

Abstract
Signalling by IFN-y and CD40 is known to trigger anti-microbial activity in macrophages infected with Toxoplasma gondii, but their effects on infected neurons are less well known. Here, we compared how stimulation with IFN-y and an agonistic anti-CD40 mAb impacts infection and cyst formation in the mouse neuroblastoma cell line Neuro-2a relative to bone marrow-derived macrophages. Both IFN-y and CD40 mAb decreased cyst emergence in Neuro-2a cells. In macrophages, these stimuli decreased infection, but had no impact on infection in the neuroblastoma cell line. Resistance to killing in Neuro-2a cells may explain why neurons preferentially harbour parasites during chronic infection in the brain.
Keywords: cyst, macrophage, neuroblastoma, Toxoplasma gondii
1 |. INTRODUCTION
A globally prevalent parasite, Toxoplasma gondii is an intracellular protozoan that owes its success largely to a remarkable ability to manipulate host immunity.1,2 Infection with T. gondii begins with an acute phase characterized by actively invading tachyzoites that disseminate widely throughout host tissues.3,4 After 10–14 days, parasites become preferentially localized in tissues of the central nervous system. Once in neuronal tissue, T. gondii establishes latency, which is characterized by slow-growing bradyzoites contained within tissue cysts. While healthy individuals are normally asymptomatic during latent T. gondii infection, immunocompromised individuals are at serious risk of parasite recrudescence that may result in lethal toxoplasmic encephalitis.5 In the brain, two critical immune mediators that are involved in preventing parasite reactivation include the proinflammatory cytokine IFN-y and the costimulatory molecule CD40.6,7
Production of IFN-y has long been known to play a critical role in host immune defence during Toxoplasma infection.8,9 In mice, this is in large part due to the induction of immune-related GTPases (IRGs) that mediate disassembly of the parasitophorous vacuole membrane.3,10–12 In humans, IFN-y contributes to host immunity through tryptophan degradation, production of reactive oxygen species and triggering of cell death.13–15 Ultimately, the role of IFN-y in host immunity is to facilitate host control of both acute and latent T. gondii infection by controlling parasite expansion. Signalling through CD40 also plays a role in macrophage control of infection. In this case, CD40 stimulates autophagy-dependent vacuole-lysosomal fusion and parasite killing dependent upon host proteins such as ATG5, ATG7 and Beclin-1.16,17 CD40 signalling appears to trigger multiple pathways leading to autophagic elimination of Toxoplasma. These pathways include AMPK-mediated ULK1 phosphorylation, autocrine TNF-α secretion leading to Beclin-1 activation, regulation of Beclin-1 protein and activation of PKR.16,18–21
Although the effects of IFN-y and CD40 signalling in macrophages are well known, less is known about how these mediators impact Toxoplasma infection in neurons. This is critical because neurons are thought to preferentially harbour T. gondii during chronic infection.22–25 To gain insight into this issue, we used the mouse neuroblastoma cell line Neuro-2a (N2a) to examine how infection and cyst formation are impacted by signalling through IFN-y and CD40 in comparison with infection in mouse macrophages.
2 |. MATERIALS AND METHODS
2.1 |. Ethics statement
All experiments were performed in strict accordance with the recommendations set forth by the National Institutes of Health Guide for the Care and Use of Laboratory Animals (8th Edition). Protocols were approved by the Institutional Animal Care and Use Committee at the University of New Mexico (Animal Welfare Assurance Number A4023-01). All efforts were made to minimize animal suffering and distress over the course of the studies.
2.2 |. Mice
Swiss Webster and C57BL/6 mouse strains were obtained from The Jackson Laboratory. Both male and female mice (6–12 weeks of age) were used in these studies.
2.3 |. Parasites and infections
Type II (PTG) parasites were purchased from the American Type Culture Collection (ATCC) and were maintained in vitro by passage on confluent monolayers of human foreskin fibroblasts (ATCC). ME49 cysts were originally provided by C. A. Hunter (University of Pennsylvania). A colony of chronically infected Swiss Webster mice was used to maintain ME49 cysts. Infection was established by intraperitoneal injection of 20 cysts. After 4–8 weeks, brains were isolated and homogenized to obtain cysts for passage in mice and for experimental studies.
2.4 |. Cell culture
Bone marrow from C57BL/6 mice was collected using a 10-mL syringe and a 27-Ga needle to flush cells from tibia. Cells were differentiated into bone marrow-derived macrophages (BMDM) using media composed of Dulbecco’s modified Eagle media (DMEM; Cat#10-017-CV), 10% bovine growth serum (BGS; HyClone Laboratories, Cat# SH30541.03), nonessential amino acids (ThermoFisher Scientific, Cat#15630-080), 100 U/mL penicillin (ThermoFisher Scientific, Cat#15140-122), 0.1 mg/mL streptomycin (ThermoFisher Scientific, Cat #15140-122) and L929 culture supernatant as previously described.26 The BMDM were used 5 days after culture initiation. N2a cells (ATCC, Cat#CCL-131) were maintained in media composed of DMEM containing 10% BGS, 100 U/mL penicillin and 0.1 mg/mL streptomycin. At 24 h prior to infection with PTG, N2a cells were differentiated with the addition of 10 μM al-trans retinoic acid (Sigma-Aldrich, Cat#302-79-4). At time of infection, cells were treated with either 100 U/mL recombinant IFN-y (Peprotec, Cat#315-05), 50 mg/mL agonistic anti-CD40 mAb (BioXCell, Cat#BE0016-2) or left untreated.
