

Abstract
Hepatocyte ferroptosis promotes the pathogenesis and progression of liver fibrosis. Salvianolic acid B (Sal B) exerts antifibrotic effects. However, the pharmacological mechanism and target has not yet been fully elucidated. In this study, liver fibrosis was induced by CCl4 in wild-type mice and hepatocyte-specific extracellular matrix protein 1 (Ecm1)-deficient mice, which were separately treated with Sal B, ferrostatin-1, sorafenib or cilengitide. Erastin- or CCl4-induced hepatocyte ferroptosis models with or without Ecm1 gene knockdown were evaluated in vitro. Subsequently, the interaction between Ecm1 and xCT and the binding kinetics of Sal B and Ecm1 were determined. We found that Sal B significantly attenuated liver fibrosis in CCl4-induced mice. Ecm1 deletion in hepatocytes abolished the antifibrotic effect of Sal B. Mechanistically, Sal B protected against hepatocyte ferroptosis by upregulating Ecm1. Further research revealed that Ecm1 as a direct target for treating liver fibrosis with Sal B. Interestingly, Ecm1 interacted with xCT to regulate hepatocyte ferroptosis. Hepatocyte ferroptosis in vitro was significantly attenuated by Sal B treatment, which was abrogated after knockdown of Ecm1 in LO2 cells. Therefore, Sal B alleviates liver fibrosis in mice by targeting up-regulation of Ecm1 and inhibiting hepatocyte ferroptosis. The interaction between Ecm1 and xCT regulates hepatocyte ferroptosis.
Keywords: Salvianolic acid B, Liver fibrosis, Ecm1, xCT, Hepatocyte ferroptosis
Graphical abstract
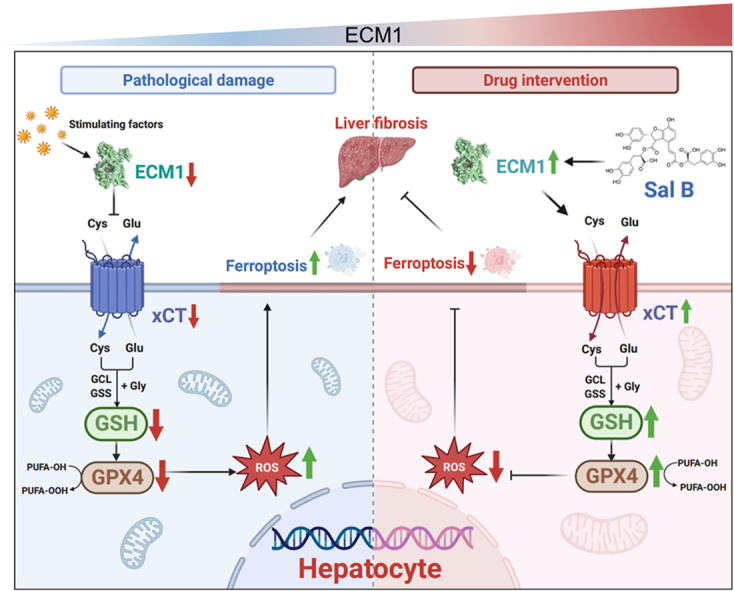
Highlights
-
•
Sal B alleviates liver fibrosis in mice by upregulating Ecm1 and inhibiting hepatocyte ferroptosis.
-
•
The interaction between Ecm1 and xCT regulates hepatocyte ferroptosis and is a hitherto underscribed mechanism of Sal B.
-
•
Ecm1 is a direct pharmacological target of Sal B to protect against liver fibrosis.
1. Introduction
Liver fibrosis is a prevalent pathological process during the progression of various chronic liver diseases that ultimately lead to cirrhosis, hepatocellular carcinoma, and even liver failure, posing a severe threat to human life and health [1,2]. Liver transplantation remains the sole efficacious treatment for end-stage liver disease, but it is beset with a plethora of challenges, such as high risk, exorbitant cost, and a paucity of donors [3]. Alleviating liver fibrosis is considered a promising strategy for preventing the malignant progression of chronic liver diseases. However, despite the emergence of small-molecule targeted drugs in recent years, their efficacy in treating liver fibrosis remains uncertain. Currently, there are no FDA-approved biological or chemical drugs available for liver fibrosis [4,5]. The urgent and critical clinical needs necessitate the investigation of novel therapeutic targets and candidate drugs that can significantly contribute to the prevention and treatment of liver fibrosis.
The process of liver fibrosis is complex and involves multiple factors, and the activation of hepatic stellate cells (HSCs) is a key factor that has been extensively studied in previous research [6]. In recent years, increasing research has focused on the contribution of hepatocytes and extracellular matrix to the progression of liver fibrosis [[7], [8], [9]]. Liver fibrosis is initiated by hepatocyte injury, while excessive deposition and remodeling of the extracellular matrix leads to fibrotic scarring. Our previous study demonstrated that extracellular matrix protein 1 (Ecm1) is a naturally occurring matrix component in the liver that inhibits liver fibrosis [9]. Ecm1 is primarily secreted by hepatocytes and binds to cell surface receptor molecules such as integrin αv to inhibit the maturation and activation of transforming growth factor-β1 (TGF-β1), ultimately inhibiting liver fibrosis. Knockout of the Ecm1 gene in mice can result in spontaneous liver fibrosis, indicating that Ecm1 plays a crucial role in maintaining liver tissue homeostasis and cellular function. In addition, it has been demonstrated that the pathogenesis and progression of liver fibrosis is attributed to hepatocyte ferroptosis, while the inhibition of hepatocyte ferroptosis has been shown to alleviate liver fibrosis [10]. However, the precise mechanism that triggers hepatocyte ferroptosis during the progression of liver fibrosis remains unclear. Ferroptosis is a recently discovered form of programmed cell death that is driven by iron-dependent lipid peroxidation-induced damage [11]. Hepatocytes, which are primarily responsible for iron storage and hepcidin production, play a crucial role in regulating iron metabolism within the body. Clinical studies have demonstrated that patients with chronic hepatitis exhibit mild to moderate iron deposition, which can stimulate the proliferation of type IV collagen and lead to the formation of fibrous nodules surrounded by iron in the liver [12]. These findings suggest that the inhibition of hepatocyte ferroptosis holds potential as a therapeutic strategy for liver fibrosis.
