

Abstract

Protecting groups (PGs) in peptide synthesis have inspired advanced design principles that incorporate “orthogonality” for selective C- and N-terminus and side-chain deprotections. The conventionally acid-stable 9-fluorenylmethoxycarbonyl (Fmoc) group is one of the most widely used N-protection groups in solid- and solution-phase synthesis. Despite the versatility of Fmoc, deprotection by the removal of the Fmoc group to unmask primary amines requires the use of a basic secondary amine nucleophile, but this stratagem poses challenges in sensitive molecules that bear reactive electrophilic groups. An expansion of PG versatility, a tunable orthogonality, in the late-stage synthesis of peptides would add flexibility to the synthetic design and implementation. Here, we report a novel Fmoc deprotection method using hydrogenolysis under mildly acidic conditions for the synthesis of Z-Arg-Lys-acyloxymethyl ketone (Z-R-K-AOMK). This new method is not only valuable for Fmoc deprotection in the synthesis of complex peptides that contain highly reactive electrophiles, or other similar sensitive functional groups, that are incompatible with traditional Fmoc deprotection conditions but also tolerant of N-Boc groups present in the substrate.
1. Introduction
The 9-fluorenylmethoxycarbonyl (Fmoc) group represents one of the most widely used groups for the protection of amines in organic synthesis.1,2 This protecting group (PG) has found extensive application due to its easy removal under mildly basic conditions such as with piperidine, diethylamine, or morpholine. Fmoc also has the advantage of being tolerant of acidic conditions. The Fmoc PG is resistant to many other reaction conditions such as oxidation and reduction in multistep total synthesis of natural products.3 The highly electrophilic byproduct of Fmoc deprotection, dibenzofulvene (Dbf), can induce a variety of side reactions by the capture of incipient nucleophiles;3 conditions that limit Fmoc use in peptide synthesis where an excess of the nitrogenous base is needed to achieve both complete removal of the Fmoc group and quenching of the resulting Dbf.3 An additional burden is the need to remove excess nitrogenous base before proceeding to the next peptide coupling step. Many peptide synthetic schemes have faced the problems described above; in essence, unmasking of reactive groups while maintaining orthogonality of protection groups for NH2, COOH, and side-chain functional groups. In particular, the exposure of a nucleophilic primary amine by Fmoc deprotection in the presence of a reactive electrophile can lead to unwanted side reactions.4,5 Similarly, the secondary amine employed in the deprotection step (e.g., diethylamine or piperidine) can readily engage reactive electrophiles to produce undesired side-products including polymeric materials. Paradoxically, the reagents used for Fmoc deprotection and the exposed primary amine product are liabilities when the deprotected peptide has electrophilic functionalities.
The response of the base labile Fmoc group to acidic deprotection has not been widely studied. Lewis acid catalyzed Fmoc deprotection utilized AlCl3 with toluene as a Friedel–Crafts scavenger;6 however, this method is limited by the requirement for deactivation of the unmasked primary amine by acidification in a separate step. This poses a risk for side reactions between the free amine and electrophilic functional groups in the product molecule. Exposure to a strong Lewis acid may result in undesired side reactions in sensitive molecules or premature deprotection of acid-sensitive PGs used in peptide synthesis (e.g., Boc). It was reported that Pd/C-catalyzed hydrogenolysis of the Fmoc group was employed in the presence of acetonitrile to produce ethylamine (the reduction product of acetonitrile), which subsequently aids in scavenging the Dbf side-product after Fmoc removal.7 Despite the claim of neutrality for this method, the in situ generation of nucleophilic EtNH2 would promote capricious side reactions with electrophiles.
In more complex peptide syntheses that require maximum flexibility in their “end-game strategies”, the following unmet need arises: alternative methods for Fmoc removal orthogonal to conventional treatment with secondary amines that mitigate both the liabilities of nucleophilic secondary-amine reagents and the revealed exposure of primary-amines within peptides containing electrophilic functionalities. Here, we show that deprotection of Fmoc-protected peptides under acidic hydrogenolysis conditions not only suppresses the nucleophilic reactivity of the amine product but is also tolerant of N-Boc protected amino groups within the same molecule.
