

Abstract
BACKGROUND:
3’, 5’-cyclic AMP (cAMP) regulates numerous cardiac functions. Various hormones and neurotransmitters elevate intracellular cAMP (i[cAMP]) in cardiomyocytes (CMs) through activating stimulatory G protein (Gs)-coupled receptors (GsPCRs) and membrane-bound adenylyl cyclases (ACs). Increasing evidence has indicated that stimulating different GsPCRs and ACs exhibits distinct, even opposite effects, on CM viability. However, the underlying mechanisms are not fully understood.
METHODS:
We used molecular and pharmacological approaches to investigate how different GsPCR/cAMP signaling differentially regulate CM viability with in vitro, ex vivo, and in vivo models.
RESULTS:
For pro-death GsPCRs, we explored beta1-adrenergic-receptor (β1AR) and histamine-H2-receptor (H2R). We found that their pro-death effects were similarly dependent on AC5 activation, ATP release to the extracellular space via pannexin-1 (PANX1) channel, and extracellular ATP (e[ATP])-mediated signaling involving in P2X purinoceptor 7 (P2X7R) and Ca2+/calmodulin-dependent protein kinase II (CaMKII). PANX1 phosphorylation at Serine-206 by cAMP-dependent-protein-kinase-A (PKA) promoted PANX1 activation, which was critical in β1AR- or H2R-induced CM death in vitro and/or in vivo. β1AR or H2R was localized proximately to PANX1, which permits ATP release. For pro-survival GsPCRs, we explored adenosine-A2-receptor (A2R), calcitonin-gene-related-peptide-receptor (CGRPR) and relaxin-family-peptide-receptor 1 (RXFP1). Their pro-survival effects were dependent on AC6 activation, cAMP efflux via multidrug-resistance-protein-4 (MRP4), extracellular cAMP metabolism to adenosine (e[cAMP]-to-e[ADO]), and e[ADO]-mediated signaling. A2R, CGRPR, or RXFP1 was localized proximately to MRP4, which enables cAMP efflux. Interestingly, exogenously increasing e[cAMP] levels by membrane-impermeable cAMP protected against CM death in vitro and in ex vivo and in vivo mouse hearts with ischemia-reperfusion (IR) injuries.
CONCLUSIONS:
Our findings indicate that the functional diversity of different GsPCRs in CM viability could be achieved by their ability to form unique signaling complexes (signalosomes) that determine the fate of cAMP: either stimulate ATP release by activating PKA or directly efflux to be e[cAMP].
Keywords: GsPCR, Cardiomyocyte viability, Extracellular cAMP, Extracellular ATP, PANX1, Basic Science Research, Cell Signaling/Signal Transduction, Ischemia, Mechanisms, Pathophysiology
Graphical Abstract
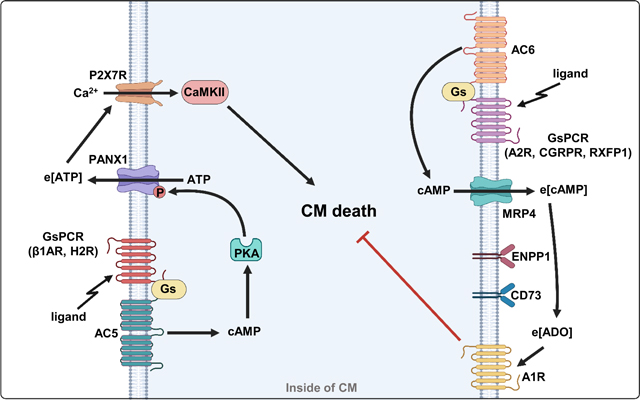
INTRODUCTION
cAMP regulates numerous biological processes in the heart, including cardiac inotropic/chronotropic function, metabolism, viability, and remodeling. i[cAMP] can be generated by ACs via stimulating various GsPCRs. Many different GsPCRs are expressed in CMs 1. There are nine membrane ACs associated with different GsPCRs and expressed in different cell types. i[cAMP] degradation is catalyzed by a large family of phosphodiesterases (PDEs), and a few of them are expressed in each cell 2. The canonical downstream effectors of i[cAMP] include PKA, exchange protein activated by cAMP (Epac), and cyclic nucleotide-gated channel (CNG). Under certain conditions, i[cAMP] is pumped out through MRPs, where e[cAMP] is sequentially converted to extracellular adenosine (e[ADO]) by ecto-nucleotidases 2. e[ADO] in turn triggers downstream signaling by activating one or more of 4 adenosine receptors: A1R, A2AR, A2BR, and A3R 2. e[ADO] has a short half-life due to multiple terminating mechanisms, including degradation by ADO deaminase, conversion to AMP by ADO kinase, and cellular uptake by ADO transporters 3. However, it remains unknown whether all GsPCR-derived cAMP could efflux.
Numerous hormones and neurotransmitters can elevate i[cAMP] levels in CMs by stimulating specific GsPCRs and activating unique ACs. However, stimulating different cAMP signaling exhibits distinct functions in CMs. For example, chronic activation of β1AR/cAMP signaling elicits detrimental effects such as promoting CM hypertrophy and apoptosis 4, while cAMP signaling through activation of A2Rs is protective 5. cAMP produced by AC5 and AC6 have different cardiac effects: AC5/cAMP is detrimental 6, while AC6/cAMP is protective against pathological cardiac remodeling 7. Promoting cAMP signaling via inhibition of different cAMP-hydrolyzing PDEs also elicits distinct effects on CM viability: PDE3A/cAMP promotes while PDE1C/cAMP attenuates CM death 8,9. However, the molecular basis for the functionally distinct cAMP signaling remains elusive. Therefore, there has been increasing interest and effort to understand how the versatility/specificity of diverse cAMP-mediated functions are achieved in CMs.
We aim to uncover different functions and underlying mechanisms of distinct GsPCR/cAMP signaling activation in CMs. The current study focused on CM viability since CM death is associated with many cardiac diseases, including myocardial infarction, ischemia-reperfusion (IR) injury, chemotherapy-induced cardiotoxicity, myocarditis, and heart failure. We used various in vitro, ex vivo, and in vivo models. We also utilized a variety of pharmacological activators and inhibitors and genetically engineered molecular tools to change the expression and activity of proteins of interest experimentally. We explored pro-death GsPCRs (such as β1AR and H2R) and pro-survival GsPCRs (such as A2R, CGRPR, and RXFP1). We found that activating β1AR and H2R promoted CM death via a mechanism dependent on ATP release and e[ATP]-mediated signaling. In contrast, activating A2R, CGRPR, or RXFP1 suppressed H2O2-induced CM death via a mechanism dependent on i[cAMP] efflux and e[cAMP]-mediated signaling. The protective effect of e[cAMP] on myocardium viability was further elucidated using exogenous membrane-impermeable cAMP in ex vivo and in vivo models. The fact that pro-death or pro-survival GsPCRs share the identical e[ATP] or e[cAMP] signaling suggests a certain commonality of these signaling mechanisms in GsPCR-mediated regulation CM viability. Thus, findings from our studies significantly advance the biological insights into how individual GsPCRs can differentially regulate CM viability through discrete cAMP signalosomes. The results may reveal novel molecular targets and pharmacological strategies to increase CM viability.
METHODS
Data Availability
The authors declare that all data and materials within the article and the Data Supplement are available publicly.
Detailed descriptions of experimental methods, materials, and statistical analysis are presented in the Materials of the Data Supplement. The Major Resource Table is also included in the Supplemental Materials.
RESULTS
Stimulation of pro-death GsPCRs such as β1AR and H2R exhibit detrimental effects on CM death, depending on AC5-synthesized cAMP.
Our goal is to understand how different GsPCRs differentially regulate CM viability. We focused on overall CM viability, which was evaluated by two methods. We primarily used trypan blue exclusion assay, a widely used, reproducible, and cost-effective method for studying adult CM viability 9,10. To ensure scientific rigor and transparency, the trypan blue assay was conducted without bias, in which cell numbers from entire fields were recorded by two individuals independently, with one person counting blindly (Figures 1A and S1). In addition, CM viability was also further confirmed by measuring lactate dehydrogenase (LDH) released from injured cells 10. Among various GsPCRs, β1AR and H2R appear detrimental to CM viability 4,11,12. Indeed, we found that β-AR agonist ISO stimulated CM death dose-dependently (Figures S2A). There are two GsPCR βARs in mouse CMs, β1AR and β2AR. β1AR mediates the effect of ISO on CM death because the antagonist for β1AR (CGP20712) but not β2AR (ICI118,551) abolished the effect of ISO (Figures 1A–1C). The results are consistent with previous reports of β1AR but not β2AR in CM apoptosis induced by βAR agonists 13,14. We also found that H2R-selective agonist amthamine dihydrobromide (AMT) caused CM death dose-dependently, which was largely abolished by H2R antagonist famotidine (FAM) (Figures 1D, 1E, and S2B).
Figure 1. Stimulating β1AR or H2R induces CM death, depending on AC5-synthesized cAMP.

A and B, Images and quantitative results of trypan blue staining in CM death induced by βAR agonist ISO (10 μM for 24 h) with or without β1AR antagonist CGP20712 (0.5 μM) or β2-AR antagonist ICI118,551 (0.5 μM). Black arrows indicate the trypan blue stained CMs (dead CM), n = independent CM isolations from 4 mice. C, Results of LDH leakage in CM death induced by ISO (10 μM for 24 h) with or without CGP20712 (0.5 μM) or ICI118,551 (0.5 μM), n=4. D and E, Results of trypan blue staining and LDH leakage in CM death induced by H2R agonist Amthamine dihydrobromide (AMT, 10 μM for 24 h) with or without H2R antagonist Famotidine (FAM, 1 μM), n=4. F, Effects of AC5 shRNA on ISO-induced CM death, n=5. G, Effects of AC5 shRNA on i[cAMP] induced by ISO (10 μM for 5 min) in the presence of ICI118,551, n=4. H, Effects of PKA inhibitor PKI 14–22 (PKI, 5 μM) on ISO-induced CM death, n=4. I, Effects of AC5 shRNA on AMT-induced CM death, n=4. J, Effects of AC5 shRNA on i[cAMP] induced by AMT (10 μM for 5 min), n=4. K, Effects of PKI on ISO-induced CM death, n=4. An average 1000 CMs/each isolation were counted for trypan blue staining. The representative images were chosen based on their quality and to most accurately reflect the group average across all the available data. Data were presented as mean ± SEM. Data in Figures 1B and 1C were analyzed by the Kruskal-Wallis test followed by Conover-Iman post-hoc test with Bonferroni corrections for 3 comparisons, Figures 1D–1K by the two-way Aligned Ranks Transformation (ART) ANOVA with Bonferroni post-hoc test corrections for 2 comparisons. All reported P-values have been adjusted for a predetermined number of multiple comparisons, as specified in the corresponding figures. P<0.05 was statistically significant. ns: no significant difference.
AC5 and AC6 are two major isozymes in CMs 15. Although AC5 and AC6 have shown distinct functions in the heart 16, the relationship of different GsPCRs with AC5 or AC6 in CM viability remains largely unknown. We next determined which AC isozyme is involved in β1AR- or H2R-induced CM death. We expressed AC5 and AC6 shRNA via lentivirus in CMs to knock down (KD) their expression, and the KD efficacy and specificity were then verified (Figures S3A–S3D). Interestingly, ISO-induced CM death was blocked by AC5 but not AC6 shRNA (Figures 1F and S3E, S3F). ISO increased i[cAMP] levels, with a peak of around 5 minutes (min) (Figure S3G). Stimulating β1AR by ISO (in the presence of a β2AR antagonist) increased cAMP, mediated by AC5 (Figure 1G). PKA is a primary cAMP effector, and we found that the PKA peptide inhibitor (PKI 14–22, PKI) largely abolished the effect of ISO on CM death (Figures 1H and S3H). H2R agonist AMT induced a sustained cAMP elevation (Figure S3I). Similarly, AC5 mediated H2R-induced CM death but not AC6 (Figures 1I, S3J, and S3K), and so did the cAMP elevation (Figure 1J). PKA activation was also critical for H2R-induced CM death (Figures 1K and S3L). These results suggest that the pro-death effect of β1AR or H2R on CM death depends on AC5-produced i[cAMP] and PKA activation.