Salvianolic acid B (Sal B), a natural and potent antioxidant, exerts hepatoprotective effects and reverses liver fibrosis [[13], [14], [15]]. Studies indicate that Sal B exerts a protective effect on hepatocytes by inhibiting the death receptor pathway and stabilizing mitochondrial membranes [14,16,17]; however, its role in regulating hepatocyte ferroptosis remains unknown. In this study, the results revealed that Sal B effectively modulates the solute carrier family 7 member 11 (SLC7A11, xCT) signaling pathway by targeting Ecm1, resulting in the inhibition of hepatocyte ferroptosis and alleviation of liver fibrosis in mice. Furthermore, our investigation revealed that Ecm1 interacts with xCT to regulate hepatocyte ferroptosis. These findings provide valuable insights into a novel mechanism and target for Sal B-mediated inhibition of liver fibrosis.
2. Materials and methods
2.1. Natural compound
Sal B (purity ≥98.0 %) was provided from Professor Lijiang Xuan (Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China). The structural identification spectra of Sal B is showed in Supporting Information Fig. S1.
2.2. Mice
Specific-pathogen-free-grade male wild-type (WT) C57/BL6, weighing 18–20 g, were purchased from Shanghai Southern Model Biotechnology Co., Ltd. (Shanghai, China). Ecm1−/−, Ecm1flox/flox (WT), and AlbcreEcm1flox/flox (Ecm1Δhep) mice were generated and maintained by Shanghai Southern Model Biotechnology Co., Ltd. (Shanghai, China). The mice were housed under standardized conditions, including an ambient temperature of 25 ± 2 °C, relative humidity of 40–60 %, and a 12-h light/dark cycle. The mice were provided ad libitum access to standard diet and water. All mouse experiments were approved by the Institutional Animal Care and Use Committee at Shanghai Research Center of the Southern Model Organisms (Approval No. 2019–0031).
2.3. Preparation of animal models and drug administration
Liver fibrosis was induced in mice by carbon tetrachloride (CCl4) as described in our previous study [18].
CCl4-induced liver fibrosis mouse model treated with different doses of Sal B: A total of 48 C57/BL6 mice were randomly divided into the control group (n = 8) and the CCl4-treated group (n = 40). The CCl4-treated mice received intraperitoneal injections of a 15 % CCl4/olive oil solution at a dose of 2 mL/kg for 6 weeks, while the control mice were intraperitoneally injected with olive oil at a dose of 2 mL/kg. On the first day of the 4th week, the CCl4-treated mice were further randomly divided into 5 groups: the CCl4 group, low-dose Sal B-treated group (5.6 mg/kg), medium-dose Sal B-treated group (11.2 mg/kg), high-dose Sal B-treated group (22.4 mg/kg), and ferrostatin-1 (Fer-1)-treated group (1 mg/kg) (n = 8 per group). Sal B-treated mice were intragastrically administered low, medium, and high doses of Sal B at a dose of 10 mL/kg for 3 weeks. Fer-1-treated mice received an intraperitoneal injection of 1 mg/kg Fer-1 dissolved in DMSO at a dose of 2 mL/kg once per day for 3 weeks. The statistical analysis was performed on a cohort of 8 mice per group for the purpose of comparison.
CCl4-induced liver fibrosis model in hepatocyte-specific Ecm1 knockout mice and WT controls treated with Sal B: We established a CCl4-induced liver fibrosis model in hepatocyte-specific Ecm1-knockout mice (Ecm1Δhep) by using littermate Ecm1flox/flox mice as WT controls according to the previously described protocols. At the beginning of the 4th week, CCl4-treated mice were randomly assigned to the following 3 groups and were treated daily for 3 weeks: the CCl4-treated group, Sal B-treated group (22.4 mg/kg), and control drug-treated group (n = 6 per group). WT mice in the control drug-treated group were orally administered sorafenib (Sora) (7 mg/kg) [18], while Ecm1Δhep mice in the control drug-treated group received an intraperitoneal injection of cilengitide (Cile) (30 mg/kg) [19]. The statistical analysis was performed on a cohort of 6 mice per group for the purpose of comparison.
2.4. Cell culture and hepatocyte ferroptosis models
The human hepatocyte cell line, LO2 cells, were cultured in RMPI 1640 high glucose medium supplemented with 10 % fetal bovine serum for this experiment. LO2 cells were seeded at a density of 1 × 105 cells/mL in the corresponding cell-culture plate. To induce hepatocyte ferroptosis models, LO2 cells were treated with either 50 μM erastin or 0.4 % CCl4 for 24 h. Additionally, the cells were treated with or without Sal B, recombinant human ECM1 protein, or Fer-1. All cell experiments were repeated three times using separate cell cultures.
2.5. Affinity kinetic determination of Sal B and Ecm1
The SA biosensor was conjugated with biotin⁃Ecm1, and the real-time kinetic parameters of Sal B and Ecm1 were measured using an Octet Red 96 molecular interaction analyzer. During the detection process, 12 SA biosensors were positioned on the sensor plate and equilibrated in PBS buffer for 10 min. The biotin⁃Ecm1 protein was then diluted to 150 μg/mL with PBS solution, while the prepared 2 mM Sal B solution was further diluted 4 times with PBS to create 5 distinct concentrations. Finally, the biotin⁃Ecm1 protein, gradient dilution of Sal B, and PBS buffer were sequentially added to the sample plate (200 μL of solution dispensed into each well). The detection temperature was set at 30 °C. The sample plate was rotated at a speed of 400 r/min. Following the fixation of the sample and the sensor plates, the established program (equilibration for 60 s, sample loading for 480 s, cleaning for 120 s, binding for 240 s, and dissociation for 240 s) was run. The data were subjected to affinity kinetic analysis.
2.6. Statistical analysis
The data were analyzed using SPSS 21.0 software. The results are presented as the mean ± standard deviation. Comparisons between multiple groups were performed by one-way ANOVA, followed by the least significant difference test, while comparisons between two groups were performed by Student's t-test. P < 0.05 was considered statistically significant.
The detailed materials and methods were provided in the supplementary material.