2. Results and Discussion
The peptide derivative Z-Arg-Lys-AOMK (1) was developed in our laboratories as a neutral pH-selective inhibitor of cathepsin B (Cat.B).5 Cat.B is believed to be released upon cellular injury from acidic lysosomes into the neutral pH cytoplasm and initiate and mediate inappropriate proteolytic degradation. Such sequelae are thought to be relevant to the pathogenesis observed with traumatic brain injury and neuropathies such as Alzheimer’s Disease. Our ongoing in vivo evaluations of Z-Arg-Lys-AOMK for its potential neurotherapeutic potential necessitated the scaled-up synthesis of this inhibitor.5 Upon embarking on this endeavor, we encountered several problems with the methodology employed in the previously published synthesis (Figure 1), which utilized solid-phase peptide synthesis and produced insufficient amounts of Z-Arg-Lys-AOMK (1, Z-R-K-AOMK).5 These problems included the irreproducibility of some steps and very low yields for others. The step involving resin-loading upon a poly(aminomethyl-styrene semicarbazide) posed significant challenges due to sensitivity of the semicarbazide linker to moisture, resulting in a very low yield at best.5,8 The biggest challenge in this synthetic route was the required removal of the Fmoc PG during the resin loading step, a deprotection that resulted in the formation of multiple undesired products from the reaction of the newly liberated free amine with the highly reactive electrophilic AOMK group (Figure 2A). Additionally, the published synthetic procedure required a final cumbersome preparative high-performance liquid chromatography (HPLC) purification.9 In our hands, multiple attempts to resynthesize the desired compound Z-R-K-AOMK using these published procedures were unsuccessful. Therefore, we reverted to a solution-phase synthesis, as described below.
Figure 1.
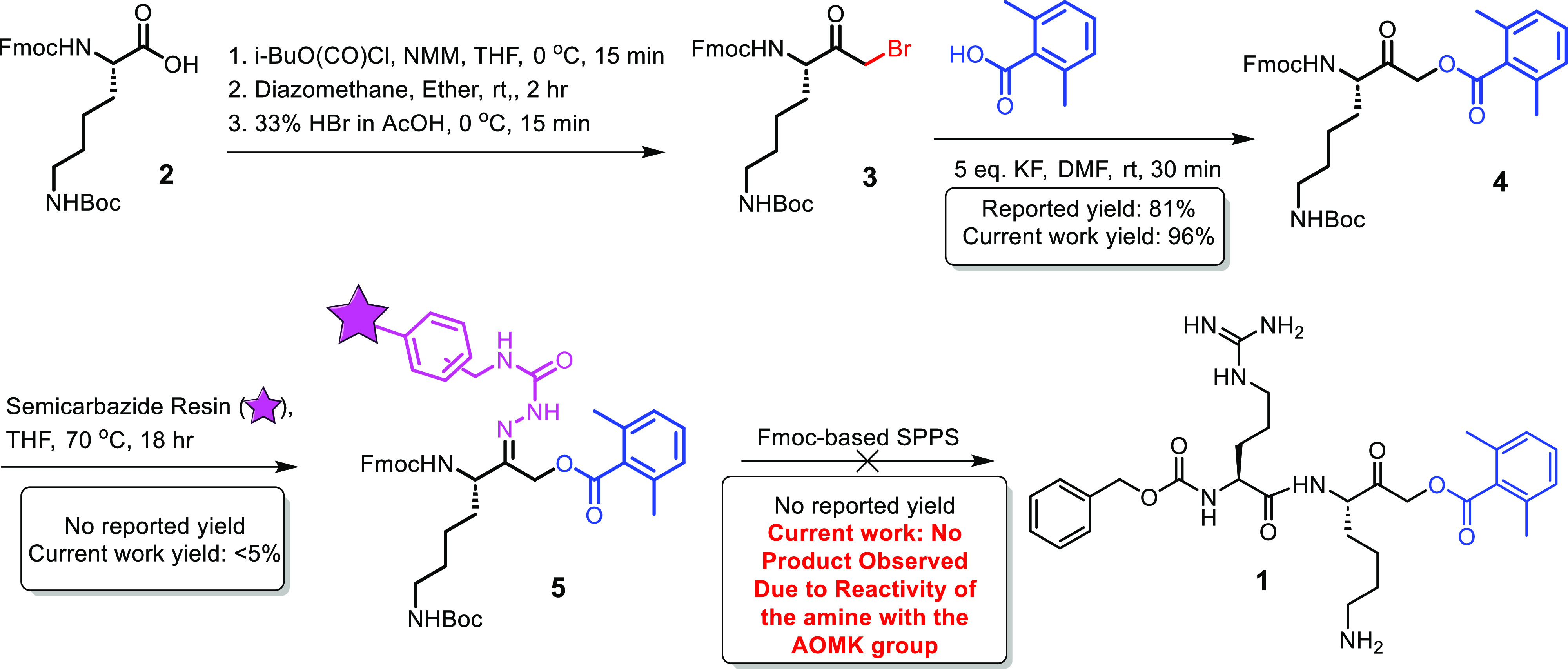
Reported literature route for Z-R-K-AOMK synthesis5 during scale-up resulted in no product.5
Figure 2.

