

Abstract
Immunoglobulin A nephropathy (IgAN) is the most common primary glomerulonephritis worldwide and a frequent cause of kidney failure. Currently, the diagnosis necessitates a kidney biopsy, with routine immunofluorescence microscopy revealing IgA as the dominant or co-dominant immunoglobulin in the glomerular immuno-deposits, often with IgG and sometimes IgM or both. Complement protein C3 is observed in most cases. IgAN leads to kidney failure in 20–40% of patients within 20 years of diagnosis and reduces average life expectancy by about 10 years. There is increasing clinical, biochemical, and genetic evidence that the complement system plays a paramount role in the pathogenesis of IgAN. The presence of C3 in the kidney immuno-deposits differentiates the diagnosis of IgAN from subclinical glomerular mesangial IgA deposition. Markers of complement activation via the lectin and alternative pathways in kidney-biopsy specimens are associated with disease activity and are predictive of poor outcome. Levels of select complement proteins in the circulation have also been assessed in patients with IgAN and found to be of prognostic value. Ongoing genetic studies have identified at least 30 loci associated with IgAN. Genes within some of these loci encode complement-system regulating proteins that can interact with immune complexes. The growing appreciation for the central role of complement components in IgAN pathogenesis highlighted these pathways as potential treatment targets and sparked great interest in pharmacological agents targeting the complement cascade for the treatment of IgAN, as evidenced by the plethora of ongoing clinical trials.
1. Introduction
Immunoglobulin A nephropathy (IgAN) was originally described in 1968 by Drs Jean Berger and Nicole Hinglais as “intercapillary deposits of IgA-IgG” in the glomeruli [1]. Since then, IgAN has been recognized as the most common primary glomerular disease in many countries and remains a leading cause of chronic kidney disease (CKD). About 20–40% of patients progress to kidney failure within 20 years of diagnosis [2] that reduces life expectancy by about 10 years [3]. Currently, the diagnosis necessitates a kidney biopsy that shows IgA as the dominant or co-dominant immunoglobulin in the mesangial immune deposits by routine immunofluorescence microscopy evaluation [4]. The IgA is often accompanied by IgG and sometimes IgM or both [4]. The IgA in the immuno-deposits is exclusively of the IgA1 subclass [5]. Complement component C3 is detected in the immuno-deposits in up to 90% of IgAN kidney biopsies [4].
The clinical presentation of IgAN varies and encompasses a wide spectrum of manifestations that include isolated microscopic hematuria, episodic macroscopic hematuria (usually coinciding with an upper-respiratory or gastrointestinal tract infection), proteinuria (which can reach the nephrotic range), acute kidney injury (AKI), and established CKD [6, 7]. IgAN is categorized broadly as being primary or secondary, the latter referring to cases that occur in the context of systemic diseases including infectious, inflammatory, or autoimmune disorders [8]. IgAN can be limited to the kidneys or be part of a systemic vasculitic process referred to as IgA vasculitis with nephritis (previously known as Henoch-Schönlein purpura nephritis) [9]. For this manuscript, we limit our review to primary IgAN.
The true incidence and prevalence of IgAN are unknown, as screening for asymptomatic kidney disease through blood and urine tests as well as the indications for and access to a kidney biopsy vary widely across the world. With these caveats, IgAN incidence is highest in East Asia, accounting for 31 to 54% of native-kidney biopsies performed, followed by 25% in Europe and 12% in North America, and is relatively rare (< 5%) in sub-Saharan Africa [10–15]. While the disease appears to have equal gender distribution in Asia, there is a 2–3:1 male to female predominance in the USA. Most cases are sporadic; however, the first description of a large pedigree from central and eastern Kentucky with multiple affected family members strongly supported the notion that the disease can be inherited [16]. Since then, studies have confirmed that about 5–8% of patients have a first- or second-degree relative with biopsy-proven IgAN or urinary abnormalities, consistent with a familial or genetic predisposition for IgAN [16, 17].
A sizeable subset of patients will progress over time to kidney failure. Clinical and pathologic parameters ascertained at the time of the kidney biopsy have been shown to predict adverse outcomes, including sustained proteinuria ≥ 1 g/day, arterial hypertension, and severity of kidney pathologic involvement [18–20]. The updated Oxford classification of IgAN (also known as the MEST-C score) is a structured and reproducible scoring of kidney biopsy light-microscopy features (Table 1) that was derived from and validated in large international cohorts. Higher scores for each parameter portended worse kidney prognosis [20]. Consequently, aggressive management of hypertension and reduction of proteinuria via lifestyle changes and therapeutic interventions including use of maximally tolerated renin-angiotensin-aldosterone system inhibitors (RAASI) are paramount for the management of patients with IgAN. Recently, review of existing scientific data suggested that there is a consistent negative relationship between the magnitude and duration of proteinuria and kidney survival. Additionally, treatment-induced reduction of proteinuria correlates with slowing the loss of kidney function [21]. These findings led the US Food and Drug Administration (US FDA) to support the use of proteinuria as a valid surrogate outcome for clinical trials testing new therapeutic agents in the treatment of IgAN [22]. Recently, two new drugs have been conditionally approved by the US FDA as treatment for IgAN on the basis of proteinuria reduction: Tarpeyo, a targeted-release formulation of oral budesonide [23], and Filspari (sparsentan), a non-immunosuppressive, single-molecule dual-acting, highly selective antagonist of endothelin type A receptor and angiotensin II subtype 1 receptor [24]. This approach ushers in a new era of clinical trials in IgAN and opens the door for other potential therapies, including complement inhibitors.
Table 1.
Updated Oxford classification of primary immunoglobulin A nephropathy
| Histological feature | Definition | Score |
|---|---|---|
| Mesangial hypercellularity | Percentage of glomeruli with > 3 mesangial cells per mesangial area | M0: ≤ 50% M1: > 50% |
| Endocapillary hypercellularity | Increased number of cells within glomerular capillary lumina causing narrowing of the lumina | E0: Absent E1: Present |
| Segmental glomerulosclerosis | Any amount of the glomerular tuft involved in sclerosis (scarring), but not involving the whole tuft or the presence of an adhesion | S0: Absent S1: Present |
| Tubular atrophy/interstitial fibrosis | Percentage of cortical area involved by tubular atrophy or interstitial fibrosis, whichever is greater | T0: 0–25% of cortical area T1: 26–50% of cortical area T2: > 50% of cortical area |
| Cellular or fibrocellular crescent | Percentage of glomeruli with cellular or fibrocellular crescent | C0: Absent C1: < 25% of glomeruli C2: ≥ 25% of glomeruli |
M mesangial hypercellularity, E endocapillary hypercellularity, S segmental glomerulosclerosis, T tubular atrophy/interstitial fibrosis, C cellular or fibrocellular crescents
2. Pathogenesis of Immunoglobulin A Nephropathy (IgAN)
Immunoglobulin A is the most abundant antibody isotype in humans and exists in two subclasses, IgA1 and IgA2, with distinctly different heavy-chain hinge regions [25]. The IgA1 heavy-chain hinge region is longer due to extra amino acids. Some of these amino acids have an attached short carbohydrate side chain comprised of N-acetylgalactosamine to which galactose, sialic acid, or both may be attached. These carbohydrates are designated O-glycans. Breakthroughs in understanding the pathogenesis of IgAN came from four key discoveries: (1) IgA in the glomerular mesangial deposits is restricted to the IgA1 subclass [5], (2) the IgA1 in the glomerular deposits is enriched for molecules with less galactose in the hinge-region than is present in normal serum IgA1 (galactose-deficient IgA1, Gd-IgA1) [26, 27], (3) circulating Gd-IgA1 is predominantly in immune complexes bound by IgG specific for Gd-IgA1 [28–30], and (4) immune complexes composed of Gd-IgA1 and autoantibodies isolated from sera of patients with IgAN activate human mesangial cells in culture [31]. IgAN is now considered an autoimmune disease and a ‘four-hit’ hypothesis is accepted as the blueprint for its pathogenesis [32] (Fig. 1). Hit 1 is an increased circulating level of polymeric Gd-IgA1. Hit 2 entails the presence of IgG or IgA autoantibodies specific for Gd-IgA1, i.e., glycoforms of IgA1 with some exposed N-acetylgalactosamine moieties in the hinge region [29]. Hit 3 is formation of immune complexes in the circulation composed of Gd-IgA1 bound by mostly IgG autoantibodies (IgG-Gd-IgA1). Other proteins, such as the soluble Fcα (sCD89) or complement proteins, can bind to these circulating immune complexes; however, it is not clear whether sCD89-associated IgA1 can activate complement [33]. Some of the circulating nephritogenic IgG-Gd-IgA1 complexes traverse the glomerular fenestrae to enter the mesangium where they activate mesangial cells to induce cellular proliferation and increase production of extracellular matrix, chemokines, and cytokines (Hit 4), events that likely lead to kidney injury. This sequential multi-step process is supported by multiple lines of evidence. As mentioned earlier, glomerular IgA immuno-deposits are enriched for Gd-IgA1 [26, 27]. More recently, our group used a very sensitive technique and a nanobody that binds to IgG to show that all kidney biopsy specimens of IgAN patients had IgG, even those without IgA by routine immunofluorescence microscopy using less-sensitive reagents [34]. The IgG extracted from the glomeruli of IgAN biopsies exhibited binding specificity to Gd-IgA1. In contrast, IgG extracted from glomeruli of kidney biopsies of patients with other glomerular diseases used as controls (lupus nephritis, membranous nephropathy) did not bind to Gd-IgA1. Furthermore, confocal microscopy indicated co-localization of IgG and IgA in the glomerular immuno-deposits, thus supporting the role of IgG in disease pathogenesis [34]. The IgG co-deposits in the glomeruli are of the IgG1 and IgG3 subclasses, as is the IgG in the circulating IgG-Gd-IgA1 immune complexes [29, 35]. Observational studies of IgAN patients who underwent kidney transplantation document frequent recurrence of the disease, supporting the concept that the immune complexes in the immuno-deposits originate from the circulation [36–39].
Fig. 1.
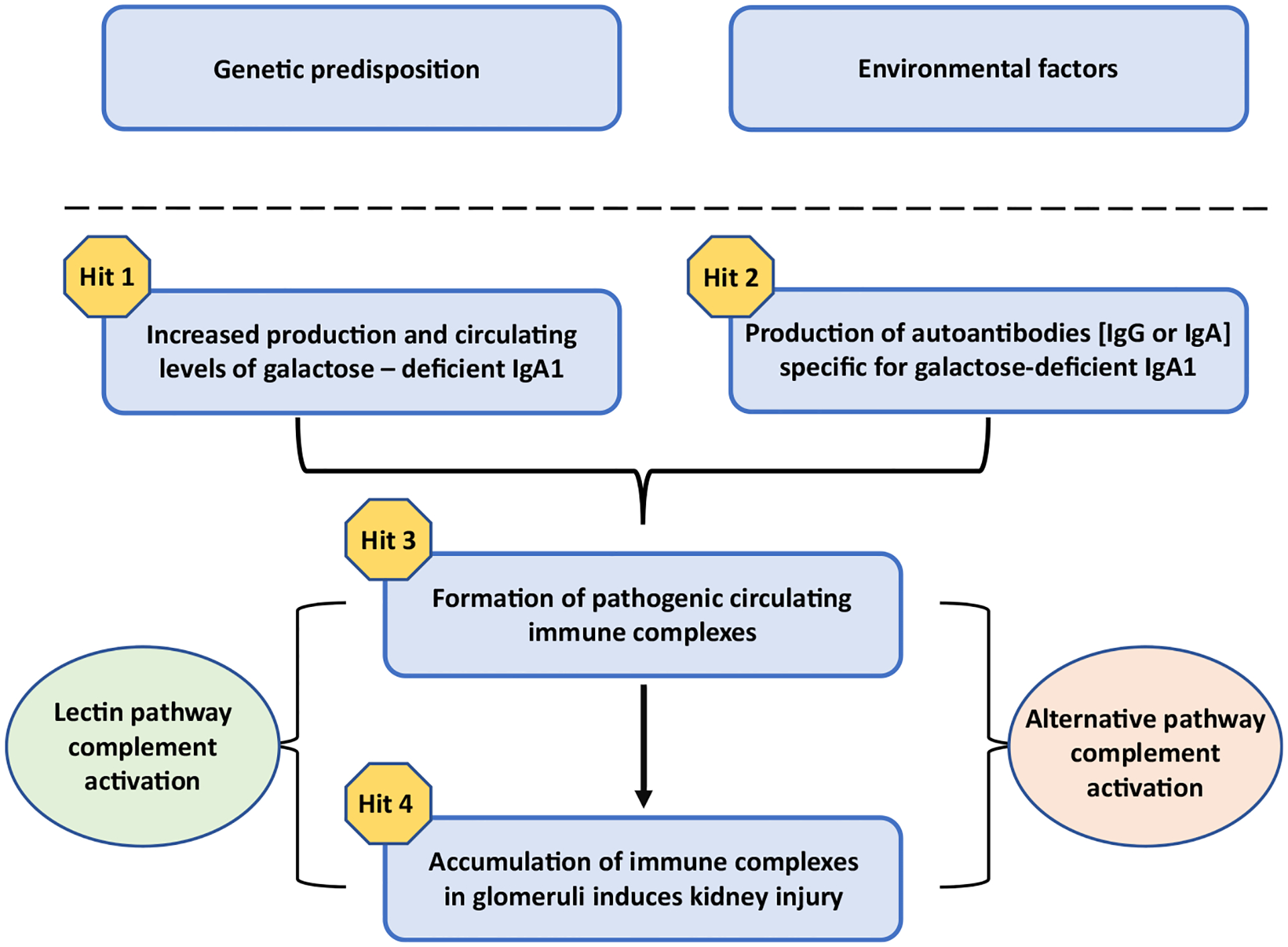
Proposed four-hit hypothesis for pathogenesis of immunoglobulin A nephropathy (IgAN). In genetically susceptible individuals, environmental factors are thought to trigger the onset of IgAN. A four-hit hypothesis has been proposed for the disease pathogenesis: increased levels of circulatory galactose-deficient IgA1 (Hit 1) lead to production of autoantibodies (either IgG or IgA, but mostly of the IgG isotype) (Hit 2). This process results in the formation of circulating nephritogenic immune complexes (Hit 3), some of which deposit in the glomeruli and induce kidney injury via mesangial cell activation and proliferation (Hit 4) [32]. There is evidence that the alternative complement pathway and, at least in some patients, the lectin pathway are involved in the pathogenesis of IgAN
3. Overview of the Complement System
The complement cascade plays a pivotal role in the innate immune response and serves as a first line of defense against a broad range of pathogens, including bacteria, parasites, and viruses. Activation of the cascade aids the acute phase response and restricts immune-mediated damage to host tissues; however, the process does not include an immunological memory that operates in the adaptive immune system. The complement system is a key link between innate and adaptive immunity given its engagement in immunosurveillance and maintenance of tissue homeostasis [40, 41]. The complement system was discovered by Nuttall and Buchner in the late 1880s; they recognized that a heat-labile component of blood depleted of white blood cells retained a capacity to kill bacteria. Initially, complement was known as “alexin,” which translates as “to ward off” in Greek, in this case implying warding off infection. Later, the term “alexin” was changed to “complement” after it was recognized that it “complemented” the bactericidal activity of serum antibodies. Complement has since been characterized as a group of proteins that react in succession and, when activated, mediate numerous biological reactions principal to host defense [41]. There are about 60 proteins in cells, tissues, and plasma within the complement system that depends on several tightly regulated activation steps [42–44].
Although complement activity was discovered in blood, hepatocytes were identified many decades later as the primary site for synthesis of complement proteins [45–47]. Ensuing studies showed that various other cell types, including macrophages, adipocytes, fibroblasts, endothelial and epithelial cells, T cells, neurons, astrocytes, and oligodendrocytes, also produce most of these proteins constitutively, upon induction, or both [48]. Many proteins associated with the complement system are proteases that are activated by proteolytic cleavage. This cascade of proteolytic reactions must be tightly controlled for this system to work—not only effectively but also safely. Broadly speaking, complement proteins are therefore characterized as either activators or regulators of complement pathways. Defects in either group may lead to immunodeficiency or autoimmune manifestations. The regulatory components of the complement system can be sub-stratified into those that are soluble, membrane-bound, or both [49–51].