3. Results
3.1. Sal B attenuates CCl4-induced mouse liver fibrosis, which is associated with the upregulation of Ecm1
To observe the pharmacological effect of Sal B on liver fibrosis, we established CCl4-induced liver fibrosis in mice according to previous studies [18], followed by treatment with different doses of Sal B for 3 weeks with continuous CCl4 administration (Fig. 1A). Serum biochemical analyses demonstrated that the activities of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) in mice in the Sal B-treated groups and Fer-1-treated group were significantly lower than those in mice in the CCl4 group (P < 0.05, P < 0.01) (Fig. 1B). As shown by hematoxylin & eosin (H&E) staining, hepatocyte cytoplasm in the CCl4-treated group was diffuse, which was accompanied by extensive infiltration of inflammatory cells around the portal areas and the formation of inflammatory zonules extending into the liver parenchyma. In contrast, the Sal B-treated groups and Fer-1-treated group exhibited a significant reduction in hepatic inflammatory cells infiltration (Fig. 1C). Sirius red (SR) staining revealed that the collagen fibers extended from the portal area in a reticular pattern with wider fiber spacing. The SR-positive area and hepatic hydroxyproline (Hyp) content were significantly increased in the CCl4-treated group compared to the Oil group (P < 0.001). The collagen levels in the liver tissues of mice treated with low, medium, and high doses of Sal B and Fer-1 were significantly reduced (P < 0.01, P < 0.001) (Fig. 1C and D). The best curative effect was observed in the high-dose Sal B group.
Fig. 1.

Sal B attenuated CCl4-induced liver fibrosis and increased hepatic Ecm1 expression. (A) Scheme of the experimental procedure for C57BL/6 mice injected with CCl4 and treated with Sal B or Fer-1. (B) Serum ALT and AST activity. (C) Representative images of mouse liver tissue stained with H&E and SR (bar = 100 μm), and semiquantitative analysis was performed to evaluate the extent of the SR-positive staining area. (D) Hepatic hyp content. (E) Representative images of IHC staining of Ecm1 (bar = 100 μm) and TEM images (bar = 2 μm) are presented. (F) Western blot analysis and quantification of Ecm1 protein expression using gray value analysis, Ecm1 mRNA expression, and detection of Ecm1 in liver tissue by ELISA. Data are represented as mean ± SD (n = 6–8/group). *P < 0.05; **P < 0.01; ***P < 0.001.
Our previous study demonstrated that Ecm1 was an endogenous matrix component that is naturally present in the liver, inhibits liver fibrosis and plays a crucial role in maintaining hepatic homeostasis and cellular functionality [9]. Immunohistochemical (IHC) staining of Ecm1 revealed substantial secretion of Ecm1 (dark brown) within the hepatic sinusoids of mice in the oil group. However, following CCl4 administration, a significant reduction was observed in the secretory levels of Ecm1 protein. In contrast, the Sal B-treated groups and the Fer-1-treated group exhibited a significant increase in secreted Ecm1 protein (Fig. 1E). Similar effects were observed on Ecm1 mRNA and protein levels, as shown by qRT‒PCR, Western blotting, and ELISA (Fig. 1F). Studies have shown that Sal B protects hepatocytes from peroxidative damage [14,16,17]. Transmission electron microscopy (TEM) was used to observe the ultrastructure of hepatocytes. The results showed that the administration of CCl4 significantly decreased mitochondrial ridges, accompanied by an increase in membrane density and a reduction in volume, which are consistent with the characteristics of hepatocyte ferroptosis, and the aberrant alterations in hepatocyte mitochondria in the Sal B-treated groups and the Fer-1-treated group were significantly ameliorated compared to those in the CCl4 group (Fig. 1E). These results suggest that Sal B significantly inhibits liver fibrosis by upregulating hepatic Ecm1 and resisting hepatocyte ferroptosis.
3.2. Ecm1 deletion in hepatocytes disrupts the pharmacological effects of Sal B on liver fibrosis
To clarify whether Sal B inhibits liver fibrosis by targeting hepatocyte Ecm1, we established Ecm1Δhep mice as described in previous studies [9], and Ecm1flox/flox mice of the same age and the same gene background (WT) were used as parallel controls (Fig. 2A). There was a significant increase in serum AST activity in Ecm1Δhep mice compared to WT controls after CCl4 administration (P < 0.01). After the administration of Sal B and Sora, a significant decrease in serum ALT and AST activities was observed in WT mice compared to mice in the CCl4 group (P < 0.05, P < 0.01). Interestingly, under the same experimental conditions, there were no significant differences in serum ALT and AST activities in the Sal B-treated group and the CCl4 group when Ecm1Δhep mice were used (Fig. 2B). Following exposure to CCl4, Ecm1Δhep mice developed worse inflammatory injury and collagen deposition in the liver than WT mice, as demonstrated by H&E and SR staining (P < 0.05) (Fig. 2C). It was obvious that WT mice in the Sal B-treated and Sora-treated groups exhibited significant reductions in inflammatory injury, collagen deposition, and Hyp content compared to the CCl4-treated group (P < 0.05, P < 0.01) (Fig. 2C and D). Conversely, Sal B treatment did not significantly reduce these indices when administered to Ecm1Δhep mice compared to mice in the CCl4-treated group. However, the Cile-treated group exhibited sustained reductions in serum ALT and AST activities, inflammatory damage and collagen deposition compared to the CCl4-treated group (Fig. 2B–D).
Fig. 2.

Ecm1 deletion in hepatocytes weakened the therapeutic effects of Sal B on liver fibrosis. (A) Scheme of the experimental procedure for WT and Ecm1Δhep mice injected with CCl4 and treated with Sal B, Sora or Cile. (B) Serum ALT and AST activity. (C) Representative images of H&E and SR staining (bar = 100 μm), and semiquantitative analysis of the SR-positive staining area. (D) Hyp levels in liver tissue. (E) Western blot analysis and quantification of α-SMA protein expression using gray value analysis, Acta2 and Col1a1 mRNA expressions, and IHC staining of α-SMA and Col-I (bar = 100 μm). (F) TGF-β1 activity in liver tissue. Data are represented as mean ± SD (n = 4–6/group). *P < 0.05; **P < 0.01; ***P < 0.001.
Our previous studies showed that Ecm1 inhibited the maturation and activation of TGF-β1, thereby inhibiting the activation of HSCs and inhibiting liver fibrosis [9]. As expected, CCl4 markedly upregulated the expression of α-SMA and Col-I, as well as the activity of TGF-β1, in Ecm1Δhep mice compared to WT mice (P < 0.05, P < 0.01) (Fig. 2E and F). In WT mice, the Sal B-treated group and Sora-treated group exhibited significant reductions in the expression levels of α-SMA and Col-I, as well as the activity of TGF-β1, compared to those in the CCl4-treated group (P < 0.05, P < 0.01). However, there was no significant difference in these profibrotic factors between the Sal B-treated and CCl4-treated groups of Ecm1Δhep mice. In contrast, Ecm1Δhep mice treated with Cile exhibited significant reductions in these profibrotic factors (P < 0.05, P < 0.01) (Fig. 2E and F). These findings indicate that the therapeutic effects of Sal B against liver fibrosis depend on hepatocyte Ecm1, while those of Sora and Cile do not rely on hepatocyte Ecm1.