(A) Failed synthesis of Z-R-K-AOMK (1) due to the basic conditions of the Fmoc removal step or due to the reactivity of the deprotected amine from compound 4. (B) Successful synthesis of Z-R-K-AOMK (1) using the novel Fmoc deprotection strategy by acidic dehydrogenation described in this paper.
In the revamped synthesis (Figure 2), commercially purchased Fmoc-Lys(Boc)-COCH2Cl was acylated with 2,6-dimethylbenzoic acid to produce Fmoc-Lys(Boc)-AOMK (4). Multiple attempts to remove the Fmoc group from 4 using different bases (diethylamine, triethylamine, and piperidine), under different organic solvent regimes (e.g., DMF or DCM), were unsuccessful (Figure 2A). Similarly, the reported “neutral” H2/Pd deprotection of Fmoc was unproductive, presumably due to unwanted reactions of the revealed nucleophilic primary-amine (Figure 2A).7 A careful analysis of the byproducts resulting from H2/Pd removal of the Fmoc group revealed that the deprotected α-amino group of Lys engaged in a nucleophilic attack on the electron-deficient carbon between the carbonyl ketone and the AOMK moiety, leading to the formation of complex polymeric side products (Figure 2A).
To address this issue, we explored Fmoc-removal by Pd-promoted catalytic hydrogenation under mildly acidic conditions (Figure 2B), which should produce a non-nucleophilic ammonium salt immediately upon Fmoc deprotection, thereby suppressing undesired side products. Indeed, the inclusion of acid within this hydrogenation process not only led to the creation of a masked salt derivative of the deprotected amine but also effectively deterred the unwanted reduction of the ketone to a secondary alcohol. This improved synthetic procedure for Z-R-K-AOMK employs a solution phase synthetic strategy (Figure 2B) in which the resulting deprotected ammonium salt (10) was coupled with Z-Arg(Boc2)–OH to give compound 11 in high yield. Finally, the target compound Z-R-K-AOMK (1) was obtained as the TFA salt by simultaneous deprotection of all three Boc PGs using TFA/CH2Cl2 in a near quantitative yield, without the need for further purification (Figure 2B). The overall yield for the synthesis of Z-R-K-AOMK using our newly developed route is 40.5% while in the previous synthetic route to Z-R-K-AOMK (Figure 1) yields were not reported for the final steps.5
The Cat.B inhibitory activity of the newly synthesized Z-R-K-AOMK (1) was assessed by IC50 and compared to the compound that was previously synthesized and reported.5 Similar levels of Cat.B inhibitory activity were observed at pH 4.6 and 7.2 for both newly synthesized compound 1 from this study and an original compound developed from the previous work (Figure 3).5
Figure 3.

pH-dependent inhibition of Cat.B cleavage activity in the presence of serial diluted inhibitor comparing newly synthesized Z-R-K-AOMK (1) vs an authentic sample of Z-R-K-AOMK from previous studies.5 Inhibitory potencies were evaluated by IC50 (n = 4) without preincubation of inhibitor and enzyme, as described in the methods.
The success of this new route to 1 inspired us to explore the scope and limitations of these new Fmoc-removal reaction conditions, including the structure of the Brønsted acid, acid concentration, and the molar equivalents of Pd (Table 1). One molecule we used in this study, compound 4, contained the acid-sensitive Boc PG that we desired to not remove in this reaction sequence, as well as the potentially reactive electrophilic α-chloroketone functionality (Figure 2A). Hence, we avoided the use of strongly acidic conditions that might cause this Boc deprotection. Optimal conditions included 2–3 equiv of HCl in MeOH, which was sufficient to achieve the efficient in situ Fmoc removal and conversion of the liberated amine to its deactivated ammonium salt. The number of Pd/C equivalents did not significantly impact the yield; however, a 20% mol equivalent to the starting material was slightly more efficient. In summary, Fmoc-deprotection in the presence of a Brønsted acid yielded the desired amine salts in high yield while preserving the Boc PG and α-chloroketone functionality (Table 1, Entry 8).