β1AR- or H2R-induced CM death depends on PANX1-medaited ATP release and P2X7R activation.
Various mechanical and biological stressors can induce high levels of ATP release into the extracellular milieu, acting as a damage-associated molecular pattern (DAMP) and contributing to cell/tissue damage 17. We thus hypothesize that neurohormonal over-stimulation acts as a pathological stressor that stimulates ATP release from CMs. Indeed, we found that ISO induced a transient increase of e[ATP] levels with a peak of around 30 min (Figure S4A). Interestingly, e[ATP] elevation was not seen with A2R agonist CV1808 which protects against CM death (Figures S4B and S4C), or with TCS2510, an agonist of the GsPCR EP4 (prostaglandin E2 receptor 4) that does not affect CM viability (Figures S4D and S4E). ISO-induced e[ATP] elevation was mediated by β1AR specifically (Figure 2A) and dependent on PKA (Figure 2B), consistent with its effect on CM death. H2R agonist AMT also induced a sustained elevation of e[ATP] (Figure S4F), which was also dependent on PKA (Figure 2C). These results suggest that e[ATP] elevation is associated explicitly with pro-death GsPCRs.
Figure 2. β1AR- or H2R-induced CM death depends on ATP release and P2X7R activation.

A, Effects of CGP20712 (0.5 μM) or ICI118,551 (0.5 μM) on e[ATP] levels induced by ISO (10 μM for 30 min), n=4. B and C, Effects of PKI (5 μM) on e[ATP] levels induced by ISO or AMT (10 μM for 30 min), n=5 for B and 4 for C. D and H, Quantitative results of trypan blue staining of CM death induced by ISO (10 μM for 24 h) or AMT (10 μM for 24 h) with PANX1 peptide inhibitor 10PANX (100 μM) or scramble peptide (PANX scr, 100 μM), n=5 for D and 4 for H. E and G, Effects of 10PANX (100 μM) or PANX scr (100 μM) on e[ATP] induced by ISO and AMT, n=5 for E and 4 for G. F, Effects of PANX1 shRNA on ISO-induced CM death, n=5. I and J, z-stacked images of PLA (red dots) performed with antibodies for PANX1 with β1AR or PANX1 with H2R in CMs. Insets are zoomed areas with white dash-lines. K and L, Effects of P2X7R antagonist A804598 (1 μM) or shRNA on ISO-induced CM death, n=5. M, Effects of A804598 on AMT-induced CM death, n=4. N, z-stacked images of PLA (red dots) performed with antibodies for PANX1 with P2X7R in CMs. Insets are zoomed areas with white dash-lines. O, The representative images of the co-immunoprecipitation of PANX1 with P2X7R, β1AR, and H2R in CMs. P and Q, Effects of CaMKII inhibitor KN93 (2 μM) or control inhibitor KN92 (2 μM) on CM death induced by ISO or AMT, n=4. R and S, Effects of A804598, KN93 or calcium chelate EGTA (1 μM) on ISO- or AMT-induced death in CMs ectopically expressing Laz or human P2X7R via the adenoviral vector, n=6. Data were presented as mean ± SEM. Data in Figures 2R and 2S were analyzed by the one-way ANOVA followed by post-hoc comparisons with Bonferroni corrections for 5 comparisons, Figure 2A by the Kruskal-Wallis test followed by Conover-Iman post-hoc test with Bonferroni corrections for 3 comparisons, Figures 2B–2H, 2K–2M, 2P and 2Q by the two-way ART ANOVA with Bonferroni post-hoc test corrections for 2 comparisons. All reported P-values have been adjusted for a predetermined number of multiple comparisons, as specified in the corresponding figures. P<0.05 was statistically significant. ns: no significant difference.
ATP release can be mediated by distinct mechanisms, including but not limited to ATP-permeable channels/pores 18. Pannexin and connexin channels have been linked to ATP release and play important roles in various pathologies 18. Connexin 43 (Cx43) is a well-known protein for gap-junction and no-junctional hemichannel in the myocardium; the non-junctional Cx43 has been shown to mediate ATP release from the cytosol 19. Thus, we first examined the role of Cx43 activation in ISO-induced CM death. We found that inhibiting Cx43 with selective inhibitor Gap 26 increased CM death and did not alter ISO-induced CM death (Figure S5A), suggesting Cx43 activation is protective in CM survival. We next focused on the role of pannexin-1 (PANX1) because it has been described in the canine CM membrane 20. Interestingly, we found that PANX1-mimetic-inhibitory-peptide (10PANX) but not the control-scramble-peptide (PANX scr) largely abolished ISO-induced CM death (Figures 2D and S5B). Consistently, PANX1 inhibition also abolished the effects of ISO on e[ATP] elevations (Figure 2E). Similar results for ISO-induced CM death were obtained with PANX1 shRNA (Figures 2F, S5C, and S5D). These results suggest that PANX1 is a predominant ATP release channel mediating CM death. e[ATP] can be quickly hydrolyzed by ecto-ATPase 21. To further determine the involvement of e[ATP], we forced change of e[ATP] levels by inhibiting e[ATP] hydrolysis with POM1 (an inhibitor of ecto-ATPase, CD39), or by accelerating e[ATP] hydrolysis with an exogenous ATPase (apyrase). We found that ISO-induced CM death was exacerbated by POM1 (Figures S5E and S5F) while abolished by active apyrase but not the heat-inactivated one (Figures S5G and S5H). Very similarly, AMT-induced e[ATP] elevation (Figure 2G) or CM death (Figures 2H and S5I) was largely blocked by PANX1 inhibition. Next, we examined whether PANX1 is localized with β1AR or H2R in close vicinity via PLA. We detected positive PLA signals of PANX1 with β1AR or H2R in isolated CMs (Figures 2I and 2J). Their negative controls with individual antibodies showed no or limited PLA signals in isolated CMs (Figure S6). We also detected the proximate localization of PANX1 with β1AR or H2R in heart tissues (Figures S7A–S7C). However, we failed to detect the interaction of PANX1 with GsPCRs such as A2AR (Figures S8A–S8C), CGRPR (Figures S8D and S8E), and RXFP1 (Figures S8F and S8G) that do not stimulate CM death.
In healthy tissues, e[ATP] levels in interstitial or pericellular areas are within the low nanomolar range. e[ATP] levels can be drastically increased to the hundred micromolar range near the cell surface under pathological states, e.g., hypoxia, ischemia, trauma, or inflammation 22. Thus, multiple e[ATP] receptors with a wide range of different ATP affinities exist. They primarily belong to two classes of membrane purine type 2 receptor subtypes, P2XRs and P2YRs. In the P2YR family, three members (P2Y2R, P2Y4R, and P2Y13R) can be activated by e[ATP] with relatively high affinities (EC50≈0.1–1 μM), thus functioning mostly under physiological conditions 23. All P2XR family members, P2X(1–7)R, are ATP-gated Ca2+-permeable non-selective cation channels 24. Among them, P2X7R has the lowest ATP affinity (EC50≈0.1–1 mM) compared to other P2XRs (EC50≈0.1–10 μM) and is particularly important under pathological conditions with high e[ATP] levels. High doses of exogenous ATP stimulated death/apoptosis of HL-1 cells (a mouse CM cell line) via P2X7R 25. P2X7R and PANX1 form a complex in neurons 26. As such, we proposed that P2X7R acts as an e[ATP] receptor. Indeed, we found that ISO-induced CM death was largely blocked by P2X7R antagonist A804598 (Figures 2K and S9A) and P2X7R shRNA (Figures 2L, S9B, and S9C). Similarly, AMT-induced CM death was blocked by P2X7R inhibition (Figures 2M and S9D). We also detected the interaction between PANX1 and P2X7R by PLA in isolated CMs (Figures 2N and S10A) and heart tissues (Figures S10B and S10C). To further confirm the signaling complexes, we performed co-immunoprecipitation (Co-IP) studies using TrueBlot® products that eliminate IgG contamination from IP-Western blots. We found that the IP of PANX1 can pull down β1AR, H2R, and P2X7R (Figure 2O), indicating that β1AR or H2R is associated with the PANX1/P2X7R signaling complex.
Because P2X7R activation can lead to Ca2+ elevation, we assessed the change of intracellular Ca2+ upon β1AR or H2R stimulation in CMs. We found that ISO increased Ca2+ levels, and around 50% of Ca2+ elevation was blocked by P2X7R inhibitor A804598 or PANX1 inhibitor 10PANX (Figures S11A and S11B). We obtained a similar observation for H2R stimulation with ATM (Figures S11C and S11D). These results suggest that β1AR or H2R stimulated Ca2+ elevation, partially mediated by e[ATP] activation of P2X7R. CaMKII has been reported in CM apoptosis 27,28. We found that CaMKII inhibitor KN93 but not the control KN92 blocked ISO-induced CM death (Figures 2P and S12A). A different CaMKII peptide inhibitor Myr-AIP elicited similar effects (Figures S12B and S12C). Similarly, CaMKII inhibition blocked the effect of AMT-induced CM death (Figures 2Q and S12D). To further determine the relationship of P2X7R with Ca2+ and CaMKII in CM death, we overexpressed P2X7R in WT CMs (Figure S12E). We found that ISO- or AMT-induced CM death was further increased upon P2X7R overexpression, which was abolished by P2X7R antagonist A804598, CaMKII inhibitor KN93, or extracellular Ca2+ chelator EGTA (Figures 2R, 2S, S12F, and S12G). CaMKIIδ has been shown to mediate CM apoptosis by diverse stimuli 28. We assessed CaMKIIδ activation by measuring CaMKIIδ and Calmodulin (CaM) binding (the mechanism of CaMKII activation) via PLA (Figure S13A). We found that ISO significantly increased CaMKIIδ and Calmodulin (CaM) binding, and ≈70–80% of binding was diminished by the inhibition of PANX1 or P2X7R (Figure S13B–S13D). Similar results were obtained with AMT (Figures S13E and S13F). These results suggest that β1AR or H2R stimulated CaMKIIδ activation, partially mediated by e[ATP] activation of P2X7R. Their negative controls with individual antibodies showed no significant PLA signals in isolated CMs (Figure S13G). Taken together, these results support the critical roles of PANX1-mediated ATP release, P2X7R activation, Ca2+ entry, and CaMKII activation in β1AR- and H2R-induced CM death.
β1AR or H2R activation induces ATP release and CM death through PKA-mediated phosphorylation of PANX1 at Ser206.