2.5 |. Western blot analysis
Differentiated N2a cells and BMDM were plated on 24-well tissue culture plates (Corning, Cat#3524) and allowed to become confluent by overnight incubation at 37°C in 5% CO2. For the case of BMDM, cells were switched to medium composed of DMEM supplemented with 1% BGS 18 h prior to infection. The following day, cells were infected with PTG tachyzoites and stimulated with either recombinant IFN-y, anti-CD40 mAb or left untreated. Lysates were prepared with the addition of reducing SDS lysis buffer and sample DNA was sheared by 3× passage through a 27-Ga needle. The samples were boiled for 3 min and then loaded onto a 10% acrylamide protein gel (Bio-Rad Laboratories, Cat#4561033). After electrophoresis, proteins were electrotransferred onto a nitrocellulose membrane (Bio-Rad, Cat#1620115). The membranes were blocked using 5% non-fat dry milk (Bio-Rad, Cat#1706404) in 20 mM Tris-buffered 150 mM NaCl (pH 7.4) with 0.05% Tween 20 (TBST) for 2 h at 25°C. Primary antibodies diluted in TBST were added and membranes were incubated overnight at 4°C on a rotating platform. Primary antibodies included anti-phospho-ERK1/2 (Cell Signaling Technologies, Cat#4511), total-ERK1/2 (Cell Signaling, Cat#4695), anti-phospho-STAT1 (Cell Signaling, Cat#9167), anti-total-STAT1 (Cell Signaling, Cat#14994), anti-phospho-STAT3 (Cell Signaling, Cat#9145) and anti-total-STAT3 (Cell Signaling, Cat#9139). After washing membranes in TBST, secondary horseradish peroxidase-linked anti-rabbit antibody (Invitrogen, Cat #A16035) diluted in TBST was added and blots were incubated on a rotating platform (2 h, room temperature). Membranes were washed in TBST, and then developed using an enhanced chemiluminescent substrate system (ThermoFisher Scientific, Cat#34580).
2.6 |. Immunofluorescence microscopy
Differentiated N2a cells and BMDM were plated in DMEM containing 10% BGS, 100 U/mL penicillin and 0.1 mg/mL streptomycin on glass coverslips and allowed to become confluent by overnight culture (37°C, 5% CO2). Following a 2:1 infection with PTG, coverslips were collected and processed for microscopy. After rinsing with PBS, cells on coverslips were fixed in 3.7% formaldehyde (MilliporeSigma, Cat#FX0410-5) for 20 min at room temperature. Following fixation, coverslips were blocked for 1 h at room temperature using 5% normal mouse serum (Invitrogen, Cat#10410) in permeabilization buffer (0.1% saponin in phosphate-buffered saline). Staining was accomplished in permeabilization buffer using fluorescein isothiocyanate (FITC)-conjugated anti-Toxoplasma polyclonal antibody (Invitrogen, Cat#PA17253) and rhodamine-conjugated Dolichos biflorus agglutinin (DBA; Vector Laboratories, Cat#RL-1032). Coverslips were mounted onto microscope slides using permount solution containing 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen, Cat#P36962). Imaging was performed using a BX53 fluorescence microscope (Olympus America, Inc) and DP manager software (Olympus). Each slide was imaged over 5 different fields each containing approximately 100 DAPI-positive cells.
2.7 |. Flow cytometry
Single cell suspensions of differentiated N2a cells and BMDM were stained with allophycocyanin-conjugated anti-CD40 Ab (BioLegend, San Diego, CA, Cat #124611) for 20 min at 4°C in FACS buffer composed of 1% bovine growth serum, 0.01% sodium azide (VWR, Cat#0639) in phosphate-buffered saline. Controls remained un-stained. Cells were then fixed in 3.7% formaldehyde. Samples were interrogated on a four laser (violet, blue, yellow, red) Attune NxT flow cytometer (ThermoFisher Scientific) and the data were processed using FlowJo v.10 software (FlowJo).
2.8 |. Statistical analyses
Statistical analyses were performed using GraphPad Prism v.9 (GraphPad). Two-tailed paired t-tests were used to compare the normally distributed data of the two groups. A confidence interval of 95% (a = 0.05) was used as the cut-off to denote significant changes between groups.
3 |. RESULTS AND DISCUSSION
3.1 |. Retinoic acid-differentiated N2a cells support generation of Toxoplasma cysts
We used PTG tachyzoites, a low-virulence Toxoplasma strain capable of establishing chronic infection in mice, to examine in vitro cyst formation in N2a mouse neuroblastoma cells relative to mouse bone marrow-derived macrophages (BMDM). Cysts were visualized by staining with rhodamine-conjugated DBA, a lectin that binds carbohydrate moieties present in the cyst wall.27 Tachyzoites were counter-stained with a FITC-labelled anti-Toxoplasma polyclonal antibody. As shown in Figure 1A, cysts spontaneously formed in both retinoic acid (RA)-differentiated N2a cells and BMDM. The cysts generated in vitro were overall approximately three-fold decreased in diameter relative to cysts isolated from latently infected mouse brain (Figure 1A). This is likely not surprising given that the in vivo generated cysts were isolated 30–60 days following infection, whereas cysts generated in vitro were visualized 72 h after infection. Overall, there was no significant difference in diameter of cysts generated in N2a cells vs. BMDM (data not shown). In contrast, cyst formation in human fibroblast monolayers was extremely rare (Figure 1B). Next, we determined if non-differentiated vs. RA-differentiated N2a cells differed in ability to support cyst formation. While the percent infection in differentiated vs. nondifferentiated N2a cells did not differ over multiple experiments (data not shown), N2a were significantly more permissive to cyst generation following RA differentiation (Figure 1C,D). We also examined the kinetics of cyst formation over 72 h in BMDM compared with RA-differentiated N2a cells. While there was a significant increase in cyst formation in BMDM compared with N2a cells at early time points, by 72 h post-infection, the percentage of infected cells harbouring cysts was equivalent in both cell types (Figure 1E). Finally, we examined percent infection in BMDM and RA-differentiated N2a cells over a range of MOI. While percent infection was significantly higher in N2A cells at intermediate MOI, at high MOI (10), infection rates converged at 100% (Figure 1F).