The Complement Nomenclature Committee, under the auspices of the International Complement Society and the European Complement Network, established a system for the naming of complement proteins [52]. Eleven proteins start with the designation “C”: C1 (a complex of three proteins: C1q, C1r, and C1s) through C9. Many proteins in this system are called “factors” referring to substances in circulation. Some proteins fall outside these two designations and are named based on their function (e.g., decay accelerating factor [DAF]). When a complement protein is activated, the resulting larger cleavage fragment is termed the “b” fragment and the smaller one the “a” fragment, with one notable exception: C2a is larger than the C2b fragment of C2 as the original paper describing these fragments antedated the current nomenclature system [41, 53]. Complement activation also facilitates uptake and destruction of microbes by phagocytic cells, due to the specific recognition of activated complement products by complement receptors on phagocytes. There are six types of complement receptors (CR): CR1–4, C3a, and C5a receptors [54, 55].
The classical, alternative, and lectin pathways are the three means of activation of the complement system (Fig. 2). Each pathway has a unique triggering mechanism; nonetheless, after the formation of the C3-activating proteolytic enzymes (C3 convertases), all pathways converge into the terminal pathway that starts with the formation of the C5 convertase and culminates in assembly of the membrane-attack complex (MAC), also called terminal complement complex [56]. Activation of the classical and lectin complement pathways is calcium-dependent, while activation of the alternative complement pathway is magnesium-dependent [41]. Renin, also known as angiotensinogenase, is an aspartyl protease secreted by the juxtaglomerular cells in the kidney that can cleave C3 into C3b and C3a, in a manner similar to the C3 convertase, thus triggering the alternative pathway [57].
Fig. 2.
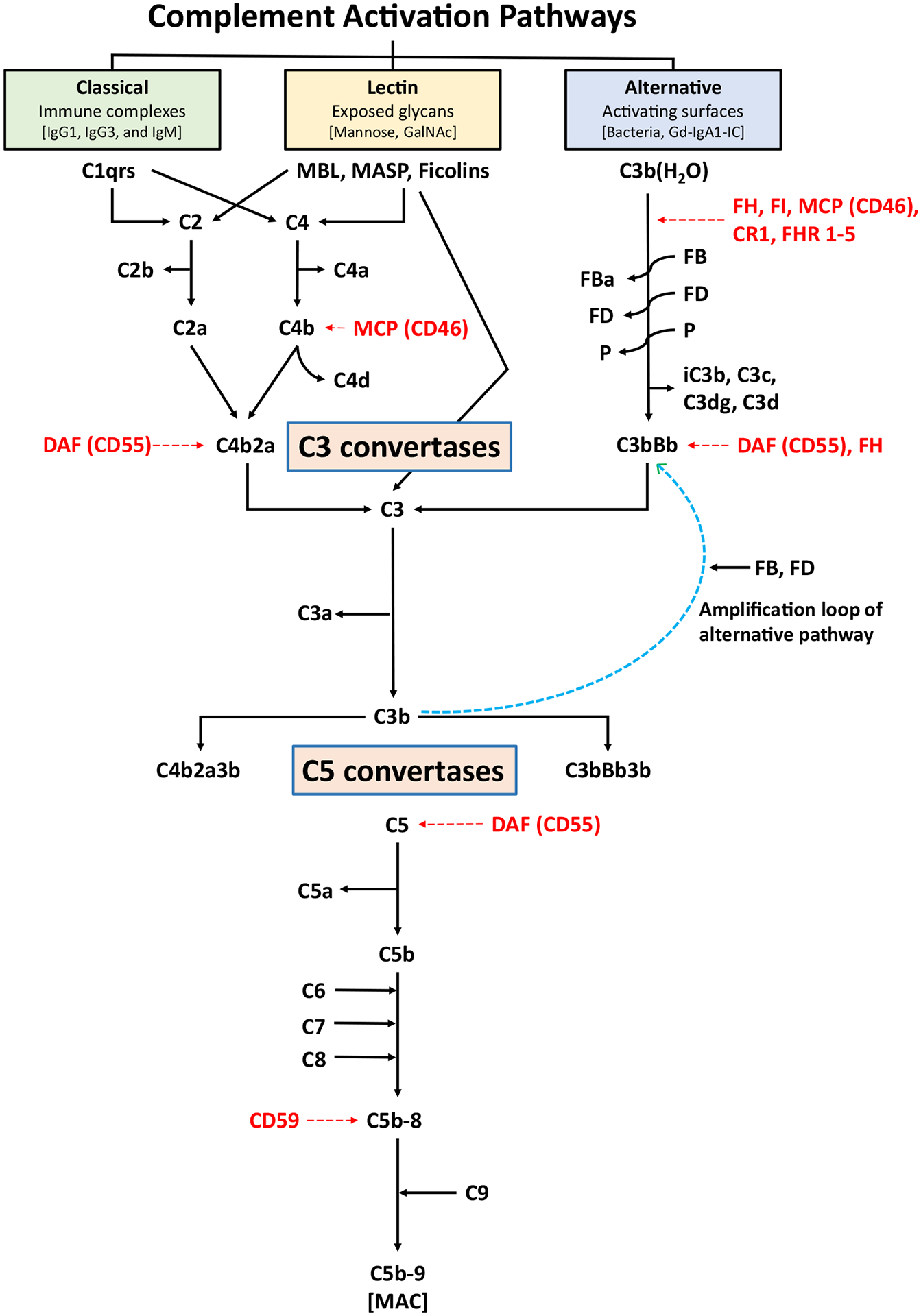
Simplified schema of the complement pathways. The three pathways of complement activation, classical, lectin, and alternative, are initiated by interactions of the complement proteins with distinct structures. Antigen-antibody complexes can activate the classical pathway. Mannose-binding lectin (MBL) recognizes carbohydrate structures, and upon association with serine proteases (MASP, mannose-associated serine proteases), can activate the lectin pathway. Complement C3 that is covalently bound to pathogen surfaces via a thioester bond as C3b initiates the alternative complement pathway. Each pathway ultimately generates an active C3 convertase, resulting in cleavage of C3 into C3a and C3b fragments. C3b can interact with C4b2a of the classical pathway or C3bBb of the alternative pathway to produce C5 convertase that cleaves C5 into C5a and C5b fragments. In the alternative complement pathway, every C3b molecule generated serves as the initiating point for a new C3 convertase (C3bBb), thereby generating numerous C3b molecules via the “amplification loop” process. In all complement pathways, C5b binds to the cell membrane of the pathogen and serves as a platform for assembly of the membrane attack complex (MAC). The formation of MAC can be inhibited by membrane-bound CD59. Several other regulatory proteins of the complement-activation pathways are shown in red. FB and FD perpetuate the alternative pathway “amplification loop”. CR1 complement receptor 1, DAF decay-accelerating factor, FB factor B, FD factor D, FHR 1–5 factor H-related proteins 1–5, FI factor I, GalNAc N-acetylgalactosamine, Gd-IgA1 galactose-deficient IgA1, Gd-IgA1-IC galactose-deficient IgA1-containing immune complexes, MCP membrane cofactor protein, P properdin
The classical pathway plays an important role in antimicrobial defense and opsonophagocytosis and is activated by C1q (a pattern-recognition molecule) bound to bacteria or other pathogens or immune complexes. C1q subsequently binds to its closely associated proteins C1r and C1s to form a complex protein C1 (C1q:C1r2:C1s2) [58, 59]. In addition to antibody-mediated activation, pathogens bound to C-reactive protein (CRP) and damaged tissue also can activate the classical pathway. Classical pathway activity in tissue can be detected based on immunostaining for C1q [60]. C1 formation leads to cleavage of C4 to C4a and C4b and cleavage of C2 to C2a and C2b. The C2a remains associated with C4b to form C4b2a, the C3 convertase complex in the classical pathway that cleaves C3 into C3a (a potent anaphylatoxin) and C3b. Structural aspects of C3 and its processing are further detailed below. As part of the next step of the cascade, C5 convertase of the classical pathway is formed by association of C3b with C4b2a to cleave C5 into C5a, a potent anaphylatoxin and chemoattractant, and C5b. Generation of C5b initiates the terminal pathway wherein complement components C6, C7, and C8 associate with the C5b protein in a sequential and concentration-dependent manner to create the C5b-8 complex that is incorporated into the host cell membrane. Subsequently, 10–16 units of the C9 component arranged in a ring-like manner are added onto the C5b-8 complex to form the C5b-9 MAC. This structure creates pores in the cell membrane leading to osmotic lysis, activates pro-inflammatory signaling pathways, and produces ion fluxes that eventually lead to cell death via cellular lysis and necrosis [40, 61]. CD59 is a major regulatory protein of the terminal complement pathway. CD59 binds to C5b-8 complex and inhibits the binding of C9 protein to this complex. If C9 molecules have already started binding to the complex, CD59 will hinder binding of additional C9 moieties, thereby restricting the pore size on cell surfaces [62, 63]. Clusterin and vitronectin are regulatory proteins that solubilize immune complexes and, thus, reduce their size. When clusterin and/or vitronectin are associated with the C5b-9 MAC moiety, they form a soluble MAC (sMAC) complex, which is present in the circulation but does not insert into the cellular membranes [64]. The exact biological roles of sMAC in homeostatic conditions and disease pathophysiology are currently not fully understood, although sMAC levels change during infection and autoimmune conditions [64, 65].
The lectin pathway is activated by pathogen-specific carbohydrates such as terminal d-mannose residues or N-acetyl-d-glucosamine on the outer surface of certain bacteria, fungi, viruses, and protozoans, and by acetylated moieties including N-acetyl-glycine, N-acetyl-cysteine, and acetylcholine on cellular motifs [49, 66]. The recognition and initiating proteins for this pathway include mannose-binding protein (MBP), ficolins, and collectins that couple with MBP-associated serine proteases-1 and −2 (MASP-1 and MASP-2). Upon activation, these complexes cleave C4 and C2 to form the C3 convertase in the lectin pathway (C4b2a) that is identical to the C3 convertase in the classical pathway. Further downstream sequences, including generation of C3b, lead to formation of the C5 convertase complex and end in the terminal pathway activation described above [40, 41, 60]. Detection of C4d, a cleaved fragment of C4 activation, in the absence of C1q confirms activation of the lectin pathway.
The alternative pathway provides protection from a wide variety of extracellular pathogens and is activated by pathogen-associated molecular patterns (PAMPs) such as lipopolysaccharide in the cell wall of Gram-negative bacteria and carbohydrates in the cell wall of certain fungi (zymosan). Unlike the classical and lectin pathways, the alternative pathway is constitutively active at a low level due to the “tick-over” mechanism; hence, it is constantly primed to react swiftly and vigorously to pathogens or injury. The “tick-over” mechanism is a multi-step process that results in the hydrolysis of C3 to C3(H2O). Factor B (FB) then binds to the hydrolyzed C3 and the complex is cleaved by factor D (FD) leading to the generation of C3(H2O)Bb, the “initiation” C3 convertase of the alternative pathway. This C3 convertase cleaves C3 molecules into C3a and C3b. C3b binds to FB that is then cleaved by FD to form C3bBb, the alternative-pathway C3 convertase. Binding of properdin to this C3 convertase stabilizes this protein, thereby extending its enzymatic half-life. The C3bBb complex cleaves C3; thus, every generated C3b molecule serves as the initiating point for a new alternative-pathway C3 convertase. Via this “amplification loop” process, numerous C3b molecules are quickly available to target and abrogate pathogen-mediated cellular injury. C3b can then bind to the alternative-pathway C3 convertase to create C3bnBb, the alternative-pathway C5 convertase converging into the terminal pathway that leads to the formation of C5b-9 (MAC) [40, 41].
To prevent complement-mediated damage to the host, the system is controlled at numerous levels by key regulatory proteins [49]. The classical pathway is regulated by C1 esterase-inhibitor that binds to the active site of serine proteases in C1r and C1s, thereby abrogating spontaneous activation [67]. C1 esterase-inhibitor also inhibits activity of the MASP proteins in the lectin pathway [68]. Further downstream, C4b-binding protein serves as a cofactor in the factor I (FI)-mediated cleavage of C4b to restrict formation and function of the C3 convertases of the classical and lectin pathways [69]. The alternative-pathway C3 convertase C3(H2O)Bb is tightly regulated by key blood regulatory and processing proteins that include factors H (FH) and FI, membrane cofactor protein (MCP, CD46), complement receptor 1 (CR1, CD35), DAF (CD55), and complement factor H-related proteins (FHRs) [70–74]. These regulators prevent production of the alternative-pathway C3 convertase or intensify its dissociation (FH, DAF, CR1) or serve as cofactors for FI-mediated inactivation of C3b to inactive C3b denoted as iC3b (FH, CR1, and MCP). iC3b is unable to participate in further complement activation and is rapidly cleaved to C3c and surface-bound C3dg [60]. The FHRs show a high degree of sequence identity with FH, particularly at the domains binding to cell surfaces or C3b; however, they lack the complement-regulating domains of FH. This difference results in the potential competition by FHRs for C3b binding and subsequent impairment of FH-mediated complement regulation [75]. Properdin is an important positive regulator of the alternative complement pathway; its main function is to stabilize C3 convertase and, to a lesser extent, C5 convertase [54, 55].
4. Role of Complement Proteins in IgAN
The pivotal role of the complement system in the pathogenesis of IgAN is increasingly recognized. Here we describe the existing pathologic, biochemical, and genetic evidence that supports its contribution to the disease onset and progression. This involvement serves as the basis for potentially immunomodulating the complement system in an attempt to change the course of IgAN.
Although more details will follow below, we provide here examples that illustrate that an imbalance in amounts or activities of specific regulators and cofactors of complement may be directly related to IgAN development and progression. For example, higher circulating levels of several complement proteins are considered potential risk factors for the development of IgAN. Higher circulating FHR-5 levels were associated with IgAN development and progression in a study of 1126 IgAN patients [76]. Elevated plasma levels of FH antagonist FHR-1 and a high FHR-1/FH ratio were associated with disease progression [77], although plasma FHR-5 levels and the FHR-5/FH ratio were not associated with disease progression in another study [78]. Of note, higher FHR-5 levels portended a poorer response to immunosuppression treatment [78]. FHR-5 in kidney biopsy specimens in IgAN patients has been associated with progressive disease, providing further evidence of its involvement in the pathogenesis of IgAN at the tissue level [79–82]. Also, gene deletions of the protective alleles of CFHR1,3 and the presence of certain rare CFHR5 variants have been associated with an increased susceptibility for development of IgAN [40].
4.1. Activation of the Complement System in the Kidneys in IgAN
The initial description of IgAN as “intercapillary deposits of IgA-IgG” by Berger and Hinglais in 1968 [1] also noted the presence of complement C3 (that was called β1C) [83]. These findings were based on using a then new technique of immunofluorescence staining of kidney biopsy specimens with antisera raised in animals against various human proteins, such as IgA, IgG, IgM, and complement components. This observation has since been reproduced in many cohorts. Immuno-histological findings of C3, C4d, properdin, mannose-binding lectin (MBL), and C5b-9 deposits in the glomerular immuno-deposits in IgAN kidney tissue confirms activation of the lectin and alternative pathways of complement [84–86]. On the other hand, the absence of C1q in the vast majority of IgAN kidney specimens suggests that the classical pathway does not significantly contribute to the tissue injury [85]. Several studies correlated the presence of MBL in glomerular immuno-deposits [87–93] with more severe kidney disease and a worse prognosis [92, 93]. These observations appear to agree with the study that assessed immunohistochemical staining for MBL, C4d, C5b-9, and other proteins in the glomerular immuno-deposits, and correlated those data with the analyses of urine samples [94]. Notably, urinary concentrations of C4d and MBL were associated with their presence in the mesangial immuno-deposits and, to a lesser degree, with urinary protein excretion [94].