3.3. Sal B physically binds to the ECM1 protein as a direct therapeutic target
To further determine whether Ecm1 is a direct pharmacological target of Sal B, we performed molecular docking simulations to investigate the binding mode of the Ecm1-Sal B complex. We found that Sal B may form a hydrogen bond network with K377, R407, I412, N500, N502, and Y503 of Ecm1 (Fig. 3A), suggesting that Ecm1 was a direct pharmacological target of Sal B. To investigate the real-time kinetics of Sal B and Ecm1 protein interactions, Octet Red 96, which is a small molecule protein interaction system based on biolayer interferometry, was used. Our results demonstrated that Sal B could directly bind to the ECM1 protein with a KD value of 6.30E-05 (Fig. 3B). These findings indicate that Ecm1 is a direct pharmacological target of Sal B in the treatment of liver fibrosis.
Fig. 3.

Molecular docking and affinity kinetic determination of Sal B/Ecm1. (A) A molecular docking simulation was performed to investigate the interaction between Sal B and Ecm1. (B) The binding kinetics between Sal B and Ecm1 were evaluated using the Octet system based on BLI technology.
3.4. Ecm1 knockout induces hepatocyte ferroptosis in mice, which is associated with xCT levels
We previously observed typical morphological features of hepatocyte ferroptosis in CCl4-treated mice, which were accompanied by a decrease in Ecm1 expression, as shown in Fig. 1E. To investigate the role of Ecm1 in hepatocyte ferroptosis, we constructed systemic Ecm1 gene knockout (Ecm1−/−) mice. Consistent with our previous results, knockout of the Ecm1 gene resulted in spontaneous liver fibrosis in mice, as evidenced by H&E, Masson and SR staining (Fig. 4A). TEM was used to investigate the mitochondrial morphology of mouse hepatocytes and revealed that the mitochondrial structure of hepatocytes remained intact in WT mice. However, in Ecm1−/− mice, hepatocytes exhibited a decrease in mitochondrial volume, an increase in membrane density, a reduction or disappearance of mitochondrial ridges, and damage to the outer mitochondrial membrane (Fig. 4A). These findings indicate that deletion of Ecm1 triggers ferroptosis in hepatocytes, thereby contributing to the progression of liver fibrosis.
Fig. 4.

Ecm1 interacted with xCT to affect hepatocyte ferroptosis. (A) Representative images of H&E, Masson and SR staining (bar = 50 μm) and TEM images (bar = 0.5 μm) of WT and Ecm1-KO mice. (B) Western blot analysis and quantification of xCT protein expression in WT and Ecm1-KO mice using gray value analysis. (C) Representative images of immunofluorescence co-staining of Ecm1 and xCT (bar = 50 μm). (D) Ecm1 interacts with xCT, as shown by co-IP. Data are represented as mean ± SD (n = 3/group). ***P < 0.001.
It is known that the xCT transporter is the primary active subunit of the cystine/glutamate antiporter (System Xc−) and plays a pivotal role in regulating cell ferroptosis [20]. Interestingly, the results revealed a significant decrease in the protein expression of xCT in Ecm1−/− mice compared to WT mice (P < 0.001) (Fig. 4B). We hypothesized that Ecm1 affected the expression of xCT, which was further confirmed by co-IF and co-IP analysis. The results revealed colocalization between Ecm1 and xCT on the hepatocyte membrane (Fig. 4C). In addition, the interaction between Ecm1 and xCT was detected by co-IP (Fig. 4D). These findings indicate that Ecm1 regulates hepatocyte ferroptosis by interacting with the xCT protein.
3.5. Ecm1 deletion in hepatocytes reduces the Sal B-mediated increase in xCT and suppression of hepatocyte ferroptosis in mice
To further determine the potential molecular mechanism by which Sal B protects against liver fibrosis, we investigated the impact of Sal B on hepatocyte ferroptosis in mice. The biochemical characteristics of ferroptosis primarily involve the dysregulation of iron metabolism and damage caused by lipid peroxidation [21]. Following CCl4 administration, there was a significant reduction in the mRNA expression of ferritin light chain (Ftl) and a marked increase in acyl-CoA synthetase long-chain family member 4 (Acsl4) mRNA expression in the liver tissue of Ecm1Δhep mice compared to WT controls (P < 0.05, P < 0.01). Ferritin heavy chain (Fth1) and Ftl mRNA levels were significantly increased in WT mice treated with Sal B compared to those in the CCl4 group (P < 0.05, P < 0.01). Correspondingly, the mRNA expression levels of Acsl4 and Ptgs2 were significantly reduced in the Sal B-treated group (P < 0.05) (Fig. 5A). Hepatic iron levels and the lipid peroxidation products 4-HNE and MDA were increased by CCl4 in Ecm1Δhep mice compared to WT controls (P < 0.05). In Sal B-treated WT mice, there was a significant reduction in hepatic iron, 4-HNE and MDA levels compared with those in the CCl4 group; furthermore, GSH levels were significantly increased (P < 0.05, P < 0.01) (Fig. 5B). Interestingly, when Ecm1Δhep mice were used, there were no significant differences in the aforementioned indicators between the Sal B-treated group and the CCl4 group. In contrast, the Cile-treated group exhibited a notable reduction in hepatic iron accumulation and lipid peroxidation damage (P < 0.05, P < 0.01) (Fig. 5A and B).
Fig. 5.

Deleting Ecm1 in hepatocytes reduced Sal B-mediated upregulation of xCT transporters and inhibition of iron-dependent lipid peroxidative injury. In WT and Ecm1Δhep mice, (A) Fth1, Ftl, Acsl4 and Ptgs2 mRNA expressions. (B) Hepatic iron, 4-HNE, MDA and GSH levels. (C) Representative images of IHC staining of Fth1, Tfrc, and Gpx4 (bar = 100 μm). (D) Western blot analysis and quantification of Tfrc, xCT, and Gpx4 protein expression using gray value analysis. Data are represented as mean ± SD (n = 4–6/group). *P < 0.05; **P < 0.01; ***P < 0.001.