Table 1. Optimization of Removal Conditions for the Fmoc PG Was Performed Using Acidic Hydrogenolysise.
| entry | acidc | acid equivalents to starting material | molar Pd/C equivalents to starting material (%)a | yieldb |
|---|---|---|---|---|
| 1 | no acid | 10 | NP | |
| 2 | CF3COOH | 10 | 10 | NP |
| 3 | CF3COOH | 2 | 10 | <5% |
| 4 | HCl/MeOH | 20 | 10 | <5% |
| 5 | HCl/MeOH | 10 | 10 | 18% |
| 6 | HCl/MeOH | 5 | 10 | 42% |
| 7 | HCl/MeOH | 4 | 10 | 68% |
| 8 | HCl/MeOH | 2 | 10 | 75% |
| 9d | HCl/MeOH | 2 | 10 | 45% |
| 10 | HCl/MeOH | 2 | 20 | 79% |
| 11 | HCl/MeOH | 2 | 30 | 78% |
Commercial 10% w/w Pd on C and 1 atm of H2.
Isolated yields when greater than 10%.
For HCl/MeOH, a 3 M stock solution was used.
In all entries the reactions were run for 12 h, except for entry #9 for which the reaction time was 4 h.
NP: no product was observed.
The scope and limitations of the method in the presence of other electrophiles were then explored. Michael acceptors are defined by carbon double bonds conjugated to powerful electron-withdrawing substituents.10−12 However, in many cases, an undesired byproduct results from the reaction of the amines used for the deprotection of Fmoc (or the resulting deprotected amine) and the enone in another molecule. Deprotection of Fmoc using acidic hydrogenation offers a nonbasic option for the deprotection and in situ protection of the resulting deprotected amine. Because no secondary amines have been used in the deprotection sequence, after the removal of the Fmoc group, the reaction can be done in a single pot by the addition of the enone to achieve the Michael addition product (Figure 4B). We envision that various intramolecular Michael addition reactions can be employed using this method. Similarly, the Mannich reaction involving a three-component acid-catalyzed reaction of aldehydes or ketones with primary or secondary amines to produce β-amino-carbonyl compounds is conveniently performed following Fmoc deprotection.13 We removed the Fmoc group of compound 17 under acid catalyzed hydrogenolysis conditions to produce the amine salt. This amine was neutralized in situ and reacted with formaldehyde and furan to produce product 19 in quantitative yield (Figure 4C). To date, amine protection for this type of reaction has been limited to the Boc group due to the traditional incompatibility of the deprotection conditions for Fmoc which require the use of a secondary amine.
Figure 4.

(A) Example of successful Fmoc removal from a molecule containing a Boc group and an α-chloroketone moiety. (B) Michael addition after Fmoc removal using acidic hydrogenolysis. (C) Mannich reaction after successful Fmoc removal using acidic hydrogenolysis.
3. Conclusions
Fmoc removal using H2/Pd in acidic media directly delivers the ammonium salt, preventing engagement in adverse nucleophilic reactions with electrophiles. In the case of Z-R-K-AOMK (1), we utilized this reaction to achieve a clean removal of the Fmoc group, providing the deactivated HCl salt form of the resulting amine. This approach effectively avoided the undesired nucleophilic side reaction with the AOMK moiety. Furthermore, we expanded the application of these conditions to the deprotection of amines in the presence of α-chloroketone moieties; this reaction yielded the desired deprotected amines in sufficiently high yield. The use of H2/Pd in acidic media for Fmoc-protected amine deprotection, along with the concurrent in situ deactivation of the resulting free amine, represents a promising and versatile method that can find broad applicability in amine deprotection strategies to mitigate undesired side reactions in the presence of highly reactive electrophiles.
4. Experimental Section
4.1. Biology
4.1.1. General
Materials for inhibitors, enzymes, substrates, and reagents. Z-Arg-Lys-AOMK (Z-R-K-AOMK) that was used previously in our studies was obtained from the Wolan lab.5 Recombinant human procathepsin B was purchased from R&D Systems, Minneapolis, MN (#953-CY-010). Z-Phe-Arg–AMC (Z-FR-AMC) was purchased from Anaspec, Fremont, CA (no. AS-24096). Cat.B inhibition assays contained buffer components of sodium phosphate dibasic anhydrous (EMD #SX-072305, Burlington, MA), citric acid monohydrate (Merck #1.00244.0500, Burlington, MA), EDTA (Calbiochem #324503, Burlington, MA), sodium chloride (Fisher Chemical #S271-1, Pittsburgh, PA), sodium acetate (Fisher Scientific #BP-333-500, Fair Lawn, NJ), and dithiothreitol (DTT) (Promega #V351, Madison, WI).