FIGURE 1.
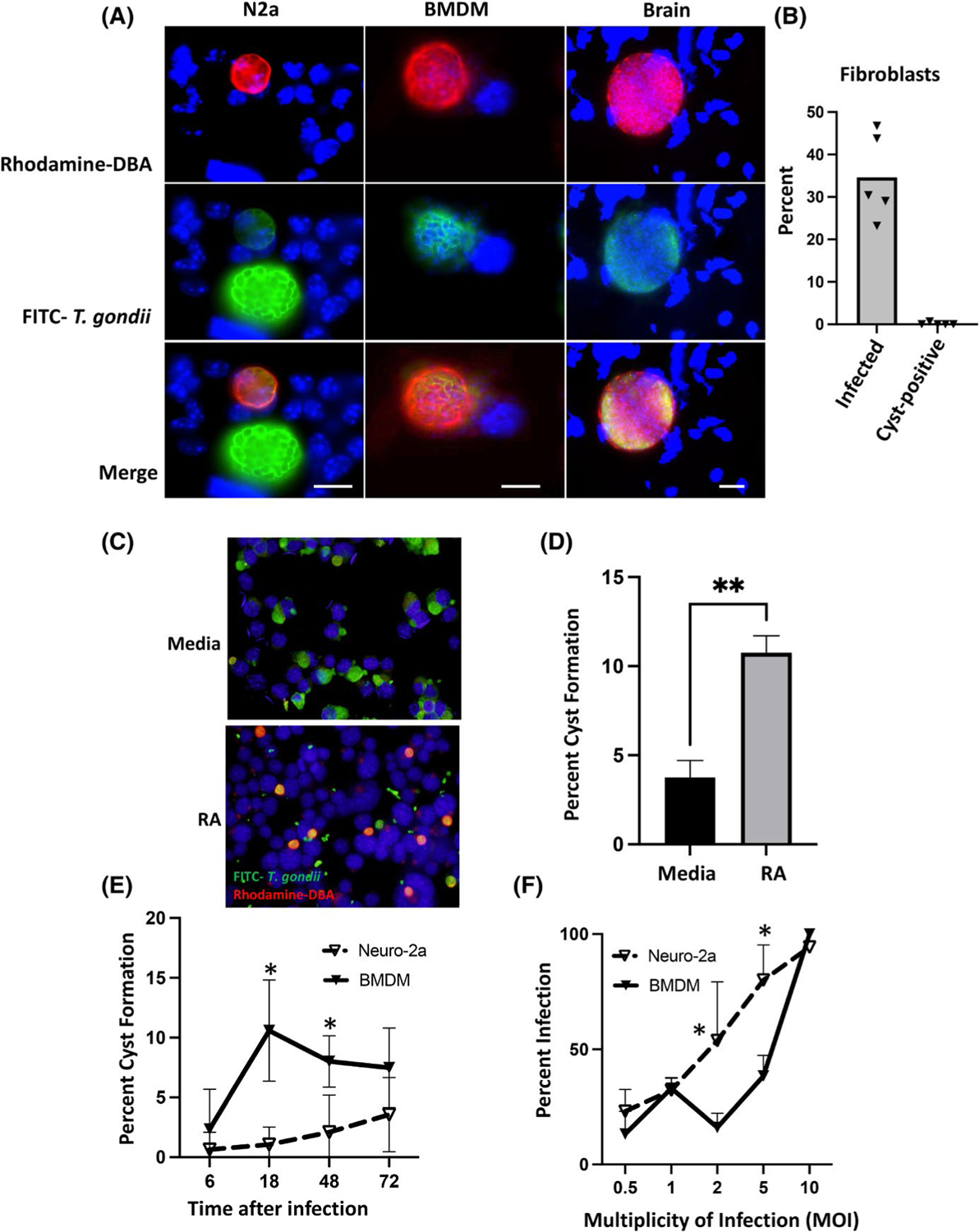
RA-differentiated N2a cells and BMDM support rapid T. gondii cyst formation. A, Immunofluorescence staining of RA-differentiated N2a and BMDM 72 h post-infection with PTG strain tachyzoites. Staining of an ME49 cyst in mouse brain homogenate 30 days post-infection is shown for comparison. Parasite cyst walls were stained using rhodamine-conjugated DBA (red), and individual parasites were labelled with FITC-conjugated anti-T. gondii Ab (green). Nuclei are stained with DAPI (blue). The scale bars in merged images of N2a cells and BMDM indicate 10 mm. Scale bar in merged image of the brain cyst indicates 20 mm. B, percent infection and percent cyst formation in human foreskin fibroblasts. C, Cyst formation in non-differentiated vs RA-differentiated N2a cells. Cysts and individual parasites were imaged as in A. Scale bar indicates 10 mm. D, Quantification of percent cyst formation in differentiated vs non-differentiated N2a cells. Percent Cyst Formation, percent of infected cells staining with rhodamine-DBA. E, Kinetics of cyst formation in RA-differentiated N2a cells and BMDM. F, Comparison of infection over a range of MOI in N2a vs. BMDM. In D, an unpaired t test with a Welch’s correction was employed to determine statistical significance where ** indicates p < .01. In E and F, a Multiple Mann-Whitney test was employed to determine statistical significance where * indicates p < .05. Each experiment was repeated three times with the same essential result
3.2 |. N2a cells and BMDM respond strongly but differently to agonistic anti-CD40 mAb
N2a cells are known to express CD40, a costimulatory molecule important in control of chronic T. gondii infection.28,29 Signalling through CD40 is also known to be capable of promoting parasite destruction in an autophagy-related manner.16 The cytokine IFN-y and its widely expressed receptor are well known to be involved in Toxoplasma killing through multiple mechanisms including the IRG system of GTPases.30 