006. Isolation of Saccharomyces cerevisiae strains producing higher levels of flavoring compounds for production of "cachaça" the Brazilian sugarcane spirit. Int J Food Microbiol 108:51–59. doi: 10.1016/j.ijfoodmicro.2005.10.018 [DOI] [PubMed] [Google Scholar]
- 9. de Souza PP, Cardeal Z de L, Augusti R, Morrison P, Marriott PJ. 2009. Determination of volatile compounds in Brazilian distilled cachaca by using comprehensive two-dimensional gas chromatography and effects of production pathways. J Chromatogr A 1216:2881–2890. doi: 10.1016/j.chroma.2008.10.061 [DOI] [PubMed] [Google Scholar]
- 10. Alvarez F, Correa LF da M, Araújo TM, Mota BEF, da Conceição LEFR, Castro I de M, Brandão RL. 2014. Variable flocculation profiles of yeast strains isolated from cachaça distilleries. Int J Food Microbiol 190:97–104. doi: 10.1016/j.ijfoodmicro.2014.08.024 [DOI] [PubMed] [Google Scholar]
- 11. de Souza APG, Vicente M de A, Klein RC, Fietto LG, Coutrim MX, de Cássia Franco Afonso RJ, Araújo LD, da Silva PHA, Bouillet LEM, Castro IM, Brandão RL. 2012. Strategies to select yeast starters cultures for production of flavor compounds in cachaça fermentations. Antonie Van Leeuwenhoek 101:379–392. doi: 10.1007/s10482-011-9643-5 [DOI] [PubMed] [Google Scholar]
- 12. da Conceição LEFR, Saraiva MAF, Diniz RHS, Oliveira J, Barbosa GD, Alvarez F, Correa LF da M, Mezadri H, Coutrim MX, Afonso RJ de CF, Lucas C, Castro IM, Brandão RL. 2015. Biotechnological potential of yeast isolates from cachaça: the Brazilian spirit. J Ind Microbiol Biotechnol 42:237–246. doi: 10.1007/s10295-014-1528-y [DOI] [PubMed] [Google Scholar]
- 13. Figueiredo BIC, Saraiva MAF, Souza Pimenta PP, Souza Testasicca MC, Sampaio GMS, Cunha AC, Afonso LCC, Queiroz M, Miranda Castro I, Brandão RL. 2017. New lager brewery strains obtained by crossing techniques using. Appl Environ Microbiol 83. doi: 10.1128/AEM.01582-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Araújo TM, Souza MT, Diniz RHS, Yamakawa CK, Soares LB, Lenczak JL, de Castro Oliveira JV, Goldman GH, Barbosa EA, Campos ACS, Castro IM, Brandão RL. 2018. Cachaça yeast strains: alternative starters to produce beer and bioethanol. Antonie Van Leeuwenhoek 111:1749–1766. doi: 10.1007/s10482-018-1063-3 [DOI] [PubMed] [Google Scholar]
- 15. Oliveira VA, Vicente MA, Fietto LG, Castro I de M, Coutrim MX, Schüller D, Alves H, Casal M, Santos J de O, Araújo LD, da Silva PHA, Brandão RL. 2008. Biochemical and molecular characterization of Saccharomyces cerevisiae strains obtained from sugar-cane juice fermentations and their impact in cachaça production. Appl Environ Microbiol 74:693–701. doi: 10.1128/AEM.01729-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Barbosa EA, Souza MT, Diniz RHS, Godoy-Santos F, Faria-Oliveira F, Correa LFM, Alvarez F, Coutrim MX, Afonso R, Castro IM, Brandão RL. 2016. Quality improvement and geographical indication of cachaça (Brazilian spirit) by using locally selected yeast strains. J Appl Microbiol 121:1038–1051. doi: 10.1111/jam.13216 [DOI] [PubMed] [Google Scholar]
- 17. Badotti F, Vilaça ST, Arias A, Rosa CA, Barrio E. 2014. Two interbreeding populations of Saccharomyces cerevisiae strains coexist in cachaça fermentations from Brazil. FEMS Yeast Res 14:289–301. doi: 10.1111/1567-1364.12108 [DOI] [PubMed] [Google Scholar]