Next, we examined whether cAMP/PKA could regulate PANX1 phosphorylation, activation, and ATP release by multiple experimental approaches. We performed PLA to detect phosphorylated PANX1 (p-PANX1) in situ in CMs with combined PANX1 and PKA-substrate antibodies (Figure S14A). We found that ISO increased p-PANX1 PLA signals, which were blocked by the antagonist of β1AR but not β2AR (Figures S14B and S14C). This was consistent with the role of β1AR in CM death (Figures 1A and 1B) and e[ATP] elevation (Figure 2A). ISO-induced increase of p-PANX1 PLA signal was dependent on PKA (Figures 3A, 3B, S14D, and S14E). Additionally, we performed immunoprecipitation (IP) of PANX1 and immunoblotting (IB) of p-PANX1 with a PKA-substrate antibody. ISO increased p-PANX1, which was blocked by PKI (Figure 3C). Increased p-PANX1 measured by PLA was also detected in mouse heart tissues with acute ISO treatment (Figures 3D, 3E, and S14F). Like β1AR, H2R stimulation increased p-PANX1 seen by PLA, blocked by PKI in CMs (Figures 3F and S14G).
Figure 3. β1AR or H2R activation induces ATP release and CM death through PKA-mediated phosphorylation of PANX1 at Ser206.

A and B, PLA images and quantitative results showing PANX1 phosphorylation in CMs treated with ISO (10 μM for 30 min) with or without PKI (5 μM). PANX1 phosphorylation was detected by PLA (red) using PANX1 plus PKA-substrate antibodies. Insets are zoomed areas with white dash-lines, n=6. C, Western blots and quantitative results showing ISO-induced and PKA-mediated PNAX1 phosphorylation assessed by Immunoprecipitating (IP) PANX1 and immunoblotting (IB) PKA-mediated phosphorylation with PKA-substrate antibody, n=4. The phosphorylated PANX1 level was normalized to the total PANX1. D and E, PLA images and quantitative results showing the effect of ISO on PKA-mediated PANX1 phosphorylation in mouse hearts treated with ISO injection (100 mg/kg, every 8 h twice s.c. in 24 h, n=6 mice). F, Quantitative results of PLA showing the effects of PKI on PKA-mediated PANX1 phosphorylation in CMs treated with AMT (10 μM for 30 min), n=6. G, Western blots and quantitative results showing the effect of PANX1 phosphorylation site mutation on ISO-induced PANX1 phosphorylation in CMs ectopically expressing PANX1(WT) and PANX1(S206A) via lentivirus, n=4. The phosphorylated PANX1 level was normalized to the total PANX1. H and I, Effects of PANX1 phosphorylation site mutation on ISO-induced CM death and e[ATP] elevation, n=5. J and K, Effects of PANX1 phosphorylation site mutation on AMT-induced CM death and e[ATP] elevation, n=4. L and M, Effects of PANX1 phosphorylation site mutation on ISO- or AMT-induced PANX1 activation measured by YO-PRO3 cellular influx, n=6. PLA quantification for isolated CMs was performed on an average of 40–50 CMs/each isolation. Quantification of YO-PRO3 influx was performed in ≈200 CMs/each isolation. The representative images were chosen based on their quality and to most accurately reflect the group average across all the available data. Data were presented as mean ± SEM. Data in Figures 3B, 3F, 3L and 3M were analyzed by the two-way ANOVA followed by post-hoc comparisons with Bonferroni corrections for 2 comparisons, Figures 3C and 3G–3K by the two-way ART ANOVA with Bonferroni post-hoc test corrections for 2 comparisons, and Figure 3E by the unpaired t-test. All reported P-values have been adjusted for a predetermined number of multiple comparisons, as specified in the corresponding figures, except in Figure 3E where the raw P-value is reported. P<0.05 was statistically significant.
To understand how cAMP/PKA regulates PANX1 phosphorylation and ATP release, we performed PANX1 protein sequence analysis. We found a PKA phosphorylation consensus site at Serine 206 of human PANX1, which is conserved among many species (Figure S15A). Serine 206 of human PANX1 is equal to Serine 205 of mouse PANX1. To confirm the functional role of PANX1 phosphorylation at Ser206, we mutated Ser206 to Alanine (S206A). We created a lentivirus to express PANX1(WT) (wild-type PANX1) or PANX1(S206A) (a phosphor-dead mutant) in CMs at equivalent levels (Figure S15B). Interestingly, we observed that ISO significantly increased p-PANX1 in CMs expressing PANX1(WT), which was not seen in CMs expressing PANX1(S206A) (Figure 3G). ISO-induced CM death was equivalent in CMs expressing PANX1(WT) or GFP (Figures S15C and S15D). However, PANX1(S206A) functioned as a dominant negative mutant and significantly blocked the ability of ISO to induce CM death (Figures 3H and S11D) and to elevate e[ATP] (Figure 3I). Like β1AR, PANX1(S206A) also significantly blocked the ability of H2R to induce CM death (Figures 3J and S15E) and e[ATP] elevation (Figure 3K). Moreover, we evaluated the effect of PANX1 phosphorylation on PANX1 channel activity by measuring the cellular uptake of YO-PRO3 dye as previously described 29. We found that ISO or AMT significantly increased the channel activity in CMs expressing PANX1(WT) but not PANX1(S206A) (Figures 3L, 3M, and S15F). We also created PANX1(S206D) (a phosphor-mimic mutant with S206 mutated to aspartic acid). We found that expressing PANX1(S206D) is sufficient to induce CM death, and ISO stimulation did not have an additional effect (Figures S15G and S15H). These results together suggest that β1AR/cAMP or H2R/cAMP stimulates PKA-mediated p-PANX1 at S206, which activates PANX1, promotes ATP release, and subsequently induces CM death.
Role of PANX1 Ser206 phosphorylation in the acute cardiac injury induced by β-AR overactivation.
Next, we confirmed the role of PKA-mediated PANX1 phosphorylation at S206 in vivo. We used an established acute heart injury mouse model induced by ISO 30,31. Acute treatments with high doses of ISO in rodents have been used to induce cardiac injuries under catecholamine overdose or surge. We created an AAV9 virus expressing GFP, PANX1(WT), or PANX1(S206A) under the control of a cardiac troponin T (cTnT) promoter 32. As shown in Figure 4A, different AAV9 vectors (2 ×1011 vg/mouse) were administrated to C57BL/6J neonatal mice at 6–7 days of age via i.p. injection, an efficient route for systemic gene delivery with high levels of expression but low doses of virus 32,33. Upon successful expression of PANX1 proteins in myocardium via AAV9, mice (16–18 weeks) were subjected to saline or two high doses of ISO injection (100 mg/kg, s. c.) every 8 hours (h) within 24 h to recapitulate catecholamine overactivation (Figure 4A), according to previous studies with modifications 30,31. Mice were divided into six groups, including GFP/Saline, GFP/ISO, PANX1(WT)/Saline, PANX1(WT)/ISO, PANX1(S206A)/Saline, and PANX1(S206A)/ISO. We found that ISO-induced p-PANX1 in mouse hearts was abolished by expressing PANX1(S206A) but not PANX1(WT) when they were expressed at equivalent levels (Figure 4B). We did not observe any abnormality in mouse hearts expressing PANX1(WT) and PANX1(S206A), which is consistent with the fact that PANX1 is not involved in the cardiac contractility regulated by β1AR (data not shown). ISO-induced myocardial damage was assessed by myocardial IgG accumulation through IgG immunostaining (Figure 4C). IgG penetration occurs when the CM membrane is disrupted and permeable to large molecules like IgG 34. We found that IgG accumulation in the myocardium was significantly increased by ISO treatment in GFP and PANX1(WT) groups, which was largely attenuated in the PANX1(S206A) group (Figures 4C and 4D). In addition, we found that ISO increased TUNEL-positive myocardial nuclei in the GFP or PANX1(WT) group, which was diminished in the PANX1(S206A) group (Figures S16A and S16B). The plasma cTnI level was significantly increased in the GFP/ISO group compared to that in the GFP/Saline group (Figure 4E), indicating cardiac injury occurred. Notably, the cTnI level in the PANX1(S206A)/ISO group was significantly lower than that in the PANX1(WT)/ISO group (Figure 4E). We only used male mice because our pilot study showed that female mice were resistant to acute ISO-induced cardiac injury in this mode (Figures S16C and S16D). These results are consistent with previous studies showing excessive β-AR stimulation with ISO caused CM apoptosis and necroptosis in mice 35. The sex/gender difference has been described in βAR-mediated signaling in CMs, partly contributed by augmented expressions and/or activities of βAR signaling molecules in the male CMs 36. Collectively, our findings suggest that PANX1 phosphorylation at S206 is critical in myocardial death after catecholamine surges in vivo.
Figure 4. PANX1 Ser206 phosphorylation is critical in ISO-induced acute cardiac injury in vivo.

A, Diagram showing the experiment design: Male mice (16–18 weeks) expressed GFP, PANX1(WT), or PANX1(S206A) by AAV-9 virus in the myocardium are treated with saline or ISO injection (100 mg/kg, every 8 h twice s.c. in 24 h). B, Western blots and quantitative results showing the effect of PANX1 phosphorylation site mutation on ISO-induced and PKA-mediated PANX1 phosphorylation in mouse hearts with CM-specific expression of GFP, PANX1(WT) or PANX1(S206A) via AAV9, n=3 (Vehicle/GFP), 4 (ISO/GFP), 4 (Vehicle/PANX1(WT)), 4 (ISO/PANX1(WT)), 4 (Vehicle/PANX1(S206A)), and 4 (ISO/PANX1(S206A)). The phosphorylated PANX1 level was normalized to the total PANX1. C and D, Images and quantitative results of cardiac IgG immunostaining (red signals) showing the effect of PANX1 phosphorylation site mutation on IgG accumulation (an indicator of cardiac injury) induced by ISO in mouse hearts. Images with the blue border are corresponding zoomed areas in the whole heart sections, n=3 (Vehicle/GFP), 7 (ISO/GFP), 6 (Vehicle/PANX1(WT)), 14 (ISO/PANX1(WT)), 6 (Vehicle/PANX1(S206A)), and 14 (ISO/PANX1(S206A)). E, Effects of PANX1 phosphorylation site mutation on ISO-induced acute cardiac injury assessed by plasma cTnI, n=3 (Vehicle/GFP), 7 (ISO/GFP), 6 (Vehicle/PANX1(WT)), 14 (ISO/PANX1(WT)), 6 (Vehicle/PANX1(S206A)), and 14 (ISO/PANX1(S206A)). The representative images were chosen based on their quality and to most accurately reflect the group average across all the available data. Data were presented as mean ± SEM. Data in Figures 4B, 4D and 4E were analyzed by the Kruskal-Wallis test followed by Conover-Iman post-hoc test with Bonferroni corrections for 3 comparisons. All reported P-values have been adjusted for a predetermined number of multiple comparisons, as specified in the corresponding figures. P<0.05 was statistically significant.
Activation of pro-survival GsPCRs such as A2R, CGRPR, and RXFP1 exhibits protective effects on CM survival and stimulates AC6-synthesized cAMP.