4.1.2. Cat.B Activity and Inhibition
Prior to the start of the assay, the recombinant proCat.B was activated to mature Cat.B by incubation at 37 °C for 30 min in 20 mM Na-acetate pH 5.5, 1 mM EDTA, 5 mM DTT, and 100 mM NaCl and then stored at −80 °C. To directly compare the Z-R-K-AOMK inhibitor synthesized by our new method and the Z-R-K-AOMK previously synthesized by the Wolan lab, assays were simultaneously conducted with both inhibitors at pH 4.6 and pH 7.2 in a 384-well plate with four replicates per condition at room temperature (25 °C) in a total volume of 30 μL. Inhibitor and substrate solutions were combined in each well prior to the addition of Cat.B to determine the activity without preincubation of inhibitor. The final buffer conditions consisted of 40 μM Z-Phe-Arg-AMC, 0.04 ng/μL Cat.B, 40 mM citrate phosphate (pH 4.6 or pH 7.2), 5 mM DTT, 100 mM NaCl, 1 mM EDTA, 1.5% DMSO, and 0.01% Tween-20. The inhibitor concentration ranged from 8304 to 2.5 nM using 1.5-fold serial dilutions. Relative fluorescence readings (RFU) (excitation 360 nm, emission 460 nm) were recorded every 46 s over a period of 30 min in a BioTek HTX microplate reader, and Cat.B activity was determined via relative fluorescent units per second (RFU/s) which was calculated using the highest slope detected for 10 consecutive readings. Graphpad Prism software was used to determine the IC50 and standard deviation from the Cat.B activity from the four replicates for each pH condition in the presence of serially diluted Z-R-K-AOMK concentrations from the two different synthetic methods.
4.2. Chemistry
4.2.1. General
All chemicals and solvents were purchased from commercial suppliers and used without further purification, unless stated otherwise. A Jasco P-2000 polarimeter 314 was used to measure optical rotations. NMR spectra were recorded on a Varian 500 MHz spectrometer (500 and 125 MHz for the 1H and 13C nuclei, respectively) using CDCl3, (CD3)2SO, or CD3OD as solvents from Cambridge Isotope Laboratories, Inc. Spectra were referenced to residual CDCl3 or CD3OD solvent as the internal standard [for CDCl3 δH 7.26, and δC 77.1; and for CD3OD δH 3.31 and δC 49.2; and for (CD3)2SO δH 2.54, and δC 40.45]. LC-high-resolution mass spectrometry (HRMS) data for the analysis of compounds 1, 12, 16, and 19 were obtained on an Agilent 6239 HR-ESI-TOFMS equipped with a Phenomenex Luna 5 μm C18 100 Å column (4.6 × 250 mm). LCMS data for purity analysis of the synthesized compounds 1, 12, 16, and 19 were obtained with a Thermo Finnigan Surveyor Autosampler-Plus/LC-PumpPlus/PDA-Plus system and a Thermo Finnigan LCQ Advantage Max mass spectrometer (monitoring 200–600 nm and m/z 150–2000 in the positive ion mode) using a linear gradient of 20–100% H2O/acetonitrile over 15–20 min; flow rate of 1 mL/min. Semipreparative HPLC purification was carried out using a Waters 515 with a Waters 996 photodiode array detector using Empower Pro software. Structural integrity and purity of the test compounds were determined from the composite of 1H and 13C NMR, HRMS, and HPLC, and all compounds were found to be >90% pure. Chemical shifts (δ) are given in parts per million (ppm) and coupling constants (J) are reported in Hertz (Hz). The compounds are named in accordance with IUPAC rules as applied by ChemBioDraw Ultra (version 16.0).
4.2.2. General Synthetic Procedure for Fmoc Acidic Hydrogenolysis
To a solution of Fmoc protected compound (10 mmol) in MeOH (10 mM) was added 10% Pd/C (20% equiv, 10 wt % on carbon) and HCl/ether (3 M solution, 3 equiv). The reaction was stirred under hydrogen balloon pressure for 5–8 h, until the thin-layer chromatography (TLC) showed complete disappearance of the starting material. The mixture was filtered through a pad of Celite and evaporated to give the crude product, which was used in the next step without further purification.