After exposure to CCl4, the protein expression of Tfrc was significantly upregulated, while the protein expression of Fth1, xCT and Gpx4 was significantly downregulated (P < 0.01, P < 0.001). However, the protein expression of Tfrc was significantly reduced in WT mice in the Sal B-treated group and Sora-treated group compared to those in the CCl4 group. Moreover, the protein expression of Fth1, xCT, and Gpx4 was significantly increased in these groups (P < 0.05, P < 0.01). However, no significant differences in protein expression were observed between the Sal B-treated group and CCl4 group when Ecm1Δhep mice were used. In contrast, the Cile-treated group exhibited a significant reduction in Tfrc protein expression and a significant increase in the expression of Fth1, xCT, and Gpx4 compared to the CCl4 group (P < 0.05, P < 0.01) (Fig. 5C and D). These results suggest that Ecm1 deletion in hepatocytes attenuates Sal B-mediated increases in xCT and decreases in hepatocyte ferroptosis in mice.
3.6. Sal B suppresses hepatocyte ferroptosis, which partially depends on Ecm1
Based on the previous findings, we hypothesized that the mechanism by which Sal B protects against liver fibrosis involves the inhibition of hepatocyte ferroptosis by targeting Ecm1. Therefore, an in vitro model of erastin-induced hepatocyte ferroptosis was established. The cell viability assay showed that Sal B and erastin exhibited significant cytotoxicity toward LO2 cells at concentrations above 50 μM and 30 μM, respectively (P < 0.01, P < 0.001) (Figs. S2A–B). Subsequently, we determined that the optimal inhibitory concentration at which erastin induced ferroptosis in LO2 cells was at 50 μM, and the effective concentration range of Sal B was 0.05 to 20 μM (P < 0.001) (Fig. S2C). Next, LO2 cells induced by erastin were treated with different doses of Sal B (0.1 μM, 1 μM, 10 μM) and recombinant human ECM1 protein (10 μg/mL) for 24 h (Fig. 6A). The erastin-induced increase in hepatocyte ROS levels and Ptgs2 and Acsl4 mRNA expression were significantly reversed by Sal B and ECM1 treatment (P < 0.01, P < 0.001) (Fig. 6B–D). In addition, the low protein expression of Ecm1, xCT and Gpx4 caused by erastin was significantly restored in the Sal B-treated groups and ECM1-treated group (P < 0.05, P < 0.01, P < 0.001), as shown by Western blotting (Fig. 6E and F). Similarly, we obtained consistent results in the CCl4-induced hepatocyte injury model (Fig. S3). These results suggest that Sal B can inhibit hepatocyte ferroptosis by upregulating Ecm1.
Fig. 6.

Both Sal B and ECM1 treatment inhibited hepatocyte ferroptosis. (A) Scheme of the experimental procedure for LO2 cells stimulated with erastin and treated with Sal B or ECM1. (B) ROS intensity in hepatocytes. (C) ROS staining in hepatocytes (bar = 50 μm). (D) Ptgs2 and Acsl4 mRNA expression. (E) and (F) Western blot analysis and quantification of Ecm1, xCT, and Gpx4 protein expression using gray value analysis. Data are represented as mean ± SD (n = 3/group). *P < 0.05; **P < 0.01; ***P < 0.001.
To further clarify the role of Ecm1 in Sal B-mediated protection against hepatocyte ferroptosis, the Ecm1 gene was silenced by lentiviral interference technology in LO2 cells (Sh-Ecm1), and a lentiviral vector was the experimental control (Sh-NC) (Fig. 7A). The results indicated that following erastin administration, LO2 cells in the Sh-Ecm1 group exhibited a significant increase in ROS activity and MDA levels, as well as a reduction in GSH levels, compared to those in the Sh-NC group (P < 0.05, P < 0.01). It was evident that ROS activity and MDA levels were significantly decreased in Sh-NC cells in the Sal B-treated group and ECM1-treated group, and there was a significant increase in GSH levels (P < 0.05, P < 0.01). Conversely, the pharmacological effects of Sal B in Sh-Ecm1 cells were markedly attenuated. Intriguingly, ECM1 treatment remained effective (P < 0.01) (Fig. 7B and C). Additionally, there was a significant reduction in the protein expression of Ecm1, xCT, and Gpx4 in LO2 cells treated with erastin in the Sh-Ecm1 group compared to the Sh-NC group (P < 0.05). However, there was no significant difference in the mRNA expression of Acsl4 or Ptgs2. The mRNA expression of Acsl4 and Ptgs2 in Sh-NC cells was significantly increased following erastin administration (P < 0.001), while there was a notable decrease in the Sal B-treated and ECM1-treated groups compared to the erastin group (P < 0.01, P < 0.001). In contrast, in Sh-Ecm1 cells, no significant difference was observed in these indicators in the Sal B-treated group and the erastin group, and a substantial reduction was observed in the ECM1-treated group (P < 0.01, P < 0.001) (Fig. 7D). The expression of Ecm1, xCT, and Gpx4 proteins was significantly increased in Sh-NC cells in the Sal B-treated and ECM1-treated groups compared to the Erastin group (P < 0.05, P < 0.01). However, no significant difference was observed in the protein expression of Ecm1, xCT, or Gpx4 in the Sal B-treated and Erastin groups of Sh-Ecm1 cells. The ECM1-treated group exhibited a significant increase in these proteins (P < 0.05) (Fig. 7E and F). These findings indicate that Sal B suppresses hepatocyte ferroptosis and protects hepatocytes in a manner that is partially dependent on Ecm1.
Fig. 7.

Ecm1 deletion abrogated the inhibitory effect of SalB on hepatocyte ferroptosis. (A) Scheme of the experimental procedure for Sh-NC and Sh-Ecm1 LO2 cells stimulated with erastin and treated with Sal B or ECM1. (B) ROS activity (bar = 50 μm). (C) MDA and GSH levels. (D) Acsl4 and Ptgs2 mRNA expression. (E) and (F) Western blot analysis and quantification of Ecm1, xCT, and Gpx4 protein expression using gray value analysis. Data are represented as mean ± SD (n = 3/group). *P < 0.05; **P < 0.01; ***P < 0.001.