There is another group of GsPCRs showing protective effects on CMs. It has been shown that A2R activation mediates a protective effect on CM survival and anti-hypertrophy 5,37. CGRP, a neuropeptide, exerts a cardiac protective effect via activating CGRPR 38,39. Relaxin, a peptide pregnancy hormone, has anti-fibrotic, anti-hypertrophic, anti-apoptotic, and anti-inflammatory effects in heart failure through activating RXFP1 (a GsPCR) 40,41. As such, we selected these three GsPCRs as pro-survival ones. As anticipated, stimulating A2R by A2R agonist CV1808 did not affect CM basal viability but attenuated H2O2-induced CM death dose-dependently (Figures 5A, 5B, and S17A). The effect of CV1808 was abolished by A2R antagonist ZM241385 (Figures 5A and 5B). CGRPR agonist CGRP also protected against H2O2-induced CM death dose-dependently, which was abolished by CGRPR antagonist CGRP8–37 (Figures 5C, 5D, and S17B). Similarly, RXFP1 agonist relaxin-2 significantly attenuated H2O2-induced CM death dose-dependently (Figures 5E, 5F, and S17C). Next, we examined the specific AC isozyme activated by A2R stimulation. Interestingly, AC6 but not AC5 shRNA abolished the protective effects of CV1808 (Figures 5G, S18A, and S18B), CGRP (Figures 5H, S18C, and S18D), or relaxin-2 (Figures 5I, S18E, and S18F). As anticipated, stimulating A2R, CGRPR, or RXFP1 increased i[cAMP] levels, with a peak of around 5 min (Figures S19A–S19C). Consistently, CV1808-, CGRP- or relaxin-2-induced i[cAMP] elevation was dependent on AC6. (Figures 5J–5L). Interestingly, their i[cAMP] elevations were enhanced by MK-571, a pan inhibitor of MRPs that act as cAMP efflux pumps (Figures S19D–S19F). However, β1AR- or H2R-induced i[cAMP] was not affected by MK-571 (Figures S19G and S19H). Thus, e[cAMP] levels were evaluated in the CM supernatants. We found that e[cAMP] levels were sustainedly elevated by CV1808, CGRP, or relaxin-2 (Figures 5M–5O). These results together suggest that these pro-survival GsPCRs stimulate AC6-derived i[cAMP] that may enable it to be pumped out.
Figure 5. Stimulation of GsPCRs such as A2R, CGRP-R, or RXFP1 protects CM survival, depending on AC6-synthesized cAMP.

A and B, Quantified results of trypan blue staining and LDH leakage in CM death induced by H2O2 (5 μM for 24 h) with or without A2R agonist CV1808 (1 μM) and A2R antagonist ZM24183 (200 nM) as indicated, n=4. C and D, Quantified results of trypan blue staining and LDH leakage in CM death induced by H2O2 with or without CGRP-R agonist CGRP (10 nM) and CGRP-R antagonist CGRP8–37 (100 nM) as indicated, n=4. E and F, Quantified results of trypan blue staining and LDH leakage in CM death induced by H2O2 with or without RXFP1 agonist relaxin-2 (20 nM) as indicated, n=4. G-I, Effects of AC6 shRNA on CV1808, CGRP or Relaxin-2 against H2O2-induced CM death, n=5 for G, 4 for H and I. J-L, Effects of AC6 shRNA on i[cAMP] stimulated by CV1808 (1 μM), CGRP (10 nM), or relaxin-2 (20 nM) for 5 min, n=4. M-O, Time course of e[cAMP] levels in supernatants of CMs stimulated by CV1808 (1 μM), CGRP (10 nM), or relaxin-2 (20 nM) as indicated, n=5 for M, 4 for N and O. The statistics were performed by comparing to the 0-min time point. Data were mean ± SEM. Data in Figures 5A–5D and 5G–5I were analyzed by the Kruskal-Wallis test followed by Conover-Iman post-hoc test with Bonferroni corrections for 3 comparisons, Figures 5M–5O by the Kruskal-Wallis test with Conover-Iman post-hoc test and Bonferroni corrections for 5 comparisons, Figures 5E, 5F and 5J–5L by the two-way ART ANOVA with Bonferroni post-hoc test corrections for 2 comparisons. All reported P-values have been adjusted for a predetermined number of multiple comparisons, as specified in the corresponding figures. P<0.05 was statistically significant. ns: no significant difference.
The protective effects of A2R, CGRPR, or RXFP1 stimulation on CM survival are dependent on cAMP efflux and e[cAMP]-e[ADO] signaling.
Since i[cAMP]-induced by these pro-survival GsPCRs might be subjected to efflux in CMs, we hypothesized that cAMP efflux is important for their pro-survival effects in CMs. We examined the effect of cAMP efflux blockade on their effects on CM survival and characterized the specific cAMP efflux pump. Among three human cyclic nucleotide transporters (MRP4, 5, and 8), MRP4 and 5 but not 8 are reported in human heart 42. Since the mouse does not have the human homolog of MRP8 43, we thus focused on MRP4 and 5 and found both expressed in mouse CMs (Figure S20A). MRP4 or 5 KD was performed with a lentivirus-mediated expression of specific shRNA (Figures S20B–S20E). The effect of CV1808 against H2O2-induced CM death was largely blocked by MRP4 but not MRP5 shRNA (Figures 6A and S20F–S20H) and similarly abolished by MRP4-selective inhibitor ceefourin1 (Figures 6B and S20I). Consistently, CV1808-induced e[cAMP] elevation was significantly abolished by ceefourin1 (Figure 6C). The effects of CGRP or relaxin-2 on CM survival and e[cAMP] elevation were also abolished by ceefourin1 (Figures 6D–6G, S20J, and S20K). To further confirm the involvement of e[cAMP], we depleted e[cAMP] with a neutralizing cAMP antibody applied in the culture medium. We found that the active cAMP antibody but not the heat-inactivated one significantly abolished the protective effect of CV1808, CGRP, or relaxin-2 on CMs (Figures 6H–6J and S20L–S20N). We next examined whether MRP4 and A2AR, CGRPR, or RXFP1 are localized in close vicinity using Proximity Ligation Assay (PLA). There are two A2Rs (A2AR and A2BR), and both contribute to CM survival equivalently 9. We thus focused on A2AR due to the available A2AR antibody. We detected a high level of PLA signals with combined MRP4 plus A2AR, CGRPR, or RXFP1 antibodies (Figures 6K–6M), suggesting the interaction of MRP4 with these GsPCRs. Negative controls were performed with single antibodies individually (Figures S21A–S21D). Interestingly, we failed to detect the interaction of MRP4 with β1AR (Figures S21E and S21F) or H2R (Figures S21G and S21H). To further confirm the signaling complexes, we performed Co-IP studies using TrueBlot® products. We found that the IP of MRP4 can pull down A2AR, CGRPR, or RXFP1 (Figure 6N), indicating that A2AR, CGRPR, or RXFP1 is associated with the MRP4 signaling complex. These results together suggest that i[cAMP] produced from A2R, CGRPR, or RXFP1 activation is allowed to efflux through the nearby MRP4 in CMs, which is responsible for A2R-, CGRPR- or RXFP1-mediated CM survival.
Figure 6. The protective effect of A2R, CGRP-R, or RXFP1 stimulation on CM survival depends on cAMP efflux.

A and B, Effects of MRP4 shRNA or MRP4-selective inhibitor ceefourin1 (20 μM) on the protective effect of A2R agonist CV1808 (1 μM) against CM death induced by H2O2 (5 μM for 24 h), n=5. C, Effects of ceefourin1 on e[cAMP] levels induced by CV1808 (1 μM) for 30 min, n=5. D and F, Effects of ceefourin1 on CGRP (10 nM) or relaxin-2 (20 nM) against CM death, n=4. E and G, Effects of ceefourin1 on e[cAMP] levels induced by CGRP (10 nM) or relaxin-2 (20 nM) for 30 min, n=4. H-J, Effects of blocking e[cAMP] with cAMP antibody (0.25 μg/mL) on CV1808, CGRP or relaxin-2 against CM death. The heat-inactivated cAMP antibody was used as a negative control, n=4. K-M, Images showing PLA signals (red dots) performed with MRP4 plus A2AR, CGRP-R, or RXFP-1 antibodies in CMs, as indicated. The inset is a zoomed area with a white dashboard. Data were mean ± SEM. N, The representative images of co-immunoprecipitation and input of MRP4 with A2AR, CGRPR, and RXFP1 in CMs. Data in Figures 6A, 6B, 6D, 6F, and 6H–6J were analyzed by the Kruskal-Wallis test followed by Conover-Iman post-hoc test with Bonferroni corrections for 3 comparisons, Figures 6C, 6E, and 6G by the two-way ART ANOVA with Bonferroni post-hoc test corrections for 2 comparisons. All reported P-values have been adjusted for a predetermined number of multiple comparisons, as specified in the corresponding figures. P<0.05 was statistically significant.
e[cAMP] can be sequentially converted to e[ADO] via two steps: 1) e[cAMP] to e[5’AMP] catalyzed by ecto-PDE (a rate-limiting step), and 2) e[5’AMP] to e[ADO] catalyzed by 5’-nucleotidase (5’-NT) 2. It remains unknown whether the e[cAMP]-e[ADO] signaling is responsible for A2R, particularly CGRPR and RXPR1 protection of CM survival. We, therefore, determined the role of e[cAMP] metabolism in their protection of CMs and characterized the specific enzymes involved in the e[cAMP] metabolism. ecto-PDEs belong to the ectonucleotide pyrophosphatase/phosphodiesterase (ENPP) family consisting of 7 members. Among them, only ENPP1–3 can hydrolyze nucleotides 44,45. We observed all three ENPPs expressed in mouse CMs (Figure S22A). We then tested the roles of these three ENPPs by KD their expressions individually via specific shRNAs (Figures S22B–S22J). We found that ENPP1 shRNA, but not ENPP2 or ENPP3 shRNA, significantly abolished the protective effect of CV1808 (Figures 7A and S23A–S23E). Consistently, ENPP1-selective inhibitor (ENPP1 inhibitor C) but not ENPP2-selective inhibitor (S2368) abolished the protective effect of CV1808 (Figures 7B and S23F–S23H). There is no ENPP3 inhibitor available. Next, we determined the role of 5’-NT, which has only one isozyme named CD73. The effect of CV1808 was abolished by CD73 shRNA (Figures S24A–S24C) or inhibitor PSB12379 (Figures S24D and S24E). Similarly, the protective effect of CGRP or relaxin-2 was also blocked by ENPP1 inhibitor C (Figures 7C, 7D, S25A, and S25B) or CD73 inhibitor PSB12379 (Figures S25C–S25F). These findings together suggest that ENPP1 and CD73 are primarily responsible for the metabolism of e[cAMP]-to-e[ADO] in CMs, which is critical in the A2R- or CGRPR-mediated protection of CM survival.
Figure 7. The protective effect of A2R, CGRP-R, or RXFP1 stimulation on CM survival depends on e[cAMP]-e[ADO] signaling.

A and B, Effects of ENPP1 shRNA or ENPP1 inhibitor (ENPP1 inhibitor C, 10 μM) on CV1808 (1 μM) against CM death induced by H2O2 (5 μM for 24 h), n=5. C and D, Effects of ENPP1 inhibition on CGRP (10 nM) or relaxin-2 (20 nM) against CM death, n=4. E-G, Effects of depleting e[ADO] with ADA (1.2 U/mL) on CV1808, CGRP, or relaxin-2 against CM death. Heat-inactivated ADA was used as a negative control, n=5 for E, 4 for F and G. H and I, Effects of A1R shRNA or A1R antagonist PSB36 (10 nM) on CV1808 against CM death, n=5. J and K, Effects of PSB36 on CGRP or relaxin-2 against CM death, n=4. L, Effects of exogenous membrane impermeable cAMP (cAMP, 10 μM) on H2O2-induced CM death, n=5. M, Effects of ENPP1 inhibition on cAMP against CM death, n=4. N, Effects of e[ADO] depletion with ADA on cAMP against CM death, n=4. O, Effects of A1R inhibition on cAMP against H2O2-induced CM death, n=4. Data were mean ± SEM. Data in Figures 7A–7K, 7M–7O were analyzed by the Kruskal-Wallis test followed by Conover-Iman post-hoc test with Bonferroni corrections for 3 comparisons, and Figure 7L by the two-way ART ANOVA with Bonferroni post-hoc test corrections for 2 comparisons. All reported P-values have been adjusted for a predetermined number of multiple comparisons, as specified in the corresponding figures. P<0.05 was statistically significant.