4.2.2.1. Synthesis of Fmoc-Lys(Boc)-AOMK (4)
Fmoc-Lys(Boc)-COCH2Cl was synthesized according to the published procedure.14 To a solution of Fmoc-Lys(Boc)-COCH2Cl (6) (500 mg, 1 mmol, 1 equiv) and 2,6-dimethylbenzoic acid (225 mg, 1.5 mmol, 1.5 equiv) in anhydrous dimethylformamide (10 mL) at 0 °C was added potassium fluoride (174 mg, 3 mmol, 3 equiv). The reaction mixture was stirred overnight at room temperature. After the reaction showed the disappearance of the starting material by TLC, the mixture was diluted with ethyl acetate and washed with saturated NaHCO3, sat aq. NaCl and dried over anhydrous sodium sulfate. The crude oil was purified by flash chromatography using 20–30% ethyl acetate/hexanes to give product 4 in 96% yield (589 mg). [α]22D +5.6 (c 0.2, CHCl3). 1H NMR (500 MHz, CDCl3): δ 7.75 (d, J = 7.6 Hz, 2H), 7.59 (d, J = 7.5 Hz, 2H), 7.39 (t, J = 7.5 Hz, 2H), 7.31 (t, J = 7.4 Hz, 2H), 7.20 (t, J = 7.6 Hz, 1H), 7.04 (d, J = 7.6 Hz, 2H), 5.58 (d, J = 7.4 Hz, 1H), 5.06–4.91 (m, 2H), 4.64 (t, J = 6.2 Hz, 1H), 4.51–4.36 (m, 2H), 4.20 (t, J = 6.6 Hz, 1H), 3.13–3.10 (m, 2H), 2.39 (s, 6H), 1.98–1.90 (m, 2H), 1.58–1.36 (m, 4H). 1.40 (s, 9H). 13C NMR (126 MHz, CDCl3): δ 202.21, 169.08, 156.29, 156.20, 143.70, 141.34, 135.70, 129.78, 127.78, 127.71, 127.12, 125.09, 125.04, 120.02, 79.27, 66.97, 66.62, 57.37, 47.22, 39.66, 30.76, 29.69, 28.41, 21.94, 19.93. HRMS: (ESI) calcd for C36H42N2O7: [M + H]+, 614.2992; found, 614.2990.
4.2.2.2. Synthesis of Z-R-K-AOMK (1)
To a solution of compound 4 (200 mg, 0.41 mmol) in methanol (10 mM) was added 10% Pd/C (20% equiv, 10% wt on carbon) and HCl/ether (3 M solution, 3 equiv). The reaction was stirred under hydrogen balloon pressure for 5 h, until the TLC showed disappearance of starting material. The mixture was filtered through a pad of Celite and evaporated to give crude product 10 in 88% yield (154 mg), which was used in the next step without further purification. [α]22D +12.1 (c 0.6, CHCl3). 1H NMR (500 MHz, DMSO-d6): δ 8.45 (s, 2H), 7.25 (dd, J = 14.9, 7.2 Hz, 1H), 7.08 (d, J = 7.6 Hz, 2H), 6.76 (t, J = 5.7 Hz, 1H), 5.34 (d, J = 17.6 Hz, 1H), 5.17 (d, J = 17.6 Hz, 1H), 4.30–4.28 (s, 1H), 2.87 (p, J = 6.7 Hz, 2H), 2.28 (s, 6H), 1.40–1.33 (m, 6H), 1.32 (s, 9H). 13C NMR (126 MHz, CD3OD): δ 199.44, 169.09, 157.31, 135.31, 129.63, 127.36, 127.31, 78.67, 65.87, 56.12, 29.41, 29.12, 27.35, 21.41, 18.58. HRMS: (ESI) calcd for C21H32N2O5: [M + H]+, 393.2389; found, 393.2386.
Commercially obtained Z-Arg(Boc)2-OH cyclohexylammonium salt was suspended in ethyl acetate and washed with 1 M HCl three times. The organic layer was washed with brine, dried over anhydrous sodium sulfate, and evaporated to afford the free carboxylic acid Z-Arg(boc)2-OH. A solution of product 10 in ethyl acetate was slowly added dropwise over 30 min to a solution of Z-Arg(Boc)2-OH, DIPEA, HBTU, and HOBt in DCM, and this was stirred overnight at RT at which time the solvents were evaporated under vacuum. The crude oily mixture was redissolved in EtOAc, washed 3× with 1 M HCl, and washed 3× with sat. NaHCO3 and then sat. NaCl. The organic layer was dried over anhydrous sodium sulfate and evaporated under reduced pressure. The crude oil was purified on flash chromatography using 30–100% ethyl acetate/hexanes to give product 11 in 48% yield. [α]22D +9.2 (c 0.3, MeOH). 1H NMR (500 MHz, DMSO-d6): δ 8.37 (d, J = 7.4 Hz, 1H), 8.26 (s, 1H), 7.50 (d, J = 7.8 Hz, 1H), 7.34–7.19 (m, 5H), 7.06 (d, J = 7.6 Hz, 2H), 6.71 (t, J = 5.7 Hz, 1H), 5.05 (s, 2H), 4.99 (s, 2H), 4.33 (dt, J = 10.4, 5.2 Hz, 1H), 4.07–4.01 (m, 1H), 3.27–3.22 (m, 2H), 2.86 (q, J = 6.5 Hz, 2H), 2.26 (s, 6H), 1.57–1.48 (m, 4H), 1.40 (s, 9H), 1.38–1.30 (m, 4H), 1.34 (s, 9H), 1.32 (s, 9H), 1.23–1.20 (m, 2H). 13C NMR (126 MHz, CDCl3): δ 205.79, 177.74, 173.10, 161.06, 161.00, 160.13, 156.60, 140.68, 139.23, 136.69, 133.32, 132.03, 131.57, 131.40, 131.21, 87.03, 83.11, 82.41, 70.66, 70.26, 59.82, 58.65, 43.85, 43.59, 33.18, 32.95, 32.86, 31.35, 31.10, 30.75, 29.21, 26.47, 22.61. HRMS: (ESI) calcd for C45H66N6O12: [M + H]+, 883.4817; found, 883.4810.