- 18. Barbosa R, Almeida P, Safar SVB, Santos RO, Morais PB, Nielly-Thibault L, Leducq J-B, Landry CR, Gonçalves P, Rosa CA, Sampaio JP. 2016. Evidence of natural hybridization in Brazilian wild lineages of Saccharomyces cerevisiae. Genome Biol Evol 8:317–329. doi: 10.1093/gbe/evv263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Barbosa R, Pontes A, Santos RO, Montandon GG, de Ponzzes-Gomes CM, Morais PB, Gonçalves P, Rosa CA, Sampaio JP. 2018. Multiple rounds of artificial selection promote microbe secondary domestication-the case of cachaça yeasts. Genome Biol Evol 10:1939–1955. doi: 10.1093/gbe/evy132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gibson B, Geertman J-M, Hittinger CT, Krogerus K, Libkind D, Louis EJ, Magalhães F, Sampaio JP. 2017. New yeasts-new brews: modern approaches to brewing yeast design and development. FEMS Yeast Res 17. doi: 10.1093/femsyr/fox038 [DOI] [PubMed] [Google Scholar]
- 21. Saerens SMG, Delvaux FR, Verstrepen KJ, Thevelein JM. 2010. Production and biological function of volatile esters in Saccharomyces cerevisiae. Microb Biotechnol 3:165–177. doi: 10.1111/j.1751-7915.2009.00106.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mendes I, Franco-Duarte R, Umek L, Fonseca E, Drumonde-Neves J, Dequin S, Zupan B, Schuller D. 2013. Computational models for prediction of yeast strain potential for winemaking from phenotypic profiles. PLoS One 8:e66523. doi: 10.1371/journal.pone.0066523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Franco-Duarte R, Mendes I, Umek L, Drumonde-Neves J, Zupan B, Schuller D. 2014. Computational models reveal genotype-phenotype associations in Saccharomyces cerevisiae. Yeast 31:265–277. doi: 10.1002/yea.3016 [DOI] [PubMed] [Google Scholar]
- 24. Fleet GH. 2003. Yeast interactions and wine flavour. Int J Food Microbiol 86:11–22. doi: 10.1016/s0168-1605(03)00245-9 [DOI] [PubMed] [Google Scholar]
- 25. Schmieder R, Edwards R. 2011. Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 6:e17288. doi: 10.1371/journal.pone.0017288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, Subgroup GPDP. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. doi: 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rosse IC, Assis JG, Oliveira FS, Leite LR, Araujo F, Zerlotini A, Volpini A, Dominitini AJ, Lopes BC, Arbex WA, Machado MA, Peixoto MGCD, Verneque RS, Martins MF, Coimbra RS, Silva MVGB, Oliveira G, Carvalho MRS. 2017. Whole genome sequencing of Guzerá cattle reveals genetic variants in candidate genes for production, disease resistance, and heat tolerance. Mamm Genome 28:66–80. doi: 10.1007/s00335-016-9670-7 [DOI] [PubMed] [Google Scholar]
- 30. Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, 1000 Genomes Project Analysis Group . 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. doi: 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GRS, Thormann A, Flicek P, Cunningham F. 2016. The ensembl variant effect predictor. Genome Biol 17:122. doi: 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, Ma’ayan A. 2013. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14:128. doi: 10.1186/1471-2105-14-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, McDermott MG, Monteiro CD, Gundersen GW, Ma’ayan A. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44:W90–7. doi: 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene ontology: tool for the unification of biology. Nat Genet 25:25–29. doi: 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kanehisa M, Goto S. 2000. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30. doi: 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guala D, Ogris C, Müller N, Sonnhammer ELL. 2020. Genome-wide functional association networks: background, data & state-of-the-art resources. Brief Bioinform 21:1224–1237. doi: 10.1093/bib/bbz064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cherry JM, Hong EL, Amundsen C, Balakrishnan R, Binkley G, Chan ET, Christie KR, Costanzo MC, Dwight SS, Engel SR, Fisk DG, Hirschman JE, Hitz BC, Karra K, Krieger CJ, Miyasato SR, Nash RS, Park J, Skrzypek MS, Simison M, Weng S, Wong ED. 2012. Saccharomyces genome database: the genomics resource of budding yeast. Nucleic Acids Res 40:D700–D705. doi: 10.1093/nar/gkr1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Damasceno S, Fonseca PAS, Rosse IC, Moraes MFD, Oliveira JAC, Garcia-Cairasco N, Brunialti Godard AL. 2021. Putative causal variant on. Front Neurol 12:647859. doi: 10.3389/fneur.2021.647859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Peter J, De Chiara M, Friedrich A, Yue J-X, Pflieger D, Bergström A, Sigwalt A, Barre B, Freel K, Llored A, Cruaud C, Labadie K, Aury J-M, Istace B, Lebrigand K, Barbry P, Engelen S, Lemainque A, Wincker P, Liti G, Schacherer J. 2018. Genome evolution across 1,011 Saccharomyces cerevisiae isolates. Nature 556:339–344. doi: 10.1038/s41586-018-0030-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Estruch F. 2000. Stress-controlled transcription factors, stress-induced genes and stress tolerance in budding yeast. FEMS Microbiol Rev 24:469–486. doi: 10.1111/j.1574-6976.2000.tb00551.x [DOI] [PubMed] [Google Scholar]
- 41. Alexandre H, Ansanay-Galeote V, Dequin S, Blondin B. 2001. Global gene expression during short-term ethanol stress in Saccharomyces cerevisiae. FEBS Lett 498:98–103. doi: 10.1016/s0014-5793(01)02503-0 [DOI] [PubMed] [Google Scholar]
- 42. Abe F, Horikoshi K. 2005. Enhanced production of isoamyl alcohol and isoamyl acetate by ubiquitination-deficient Saccharomyces cerevisiae mutants. Cell Mol Biol Lett 10:383–388. [PubMed] [Google Scholar]
- 43. Kobayashi M, Shimizu H, Shioya S. 2008. Beer volatile compounds and their application to low-malt beer fermentation. J Biosci Bioeng 106:317–323. doi: 10.1263/jbb.106.317 [DOI] [PubMed] [Google Scholar]
- 44. Goossens K, Willaert R. 2010. Flocculation protein structure and cell-cell adhesion mechanism in Saccharomyces cerevisiae. Biotechnol Lett 32:1571–1585. doi: 10.1007/s10529-010-0352-3 [DOI] [PubMed] [Google Scholar]
- 45. Duong CT, Strack L, Futschik M, Katou Y, Nakao Y, Fujimura T, Shirahige K, Kodama Y, Nevoigt E. 2011. Identification of Sc-type ILV6 as a target to reduce diacetyl formation in lager brewers' yeast. Metab Eng 13:638–647. doi: 10.1016/j.ymben.2011.07.005 [DOI] [PubMed] [Google Scholar]
- 46. Abt TD, Souffriau B, Foulquié-Moreno MR, Duitama J, Thevelein JM. 2016. Genomic saturation mutagenesis and polygenic analysis identify novel yeast genes affecting ethyl acetate production, a non-selectable polygenic trait. Microb Cell 3:159–175. doi: 10.15698/mic2016.04.491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Trindade de Carvalho B, Holt S, Souffriau B, Lopes Brandão R, Foulquié-Moreno MR, Thevelein JM, Winston FM. 2017. Identification of novel alleles conferring superior production of rose flavor phenylethyl acetate using polygenic analysis in yeast. mBio 8:e01173-17. doi: 10.1128/mBio.01173-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dzialo MC, Park R, Steensels J, Lievens B, Verstrepen KJ. 2017. Physiology, ecology and industrial applications of aroma formation in yeast. FEMS