Therefore, we sought to determine how CD40 and IFN-y influenced cell signalling in RA-differentiated N2a cells compared with BMDM. As shown in Figure 2A, stimulation of N2a cells with exogenous IFN-y triggered rapid signal transducer and activator of transcription (STAT)-1 activation followed by deactivation in a manner kinetically similar to that occurring in BMDM. We then examined the ability of an agonistic CD40 mAb to trigger signalling in N2a cells compared with BMDM. Signalling through CD40 has previously been shown to activate a p42/p44 mitogen-activated protein kinase (MAPK) signalling cascade.28 Indeed, in RA-differentiated N2a cells, p42/p44 MAPK was strongly activated by CD40 mAb (Figure 2B). In contrast to STAT-1 activation, the p42/p44 MAPK phosphorylation response was both delayed and sustained (Figure 2A vs. B) in the RA-differentiated N2a cells. In parallel BMDM cultures, we did not detect activation of p42/p44 MAPK in response to agonistic anti-CD40 mAb. Previous studies have reported STAT3 activation in response to CD40 signalling.31 Interestingly, we found that this STAT molecule was phosphorylated in anti-CD40 stimulated BMDM, but not N2a cells (Figure 2C). In Figure 2D, we compared expression of CD40 RA-differentiated N2a cells and BMDM. Levels of expression were overall similar in both cell types (Figure 2D). While expression of CD40 was relatively low, this is in line with previous studies that examined CD40 expression in BMDM.32,33
FIGURE 2.
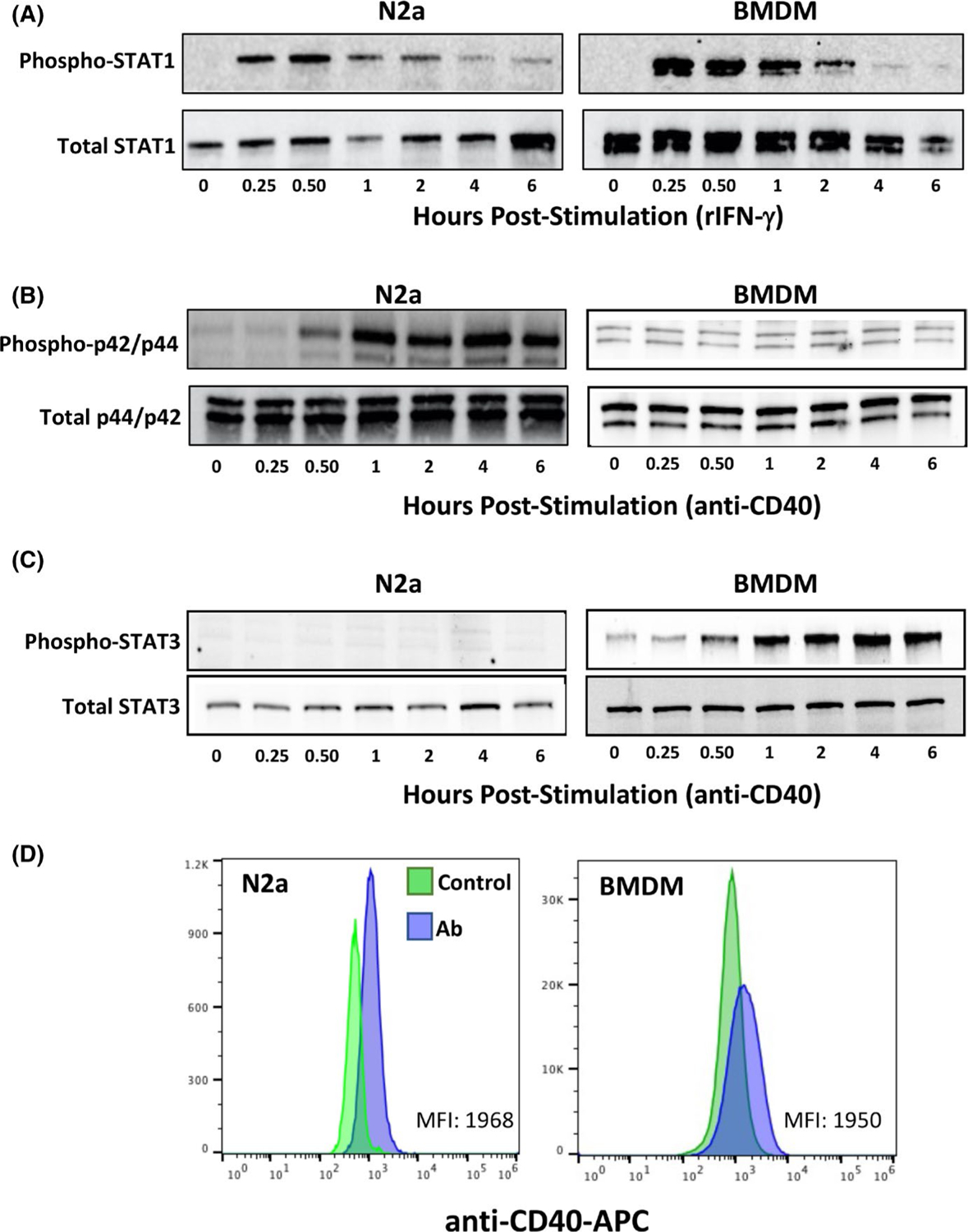
Effect of recombinant IFN-y and agonistic anti-CD40 mAb on signalling in N2a cells and BMDM. A, STAT-1 tyrosine phosphorylation in response to rIFN-y in RA-differentiated N2a cells and BMDM. B, Ser-Thr phosphorylation of p42/p44 MAPK in RA-differentiated N2a cells and BMDM after stimulation with agonistic anti-CD40 antibody. C, Tyr phosphorylation of STAT3 in responses to anti-CD40 antibody treatment. D, Flow cytometric analysis of cell surface CD40 expression on differentiated N2a cells and BMDM. Control, unstained cells. Ab, anti-CD40 Ab stained. These experiments were repeated three times with the same result