The product was dissolved in CH2Cl2, TFA was added, and the mixture was stirred under nitrogen. The reaction was monitored by LCMS, and once the reaction showed completion, the mixture was evaporated to dryness under reduced pressure to afford the pure product 1 in near quantitative yield. [α]22D −3.6 (c 0.1, MeOH). 1H NMR (500 MHz, DMSO-d6): δ 8.43 (d, J = 7.5 Hz, 1H), 7.68–7.61 (m, 4H), 7.56 (d, J = 7.5 Hz, 1H), 7.52 (s, 1H), 7.37–7.28 (m, 5H), 7.25 (t, J = 7.6 Hz, 1H), 7.09 (d, J = 7.6 Hz, 2H), 5.09 (d, J = 1.8 Hz, 2H), 5.00 (d, J = 7.6 Hz, 2H), 4.40 (t, J = 10.9 Hz, 1H), 4.03–3.96 (m, 1H), 3.3.08–3.03 (s, 2H), 2.77–2.70 (m, 2H), 2.28 (s, 6H), 1.85–1.81 (m, 2H), 1.69–1.62 (m, 2H), 1.57–1.46 (m, 4H), 1.36–1.30 (m, 2H). 13C NMR (126 MHz, CD3OD): δ 201.83, 173.75, 169.38, 165.80, 157.19, 136.63, 135.27, 134.14, 132.59, 129.51, 129.18, 128.15, 127.73, 127.41, 127.33, 67.34, 66.41, 55.59, 54.61, 40.57, 38.89, 30.29, 28.72, 24.97, 23.64, 22.61, 18.59. HRMS: (ESI) calcd for C30H42N6O6: [M + H]+, 583.3244; found, 583.3239.
4.2.2.3. Synthesis of tert-Butyl (5-Amino-7-chloro-6-oxoheptyl)carbamate Hydrochloride (12)
Compound 12 was synthesized according to the general procedure for Fmoc acidic hydrogenolysis.
4.2.2.4. Synthesis of 1-(Cyclohexylamino)octan-3-one (16)
Compound 14 was synthesized according to the general procedure for the Fmoc acidic hydrogenolysis. To a stirred solution of compound 14 (0.27 g, 2.0 mmol) in DCM (35 mL) was added vinyl ketone 15 (0.71 g, 2.0 mmol) in one portion. The reaction was stirred for 2 h and then evaporated under reduced pressure. The crude oil was purified by flash chromatography using 0–100% ethyl acetate/hexanes to give product 16 in 65% yield (293 mg).
4.2.2.5. Synthesis of 1-(Furan-2-ylmethyl)piperidine (19)
Compound 18 was synthesized from commercial 17 according to the general procedure for the Fmoc acidic hydrogenolysis. To a 10 mL oven-dried round bottomed flask were added 4 Å molecular sieves. The flask was degassed and refilled with N2 before anhydrous MeCN (6 mL) and amine 18 (157 mg, 1.3 mmol) were added. Formaldehyde solution (35 wt %, 0.1 mL, 1.34 mmol) was added followed by furan (88 mg, 1.3 mmol). The reaction mixture was stirred for 16 h and then evaporated under reduced pressure. The crude oil was purified by flash chromatography using 10–100% ethyl acetate/hexanes to give product 19 in 48% yield (103 mg).
Acknowledgments
J.A. is grateful to the dean of scientific research at the University of Jordan for the research fund and would also like to acknowledge the University of Jordan for the scientific leave. J.A. is also grateful for the Saint Baldrick’s Foundation for the international scholar ward. We acknowledge funding under NIH grant R01NS109075 (awarded to VH).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.3c08629.