To further confirm the involvement of e[ADO] production, we depleted e[ADO] by adding ADO deaminase (ADA) that degrades e[ADO] 46. We found that the active ADA applied in the culture medium, but not the heat-inactivated one, abolished the effect of CV1808, CGRP, or relaxin-2 against CM death (Figures 7E–7G and S26A–S26C). It has been suggested that the protective effect of e[ADO] on CM survival is mediated by stimulating A1R and subsequent activation of PI3K and AKT signaling 47. Indeed, the effect of CV1808 on CM survival was blocked by A1R shRNA or antagonist PSB36 (Figures 7H, 7I, and S26D–S26F) as well as by PI3K inhibitor LY294002 (Figures S26G and S26H). Similarly, the protective effect of CGRP or relaxin-2 was abolished by A1R antagonism with PSB36 (Figures 7J, 7K, S26I, and S26J). These results support the role of e[ADO] and A1R-dependent signaling in A2R or CGRPR-mediated protective effect on CM survival.
Exogenously increased e[cAMP] protects against CM death and IR-induced myocardial injury in ex vivo and in vivo mouse hearts.
Since e[cAMP] elevation is critical for the protective effects of pro-survival GsPCRs such as A2R, CGRPR, and RXFP1, we examined the effects of the forced increase of e[cAMP] in CM protection using exogenous cAMP (hydrophilic and membrane-impermeable). In cultured CMs, exogenous cAMP did not affect CM basal viability but significantly inhibited H2O2-induced CM death (Figures 7L and S27A). The protective effect of e[cAMP] on CMs was largely inhibited by enpp1 inhibition (Figures 7M and S27B), CD73 inhibition (Figures S27C and S27D), e[ADO] depletion by ADA (Figures 7N and S27E), and A1R inhibition (Figures 7O and S27F). These results further confirm the critical role of e[cAMP] elevation and e[cAMP]-e[ADO] signaling in CM survival.
We next confirmed the protective effect of e[cAMP] in the intact heart using ex vivo Langendorff perfusion model 48, a suitable method for studying signaling molecules and pharmacological agents without influences from other organs but with the structure of an intact heart preserved. As shown in Figure 8A, isolated C57BL/6J mouse hearts were connected to a Langendorff perfusion system and subjected to equilibration perfusion for 30 min with vehicle or 20 μM cAMP. Ischemia/reperfusion (IR) was induced by subjecting hearts to 40 min global ischemia with no flow, followed by 50 min reperfusion in the presence of indicated treatment. Finally, the coronary outflow was collected to assess creatine kinase (CK) activity, a hallmark of myocardium injury 49. The heart was further perfused with FITC-Dextran (molecular weight 40 kDa) and finally harvested for cryosection. FITC-Dextran molecules entering the myocardium can be used to assess myocardial membrane damage. We found that IR significantly increased the FITC-Dextran positive myocardial area and CK activity, which were reduced considerably by cAMP (Figures 8B–8D). The in vitro studies showed that the protective effect of exogenous cAMP on CM depends on e[cAMP]-to-e[ADO] metabolism and e[ADO]-mediated activation of A1R. We thus examined if A1R antagonist PSB36 can block the protective effect of exogenous cAMP against ex vivo IR-induced heart injury. As shown in Figure S28A, isolated C57BL/6J mouse hearts were connected to the Langendorff perfusion system and subjected to equilibration perfusion with vehicle or 20 μM cAMP in the presence of PSB36. The global ischemia and reperfusion were performed similarly in the presence of indicated treatments. We found that with A1R antagonism, exogenous cAMP failed to exhibit any protective effects on the heart (Figure S28B and S28C), suggesting that A1R activation in the heart is a critical downstream event in exogenous cAMP-mediated cardiac protection.
Figure 8. e[cAMP] protects against myocardial IR injury in ex vivo and in vivo hearts.

A, Diagram of the ex vivo experiment design: Isolated mouse hearts were connected to a Langendorff perfusion system and subjected to 30 min equilibration, 40 min global ischemia with no flow, followed by 50 min reperfusion with or without cAMP (20 μM). B and C, Images and quantified results of myocardial FITC-dextran accumulation (an indicator of cardiac injury) showing the effect of exogenous membrane impermeable cAMP on IR-induced myocardial injury ex vivo, n=6. D, Effects of cAMP on CK activity from mouse hearts with ex vivo IR, n=6. E, Diagram of the in vivo experiment design: Mice (10–12 weeks) were subjected to 45 min cardiac ischemia by LAD ligation, and then subjected to 24 h reperfusion. At the beginning of reperfusion, mice were subcutaneously injected with cAMP (10 mg/kg) or saline. F, Effects of cAMP against IR-induced cardiac dysfunction (ejection fraction), n=6 (Sham/Vehicle), 10 (IR/Vehicle), 6 (Sham/cAMP), and 10 (IR/cAMP). G, Effects of cAMP against cardiac injury assessed by plasma cTnI, n=6 (Sham/Vehicle), 10 (IR/Vehicle), 6 (Sham/cAMP), and 10 (IR/cAMP). H-J, Images and quantitative results showing the effect of cAMP against IR-induced myocardial infarction by TTC staining (red indicates TTC positive staining). The area at risk (red and white, unstained by Evans blue dye) and the infarct area (white, unstained by TTC) were analyzed by Image J, n=10. K and L, Images and quantified results of cardiac IgG immunostaining (red signals) showing the effect of cAMP on IR-induced cardiac IgG accumulation (an indicator of cardiac injury), n=6. Data were mean ± SEM. The representative images were chosen based on their quality and to most accurately reflect the group average across all the available data. Data in Figure 8G were analyzed by the one-way ANOVA followed by post-hoc comparisons with Bonferroni corrections for 2 comparisons, Figures 8F and 8L by the Welch ANOVA with Dunnett T3 post hoc tests for 2 comparisons, Figure 8C by the Welch’s t-test, Figure 8D by the unpaired t-test, Figures 8I and 8J by the Mann-Whitney test. All reported P-values are raw P-values, except in Figures 8F, 8G, and 8L where P-values have been adjusted for a predetermined number of multiple comparisons, as specified in the corresponding figures. P<0.05 was statistically significant. ns: no significant difference.
We further confirmed the protective effect of e[cAMP] in the intact heart using in vivo mouse cardiac IR model 48. As shown in Figure 8E, C57BL/6J mice (10–12 weeks) were subjected to 45 min cardiac ischemia by LAD ligation and then to 24 h reperfusion. The sham group mice were subjected to the same surgery without LAD ligation. At the beginning of reperfusion, mice were subcutaneously injected with cAMP (10 mg/kg) or saline. At the end of reperfusion, the mouse cardiac function was assessed by an echocardiogram. We found that IR significantly reduced cardiac systolic function (such as ejection fraction) for male mice, and cAMP partially attenuated IR-induced cardiac dysfunction (Figure 8F). Myocardial injury was evaluated by plasma cardiac troponin I (cTnI) level, 2,3,5-Triphenyltetrazolium chloride (TTC) staining and immunostaining for IgG that can be accumulated in damaged myocardium 34. IR significantly increased plasma cTnI levels, myocardial infarction areas, and IgG-positive myocardium, partially alleviated by cAMP administration (Figures 8G–8L). In addition, IR largely increased TUNEL-positive myocardial nuclei, which was partially reduced by cAMP treatment (Figures S29A and S29B). The endpoint plasma cAMP levels were also assessed (Figure S29C). IR mice had significantly decreased plasma cAMP levels in control mice without cAMP administration compared to sham mice. Exogenous cAMP administration significantly increased plasma cAMP levels in both sham and IR mice. After cAMP treatment, plasma cAMP levels of IR mice were almost equivalent to those of normal mice (sham mice without cAMP treatment). We also examined female mice and found that cAMP partially inhibited IR-induced plasma cTnI elevation and myocardial infarction (Figures S29D–S29F). The endpoint plasma cAMP levels were also evaluated in female mice, like in male mice (Figure S29G). However, we did not detect significant effects of cAMP on IR-induced cardiac dysfunction in female mice (Figure S29H). Collectively, our findings from in vitro, ex vivo, and in vivo studies strongly suggest a protective effect of exogenously elevated e[cAMP] on CMs. However, e[cAMP] actions on other cell types may also partially contribute to the myocardial impacts in the ex vivo and in vivo hearts.
DISCUSSION
The activation of various GsPCRs and elevation of i[cAMP] could exert different functions in a single cell. However, the underlying molecular mechanisms remain largely unknown. In the current study, we provided experimental evidence to elucidate that different GsPCRs/i[cAMP] differentially regulate CM viability via forming unique signaling complexes that control their ability to promote the pro-death e[ATP] signaling or pro-survival e[cAMP] signaling (Graphical abstract). Among five GsPCRs examined, stimulating β1AR or H2R promotes CM death via AC5-derived i[cAMP], while stimulating A2R, CGRPR, or RXFP1 promotes CM survival via AC6-derived i[cAMP]. Importantly, these i[cAMP] molecules have distinct fates, either to stimulate PKA-dependent PANX1 activation and release pro-death e[ATP] or efflux via MRP4 and produce pro-survival e[ADO]. We further demonstrated that β1AR and H2R are spatially close to ATP-releasing channel PANX1, thus enabling ATP release, subsequently activating e[ATP]-P2X7R-CaMKII signaling and leading to CM death upon β1AR or H2R activation. We also defined a crucial role of PKA-dependent p-PANX1 at Ser206 in ATP release and CM death in vitro and/or in vivo. Very differently, A2R, CGRPR, or RXFP1 are localized spatially close to the efflux pump MRP4, thus enabling i[cAMP] efflux. The subsequent e[cAMP] metabolism and the e[cAMP]-e[ADO]-A1R signaling mediate CM survival upon A2R, CGRPR, or RXFP1 activation. We also demonstrated that the protective effect of administrating exogenous cAMP to increase e[cAMP] levels against CM death is dependent on the e[cAMP]-e[ADO] pro-survival signaling. Exogenous cAMP application protected against myocardial IR injury in mouse hearts ex vivo and in vivo. Taken together, our findings suggest that e[ATP]- and e[cAMP]-mediated signaling could be common mechanisms for the pro-death and pro-survival GsPCRs in CMs, respectively, which could be achieved by forming distinct signalosomes that determine the fate and efflux-ability of i[cAMP]. The critical molecular components of the e[ATP]- or e[cAMP]-signaling may represent novel molecular targets for combating CM death in cardiac disorders.
We defined a novel mechanism by which the cAMP/PKA signaling from pro-death GsPCRs, such as β1AR and H2R, stimulates the phosphorylation and activation of ATP-releasing channel PANX1, promotes ATP release, activates the e[ATP]-P2X7R-Ca2+-CaMKII signaling axis, and ultimately leads to CM death (Graphical abstract). In experimental mouse models, we also confirmed the roles of crucial molecules involved in ATP release (i.e., p-PANX1). We further identified β1AR or H2R proximate PANX1 in multiprotein complexes, which likely warrants β1AR/cAMP/PKA or H2R/cAMP/PKA signaling to induce ATP release specifically and effectively through PANX1 and subsequently activate the e[ATP]/P2X7R-dependent death pathway. The specific feature of these pro-death GsPCRs in ATP release is further supported by our experimental evidence that there was no detectable e[ATP] elevation seen by the activation of β2AR, A2R, or EP4 that did not promote CM death or had no effect on CM viability. The fact that two different pro-death GsPCRs, β1AR and H2R, stimulate CM death via the same e[ATP]-dependent mechanism suggests a commonality of this mechanism for other pro-death GsPCRs. The unique role of ATP release in CM death is consistent with the notion that e[ATP], particularly at high levels, serves as a danger signal involved in cell/tissue injuries 17. The catecholamine and histamine signaling are reported to be overactivated in human and animal heart failure with different etiologies 50,51. Thus, the e[ATP]-dependent pro-death signaling may also be critical for other cardiac disorders associated with CM death.