A greater degree of granularity emerged from a study that evaluated multiple proteins associated with the lectin and alternative pathways of complement activation [79]. Specifically, serum levels of MASP-3 were low in IgAN patients with progressive disease. Their kidney biopsies exhibited increased glomerular staining for C3b/iC3b/C3c, C3d, C4d, C5b-9, and FHR5 and, conversely, reduced glomerular deposition of FH. These findings are consistent with the antagonistic role of FHR-5 in the binding of FH to C3/C3b and, more generally, with the role of FHR-5 in FH deregulation [79–82].
In summary, most patients with IgAN have some C3 in glomerular IgA-containing immuno-deposits, indicating involvement of the alternative pathway of complement. There may be a complex interplay involved in the regulation of C3 processing in situ, as evidenced by co-deposits of FHR-5, especially in the patients with progressive disease. Moreover, some patients also have co-deposits of complement proteins from the lectin pathway. Thus, there is a need for developing a better understanding of the regulation of complement activation pathways and various processed products in patients with more or less severe pathologic findings.
4.2. Activation of the Complement System in the Circulation in IgAN
As the original observations indicated that the glomerular IgA immuno-deposits originate from circulating immune complexes, multiple groups assessed the presence and composition of IgA1-containing immune complexes in serum. Initial attempts to isolate these complexes used polyethylene glycol precipitation [95], although it was later shown not to be specific for immune complexes [96]. These early data showed that the polyethylene glycol-precipitated circulating immune complexes contained IgA and C3 [95]. Serum levels of these complexes were elevated in 48% of patients with IgAN with IgA1 as the predominant IgA subclass. IgA1-IgG complexes were detected in 44% of patients with IgAN, whereas IgA2-IgG complexes were detected in only 7% of patients and IgM-IgA1 complexes in 16% of patients [95].
High serum levels of C3, FB, FH, and FI may be characteristic for IgAN patients with stable normal glomerular filtration rate (GFR) [97]. Furthermore, complement deficiencies may not be infrequent in IgAN [97]. Consistent with these findings, other studies postulated that high serum IgA levels and the serum IgA/C3 ratio distinguished IgAN from other primary kidney diseases [98–101]. Additional studies went even further and suggested the serum IgA/C3 ratio reflects the histological severity of IgAN and predicts disease progression [102–105]. In a cohort of pediatric IgAN patients, the serum IgA/C3 ratio and glomerular C3 staining predicted disease progression but did not correlate with kidney-biopsy findings at the onset of disease [106]. Conversely, another study of pediatric IgAN patients correlated the Oxford MEST-C classification scores of the light-microscopy features of kidney biopsies with serum IgA/C3 ratios; the mean IgA/C3 ratio was higher in patients with greater kidney injury, as indicated by M1, S1, and T1 scores, compared with patients with M0, S0, and T0 scores [107].
The interest in urinary biomarkers to monitor the course of IgAN has grown recently, stemming in part from previously documented association of MBL excess in the circulation with the disease progression in patients with IgAN [108]. A study of a Chinese cohort showed that urinary MBL and C4d concentrations were associated with the proportion of glomerular crescents (a histologic feature indicative of severe kidney injury) and glomerular C4d deposition [109]. A proteomic study extended these data by confirming the abundance of C3, MAC, complement regulatory proteins of the alternative pathway, MBL, and MASP-2 in the urine of IgAN patients with progressive disease [110].
Due to the prominent role of C3 in IgAN, we will provide more details on the structure and processing of C3. C3 is present not only in the glomerular immuno-deposits but also within the IgA-containing circulating immune complexes [111]. The labile thioester bond [112] likely plays a role in this association, as this bond can attach C3 to immunoglobulins in immune complexes [113–116]. A developing understanding of the processes involved in the activation and processing of C3 in the alternative pathway of complement has emerged during the past two decades. Mature C3 is formed from two chains [117], β (amino acid residues 1–645) and α (amino acid residues 650–1641) that are connected by disulfide bonds. Together, these two chains fold to form 13 domains (Fig. 3A–C) [118]. Among these domains are eight macroglobulin (MG) domains (MG1–MG8), a linker domain (LNK), an anaphylatoxin domain (ANA, the predecessor of C3a), an extended connector loop, α’NT, CUB (complement C1r/C1s, Uegf, Bmp1) domain, a thioester-containing domain (TED), a short linker designated as anchor, and a carboxy-terminal domain (C345C) with a netrin-like fold. C3 function is controlled by cleavage events and large conformational rearrangements of these domains that yield different tertiary structures [119]. The normal processing of C3 to C3b (Fig. 3D) is catalyzed by C3 convertase [120]. Conversely, the inactivation of C3b requires another processing step, cleavage by FI with the assistance of FH. The resultant cleavage of the α chain releases C3f (a dodecapeptide fragment of C3) and yields inactivated C3 (i.e., iC3b, Fig. 3E) attached to the activating surface (e.g., bacteria, immune complexes). Further cleavage produces a soluble component C3c and C3dg, the latter of which remains attached to the activating surface (Fig. 3F). Notably, inactivation of C3 (production of iC3b from C3b) is dependent on binding of FH to C3b, a process that can be countered by FHR 1–5 polypeptides [60, 121, 122].
Fig. 3.
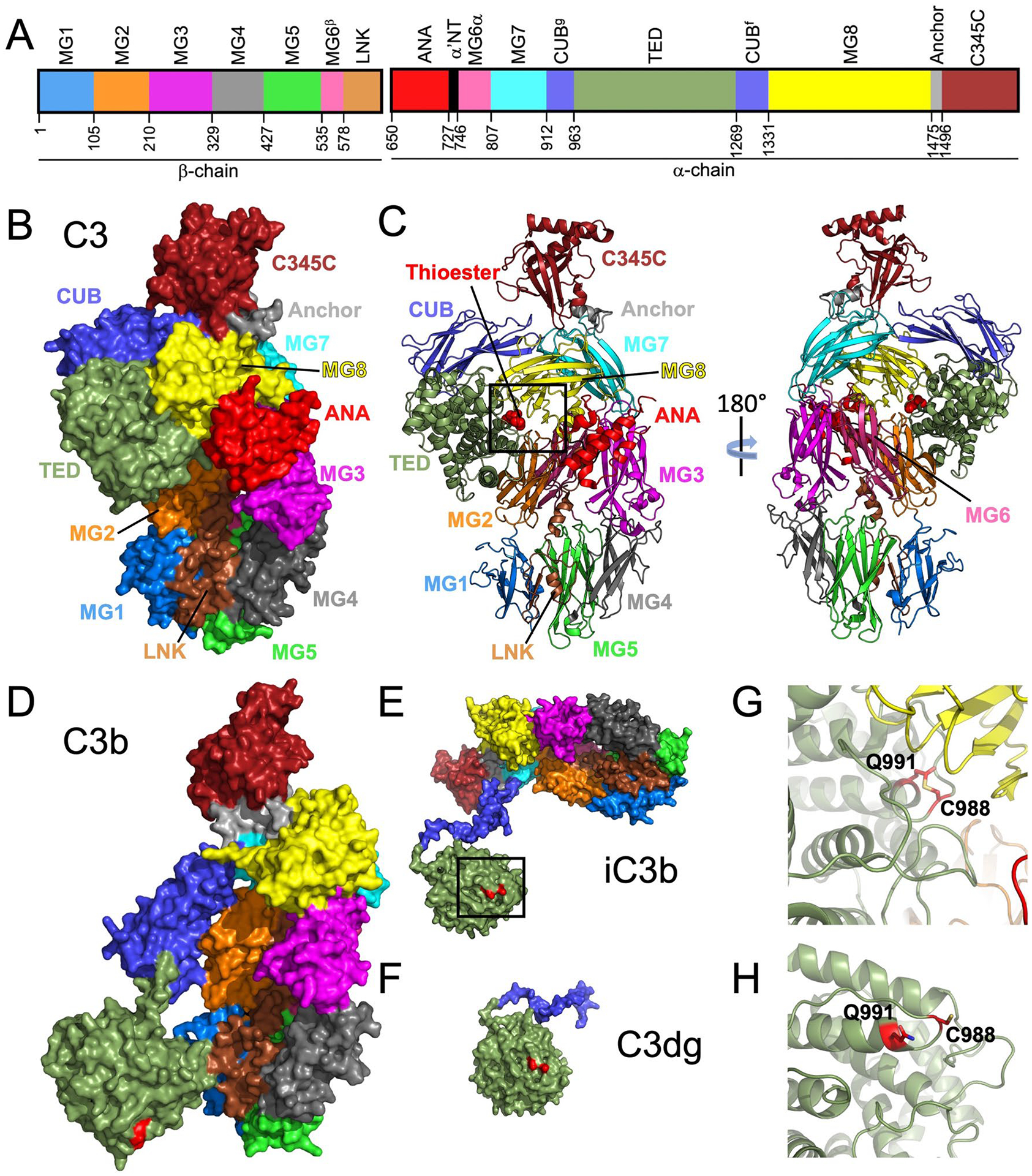
Structures of human complement C3 and C3 cleavage products and the thioester bond. IgA1-containing circulating immune complexes in IgAN patients have C3, presumably covalently attached through the thioester bond to IgA1 and/or IgG, as reported for other types of immune complexes [113–116]. A Domain arrangement of C3 β and α chains. Domains are individually colored and labeled. Domain-starting residues are noted below the schematic. Residue numbering and colors follow those in the publication of Janssen et al [118]. B, C Surface rendering and cartoon models of C3 (PDB ID: 2A73 [118]) are shown. The conformation of C3 protects the thioester bond (red spheres boxed in [C]) from nucleophilic activation. D–F Cleavage products of C3 are shown in surface representation. (D) C3b (PDB ID: 2I07 [190]) undergoes considerable conformational change, most notable is the movement of the thioester-containing domain (TED). This change exposes and activates residues C988 and Q991 (labeled and shaded in red on structures in D–F), which are involved in formation of a thioester bond. E iC3b (PDB ID: 7AKK [191]) and F C3dg (derived from PDB ID: 7AKK) are shown. G A close-up view of the boxed area in C is shown with the thioester bond represented with stick model. The thioester bond in the structure of iC3b is broken and shown as the amino acid residues C988 and Q991. This view is from the boxed area in E. IgAN immunoglobulin A nephropathy, PDB ID Protein Data Bank identification code
The ability of C3 to covalently bind to carbohydrates and proteins is facilitated by the activation of a thioester formed between two amino acid residues, C988 and Q991, within the TED (Fig. 3B, C, G). The covalent linkage between these two residues is hidden in the closed conformation of C3 (Fig. 3B, C, G). To activate, the thioester bond can be cleaved by nucleophiles (N) [119], such as water (H2O) [123], ammonia (NH3) [124], or CH3NH2 [125] to form an unstable intermediate termed C3(N*) (Fig. 3) [119]. This nucleophilic attack is commonly referred to as the tick-over of C3 (for a comprehensive review, please see [126]), and the reaction can either be reversed [124] or progress with an irreversible conformational transition to yield C3(N), a form of C3 that functionally resembles C3b. This conformational change makes C3 available to bind other complement components described above, enabling progressive cleavage. The activated thioester bond can then be used for binding to cells, foreign components (e.g., bacteria, viruses), or immune complexes through covalent binding to hydroxyl and amine groups. C3b has been shown to covalently bind to IgG and IgA [114–116], which in the context of IgAN are represented by IgG autoantibodies and Gd-IgA1, respectively [127]. The binding site of C3b on IgG has been mapped to heavy-chain constant domain 1 (CH1), localizing to an amino acid residue between positions 131 and 156 [115, 116].
With the central role of IgA1-containing immune complexes in the pathogenesis of IgAN, several groups have sought to delineate the biological activities of these immune complexes in the circulation of patients with IgAN (for review, see [128–130]). These complexes can be purified by size-exclusion chromatography and then tested for biological activity using cultured primary human mesangial cells [131]. Correlation of activity with protein composition showed the complexes that stimulated proliferation of the mesangial cells contained IgA1 that was enriched for Gd-IgA1. Other components of these biologically active immune complexes included IgG autoantibodies specific for Gd-IgA1 and complement C3 [111]. Pull-down experiments with IgA1 O-glycan targeting reagents removed the biologically active IgA1-containing complexes [132]. At the same time, other components were also removed, such as IgG and C3, suggesting that IgA1, IgG, and C3 were physically associated in these circulating complexes [111]. Presumably, the association of C3 with IgA1 and/or IgG is covalent, through the thioester bond, as reported for C3 binding to other types of immune complexes [113–116]. Notably, it is not known whether any current complement-targeting reagents could prevent the covalent association of C3 to IgA1-containing immune complexes. Consistent with the activities of the IgA1-IgG-containing complexes, ELISA tests showed that serum levels of Gd-IgA1 and IgG autoantibodies specific for Gd-IgA1 correlated in patients with IgAN [133] and that their levels predicted disease progression and disease recurrence after transplantation [134–138].
It has been postulated that complement components and complement activation are relevant for the nephritogenic circulating immune complexes as well as for immuno-deposits in the glomeruli [40, 85, 130]. In addition to multiple complement regulatory proteins that are known to participate in the fine-tuning of complement activation and inactivation, there are emerging roles for other blood components, such as heme generated by hemolysis during episodes of macroscopic hematuria and acute kidney injury in patients with IgAN [139]. Specifically, heme interferes with the regulatory capacity of FI and thus enhances amplification of the complement alternative pathway [140]. The inhibitory effects of free heme on FI can be prevented by its removal, for example through heme-scavenging proteins in the circulation, such as hemopexin [140].
Direct experimental proof of the four-hit hypothesis for pathogenesis of IgAN detailed in the sections above requires recapitulation in an animal model. Development of small-animal models that would mimic human pathogenetic processes has been difficult. Notably, this challenge is due in part to the lack of O-glycans on IgA of small experimental animals, such as rodents. A transgenic mouse model has been developed that expressed human IgA1 and human CD89 that formed complexes that induced glomerular injury [33]. However, IgG autoantibodies specific for Gd-IgA1 have not been tested in this model. Our group at the University of Alabama at Birmingham with collaborators developed an approach that circumvented these shortcomings in animal models. We formed immune complexes in vitro from human Gd-IgA1 and human IgG autoantibodies specific for Gd-IgA1 and intravenously injected them into immunodeficient mice. These complexes deposited in the glomeruli and induced histological changes consistent with the features of IgAN; immunofluorescence staining revealed that the glomerular immuno-deposits contained human IgA and IgG and murine C3 [141]. Moreover, injection of un-complexed Gd-IgA1 or IgG autoantibodies alone did not have these pathological effects, confirming the nephritogenic potential of these Gd-IgA1-IgG immune complexes as well as the role of C3.
4.3. Genetic Evidence for the Role of the Complement System in IgAN
Since the discovery of the familial form of IgAN that suggested genetic components in the pathogenesis of IgAN [16], new tools have been developed and applied to genetic/genomic studies of IgAN (for review, see [142, 143]). Genome-wide association studies (GWAS) have identified approximately 30 independent risk loci for IgAN [144–152]. These studies implicated defects in several pathways, including intestinal network of IgA production, innate immunity against mucosal pathogens, and the complement cascade (for review, see [151, 152]).
A 2011 GWAS paper revealed five IgAN-associated loci that explain up to 7% of the disease variance [145]. Notably, some alleles in these loci were protective whereas others were risk-associated. The IgAN-protective alleles carried an increased risk for other autoimmune or infectious diseases. A common deletion of CFHR1 and CFHR3 at chromosome 1q32, the locus that encodes complement regulating proteins FH and FHR1–5, was among the protective alleles at this locus [145]. As noted in the section describing the complement pathways, FHRs 1–5 are involved in regulation of FH activity, specifically by competing for binding to C3b [79–82]. FHR-1 and FHR-5 behave as competitive antagonists and this interference is enhanced by dimerization of FHR proteins [82]. When FHR protein is associated with C3, FH protein does not bind to C3 to function as a cofactor for FI. This effect prevents FI from cleaving the C3f peptide from C3b, leading to inactivation of C3b by forming iC3b [40, 85, 130, 151, 152].