3.3 |. rIFN-y and agonistic anti-CD40 mAb differentially impact infection and cyst formation in N2a cells and BMDM
We then investigated how infection and cyst formation were influenced by IFN-y added at the time of infection. The presence of IFN-y had no impact on infection at 72 h in RA-differentiated N2a cells (Figure 3A). However, stimulation with IFN-y impeded emergence of cysts in N2a cells (Figure 3B). In BMDM, and as expected, IFN-y triggering at the time of infection decreased the percent of parasite-positive cells at 72 h (Figure 3C). Emergence of cysts in BMDM was not significantly impacted by the presence of IFN-y (Figure 3D).
FIGURE 3.
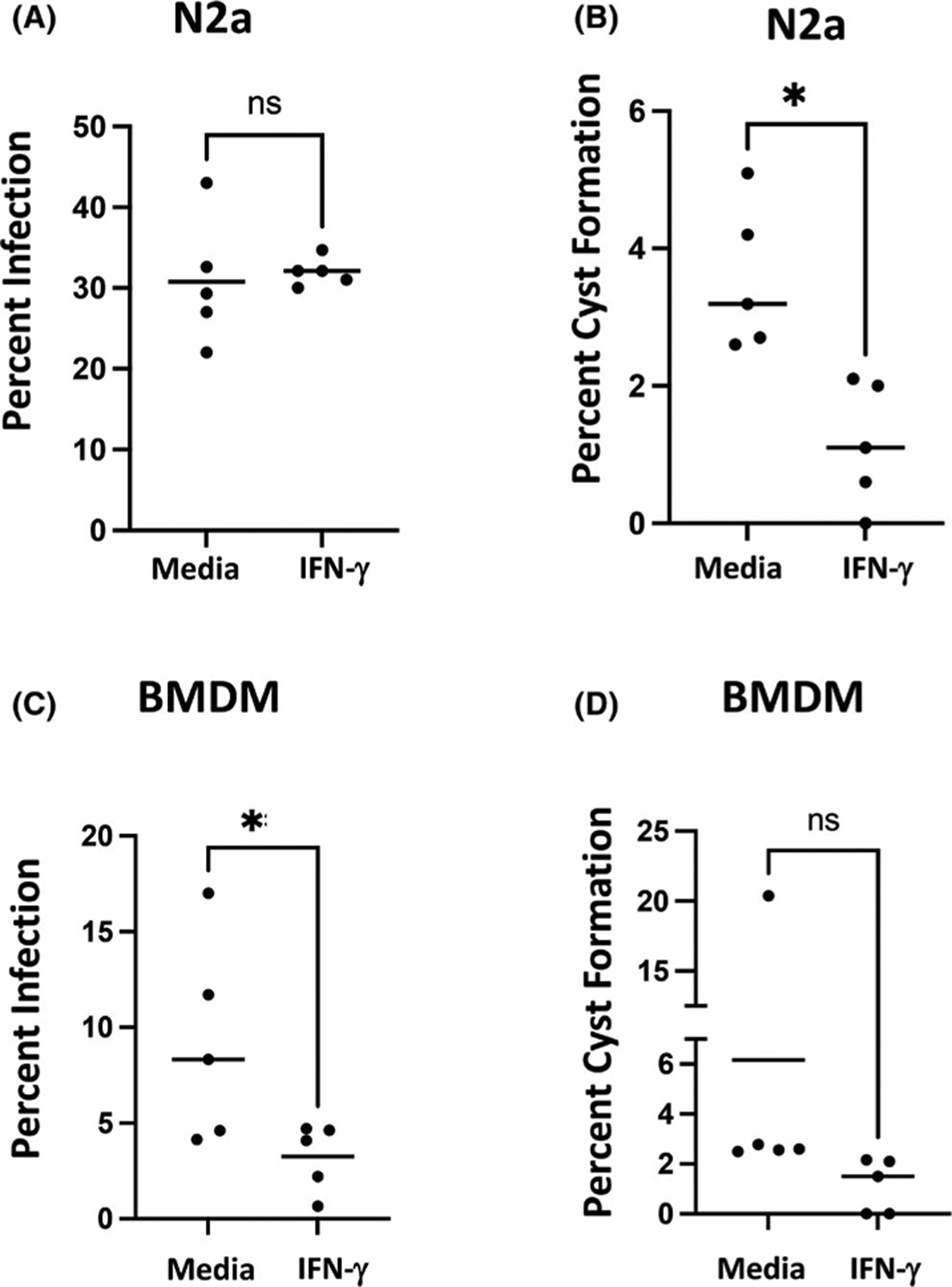
Disparate effects of rIFN-y on percent infection and cyst formation in BMDM and RA-differentiated N2a cells. RA-differentiated N2a cells (A and B) and BMDM (C and D) were infected with PTG tachyzoites simultaneously with the addition of rIFN-y at a 2:1 MOI. At 72 h post-infection, cells were fixed and stained with FITC-labelled anti-Toxoplasma Ab and rhodamine-conjugated DBA as described in Figure 1 legend. Percent infection (A and C) and percent cyst formation in infected cells (B and D) are shown. Each symbol represents an independent experiment (n = 5) quantifying approximately 500 cells over 5 imaging fields. *p < .05 (unpaired t test). ns, not significant
We next wanted to determine if signalling through CD40 affected persistence of infection or cyst emergence in N2a cells compared with BMDM. Accordingly, agonistic anti-CD40 mAb was added to RA-differentiated N2a cells concurrently with parasite inoculation, and then 72 h later, percent infection and percent of infected cells harbouring cysts were evaluated. Similar to the stimulation with IFN-y, signalling through CD40 had no significant effect on the degree of infection in N2a cells (Figure 4A). However, emergence of cysts was