4. Discussion
Hepatocytes, which constitute more than 80 % of the liver mass, represent the predominant cellular population in the liver and play a pivotal role in its physiological and pathological processes. Ecm1, which is a protein component of the extracellular matrix in the healthy liver, is primarily secreted by hepatocytes. However, in the presence of pathogenic factors leading to liver disease and the subsequent impairment of hepatocyte function, the secretion of Ecm1 is reduced. It has been shown that decreased expression of Ecm1 accompanies the progression of liver fibrosis at different stages [9]. Further studies have shown that administering Ecm1 via adreno-associated virus effectively mitigates CCl4-induced liver fibrosis in mice. This indicates an inverse correlation between Ecm1 expression and liver fibrosis, suggesting that upregulating hepatic Ecm1 expression partially reverses liver fibrosis.
As expected, a reduction in hepatic Ecm1 expression was observed in CCl4-induced liver fibrosis. However, Sal B could increase hepatic Ecm1 expression, ultimately relieving liver fibrosis in mice. To investigate whether Sal B inhibits liver fibrosis by acting on Ecm1 in hepatocytes, liver fibrosis was induced in Ecm1Δhep mice by CCl4. We administered 22.4 mg/kg Sal B and used Ecm1flox/flox mice (WT) of matched age and syngeneic backgrounds as parallel controls. As hypothesized, the deletion of Ecm1 in hepatocytes not only worsened liver fibrosis induced by CCl4 in mice but also nullified the therapeutic benefits of Sal B against liver fibrosis. These findings suggest that Ecm1 plays a crucial role in protecting hepatocytes and serves as a vital effector molecule mediating the protective effect of Sal B on hepatocytes. In contrast to Sal B, the absence of Ecm1 in hepatocytes did not impact the pharmacological effects of Sora and Cile, which are a kinase inhibitor and integrin αv inhibitor, respectively. Sora and Cile can directly inhibit the TGF-β1/Smad signaling pathway [22,23] without being affected by Ecm1 in hepatocytes, thereby influencing HSCs activation. However, the inhibitory effect of Sal B on hepatic TGF-β1 activity was attenuated due to the loss of hepatocyte Ecm1 in mice. Our previous studies showed that Ecm1 interacted with αv integrins, thereby inhibiting the maturation and activation of latent TGF-β1 and preventing the activation of HSCs [9]. This suggests that the inhibitory effect of SalB on TGF-β1 activity depends on hepatocyte Ecm1, which is an antifibrotic target of Sal B. Furthermore, molecular docking simulations and BLI technology were used to observe the binding mode and real-time kinetics of Sal B and Ecm1. Our findings confirmed that Ecm1 was a direct pharmacological target of Sal B.
Ferroptosis is triggered by iron-dependent lipid peroxidative damage, which is characterized by an increase in mitochondrial membrane density and a reduction or disappearance of mitochondrial cristae [21]. The xCT/Gpx4 signaling pathway plays a crucial role in regulating ferroptosis. xCT, as the primary active subunit of the cystine/glutamate antiporter (System Xc−), collaborates with accessory protein solute carrier family 3 member 2 (SLC3A2) to facilitate the uptake of cystine and release of glutamate, leading the synthesis of cysteine and GSH [20]. Subsequently, Gpx4 effectively converts toxic lipid peroxides into nontoxic lipid alcohols by using GSH, thereby protecting cells from lipid peroxidation damage and ferroptosis [24]. Inhibiting xCT/Gpx4 activity or impairing GSH synthesis results in the occurrence of ferroptosis. Existing studies [[25], [26], [27]] have demonstrated the significant contribution of ferroptosis to the pathogenesis of acute and chronic liver diseases, including viral hepatitis, alcoholic liver disease, nonalcoholic fatty liver disease, and hepatocellular carcinoma. Additionally, it is reported that ferroptosis is also involved in the process of diabetes-related liver pathological damage and plays a pathogenic role, thereby presenting a promising therapeutic avenue [[28], [29], [30]]. In specific pathological liver conditions or disease stages, such as virus infection, hereditary disease and metabolic associated liver diseases, the cell types in which ferroptosis occurs in the liver and their respective roles in the pathological progression are different [31]. Based on well-established targets of ferroptosis (xCT, transferrin, ferritin, Nrf2, etc) and their specificity in different liver pathologies, significant efforts have been devoted to drug development [10,[32], [33], [34], [35]]. Despite substantial progress in understanding the role of ferroptosis in liver pathology, further clarification is needed regarding the clinical response to therapies targeting ferroptosis. Moreover, although limited reports are available on hepatocyte ferroptosis, its role in liver fibrosis has also been established.
Our study revealed that CCl4-induced liver fibrosis was characterized by the occurrence of hepatocyte ferroptosis, as indicated by hepatic iron accumulation and lipid peroxidation damage. Additionally, typical morphological characteristics associated with ferroptosis were also observed. Sal B could ameliorate mitochondrial morphological abnormalities to inhibit ferroptosis in hepatocytes, and the ferroptosis inhibitor Fer-1 could reduce liver fibrosis in mice. These findings suggest that ferroptosis in hepatocytes contributes to the progression of liver fibrosis. Additionally, the reduction in liver fibrosis in mice mediated by Ecm1 upregulation by Sal B may involve the inhibition of ferroptosis in hepatocytes. Previous studies [14,16,17] have demonstrated that Sal B protects hepatocytes from apoptosis by inhibiting the death receptor pathway and stabilizing mitochondrial membranes. However, the relationship between Sal B and hepatocyte ferroptosis remains unclear.
To gain insight into the mechanism by which Sal B inhibits hepatocyte ferroptosis and inhibits liver fibrosis by targeting Ecm1, we further evaluated the effect of Sal B on hepatocyte ferroptosis in relation to Ecm1. In this study, we established an in vitro model of erastin-induced ferroptosis using LO2 cells. The results revealed that the occurrence of ferroptosis in hepatocytes was associated with a reduction in Ecm1 protein levels. Sal B treatment increased Ecm1 expression and inhibited ferroptosis by suppressing ROS activity, reducing the mRNA expression of the ferroptosis marker genes Ptgs2 and Acsl4 and upregulating the key ferroptosis regulatory proteins xCT and Gpx4 in hepatocytes. In addition, exogenous administration of recombinant human ECM1 protein mitigated lipid peroxidation-induced damage in hepatocytes and enhanced the activity of the xCT/Gpx4 signaling pathway. This phenomenon was further investigated by using lentivirus interference technology to establish an LO2 cell line with Ecm1 gene knockdown. Subsequently, erastin was used to induce hepatocyte ferroptosis. Our study showed that Ecm1 knockdown in hepatocytes exacerbated erastin-induced lipid peroxidation damage and ferroptosis in LO2 cells, as evidenced by increases in ROS activity and MDA levels, decreases in GSH levels, upregulated mRNA expression of Ptgs2 and Acsl4, and downregulated protein expression of xCT and Gpx4. In contrast, knockdown of Ecm1 in hepatocytes eliminated the protective effects of Sal B against lipid peroxidation damage and ferroptosis and disrupted Sal B-mediated regulation of the xCT/Gpx4 signaling pathway. These findings are consistent with the results obtained in the Ecm1Δhep mouse experiment. The addition of exogenous recombinant human ECM1 protein replenished ECM1 and partially attenuated erastin-induced lipid peroxidative injury and ferroptosis in LO2 cells. This suggests that Ecm1 plays a pivotal role in protecting hepatocytes against ferroptosis and that Sal B suppresses hepatocyte ferroptosis in a manner partially dependent on Ecm1.