Several follow-up studies evaluated clinical effects of genomic variations in CFH, CFHR3, and CFHR1 on IgAN susceptibility and progression [153–155]. One study showed that serum levels of FH correlated with those of C3 [153]. Furthermore, the presence of mesangial C3 was negatively associated with serum FH levels but positively associated with serum Gd-IgA1 levels. Another study assessed copy number of CFHR3 and CFHR1 genes to determine the impact of heterozygous and homozygous deletions of the CFHR3-1 genes. IgAN patients with heterozygous or homozygous gene deletions exhibited less IgA, IgG, and C3 in the glomerular immuno-deposits as compared with patients with no deletion. The individuals with this genetic deletion did not progress to kidney failure or death [154]. Another study assessed genetic variants in the CFH–CFHR1–5 locus and found variants in CFHR5 that may contribute to the genetic susceptibility to IgAN [156]. Specifically, the observed variants in CFHR5 gene included 28 rare and 4 common alleles, of which several were potentially functional but with predicted amino-acid changes in the protein-coding segments. Follow-up functional assays revealed that some of these amino-acid changes impacted the affinity of FHR-5 for C3b [156]. Other clinical studies, however, indicated that the circulating serum FHR-5 level is an independent risk factor for IgAN progression [76] and that CFHR5 genes may indeed exist as rare variants [157]. Concerning FH, a recent study showed that IgAN patients with thrombotic microangiopathy exhibited more severe clinical conditions and higher serum levels of FH [158]. However, the CFH gene variants discovered in this study did not correlate with serum FH or serum C3 levels or the intensity of C3 glomerular immuno-deposits.
A 2014 GWAS paper revealed six new genome-wide significant associations with IgAN, including the ITGAM-ITGAX locus [147]. The IgAN risk alleles were associated with risk of inflammatory bowel disease or response to mucosal pathogens and maintenance of the intestinal epithelial barrier. An increasing number of risk alleles was associated with progressively younger age at disease onset. The geospatial distribution of risk alleles indicated multi-locus adaptation, perhaps being shaped by host-intestinal pathogen interactions [147]. ITGAM and ITGAX genes encode integrins alphaM (CD11b) and alphaX (CD11c). These proteins, together with integrin β2 chain (CD18), form CR3 and CR4, respectively. These complement receptors expressed on leukocytes bind iC3b, further underscoring the role for complement in IgAN. ITGAM-ITGAX genetic variants were also confirmed in other studies of Han Chinese populations [149, 159]. Despite the obvious functional connection with C3 binding to these C3 receptors, a better delineation is needed of these ITGAM-ITGAX polymorphisms and their impact on the biological mechanisms associated with the risk-imparting or protective effects in IgAN. It is hoped that the efforts will also expand genetic discoveries in more diverse populations worldwide [160].
A GWAS of serum IgA levels found 20 loci and determined positive genetic correlations of serum IgA levels with IgAN, type 2 diabetes mellitus, and body mass index, and negative correlations with celiac disease, inflammatory bowel disease, and several infections [161]. Mendelian randomization supported elevated serum levels of IgA as a causal factor in IgAN. Another study confirmed that inflammatory bowel disease, ulcerative colitis, and Crohn’s disease were causally associated with the increased risk of IgAN [162].
In summary, there is accumulating genetic evidence for complement involvement in IgAN. Genome-wide association studies are a powerful approach for identification of loci of interest, whereas post-GWAS studies validate these findings and identify specific genes and gene products and their biological roles and functions [163, 164]. Various experimental systems and platforms are then utilized and optimized to test the hypotheses generated by GWAS and post-GWAS approaches [165–169].
5. Treatment Targets and Ongoing Clinical Trials in IgAN
Our expanding knowledge of the pathways involved in complement activation in the pathogenesis and progression of IgAN has allowed the identification of potential therapeutic targets. The advent of drugs that can block different stages of the complement cascade will likely lead to new attractive treatment options.
Initial data were limited to case reports. The first publication from Sweden described a 16-year-old male with crescentic IgAN who failed to respond to corticosteroids and mycophenolate mofetil but subsequently stabilized after treatment with eculizumab, which binds to C5, thereby inhibiting its cleavage into C5a and C5b. Unfortunately, the response was not sustained [170]. Shortly thereafter, another 16-year-old male with crescentic IgAN who had failed corticosteroids, cyclophosphamide, and plasma exchange, was reported to transiently respond to eculizumab [171]. In a third report, the attempt to salvage a kidney allograft from recurrent crescentic IgAN in a 28-year-old male failed despite the use of eculizumab; however, the terminal-pathway inhibition was admittedly instituted late in the disease course [172]. Despite these encouraging attempts at targeting the complement system, clinical trials in patients with earlier and less aggressive IgAN presentations had been hindered by the relatively slow progression of disease. As such, use of hard outcomes (doubling of serum creatinine, reaching kidney failure, etc.) rendered the size and cost of such trial designs prohibitive. In 2019, the Kidney Health Initiative, a private-public partnership between the American Society of Nephrology and the US FDA, recognized proteinuria as a “reasonably likely” adequate surrogate end-point to test the effect of therapeutics for patients with progressive IgAN. Furthermore, a beneficial outcome on proteinuria became an acceptable criterion for accelerated approval of the drug being tested [22]. This policy announcement led to the initiation of multiple clinical trials (including those evaluating complement inhibitors) for IgAN patients at high risk of disease progression [173].
The updated Kidney Disease Improving Global Outcomes guidelines for treatment of patients with IgAN highlight the importance of implementing aggressive supportive care directed at lifestyle and cardiovascular risk modifications, optimization of blood pressure control and maximally tolerated use of RAASI with the intent to reduce proteinuria [174]. In patients at high risk for disease progression, namely those with proteinuria persistently > 1 g/day and relatively preserved kidney function (estimated GFR [eGFR] ≥ 30 mL/min/1.73 m2), the guidelines, for the first time, recommend offering patients participation in ongoing clinical trials, which include several targeting the complement system [174]. Among those clinical trials are studies targeting the alternative and lectin pathways as well as the formation of terminal complement complex (i.e., MAC). A full listing of ongoing trials is included in Table 2. Pegcetacoplan (APL-2) is a synthetic peptide derivative of compstatin that binds to C3 and C3b. A Phase II, open-label, basket trial is evaluating the safety and preliminary efficacy of its use in several glomerulo-nephritides, including IgAN (ClinicalTrials.gov Identifier NCT03453619). Cemdisiran (ALN-CC5), a subcutaneously administered ribonucleic acid (RNA) inhibitor targeting C5, reduced mean proteinuria by 37% versus placebo therapy at 32 weeks in a Phase II randomized placebo-controlled study of IgAN patients with proteinuria > 1 g/day and was generally well tolerated; full results are awaited (NCT03841448) [175]. Ravulizumab is a FDA-approved monoclonal antibody that binds C5 and prevents its activation. In an ongoing Phase II, randomized, placebo-controlled study (NCT04564339), the efficacy of ravulizumab administered intravenously is being tested in patients with IgAN. Gefurulimab is a small molecule inhibitor of C5 that is administered subcutaneously (anti-C5 albumin-binding humanized biospecific VHH antibody) and is being tested for IgAN in a single-arm Phase I study (NCT05314231). Avacopan (CCX168), an oral small-molecule antagonist of the inflammatory response, binds to the surface of C5aR to block C5a binding through allosteric effects on the C5a-binding pocket. In a recent publication, avacopan was found to be non-inferior to corticosteroids in the treatment of anti-neutrophilic cytoplasmic antibody (ANCA)-associated vasculitis at 26 weeks and superior at 52 weeks [176]. A short-term open-label pilot study evaluated the efficacy of avacopan in patients with IgAN. Avacopan 30 mg twice daily (BID) reduced proteinuria by about 50% in three of seven IgAN patients (eGFR > 60 mL/min/1.73 m2 or > 45 mL/min/1.73 m2 if eGFR had not declined > 10 mL/min/1.73 m2 over the previous 24 weeks) with a urine protein-to-creatinine ratio (UPCR) > 1 g/g despite 8 weeks of maximally tolerated RAASI use. The urinary monocyte chemoattractant protein-1-to-creatinine ratio decreased by 30% after 8 weeks of avacopan use, possibly due to its anti-inflammatory property, and therapy was well tolerated (NCT02384317) [177].
Table 2.
Ongoing clinical trials targeting a complement pathway for treatment of primary immunoglobulin A nephropathy
| Intervention | Mechanism | Phase | Design | Study population* | Outcomes* | Identifier |
|---|---|---|---|---|---|---|
| ARO-C3 | Ribonucleic acid interference [RNAi] molecule that inhibits hepatic complement component 3 [C3] production | I, IIa | Randomized, doseescalating |
Inclusion criteria: Age 18–75 y Vaccinated or willing to undergo vaccination with a meningococcal, pneumococcal, and Haemophilus influenzas vaccine Significant proteinuria eGFR ≥ 40 mL/min/1.73 m2 Maximally recommended or tolerated dose of ACEi/ARB Exclusion criteria: History of recurrent or chronic infections History of meningococcal infection History of asplenia or splenectomy |
Primary outcome: Adverse events and/or serious adverse events up to Day 169 Secondary outcomes: Pharmacokinetics of investigational compound up to 48 h post-dose Change from baseline in serum C3 level from baseline up to Day 169 Percent change in serum C3 levels from baseline up to Day 169 |
NCT05083364 |
| Pegcetacoplan [APL-2] | Cyclic peptide inhibitor of complement component C3 | II | Single group, open-label |
Inclusion criteria: Age ≥ 18 y Prior kidney-biopsy results for C3 and C4d staining Proteinuria >750 mg/g eGFR ≥ 30 mL/min/1.73 m2 Optimized treatment with immunosuppressants, anti-hypertensives and/or anti-proteinuric agents for 2 mo Vaccination or being willing to be vaccinated for pneumococcal, meningococcal, and Haemophilus influenzae type b Exclusion criteria: Absolute neutrophil count < 1000 cells/mm3 ALT or AST ≥ 3 times the upper limit of normal History of solid-organ transplant Diagnosis of HIV, HBV, or HCV infection, or positive serology [previous HBV or HCV cleared by treatment is allowed] |
Primary outcome: Reduction in UPCR from baseline to Week 48 Secondary outcomes: Changes in serum C3 levels, AH50 levels, C3a concentrations, and serum albumin levels up to Week 48 UPCR < 200 mg/g at Week 48 Stabilization or improvement in eGFR from baseline to Week 48 |
NCT03453619 |
| IONIS-FB-LRx | Antisense oligonucleotide [ASO] inhibitor of complement factor B | II | Single group, open-label |
Inclusion criteria: Kidney-biopsy diagnosis of IgAN eGFR > 40 mL/min/1.73 m2 Exclusion criteria: Immunodeficiencies of B-lymphocyte function, splenectomy, or history of recurrent meningococcal disease History of kidney or other transplant Immunosuppressant use within 12 mo of study drug administration |
Primary outcome: Percent reduction in 24-h proteinuria from baseline to Week 29 Secondary outcomes: Reduction in 24-h proteinuria from baseline to Week 29 Reduction in UACR and UPCR from baseline to Week 29 % change in plasma FB level from baseline to Week 29 % change in plasma AH50 from baseline to Week 29 |
NCT04014335 |
| R07434656 [Previously IONIS-FB-LRx] | Antisense oligonucleotide [ASO] inhibitor of complement factor B | III | Multicenter, randomized, double-blind, placebo-controlled |
Inclusion criteria: Kidney biopsy diagnosis of IgAN within 7 y Maximum tolerated doses of ACEi/ARB for ≥ 90 d prior to screening UPCR ≥ 1 g/g or proteinuria ≥ 1 g/d eGFR ≥ 20 mL/min/1.73 m2 Exclusion criteria: ≥ 50% crescents on kidney biopsy, sustained doubling of serum creatinine within 3 mo, or rapidly progressive glomerulonephritis |
Primary outcome: Change in proteinuria from baseline to Week 37 Secondary outcomes: eGFR slope from baseline to Week 105 Time to the composite kidney failure endpoint [up to 7 y]; defined as dialysis or transplantation, or a sustained decline in eGFR ≥ 30%, without use of other immunosuppressives for IgAN |
NCT05797610 |
| Iptacopan [LNP023] | Oral complement factor B inhibitor | III | Randomized, double-blind, placebo-controlled, parallel group |
Inclusion criteria: Age ≥ 18 y For patients with eGFR ≥ 45 mL/min/1.73 m2, biopsy within preceding 5 y For patients with eGFR ≥ 30 but < 45 mL/min/1.73 m2, biopsy within 2 y with < 50% tubulointerstitial fibrosis For patients with eGFR ≥ 20 but < 30 mL/min/1.73 m2, biopsy at any time UPCR ≥ 1 g/g Vaccination against Neisseria meningitides, Streptococcus pmum/miaz and Haemophilus influenza, prior to study treatment Stable dose of maximal labelled or tolerated ACEi/ARB for 90 d prior to study treatment Exclusion criteria: Uncontrolled hypertension Prior treatment with immunosuppressive or immunomodulatory agents History of recurrent invasive infections caused by encapsulated organisms, such as meningococcus and pneumococcus Active systemic bacterial, viral, or fungal infection < 14 d prior starting study drug |
Primary outcomes: Ratio [UPCR 9-month]/[UPCR baseline] Annualized slope of eGFR over 24 months Secondary outcomes: Change in eGFR from baseline to 9 months Proportion of participants reaching UPCR < 1 g/g at 9 months Annualized slope of eGFR over 12 months |
NCT04578834 |
| Vemircopan [ALXN2050] | Oral complement factor D inhibitor | II | Randomized, double-blind, placebo-controlled |
Inclusion criteria: Age 18–75 y eGFR > 30 mL/min/1.73 m2 Proteinuria ≥ 1 g/d Hematuria on urinalysis [only applicable if IgAN biopsy > 2 y prior to screening] Maximum allowed or tolerated ACEi and/or ARB dose for ≥ 3 mo Stable BP control Exclusion criteria: ≥ 50% interstitial fibrosis, tubular atrophy, glomerular sclerosis, or crescent formation in glomeruli on most recent kidney biopsy Splenectomy or functional asplenia Known or suspected complement deficiency, unless attributable to underlying IgAN Absolute neutrophil count < 1.3 × 103/μL or platelet count < 50,000/mm3 eGFR loss ≥ 30% over 3 mo Prednisone or prednisone equivalent > 20 mg/d for > 14 consecutive d or any other immunosuppression within 6 mo |
Primary outcome: Percentage change in proteinuria from baseline to Week 26 Secondary outcomes: Percentage change in proteinuria from baseline to Week 50 Participants achieving > 30% and > 50% reduction in proteinuria at Week 26 and Week 50 compared to baseline proteinuria Change in eGFR from baseline to Week 26 and Week 50 Absolute values and change from baseline in plasma Bb level to Week 50 Absolute values and change from baseline in serum alternative pathway activity at Week 50 |