Microbiol Rev 41:S95–S128. doi: 10.1093/femsre/fux031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Holt S, Trindade de Carvalho B, Foulquié-Moreno MR, Thevelein JM, Winston FM. 2018. Polygenic analysis in absence of major effector. mBio 9:e01279-18. doi: 10.1128/mBio.01279-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sariki SK, Kumawat R, Singh V, Tomar RS. 2019. Flocculation of Saccharomyces cerevisiae is dependent on activation of Slt2 and Rlm1 regulated by the cell wall integrity pathway. Mol Microbiol 112:1350–1369. doi: 10.1111/mmi.14375 [DOI] [PubMed] [Google Scholar]
- 51. Jhariya U, Dafale NA, Srivastava S, Bhende RS, Kapley A, Purohit HJ. 2021. Understanding ethanol tolerance mechanism in Saccharomyces cerevisiae to enhance the bioethanol production: current and future prospects. Bioenerg Res 14:670–688. doi: 10.1007/s12155-020-10228-2 [DOI] [Google Scholar]
- 52. Roy A, Lu CF, Marykwas DL, Lipke PN, Kurjan J. 1991. The AGA1 product is involved in cell surface attachment of the Saccharomyces cerevisiae cell adhesion glycoprotein a-agglutinin. Mol Cell Biol 11:4196–4206. doi: 10.1128/mcb.11.8.4196-4206.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. van Dyk D, Pretorius IS, Bauer FF. 2005. Mss11p is a central element of the regulatory network that controls FLO11 expression and invasive growth in Saccharomyces cerevisiae. Genetics 169:91–106. doi: 10.1534/genetics.104.033704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bester MC, Pretorius IS, Bauer FF. 2006. The regulation of Saccharomyces cerevisiae FLO gene expression and Ca2+ -dependent flocculation by Flo8p and Mss11p. Curr Genet 49:375–383. doi: 10.1007/s00294-006-0068-z [DOI] [PubMed] [Google Scholar]
- 55. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102:15545–15550. doi: 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gustin MC, Albertyn J, Alexander M, Davenport K. 1998. MAP kinase pathways in the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev 62:1264–1300. doi: 10.1128/MMBR.62.4.1264-1300.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Saito H, Tatebayashi K. 2004. Regulation of the osmoregulatory HOG MAPK cascade in yeast. J Biochem 136:267–272. doi: 10.1093/jb/mvh135 [DOI] [PubMed] [Google Scholar]
- 58. Chen RE, Thorner J. 2007. Function and regulation in MAPK signaling pathways: lessons learned from the yeast Saccharomyces cerevisiae. Biochim Biophys Acta 1773:1311–1340. doi: 10.1016/j.bbamcr.2007.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vilella F, Herrero E, Torres J, de la Torre-Ruiz MA. 2005. Pkc1 and the upstream elements of the cell integrity pathway in Saccharomyces cerevisiae, Rom2 and Mtl1, are required for cellular responses to oxidative stress. J Biol Chem 280:9149–9159. doi: 10.1074/jbc.M411062200 [DOI] [PubMed] [Google Scholar]
- 60. Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, Jensen LJ, von Mering C. 2021. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 49:10800. doi: 10.1093/nar/gkab835 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of the results of the sequencing and mapping of the genomes of the 11 strains.
Sequencing coverage.
Alleles observed in the 11 LBCM strains related to the new variants in relation to S. cerevisiae strain S288c.
Reference list of genes involved in the production of flavoring compounds; flocculation; stress resistance and ethanol production.
Genes carrying missense variants shared by the 11 strains involved with the phenotypes of interest.