significantly hindered in the presence of anti-CD40 mAb (Figure 4B). In BMDM, signalling through CD40 decreased the percentage of infected cells at 72 h (Figure 4C). Signalling through CD40 appeared to promote emergence of cysts, although in this case the results did not attain statistical significance (Figure 4D; p = .19). Overall, neither CD40 nor IFN-y signalling impacted short-term parasite persistence, but both pathways impeded cyst formation in differentiated N2a cells. In contrast, both pathways decreased infection in BMDM, but the effects of IFN-y and CD40 signalling may diverge in influencing cyst emergence in this cell type. Divergent activation of p42/44 MAPK and STAT3 may underpin the differential effects of CD40 triggering in BMDM compared with N2a cells.
FIGURE 4.
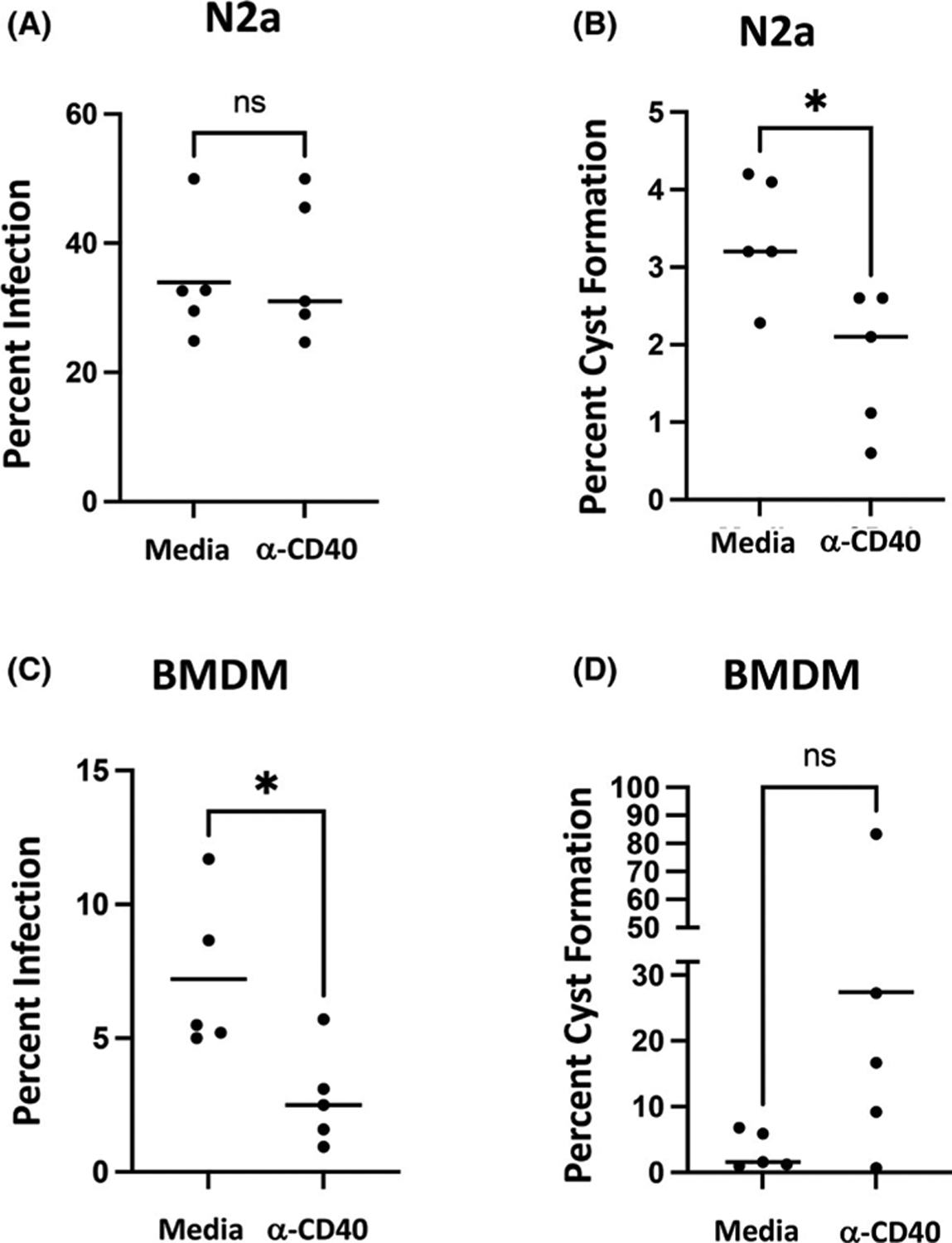
Signalling through CD40 decreases infection in BMDM and decreases cysts formation in N2a cells. Cells were infected with PTG tachyzoites and agonistic anti-CD40 mAb was added at Day 0. PTG tachyzoites were added in a 2:1 MOI to host cells. On Day 3, cells were fixed and stained, and then, percent infection and percent cyst formation were determined as in Figure 3. *p < .05; ns, not significant
In Figures 3A and 4A, we observed that activation of IFN-y and CD40 signalling did not impact infection in RA-differentiated N2a cells. This may be of relevance to in vivo infection insofar as neurons in the central nervous system are preferentially used as host cells during chronic T. gondii infection.22 Lack of inherent killing mechanisms may make these cells attractive targets of infection when the parasite reaches the brain. In contrast, other brain-resident cells such as astrocytes and microglia are capable of toxoplasmacidal activity in humans and mice.34–38 An alternative view is that neurons are hypersusceptible to infection.22 Indeed, inoculation of parasites in our experiments routinely led to greater infection in N2a cells relative to BMDM (Figure 3A vs. C; Figure 4A vs. C).