NCT05097989 |
| KP104 | Fusion protein of an IgG4 monoclonal antibody directed against complement component C5 fused to complement factor H 1–5 domain [FH 1–5] | II | Randomized, sequential assignment, open-label |
Inclusion criteria: Age 18–75 y eGFR > 30 mL/min/1.73 m2 UPCR > 1.5 g/g Kidney-biopsy diagnosis in past 12 mo Stable regimen of ACEi/ARB for 12 wk and/or SGLT2 inhibitors for 6 wk Exclusion criteria: Treatment of any infection with IV [within 30 d of screening] or oral [within 14 d of screening] antibiotics, antivirals, or antifungals History of infections with encapsulated organisms or tuberculosis Absolute neutrophil count < 500 cells/μL Crescents > 50% of glomeruli on kidney biopsy Nephrotic syndrome Decline in eGFR > 30 mL/min/1.73 m2 in 24 wk Receiving or anticipated dialysis or transplantation during study |
Primary outcomes: Percent change in proteinuria from baseline to Week 48 with weekly dosing Percent change in proteinuria from baseline to Week 47 with biweekly dosing Secondary outcomes: Adverse events up to Week 56 Change from baseline in rabbit red blood cell assay at baseline and at Week 48 Change in C3b activity assay from baseline to Week 48 Change in free serum C5 levels from baseline to Week 48 Change in eGFR from baseline to Week 48 for weekly maintenance dosing Change in eGFR from baseline to Week 47 for biweekly maintenance dosing |
NCT05517980 |
| Gefurulimab [ALXN1720] | Human-derived bispecific antibody against complement component C5 and albumin | lb | Open-label, single-dose |
Inclusion criteria: Age 19–65 y eGFR > 30 mL/min/1.73 m2 Proteinuria > 1 g/d Exclusion criteria: Kidney transplant Treatment with a complement inhibitor at any time Prior treatment with rituximab |
Primary outcome: Serum concentration of ALXN1720 Secondary outcomes: Treatment-emergent adverse events Serum C5 concentration [free and total] Number of participants with anti-ALXN1720 antibodies |
NCT05314231 |
| Ravulizumab | Humanized monoclonal antibody against complement component C5 | II | Randomized, double-blind, placebo-controlled |
Inclusion criteria: Age 18–75 y eGFR > 30 mL/min/1.73 m2 Proteinuria ≥ 1 g/d or ≥ 1 g/g Vaccinated against meningococcal infection Vaccinated against Haemophilus influenzae type b and Streptococcus pneumoniae Stable and optimal dose of RAAS inhibitor ≥ 3 mo Exclusion criteria: Previous treatment with a complement inhibitor History of other solid-organ or bone-marrow transplant Rapidly progressive glomerulonephritis Prednisone or prednisone equivalent > 20 mg/d for > 14 consecutive d or any other immunosuppression within 6 mo |
Primary outcome: Change in proteinuria from baseline to Week 26 Secondary outcomes: Change in proteinuria from baseline to Week 50 Change in eGFR from baseline to Weeks 26 & 50 Partial remission of proteinuria at Weeks 26 & 50 |
NCT04564339 |
| Avacopan [CCX168] | Oral small-molecule C5a receptor antagonist | II | Single group, open-label |
Inclusion criteria: Age ≥ 18 y eGFR > 60 mL/min/1.73 m2 UPCR > 1 g/g Exclusion criteria: Proteinuria > 8 g/d or UPCR > 8 g/g Systemic manifestations of IgA vasculitis within prior 2 y Severe crescentic IgAN Treatment with immunosuppressants within prior 24 wk Active infection |
Primary outcome: Number of patients with adverse events up to 169 days Secondary outcome: Change in proteinuria from baseline up to 169 days |
NCT02384317 |
| Cemdisiran [ALN-CC5] | N-acetylgalactosamine-conjugated small interfering RNA [siRNA] that suppresses liver production of complement component C5 | II | Randomized, double-blind, placebo-controlled |
Inclusion criteria: Age 18–75 y eGFR > 30 m\L/min/1.73 m2 Stable optimal conservative therapy with ACEi/ARB/direct renin-inhibitor Proteinuria > 1 g/d Exclusion criteria: Rapidly progressive glomerulonephritis IgA vasculitis [Henoch-Schönlein purpura] Diagnosis of HIV, HBV, or HCV infection Treatment with systemic corticosteroids ≥ 7 d, or other immunosuppressant agents in past 6 mo Organ transplant recipient |
Primary outcome: Change in UPCR from baseline to Week 32 Secondary outcomes: Change in proteinuria g/d from baseline to Week 32 Partial clinical remission at Week 32 > 50% reduction in proteinuria g/d at Week 32 Change in UPCR from baseline to Week 32 |
NCT03841448 |
| CM338 | Monoclonal antibody targeting mannose-binding lectin-associated serine protease-2 [MASP-2] | II | Randomized, parallel assignment, open-label |
Inclusion criteria: Age 18–75 y Exclusion criteria: Active HIV or mycobacterium tuberculosis infection |
Primary outcome: Incidence of adverse events |
NCT05775042 |
| Narsoplimab [OMS721] | Humanized monoclonal antibody against MASP-2 | III | Randomized, double-blind, placebo-controlled |
Inclusion criteria: Age ≥ 18 y Biopsy diagnosis of IgAN within 8 y Proteinuria > 1 g/d within 6 mo or UPCR >750 mg/g Proteinuria > 1 g/d eGFR ≥ 30 mL/min/1.73 m2 Exclusion criteria: Treatment with immunosuppressive or cytotoxic drugs for IgAN within 8 weeks Type 1 diabetes mellitus, HbA1c > 7.5, diabetic nephropathy on biopsy, IgA vasculitis, secondary IgAN, or other kidney disease History of kidney transplantation Rapidly progressive glomerulonephritis |
Primary outcome: Change in proteinuria after 36 wk on treatment Secondary outcomes: Number of patients with treatment-related adverse events up to 168 wk Change from baseline in kidney function as determined by the rate of change in eGFR up to 144 wk on treatment Change from baseline in proteinuria g/day at 36 wk on treatment in the subset of patients with baseline high proteinuria [defined as proteinuria ≥ 2 g/d] Time-averaged change in UPCR through 36 wk |
NCT03608033 |
ACEi angiotensin converting enzyme inhibitor, AH-50 hemolytic complement activity of the alternative complement pathway, ALT alanine transaminase, ARB angiotensin II receptor blocker, AST aspartate transaminase, BP blood pressure, eGFR estimated glomerular filtration rate, HbA1c hemoglobin A1c, HBV hepatitis B virus, HCV hepatitis C virus, HIV human immunodeficiency virus, IV intravenous, RAAS renin-angiotensin-aldosterone system, SGLT2 sodium glucose cotransporter 2, UACR urine albumin-to-creatinine ratio, UPCR urine protein-to-creatinine ratio
Complete inclusion and exclusion criteria and secondary outcome measures are available upon review of the study protocol by entering the Identifier at the www.clinicaltrials.gov website
Beyond the terminal complement pathway, other strategies have focused on upstream targeting of the alternative and lectin pathways. Iptacopan (LNP023) is an oral, highly potent, selective inhibitor of FB that effectively blocks the alternative pathway by suppressing activity of its C3 convertase, thereby preventing cleavage of C3 and activation of the amplification loop. In a recent Phase II study, iptacopan reduced proteinuria by 23% at 3 months and led to further reduction, up to 40%, at 6 months [178]. With these encouraging results, Novartis launched its Phase III APPLAUSE-IgAN trial (NCT04578834) testing iptacopan 200 mg BID versus placebo in IgAN patients at high risk of disease progression [179]. IONIS-FB-LRx is an antisense inhibitor of FB messenger RNA that disrupts FB translation in hepatocytes. In an exploratory single-arm open-label Phase II study involving 10 IgAN patients with eGFR > 45 mL/min/1.73 m2 and proteinuria > 1 g/day despite maximal tolerated RAASI for at least 60 days reduced proteinuria by 44% with no change in eGFR at 29 weeks compared to baseline and was well tolerated (NCT04014335) [180]. Another trial targeting the alternative complement pathway is testing the efficacy and safety of vemircopan [ALXN2050), an oral FD inhibitor, in patients with proliferative lupus nephritis or IgAN. Recruitment for this study is still underway (NCT05097989).
Mannose-binding protein-associated serine proteases (MASP)-2 is an important component of the lectin pathway that, with MASP-1, cleaves C4 and C2 into active fragments. Subsequently MASP-2 triggers formation of C3 convertase and leads to downstream inflammatory effects. Mannose-binding protein-associated serine proteases (MASP)-2 inhibition can thereby curtail activation of the lectin pathway while preserving the capacity to generate C3 convertase via the classical and alternative pathways. Narsoplimab (OMS721) is a humanized monoclonal IgG4 antibody selectively targeting MASP-2. In a Phase II multicenter clinical trial, IgAN patients with proteinuria > 1 g/day despite maximally tolerated RAASI therapy and baseline eGFR > 30 mL/min/1.73 m2 were enrolled into two sub-studies based on whether they were receiving low-dose corticosteroid or not at baseline. Interim analysis of both groups revealed the drug was safe, well-tolerated, and decreased proteinuria while maintaining a stable eGFR [181]. Based on these preliminary results, a randomized, double-blind, phase III trial testing weekly intravenous infusions of narsoplimab versus placebo was launched (ARTEMIS-IGAN; NCT03608033).
Besides evaluating the efficacy of complement inhibitors, a major goal of all these trials is to assess the risks (particularly infections) associated with their use. Most safety data available to date are derived from the use of inhibitors of the terminal pathway in other disease states. Eculizumab substantially increases the risk of infections with encapsulated organisms. While rare, the frequency of meningococcal infection is increased nearly 2000-fold compared to that in the general population, according to the package insert for the drug. Fortunately, this risk can be mitigated by appropriate vaccination practices and patients prescribed eculizumab or ravulizumab are advised to undergo vaccination against meningococcal serogroups A, B, C, Y, and W-135 at least two weeks prior to therapy initiation. Alternatively, patients who need urgent treatment with C5 inhibitors are encouraged to be vaccinated against encapsulated organisms, and are given antibiotic prophylaxis in the interim [182]. Inhibitors of the lectin or alternative pathway do not prevent downstream activation of terminal complement pathway and formation of MAC and, thus, are likely to confer a better safety profile. Nonetheless, vaccination against Neisseria and pneumococcal infections is recommended for patients in clinical trials assessing the efficacy of inhibition of the alternative complement pathway is recommended as this approach mitigates the risk of infection [183, 184].
Because therapeutic targeting of the complement system is a potential treatment option for IgAN, further efforts to develop and validate clinical markers of complement activation are needed. These markers should enable providers to select appropriate patients for treatment with complement inhibitors and to assess the response to and duration of therapy [185]. Safety data, however, remain paramount before complement inhibitors can be widely used in clinical practice [186]. Ultimately, the risk/benefit ratio of immunomodulation with complement inhibitors in IgAN will need to be addressed by these ongoing and future clinical trials. The balance will depend on the pathway targeted and treatment duration; with other potential therapies also on the horizon, the optimal combinations will need to be defined.
It is noteworthy that IgAN could be the first disease with significant prevalence to be treated with drugs targeting the complement system: to date, such therapy has been approved for only for ultra-rare and orphan diseases, including atypical hemolytic uremic syndrome and paroxysmal nocturnal hemoglobinuria [187, 188]. The cost of complement-based therapies in IgAN warrants consideration. Currently available inhibitors of the complement system, such as eculizumab and ravulizumab, are expensive (> 500,000 USD per year per patient] [189]. Cheaper prescription could potentially be achieved by extending the applications of complement-targeted therapies to a larger patient population, such as those with IgAN. Furthermore, joint support from the pharmaceutical industry, medical insurance providers, and governmental authorities would be pivotal in the large-scale implementation of complement-based therapeutics.
6. Conclusion
Tremendous progress has been made in understanding the role of the complement system in IgAN pathogenesis and prognosis over the last few decades. Translational data from pathologic, biochemical, and genetic studies as well as animal models provide confirmatory evidence regarding the involvement of the lectin and alternative complement pathways. Markers of complement activation provide valuable diagnostic information and more recently have emerged as key prognostic tools to risk-stratify disease severity. Complement components contribute significantly in amplifying the inflammatory signals responsible for the formation of nephritogenic immune complexes and their deposition in the glomeruli. These findings have rekindled a marked interest in targeting the complement cascade at various levels in an effort to arrest or slow kidney disease progression. The plethora of clinical trials in IgAN will hopefully shed further insight regarding the optimal timing, intensity, and duration of the use of complement inhibitors along with key efficacy and safety data on another new class of drugs available in our armamentarium for the treatment of this chronic disease.
Key Points.
The alternative and lectin pathways of complement system play an important role in the pathogenesis of IgAN, and their components in the circulation and the immune deposits in the kidneys in patients with IgAN are of prognostic value.
Therapeutic agents targeting the alternative and lectin complement pathways are increasingly being tested in clinical trials as potential treatment options for patients with IgAN.
Biomarkers of activation of the complement-system should enable providers to select appropriate IgAN patients who may benefit from personalized complement-directed therapy.
Biomarkers that can assess the effects of inhibitors of the complement system are needed to guide treatment duration and monitor the response to therapy.
Funding
The authors of this manuscript have been supported in part by Grants AI149431, DK078244, and DK082753 from the National Institutes of Health, and a gift from the IGA Nephropathy Foundation.
Conflict of interest
JN is a co-founder and co-owner of and consultant for Reliant Glycosciences, LLC. JN is a co-inventor on US patent application 14/318,082 (assigned to UAB Research Foundation). JN has a sponsored research agreement with Travere Therapeutics. BAJ is a co-founder and co-owner of Reliant Glycosciences, LLC and a co-inventor on US patent application 14/318,082 (assigned to UAB Research Foundation). MBR is a co-founder and co-owner of and consultant for Reliant Glycosciences, LLC. MBR is a co-inventor on US patent application 14/318,082 (assigned to UAB Research Foundation). DVR received research funding from Reata Pharmaceuticals, Travere Therapeutics (Retrophin), Pfizer Pharmaceuticals, Calliditas Therapeutics (Pharmalink), Otsuka Pharmaceuticals (Visterra), Vertex Pharmaceuticals, Chinook Pharmaceuticals, consultancy fees from Novartis, GSK, George Clinical, Eledon Pharmaceuticals, Otsuka Pharmaceuticals (Visterra), Calliditas Therapeutics (Pharmalink), Chinook Pharmaceuticals. DVR is co-founder and co-owner of Reliant Glycosciences, LLC. None of the listed commercial entities contributed to this study. AR and TJG have nothing to disclose.