ATP can be released in multiple ways, varied by cell types, stimuli, and cellular conditions 18. Our results showed that blocking PANX1 but not Cx43 abolished ≈80% of the effects of β1AR and H2R stimulation on ATP release and CM death, suggesting that PANX1 is a predominant ATP-releasing channel. Post-translational modifications of PANX1 by tyrosine phosphorylation and/or serine/threonine phosphorylation are important in channel activity regulation. For example, TNFα stimulated Src-dependent phosphorylation of tyrosine 198 (Y198) PANX1, leading to PANX1 activation and increased inflammation in endothelial cells 52. N-methyl-D-aspartate receptor (NMDAR) activated PANX1 by Src-mediated phosphorylation at Y308 of PANX1 in neurons 53. In HEK-293 cells with ectopically expressed PANX1, nitric oxide (NO) donors decreased PANX1 activity via cGMP/PKG-mediated phosphorylation of PANX1 at S206 54. This appears to conflict with our finding that PANX1-S206 phosphorylation by PKA increased PANX1 activity. The discrepancy of S206 phosphorylation in PANX1 activity regulation may be due to additional modification(s) of PANX1 by NO. For example, PANX1 channels were inhibited by NO-induced S-nitrosylation on cysteine residues 55. Our finding is supported by a previous study showing that stimulating the thromboxane receptor increased PANX1 activation by cAMP/PKA in macrophages 56. Taken together, the regulation of PANX1 activation/inactivation could be a complex interplay between diverse post-translational modifications and/or interacting partners in a context-dependent manner 57.
Ca2+ plays a fundamental role in cell death 58. Ca2+ molecules from multiple sources have been implicated in apoptotic and non-apoptotic CM death 59. These primarily include external Ca2+ influx via various Ca2+-channels (e.g., L-type-calcium-channels (LTCC) and transient-receptor-potential-canonical (TRPC) channels) as well as internal Ca2+ mobilization from various intracellular organelles (e.g., endoplasmic reticulum and mitochondria). The mutual interplay between different sources of Ca2+ has been documented, i.e., Ca2+ entry can trigger Ca2+ release (a mechanism referred to as Ca2+-induced Ca2+ release). In the current study, we reported a critical role of P2X7R-derived Ca2+ in β1AR- or H2R-induced CM death, although we do not exclude the contribution of other Ca2+ sources. Ca2+ entry from P2X7R has also been shown to stimulate other sources of Ca2+, e.g., P2X7R-derived Ca2+ induced mitochondrial Ca2+ release and neuron death 60. LTCC-dependent and -independent mechanisms have been reported for β1AR-induced CM death 61. Differently, we failed to detect the contribution of LTCC in β1AR-induced death of CMs when the death studies were performed in the presence of blebbistatin (a specific myosin II inhibitor) to block CM contraction for extending CM survival during the culture (Figure S31A) 9,62. Thus, one possibility is that β1AR-induced CM contraction decreases CM viability when CM is cultured in vitro, which is LTCC-dependent. Taken together, we believe that the upstream eATP-P2X7R-Ca2+-CaMKII signaling axis can activate different downstream death pathways and contributes to CM death. In addition, the role of PKA in β1AR-mediated CM viability appears to be still controversial. For example, one previous study showed that CM apoptosis induced by β1AR activation depended on CaMKII but not PKA 27. However, another study reported that both PKA and CaMKII were required in CM apoptosis induced by β1AR activation, which is in line with our findings in the current study. The discrepancy might be due to different CM culture conditions, e.g., with and without blocking CM contraction during CM culture. The causes for the PKA discrepancy remain unclear and require more investigation.
i[cAMP] efflux, particularly A2R-derived i[cAMP], and e[cAMP]-e[ADO] signaling, have been previously reported in several other tissues, such as kidney 2, skeletal muscle 2, lung 63, and adipose tissue 64. However, the e[cAMP] signaling in CMs remains poorly characterized. Herein, we do not only fully depict cAMP-efflux and e[cAMP]-e[ADO] signaling for A2R in CMs but also CGRPR and RXFP1 for the first time (Graphical abstract), suggesting that this may represent a common mechanism for more GsPCRs in CM survival. β2AR is a unique GsPCR that couples to both Gs and Gi 65. It has been shown that β2AR stimulation protects CM survival via Gi-mediated activation of PI3K/AKT signaling 13,66. We also found that the β2AR agonist Zinterol attenuated H2O2-induced CM death, which was blocked by the β2AR antagonist ICI118,551 (Figure S30A). Unlike A2R, CGRPR, and RXFP1, the protective effect of β2AR was independent of MRP4, ENPP1, or A1R function (Figure S30B). However, the effect of β2AR depended on PI3K (Figure S30C), consistent with the report of β2AR/Gi/PI3K signaling in CM survival. We also showed that exogenously elevating e[cAMP] using membrane-impermeable cAMP exhibited protection for CM survival, which is dependent on e[cAMP]-e[ADO] pro-survival signaling. These findings further confirmed the crucial role of e[cAMP]-e[ADO] signaling in the pro-survival effect of GsPCRs on CMs. We also found that exogenous cAMP application elicited protective effects against IR injury ex vivo and in vivo (Figures 8B–8D, 8F–8L, S29A, S29B, and S29D–S29F). Chronic infusion of exogenous cAMP has also been shown to attenuate cardiac hypertrophy and fibrosis induced by phenylephrine (PE)/ISO infusion in mice 67, and in lung remodeling associated with pulmonary hypertension in rats and mice 63. These findings imply potential protective effects of exogenous cAMP application in different tissues. It is noteworthy that besides CMs, other cell types may contribute to the protective effects of exogenous cAMP in the heart. This is because non-CM cells in the heart, such as CF, EC, SMC, and immune cells, can also convert e[cAMP] to e[ADO]. In addition, many cells/tissues can release cAMP into circulation or urine 68. The released cAMP may be important for the functional crosstalk between different tissues/organs or act as a biomarker of diseases.
In 2014, Sassi et al. reported that ISO/β-AR elevated e[cAMP] levels due to the direct efflux of ISO/β-AR-produced i[cAMP] 67. Indeed, we also detected an ISO-induced increase of e[cAMP] in CMs (Figure S31C). The major question is whether ISO/β1AR-derived i[cAMP] can be directly pumped out. To address this question, we performed a series of experiments. We demonstrated that the elevation of e[cAMP] induced by ISO/β1AR stimulation is not due to direct i[cAMP] efflux but instead a result of ATP release, e[ATP] metabolism to e[ADO], e[ADO] stimulation of A2R, and A2R-derived i[cAMP] efflux (Figure S31B). Our key supporting data are summarized in supplementary Figure S31, including that e[cAMP] elevation induced by ISO was significantly abolished by blocking the function of ecto-ATPase CD39 with inhibitor POM1 or shRNA (Figures S31C–S31E). However, CV1808/A2R induced e[cAMP] elevation was not affected by CD39 inhibition (Figure S31F), suggesting that the sources of e[cAMP] are different between ISO/βAR and CV1808/A2R. As anticipated, ISO-induced e[cAMP] elevation was also abolished by A2R selective antagonist (Figure S31G). Moreover, the PLA performed with β1AR and MRP4 antibodies indicated no proximity of β1AR and MRP4 in CMs (Figure S21E). Unlike Sassi et al.’s report, our results suggest that β1AR-derived i[cAMP] is not directly pumped out. ISO-induced e[cAMP] elevation is dependent on ATP release, e[ATP] metabolism to e[ADO], and A2R activation. The formation of e[cAMP] from exogenous ATP was previously reported in isolated hepatocytes and adipocytes 69.
There are several limitations to our study. The current study provides insight into molecular mechanisms by which β1AR/H2R and A2R/CGRPR/RXFP1 could conversely regulate CM viability by stimulating pro-death e[ATP] signaling and the pro-survival e[cAMP] signaling, respectively. It remains unknown whether these two mechanisms are common for all other GsPCRs that regulate CM viability. Also, understanding the fate of i[cAMP] derived from a set of GsPCRs that do not regulate CM viability is of great interest. Moreover, it deserves to determine whether similar GsPCR-mediated signaling is present in other cell types. In the current study, we have focused on CM viability. GsPCR/cAMP signaling pathways often also regulate other functions of CMs, such as contractility and/or hypertrophy. It remains unknown whether these distinctive functions share similar signaling mechanisms or not. Moreover, the paracrine functions of e[cAMP], e[ADO], and e[ATP] between CM and CM or between CM and non-CM also deserve to be examined in the future. e[ATP] may also activate other P2X and P2Y receptors 70 and exhibit distinct functions that remain characterized. Finally, the roles of these GsPCR-mediated signaling pathways need to be tested in other models of cardiac diseases in which CM viability is important.
Supplementary Material
Novelty and Significance.
What is known?
Elevating intracellular cAMP, through targeting different stimulatory-G-protein-coupled-receptors (GsPCRs), adenylyl cyclases (Acs), or phosphodiesterases (PDEs), exhibits distinct, even opposing effects on cardiomyocyte (CM) viability.
Adenosine-A2-receptor (A2R)-stimulated intracellular cAMP can be exported and metabolized to ADO in some cell types.
What new information does this article contribute?
Pro-death GsPCRs, such as β1AR or H2R, induce CM death via cAMP/PKA-mediated phosphorylation and activation of pannexin-1 (PANX1), ATP release to the extracellular space, and e[ATP]-mediated activation of P2X purinoceptor 7 (P2X7R)-Ca2+-CaMKII signaling. This signaling cascade likely relies on the unique multiprotein complex with GsPCR, PANX1, and P2X7R.
Pro-survival GsPCRs, such as A2R, CGRPR, or RXFP1, protect against CM death via multidrug resistance protein (MRP4)-facilitated cAMP efflux, e[cAMP] metabolism into e[ADO], and e[ADO]-triggered activation of A1R/PI3K/AKT signaling. The effectiveness of this signaling cascade is dependent on the proximity of the pro-survival GsPCR to MRP4.
Exogenous cAMP administration mimics e[cAMP] elevation, which elicits protective effects against cardiac IR injury.
We aim to explore how distinct GsPCR/cAMP signaling differentially regulates CM viability. We found that stimulating pro-death GsPCRs (such as β1AR and H2R) promoted CM death via a mechanism depending on ATP release and e[ATP]-mediated signaling. In contrast, stimulating pro-survival GsPCRs (such as A2R, CGRPR, and RXFP1) suppressed H2O2-induced CM death via a mechanism depending on cAMP efflux and e[cAMP]-mediated signaling. The protective effect of e[cAMP] on CM viability was further elucidated using the exogenous application of membrane-impermeable cAMP in ex vivo and in vivo heart injury models. The fact that pro-death or pro-survival GsPCRs share identical e[ATP] or e[cAMP] signaling, respectively, suggests a certain commonality of these signaling mechanisms in GsPCR regulation of CM viability. These findings provide biological insight into how GsPCRs can differentially regulate CM viability through discrete cAMP signalosomes. The critical molecular components of the e[ATP]- or e[cAMP]-signaling may represent novel molecular targets for combating CM death in various cardiac disorders.
ACKNOWLEDGEMENTS
We thank Linda Marie Callahan for the image acquisition consultation. The graphical abstract was created with BioRender.com.