While IFN-y or CD40 signalling did not affect persistence of infection in N2a cells, we observed an inhibitory effect of both on cyst differentiation over 72 h (Figures 3B and 4B). This may have relevance to in vivo infection, since it was long ago established that both IFN-y and CD40/CD40L are required to prevent emergence of toxoplasmic encephalitis (TE) and concomitant uncontrolled parasite growth in mouse models of infection.11,29,39–41 CD4+ and CD8+ T lymphocytes are also required to avoid TE.39 Therefore, a plausible model is that T cell CD40L-neuronal CD40 interactions are required to prevent establishment of high numbers of cysts that would otherwise lead to inflammatory pathology.
Funding information
This works was funded by grants from the National Institute of Allergy and Infectious Disease (AI139628, EYD) and the National Institute of General Medical Sciences (P30 GM110907).
Footnotes
CONFLICT OF INTEREST
Authors have no conflicts of interest to declare.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Mendez OA, Koshy AA. Toxoplasma gondii: entry, association, and physiological influence on the central nervous system. PLoS Pathog. 2017;13(7):e1006351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laliberte J, Carruthers VB. Host cell manipulation by the human pathogen Toxoplasma gondii. Cell Mol Life Sci. 2008;65(12):1900–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao XY, Ewald SE. The molecular biology and immune control of chronic Toxoplasma gondii infection. J Clin Invest. 2020;130(7):3370–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Valle-Noguera A, Gomez-Sanchez MJ, Girard-Madoux MJH, Cruz-Adalia A. Optimized protocol for characterization of mouse gut innate lymphoid cells. Front Immunol. 2020;11:563414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luft BJ, Remington JS. Toxoplasmic encephalitis in AIDS. Clin Infect Dis. 1992;15:211–222. [DOI] [PubMed] [Google Scholar]
- 6.Subauste CS, Wessendarp M, Sorensen RU, Leiva LE. CD40-CD40 ligand interaction is central to cell-mediated immunity against Toxoplasma gondii: patients with hyper IgM syndrome have a defective Type I immune response that can be restored by soluble CD40 ligand trimer. J Immunol. 1999;162:6690–6700. [PubMed] [Google Scholar]
- 7.Meira CS, Pereira-Chioccola VL, Vidal JE, et al. Cerebral and ocular toxoplasmosis related with IFN-γ, TNF-α, and IL-10 levels. Front Microbiol. 2014;5:492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Suzuki Y, Orellana MA, Schreiber RD, Remington JS. Interferon-g: The major mediator of resistance against Toxoplasma gondii. Science. 1988;240(4851):516–518. [DOI] [PubMed] [Google Scholar]
- 9.Sher A, Denkers EY, Gazzinelli RT. Induction and regulation of host cell-mediated immunity by Toxoplasma gondii. Ciba Found Symp. 1995;195:95–104; discussion 104–109. [DOI] [PubMed] [Google Scholar]
- 10.Yamamoto M, Okuyama M, Ma JS, et al. A cluster of interferon-gamma-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity. 2012;37(2):302–313. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki Y, Conley FK, Remington JS. Importance of endogenous IFN-g for the prevention of toxoplasmic encephalitis in mice. J Immunol. 1989;143:2045–2050. [PubMed] [Google Scholar]
- 12.Zhao Y, Ferguson DJ, Wilson DC, Howard JC, Sibley LD, Yap GS. Virulent Toxoplasma gondii evade immunity-related GTPase-mediated parasite vacuole disruption within primed macrophages. J Immunol. 2009;182(6):3775–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krishnamurthy S, Konstantinou EK, Young LH, Gold DA, Saeij JP. The human immune response to Toxoplasma: autophagy versus cell death. PLoS Pathog. 2017;13(3):e1006176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fisch D, Clough B, Frickel EM. Human immunity to Toxoplasma gondii. PLoS Pathog. 2019;15(12):e1008097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lima TS, Lodoen MB. Mechanisms of human innate immune evasion by Toxoplasma gondii. Front Cell Infect Microbiol. 2019;9:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Portillo JA, Okenka G, Reed E, et al. The CD40-autophagy pathway is needed for host protection despite IFN-Gamma-dependent immunity and CD40 induces autophagy via control of P21 levels. PLoS One. 2010;5(12):e14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andrade RM, Wessendarp M, Gubbels MJ, Striepen B, Subauste CS. CD40 induces macrophage anti-Toxoplasma gondii activity by triggering autophagy-dependent fusion of pathogen-containing vacuoles and lysosomes. J Clin Invest. 2006;116(9):2366–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu E, Lopez Corcino Y, Portillo JA, Miao Y, Subauste CS. Identification of signaling pathways by which CD40 stimulates autophagy and antimicrobial activity against Toxoplasma gondii in macrophages. Infect Immun. 2016;84(9):2616–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Subauste CS, Andrade RM, Wessendarp M. CD40-TRAF6 and autophagy-dependent anti-microbial activity in macrophages. Autophagy. 2007;3(3):245–248. [DOI] [PubMed] [Google Scholar]