References
- 1.Berger J, Hinglais N. intercapillary deposits of IgA-IgG. J Urol Nephrol (Paris). 1968;74(9):694–5. [PubMed] [Google Scholar]
- 2.Berthoux FC, Mohey H, Afiani A. Natural history of primary IgA nephropathy. Semin Nephrol. 2008;28(1):4–9. 10.1016/j.semnephrol.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 3.Hastings MC, Bursac Z, Julian BA, Villa Baca E, Featherston J, Woodford SY, et al. Life expectancy for patients from the southeastern United States with IgA nephropathy. Kidney Int Rep. 2018;3(1):99–104. 10.1016/j.ekir.2017.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jennette JC. The immunohistology of IgA nephropathy. Am J Kidney Dis. 1988;12(5):348–52. 10.1016/s0272-6386(88)80022-2. [DOI] [PubMed] [Google Scholar]
- 5.Conley ME, Cooper MD, Michael AF. Selective deposition of immunoglobulin A1 in immunoglobulin A nephropathy, anaphylactoid purpura nephritis, and systemic lupus erythematosus. J Clin Invest. 1980;66(6):1432–6. 10.1172/jci109998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rajasekaran A, Julian BA, Rizk DV. IgA nephropathy: an interesting autoimmune kidney disease. Am J Med Sci. 2021;361(2):176–94. 10.1016/j.amjms.2020.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Amico G Natural history of idiopathic IgA nephropathy and factors predictive of disease outcome. Semin Nephrol. 2004;24(3):179–96. 10.1016/j.semnephrol.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 8.Saha MK, Julian BA, Novak J, Rizk DV. Secondary IgA nephropathy. Kidney Int. 2018;94(4):674–81. 10.1016/j.kint.2018.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hastings MC, Rizk DV, Kiryluk K, Nelson R, Zahr RS, Novak J, et al. IgA vasculitis with nephritis: update of pathogenesis with clinical implications. Pediatr Nephrol. 2022;37(4):719–33. 10.1007/s00467-021-04950-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Simon P, Ramee MP, Boulahrouz R, Stanescu C, Charasse C, Ang KS, et al. Epidemiologic data of primary glomerular diseases in western France. Kidney Int. 2004;66(3):905–8. 10.1111/j.1523-1755.2004.00834.x. [DOI] [PubMed] [Google Scholar]
- 11.Donadio JV, Grande JP. IgA nephropathy. N Engl J Med. 2002;347(10):738–48. 10.1056/NEJMra020109. [DOI] [PubMed] [Google Scholar]
- 12.Utsunomiya Y, Koda T, Kado T, Okada S, Hayashi A, Kanzaki S, et al. Incidence of pediatric IgA nephropathy. Pediatr Nephrol. 2003;18(6):511–5. 10.1007/s00467-003-1127-z. [DOI] [PubMed] [Google Scholar]
- 13.Shibano T, Takagi N, Maekawa K, Mae H, Hattori M, Takeshima Y, et al. Epidemiological survey and clinical investigation of pediatric IgA nephropathy. Clin Exp Nephrol. 2016;20(1):111–7. 10.1007/s10157-015-1129-8. [DOI] [PubMed] [Google Scholar]
- 14.McQuarrie EP, Mackinnon B, McNeice V, Fox JG, Geddes CC. The incidence of biopsy-proven IgA nephropathy is associated with multiple socioeconomic deprivation. Kidney Int. 2014;85(1):198–203. 10.1038/ki.2013.329. [DOI] [PubMed] [Google Scholar]
- 15.Schena FP, Nistor I. Epidemiology of IgA nephropathy: a global perspective. Semin Nephrol. 2018;38(5):435–42. 10.1016/j.semnephrol.2018.05.013. [DOI] [PubMed] [Google Scholar]
- 16.Julian BA, Quiggins PA, Thompson JS, Woodford SY, Gleason K, Wyatt RJ. Familial IgA nephropathy. Evidence of an inherited mechanism of disease. N Engl J Med. 1985;312(4):202–8. 10.1056/nejm198501243120403. [DOI] [PubMed] [Google Scholar]
- 17.Hsu SI, Ramirez SB, Winn MP, Bonventre JV, Owen WF. Evidence for genetic factors in the development and progression of IgA nephropathy. Kidney Int. 2000;57(5):1818–35. 10.1046/j.1523-1755.2000.00032.x. [DOI] [PubMed] [Google Scholar]
- 18.Cattran DC, Coppo R, Cook HT, Feehally J, Roberts IS, Troyanov S, et al. The Oxford classification of IgA nephropathy: rationale, clinicopathological correlations, and classification. Kidney Int. 2009;76(5):534–45. 10.1038/ki.2009.243. [DOI] [PubMed] [Google Scholar]
- 19.Trimarchi H, Barratt J, Cattran DC, Cook HT, Coppo R, Haas M, et al. Oxford classification of IgA nephropathy 2016: an update from the IgA nephropathy classification working group. Kidney Int. 2017;91(5):1014–21. 10.1016/j.kint.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Berthoux F, Mohey H, Laurent B, Mariat C, Afiani A, Thibaudin L. Predicting the risk for dialysis or death in IgA nephropathy. J Am Soc Nephrol. 2011;22(4):752–61. 10.1681/asn.2010040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inker LA, Heerspink HJL, Tighiouart H, Chaudhari J, Miao S, Diva U, et al. Association of treatment effects on early change in urine protein and treatment effects on GFR slope in IgA nephropathy: an individual participant meta-analysis. Am J Kidney Dis. 2021;78(3):340–9.e1. 10.1053/j.ajkd.2021.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thompson A, Carroll K, Inker LA, Floege J, Perkovic V, Boyer-Suavet S, et al. Proteinuria reduction as a surrogate end point in trials of IgA nephropathy. Clin J Am Soc Nephrol. 2019;14(3):469–81. 10.2215/cjn.08600718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barratt J, Lafayette R, Kristensen J, Stone A, Cattran D, Floege J, et al. Results from part A of the multi-center, double-blind, randomized, placebo-controlled NefIgArd trial, which evaluated targeted-release formulation of budesonide for the treatment of primary immunoglobulin A nephropathy. Kidney Int. 2023;103(2):391–402. 10.1016/j.kint.2022.09.017. [DOI] [PubMed] [Google Scholar]
- 24.Heerspink HJL, Radhakrishnan J, Alpers CE, Barratt J, Bieler S, Diva U, et al. Sparsentan in patients with IgA nephropathy: A prespecified interim analysis from a randomised, double-blind, active-controlled clinical trial. Lancet. 2023;401(10388):1584–94. 10.1016/s0140-6736(23)00569-x. [DOI] [PubMed] [Google Scholar]
- 25.Fagarasan S. Evolution, development, mechanism and function of IgA in the gut. Curr Opin Immunol. 2008;20(2):170–7. 10.1016/j.coi.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 26.Allen AC, Bailey EM, Brenchley PE, Buck KS, Barratt J, Feehally J. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: observations in three patients. Kidney Int. 2001;60(3):969–73. 10.1046/j.1523-1755.2001.060003969.x. [DOI] [PubMed] [Google Scholar]
- 27.Hiki Y, Odani H, Takahashi M, Yasuda Y, Nishimoto A, Iwase H, et al. Mass spectrometry proves under-O-glycosylation of glomerular IgA1 in IgA nephropathy. Kidney Int. 2001;59(3):1077–85. 10.1046/j.1523-1755.2001.0590031077.x. [DOI] [PubMed] [Google Scholar]
- 28.Tomana M, Matousovic K, Julian BA, Radl J, Konecny K, Mestecky J. Galactose-deficient IgA1 in sera of IgA nephropathy patients is present in complexes with IgG. Kidney Int. 1997;52(2):509–16. 10.1038/ki.1997.361. [DOI] [PubMed] [Google Scholar]
- 29.Suzuki H, Fan R, Zhang Z, Brown R, Hall S, Julian BA, et al. Aberrantly glycosylated IgA1 in IgA nephropathy patients is recognized by IgG antibodies with restricted heterogeneity. J Clin Invest. 2009;119(6):1668–77. 10.1172/jci38468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomana M, Novak J, Julian BA, Matousovic K, Konecny K, Mestecky J. Circulating immune complexes in IgA nephropathy consist of IgA1 with galactose-deficient hinge region and anti-glycan antibodies. J Clin Invest. 1999;104(1):73–81. 10.1172/jci5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Novak J, Tomana M, Matousovic K, Brown R, Hall S, Novak L, et al. IgA1-containing immune complexes in IgA nephropathy differentially affect proliferation of mesangial cells. Kidney Int. 2005;67(2):504–13. 10.1111/j.1523-1755.2005.67107.x. [DOI] [PubMed] [Google Scholar]
- 32.Suzuki H, Kiryluk K, Novak J, Moldoveanu Z, Herr AB, Renfrow MB, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22(10):1795–803. 10.1681/asn.2011050464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Launay P, Grossetête B, Arcos-Fajardo M, Gaudin E, Torres SP, Beaudoin L, et al. Fc α receptor (CD89) mediates the development of immunoglobulin A (IgA) nephropathy (Berger’s disease). Evidence for pathogenic soluble receptor-IgA complexes in patients and CD89 transgenic mice. J Exp Med. 2000;191(11):1999–2009. 10.1084/jem.191.11.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rizk DV, Saha MK, Hall S, Novak L, Brown R, Huang ZQ, et al. Glomerular immunodeposits of patients with IgA nephropathy are enriched for IgG autoantibodies specific for galactose-deficient IgA1. J Am Soc Nephrol. 2019;30(10):2017–26. 10.1681/asn.2018111156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Aucouturier P, Monteiro RC, Noël LH, Preud’homme JL, Lesavre P. Glomerular and serum immunoglobulin G subclasses in IgA nephropathy. Clin Immunol Immunopathol. 1989;51(3):338–47. 10.1016/0090-1229(89)90032-9. [DOI] [PubMed] [Google Scholar]
- 36.Berger J, Yaneva H, Nabarra B, Barbanel C. Recurrence of mesangial deposition of IgA after renal transplantation. Kidney Int. 1975;7(4):232–41. 10.1038/ki.1975.35. [DOI] [PubMed] [Google Scholar]
- 37.Berger J, Noël LH, Nabarra B. Recurrence of mesangial IgA nephropathy after renal transplantation. Contrib Nephrol. 1984;40:195–7. 10.1159/000409749. [DOI] [PubMed] [Google Scholar]
- 38.Berger J Recurrence of IgA nephropathy in renal allografts. Am J Kidney Dis. 1988;12(5):371–2. 10.1016/s0272-6386(88)80027-1. [DOI] [PubMed] [Google Scholar]
- 39.Chandrakantan A, Ratanapanichkich P, Said M, Barker CV, Julian BA. Recurrent IgA nephropathy after renal transplantation despite immunosuppressive regimens with mycophenolate mofetil. Nephrol Dial Transplant. 2005;20(6):1214–21. 10.1093/ndt/gfh773. [DOI] [PubMed] [Google Scholar]
- 40.Rizk DV, Maillard N, Julian BA, Knoppova B, Green TJ, Novak J, et al. The emerging role of complement proteins as a target for therapy of IgA nephropathy. Front Immunol. 2019;10:504. 10.3389/fimmu.2019.00504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barnum SR. Complement: a primer for the coming therapeutic revolution. Pharmacol Ther. 2017;172:63–72. 10.1016/j.pharmthera.2016.11.014. [DOI] [PubMed] [Google Scholar]
- 42.Hajishengallis G, Reis ES, Mastellos DC, Ricklin D, Lambris JD. Novel mechanisms and functions of complement. Nat Immunol. 2017;18(12):1288–98. 10.1038/ni.3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nesargikar PN, Spiller B, Chavez R. The complement system: history, pathways, cascade and inhibitors. Eur J Microbiol Immunol (Bp). 2012;2(2):103–11. 10.1556/EuJMI.2.2012.2.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–97. 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alper CA, Johnson AM, Birtch AG, Moore FD. Human C3: evidence for the liver as the primary site of synthesis. Science. 1969;163(3864):286–8. 10.1126/science.163.3864.286. [DOI] [PubMed] [Google Scholar]
- 46.Hauptmann G, Tongio MM, Klein J, Mayer S, Cinqualbre J, Jeanblanc B, et al. Change in serum properdin factor B phenotype following human orthoptic liver transplantation. Immunobiology. 1980;158(1–2):76–81. 10.1016/s0171-2985(80)80043-x. [DOI] [PubMed] [Google Scholar]
- 47.Wölpl A, Robin-Winn M, Pichlmayr R, Goldmann SF. Fourth component of complement (C4) polymorphism in human orthotopic liver transplantation. Transplantation. 1985;40(2):154–7. 10.1097/00007890-198508000-00009. [DOI] [PubMed] [Google Scholar]
- 48.Colten HR. Biosynthesis of complement. Adv Immunol. 1976;22:67–118. 10.1016/s0065-2776(08)60548-9. [DOI] [PubMed] [Google Scholar]
- 49.Bajic G, Degn SE, Thiel S, Andersen GR. Complement activation, regulation, and molecular basis for complement-related diseases. Embo J. 2015;34(22):2735–57. 10.15252/embj.201591881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ricklin D, Reis ES, Lambris JD. Complement in disease: a defence system turning offensive. Nat Rev Nephrol. 2016;12(7):383–401. 10.1038/nrneph.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wouters D, Zeerleder S. Complement inhibitors to treat IgM-mediated autoimmune hemolysis. Haematologica. 2015;100(11):1388–95. 10.3324/haematol.2015.128538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kemper C, Pangburn MK, Fishelson Z. Complement nomenclature 2014. Mol Immunol. 2014;61(2):56–8. 10.1016/j.molimm.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 53.Kerr MA. Limited proteolysis of complement components C2 and factor B. Structural analogy and limited sequence homology. Biochem J. 1979;183(3):615–22. 10.1042/bj1830615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dobó J, Kocsis A, Gál P. Be on target: strategies of targeting alternative and lectin pathway components in complement-mediated diseases. Front Immunol. 2018;9:1851. 10.3389/fimmu.2018.01851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I—molecular mechanisms of activation and regulation. Front Immunol. 2015;6:262. 10.3389/fimmu.2015.00262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Noris M, Remuzzi G. Overview of complement activation and regulation. Semin Nephrol. 2013;33(6):479–92. 10.1016/j.semnephrol.2013.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Békássy ZD, Kristoffersson AC, Rebetz J, Tati R, Olin AI, Karpman D. Aliskiren inhibits renin-mediated complement activation. Kidney Int. 2018;94(4):689–700. 10.1016/j.kint.2018.04.004. [DOI] [PubMed] [Google Scholar]
- 58.Almitairi JOM, Venkatraman Girija U, Furze CM, Simpson-Gray X, Badakshi F, Marshall JE, et al. Structure of the C1r–C1s interaction of the C1 complex of complement activation. Proc Natl Acad Sci USA. 2018;115(4):768–73. 10.1073/pnas.1718709115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaboriaud C, Thielens NM, Gregory LA, Rossi V, Fontecilla-Camps JC, Arlaud GJ. Structure and activation of the C1 complex of complement: Unraveling the puzzle. Trends Immunol. 2004;25(7):368–73. 10.1016/j.it.2004.04.008. [DOI] [PubMed] [Google Scholar]
- 60.Medjeral-Thomas NR, Cook HT, Pickering MC. Complement activation in IgA nephropathy. Semin Immunopathol. 2021;43(5):679–90. 10.1007/s00281-021-00882-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Morgan BP. The membrane attack complex as an inflammatory trigger. Immunobiology. 2016;221(6):747–51. 10.1016/j.imbio.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 62.Meri S, Waldmann H, Lachmann PJ. Distribution of protectin (CD59), a complement membrane attack inhibitor, in normal human tissues. Lab Invest. 1991;65(5):532–7. [PubMed] [Google Scholar]
- 63.Morgan BP, Harris CL. Complement, a target for therapy in inflammatory and degenerative diseases. Nat Rev Drug Discov. 2015;14(12):857–77. 10.1038/nrd4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barnum SR, Bubeck D, Schein TN. Soluble membrane attack complex: biochemistry and immunobiology. Front Immunol. 2020;11: 585108. 10.3389/fimmu.2020.585108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zipfel PF, Skerka C. Complement regulators and inhibitory proteins. Nat Rev Immunol. 2009;9(10):729–40. 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 66.Fujita T, Matsushita M, Endo Y. The lectin-complement pathway—its role in innate immunity and evolution. Immunol Rev. 2004;198:185–202. 10.1111/j.0105-2896.2004.0123.x. [DOI] [PubMed] [Google Scholar]
- 67.Davis AE 3rd, Lu F, Mejia P. C1 inhibitor, a multi-functional serine protease inhibitor. Thromb Haemost. 2010;104(5):886–93. 10.1160/th10-01-0073. [DOI] [PubMed] [Google Scholar]