SOURCES OF FUNDING
This work was financially supported by the National Institute of Health HL154318 and HL162259 (to C.Y.).
Nonstandard Abbreviations and Acronyms
- A1R
adenosine-A1-receptor
- A2R
adenosine-A2-receptor
- AC
adenylyl cyclase
- AMT
amthamine dihydrobromide
- β1AR
beta1-adrenergic-receptor
- CaMKII
calcium/calmodulin-dependent protein kinase II
- CGRPR
calcitonin-gene-related-peptide-receptor
- CK
creatine kinase
- CM
cardiomyocyte
- cTNI
cardiac troponin I
- e[ADO]
extracellular adenosine
- e[ATP]
extracellular ATP
- e[cAMP]
extracellular cAMP
- GsPCR
stimulatory-G-protein-coupled-receptors
- H2R
histamine-H2-receptor
- i[cAMP]
intracellular cAMP
- IR
ischemia-reperfusion
- ISO
isoproterenol
- KD
knock-down
- LDH
lactate dehydrogenase
- MRP
multidrug resistance protein
- PANX1
pannexin-1
- P2X7R
P2X purinoceptor 7
- PKA
cAMP-dependent-protein-kinase-A
- PKI
PKA inhibitor
- PLA
proximity Ligation Assay
- RXFP1
relaxin-family-peptide-receptor 1
Footnotes
DISCLOSURES
There is no conflict of interest.
REFERENCES
- 1.Wang J, Gareri C, Rockman HA. G-Protein-Coupled Receptors in Heart Disease. Circ Res. 2018;123:716–735. doi: 10.1161/CIRCRESAHA.118.311403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Godinho RO, Duarte T, Pacini ES. New perspectives in signaling mediated by receptors coupled to stimulatory G protein: the emerging significance of cAMP e ffl ux and extracellular cAMP-adenosine pathway. Front Pharmacol. 2015;6:58. doi: 10.3389/fphar.2015.00058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hasko G, Antonioli L, Cronstein BN. Adenosine metabolism, immunity and joint health. Biochem Pharmacol. 2018;151:307–313. doi: 10.1016/j.bcp.2018.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. 2003;93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA [DOI] [PubMed] [Google Scholar]
- 5.Peart JN, Headrick JP. Adenosinergic cardioprotection: multiple receptors, multiple pathways. Pharmacol Ther. 2007;114:208–221. doi: 10.1016/j.pharmthera.2007.02.004 [DOI] [PubMed] [Google Scholar]
- 6.Okumura S, Vatner DE, Kurotani R, Bai Y, Gao S, Yuan Z, Iwatsubo K, Ulucan C, Kawabe J, Ghosh K, et al. Disruption of type 5 adenylyl cyclase enhances desensitization of cyclic adenosine monophosphate signal and increases Akt signal with chronic catecholamine stress. Circulation. 2007;116:1776–1783. doi: 10.1161/CIRCULATIONAHA.107.698662 [DOI] [PubMed] [Google Scholar]
- 7.Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, Lew WY, Clopton P, Hammond HK. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation. 2006;114:388–396. doi: 10.1161/CIRCULATIONAHA.106.632513 [DOI] [PubMed] [Google Scholar]
- 8.Ding B, Abe JI, Wei H, Huang Q, Walsh RA, Molina CA, Zhao A, Sadoshima J, Blaxall BC, Berk BC, et al. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation. 2005;111:2469–2476. doi: 10.1161/01.CIR.0000165128.39715.87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang Y, Knight W, Chen S, Mohan A, Yan C. Multiprotein Complex With TRPC (Transient Receptor Potential-Canonical) Channel, PDE1C (Phosphodiesterase 1C), and A2R (Adenosine A2 Receptor) Plays a Critical Role in Regulating Cardiomyocyte cAMP and Survival. Circulation. 2018;138:1988–2002. doi: 10.1161/CIRCULATIONAHA.118.034189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knight WE, Chen S, Zhang Y, Oikawa M, Wu M, Zhou Q, Miller CL, Cai Y, Mickelsen DM, Moravec C, et al. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci U S A. 2016;113:E7116–E7125. doi: 10.1073/pnas.1607728113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hill SJ, Ganellin CR, Timmerman H, Schwartz JC, Shankley NP, Young JM, Schunack W, Levi R, Haas HL. International Union of Pharmacology. XIII. Classification of histamine receptors. Pharmacol Rev. 1997;49:253–278. [PubMed] [Google Scholar]
- 12.Luo T, Chen B, Zhao Z, He N, Zeng Z, Wu B, Fukushima Y, Dai M, Huang Q, Xu D, et al. Histamine H2 receptor activation exacerbates myocardial ischemia/reperfusion injury by disturbing mitochondrial and endothelial function. Basic Res Cardiol. 2013;108:342. doi: 10.1007/s00395-013-0342-4 [DOI] [PubMed] [Google Scholar]
- 13.Communal C, Singh K, Sawyer DB, Colucci WS. Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis : role of a pertussis toxin-sensitive G protein. Circulation. 1999;100:2210–2212. doi: 10.1161/01.cir.100.22.2210 [DOI] [PubMed] [Google Scholar]
- 14.Zaugg M, Xu W, Lucchinetti E, Shafiq SA, Jamali NZ, Siddiqui MA. Beta-adrenergic receptor subtypes differentially affect apoptosis in adult rat ventricular myocytes. Circulation. 2000;102:344–350. doi: 10.1161/01.cir.102.3.344 [DOI] [PubMed] [Google Scholar]
- 15.Defer N, Best-Belpomme M, Hanoune J. Tissue specificity and physiological relevance of various isoforms of adenylyl cyclase. Am J Physiol Renal Physiol. 2000;279:F400–416. doi: 10.1152/ajprenal.2000.279.3.F400 [DOI] [PubMed] [Google Scholar]
- 16.Brand CS, Hocker HJ, Gorfe AA, Cavasotto CN, Dessauer CW. Isoform selectivity of adenylyl cyclase inhibitors: characterization of known and novel compounds. J Pharmacol Exp Ther. 2013;347:265–275. doi: 10.1124/jpet.113.208157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao H, Kilgas S, Alam A, Eguchi S, Ma D. The Role of Extracellular Adenosine Triphosphate in Ischemic Organ Injury. Crit Care Med. 2016;44:1000–1012. doi: 10.1097/CCM.0000000000001603 [DOI] [PubMed] [Google Scholar]
- 18.Taruno A. ATP Release Channels. Int J Mol Sci. 2018;19. doi: 10.3390/ijms19030808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boengler K, Schulz R, Heusch G. Connexin 43 signalling and cardioprotection. Heart. 2006;92:1724–1727. doi: 10.1136/hrt.2005.066878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dolmatova E, Spagnol G, Boassa D, Baum JR, Keith K, Ambrosi C, Kontaridis MI, Sorgen PL, Sosinsky GE, Duffy HS. Cardiomyocyte ATP release through pannexin 1 aids in early fibroblast activation. Am J Physiol Heart Circ Physiol. 2012;303:H1208–1218. doi: 10.1152/ajpheart.00251.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yegutkin GG. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim Biophys Acta. 2008;1783:673–694. doi: 10.1016/j.bbamcr.2008.01.024 [DOI] [PubMed] [Google Scholar]
- 22.Falzoni S, Donvito G, Di Virgilio F. Detecting adenosine triphosphate in the pericellular space. Interface Focus. 2013;3:20120101. doi: 10.1098/rsfs.2012.0101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, et al. International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic SS. Activation and regulation of purinergic P2X receptor channels. Pharmacol Rev. 2011;63:641–683. doi: 10.1124/pr.110.003129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazzola A, Amoruso E, Beltrami E, Lecca D, Ferrario S, Cosentino S, Tremoli E, Ceruti S, Abbracchio MP. Opposite effects of uracil and adenine nucleotides on the survival of murine cardiomyocytes. J Cell Mol Med. 2008;12:522–536. doi: 10.1111/j.1582-4934.2007.00133.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bravo D, Maturana CJ, Pelissier T, Hernandez A, Constandil L. Interactions of pannexin 1 with NMDA and P2X7 receptors in central nervous system pathologies: Possible role on chronic pain. Pharmacol Res. 2015;101:86–93. doi: 10.1016/j.phrs.2015.07.016 [DOI] [PubMed] [Google Scholar]
- 27.Zhu WZ, Wang SQ, Chakir K, Yang D, Zhang T, Brown JH, Devic E, Kobilka BK, Cheng H, Xiao RP. Linkage of beta1-adrenergic stimulation to apoptotic heart cell death through protein kinase A-independent activation of Ca2+/calmodulin kinase II. J Clin Invest. 2003;111:617–625. doi: 10.1172/JCI16326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu W, Woo AY, Yang D, Cheng H, Crow MT, Xiao RP. Activation of CaMKIIdeltaC is a common intermediate of diverse death stimuli-induced heart muscle cell apoptosis. J Biol Chem. 2007;282:10833–10839. doi: 10.1074/jbc.M611507200 [DOI] [PubMed] [Google Scholar]
- 29.Burma NE, Bonin RP, Leduc-Pessah H, Baimel C, Cairncross ZF, Mousseau M, Shankara JV, Stemkowski PL, Baimoukhametova D, Bains JS, et al. Blocking microglial pannexin-1 channels alleviates morphine withdrawal in rodents. Nat Med. 2017;23:355–360. doi: 10.1038/nm.4281 [DOI] [PubMed] [Google Scholar]
- 30.Park SW, Persaud SD, Ogokeh S, Meyers TA, Townsend D, Wei LN. CRABP1 protects the heart from isoproterenol-induced acute and chronic remodeling. J Endocrinol. 2018;236:151–165. doi: 10.1530/JOE-17-0613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wallner M, Duran JM, Mohsin S, Troupes CD, Vanhoutte D, Borghetti G, Vagnozzi RJ, Gross P, Yu D, Trappanese DM, et al. Acute Catecholamine Exposure Causes Reversible Myocyte Injury Without Cardiac Regeneration. Circ Res. 2016;119:865–879. doi: 10.1161/CIRCRESAHA.116.308687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prasad KM, Xu Y, Yang Z, Acton ST, French BA. Robust cardiomyocyte-specific gene expression following systemic injection of AAV: in vivo gene delivery follows a Poisson distribution. Gene Ther. 2011;18:43–52. doi: 10.1038/gt.2010.105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, Chen C, Li J, Xiao X. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005;23:321–328. doi: 10.1038/nbt1073 [DOI] [PubMed] [Google Scholar]
- 34.Meyers TA, Heitzman JA, Krebsbach AM, Aufdembrink LM, Hughes R, Bartolomucci A, Townsend D. Acute AT(1)R blockade prevents isoproterenol-induced injury in mdx hearts. J Mol Cell Cardiol. 2019;128:51–61. doi: 10.1016/j.yjmcc.2019.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, Szeto C, Gao E, Tang M, Jin J, Fu Q, Makarewich C, Ai X, Li Y, Tang A, et al. Cardiotoxic and cardioprotective features of chronic beta-adrenergic signaling. Circ Res. 2013;112:498–509. doi: 10.1161/CIRCRESAHA.112.273896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vizgirda VM, Wahler GM, Sondgeroth KL, Ziolo MT, Schwertz DW. Mechanisms of sex differences in rat cardiac myocyte response to beta-adrenergic stimulation. Am J Physiol Heart Circ Physiol. 2002;282:H256–263. doi: 10.1152/ajpheart.2002.282.1.H256 [DOI] [PubMed] [Google Scholar]