- 20.Ogolla PS, Portillo JA, White CL, et al. The protein kinase double-stranded RNA-dependent (PKR) enhances protection against disease cause by a non-viral pathogen. PLoS Pathog. 2013;9(8):e1003557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Grol J, Muniz-Feliciano L, Portillo JA, Bonilha VL, Subauste CS. CD40 induces anti-Toxoplasma gondii activity in nonhematopoietic cells dependent on autophagy proteins. Infect Immun. 2013;81(6):2002–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cabral CM, Tuladhar S, Dietrich HK, et al. Neurons are the primary target cell for the brain-tropic intracellular parasite Toxoplasma gondii. PLoS Pathog. 2016;12(2):e1005447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ferguson DJ, Hutchison WM. The host-parasite relationship of Toxoplasma gondii in the brains of chronically infected mice. Virchows Arch A Pathol Anat Histopathol. 1987;411(1):39–43. [DOI] [PubMed] [Google Scholar]
- 24.Ferguson DJ, Hutchison WM. An ultrastructural study of the early development and tissue cyst formation of Toxoplasma gondii in the brains of mice. Parasitol Res. 1987;73(6):483–491. [DOI] [PubMed] [Google Scholar]
- 25.Melzer TC, Cranston HJ, Weiss LM, Halonen SK. Host cell preference of Toxoplasma gondii cysts in murine brain: a confocal study. J Neuroparasitology. 2010;1:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim L, Butcher BA, Denkers EY. Toxoplasma gondii interferes with lipopolysaccharide-induced mitogen-activated protein kinase activation by mechanisms distinct from endotoxin tolerance. J Immunol. 2004;172(5):3003–3010. [DOI] [PubMed] [Google Scholar]
- 27.Boothroyd JC, Black M, Bonnefoy S, et al. Genetic and biochemical analysis of development in Toxoplasma gondii. Philos Trans R Soc Lond B Biol Sci. 1997;352(1359):1347–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan J, Town T, Mori T, et al. CD40 is expressed and functional on neuronal cells. EMBO J. 2002;21(4):643–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reichmann G, Walker W, Villegas EN, et al. The CD40/CD40 ligand interaction is required for resistance to toxoplasmic encephalitis. Infect Immun. 2000;68:1312–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taylor GA. IRG proteins: key mediators of interferon-regulated host resistance to intracellular pathogens. Cell Microbiol. 2007;9(5):1099–107. [DOI] [PubMed] [Google Scholar]
- 31.Hanissian SH, Geha RS. Jak3 is associated with CD40 and is critical for CD40 induction of gene expression in B cells. Immunity. 1997;6(4):379–387. [DOI] [PubMed] [Google Scholar]
- 32.Morgado P, Sudarshana DM, Gov L, et al. Type II Toxoplasma gondii induction of CD40 on infected macrophages enhances interleukin-12 responses. Infect Immun. 2014;82(10):4047–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Suttles J, Stout RD. Macrophage CD40 signaling: a pivotal regulator of disease protection and pathogenesis. Semin Immunol. 2009;21(5):257–264. [DOI] [PubMed] [Google Scholar]
- 34.Halonen SK, Taylor GA, Weiss LM. Gamma interferon-induced inhibition of Toxoplasma gondii in astrocytes is mediated by IGTP. Infect Immun. 2001;69:5573–5576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hidano S, Randall LM, Dawson L, et al. STAT1 signaling in astrocytes is essential for control of infection in the central nervous system. MBio. 2016;7(6):e01881–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peterson PK, Gekker G, Hu S, Chao CC. Human astrocytes inhibit intracellular multiplication of Toxoplasma gondii by a nitric oxide-mediated mechanism. J Infect Dis. 1995;171(2):516–518. [DOI] [PubMed] [Google Scholar]
- 37.Chao CC, Anderson WR, Hu S, Gekker G, Martella A, Peterson PK. Activated microglia inhibit multiplication of Toxoplasma gondii via a nitric oxide mechanism. Clin Immunol Immunopathol. 1993;67(2):178–183. [DOI] [PubMed] [Google Scholar]
- 38.Chao CC, Gekker G, Hu S, Peterson PK. Human microglial cell defense against Toxoplasma gondii. The role of cytokines. J Immunol. 1994;152(3):1246–1252. [PubMed] [Google Scholar]
- 39.Gazzinelli R, Xu Y, Hieny S, Cheever A, Sher A. Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii. J Immunol. 1992;149:175–180. [PubMed] [Google Scholar]
- 40.Denkers EY, Gazzinelli RT. Regulation and function of T cell-mediated immunity during Toxoplasma gondii infection. Clin Microbiol Rev. 1998;11:569–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki Y Immunopathogenesis of cerebral toxoplasmosis. J Infect Dis. 2002;186(Suppl 2):S234–S240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.