- 68.Beinrohr L, Dobó J, Závodszky P, Gál P. C1, MBL-MASPs and C1-inhibitor: Novel approaches for targeting complement-mediated inflammation. Trends Mol Med. 2008;14(12):511–21. 10.1016/j.molmed.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 69.Ermert D, Blom AM. C4b-binding protein: The good, the bad and the deadly. Novel functions of an old friend. Immunol Lett. 2016;169:82–92. 10.1016/j.imlet.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 70.Nilsson B, Nilsson EK. The tick-over theory revisited: is C3 a contact-activated protein? Immunobiology. 2012;217(11):1106–10. 10.1016/j.imbio.2012.07.008. [DOI] [PubMed] [Google Scholar]
- 71.Schmidt CQ, Herbert AP, Hocking HG, Uhrín D, Barlow PN. Translational mini-review series on complement factor H: structural and functional correlations for factor H. Clin Exp Immunol. 2008;151(1):14–24. 10.1111/j.1365-2249.2007.03553.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim DD, Song WC. Membrane complement regulatory proteins. Clin Immunol. 2006;118(2–3):127–36. 10.1016/j.clim.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 73.Liszewski MK, Atkinson JP. Complement regulator CD46: genetic variants and disease associations. Hum Genomics. 2015;9(1):7. 10.1186/s40246-015-0029-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lublin DM. Review: Cromer and DAF: role in health and disease. Immunohematology. 2005;21(2):39–47. [PubMed] [Google Scholar]
- 75.Medjeral-Thomas N, Pickering MC. The complement factor H-related proteins. Immunol Rev. 2016;274(1):191–201. 10.1111/imr.12477. [DOI] [PubMed] [Google Scholar]
- 76.Zhu L, Guo WY, Shi SF, Liu LJ, Lv JC, Medjeral-Thomas NR, et al. Circulating complement factor H-related protein 5 levels contribute to development and progression of IgA nephropathy. Kidney Int. 2018;94(1):150–8. 10.1016/j.kint.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 77.Tortajada A, Gutiérrez E, Goicoechea de Jorge E, Anter J, Segarra A, Espinosa M, et al. Elevated factor H-related protein 1 and factor H pathogenic variants decrease complement regulation in IgA nephropathy. Kidney Int. 2017;92(4):953–63. 10.1016/j.kint.2017.03.041. [DOI] [PubMed] [Google Scholar]
- 78.Medjeral-Thomas NR, Lomax-Browne HJ, Beckwith H, Willicombe M, McLean AG, Brookes P, et al. Circulating complement factor H-related proteins 1 and 5 correlate with disease activity in IgA nephropathy. Kidney Int. 2017;92(4):942–52. 10.1016/j.kint.2017.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Medjeral-Thomas NR, Troldborg A, Constantinou N, Lomax-Browne HJ, Hansen AG, Willicombe M, et al. Progressive IgA nephropathy is associated with low circulating mannan-binding lectin-associated serine protease-3 (MASP-3) and increased glomerular factor H-related protein-5 (FHR5) deposition. Kidney Int Rep. 2018;3(2):426–38. 10.1016/j.ekir.2017.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tortajada A, Gutierrez E, Pickering MC, Praga Terente M, Medjeral-Thomas N. The role of complement in IgA nephropathy. Mol Immunol. 2019;114:123–32. 10.1016/j.molimm.2019.07.017. [DOI] [PubMed] [Google Scholar]
- 81.Daina E, Cortinovis M, Remuzzi G. Kidney diseases. Immunol Rev. 2023;313(1):239–61. 10.1111/imr.13167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Goicoechea de Jorge E, Caesar JJ, Malik TH, Patel M, Colledge M, Johnson S, et al. Dimerization of complement factor H-related proteins modulates complement activation in vivo. Proc Natl Acad Sci USA. 2013;110(12):4685–90. 10.1073/pnas.1219260110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Intercapillary deposits of IgA-IgG. J Am Soc Nephrol. 2000;11(10):1957–9 10.1681/asn.V11101957. [DOI] [PubMed] [Google Scholar]
- 84.Floege J, Daha MR. IgA nephropathy: new insights into the role of complement. Kidney Int. 2018;94(1):16–8. 10.1016/j.kint.2018.03.009. [DOI] [PubMed] [Google Scholar]
- 85.Maillard N, Wyatt RJ, Julian BA, Kiryluk K, Gharavi A, Fremeaux-Bacchi V, et al. Current understanding of the role of complement in IgA nephropathy. J Am Soc Nephrol. 2015;26(7):1503–12. 10.1681/asn.2014101000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wyatt RJ, Kanayama Y, Julian BA, Negoro N, Sugimoto S, Hudson EC, et al. Complement activation in IgA nephropathy. Kidney Int. 1987;31(4):1019–23. 10.1038/ki.1987.101. [DOI] [PubMed] [Google Scholar]
- 87.Endo M, Ohi H, Ohsawa I, Fujita T, Matsushita M, Fujita T. Glomerular deposition of mannose-binding lectin (MBL) indicates a novel mechanism of complement activation in IgA nephropathy. Nephrol Dial Transplant. 1998;13(8):1984–90. 10.1093/ndt/13.8.1984. [DOI] [PubMed] [Google Scholar]
- 88.Lhotta K, Würzner R, König P. Glomerular deposition of mannose-binding lectin in human glomerulonephritis. Nephrol Dial Transplant. 1999;14(4):881–6. 10.1093/ndt/14.4.881. [DOI] [PubMed] [Google Scholar]
- 89.Endo M, Ohi H, Satomura A, Hidaka M, Ohsawa I, Fujita T, et al. Regulation of in situ complement activation via the lectin pathway in patients with IgA nephropathy. Clin Nephrol. 2001;55(3):185–91. [PubMed] [Google Scholar]
- 90.Roos A, Bouwman LH, van Gijlswijk-Janssen DJ, Faber-Krol MC, Stahl GL, Daha MR. Human IgA activates the complement system via the mannan-binding lectin pathway. J Immunol. 2001;167(5):2861–8. 10.4049/jimmunol.167.5.2861. [DOI] [PubMed] [Google Scholar]
- 91.Daha MR, van Kooten C. Role of complement in IgA nephropathy. J Nephrol. 2016;29(1):1–4. 10.1007/s40620-015-0245-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roos A, Rastaldi MP, Calvaresi N, Oortwijn BD, Schlagwein N, van Gijlswijk-Janssen DJ, et al. Glomerular activation of the lectin pathway of complement in IgA nephropathy is associated with more severe renal disease. J Am Soc Nephrol. 2006;17(6):1724–34. 10.1681/asn.2005090923. [DOI] [PubMed] [Google Scholar]
- 93.Liu LL, Liu N, Chen Y, Wang LN, Jiang Y, Wang J, et al. Glomerular mannose-binding lectin deposition is a useful prognostic predictor in immunoglobulin A nephropathy. Clin Exp Immunol. 2013;174(1):152–60. 10.1111/cei.12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Segarra-Medrano A, Carnicer-Caceres C, Valtierra-Carmeno N, Agraz-Pamplona I, Ramos-Terrades N, Jatem Escalante E, et al. Study of the variables associated with local complement activation in IgA nephropathy. Nefrologia. 2017;37(3):320–9. 10.1016/j.nefro.2016.11.019. [DOI] [PubMed] [Google Scholar]
- 95.Czerkinsky C, Koopman WJ, Jackson S, Collins JE, Crago SS, Schrohenloher RE, et al. Circulating immune complexes and immunoglobulin A rheumatoid factor in patients with mesangial immunoglobulin A nephropathies. J Clin Invest. 1986;77(6):1931–8. 10.1172/jci112522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Crowley-Nowick PA, Campbell E, Schrohenloher RE, Mestecky J, Mestecky J, Jackson S. Polyethylene glycol precipitates of serum contain a large proportion of uncomplexed immunoglobulins and C3. Immunol Invest. 1996;25(1–2):91–101. 10.3109/08820139609059293. [DOI] [PubMed] [Google Scholar]
- 97.Julian BA, Wyatt RJ, McMorrow RG, Galla JH. Serum complement proteins in IgA nephropathy. Clin Nephrol. 1983;20(5):251–8. [PubMed] [Google Scholar]
- 98.Maeda A, Gohda T, Funabiki K, Horikoshi S, Shirato I, Tomino Y. Significance of serum IgA levels and serum IgA/C3 ratio in diagnostic analysis of patients with IgA nephropathy. J Clin Lab Anal. 2003;17(3):73–6. 10.1002/jcla.10071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ishiguro C, Yaguchi Y, Funabiki K, Horikoshi S, Shirato I, Tomino Y. Serum IgA/C3 ratio may predict diagnosis and prognostic grading in patients with IgA nephropathy. Nephron. 2002;91(4):755–8. 10.1159/000065043. [DOI] [PubMed] [Google Scholar]
- 100.Tomino Y, Suzuki S, Imai H, Saito T, Kawamura T, Yorioka N, et al. Measurement of serum IgA and C3 may predict the diagnosis of patients with IgA nephropathy prior to renal biopsy. J Clin Lab Anal. 2000;14(5):220–3. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gong WY, Liu M, Luo D, Liu FN, Yin LH, Li YQ, et al. High serum IgA/C3 ratio better predicts a diagnosis of IgA nephropathy among primary glomerular nephropathy patients with proteinuria ≤ 1 g/d: An observational cross-sectional study. BMC Nephrol. 2019;20(1):150. 10.1186/s12882-019-1331-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Komatsu H, Fujimoto S, Hara S, Sato Y, Yamada K, Eto T. Relationship between serum IgA/C3 ratio and progression of IgA nephropathy. Intern Med. 2004;43(11):1023–8. 10.2169/internalmedicine.43.1023. [DOI] [PubMed] [Google Scholar]
- 103.Karahisar Şirali S, Büberci R. Correlation between IgAC3 ratio and Oxford score in IgA nephropathy. Clin Exp Nephrol. 2022;26(10):982–7. 10.1007/s10157-022-02244-7. [DOI] [PubMed] [Google Scholar]
- 104.Kawasaki Y, Maeda R, Ohara S, Suyama K, Hosoya M. Serum IgA/C3 and glomerular C3 staining predict severity of IgA nephropathy. Pediatr Int. 2018;60(2):162–7. 10.1111/ped.13461. [DOI] [PubMed] [Google Scholar]
- 105.Zhang J, Wang C, Tang Y, Peng H, Ye ZC, Li CC, et al. Serum immunoglobulin A/C3 ratio predicts progression of immunoglobulin A nephropathy.Nephrology (Carlton). 2013;18(2):125–31. 10.1111/nep.12010. [DOI] [PubMed] [Google Scholar]
- 106.Lang Y, Song S, Zhao L, Yang Y, Liu T, Shen Y, et al. Serum IgA/C3 ratio and glomerular C3 staining predict progression of IgA nephropathy in children. Transl Pediatr. 2021;10(3):666–72. 10.21037/tp-21-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mizerska-Wasiak M, Małdyk J, Rybi-Szumińska A, Wasilewska A, Miklaszewska M, Pietrzyk J, et al. Relationship between serum IgA/C3 ratio and severity of histological lesions using the Oxford classification in children with IgA nephropathy. Pediatr Nephrol. 2015;30(7):1113–20. 10.1007/s00467-014-3024-z. [DOI] [PubMed] [Google Scholar]
- 108.Guo WY, Zhu L, Meng SJ, Shi SF, Liu LJ, Lv JC, et al. Mannose-binding lectin levels could predict prognosis in IgA nephropathy. J Am Soc Nephrol. 2017;28(11):3175–81. 10.1681/asn.2017010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang Z, Xie X, Li J, Zhang X, He J, Wang M, et al. Complement activation is associated with crescents in IgA nephropathy. Front Immunol. 2021;12: 676919. 10.3389/fimmu.2021.676919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Niu X, Zhang S, Shao C, Guo Z, Wu J, Tao J, et al. Urinary complement proteins in IgA nephropathy progression from a relative quantitative proteomic analysis. PeerJ. 2023;11: e15125. 10.7717/peerj.15125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hall S, Coffee CS, Huang ZQ, Maillard N, Moldoveanu Z, Hargett AA, et al. Biologically active circulatory immune complexes in IgA nephropathy contain polymeric IgA1, with galactose-deficient and minimally sialylated O-glycans, IgG, and complement C3b and iC3b. J Am Soc Nephrol. 2022;33:780. [Google Scholar]
- 112.Law SK, Dodds AW. The internal thioester and the covalent binding properties of the complement proteins C3 and C4. Protein Sci. 1997;6(2):263–74. 10.1002/pro.5560060201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Antón LC, Ruiz S, Barrio E, Marqués G, Sánchez A, Vivanco F. C3 binds with similar efficiency to Fab and Fc regions of IgG immune aggregates. Eur J Immunol. 1994;24(3):599–604. 10.1002/eji.1830240316. [DOI] [PubMed] [Google Scholar]
- 114.Gadd KJ, Reid KB. The binding of complement component C3 to antibody-antigen aggregates after activation of the alternative pathway in human serum. Biochem J. 1981;195(2):471–80. 10.1042/bj1950471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sahu A, Pangburn MK. Covalent attachment of human complement C3 to IgG. Identification of the amino acid residue involved in ester linkage formation. J Biol Chem. 1994;269(46):28997–9002. [PubMed] [Google Scholar]
- 116.Vidarte L, Pastor C, Mas S, Blazquez AB, de los Rios V, Guerrero R, et al. Serine 132 is the C3 covalent attachment point on the CH1 domain of human IgG1. J Biol Chem. 2001;276(41):38217–23. 10.1074/jbc.M104870200. [DOI] [PubMed] [Google Scholar]
- 117.de Bruijn MH, Fey GH. Human complement component C3: cDNA coding sequence and derived primary structure. Proc Natl Acad Sci USA. 1985;82(3):708–12. 10.1073/pnas.82.3.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Janssen BJ, Huizinga EG, Raaijmakers HC, Roos A, Daha MR, Nilsson-Ekdahl K, et al. Structures of complement component C3 provide insights into the function and evolution of immunity. Nature. 2005;437(7058):505–11. 10.1038/nature04005. [DOI] [PubMed] [Google Scholar]
- 119.Nishida N, Walz T, Springer TA. Structural transitions of complement component C3 and its activation products. Proc Natl Acad Sci USA. 2006;103(52):19737–42. 10.1073/pnas.0609791104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zarantonello A, Revel M, Grunenwald A, Roumenina LT. C3-dependent effector functions of complement. Immunol Rev. 2023;313(1):120–38. 10.1111/imr.13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Renner B, Laskowski J, Poppelaars F, Ferreira VP, Blaine J, Antonioli AH, et al. Factor H related proteins modulate complement activation on kidney cells. Kidney Int. 2022;102(6):1331–44. 10.1016/j.kint.2022.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Papp A, Papp K, Uzonyi B, Cserhalmi M, Csincsi ÁI, Szabó Z, et al. Complement factor H-related proteins FHR1 and FHR5 interact with extracellular matrix ligands, reduce factor H regulatory activity and enhance complement activation. Front Immunol. 2022;13: 845953. 10.3389/fimmu.2022.845953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med. 1981;154(3):856–67. 10.1084/jem.154.3.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pangburn MK. Spontaneous reformation of the intramolecular thioester in complement protein C3 and low temperature capture of a conformational intermediate capable of reformation. J Biol Chem. 1992;267(12):8584–90. [PubMed] [Google Scholar]
- 125.Isenman DE, Kells DI, Cooper NR, Muller-Eberhard HJ, Pangburn MK. Nucleophilic modification of human complement protein C3: correlation of conformational changes with acquisition of C3b-like functional properties. Biochemistry. 1981;20(15):4458–67. 10.1021/bi00518a034. [DOI] [PubMed] [Google Scholar]
- 126.Pangburn MK. Initiation of the alternative pathway of complement and the history of “tickover.” Immunol Rev. 2023;313(1):64–70. 10.1111/imr.13130. [DOI] [PubMed] [Google Scholar]
- 127.Hiemstra PS, Gorter A, Stuurman ME, Van Es LA, Daha MR. Activation of the alternative pathway of complement by human serum IgA. Eur J Immunol. 1987;17(3):321–6. 10.1002/eji.1830170304. [DOI] [PubMed] [Google Scholar]
- 128.Suzuki H, Novak J. IgA glycosylation and immune complex formation in IgAN. Semin Immunopathol. 2021;43(5):669–78. 10.1007/s00281-021-00883-8. [DOI] [PubMed] [Google Scholar]
- 129.Knoppova B, Reily C, King RG, Julian BA, Novak J, Green TJ. Pathogenesis of IgA nephropathy: Current understanding and implications for development of disease-specific treatment. J Clin Med. 2021. 10.3390/jcm10194501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Knoppova B, Reily C, Maillard N, Rizk DV, Moldoveanu Z, Mestecky J, et al. The origin and activities of IgA1-containing immune complexes in IgA nephropathy. Front Immunol. 2016;7:117. 10.3389/fimmu.2016.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Maillard N, Boerma L, Hall S, Huang Z, Mrug M, Moldoveanu Z, et al. Proteomic analysis of engineered IgA1-IgG immune complexes reveals association with activated complement C3. J Am Soc Nephrol. 2013;24:490A. [Google Scholar]