To investigate the direct effect of Ecm1 on xCT transporters and its role in inhibiting ferroptosis in hepatocytes, we generated mice with a systemic knockout of Ecm1 (Ecm1−/−). The loss of Ecm1 in mice resulted in liver fibrosis, which was accompanied by hepatocyte ferroptosis and a significant decrease in hepatic expression of xCT. Further investigation revealed the colocalization of Ecm1 and xCT on the surface membranes of hepatocytes, and co-IP experiments confirmed a direct interaction between them. This preliminary research elucidates the key molecular basis of the pharmacological effect of Sal B. However, further investigation is warranted to determine the specific binding site and mechanism. The unresolved molecular structure of the Ecm1 protein poses a substantial obstacle to the investigation of its functionality.
5. Conclusions
In summary, we conducted in vitro and in vivo experiments to explore the molecular mechanism by which Sal B protects against liver fibrosis based on the physiological function of Ecm1 produced by hepatocytes. Our findings suggest that Ecm1 is a direct pharmacological target of Sal B to protect against liver fibrosis. The interaction between Ecm1 and xCT regulates hepatocyte ferroptosis and is a hitherto undescribed mechanism of the inhibitory effect of Sal B on liver fibrosis. These results have further expanded our understanding of the pathogenesis of liver fibrosis and partially showed that reductions in Ecm1 levels in hepatocytes trigger the development of hepatocyte ferroptosis and liver fibrosis. Restoring hepatic ECM1 expression represents a previously unknown mechanism by which Sal B can treat liver fibrosis.
Funding
This study was supported by the National Natural Science Foundation of China (No.81530101, 82204738, 82130120, 81973613, China), Shanghai Rising-Star Program (No.19QA1408900, China), China Postdoctoral Science Foundation (No.2022M713143, China), and the Shanghai Science and Technology Innovation Action (No.20S11901800, China). The authors would like to extend the sincere gratitude to Professor Lijiang Xuan (Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China), for providing the high quality Sal B in this study.
CRediT authorship contribution statement
Yadong Fu: Data curation, Investigation, Methodology, Writing – original draft, Funding acquisition. Xiaoxi Zhou: Data curation, Investigation, Methodology, Writing – original draft. Lin Wang: Investigation. Weiguo Fan: Investigation. Siqi Gao: Investigation. Danyan Zhang: Investigation. Zhiyang Ling: Investigation. Yaguang Zhang: Investigation. Liyan Ma: Investigation. Fang Bai: Investigation. Jiamei Chen: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing. Bing Sun: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing. Ping Liu: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2024.103029.
Contributor Information
Jiamei Chen, Email: cjm0102@126.com.
Bing Sun, Email: bsun@sibs.ac.cn.
Ping Liu, Email: liuliver@vip.sina.com.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
Data availability
Data will be made available on request.
References
- 1.Younossi Z.M. Non-alcoholic fatty liver disease - a global public health perspective. J. Hepatol. 2019;70(3):531–544. doi: 10.1016/j.jhep.2018.10.033. [DOI] [PubMed] [Google Scholar]
- 2.Zhou J., Zhou F., Wang W., Zhang X.J., Ji Y.X., Zhang P., et al. Epidemiological features of NAFLD from 1999 to 2018 in China. Hepatology. 2020;71(5):1851–1864. doi: 10.1002/hep.31150. [DOI] [PubMed] [Google Scholar]
- 3.Bodzin A.S., Baker T.B. Liver transplantation today: where we are now and where we are going. Liver Transplant. 2018;24(10):1470–1475. doi: 10.1002/lt.25320. [DOI] [PubMed] [Google Scholar]
- 4.Tan Z., Sun H., Xue T., Gan C., Liu H., Xie Y., et al. Liver fibrosis: therapeutic targets and advances in drug therapy. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.730176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rupcic Rubin V., Bojanic K., Smolic M., Rubin J., Tabll A., Smolic R. An update on efficacy and safety of emerging hepatic antifibrotic agents. J Clin Transl Hepatol. 2021;9(1):60–70. doi: 10.14218/JCTH.2020.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Higashi T., Friedman S.L., Hoshida Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017;121:27–42. doi: 10.1016/j.addr.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gautheron J., Gores G.J., Rodrigues C.M.P. Lytic cell death in metabolic liver disease. J. Hepatol. 2020;73(2):394–408. doi: 10.1016/j.jhep.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramachandran P., Matchett K.P., Dobie R., Wilson-Kanamori J.R., Henderson N.C. Single-cell technologies in hepatology: new insights into liver biology and disease pathogenesis. Nat. Rev. Gastroenterol. Hepatol. 2020;17(8):457–472. doi: 10.1038/s41575-020-0304-x. [DOI] [PubMed] [Google Scholar]
- 9.Fan W., Liu T., Chen W., Hammad S., Longerich T., Hausser I., et al. ECM1 prevents activation of transforming growth factor beta, hepatic stellate cells, and fibrogenesis in mice. Gastroenterology. 2019;157(5):1352–1367. doi: 10.1053/j.gastro.2019.07.036. e13. [DOI] [PubMed] [Google Scholar]
- 10.Yu Y.Y., Jiang L., Wang H., Shen Z., Cheng Q., Zhang P., et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136(6):726–739. doi: 10.1182/blood.2019002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mancardi D., Mezzanotte M., Arrigo E., Barinotti A., Roetto A. Iron overload, oxidative stress, and ferroptosis in the failing heart and liver. Antioxidants. 2021;10(12) doi: 10.3390/antiox10121864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang X. Susceptibility-weighted imaging in evaluating iron deposition in chronic liver disease. Chin. J. Interventional Imaging Ther. 2018;15(7):424–428. [Google Scholar]