1H, 13C NMR spectra for synthetic compounds (PDF)
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare the following competing financial interest(s): Dr. Vivian Hook has an equity position at American Life Science Pharmaceuticals (ALSP) and is a founder and advisor to ALSP. V. Hook’s spouse, G. Hook, is co-founder with an equity position at ALSP. VHs conflict has been disclosed and is managed by her employer, the University of California, San Diego.
Supplementary Material
References
- Li W.; O’Brien-Simpson N. M.; Hossain M. A.; Wade J. D.; Li W.; O’Brien-Simpson N. M.; Hossain M. A.; Wade J. D. The 9-Fluorenylmethoxycarbonyl (Fmoc) Group in Chemical Peptide Synthesis - Its Past, Present, and Future. Aust. J. Chem. 2020, 73 (4), 271–276. 10.1071/CH19427. [DOI] [Google Scholar]
- Behrendt R.; White P.; Offer J. Advances in Fmoc Solid-Phase Peptide Synthesis. J. Pept. Sci. 2016, 22 (1), 4–27. 10.1002/psc.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralhan K.; Krishnakumar V. G.; Gupta S. Piperazine and DBU: A Safer Alternative for Rapid and Efficient Fmoc Deprotection in Solid Phase Peptide Synthesis. RSC Adv. 2015, 5, 104417. 10.1039/C5RA23441G. [DOI] [Google Scholar]
- Harris P. W. R.; Brimble M. A. A Comparison of Boc and Fmoc SPPS Strategies for the Preparation of C-Terminal Peptide α-Thiolesters: NY-ESO-1 39Cys-68Ala-COSR. Pept. Sci. 2013, 100 (4), 356–365. 10.1002/bip.22223. [DOI] [PubMed] [Google Scholar]
- Yoon M. C.; Solania A.; Jiang Z.; Christy M. P.; Podvin S.; Mosier C.; Lietz C. B.; Ito G.; Gerwick W. H.; Wolan D. W.; Hook G.; O’Donoghue A. J.; Hook V. Selective Neutral PH Inhibitor of Cathepsin B Designed Based on Cleavage Preferences at Cytosolic and Lysosomal PH Conditions. ACS Chem. Biol. 2021, 16 (9), 1628–1643. 10.1021/acschembio.1c00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leggio A.; Liguori A.; Napoli A.; Siciliano C.; Sindona G. New Strategies for an Efficient Removal of the 9-Fluorenylmethoxycarbonyl (Fmoc) Protecting Group in the Peptide Synthesis. Eur. J. Org Chem. 2000, 2000 (4), 573–575. . [DOI] [Google Scholar]
- Maegawa T.; Fujiwara Y.; Ikawa T.; Hisashi H.; Monguchi Y.; Sajiki H. Novel Deprotection Method of Fmoc Group under Neutral Hydrogenation Conditions. Amino Acids 2009, 36 (3), 493–499. 10.1007/s00726-008-0109-7. [DOI] [PubMed] [Google Scholar]
- Vázquez J.; Albericio F. A Convenient Semicarbazide Resin for the Solid-Phase Synthesis of Peptide Ketones and Aldehydes. Tetrahedron Lett. 2006, 47 (10), 1657–1661. 10.1016/j.tetlet.2005.12.101. [DOI] [Google Scholar]
- NMR data were not reported for the final products in the previous report (ref (5)).
- Zhang Y.; Wang W. Recent Advances in Organocatalytic Asymmetric Michael Reactions. Catal. Sci. Technol. 2012, 2 (1), 42–53. 10.1039/C1CY00334H. [DOI] [Google Scholar]
- Malkar R. S.; Jadhav A. L.; Yadav G. D. Innovative Catalysis in Michael Addition Reactions for C-X Bond Formation. Mol. Catal. 2020, 485, 110814. 10.1016/j.mcat.2020.110814. [DOI] [Google Scholar]
- Poon T.; Mundy B. P.; Shattuck T. W. The Michael Reaction. J. Chem. Educ. 2002, 79 (2), 264. 10.1021/ed079p264. [DOI] [Google Scholar]
- Shi Y.; Wang Q.; Gao S. Recent Advances in the Intramolecular Mannich Reaction in Natural Products Total Synthesis. Org. Chem. Front. 2018, 5 (6), 1049–1066. 10.1039/C7QO01079F. [DOI] [Google Scholar]
- Wood W. J. L.; Huang L.; Ellman J. A. Synthesis of a Diverse Library of Mechanism-Based Cysteine Protease Inhibitors. J. Comb. Chem. 2003, 5 (6), 869–880. 10.1021/cc034008r. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.