- 132.Novak J, Raskova Kafkova L, Suzuki H, Tomana M, Matousovic K, Brown R, et al. IgA1 immune complexes from pediatric patients with IgA nephropathy activate cultured human mesangial cells. Nephrol Dial Transplant. 2011;26(11):3451–7. 10.1093/ndt/gfr448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Placzek WJ, Yanagawa H, Makita Y, Renfrow MB, Julian BA, Rizk DV, et al. Serum galactose-deficient-IgA1 and IgG autoantibodies correlate in patients with IgA nephropathy. PLoS ONE. 2018;13(1): e0190967. 10.1371/journal.pone.0190967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhao N, Hou P, Lv J, Moldoveanu Z, Li Y, Kiryluk K, et al. The level of galactose-deficient IgA1 in the sera of patients with IgA nephropathy is associated with disease progression. Kidney Int. 2012;82(7):790–6. 10.1038/ki.2012.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Berthoux F, Suzuki H, Thibaudin L, Yanagawa H, Maillard N, Mariat C, et al. Autoantibodies targeting galactose-deficient IgA1 associate with progression of IgA nephropathy. J Am Soc Nephrol. 2012;23(9):1579–87. 10.1681/asn.2012010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Berthoux F, Suzuki H, Mohey H, Maillard N, Mariat C, Novak J, et al. Prognostic value of serum biomarkers of autoimmunity for recurrence of IgA nephropathy after kidney transplantation. J Am Soc Nephrol. 2017;28(6):1943–50. 10.1681/asn.2016060670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Maixnerova D, Ling C, Hall S, Reily C, Brown R, Neprasova M, et al. Galactose-deficient IgA1 and the corresponding IgG autoantibodies predict IgA nephropathy progression. PLoS ONE. 2019;14(2): e0212254. 10.1371/journal.pone.0212254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Berthelot L, Robert T, Vuiblet V, Tabary T, Braconnier A, Dramé M, et al. Recurrent IgA nephropathy is predicted by altered glycosylated IgA, autoantibodies and soluble CD89 complexes. Kidney Int. 2015;88(4):815–22. 10.1038/ki.2015.158. [DOI] [PubMed] [Google Scholar]
- 139.Gutiérrez E, Egido J, Rubio-Navarro A, Buendía I, Blanco Colio LM, Toldos O, et al. Oxidative stress, macrophage infiltration and CD163 expression are determinants of long-term renal outcome in macrohematuria-induced acute kidney injury of IgA nephropathy. Nephron Clin Pract. 2012;121(1–2):c42–53. 10.1159/000342385. [DOI] [PubMed] [Google Scholar]
- 140.Gerogianni A, Dimitrov JD, Zarantonello A, Poillerat V, Chonat S, Sandholm K, et al. Heme interferes with complement factor I-dependent regulation by enhancing alternative pathway activation. Front Immunol. 2022;13: 901876. 10.3389/fimmu.2022.901876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Moldoveanu Z, Suzuki H, Reily C, Satake K, Novak L, Xu N, et al. Experimental evidence of pathogenic role of IgG autoantibodies in IgA nephropathy. J Autoimmun. 2021;118: 102593. 10.1016/j.jaut.2021.102593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kiryluk K, Julian BA, Wyatt RJ, Scolari F, Zhang H, Novak J, et al. Genetic studies of IgA nephropathy: past, present, and future. Pediatr Nephrol. 2010;25(11):2257–68. 10.1007/s00467-010-1500-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kiryluk K, Gharavi AG, Izzi C, Scolari F. IgA nephropathy—the case for a genetic basis becomes stronger. Nephrol Dial Transplant. 2010;25(2):336–8. 10.1093/ndt/gfp593. [DOI] [PubMed] [Google Scholar]
- 144.Feehally J, Farrall M, Boland A, Gale DP, Gut I, Heath S, et al. HLA has strongest association with IgA nephropathy in genome-wide analysis. J Am Soc Nephrol. 2010;21(10):1791–7. 10.1681/asn.2010010076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Gharavi AG, Kiryluk K, Choi M, Li Y, Hou P, Xie J, et al. Genome-wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43(4):321–7. 10.1038/ng.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kiryluk K, Li Y, Sanna-Cherchi S, Rohanizadegan M, Suzuki H, Eitner F, et al. Geographic differences in genetic susceptibility to IgA nephropathy: GWAS replication study and geospatial risk analysis. PLoS Genet. 2012;8(6): e1002765. 10.1371/journal.pgen.1002765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kiryluk K, Li Y, Scolari F, Sanna-Cherchi S, Choi M, Verbitsky M, et al. Discovery of new risk loci for IgA nephropathy implicates genes involved in immunity against intestinal pathogens. Nat Genet. 2014;46(11):1187–96. 10.1038/ng.3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yu XQ, Li M, Zhang H, Low HQ, Wei X, Wang JQ, et al. A genome-wide association study in Han Chinese identifies multiple susceptibility loci for IgA nephropathy. Nat Genet. 2011;44(2):178–82. 10.1038/ng.1047. [DOI] [PubMed] [Google Scholar]
- 149.Li M, Foo JN, Wang JQ, Low HQ, Tang XQ, Toh KY, et al. Identification of new susceptibility loci for IgA nephropathy in Han Chinese. Nat Commun. 2015;6:7270. 10.1038/ncomms8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li M, Wang L, Shi DC, Foo JN, Zhong Z, Khor CC, et al. Genome-wide meta-analysis identifies three novel susceptibility loci and reveals ethnic heterogeneity of genetic susceptibility for IgA nephropathy. J Am Soc Nephrol. 2020;31(12):2949–63. 10.1681/asn.2019080799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kiryluk K, Novak J, Gharavi AG. Pathogenesis of immunoglobulin A nephropathy: recent insight from genetic studies. Annu Rev Med. 2013;64:339–56. 10.1146/annurev-med-041811-142014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kiryluk K, Novak J. The genetics and immunobiology of IgA nephropathy. J Clin Invest. 2014;124(6):2325–32. 10.1172/jci74475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhu L, Zhai YL, Wang FM, Hou P, Lv JC, Xu DM, et al. Variants in complement factor H and complement factor H-related protein genes, CFHR3 and CFHR1, affect complement activation in IgA nephropathy. J Am Soc Nephrol. 2015;26(5):1195–204. 10.1681/asn.2014010096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Jullien P, Laurent B, Claisse G, Masson I, Dinic M, Thibaudin D, et al. Deletion variants of CFHR1 and CFHR3 associate with mesangial immune deposits but not with progression of IgA nephropathy. J Am Soc Nephrol. 2018;29(2):661–9. 10.1681/asn.2017010019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Guo WY, Liu QZ, Zhu L, Li ZY, Meng SJ, Shi SF, et al. Coding and noncoding variants in CFH act synergistically for complement activation in immunoglobulin A nephropathy. Am J Med Sci. 2018;356(2):114–20. 10.1016/j.amjms.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 156.Zhai YL, Meng SJ, Zhu L, Shi SF, Wang SX, Liu LJ, et al. Rare variants in the complement factor H-related protein 5 gene contribute to genetic susceptibility to IgA nephropathy. J Am Soc Nephrol. 2016;27(9):2894–905. 10.1681/asn.2015010012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Guzzo G, Sadallah S, Fodstad H, Venetz JP, Rotman S, Teta D, et al. Case report: a rare truncating variant of the CFHR5 gene in IgA nephropathy. Front Genet. 2021;12: 529236. 10.3389/fgene.2021.529236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hou W, Shi S, Zhou X, Wang S, Cai Q, Chen P, et al. Complement factor H variants are associated with microangiopathy lesions in IgA nephropathy. Int Immunopharmacol. 2022;112: 109234. 10.1016/j.intimp.2022.109234. [DOI] [PubMed] [Google Scholar]
- 159.Shi D, Zhong Z, Xu R, Li B, Li J, Habib U, et al. Association of ITGAX and ITGAM gene polymorphisms with susceptibility to IgA nephropathy. J Hum Genet. 2019;64(9):927–35. 10.1038/s10038-019-0632-2. [DOI] [PubMed] [Google Scholar]
- 160.Sanchez-Rodriguez E, Southard CT, Kiryluk K. GWAS-based discoveries in IgA nephropathy, membranous nephropathy, and steroid-sensitive nephrotic syndrome. Clin J Am Soc Nephrol. 2021;16(3):458–66. 10.2215/cjn.14031119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Liu L, Khan A, Sanchez-Rodriguez E, Zanoni F, Li Y, Steers N, et al. Genetic regulation of serum IgA levels and susceptibility to common immune, infectious, kidney, and cardio-metabolic traits. Nat Commun. 2022;13(1):6859. 10.1038/s41467-022-34456-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Xiao M, Ran Y, Shao J, Lei Z, Chen Y, Li Y. Causal association between inflammatory bowel disease and IgA nephropathy: a bidirectional two-sample mendelian randomization study. Front Genet. 2022;13:1002928. 10.3389/fgene.2022.1002928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gallagher MD, Chen-Plotkin AS. The post-GWAS era: from association to function. Am J Hum Genet. 2018;102(5):717–30. 10.1016/j.ajhg.2018.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Falola O, Adam Y, Ajayi O, Kumuthini J, Adewale S, Mosaku A, et al. SysBiolPGWAS: simplifying post-GWAS analysis through the use of computational technologies and integration of diverse omics datasets. Bioinformatics. 2023. 10.1093/bioinformatics/btac791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jurrjens AW, Seldin MM, Giles C, Meikle PJ, Drew BG, Calkin AC. The potential of integrating human and mouse discovery platforms to advance our understanding of cardiometabolic diseases. eLife. 2023. 10.7554/eLife.86139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Baron C, Cherkaoui S, Therrien-Laperriere S, Ilboudo Y, Poujol R, Mehanna P, et al. Gene-metabolite annotation with shortest reactional distance enhances metabolite genome-wide association studies results. bioRxiv. 2023. 10.1101/2023.03.22.533869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kong S, Li R, Tian Y, Zhang Y, Lu Y, Ou Q, et al. Single-cell omics: a new direction for functional genetic research in human diseases and animal models. Front Genet. 2022;13:1100016. 10.3389/fgene.2022.1100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Battram T, Gaunt TR, Relton CL, Timpson NJ, Hemani G. A comparison of the genes and genesets identified by GWAS and EWAS of fifteen complex traits. Nat Commun. 2022;13(1):7816. 10.1038/s41467-022-35037-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gulbranson DR. Generating custom pooled CRISPR libraries for genetic dissection of biological pathways. Methods Mol Biol. 2022;2473:333–47. 10.1007/978-1-0716-2209-4_21. [DOI] [PubMed] [Google Scholar]
- 170.Rosenblad T, Rebetz J, Johansson M, Békássy Z, Sartz L, Karpman D. Eculizumab treatment for rescue of renal function in IgA nephropathy. Pediatr Nephrol. 2014;29(11):2225–8. 10.1007/s00467-014-2863-y. [DOI] [PubMed] [Google Scholar]
- 171.Ring T, Pedersen BB, Salkus G, Goodship TH. Use of eculizumab in crescentic IgA nephropathy: proof of principle and conundrum? Clin Kidney J. 2015;8(5):489–91. 10.1093/ckj/sfv076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Herzog AL, Wanner C, Amann K, Lopau K. First treatment of relapsing rapidly progressive IgA nephropathy with eculizumab after living kidney donation: a case report. Transplant Proc. 2017;49(7):1574–7. 10.1016/j.transproceed.2017.02.044. [DOI] [PubMed] [Google Scholar]
- 173.Rizk DV. The rapidly changing landscape of IgA nephropathy treatment. Kidney news. 2022;14(12):23. [Google Scholar]
- 174.Rovin BH, Adler SG, Barratt J, Bridoux F, Burdge KA, Chan TM, et al. Executive summary of the KDIGO 2021 guideline for the management of glomerular diseases. Kidney Int. 2021;100(4):753–79. 10.1016/j.kint.2021.05.015. [DOI] [PubMed] [Google Scholar]
- 175.Barratt J, Yeo SC, Fernstrom A, Barbour S, Sperati J, Villanueva AR, et al. FR-OR67: exploratory results from the phase 2 study of cemdisiran in patients with IgA nephropathy. J Am Soc Nephrol. 2022;33:B2. [Google Scholar]
- 176.Jayne DRW, Merkel PA, Schall TJ, Bekker P. Avacopan for the treatment of ANCA-associated vasculitis. N Engl J Med. 2021;384(7):599–609. 10.1056/NEJMoa2023386. [DOI] [PubMed] [Google Scholar]
- 177.Bruchfeld A, Magin H, Nachman P, Parikh S, Lafayette R, Potarca A, et al. C5a receptor inhibitor avacopan in immuneglobulin A nephropathy-an open-label pilot study. Clin Kidney J. 2022;15(5):922–8. 10.1093/ckj/sfab294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Barratt J, Rovin B, Zhang H, Kashihara N, Maes B, Rizk D, et al. POS-546 efficacy and safety of iptacopan in IgA nephropathy: results of a randomized double-blind placebo-controlled phase 2 study at 6 months. Kidney Int Rep. 2022;7(2, Supplement):S236. 10.1016/j.ekir.2022.01.577. [DOI] [Google Scholar]
- 179.Rizk DV, Rovin BH, Zhang H, Kashihara N, Maes B, Trimarchi H, et al. Targeting the alternative complement pathway with iptacopan to treat IgA nephropathy: design and rationale of the APPLAUSE-IgAN study. Kidney Int Rep. 2023;8(5):968–79. 10.1016/j.ekir.2023.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Barbour S, Hladunewich MA, Irvine J, Makris A, Robson RA, Tan SJ, et al. SA-PO714: an exploratory trial of an investigational RNA therapeutic, IONIS-FB-LRx, for treatment of IgA nephropathy. ASN Kidney Week 11/5/2022. J Am Soc Nephrol. 2022;33:800. [Google Scholar]
- 181.Lafayette RA, Rovin BH, Reich HN, Tumlin JA, Floege J, Barratt J. Safety, tolerability and efficacy of narsoplimab, a novel MASP-2 inhibitor for the treatment of IgA nephropathy. Kidney Int Rep. 2020;5(11):2032–41. 10.1016/j.ekir.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Barnum SR. Therapeutic inhibition of complement: well worth the risk. Trends Pharmacol Sci. 2017;38(6):503–5. 10.1016/j.tips.2017.03.009. [DOI] [PubMed] [Google Scholar]
- 183.Muri L, Ispasanie E, Schubart A, Thorburn C, Zamurovic N, Holbro T, et al. Alternative complement pathway inhibition abrogates pneumococcal opsonophagocytosis in vaccine-naïve, but not in vaccinated individuals. Front Immunol. 2021;12: 732146. 10.3389/fimmu.2021.732146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ispasanie E, Muri L, Schubart A, Thorburn C, Zamurovic N, Holbro T, et al. Alternative complement pathway inhibition does not abrogate meningococcal killing by serum of vaccinated individuals. Front Immunol. 2021;12: 747594. 10.3389/fimmu.2021.747594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Selvaskandan H, Shi S, Twaij S, Cheung CK, Barratt J. Monitoring immune responses in IgA nephropathy: biomarkers to guide management. Front Immunol. 2020;11: 572754. 10.3389/fimmu.2020.572754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Tesař V, Radhakrishnan J, Charu V, Barratt J. Challenges in IgA nephropathy management: an era of complement inhibition. Kidney Int Rep. 2023. 10.1016/j.ekir.2023.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Legendre CM, Licht C, Muus P, Greenbaum LA, Babu S, Bedrosian C, et al. Terminal complement inhibitor eculizumab in atypical hemolytic-uremic syndrome. N Engl J Med. 2013;368(23):2169–81. 10.1056/NEJMoa1208981. [DOI] [PubMed] [Google Scholar]
- 188.Gavriilaki E, de Latour RP, Risitano AM. Advancing therapeutic complement inhibition in hematologic diseases: PNH and beyond. Blood. 2022;139(25):3571–82. 10.1182/blood.2021012860. [DOI] [PubMed] [Google Scholar]
- 189.Wang Y, Johnston K, Popoff E, Myren KJ, Cheung A, Faria C, et al. A US cost-minimization model comparing ravulizumab versus eculizumab for the treatment of atypical hemolytic uremic syndrome. J Med Econ. 2020;23(12):1503–15. 10.1080/13696998.2020.1831519. [DOI] [PubMed] [Google Scholar]
- 190.Janssen BJ, Christodoulidou A, McCarthy A, Lambris JD, Gros P. Structure of C3b reveals conformational changes that underlie complement activity. Nature. 2006;444(7116):213–6. 10.1038/nature05172. [DOI] [PubMed] [Google Scholar]
- 191.Fernández FJ, Santos-López J, Martínez-Barricarte R, Querol-García J, Martín-Merinero H, Navas-Yuste S, et al. The crystal structure of iC3b-CR3 αI reveals a modular recognition of the main opsonin iC3b by the CR3 integrin receptor. Nat Commun. 2022;13(1):1955. 10.1038/s41467-022-29580-2. [DOI] [PMC free article] [PubMed] [Google Scholar]