- 37.Szentmiklosi AJ, Cseppento A, Harmati G, Nanasi PP. Novel trends in the treatment of cardiovascular disorders: site- and event- selective adenosinergic drugs. Curr Med Chem. 2011;18:1164–1187. doi: 10.2174/092986711795029753 [DOI] [PubMed] [Google Scholar]
- 38.Kee Z, Kodji X, Brain SD. The Role of Calcitonin Gene Related Peptide (CGRP) in Neurogenic Vasodilation and Its Cardioprotective Effects. Front Physiol. 2018;9:1249. doi: 10.3389/fphys.2018.01249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar A, Potts JD, DiPette DJ. Protective Role of alpha-Calcitonin Gene-Related Peptide in Cardiovascular Diseases. Front Physiol. 2019;10:821. doi: 10.3389/fphys.2019.00821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarwar M, Du XJ, Dschietzig TB, Summers RJ. The actions of relaxin on the human cardiovascular system. Br J Pharmacol. 2017;174:933–949. doi: 10.1111/bph.13523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu SY, Nakabayashi K, Nishi S, Kumagai J, Kudo M, Sherwood OD, Hsueh AJ. Activation of orphan receptors by the hormone relaxin. Science. 2002;295:671–674. doi: 10.1126/science.1065654 [DOI] [PubMed] [Google Scholar]
- 42.Bera TK, Lee S, Salvatore G, Lee B, Pastan I. MRP8, a new member of ABC transporter superfamily, identified by EST database mining and gene prediction program, is highly expressed in breast cancer. Mol Med. 2001;7:509–516. [PMC free article] [PubMed] [Google Scholar]
- 43.Shimizu H, Taniguchi H, Hippo Y, Hayashizaki Y, Aburatani H, Ishikawa T. Characterization of the mouse Abcc12 gene and its transcript encoding an ATP-binding cassette transporter, an orthologue of human ABCC12. Gene. 2003;310:17–28. doi: 10.1016/s0378-1119(03)00504-3 [DOI] [PubMed] [Google Scholar]
- 44.Stefan C, Jansen S, Bollen M. NPP-type ectophosphodiesterases: unity in diversity. Trends Biochem Sci. 2005;30:542–550. doi: 10.1016/j.tibs.2005.08.005 [DOI] [PubMed] [Google Scholar]
- 45.Stefan C, Jansen S, Bollen M. Modulation of purinergic signaling by NPP-type ectophosphodiesterases. Purinergic Signal. 2006;2:361–370. doi: 10.1007/s11302-005-5303-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cristalli G, Costanzi S, Lambertucci C, Lupidi G, Vittori S, Volpini R, Camaioni E. Adenosine deaminase: functional implications and different classes of inhibitors. Med Res Rev. 2001;21:105–128. doi: [DOI] [PubMed] [Google Scholar]
- 47.Regan SE, Broad M, Byford AM, Lankford AR, Cerniway RJ, Mayo MW, Matherne GP. A1 adenosine receptor overexpression attenuates ischemia-reperfusion-induced apoptosis and caspase 3 activity. Am J Physiol Heart Circ Physiol. 2003;284:H859–866. doi: 10.1152/ajpheart.00251.2002 [DOI] [PubMed] [Google Scholar]
- 48.Wu MP, Zhang YS, Zhou QM, Xiong J, Dong YR, Yan C. Higenamine protects ischemia/reperfusion induced cardiac injury and myocyte apoptosis through activation of beta2-AR/PI3K/AKT signaling pathway. Pharmacol Res. 2016;104:115–123. doi: 10.1016/j.phrs.2015.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Castillo OA, Herrera G, Manriquez C, Rojas AF, Gonzalez DR. Pharmacological Inhibition of S-Nitrosoglutathione Reductase Reduces Cardiac Damage Induced by Ischemia-Reperfusion. Antioxidants (Basel). 2021;10. doi: 10.3390/antiox10040555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Patella V, Marino I, Arbustini E, Lamparter-Schummert B, Verga L, Adt M, Marone G. Stem cell factor in mast cells and increased mast cell density in idiopathic and ischemic cardiomyopathy. Circulation. 1998;97:971–978. doi: 10.1161/01.cir.97.10.971 [DOI] [PubMed] [Google Scholar]
- 51.Frangogiannis NG, Perrard JL, Mendoza LH, Burns AR, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, Entman ML. Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation. 1998;98:687–698. doi: 10.1161/01.cir.98.7.687 [DOI] [PubMed] [Google Scholar]
- 52.Lohman AW, Leskov IL, Butcher JT, Johnstone SR, Stokes TA, Begandt D, DeLalio LJ, Best AK, Penuela S, Leitinger N, et al. Pannexin 1 channels regulate leukocyte emigration through the venous endothelium during acute inflammation. Nat Commun. 2015;6:7965. doi: 10.1038/ncomms8965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weilinger NL, Lohman AW, Rakai BD, Ma EM, Bialecki J, Maslieieva V, Rilea T, Bandet MV, Ikuta NT, Scott L, et al. Metabotropic NMDA receptor signaling couples Src family kinases to pannexin-1 during excitotoxicity. Nat Neurosci. 2016;19:432–442. doi: 10.1038/nn.4236 [DOI] [PubMed] [Google Scholar]
- 54.Poornima V, Vallabhaneni S, Mukhopadhyay M, Bera AK. Nitric oxide inhibits the pannexin 1 channel through a cGMP-PKG dependent pathway. Nitric Oxide. 2015;47:77–84. doi: 10.1016/j.niox.2015.04.005 [DOI] [PubMed] [Google Scholar]
- 55.Lohman AW, Weaver JL, Billaud M, Sandilos JK, Griffiths R, Straub AC, Penuela S, Leitinger N, Laird DW, Bayliss DA, et al. S-nitrosylation inhibits pannexin 1 channel function. J Biol Chem. 2012;287:39602–39612. doi: 10.1074/jbc.M112.397976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.da Silva-Souza HA, de Lira MN, Patel NK, Spray DC, Persechini PM, Scemes E. Inhibitors of the 5-lipoxygenase pathway activate pannexin1 channels in macrophages via the thromboxane receptor. Am J Physiol Cell Physiol. 2014;307:C571–579. doi: 10.1152/ajpcell.00087.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Penuela S, Simek J, Thompson RJ. Regulation of pannexin channels by post-translational modifications. FEBS Lett. 2014;588:1411–1415. doi: 10.1016/j.febslet.2014.01.028 [DOI] [PubMed] [Google Scholar]
- 58.Feng N, Anderson ME. CaMKII is a nodal signal for multiple programmed cell death pathways in heart. J Mol Cell Cardiol. 2017;103:102–109. doi: 10.1016/j.yjmcc.2016.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dorn GW 2nd. Apoptotic and non-apoptotic programmed cardiomyocyte death in ventricular remodelling. Cardiovasc Res. 2009;81:465–473. doi: 10.1093/cvr/cvn243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nishida K, Nakatani T, Ohishi A, Okuda H, Higashi Y, Matsuo T, Fujimoto S, Nagasawa K. Mitochondrial dysfunction is involved in P2X7 receptor-mediated neuronal cell death. J Neurochem. 2012;122:1118–1128. doi: 10.1111/j.1471-4159.2012.07868.x [DOI] [PubMed] [Google Scholar]
- 61.Wang W, Zhang H, Gao H, Kubo H, Berretta RM, Chen X, Houser SR. beta1-Adrenergic receptor activation induces mouse cardiac myocyte death through both L-type calcium channel-dependent and -independent pathways. Am J Physiol Heart Circ Physiol. 2010;299:H322–331. doi: 10.1152/ajpheart.00392.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kabaeva Z, Zhao M, Michele DE. Blebbistatin extends culture life of adult mouse cardiac myocytes and allows efficient and stable transgene expression. Am J Physiol Heart Circ Physiol. 2008;294:H1667–1674. doi: 10.1152/ajpheart.01144.2007 [DOI] [PubMed] [Google Scholar]
- 63.Jones C, Bisserier M, Bueno-Beti C, Bonnet G, Neves-Zaph S, Lee SY, Milara J, Dorfmuller P, Humbert M, Leopold JA, et al. A novel secreted-cAMP pathway inhibits pulmonary hypertension via a feed-forward mechanism. Cardiovasc Res. 2020;116:1500–1513. doi: 10.1093/cvr/cvz244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Strouch MB, Jackson EK, Mi Z, Metes NA, Carey GB. Extracellular cyclic AMP-adenosine pathway in isolated adipocytes and adipose tissue. Obes Res. 2005;13:974–981. doi: 10.1038/oby.2005.114 [DOI] [PubMed] [Google Scholar]
- 65.Xiao RP. Beta-adrenergic signaling in the heart: dual coupling of the beta2-adrenergic receptor to G(s) and G(i) proteins. Sci STKE. 2001;2001:re15. doi: 10.1126/stke.2001.104.re15 [DOI] [PubMed] [Google Scholar]
- 66.Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci U S A. 2001;98:1607–1612. doi: 10.1073/pnas.98.4.1607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sassi Y, Ahles A, Truong DJ, Baqi Y, Lee SY, Husse B, Hulot JS, Foinquinos A, Thum T, Muller CE, et al. Cardiac myocyte-secreted cAMP exerts paracrine action via adenosine receptor activation. J Clin Invest. 2014;124:5385–5397. doi: 10.1172/JCI74349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bankir L, Ahloulay M, Devreotes PN, Parent CA. Extracellular cAMP inhibits proximal reabsorption: are plasma membrane cAMP receptors involved? Am J Physiol Renal Physiol. 2002;282:F376–392. doi: 10.1152/ajprenal.00202.2001 [DOI] [PubMed] [Google Scholar]
- 69.Crooke MJ, Allan E, Pattinson N, Sneyd JG. Formation of cyclic AMP from exogenous ATP by isolated hepatocytes and adipocytes. Biochim Biophys Acta. 1980;631:28–39. doi: 10.1016/0304-4165(80)90050-1 [DOI] [PubMed] [Google Scholar]
- 70.Kukulski F, Levesque SA, Sevigny J. Impact of ectoenzymes on p2 and p1 receptor signaling. Adv Pharmacol. 2011;61:263–299. doi: 10.1016/B978-0-12-385526-8.00009-6 [DOI] [PubMed] [Google Scholar]
- 71.Rellos P, Pike AC, Niesen FH, Salah E, Lee WH, von Delft F, Knapp S. Structure of the CaMKIIdelta/calmodulin complex reveals the molecular mechanism of CaMKII kinase activation. PLoS Biol. 2010;8:e1000426. doi: 10.1371/journal.pbio.1000426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guatimosim S, Guatimosim C, Song LS. Imaging calcium sparks in cardiac myocytes. Methods Mol Biol. 2011;689:205–214. doi: 10.1007/978-1-60761-950-5_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chen S, Zhang Y, Lighthouse JK, Mickelsen DM, Wu J, Yao P, Small EM, Yan C. A Novel Role of Cyclic Nucleotide Phosphodiesterase 10A in Pathological Cardiac Remodeling and Dysfunction. Circulation. 2020;141:217–233. doi: 10.1161/CIRCULATIONAHA.119.042178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen S, Chen J, Du W, Mickelsen DM, Shi H, Yu H, Kumar S, Yan C. PDE10A Inactivation Prevents Doxorubicin-Induced Cardiotoxicity and Tumor Growth. Circ Res. 2023;133:138–157. doi: 10.1161/CIRCRESAHA.122.322264 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data and materials within the article and the Data Supplement are available publicly.
Detailed descriptions of experimental methods, materials, and statistical analysis are presented in the Materials of the Data Supplement. The Major Resource Table is also included in the Supplemental Materials.