- 13.Xiao Z., Liu W., Mu Y.P., Zhang H., Wang X.N., Zhao C.Q., et al. Pharmacological effects of salvianolic acid B against oxidative damage. Front. Pharmacol. 2020;11 doi: 10.3389/fphar.2020.572373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan X., Zhou T., Tao Y., Wang Q., Liu P., Liu C. Salvianolic acid B attenuates hepatocyte apoptosis by regulating mediators in death receptor and mitochondrial pathways. Exp. Biol. Med. (Maywood) 2010;235(5):623–632. doi: 10.1258/ebm.2009.009293. [DOI] [PubMed] [Google Scholar]
- 15.Liu P., Hu Y.Y., Liu C., Zhu D.Y., Xue H.M., Xu Z.Q., et al. Clinical observation of salvianolic acid B in treatment of liver fibrosis in chronic hepatitis B. World J. Gastroenterol. 2002;8(4):679–685. doi: 10.3748/wjg.v8.i4.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang R., Yu X.Y., Guo Z.Y., Wang Y.J., Wu Y., Yuan Y.F. Inhibitory effects of salvianolic acid B on CCl(4)-induced hepatic fibrosis through regulating NF-kappaB/IkappaBalpha signaling. J. Ethnopharmacol. 2012;144(3):592–598. doi: 10.1016/j.jep.2012.09.048. [DOI] [PubMed] [Google Scholar]
- 17.Yan X.F., Zhao P., Ma D.Y., Jiang Y.L., Luo J.J., liu L., et al. Salvianolic acid B protects hepatocytes from H2O2 injury by stabilizing the lysosomal membrane. World J. Gastroenterol. 2017;23(29):5333–5344. doi: 10.3748/wjg.v23.i29.5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu Y., Xiao Z., Tian X., Liu W., Xu Z., Yang T., et al. The novel Chinese medicine JY5 formula alleviates hepatic fibrosis by inhibiting the notch signaling pathway. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.671152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patsenker E., Popov Y., Stickel F., Schneider V., Ledermann M., Sagesser H., et al. Pharmacological inhibition of integrin alphavbeta3 aggravates experimental liver fibrosis and suppresses hepatic angiogenesis. Hepatology. 2009;50(5):1501–1511. doi: 10.1002/hep.23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koppula P., Zhuang L., Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12(8):599–620. doi: 10.1007/s13238-020-00789-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tang D., Chen X., Kang R., Kroemer G. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi: 10.1038/s41422-020-00441-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen Y.L., Lv J., Ye X.L., Sun M.Y., Xu Q., Liu C.H., et al. Sorafenib inhibits transforming growth factor beta1-mediated epithelial-mesenchymal transition and apoptosis in mouse hepatocytes. Hepatology. 2011;53(5):1708–1718. doi: 10.1002/hep.24254. [DOI] [PubMed] [Google Scholar]
- 23.Patsenker E., Popov Y., Wiesner M., Goodman S.L., Schuppan D. Pharmacological inhibition of the vitronectin receptor abrogates PDGF-BB-induced hepatic stellate cell migration and activation in vitro. J. Hepatol. 2007;46(5):878–887. doi: 10.1016/j.jhep.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 24.Jiang X., Stockwell B.R., Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021;22(4):266–282. doi: 10.1038/s41580-020-00324-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capelletti M.M., Manceau H., Puy H., Peoc'h K. Ferroptosis in liver diseases: an overview. Int. J. Mol. Sci. 2020;21(14) doi: 10.3390/ijms21144908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsurusaki S., Tsuchiya Y., Koumura T., Nakasone M., Sakamoto T., Matsuoka M., et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019;10(6):449. doi: 10.1038/s41419-019-1678-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pan Q., Luo Y., Xia Q., He K. Ferroptosis and liver fibrosis. Int. J. Med. Sci. 2021;18(15):3361–3366. doi: 10.7150/ijms.62903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gong Y., Liu Z., Zhang Y., Zhang J., Zheng Y., Wu Z. AGER1 deficiency-triggered ferroptosis drives fibrosis progression in nonalcoholic steatohepatitis with type 2 diabetes mellitus. Cell Death Dis. 2023;9(1):178. doi: 10.1038/s41420-023-01477-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo T., Yan W., Cui X., Liu N., Wei X., Sun Y., et al. Liraglutide attenuates type 2 diabetes mellitus-associated non-alcoholic fatty liver disease by activating AMPK/ACC signaling and inhibiting ferroptosis. Mol. Med. 2023;29(1):132. doi: 10.1186/s10020-023-00721-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stancic A., Velickovic K., Markelic M., Grigorov I., Saksida T., Savic N., et al. Involvement of ferroptosis in diabetes-induced liver pathology. Int. J. Mol. Sci. 2022;23(16) doi: 10.3390/ijms23169309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen J., Li X., Ge C., Min J., Wang F. The multifaceted role of ferroptosis in liver disease. Cell Death Differ. 2022;29(3):467–480. doi: 10.1038/s41418-022-00941-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He F., Zhang P., Liu J., Wang R., Kaufman R.J., Yaden B.C., et al. ATF4 suppresses hepatocarcinogenesis by inducing SLC7A11 (xCT) to block stress-related ferroptosis. J. Hepatol. 2023;79(2):362–377. doi: 10.1016/j.jhep.2023.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu Y.Y., Jiang L., Wang H., Shen Z., Cheng Q., Zhang P., et al. Hepatic transferrin plays a role in systemic iron homeostasis and liver ferroptosis. Blood. 2020;136(6):726–739. doi: 10.1182/blood.2019002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yi J., Wu S., Tan S., Qin Y., Wang X., Jiang J., et al. Berberine alleviates liver fibrosis through inducing ferrous redox to activate ROS-mediated hepatic stellate cells ferroptosis. Cell Death Dis. 2021;7(1):374. doi: 10.1038/s41420-021-00768-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J., Huang C., Liu J., Meng C., Gu Q., Du X., et al. Nrf2 and its dependent autophagy activation cooperatively counteract ferroptosis to alleviate acute liver injury. Pharmacol. Res. 2023;187 doi: 10.1016/j.phrs.2022.106563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data will be made available on request.